Acid-Base Balance
After reading this chapter you will be able to:
 Describe how the lungs and kidneys regulate volatile and fixed acids.
Describe how the lungs and kidneys regulate volatile and fixed acids.
 Describe how the equilibrium constant of an acid is related to its ionization and strength.
Describe how the equilibrium constant of an acid is related to its ionization and strength.
 State what constitutes open and closed buffer systems.
State what constitutes open and closed buffer systems.

 Explain how to use the Henderson-Hasselbalch equation in hypothetical clinical situations.
Explain how to use the Henderson-Hasselbalch equation in hypothetical clinical situations.
 Describe how the kidneys and lungs compensate for each other when the function of one is abnormal.
Describe how the kidneys and lungs compensate for each other when the function of one is abnormal.
 Explain how renal absorption and excretion of electrolytes affect acid-base balance.
Explain how renal absorption and excretion of electrolytes affect acid-base balance.
 Classify and interpret arterial blood acid-base results.
Classify and interpret arterial blood acid-base results.
 Explain how to use arterial acid-base information to decide on a clinical course of action.
Explain how to use arterial acid-base information to decide on a clinical course of action.

 Calculate the anion gap and use it to determine the cause of metabolic acidosis.
Calculate the anion gap and use it to determine the cause of metabolic acidosis.


Hydrogen Ion Regulation in Body Fluids
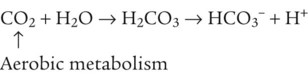
As CO2 diffuses into the blood at the tissue level, this reaction occurs primarily in the erythrocyte where it is catalyzed by carbonic anhydrase, an intracellular enzyme. In a process called isohydric buffering,1 most H+ produced in this fashion causes no change in pH because hemoglobin (Hb) in the erythrocyte immediately buffers the H+. When blood reaches the lungs, Hb releases H+ to form CO2 as shown:
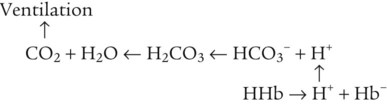
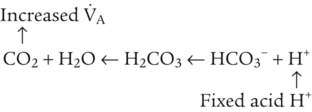
Catabolism of proteins continually produces fixed (nonvolatile) acids such as sulfuric and phosphoric acids. In addition, anaerobic metabolism produces lactic acid. In contrast to carbonic acid, these nonvolatile acids are not in equilibrium with a gaseous component. However, H+ of fixed acids can be buffered by bicarbonate ions (HCO3−) and converted to CO2 and water (H2O) (see the previous reaction); the CO2 formed is eliminated in exhaled gas. Compared with daily CO2 production, fixed acid production is small, averaging only about 50 to 70 mEq/day.2 Certain diseases, such as untreated diabetes, increase fixed acid production. Hydrogen ions produced in this way stimulate respiratory centers in the brain. The resulting increase in ventilation eliminates more CO2, pulling the hydration reaction to the left:
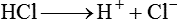
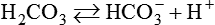
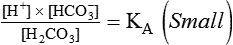
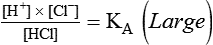
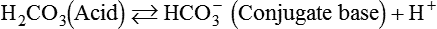
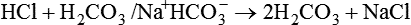
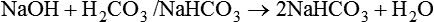
Bicarbonate and Nonbicarbonate Buffer Systems
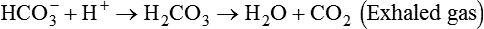
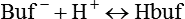
Box 13-1 summarizes the characteristics and components of bicarbonate and nonbicarbonate buffer systems.
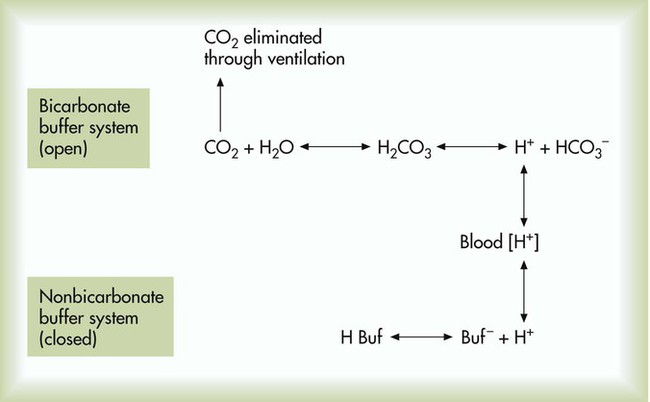
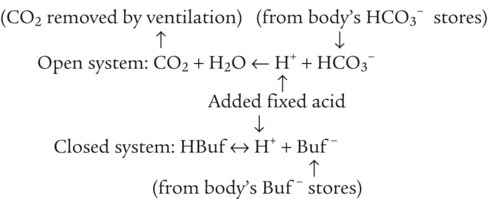
Open and closed buffer systems play different roles in buffering fixed and volatile acids, and they differ in their ability to function in wide-ranging pH environments. Volatile acid (H2CO3) accumulates only if ventilation cannot eliminate CO2 fast enough to keep up with the body’s CO2 production. In such a case, the reaction between CO2 and H2O moves continually to the right, creating more H2CO3 and, ultimately, more H+ and HCO3−. The HCO3− produced in this way is incapable of buffering the H+ with which it was coproduced. The only buffer system that can buffer the H+ of volatile acid is the nonbicarbonate buffer system. Both nonbicarbonate and bicarbonate buffer systems can buffer the H+ produced by fixed acids; this is true of the bicarbonate buffer system only if ventilation is not impaired and CO2 can be adequately eliminated. Both systems are physiologically important, each playing a unique and essential role in maintaining pH homeostasis. Table 13-1 summarizes the approximate contributions of various blood buffers to the total buffer base. Bicarbonate buffers have the greatest buffering capacity because they function in an open system.
TABLE 13-1
Individual Buffer Contributions to Whole Blood Buffering
| Buffer Type | Total Buffering (%) |
| Bicarbonate | |
| Plasma bicarbonate | 35 |
| Erythrocyte bicarbonate | 18 |
| Total bicarbonate buffering | 53 |
| Nonbicarbonate | |
| Hemoglobin | 35 |
| Organic phosphates | 3 |
| Inorganic phosphates | 2 |
| Plasma proteins | 7 |
| Total nonbicarbonate buffering | 47 |
| Total | 100 |

From Beachey W: Respiratory care anatomy and physiology: foundations for clinical practice, ed 2, St Louis, 2007, Mosby.
