CHAPTER 9 Abnormalities of the structure and synthesis of hemoglobin
Normal human hemoglobin: structure and synthesis
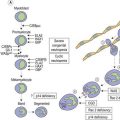
The switch from embryonic to fetal hemoglobin production begins as early as week 5 of gestation and is completed by week 10 (Fig. 9.1). β-globin expression starts as early as week 8 but the synthesis remains low, increases to approximately 10% at weeks 30–35 of gestation with a dramatic up-regulation of β-globin synthesis just before birth, coinciding with a precipitous decline in γ-globin expression. At birth, HbF (α2γ2) comprises 60–80% of the total hemoglobin, falling to ~5% at 6 months of age and eventually reaching the adult level of about 1% at 2 years, by which stage mutations affecting the adult β-globin gene should become apparent. The switch from fetal to adult hemoglobin production is not total; γ-globin production persists at a low level throughout adult life, with the residual amounts of HbF restricted to a small subset (0.2–7%) of erythrocytes termed F cells.1
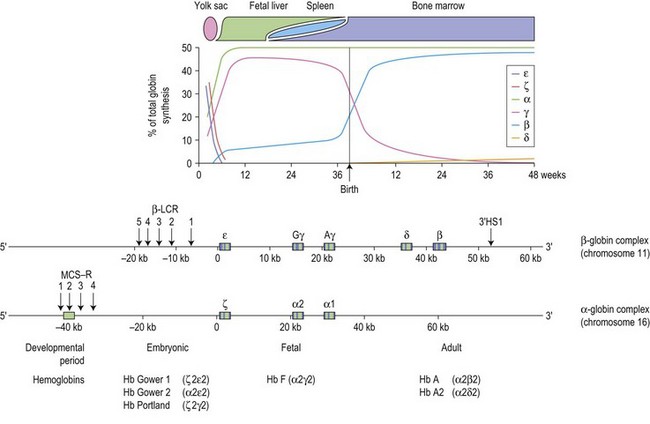
Each of the α-like and β-like globin chains are encoded by genetically distinct gene clusters, the α-like cluster on the tip of chromosome 16p (5′-ζ-α2-α1-3′), and the β-like cluster on chromosome 11p15.5 (5′-ε-Gγ-Aγ-δ-β-3′) (Fig. 9.1). In both clusters, the genes are arranged along the chromosome in the order in which they are expressed during development, suggesting that gene order may be important in the program of their expression. The coding region of each globin gene is interrupted by two intervening sequences (IVSs) or introns. In the β-like globin genes, IVS-1 (122–130 bp) interrupts the sequence between codons 30 and 31, and IVS-2 (850–900 bp), between codons 104 and 105. In the α-like globin genes, IVS-1 interrupts the coding region between codons 30 and 31, and IVS-2, between codons 99 and 100. In addition to the primary cis-determinants of individual globin gene expression which are found in the promoter region immediately upstream of each gene, there are other local regulatory elements known as enhancers which are located at variable distances from the individual genes.1
Throughout development, the appropriate genes of the two globin gene clusters are coordinately expressed, maintaining a tight balance in the production of α- and β-like globins needed for the synthesis of normal hemoglobin; imbalance between the different proteins may lead to anemia. Expression of the individual genes within each cluster is controlled by complex interactions between the local regulatory sequences within each gene and regulatory elements upstream of the cluster, mediated by a series of transcription factors. In the β cluster, the upstream element is referred to as the β locus control region (β-LCR), which consists of five DNase I hypersensitive sites (designated HSs1–5) distributed between 5 and 25 kb upstream of the ε-globin gene (Fig. 9.1). The region was originally implicated in patients with anemia due to β-globin chain deficiency despite carrying normal β-globin genes. These patients were subsequently shown to be carrying mutations that deleted sequences upstream of the ε-globin gene. These natural mutants prompted experiments that demonstrated the absolute importance of the β-LCR for high levels of globin gene expression and led to a general definition of LCRs: elements that confer high levels of tissue-specific expression to a cis-linked gene dependent on copy-number but independent of position of integration site. The corresponding region in the α globin cluster consists of the four multispecies conserved sequence regions (MCS-Rs), lying 30–70 kb upstream of the α-globin genes termed MCS-R1 to R4.2 Of these elements, only MCS-R2, which consists of a single DNase hypersensitive site, has been shown to be essential for α-globin expression. MCS-R2, which lies 40 kb upstream of the cluster, is also known as HS-40. The β-LCR establishes a transcriptionally active chromatin domain that encompasses the whole β-globin cluster and acts as a unique enhancer while the α-globin HS-40 is most similar to HS2 of the β-LCR and acts as an enhancer. In both clusters, full expression of the downstream genes is critically dependent on the presence of the upstream regulatory elements.
