160 Acid-Base Disorders
 Key Points
Key Points• Normal pH or serum bicarbonate values can mask an important, underlying acidosis in the setting of a mixed disorder.
• An elevated anion gap is a sign of metabolic acidosis and should be calculated on each chemistry sample.
• Arterial and venous blood gas sampling is a useful emergency department test because of the strong association between arterial and venous HCO3− and pH.
• Correlation between venous PCO2 and arterial PCO2 is lacking, although venous PCO2 levels may be used as a screening tool for hypercapnia.
• Admission lactate level and standard base excess are markers of illness severity that correlate with patient morbidity and mortality in the hospital.
• The urine ketone dipstick test is highly sensitive for serum ketosis.
• Venous and arterial lactate samples are equivalent.
• Indiscriminate use of sodium bicarbonate for the treatment of undifferentiated metabolic acidosis should be avoided.
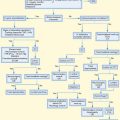
Pathophysiology
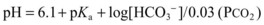
Diagnostic Interpretation
 Facts and Formulas
Facts and Formulas
pH = 6.1 + Log[HCO3−]/0.03 × PCO2
Anion gap = Unmeasured anions − Unmeasured cations = Na+ − [Cl− + HCO3−]
Delta gap = Δ Anion gap − Δ HCO3 = [Calculated anion gap − 10] − [24 − Measured serum HCO3]
Calculated Sosm (mOsm/kg) = 2 (Na+) + BUN/2.8 + Glucose/18 + Ethanol/4.8
Arterial and Venous Blood Gases
The ability to substitute venous blood gas samples for arterial samples is appealing because of the pain, difficulty, and complications associated with arterial sampling. Arterial pH and venous pH vary by less 0.04 in most situations.1–3 Patients in clinical shock are an important exception, however, because arteriovenous PCO2 (and therefore pH) can vary significantly.
Despite incomplete correlation between venous and arterial PCO2, venous PCO2 may be used to screen for arterial hypercapnia. In hemodynamically normal patients, PCO2 higher than 45 mm Hg is sensitive (but less than 50% specific) for the detection of arterial hypercapnia, which is defined as PCO2 higher than 50 mm Hg. Venous blood gas screening led to a 29% reduction in arterial sampling in one study.4 Finally, arterial blood gas analysis enables precise interpretation of respiratory compensation when needed.
Standard Base Excess
SBE has been studied extensively as a resuscitation end point in trauma and as a marker of tissue acidosis. Preresuscitation base excess values are reliably linked to the degree of tissue acidosis and serve as independent predictors of mortality in critically ill patients. Base excess has been shown to correlate with hypovolemia, length of hospital stay, and transfusion requirements, whereas the rate of normalization correlates with patient survival.5,6
Lactate
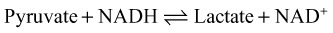
Arterial lactate sampling is considered the most reliable measure for detecting hyperlactatemia; however, venous and capillary sampling is also used. Central venous sampling is highly correlated with arterial lactate measurements. Peripheral venous samples are sufficient to screen for hyperlactatemia but retain poor specificity (57%) when compared with arterial samples.7 Elevations in venous lactate should be confirmed with arterial sampling.
Anion Gap
Within serum, the requirement for electroneutrality dictates that the net serum cation charge equal the net total anion charge. The calculated difference in commonly measured serum ions is termed the anion gap (AG). It is important to note that the AG represents anions that are present but unmeasured (at least historically) and that an AG is present during health. Fortunately, the difference between unmeasured anions and unmeasured cations may change (increased or decreased AG) and therefore provide a clue to disease states (Box 160.1).
When acids are added to the system, bicarbonate is replaced by the acid anion (X) as follows:
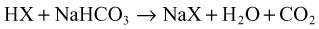
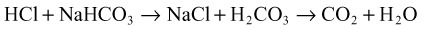
Hyperchloremia maintains electroneutrality without altering the AG. Gastrointestinal and renal losses are the most common causes of non-AG metabolic acidosis (Box 160.2).