Bicarbonate and nonbicarbonate buffer systems do not function in isolation from one another but are intermingled in the same solution (whole blood), in equilibrium with the same [H+] (Figure 13-1). Increased ventilation increases the CO2 removal rate, causing nonbicarbonate buffers (Hbuf) to release H+. Decreased ventilation ultimately causes Hbuf to accept more H+.
pH of a Buffer System: Henderson-Hasselbalch Equation
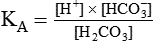
[H+] can be calculated by algebraic rearrangement of this equation, as follows:
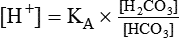
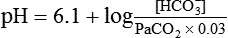
pH is a logarithmic expression of [H+], and the term 6.1 is the logarithmic expression of the H2CO3 equilibrium constant. Because dissolved carbon dioxide (Pco2 × 0.03) is in equilibrium with and directly proportional to blood [H2CO3], and because blood Pco2 is more easily measured than [H2CO3], dissolved CO2 is used in the denominator of the H-H equation. The H-H equation is specific for calculating the pH of the bicarbonate buffer system of the blood. The calculation of this pH is important because it equals the pH of blood plasma; because all buffer systems in the blood are in equilibrium with the same pH, the pH of one buffer system is the same as the pH of the entire plasma solution (the isohydric principle).1
Clinical Use of Henderson-Hasselbalch Equation
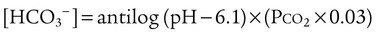
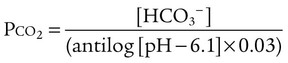
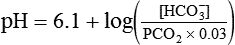
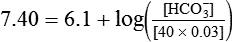
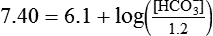
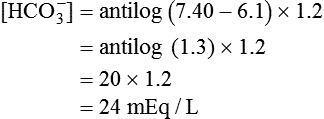
 Problem
Problem ) of 8 L/min. The patient’s PaCO2 is 55 mm Hg, pH is 7.30, and bicarbonate is 26 mEq/L, and the therapist wishes to maintain a pH of 7.35. How much does the RT need to change the PaCO2 to achieve this desired pH, and what change in the patient’s VT does this require?
) of 8 L/min. The patient’s PaCO2 is 55 mm Hg, pH is 7.30, and bicarbonate is 26 mEq/L, and the therapist wishes to maintain a pH of 7.35. How much does the RT need to change the PaCO2 to achieve this desired pH, and what change in the patient’s VT does this require?
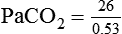
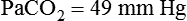
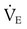 required to produce a PaCO2 of 49 mm Hg. Because
required to produce a PaCO2 of 49 mm Hg. Because 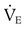 is inversely proportional to PaCO2, the following can be stated:
is inversely proportional to PaCO2, the following can be stated: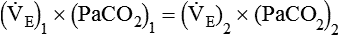
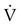 )2 as follows:
)2 as follows: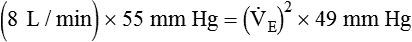
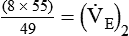
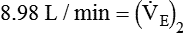
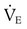 from 8 L/min to approximately 9 L/min yields a PaCO2 of 49 mm Hg and a pH of approximately 7.35. Now the RT divides the new
from 8 L/min to approximately 9 L/min yields a PaCO2 of 49 mm Hg and a pH of approximately 7.35. Now the RT divides the new 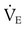 of 9 L/min by the respiratory frequency to calculate the new VT required:
of 9 L/min by the respiratory frequency to calculate the new VT required: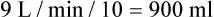
Physiologic Roles of Bicarbonate and Nonbicarbonate Buffer Systems
The functions of bicarbonate and nonbicarbonate buffer systems are summarized in Table 13-2.
TABLE 13-2
| Buffer | Type of System | Acids Buffered |
| Bicarbonate | Open | Fixed (nonvolatile) |
| Nonbicarbonate | Closed | Volatile (carbonic) |
| Fixed |
From Beachey W: Respiratory care anatomy and physiology: foundations for clinical practice, ed 2, St Louis, 2007, Mosby.
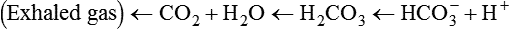
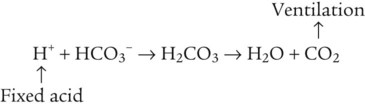
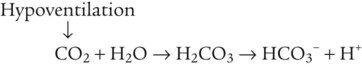
Nonbicarbonate Buffer System
Table 13-1 lists the nonbicarbonate buffers in the blood. Of these, Hb is the most important because it is the most abundant. As mentioned, these buffers are the only ones available to buffer carbonic acid. However, they can buffer H+ produced by any acid, fixed or volatile. Because nonbicarbonate buffers (Buf−/HBuf) function in closed systems, the products of their buffering activity eventually accumulate, slowing or stopping further buffering activity:
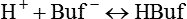
Acid Excretion
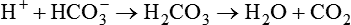
Kidneys
• Excretion is the elimination of substances from the body in the urine.
• Secretion is the process by which renal tubule cells actively transport substances into the fluid of the tubule lumen, or filtrate.
• Reabsorption is the active or passive transport of filtrate substances back into the tubule cell and into the blood of nearby capillaries.
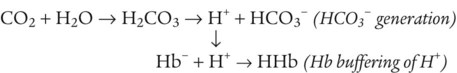
The amount of H+ the kidney tubules secrete into the filtrate depends on the blood’s pH. Secreted H+ may originate from H2CO3 (when the blood PCO2 is increased) or from fixed acids. The kidneys excrete less than 100 mEq of fixed acid per day, which is a small amount compared with volatile H2CO3 elimination by the lungs.3 In addition to excreting H+, the kidneys influence blood pH by retaining or excreting HCO3−. If the blood PCO2 is high, creating high levels of H2CO3, the kidneys excrete greater amounts of H+ and reabsorb all of the tubule filtrate’s HCO3− back into the blood. The opposite happens when the blood PCO2 is low. The kidneys excrete less H+ and more HCO3−. Compared with the ability of the lungs to change blood PCO2 in seconds, the renal process is slow, requiring hours to days.
Basic Kidney Function
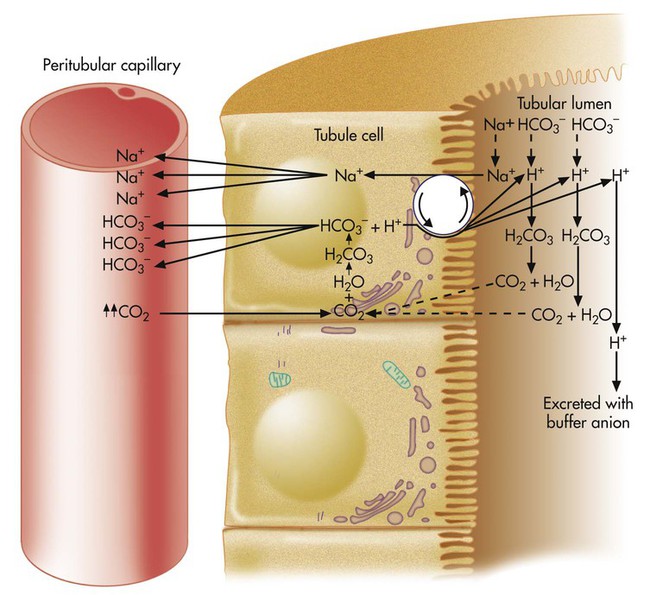
HCO3− is one of the electrolytes filtered from the blood at the glomerulus to become part of the tubular filtrate. In this way, base (HCO3−) is removed from the blood. This loss of base is offset by the simultaneous secretion of the nephron tubular epithelium of H+ into the tubular lumen and into the filtrate. Under normal conditions, the rate of H+ secretion is almost the same as the rate of HCO3− filtration.4 In this way, the kidneys titrate H+ and HCO3− against each other to form CO2 and H2O.