The precise molecular mechanisms by which the globin genes are expressed in a tissue- and developmental-stage-specific manner are still poorly understood. Each cluster contains various binding sites for both erythroid-specific and more ubiquitous DNA-binding proteins in the upstream regulatory elements as well as in the local promoters of the genes. Tissue-specific expression may be explained by the presence of the binding sites for the erythroid-specific transcription factors. These motifs include (A/T) GATA (A/G) binding the tissue-restricted zinc finger proteins (GATA-1 and GATA-2), and their cofactors (FOG1 and FOG2); ‘CACCC’ binding the erythroid Krüppel-like factors (EKLF and FLKL) and, ‘TGA(C/G)TCA’ (NF-E2/AP-1 like) elements, binding the b-Zip family of proteins (NF-E2, Nrf1, Nrf2, Nrf3, Bach1 and Bach2).1,3 These transcription factors can both activate and repress gene expression. They are modular proteins and their dual functions are mediated via the distinct domains either through direct DNA binding or interaction with other proteins. Recent work has also shown that part of the function of these transcription factors involves modification of chromatin states. It seems likely that these erythroid-specific transcription factors form part of a network of factors that commit hemopoietic cells to erythroid differentiation. Elucidation of the complexity of transcription factor function has been facilitated in recent years by a series of technological advancements that included ‘chromosome conformation capture’ (3C, 4C and 3C-sequencing), RNA TRAP (tagging and recovery of associated proteins), chromatin immunoprecipitation (ChIP), and microarray analysis to identify gene targets of the transcription factors.4 The 3C technique can be used to demonstrate interactions among chromosomal fragments and indeed, showed that the β-LCR and actively transcribed globin genes are in close spatial proximity in a cluster referred to as an active chromatin hub (ACH).5
The mechanisms by which developmental regulation is controlled are less clear and rely on two mechanisms, autonomous gene silencing and gene competition.6 It appears that the ε and ζ genes are switched on in embryonic cells and autonomously switched off in definitive cells (liver and bone marrow) in which they cannot be substantially reactivated. The regulatory sequences mediating silencing of the ε-globin gene have been mapped to its distal and proximal promoters. The second switch from γ to β gene expression is more complex and involves both autonomous silencing of the γ genes and competition between the γ and β genes for the β-LCR. The LCR up-regulates only one gene at a time, and the genes compete with each other for activation by the LCR.7 The main determinant for activation of a gene is the relative distance from the LCR. In human adult tissues, the switch leading to expression of the further downstream located β and δ genes is achieved by autonomous silencing of the fetal γ genes. The balance between the γ- and β-gene expression is thought to be mediated by changes in the repertoire and/or abundance of various nuclear factors favoring particular promoter–LCR interactions. A variety of nuclear factors involved in the transcriptional regulation and hemoglobin switching have been suggested, including BCL11A, SOX6, GATA-1, KLF1, NF-E4, COUP-TF, DRED /TR2 /TR4, Ikaros-PYR and BRG1 (SW1/SNF).8 KLF1 (Krüppel-like factor 1), also known as erythroid Krüppel-like factor (EKLF) is restricted mainly to erythroid cells; it is also a highly promoter-specific activator, binding with high affinity to the β-globin CACCC box.9 Mutations found in β-globin gene CACCC box abrogate EKLF binding result in reduced β-globin expression and β thalassemia.10
BCL11A (B cell lymphoma/leukemia 11A), also known as Evi9, CTIP1, is a zinc finger transcriptional factor, essential for normal lymphoid development.11 Its role in hemoglobin switching was originally implicated in genetic association studies on HbF levels in humans.12,13 Subsequent collective clinical and functional studies suggest that BCL11A acts as a stage-specific repressor of γ-globin expression.14,15
Inherited disorders of hemoglobin
