Acid-Base Disorders
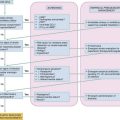
Diagnostic Strategies
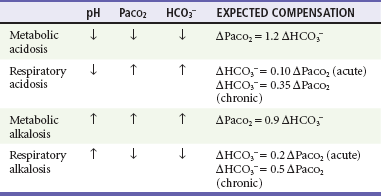
Simple acid-base disorders are categorized by the serum pH, PaCO2, and HCO3− concentrations (Table 124-1). When the primary disturbance is identified, the next step is to determine its etiology and whether an appropriate compensation has occurred. An inappropriate compensation suggests that the process underlying the primary disturbance has hindered an appropriate response or that more than one primary disturbance is present. Simple formulas allow the calculation of an appropriate response once the primary disturbance has been categorized. Compensatory processes generally return pH toward normal but not to normal and never beyond normal; such an exaggerated “compensation” actually represents a second primary disorder.
Blood gas and serum chemistry analysis allows calculation and interpretation of the serum anion gap.
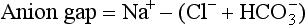
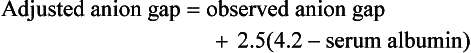
Independent of bicarbonate levels, sodium and chloride play an important role in acid-base status. When [Na+] − [Cl−], the so-called strong ion difference, is significantly less than 38, an acidosis is present. An alternative to using the anion gap to identify both the presence and cause of acid-base disorders has the clinician start with the base excess (as calculated by a blood gas analyzer), subtract the sodium chloride effect,* and subtract the albumin effect.† What remains is, in the case of metabolic acidosis, the unmeasured anions (e.g., lactate, ketones, uremic acids, toxic alcohols, or other toxins; see later).1
Painful arterial blood sampling is unnecessary for the evaluation of acid-base disturbances. PaCO2, HCO3−, and pH values taken from peripheral venous, central venous, intraosseous, and capillary blood are all suitable for acid-base assessment.2–4
Respiratory Acidosis
Respiratory acidosis occurs when hypoventilation leads to an inappropriately elevated PaCO2 and resulting acidemia. Any condition that reduces minute ventilation may cause respiratory acidosis (Box 124-1). This commonly occurs acutely in the setting of airway embarrassment, pulmonary insults, major trauma, intracranial catastrophe, or central nervous system (CNS) depressants and chronically in progressive lung disease, neurologic muscle weakness conditions, or obesity hypoventilation syndrome. Respiratory acidosis may be accompanied by hypoxemia, which, depending on its pace and severity, will cause end-organ effects such as headache, ischemic chest pain, altered mental status (usually agitation), bradycardia, and circulatory collapse. When tissue oxygenation is adequate, hypercapnia—which is better tolerated than hypoxemia—tends to produce somnolence and obtundation with cerebral vasodilation and resulting elevation of intracranial pressure.
Patients with chronic respiratory acidosis are often hypoxemic at baseline and may be susceptible to further hypoventilation if normal oxygen saturation levels are promptly restored.5 Typically, a patient with chronic obstructive pulmonary disease being treated for an acute exacerbation receives high-flow oxygen and is later found to be obtunded with profound hypercapnia. It is therefore prudent to target a lower than normal oxygen saturation in patients thought to be so habituated; SpO2 fractions in the low 90s and high 80s are usually well tolerated in this population.6 However, supplemental oxygen must not be withheld from patients with dangerously low oxygen saturation or patients with end-organ hypoxic insults, such as myocardial or cerebral ischemia. In these cases, aggressive oxygenation therapies must be instituted, with preparations made to initiate mechanical ventilation if it is needed.
Respiratory Alkalosis
Respiratory alkalosis occurs when increased minute ventilation causes a decreased PaCO2 and subsequent increase in serum pH. It is often associated with hysterical hyperventilation; however, as always, the emergency physician must consider dangerous conditions before arriving at a psychiatric diagnosis (Box 124-2).
Pregnant women hyperventilate throughout gestation and normally have a PaCO2 between 31 and 35 mm Hg, serum pH between 7.46 and 7.50, and serum bicarbonate concentration between 18 and 22 mEq/L. Clinicians should be mindful that eucapnia (PaCO2 ~40 mm Hg) in the pregnant patient may represent hypoventilation.7
As with other acid-base disorders, management of respiratory alkalosis is directed at the underlying cause. When organic causes of respiratory alkalosis have been excluded, hypocapnia symptoms can be relieved in patients with psychological hyperventilation by reassurance. The technique of using a paper bag to cause rebreathing probably works through the placebo effect rather than by changes in PaCO2 and carries the potential danger of inducing hypoxemia.8
Metabolic Acidosis
Etiology
Elevated Anion Gap Metabolic Acidosis
The causes of elevated anion gap metabolic acidosis are classically remembered by the mnemonic MUDPILES; however, it is more useful to focus on the identity of the unmeasured anion (Box 124-3). If the cause of an elevated anion gap metabolic acidosis is found to be lactate or ketones, the next step is to determine the cause of the high serum lactate or ketone concentration.