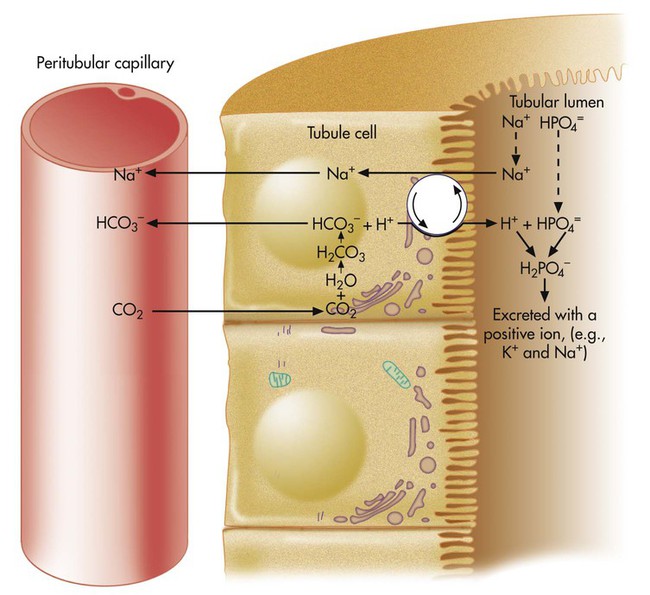
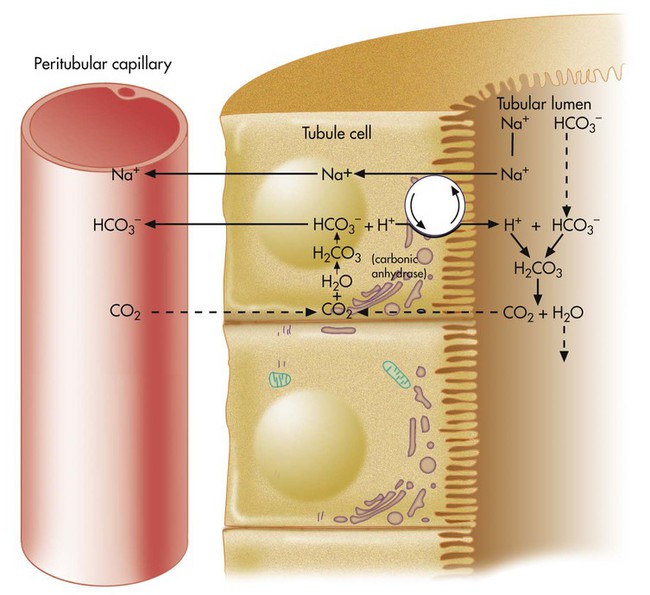
H+ secretion begins with the diffusion of blood CO2 into the tubule cell (Figure 13-2). Aided by the enzyme carbonic anhydrase, CO2 reacts with H2O to form carbonic acid, which forms HCO3− and H+. The tubule cell actively secretes H+ into the filtrate by means of countertransport, in which Na+ and H+ are simultaneously transported in opposite directions. That is, Na+ and H+ combine with opposite ends of a carrier protein in the luminal border of the tubule cell membrane. Sodium ions move into the cell down its high concentration gradient, providing the energy to secrete H+ into the tubular filtrate (see Figure 13-2).4
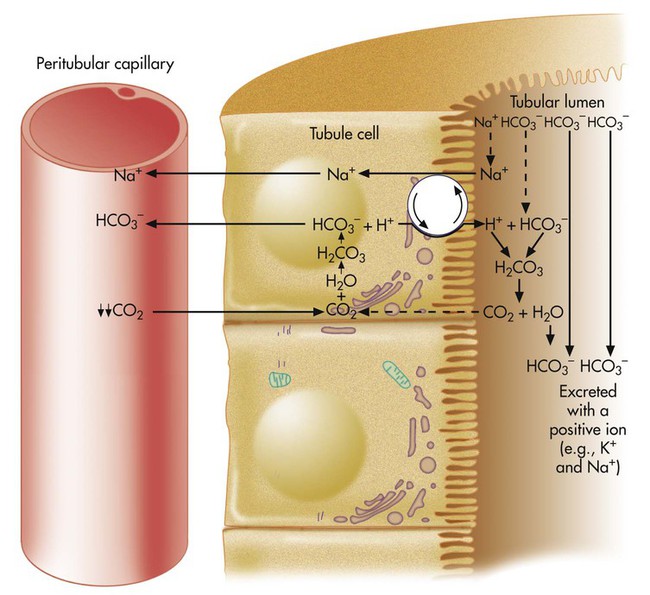
The rate of tubular H+ secretion increases if the concentration of H+ in the blood plasma increases. Conversely, the rate of H+ secretion decreases if blood plasma [H+] decreases (Figure 13-3). Any factor that increases PaCO2, such as hypoventilation, increases H+ secretion, and any factor that decreases PaCO2, such as hyperventilation, decreases H+ secretion.
HCO3− formed in the tubule cell from the reaction between CO2 and H2O (see Figure 13-2) diffuses back into the blood plasma because the luminal side of the tubule cell is relatively impermeable to HCO3−. HCO3− and Na+ are reabsorbed whenever H+ is secreted into the tubular filtrate.
Reabsorption of Bicarbonate Ion
Because the luminal side of the renal tubule cell is relatively impermeable to HCO3−, these ions are reabsorbed indirectly, as shown in Figure 13-2. The HCO3− in the filtrate reacts with the H+ secreted by the tubular cells. The resulting carbonic acid breaks down into CO2 and H2O. Because CO2 is extremely diffusible through biologic membranes, it diffuses instantly into the tubule cell. There, CO2 reacts rapidly with H2O in the presence of carbonic anhydrase, rapidly forming HCO3− and H+. The HCO3− diffuses back through the nonluminal side of the tubule cell into the blood. The reabsorbed HCO3− ion is not the same HCO3− ion that existed in the tubular fluid. If the tubule cells secrete sufficient H+, all HCO3− in the tubular fluid is reabsorbed in this manner.
The net effect of secreting H+ (caused by high blood CO2 or hypoventilation, as shown in Figure 13-2) is to reabsorb all filtrate HCO3−, increasing the quantity of HCO3− in the blood. According to the H-H equation, this brings blood pH up toward the normal range.
If blood CO2 is low, as is the case in a state of hyperventilation (see Figure 13-3), the ratio of HCO3− to dissolved CO2 molecules increases, and the renal filtrate has more HCO3− than H+. Because HCO3− cannot be reabsorbed without first reacting with H+, the excess HCO3− is excreted in the urine, carrying positive ions such as Na+ or K+ in the filtrate. The net effect of secreting less H+ is to increase the quantity of HCO3− (base) lost in the urine. According to the H-H equation, this brings blood pH down toward the normal range. These renal responses to high and low blood PCO2 are the mechanisms by which the kidneys compensate for respiratory acid-base disturbances.
Excess Hydrogen Ion Excretion and Role of Urinary Buffers
If no buffers existed in the filtrate to react with H+, the H+-secreting mechanism would soon cease to function because when the filtrate pH decreases to 4.5, H+ secretion stops.4 Buffers in the tubular fluid are essential for the secretion and elimination of excess H+ in acidotic states.