The vast majority of disorders affecting hemoglobin are inherited; it is estimated that ~7% of the world’s population are carriers for different inherited disorders of hemoglobin, making them the commonest monogenic diseases.16 The disorders can be divided into two main groups, those in which there is a structural change in a globin chain (hemoglobin variants) and the thalassemias, which result from a quantitative deficiency in one or more of the globin chains of hemoglobin. Hemoglobin variants cause a wide range of clinical problems including sickle cell disease, unstable hemoglobin, decreased oxygen affinity and increased oxygen affinity, but the majority of hemoglobin variants cause no significant change in hemoglobin properties or clinical problems. Some hemoglobin variants are synthesized at a reduced rate, resulting in a phenotype of thalassemia. The most common example is HbE, β26 (Glu→Lys), in which the substitution at β-codon 26 (GAG→AAG) that causes HbE also causes alternative splicing of the β-globin mRNA, leading to a reduction of the normally spliced β message that encodes the variant. Other hemoglobin variants result in a thalassemic phenotype due to its extreme instability leading to a functional deficiency of the globin chain variants; for example, Hb Geneva, a dominantly inherited β thalassemia,17 and Hb Constant Spring (an α thalassemia variant).1 Some hemoglobin disorders are acquired; these can also be classified into those characterized by a reduced synthesis of the globin chain (e.g. acquired HbH disease) and those which alter the structure and function of hemoglobin so that oxygen transport is affected (e.g. carboxyhemoglobinemia, methemoglobinemia).1
There is another group of β-thalassemia-like disorders referred to as δβ thalassemias and hereditary persistence of fetal hemoglobin (HPFH). These are caused by mutations that alter the switch from fetal to adult hemoglobin, and are distinguishable from β thalassemias by the substantial increases in HbF levels.1
Thalassemias
Background
Thalassemia was first recognized by Cooley and Lee in 192518 as a form of severe anemia associated with splenomegaly and bone changes in children. The term thalassemia is derived from the Greek φαλασσα (the sea) since many of the early cases came from the Mediterranean region. However, it is now clear that the disorder is not limited to the countries around the Mediterranean but occurs throughout the world, being also prevalent in the tropical and subtropical regions including the Middle East, parts of Africa, the Indian subcontinent and Southeast Asia. It appears that heterozygotes for thalassemia are protected from the severe effects of falciparum malaria and natural selection has increased and maintained their gene frequencies in these malarious regions.
The thalassemias are classified into α, β, δβ, γδβ, δ, γ and εγδβ thalassemias according to the type of globin chain(s) that is reduced. The two major categories are the α and β thalassemias while the rare forms include the δβ, γδβ and εγδβ thalassemias. Hereditary persistence of fetal hemoglobin (HPFH) refers to the group of disorders in which the switch from fetal to adult hemoglobin production is altered and high levels of fetal hemoglobin are synthesized in individuals. Because of their concomitant increase in HbF levels, the δβ and γδβ thalassemias are often considered with the HPFH syndromes. In many populations the α and β thalassemias coexist with a variety of different structural hemoglobin variants. In these populations it is quite common to inherit a combination of α and/or β thalassemia and/or structural hemoglobin variant genes; these complex interactions give rise to an extremely wide spectrum of clinical phenotypes which together constitute the thalassemia syndromes.10
Most thalassemias are inherited in a Mendelian recessive fashion. Heterozygotes are normally symptomless, although they can often be recognized by simple hematological analysis. Severely affected individuals have inherited two copies of mutant hemoglobin gene, homozygotes for α or β thalassemia, or compound heterozygotes for different molecular forms of α or β thalassemia and a hemoglobin variant. It has been estimated that about 300 000 individuals severely affected with thalassemia are born each year, posing a heavy burden on the health services.10 Due to recent population movements, these hemoglobin disorders have become an important part of clinical practice in all countries, including the UK.19
β Thalassemias
Genetic basis of disease
Molecular analysis of the β thalassemia genes has demonstrated a striking heterogeneity. Although almost 300 β thalassemia alleles (including deletions) have been characterized, population studies indicate that probably only 20 β thalassemia alleles account for >80% of the β thalassemia mutations in the whole world.1,10 This is because in each of the high-frequency areas, only a few (4–6) mutations are common, reflecting the local selection from malaria, with a varying number of rare ones. Each of these populations has its own unique group of mutations.