Increased production of lactate is a common cause of elevated anion gap metabolic acidosis, and measurement of serum lactate is indicated when another cause is not apparent. When the elevation of the anion gap is accounted for by serum lactate concentration, the clinician next considers the causes of lactic acidosis; these may be divided into hypoxic or hypoperfusion states (most commonly shock) and exogenous cellular toxins, such as cyanide and carbon monoxide. Idiosyncratic causes of lactic acidosis include short gut syndrome (usually after bariatric surgery),9 reverse transcriptase inhibitor therapy for human immunodeficiency virus (HIV) infection,10 and metformin therapy. The likelihood of metformin causing lactic acidosis is thought to be enhanced by renal failure (serum creatinine > 1.5 mg/dL), congestive heart failure, coexisting metabolic acidosis, and exposure to intravenous radiologic contrast media.
Carbon Monoxide and Cyanide.: Exposure to carbon monoxide and cyanide causes lactic acidosis through tissue hypoxia; both are important elements of the differential diagnosis of an elevated anion gap of unknown cause. Although patients extricated from fires have a variety of reasons to have a high lactate concentration, most of these patients, especially if they are obtunded or in cardiac arrest, should be empirically treated for both carbon monoxide and cyanide toxicity. In treatment of cyanide toxicity with suspected coincident carbon monoxide exposure, the nitrite portions of the antidote kit are withheld for concern that the resultant methemoglobinemia will be poorly tolerated given the carboxyhemoglobin burden. Severity of carbon monoxide exposure and treatment requirements may be better predicted by the degree of initial acidemia than by carboxyhemoglobin level.11
Diabetic Ketoacidosis.: Diabetic ketoacidosis is a common acid-base disorder defined by hyperglycemia, ketonemia, and acidemia. It may occur in type 1 insulin-dependent diabetics or type 2 non–insulin-dependent diabetics. Patients with diabetic ketoacidosis classically complain of progressive polyuria, polydipsia, and malaise, but diabetic ketoacidosis may be manifested atypically with chief complaints of vomiting, abdominal pain, or altered mental status. Sodium concentration is not corrected for hyperglycemia when the anion gap is calculated because this dilutional hyponatremia affects other ions in the serum. Diabetic ketoacidosis is often caused by medication noncompliance but may complicate any physiologic stress; when diabetic ketoacidosis is diagnosed, identification of the precipitant is a management priority. Treatment focuses on volume expansion with intravenous fluids, insulin therapy, and careful attention to and replacement of electrolytes, particularly potassium.
Alcoholic Ketoacidosis.: Alcoholic ketoacidosis causes an elevated anion gap metabolic acidosis that may be mistaken for the more common diabetic ketoacidosis. Alcoholic ketoacidosis is usually seen when a long-standing ethanol user abruptly stops drinking; ketones are generated by a combination of malnutrition and dehydration. Patients with alcoholic ketoacidosis may demonstrate a high anion gap, but a mixed acid-base disorder can be due to concomitant ethanol withdrawal, which may cause a respiratory alkalosis, and vomiting, which produces a metabolic alkalosis. The resultant serum pH can therefore be acidemic, normal, or alkalemic. Like diabetic ketoacidosis, alcoholic ketoacidosis includes the symptoms of vomiting, abdominal pain, dehydration, altered mental status, prostration, and lethargy; however, alcoholic ketoacidosis patients generally do not demonstrate hyperglycemia or glycosuria. In alcoholic ketoacidosis, the ratio of the keto acid β-hydroxybutyrate to its metabolites acetoacetate and acetone is double the ratio seen in diabetic ketoacidosis. Urine dipsticks detect acetoacetate but not β-hydroxybutyrate; this causes an apparent worsening of ketonemia as the patient metabolizes β-hydroxybutyrate to acetoacetate, whereas the patient is actually recovering.12 The treatment of alcoholic ketoacidosis is dextrose-containing fluids; insulin is contraindicated.