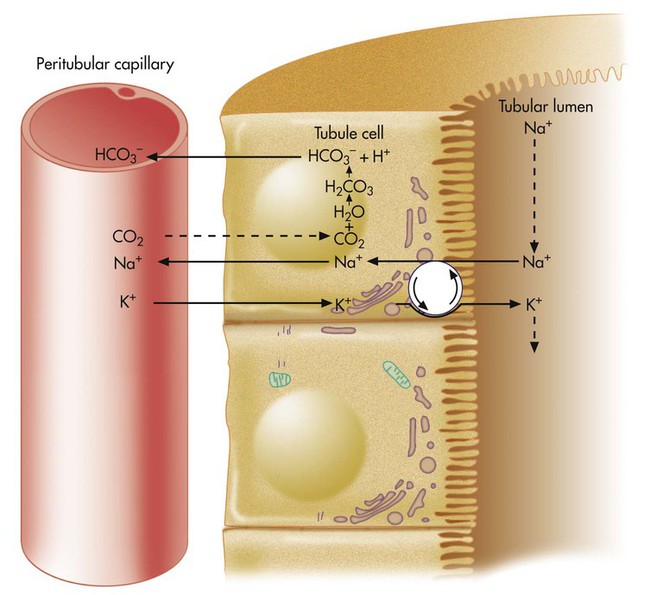
In Figure 13-2, more H+ than HCO3− is present in the filtrate. After all available HCO3− reacts with H+, the remaining H+ reacts with two other filtrate buffers, phosphate and ammonia, as illustrated in Figures 13-4 and 13-5. In Figure 13-4, phosphate and H+ react to form H2PO4−, which must be excreted with a positive ion to maintain tubular electroneutrality. Figure 13-5 shows that when urinary buffers are depleted, the resulting fall in filtrate pH stimulates the tubules to secrete ammonia. The NH3 molecule buffers H+ by reacting with it to form the ammonium ion (NH4+). To maintain electroneutrality, the kidney excretes a negatively charged ion to accompany NH4+. This negative ion is chloride (Cl−), the most abundant filtrate anion.
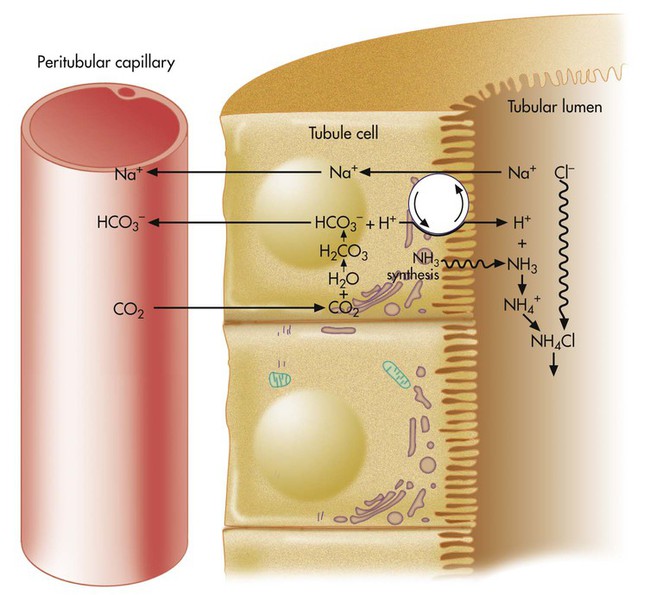
When NH4+ reacts with H+, HCO3− diffuses from the tubule cell into the blood (see Figure 13-5). The net effect of ammonia buffer activity is to cause more bicarbonate to be reabsorbed into the blood, counteracting the acidic state of the blood. Figure 13-5 shows that when Cl− is excreted in combination with NH4−, the blood gains HCO3−. Blood [Cl−] and [HCO3−] are reciprocally related (i.e., when one is high, the other is low). This relationship explains why people with chronically high blood PCO2 tend to have low blood [Cl−] or hypochloremia. Activation of the ammonia buffer system enhances Cl− loss and HCO3− gain.
Acid-Base Disturbances
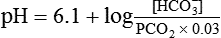
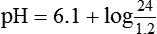
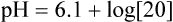
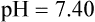

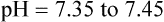
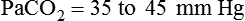
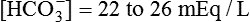
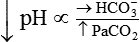
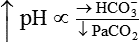
Primary Metabolic (Nonrespiratory) Disturbances
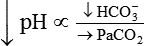
In contrast, ingesting too much alkali (e.g., NaHCO3 or other antacids) increases [HCO3−] and pH:
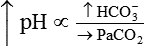
Plasma [HCO3−] can be increased by its addition, as in the previous example, or by its generation, as occurs when fixed acid is lost from the body.5 An individual may lose HCl from the body by vomiting large amounts of gastric juice. This loss generates HCO3−, as discussed later (see Figure 13-8 further on).
Processes that increase arterial pH by losing fixed acid or gaining HCO3− produce a condition called metabolic alkalosis. Table 13-3 shows the four primary acid-base disturbances causing alkalemia and acidemia.
TABLE 13-3
Primary Acid-Base Disorders and Compensatory Responses
| Acid-Base Disorder | Primary Defect | Compensatory Response |
| Respiratory acidosis | 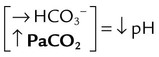 |
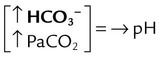 |
| Respiratory alkalosis | 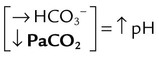 |
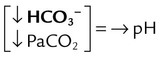 |
| Metabolic acidosis | 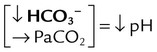 |
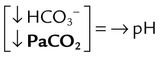 |
| Metabolic alkalosis | 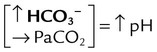 |
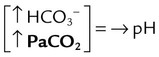 |
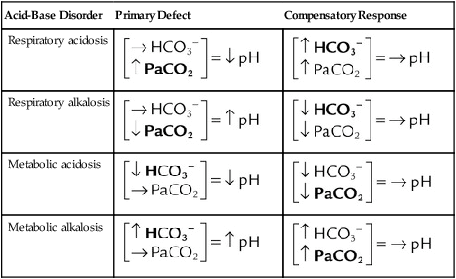
→, No change; ↓, decrease; ↑, increase.
Note: Primary defects and compensatory responses appear in boldface type.
From Beachey W: Respiratory care anatomy and physiology: foundations for clinical practice, ed 2, St Louis, 2007, Mosby.
Compensation: Restoring pH to Normal
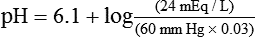
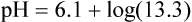
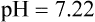
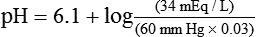
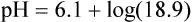
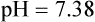
The lungs normally compensate quickly for metabolic acid-base defects because ventilation can change the PaCO2 within seconds. The kidneys require more time to retain or excrete significant amounts of HCO3− and compensate for respiratory defects at a much slower pace. Table 13-3 summarizes the four primary acid-base disturbances and the body’s compensatory responses.