Unlike α thalassemia, in which deletions in the α-globin gene cluster account for most of the mutations, the molecular defects causing β thalassemia are usually point mutations involving one (or a limited number of nucleotides) within the β gene or its immediate flanking regions.1,10,20
These point mutations involve the critical sequences that interfere with gene function at either the transcriptional or the post-transcriptional level, including translation (Fig. 9.2). Approximately half of these mutations completely inactivate the β gene with no β-globin production causing β° thalassemia. Other mutations allow the production of some β-globin, and are classified as β+ or β++ thalassemia, depending on the degree of quantitative reduction in the output of the β chains. The β-globin chains that are synthesized are usually structurally normal. Mutations affecting the conserved sequences in the 5′ promoter, i.e. TATA box, proximal CACCC and distal CACCC box, typically cause a 70–80% reduction in promoter activity and are often very mild. Mutations affecting the polyadenylation signal (AATAAA) at the 3′ end, also generally result in a mild β+ thalassemia phenotype.
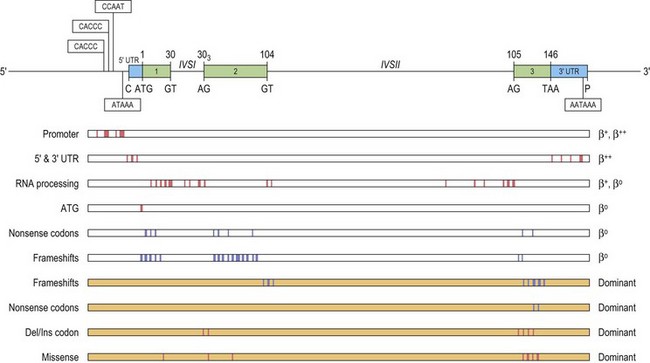
A few β thalassemia mutations are ‘silent’; carriers do not have any evident hematological phenotypes with near normal red cell indices and HbA2 levels, the only abnormality being an imbalanced globin chain synthesis. These β thalassemia mutations have usually been ascertained by finding individuals with intermediate forms of β thalassemia resulting from compound heterozygosity for one typical β thalassemia mutation in combination with a very mild β+ thalassemia allele. In this case, one parent has typical β° thalassemia trait and the other, apparently normal. Overall, the ‘silent’ β° thalassemia alleles are uncommon except for the –101 C→T mutation which has been observed fairly frequently in the Mediterranean region where it interacts with a variety of more severe β thalassemia mutations to produce milder forms of β thalassemia.21
The β° thalassemia alleles are mostly caused by premature termination of translation, either by single base substitution to a nonsense codon, or through a frameshift mutation. Studies show that the different in-phase termination mutants exhibit a ‘positional’ effect and are subjected to a surveillance mechanism (nonsense mediated RNA decay or NMD) to prevent the accumulation of mutant mRNAs coding for truncated peptides.22,23 Frameshifts and nonsense mutations that result in premature termination early in the sequence (in exon 1 and 2) are associated with minimal amounts of mutant β-mRNA.24 In such cases, no β chain is produced from the mutant allele, resulting in a phenotype of typical heterozygous β thalassemia in the carrier. However, some mutations that produce in-phase terminations later in the β sequence, in exon 3 and the 3′ half of exon 225 escape NMD, and are associated with substantial amounts of abnormal β-mRNA from the mutant allele. The mutant β-mRNA leads to a synthesis of abnormal β chain variants that are often highly unstable and non-functional, and are not able to form viable tetramers, thus effectively causing a functional deficiency of β-globin chain.1 Not only are these abnormal β-globin chain variants non-functional, but they prove to be an additional nuisance as they precipitate in the erythroid precursors, overloading the proteolytic mechanism and accentuating the ineffective erythropoiesis. Carriers for such β thalassemia alleles have fairly severe anemias, hence the term ‘dominantly inherited β thalassemia’. For a detailed review of the dominantly inherited β thalassemias, see Thein & Wood 20091 and Thein 1999.26