The historical range for the AG was 12 ± 4 (8 to 16 mEq/L). With the adoption of ion-specific electrodes, chloride is measured at a higher concentration such that the currently accepted range of AG is 7 ± 3 (4 to 10 mEq/L). More importantly, the AG must be corrected for individual patients. Albumin accounts for 80% of the AG in health. Consequently, large and important deviations may occur if serum albumin is assumed to be normal. AG is thus commonly corrected for serum albumin to improve sensitivity of the AG as a screening tool.8 The correction factor is calculated as follows:
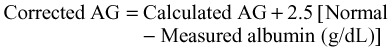
Delta-Delta Calculation
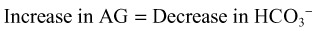
The difference between the change in the AG and the change in serum bicarbonate is called the delta gap, or delta-delta calculation. Deviations from this stoichiometric relationship indicate a mixed metabolic disturbance. When the change (delta) in the AG is greater than the change in bicarbonate, a preexisting or concomitant metabolic alkalosis is present. Alternatively, a change in AG less than the change in bicarbonate identifies a coexisting non-AG acidosis. A delta gap higher than 6 is generally considered significant.9
Serum Osmolar Gap
Sosm is elevated in the presence of osmotically active particles such as alcohols, glycols, and sugars. The difference in measured and calculated Sosm is termed the osmolar gap. An elevated osmolar gap confirms the presence of unmeasured osmotically active particles, which may be helpful when investigating the cause of unexplained AG acidosis. Osmolar gaps greater than 10 mOsm/kg are considered abnormal and may reflect the presence of a toxic alcohol as the source of the acidosis. However, delayed evaluation after the ingestion of toxic alcohol will show little to no elevation in the osmolar gap as a result of metabolism of the offending alcohol. Likewise, normal osmolar gap values are imprecisely defined, with ranges of −13 to +14.0 mOsm/kg noted in healthy patients.10,11 This wide variation in the normal range may lead to a normal osmolar gap in the presence of a significant ingestion.
Evaluation of Mixed Acid-Base Disorders
By applying the formulas for AG, delta gap, and expected physiologic compensation, a stepwise approach to the evaluation of simple and mixed acid-base problems can be developed.12 This process is summarized in Box 160.3.
Box 160.3
Five-Step Approach to Acid-Base Disorders
Rule 1: Determine the pH status (alkalemia or acidemia: >7.44 or <7.40)
Rule 2: Determine whether the primary process is respiratory, metabolic, or both
Rule 3: Calculate the anion gap*
Rule 4: Check the degree of compensation
Rule 5: Determine whether there is a 1 : 1 relationship between the change in the anion gap and the change in serum bicarbonate
Modified from Whittier WL, Rutecki GW. Primer on clinical acid-base problem solving. Dis Mon 2004;50:122–62.
Additionally the clinical history is centrally important for proper interpretation of acid-base disorders (Box 160.4).
Box 160.4 Acid-Base Interpretation Based on Clinical History
Case 1
Specific Acid-Base Disorders
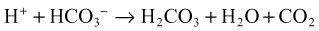
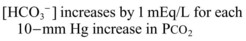

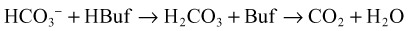
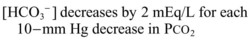
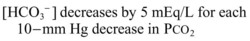
Metabolic Acidosis
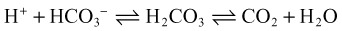
It is important to remember that compensatory responses do not fully normalize pH. If a normal pH is seen in a patient with metabolic acidosis, a second acid-base disorder must be present. The typical laboratory pattern of metabolic acidosis is decreased pH and bicarbonate with a compensatory decrease in PCO2 (Box 160.5).
Box 160.5 Diabetic Patient with Mixed Acid-Base Disorder
Laboratory evaluation shows the following:
Step 1: Arterial blood gas analysis shows that the patient is minimally acidemic
Step 2: Serum bicarbonate is less than 25 mEq/L, indicative of metabolic acidosis
Step 3: The anion gap is elevated: 134 − 94 − 20 = 20
Step 4: Respiratory compensation is appropriate: 1.5 [HCO3−] + 8 = [1.5 × 20] + 8 = 38 mm Hg
Step 5: Calculation of the delta gap reveals that the change in HCO3− (24 − 20 = 4 units) is significantly less than the change in anion gap (20 − 10 = 10 units). This indicates the presence of a concomitant metabolic alkalosis that is masking the significant anion gap acidosis. These findings are explained by vomiting-induced alkalosis in a patient with diabetic ketoacidosis

This equation assesses the adequacy of respiratory compensation. A PCO2 that is significantly higher or lower than this calculated value signals the presence of a secondary respiratory acidosis or alkalosis, which may have a profound impact on treatment decisions (Box 160.6).