Effect of Carbon Dioxide Hydration Reaction on [HCO3−]
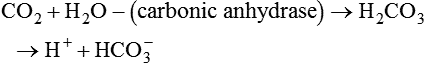
As H+ and HCO3− are rapidly produced, Hb immediately buffers H+, generating HCO3− in the process:
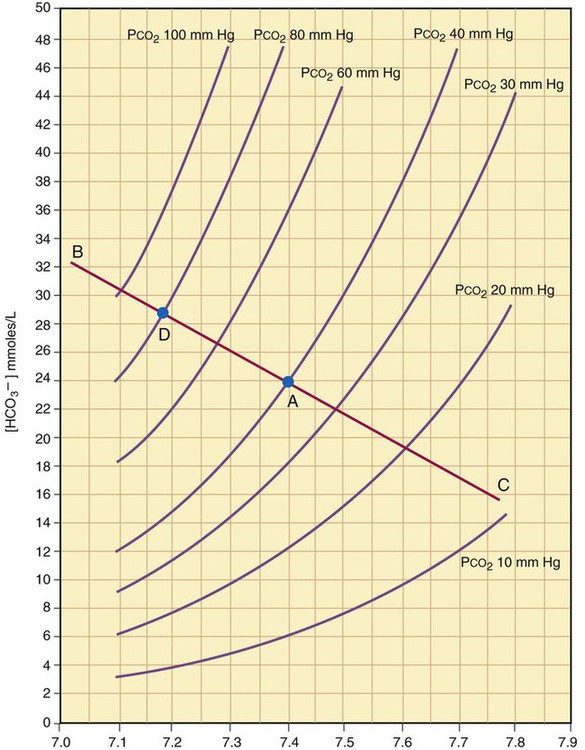
The amount of HCO3− increase depends on the amount of nonbicarbonate buffer that is available to accept the H+ produced by the hydration reaction. Generally, when the nonbicarbonate buffer concentration is normal, and the PCO2 increase is acute, the hydration reaction increases the plasma [HCO3−] approximately 1 mEq/L for every 10-mm Hg increase in PCO2 higher than 40 mm Hg. Figure 13-6 illustrates this hydration reaction effect. Normal status is represented by point A: PaCO2 of 40 mm Hg, pH of 7.40, and plasma HCO3− of 24 mEq/L. An acute increase in PaCO2 from 40 mm Hg to 80 mm Hg proceeds from point A, moving to the left, up the normal blood buffer line (line BAC) to point D, where the buffer line intersects the PaCO2 = 80 mm Hg isopleth. Point D indicates an HCO3− of approximately 28.5 mEq/L and a pH of approximately 7.18. This small change in [HCO3−] should not be erroneously interpreted as early renal compensation.
 Rule of Thumb
Rule of ThumbClinical Acid-Base States
Systematic Acid-Base Classification
In analyzing an acid-base problem, it is helpful to use a series of systematic steps. Consistently applying them to all acid-base disturbances helps avoid the tendency to jump to conclusions. Four steps in acid-base classification are outlined in Box 13-2. The order of the steps is not as important as following the same procedure for each situation.
Step 4: Assess for Compensation
Complete compensation refers to any case in which the compensatory response returns the pH to the normal range (7.35 to 7.45). Partial compensation refers to instances in which the expected compensatory response has begun but has not had sufficient time to return the pH into the normal range. For example, suppose a patient has a partially compensated respiratory acidosis. This condition is characterized by high PaCO2, pH less than 7.35, and plasma [HCO3−] greater than 26 mEq/L. The compensatory response (increased HCO3−) is not yet sufficient to return the pH into the normal range, although the expected compensatory activity has begun. By comparison, a completely compensated respiratory acidosis is shown by the same patient several hours later, when the kidneys have had enough time to retain sufficient plasma HCO3− to bring the pH into the normal range. This completely compensated respiratory acidosis is characterized by the same originally observed high PaCO2, pH that is in the 7.35 to 7.39 range, and plasma [HCO3−] that is greater than it was before complete compensation took place. The pH is on the acidic side of 7.40 because the primary disturbance (high PaCO2) originally created an acidotic environment. Generally, the body does not overcompensate for an acid-base disturbance. Table 13-4 summarizes acid-base and ventilatory classification. Table 13-5 classifies the degree of compensation for acid-base disturbances.
TABLE 13-4
Acid-Base and Ventilatory Classification
| Component | Classification | Range |
| pH (arterial) | Normal status | 7.35-7.45 |
| Acidemia | <7.35 | |
| Alkalemia | >7.45 | |
| PaCO2 (mm Hg) | Normal ventilatory status | 35-45 |
| Respiratory acidosis (hypoventilation) | >45 | |
| Respiratory alkalosis (hyperventilation) | <35 | |
| HCO3− (mEq/L) | Normal metabolic status | 22-26 |
| Metabolic acidosis | <22 | |
| Metabolic alkalosis | >26 |
From Beachey W: Respiratory care anatomy and physiology: foundations for clinical practice, ed 2, St Louis, 2007, Mosby.
TABLE 13-5
Degrees of Acid-Base Compensation
| Compensating (Noncausative Component) | pH | Classification |
| Within normal range | Abnormal | Noncompensated (acute) |
| Out of normal range in the expected direction | Abnormal | Partially compensated |
| Out of normal range in the expected direction | Normal | Compensated (chronic) |
From Beachey W: Respiratory care anatomy and physiology: foundations for clinical practice, ed 2, St Louis, 2007, Mosby.
Respiratory Acidosis
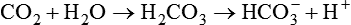
Mini Clini
Acute (Uncompensated) Respiratory Acidosis
 Problem
ProblemThe RT should follow these steps:
1. Categorize the pH. The pH is below normal, indicating the presence of acidemia.
2. Determine respiratory involvement. PaCO2 is elevated above normal, consistent with a low pH, indicating hypoventilation as a contributing factor to acidemia (respiratory acidosis).
3. Determine metabolic involvement. HCO3− is elevated slightly above normal. However, this is in the expected range for acute respiratory acidosis (1 mEq for each 10-mm Hg increase in PCO2).
4. Assess for compensation. As explained in step 3, HCO3− is within the expected range for acute respiratory acidosis. There is no evidence of metabolic compensation.
Hypercapnia is synonymous with respiratory acidosis.
Causes
Any process in which alveolar ventilation fails to eliminate CO2 as rapidly as the body produces it causes respiratory acidosis. This acidosis could occur in different ways. A person’s ventilation may be decreased from a drug-induced central nervous system depression, or a person with limited ventilatory reserve may have a normal PaCO2 at rest but cannot accommodate the increased CO2 production associated with increased physical activity. Box 13-3 summarizes causes of respiratory acidosis.
Compensation
Renal compensation for respiratory acidosis begins as soon as PaCO2 increases. The kidney reabsorbs HCO3− from the renal tubular filtrate, bringing the arterial pH into the normal range (see Figure 13-2). However, this process cannot keep pace with an acutely increasing PaCO2. Full compensation may take several days.
Correction
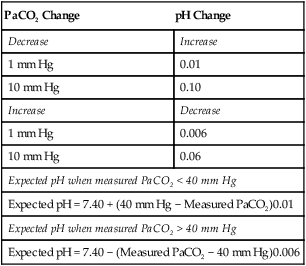
The main goal in correcting respiratory acidosis is to improve alveolar ventilation. Various respiratory care modalities may be employed ranging from bronchial hygiene and lung expansion techniques to noninvasive positive pressure ventilation to endotracheal intubation and mechanical ventilation. If hypoventilation is chronic and compensation has restored pH within the normal range, corrective action aimed at decreasing PaCO2 is inappropriate and possibly harmful. In this instance, rapidly decreasing PaCO2 to normal would induce an alkalosis because of the compensatory [HCO3−] retention by the kidneys (Table 13-6).