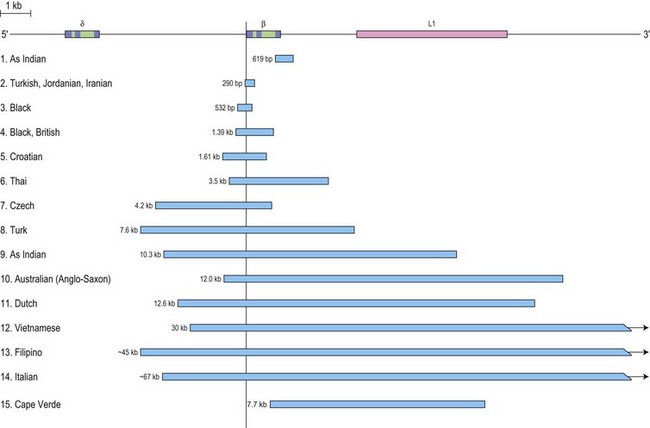
β thalassemia is rarely caused by deletions. Of these, only the 619 bp deletion at the 3′ end of the β gene is common, but even that is restricted to the Sind populations of India and Pakistan where it constitutes ~30% of the β thalassemia alleles.1,27 The other deletions, although extremely rare, are of particular clinical interest because they are associated with an unusually high level of HbA2 in heterozygotes. The mechanism underlying the markedly elevated levels of HbA2 and the variable increases in HbF in heterozygotes for these deletions is postulated to be related to the removal of the 5′ promoter region of the β-globin gene which removes competition for the upstream β-LCR and limiting transcription factors, resulting in an increased interaction of the LCR with the γ and δ genes in cis, thus enhancing their expression (Fig. 9.3). This mechanism may also explain the unusually high HbA2 levels associated with the promoter mutations at positions –88 and –29.
Unusual causes of β thalassemia, although rare, illustrate the numerous molecular mechanisms of down-regulating the β-globin gene.1 They include transposable elements which may disrupt and inactivate human genes. The insertion of such an element, a retrotransposon of the LI family into intron 2 of the β-globin gene, has been reported to cause β+ thalassemia.28 Mutations in other genes distinct from the β-globin complex (trans-acting mutations) can down-regulate β-globin expression. Such trans-acting mutations have been described affecting the XPD protein that is part of the general transcription factor TF11H,29 and the erythroid-specific GATA-1 protein.30 Somatic deletions of the β-globin gene have been reported as the contributory cause for the unusually severe anemia in three unrelated families of French and Italian origin31,32 (see intermediate forms of β thalassemia). Unipaternal isodisomy of chromosome 11p15.5 that encompassed the β-globin gene cluster contributed to thalassemia major in a Chinese patient.33
Pathophysiology
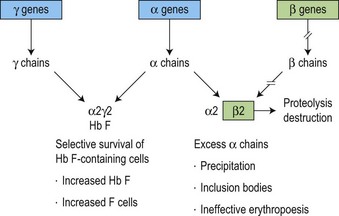
The molecular defects in β thalassemias result in absent or reduced β chain production while α chain synthesis proceeds at a normal rate. This imbalance in globin synthesis in β thalassemia gives rise to excess α chains which are extremely unstable and precipitate in the red cell precursors forming inclusion bodies (Fig. 9.4). These inclusions interfere with the red cell maturation and are responsible for the intramedullary destruction of the erythroid precursors and hence the ineffective erythropoiesis that characterizes all β thalassemias. The anemia of β thalassemia results from a combination of underproduction of hemoglobin and ineffective erythropoiesis and, ultimately, correlates well with the degree of α- to non α-globin chain imbalance and the amount of free α chains. The complications of splenomegaly, bone disease, endocrine and cardiac damage are related to the severity of anemia and the hypercatabolic state, and the subsequent degree of iron loading resulting from the increased gastrointestinal iron absorption and from repeated blood transfusion.