Box 160.6 Metabolic Acidosis with Inadequate Respiratory Compensation
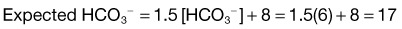
Non-AG acidoses are those that add HCl to the system. The acid anion in these cases is chloride; because of its inclusion in the AG equation, no change in the gap is noted. The most common causes of non-AG acidosis include renal and gastrointestinal bicarbonate wasting (Box 160.7).
Box 160.7 Four-Year-Old Boy with Diarrhea for 5 Days
Physical examination reveals dry mucous membranes.
Laboratory Data:
Na+ = 134; K+ = 4.8; Cl− = 114; HCO3− = 3; pH = 6.98; PCO2 = 13 mm Hg; PO2 = 110 mm Hg
Step 1: The patient is found to be acidemic on examination of arterial blood gases
Step 2: Serum bicarbonate is less than 25 mEq, indicative of the presence of metabolic acidosis
Step 3: The anion gap is elevated: 134 − 116 − 3 = 15
Step 4: Respiratory compensation is appropriate: 1.5 × [HCO3−] + 8 = [1.5 × 3] + 8 = 12.5
Step 5: Calculation of the delta gap reveals that the change in HCO3− (24 − 3 = 21 units) is significantly greater than that the change in anion gap (15 − 10 = 5 units). This indicates the presence of a concomitant non–anion gap acidosis that is overshadowing the anion gap acidosis. The mixed acidosis can be clinically explained by the presence of diarrhea (non–anion gap acidosis) with dehydration (anion gap acidosis)
Metabolic Alkalosis
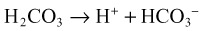
Metabolic alkalosis is the second most common acid-base disorder and is found in approximately one third of hospitalized patients. It can be caused by several processes: increased H+ loss, typically through renal or gastrointestinal wasting; increased bicarbonate resorption; infusion or ingestion of bicarbonate; intracellular shifts in H+; or contraction of extracellular fluid around a stable HCO3− pool (Box 160.8).

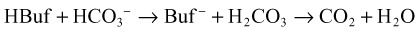
1 Kelly AM, McAlpine R, Kyle E. Venous pH can safely replace arterial pH in the initial evaluation of patients in the emergency department. Emerg Med J. 2001;18:340–342.
2 Eizadi-Mood N, Moein N, Saghaei M. Evaluation of relationship between arterial and venous blood gas values in the patients with tricyclic antidepressant poisoning. Clin Toxicol. 2005;43:357–360.
3 Gokel Y, Paydas S, Koseoglu Z, et al. Comparison of blood gas and acid-base measurements in arterial and venous blood samples in patients with uremic acidosis and diabetic ketoacidosis in the emergency room. Am J Nephrol. 2000;20:319–323.
4 Kelly AM, Kerr D, Middleton P. Validation of venous PCO2 to screen for arterial hypercarbia in patients with chronic obstructive pulmonary disease. J Emerg Med. 2005;28:377–379.
5 Rutherford E, Morris JA, Jr., Reed GW, et al. Base deficit stratifies mortality and determines therapy. J Trauma. 1992;33:417–423.
6 Davis JW, Kaups KL, Parks SN. Base deficit is superior to pH in evaluating clearance of acidosis after traumatic shock. J Trauma. 1998;44:114–118.
7 Middleton P, Kelly AM, Brown J, et al. Agreement between arterial and venous values for pH, bicarbonate, base excess and lactate. Acad Emerg Med. 2005;12(5 Suppl 1):174.
8 Hatherill M, Waggie Z, Purves L, et al. Correction of the anion gap for albumin in order to detect occult tissue anions in shock. Arch Dis Child. 2002;87:526–529.
9 Wrenn K. The delta (delta) gap: an approach to mixed acid-base disorders. Ann Emerg Med. 1990;19:1310–1313.
10 Hoffman RS, Smilkstein MJ, Howland MA, et al. Osmol gaps revisited: normal values and limitations. Clin Toxicol. 1993;31:81–93.
11 McQuillen KK, Anderson AC. Osmol gaps in the pediatric population. Acad Emerg Med. 1999;6:27–30.
12 Whittier WL, Rutecki GW. Primer on clinical acid-base problem solving. Dis Mon. 2004;50:122–162.