TABLE 13-6
Expected Effect of Acute Changes in PaCO2 on Arterial pH
| PaCO2 Change | pH Change |
| Decrease | Increase |
| 1 mm Hg | 0.01 |
| 10 mm Hg | 0.10 |
| Increase | Decrease |
| 1 mm Hg | 0.006 |
| 10 mm Hg | 0.06 |
| Expected pH when measured PaCO2 < 40 mm Hg | |
| Expected pH = 7.40 + (40 mm Hg − Measured PaCO2)0.01 | |
| Expected pH when measured PaCO2 > 40 mm Hg | |
| Expected pH = 7.40 − (Measured PaCO2 − 40 mm Hg)0.006 | |
Mini Clini
Chronic (Compensated) Respiratory Acidosis
 Problem
ProblemThe RT should follow these steps:
1. Categorize the pH. The pH is on the acidic side of the normal range, but it is still normal.
2. Determine respiratory involvement. PaCO2 is higher than normal, indicating hypoventilation as a contributing factor to the low-normal pH (respiratory acidosis).
3. Determine metabolic involvement. HCO3− is substantially elevated. By itself, this would cause alkalemia, but because pH is on the acidic side of normal, primary metabolic alkalosis is ruled out. Compensation for the respiratory acidosis has occurred.
4. Assess for compensation. HCO3− is approximately 8 to 10 mEq higher than normal. This is consistent with a compensatory response by the kidneys. In addition, the expected pH for a PaCO2 of 64 mm Hg is [7.40 − (64 mm Hg − 40 mm Hg) 0.006], or 7.26 (see Table 13-6). Because the actual pH is 7.36, metabolic compensation (retention of HCO3−) must have occurred.
Respiratory Alkalosis
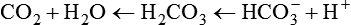
Mini Clini
Acute (Uncompensated) Respiratory Alkalosis
 Problem
ProblemThe RT should follow these steps:
1. Categorize the pH. The pH is substantially higher than normal, indicating the presence of an alkalemia.
2. Determine respiratory involvement. PaCO2 is well below normal, which is consistent with the high pH, indicating hyperventilation as a contributing factor in alkalemia (respiratory alkalosis).
3. Determine metabolic involvement. HCO3− is slightly lower than normal. However, this is within the expected range for acute respiratory alkalosis (hydrolysis effect).
4. Assess for compensation. The decrease in HCO3− is within the expected range for acute respiratory alkalosis (1 mEq for each 5-mm Hg decline in PCO2).
Hypocapnia is synonymous with respiratory alkalosis.
Causes
Any process in which ventilatory elimination of CO2 exceeds production of CO2 causes respiratory alkalosis. The most common cause of hyperventilation in patients with pulmonary disease is decreased PaO2 (hypoxemia). Hypoxemia causes specialized neural structures to signal the brain, increasing ventilation (see Chapter 14). Anxiety, fever, stimulatory drugs, pain, and central nervous system injuries are possible causes of hyperventilation. Other possible causes include stimulation of irritant receptors in the lung parenchyma, which may occur in pneumonia or pulmonary edema.
Hyperventilation and respiratory alkalosis also may be iatrogenically induced (induced by medical treatment). Iatrogenic hyperventilation is most commonly associated with overly aggressive mechanical ventilation. It may also be associated with aggressive deep breathing and lung expansion respiratory care procedures. Decreased PaCO2, increased pH, and normal-range [HCO3−] characterize acute respiratory alkalosis. A slight decrease in [HCO3−] is expected from the effect of the hydration reaction. Box 13-4 summarizes causes of respiratory alkalosis.
Compensation
The kidneys compensate for respiratory alkalosis by excreting HCO3− in the urine (bicarbonate diuresis; see Figure 13-3). This activity brings arterial pH down into the normal range. As with respiratory acidosis, renal compensation is a slow process. Complete compensation may take days.
Correction
Mini Clini
Compensated (Chronic) Respiratory Alkalosis
 Problem
ProblemThe RT should follow these steps:
1. Categorize the pH. The pH is on the alkalotic side of the normal range, but it is still normal.
2. Determine respiratory involvement. PCO2 is well below normal, indicating hyperventilation as a contributing factor to the high-normal pH (respiratory alkalosis).
3. Determine metabolic involvement. HCO3− is substantially lower than normal, but because the pH is on the alkalotic side of normal, primary metabolic acidosis is ruled out. Compensation for the respiratory alkalosis has occurred.
4. Assess for compensation. HCO3− is approximately 7 mEq below normal. This is consistent with a compensatory response by the kidneys. In addition, the expected pH for PaCO2 of 26 mm Hg is [7.40 + (40 mm Hg − 26 mm Hg) 0.01], or 7.54 (see Table 13-6). Because the actual pH is 7.44, metabolic compensation (excretion of HCO3−) must have occurred.
Metabolic (Nonrespiratory) Acidosis
Anion Gap
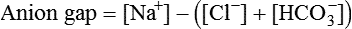
Figure 13-7, A shows that normal concentrations of these ions in the plasma are as follows: 140 mEq/L for Na+, 105 mEq/L for Cl−, and 24 mEq/L for HCO3−, yielding an anion gap of 11 mEq/L (140 mEq/L − [105 mEq/L + 24 mEq/L] = 11 mEq/L). The normal anion gap range is 9 to 14 mEq/L.6
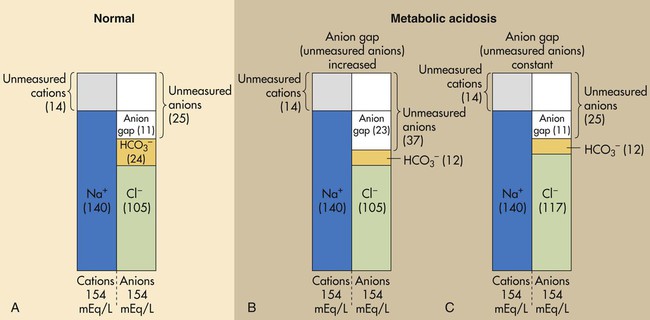
An increased anion gap (>14 mEq/L) is caused by metabolic acidosis in which fixed acids accumulate in the body. The H+ of these acids reacts with plasma HCO3−, lowering its concentration; this leads to an increased anion gap (i.e., an increase in unmeasured anions) (see Figure 13-7, B). (When the H+ of fixed acids is buffered by HCO3−, the anion portion of the fixed acid remains in the plasma, increasing unmeasured anion concentration.) A high anion gap indicates that fixed acid concentration in the body has increased.
Metabolic acidosis caused by HCO3− loss from the body does not cause an increased anion gap. Bicarbonate loss is accompanied by Cl− gain, which keeps the anion gap within normal limits (see Figure 13-7, C). The law of electroneutrality helps explain the reciprocal nature of [HCO3−] and [Cl−] in this instance. With a constant cation concentration, losing HCO3− means that another anion must be gained to maintain electroneutrality. In this case, the kidney increases its reabsorption of the most abundant anion in the tubular filtrate, the Cl−. The kind of metabolic acidosis in which HCO3− is lost from the body is sometimes called hyperchloremic acidosis because of the characteristic increase in plasma [Cl−]. Box 13-5 summarizes causes of anion gap and non–anion gap metabolic acidosis.