Genotype-phenotype correlation
The main genetic interactions that result in a phenotype of thalassemia intermedia are summarized in Table 9.1. Thalassemia intermedia can result from the inheritance of one or two β thalassemia alleles. In 60–90% of the cases34–36 the patients have inherited two β thalassemia alleles and the reduced disease severity can be explained by the inheritance of the milder forms (β++ and ‘silent’ β thalassemia alleles) that allow the production of a significant proportion of β-globin chains. Co-inheritance of a single α-globin gene deletion (αα/α−) has very little effect on β° thalassemia, but individuals with two α-globin gene deletions (α−/α− or αα/−−) and β+ thalassemia have a mild disease requiring intermittent transfusions. The role of increased HbF response as an ameliorating factor becomes evident in the group of thalassemia intermedia patients who are mildly affected despite having minimal amounts or no HbA (α2β2), and without α thalassemia. Three major QTLs – Xmn1-Gγ, HBS1L-MYB on chromosome 6q, and BCL11A on chromosome 2p – recently identified in genome-wide association studies (GWAS) have been shown to impact HbF levels, not only in healthy individuals, but also patients with β thalassemia and sickle cell disease (SCD).13,37,38 In Sardinia, BCL11A and HBS1L-MYB with α thalassemia accounts for 75% of variable phenotypic severity of β thalassemia,39 and in Thailand, the three HbF QTLs are associated with both disease severity and HbF levels in HbE/β thalassemia.40
Table 9.1 Thalassemia intermedia: the common genetic interactions that underlie the phenotype of thalassemia intermedia
| I | Homozygous or compound heterozygous state for β thalassemia |
| (a) Inheritance of mild β thalassemia alleles, β ‘silent’ and β++, in homozygous or compound heterozygous states | |
| Phenotype depends on the sum total of β-globin output | |
| (b) Co-inheritance of α thalassemia | |
| Phenotype depends on severity of imbalance between α/non-α globin reflecting severity of α and β-globin deficit | |
| (c) Increased HbF response | |
| – β-globin gene promoter mutations (deletional or non-deletional) | |
| – Co-inheritance of HbF quantitative trait loci (QTLs) for HbF on 6q (HBS1L–MYB intergenic polymorphisms, BCL11A on 2p, Xmn1-Gγ on HBB cluster) | |
| II | Heterozygous state for β thalassemia |
| (a) Co-inheritance of extra α globin genes | |
| (ααα/αα,ααα/ααα,αααα/αα,αααα/αααα, and segmental duplication of whole α-globin gene cluster) | |
| (b) Dominantly inherited β thalassemia | |
| (Hyperunstable β-globin chain variants) | |
| (c) Somatic deletion of the other β-globin locus – mosaicism | |
| III | Compound heterozygotes for β thalassemia and β chain variants |
| e.g. HbE/β thalassemia | |
| IV | Compound heterozygotes for β thalassemia and HPFH or δβ thalassemia |
| A considerable variation in clinical phenotype of these genetic interactions has been observed |
Inheritance of single copies of β thalassemia gene can also lead to thalassemia intermedia. In the majority of cases this is caused by the co-inheritance of extra α-globin genes, the severity of outcome depends on the total number of α-globin genes inherited (from one or two copies of triplicated (/ααα), or quadruplicated (/αααα) α-globin complexes), and the type of β thalassemia mutation (β° or β+).41,42 More recently, another mechanism of inheriting extra α-globin genes has been described which involved segmental duplication of the whole α-globin gene cluster.43,44 The additional α-globin genes have no phenotype in normal people but the small excess of α chains in heterozygous β thalassemia appears to tip the balance, crossing the critical threshold of α-globin excess with phenotypic consequences.
In a number of cases, the unusually severe heterozygous state is associated with a normal α-globin genotype. In such cases, the β thalassemia mutation itself leads to the synthesis of highly unstable, structurally abnormal β-globin chain variants.17 The hyperunstable β chain variants are rapidly destroyed in the erythroid precursors, giving rise to a functional deficiency of β chains and simulating a phenotype of β thalassemia. The non-functional β chain variants together with the unmatched α-globin chains aggravate the ineffective erythropoiesis causing a disease phenotype even when present in a single copy. Hence, the term ‘dominantly inherited β thalassemia’.
Finally, although rare, somatic mosaicisms of the β-globin gene in β thalassemia heterozygotes have been reported causing thalassemia intermedia. The individuals from three different families had moderately severe anemia despite being constitutionally heterozygous for a common β thalassemia mutation; they also had a normal α (αα/αα) genotype.31,32 Subsequent investigations revealed that these individuals had a somatic deletion including the β-globin complex, on the other chromosome 11p15, in a subpopulation (~80%) of erythroid cells giving rise to a somatic mosaic: about 20% of the erythroid cells were heterozygous with one normal copy of β-globin gene and the rest hemizigous (i.e. without any normal β-globin gene). The sum total of the β-globin product is thus about 80% less than the asymptomatic β thalassemia trait. These unusual cases once again illustrate that the severity of anemia in β thalassemia reflects the quantitative deficiency of β-globin production.