Methanol and Ethylene Glycol.: Methanol and ethylene glycol, the toxic alcohols, represent an important element of the emergency differential for elevated anion gap metabolic acidosis because their considerable morbidity and mortality can be prevented by timely therapy, and the presence of a wide anion gap may be the most conspicuous sign that this dangerous ingestion has occurred. Toxic alcohols are typically consumed by alcoholics seeking a less expensive drink, by children as an accidental ingestion (usually ethylene glycol, which is sweet), or in a suicide attempt. The first measurable sign of toxic alcohol poisoning is an osmol gap, which precedes the elevated anion gap; as the parent compounds are converted to their toxic metabolites, the osmol gap closes and the anion gap widens. Treatment centers on prevention of the enzymatic conversion of methanol and ethylene glycol into their toxic metabolites by use of either ethanol or fomepizole. Many laboratories will report a spuriously elevated lactate concentration in the presence of glycolate, a byproduct of ethylene glycol poisoning. Isopropyl (rubbing) alcohol ingestion causes intoxication, ketosis, and an osmol gap but does not cause acidosis.
Renal Failure.: Under normal circumstances, the body generates a net acid load that the kidneys must continuously excrete. Any condition that reduces glomerular filtration rate will undermine this function, and unmeasured anions such as sulfate, phosphate, urate, and hippurate will generate an elevated anion gap metabolic acidosis. Administration of sodium bicarbonate is used as a bridge to renal replacement therapy when uremia-associated metabolic acidosis is severe.
Isoniazid.: Grand mal seizures are associated with high lactate levels that typically resolve within 1 hour. Isoniazid (INH) toxicity is usually listed in the differential for metabolic acidosis, but consideration of INH toxicity should primarily be prompted by intractable seizures because it is the seizures that cause the lactic acidosis, not INH. INH toxicity is an important consideration in these cases because it causes seizures that do not respond to usual treatments of status epilepticus. Continuous seizure activity, in addition to cardiorespiratory embarrassment, causes a profound, life-threatening acidemia from lactate generation. The initial treatment of status epilepticus related to INH toxicity is the administration of vitamin B6 (pyridoxine). Specific therapies targeting nonepileptic causes of seizures, such as hypoglycemia, eclampsia, hyponatremia, and cyanide toxicity, should also be considered in the patient with seizures unresponsive to standard therapy.
Salicylate Toxicity.: Salicylate toxicity first produces a respiratory alkalosis, as described before. Untreated, the syndrome evolves into a complex acid-base disorder with paradoxical aciduria during the initial respiratory alkalosis, followed by an elevated anion gap metabolic acidosis and hyperthermia due to its interference with cellular metabolism. Serum pH can be normal during the initial phase of the disease—a false reassurance. Dehydration, electrolyte disturbances, fatigue, and lung injury ultimately lead to decompensation with respiratory acidosis—completing the so-called triple acid-base disturbance—followed by cardiovascular collapse. Management of aspirin toxicity includes gastrointestinal decontamination, urine alkalinization, meticulous supportive care, and hemodialysis.
Iron.: Iron ingestion is a dangerous cause of metabolic acidosis that classically is manifested with prominent vomiting and other gastrointestinal symptoms from gastric irritation, followed by systemic toxicity that includes myocardial depression, liver injury, and lactic acidosis from cellular mitochondrial poisoning. A plain radiograph of the abdomen may provide evidence of an iron ingestion while serum iron concentration is determined. Treatment measures include fluid resuscitation, gastrointestinal decontamination, and chelation with deferoxamine.
Normal Anion Gap Metabolic Acidosis
Normal anion gap metabolic acidosis is less likely to be immediately dangerous or caused by an imminently dangerous condition and is therefore of less interest to the emergency physician. The majority of cases encountered in the emergency department are caused by gastric loss of bicarbonate in the setting of diarrhea. A variety of other conditions and medications may cause a normal anion gap metabolic acidosis (Box 124-4), including renal tubular acidosis and bladder-diverting urologic procedures. An important iatrogenic cause of normal anion gap metabolic acidosis is the rapid infusion of large volumes of normal saline; 0.9% normal saline contains 154 mEq/L of sodium and chloride without bicarbonate or any other buffer and is therefore acidic compared with serum. The hyperchloremic acidosis associated with normal saline therapy is generally well tolerated and causes less physiologic derangement than in acidoses of equal magnitude caused by other mechanisms; however, in patients with existing severe acidemia who require aggressive volume resuscitation, use of a more balanced fluid (lactated Ringer’s solution, Normosol, Plasma-Lyte), especially after the first several liters of normal saline, may be prudent.
Management
• Paradoxical CNS acidosis. Because sodium bicarbonate does not readily cross the blood-brain barrier, serum alkalinizes much faster than the cerebrospinal fluid. As serum pH rises, minute ventilation slows and carbon dioxide, which readily crosses the blood-brain barrier, delivers an additional acid load to the CNS.