 Rule of Thumb
Rule of ThumbCompensation
Mini Clini
Partially Compensated Metabolic Acidosis
 Problem
ProblemThe RT should follow these steps:
1. Categorize the pH. The pH is below the normal range, indicating the presence of acidemia.
2. Determine respiratory involvement. PaCO2 is well below normal, indicating severe hyperventilation. By itself, this would cause alkalosis, but the presence of acidemia rules out primary respiratory alkalosis. The low PaCO2 is probably a compensatory response to primary metabolic acidosis, although this response is insufficient to restore pH to the normal range.
3. Determine metabolic involvement. HCO3− is severely reduced, consistent with the low pH. In the presence of low pH and low PaCO2, a low HCO3− signals primary metabolic acidosis. This is confirmed by the large BE.
4. Assess for compensation. The severe hyperventilation represents a compensatory response to primary metabolic acidosis, although compensation is far from complete. Nevertheless, the pH level would be even lower if PaCO2 were normal.
Symptoms
Mini Clini
Compensated Metabolic Acidosis
 Problem
ProblemThe RT should follow these steps:
1. Categorize the pH. The pH is on the acidic side of the normal range, but it is still normal.
2. Determine respiratory involvement. PaCO2 is below normal, indicating hyperventilation. By itself, this would cause alkalosis; however, because the pH is on the acidic side of normal, the presence of primary respiratory alkalosis is ruled out. The low PaCO2 is likely a compensatory response to a primary metabolic problem (possible metabolic acidosis).
3. Determine metabolic involvement. HCO3− level is substantially lower than normal, consistent with a low pH. Given that the pH level is on the acidic side of normal, the low HCO3− level signals a possible metabolic acidosis. This is confirmed by the large BE.
4. Assess for compensation. The hyperventilation previously described must represent a compensatory response to primary metabolic acidosis. The pH is in the normal range.
Correction
The initial goal in severe acidemia is to increase the arterial pH greater than 7.20, a level below which serious cardiac arrhythmias are likely.7 If respiratory compensation maintains the pH at or above this level, immediate corrective action is usually not indicated. Treatment of the underlying cause of acid gain or base loss is the rational approach.
Metabolic Alkalosis
Mini Clini
Metabolic Alkalosis
 Problem
ProblemThe RT should follow these steps:
1. Categorize the pH. The pH level is substantially above normal, indicating the presence of alkalemia.
2. Determine respiratory involvement. PaCO2 is slightly higher than normal, indicating mild hypoventilation. However, because alkalemia is present, the existence of primary respiratory acidosis is ruled out. The elevated PaCO2 may be a compensatory response to a primary metabolic problem (possible metabolic alkalosis).
3. Determine metabolic involvement. HCO3− is substantially higher than normal. Given the high pH, the elevated HCO3− signals a metabolic alkalosis. This is confirmed by the large BE. In addition, the low serum K+ and Cl− values indicate hypokalemic/hypochloremic metabolic alkalosis.
4. Assess for compensation. Although PaCO2 is slightly elevated, compensation for metabolic alkalosis is minimal. This lack of compensation is consistent with the presence of hypokalemic metabolic alkalosis.
Causes
Metabolic alkalosis can occur in one of the following two ways: (1) loss of fixed acids or (2) gain of blood buffer base. Both processes increase plasma [HCO3−]. To explain why losing fixed acid increases the plasma [HCO3−], consider a situation in which vomiting removes gastric HCl from the body (Figure 13-8). In response to HCl loss, H+ diffuses out of the gastric cell into the gastric fluid, where Cl− accompanies it; this forces the CO2 hydration reaction in the gastric cell to the right, which generates HCO3−. The HCO3− enters the blood in exchange for the Cl−. The plasma gains an HCO3− for each Cl− (or H+) that is lost (see Figure 13-8).7
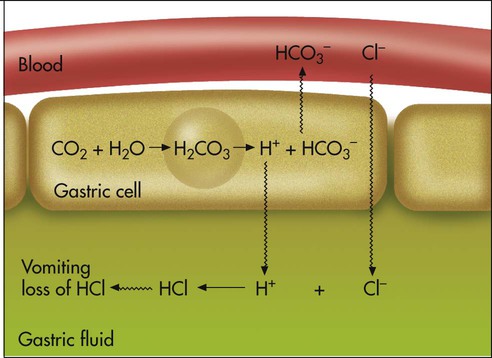
The causes of metabolic alkalosis are summarized in Box 13-6. Metabolic alkalosis is common in acutely ill patients and is probably the most complicated acid-base imbalance to treat because it involves fluid and electrolyte imbalances. Metabolic alkalosis is often iatrogenic, resulting from the use of diuretics, low-salt diets, and gastric drainage.
To understand how the loss of Cl−, K+, and fluid volume may cause alkalosis, one needs to understand how the kidney regulates Na+. Approximately 26,000 mEq of Na+ passes through the glomerular membrane daily, but the body’s daily Na+ intake averages only approximately 150 mEq.4 The kidney’s main job is to reabsorb Na+, not to excrete it. For this reason, and because Na+ has a major role in maintaining fluid balance, the kidney places a greater priority on reabsorbing Na+ than on maintaining Cl−, K+, or acid-base balance.
Normally, Na+ is reabsorbed through primary active transport (Figure 13-9), in which the sodium-potassium-adenosine triphosphatase (Na+,K+-ATPase) pump actively transports Na+ out of the renal tubule cell into the blood. This process causes Na+ to diffuse continually from the filtrate into the tubule cell. Cl− must accompany Na+ to maintain electroneutrality in the filtrate. If blood Cl− concentration is low (hypochloremia), less Cl− is present in the filtrate, which means that the kidney relies more on other mechanisms to reabsorb Na+. These mechanisms, called secondary active secretion, require the kidney to secrete H+ or K+ into the filtrate in exchange for Na+. In this way, Na+ is reabsorbed, and filtrate electroneutrality is preserved. Figures 13-10 and 13-11 illustrate the secondary active secretion process for H+ and Na+, which may lead to depletion of blood H+ (alkalemia) and K+ (hypokalemia). Preexisting hypokalemia (e.g., from inadequate K+ intake) in the presence of hypochloremia places an even greater demand on the kidney to secrete H+ to reabsorb Na+; hypokalemia produces alkalosis. Dehydration (fluid volume depletion or hypovolemia) aggravates alkalosis and hypokalemia further because hypovolemia profoundly increases the kidney’s stimulus to reabsorb Na+.
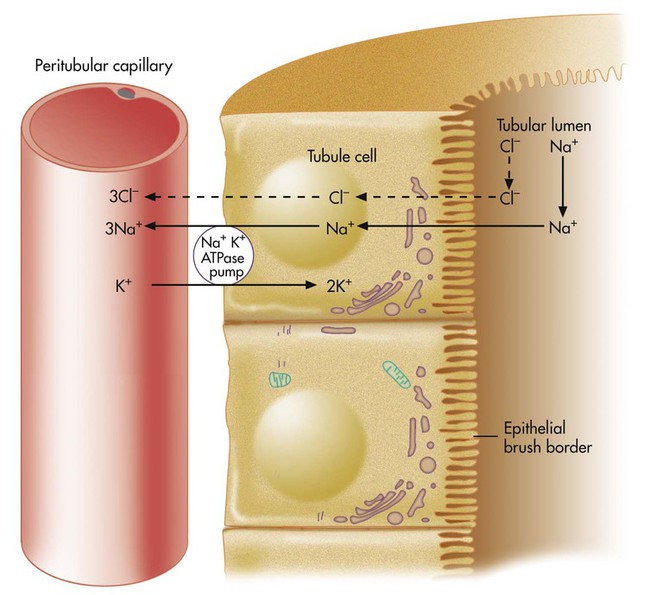
Compensation
The expected compensatory response to metabolic alkalosis is hypoventilation (CO2 retention). Traditionally, it was thought that the hypoxemia accompanying hypoventilation greatly limited respiratory compensation for metabolic alkalosis (i.e., hypoxemia stimulates ventilation and should prevent compensatory hypoventilation). However, more recent evidence does not support this theory.6 Metabolic alkalosis apparently blunts hypoxemic stimulation to ventilation. Individuals with PaO2 levels of 50 mm Hg may still hypoventilate to PaCO2 levels of 60 mm Hg to compensate for metabolic alkalosis. Nevertheless, significant CO2 retention is not seen often in cases of metabolic alkalosis, probably because metabolic alkalosis commonly coexists with other conditions that may cause hyperventilation, such as anxiety, pain, infection, fever, or pulmonary edema.
Correction
Correction of metabolic alkalosis is aimed at restoring normal fluid volume and electrolyte concentrations, especially K+ and Cl− levels. Inadequate fluid volume, especially if coupled with hypochloremia, causes excessive secretion and loss of H+ and K+ because of the great need to reabsorb Na+. In treating this type of alkalosis, it is important to supply adequate fluids containing Cl−. If hypokalemia is a primary factor, potassium chloride (KCl) is the preferred corrective agent. In cases of severe metabolic alkalosis, acidifying agents, such as dilute HCl or ammonium chloride may be infused directly into a large central vein.8
Metabolic Acid-Base Indicators
Standard Bicarbonate
Base Excess
To illustrate, consider the Mini Clini in the left-hand column on p. 304 in which the patient has respiratory acidosis for which the body has compensated by renal retention of HCO3−. If this patient’s blood were equilibrated in vitro to a PaCO2 of 40 mm Hg, the HCO3− would decrease by only 2 to 3 mEq/L to 32 to 33 mEq/L, and the pH would increase to much greater than 7.45. This patient’s BE would be well above normal. The high BE may lead the clinician to conclude incorrectly that this patient has a primary metabolic alkalosis. However, in this instance, the high BE merely reflects the fact that renal compensation has occurred.
Stewart’s Strong Ion Approach to Acid-Base Balance
In the strong ion approach, substances that affect acid-base balance in body fluids are classified into three groups, based on their degree of dissociation in an aqueous solution: (1) strong ions, (2) weak ions, (3) and nonelectrolytes. Strong ions such as Na+ and Cl− are always fully dissociated, existing only in their charged forms in aqueous solutions. This means the number of strong ions in body fluids can never be converted back to the parent compound (e.g., NaCl or KCl), as occurs with weak ions. Weak ions are produced from compounds that only partially dissociate in solution, such as volatile carbonic acid ions (HCO3− and H+) and nonvolatile acid ions such as phosphates and proteins.9 As explained earlier in this chapter, weak acid molecules dissociate until they reach equilibrium with the concentrations of their component ions, each acid in accordance with its unique equilibrium constant. Nonvolatile weak acids include protein and inorganic phosphate molecules. Nonelectrolytes are substances that never dissociate in solution but contribute to the solution’s osmotic pressure and affect the movement of ions and water across biologic membranes that separate body fluids.
Stewart distinguished between independent and dependent variables involved in acid-base regulation. Through a series of complex equations, he showed that the [H+] of a solution is a dependent variable determined solely by three independent variables10: (1) the strong ion difference, [SID]; (2) the total concentration of nonvolatile weak acids, [ATOT]; and (3) dissolved [CO2], which is a function of PCO2. The [SID] is defined as the difference between the summative concentrations of all strong positively charged ions (cations) and all strong negatively charged ions (anions) in body fluids; for clinical purposes, [SID] = ([Na+] + [K+] + [Ca++] + [Mg++]) − ([Cl−] + [other strong anions]).10,11 Normal body fluids contain more positively charged strong ions than negatively charged strong ions; [SID] represents a net positive charge. The kidneys are responsible for maintaining a normal [SID] of about 40 to 42 mEq/L, primarily by excreting or reabsorbing Na+, K+, and Cl−.
The powerful principle of electrical neutrality demands that the normal [SID] of body fluids—which represents a net positive charge—must be equal to the net negative charges of all weak anions in solution (i.e., volatile and nonvolatile weak acid anions).10 This means that normally [SID] is equal to the sum of [HCO3−] + [A−], where [A−] is the concentration of nonvolatile weak acid anions. The number of weak anions supplied by volatile acid (i.e., HCO3−) is determined by PCO2, an independent variable that is controlled by ventilation. The number of weak anions supplied by all nonvolatile weak acids (i.e., A− ions) is determined by [ATOT], an independent variable that is controlled by its temperature-dependent dissociation constant. At any given point in time, ventilation and body temperature are relatively constant, and PCO2 and the dissociation of [ATOT] are relatively constant. With these two independent variables predetermined, a change in [SID] generates powerful electrochemical forces that affect the H2O molecule’s dissociation such that electrical neutrality is maintained. In this way, changes in [SID] affect the solution’s [H+]; a fall in [SID] (a decrease in net positive charges) increases H2O dissociation, liberating more H+ to maintain electrical neutrality, whereas an increase in [SID] (an increase in net positive charges) has the opposite effect.10
Stewart’s perspective is a departure from the Henderson-Hasselbalch concept of acid-base balance in which [HCO3−] is treated as though it varies independently of dissolved [CO2], independently influencing pH or [H+]. On closer inspection, however, [HCO3−] and [CO2] cannot vary independently of each other as the CO2 hydrolysis reaction clearly shows; instead, both [HCO3−] and [H+] depend on [CO2]10:
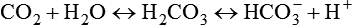
Nevertheless, clinicians generally agree that the complex nature of the equations involved in Stewart’s strong ion approach make this method unwieldy. PaCO2 and arterial pH are easy, direct measurements in the clinical setting, and [HCO3−] can be calculated with sufficient precision from these values; there is no need to calculate [HCO3−] and pH from the concentrations of electrolytes and weak acids, which are often unknown.12 Although the strong ion approach is conceptually more correct, it is not sufficiently superior to the Henderson-Hasselbalch approach to merit its universal adoption. It is reasonable and clinically appropriate to explain metabolic acid-base physiology in terms of [H+] and [HCO3−] from the Henderson-Hasselbalch perspective,9,11,12 and so the Henderson-Hasselbalch approach is retained in this chapter.