• Hypokalemia (which can cause respiratory muscle weakness, impairing respiratory compensation), hypocalcemia, hypernatremia
Many authors question the utility of bicarbonate therapy even in cases of severe acidemia, given these potential harms and the absence of demonstrated benefit.13 However, although evidence is lacking, it is commonly recommended to treat serum pH less than 7.1 empirically with sodium bicarbonate, 1 mEq/kg, unless the underlying acidosis is thought to be quickly responsive to therapy.14 Many experts will not treat pH above 6.7, however, especially in diabetic ketoacidosis, in which profound acidemia is usually well tolerated while the underlying lesion is corrected. Endpoints of therapy include pH above 7.1 and serum bicarbonate concentration above 10 mEq/L. A bicarbonate drip is prepared by adding 150 mEq sodium bicarbonate (three “crash cart ampules,” which are usually 50 mL of 8.4% solution) to 1 liter of 5% dextrose in water and infusing as slowly as the clinical situation permits. Sodium bicarbonate should generally not be added to normal saline for concern of hypertonicity.
Bicarbonate has been variously recommended and used as an empirical therapy in cardiac arrest. However, most studies show either no benefit or poorer outcomes in this context, and the 2010 American Heart Association guidelines state that routine use of sodium bicarbonate is not recommended for patients in cardiac arrest.15
Metabolic Alkalosis
Metabolic alkalosis occurs from the loss of H+ or retention of HCO3−. Emergency physicians encounter metabolic alkalosis usually as a consequence of prolonged vomiting or nasogastric suction or as a compensation for chronic respiratory acidosis. The differential diagnosis includes a variety of predominantly endocrine and electrolyte disorders (Box 124-5). Patients taking diuretic medications may reduce their plasma volume around a steady concentration of HCO3−, causing a contraction alkalosis. Contraction alkalosis is often coincident with hypokalemia, which itself causes a metabolic alkalosis as intracellular K+ is exchanged for H+.
Mixed Disorders






References
1. Story, DA, Morimatsu, H, Bellomo, R. Strong ions, weak acids and base excess: A simplifed Fencl-Stewart approach to clinical acid-base disorders. Brit J Anaesth. 2004;92:54.
2. Kissoon, N, et al. Comparison of the acid-base status of blood obtained from intraosseous and central venous sites during steady and low-flow states. Crit Care Med. 1993;21:1765.
3. Brandenburg, MA. Comparison of arterial and venous blood gas values in the initial emergency department evaluation of patients with diabetic ketoacidosis. Ann Emerg Med. 1998;31:459.
4. Harrison, AM, et al. Comparison of simultaneously obtained arterial and capillary blood gases in pediatric intensive care unit patients. Crit Care Med. 1997;25:1904.
5. Aubier, M, et al. Effects of administration of O2 on ventilation and blood gases in patients with chronic obstructive pulmonary disease during acute respiratory failure. Am Rev Respir Dis. 1980;122:747–754.
6. Stoller, JK. Acute exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002;346:988–994.
7. Landon, M. Acid-base disorders during pregnancy. Clin Obstet Gynecol. 1994;37:16.
8. van der Hout, MA, Boek, C, van der Molen, GM, Jansen, A, Griez, E. Rebreathing to cope with hyperventilation: Experimental tests of the paper bag method. J Behav Med. 1989;11:303.
9. Uribarri, J, Oh, MS, Carroll, HJ. D-Lactic acidosis: A review of clinical presentation, biochemical features and pathophysiologic mechanisms. Medicine (Baltimore). 1998;77:73.
10. Gérard, Y, et al. Symptomatic hyperlactataemia: An emerging complication of antiretroviral therapy. AIDS. 2000;14:2723.
11. Turner, M, Esaw, M, Clark, RJ. Carbon monoxide poisoning treated with hyperbaric oxygen: Metabolic acidosis as a predictor of treatment requirements. J Acad Emerg Med. 1999;16:96.
12. McGarry, JD, Foster, DW, Diabetic ketoacidosis, Diabetes Mellitus. Rifkin, H, Raskin, P, eds., Diabetes Mellitus. Bowie, Md:Robert J. Brady Co; 1981;vol 5:185.
13. Forsythe, S, Schmidt, G. Sodium bicarbonate for treatment of lactic acidsosis. Chest. 2000;117:260.
14. Kraut, JA, Madias, NE. Metabolic acidosis: Pathophysiology, diagnosis and management. Nat Rev Nephrol. 2010;6:274.
15. Neumar, RW, Otto, CW, Link, MS, et al. Part 8: Adult advanced cardiovascular life support: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122(Suppl 3):S729–S767.