Tertiary modifiers affect complications of the disease; the severity of osteopenia and osteoporosis, iron loading and jaundice may be affected by polymorphisms of genes involved in the metabolic pathways concerned with these complications.45 Bone mass is a quantitative trait under strong genetic control involving multiple quantitative trait loci (QTLs) and the QTLs implicated include estrogen receptor gene, vitamin D receptor (VDR), collagen type a1 genes and transforming growth factor β1 (TGFβ1).46 The elevations of bilirubin and incidence of gallstones in thalassemia, as in other hemolytic anemias, are influenced by a polymorphic variant (seven (TA) repeats) in the promoter of the uridine diphosphate-glucuronosyltransferase 1A (UGT1A1) gene, also referred to as Gilbert’s syndrome.47,48 Iron loading in β thalassemia is also variable and results not just from blood transfusion but also from increased iron absorption. Variants in the HFE gene have a modulating effect on iron absorption,49 and as other genes in iron homeostasis become uncovered, it is likely that there will be genetic variants in these loci that influence the different degrees of iron loading in β thalassemia.50 For instance, recent genome wide association studies have identified variants in HFE, and the TMPRSS6 (transmembrane protease serine 6 gene that regulates hepcidin expression) associated with iron status, erythrocyte volume and concentration.51–53
Diagnosis
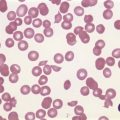
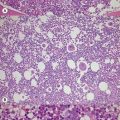
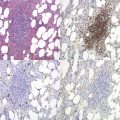
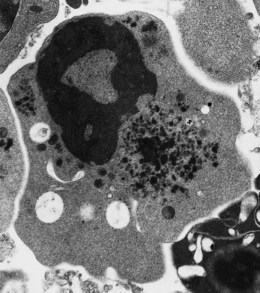
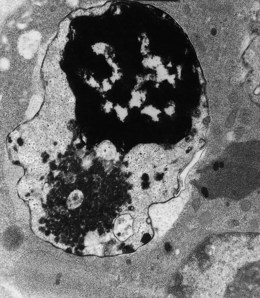
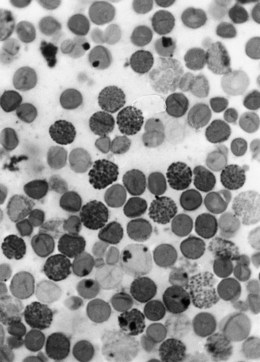
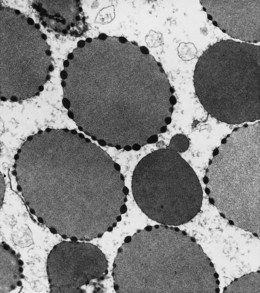
In β thalassemia major, untransfused hemoglobin levels are usually less than 5 g/dl but can be as low as 2–3 g/dl. Mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) are low, with a very wide red cell distribution width (RDW), marked anisopoikilocytosis, target cell formation, and basophilic stippling. Poorly hemoglobinized nucleated red cells are frequently found in the peripheral blood, and may reach very high levels after splenectomy. The reticulocyte count is elevated but less than expected for the degree of anemia in keeping with the ineffective erythropoiesis. A bone marrow aspirate is not essential to make the diagnosis, but if performed shows marked erythroid hyperplasia characterized by poorly hemoglobinized normoblasts and dyserythropoiesis. Supravital staining (e.g. methyl violet) shows ragged inclusions in many of the erythroid precursor cells; similar inclusions are found in the peripheral red blood cells after splenectomy. Immunoelectron microscopy confirms that these inclusions consist of precipitated α-globin chains (Figs 9.5 and 9.6). Increased iron deposition is also seen in the bone marrow; the majority of the iron granules are randomly distributed. Biochemical evidence of hemolysis such as elevated bilirubin, aspartate transaminase (AST) and lactate dehydrogenase (LDH) with a normal alanine transaminase (ALT) and progressive iron loading is observed. Other biochemical changes may include evidence of diabetes and endocrine dysfunction such as parathyroid or thyroid insufficiency.
Clinical features and management
The clinical phenotypes of β thalassemia range from very severe (major) to a completely silent carrier state, with a huge range of intermediate phenotypes (thalassemia intermedia) between the two ends of the spectrum.10
Adequately transfused children grow and develop normally until early puberty. Iron overload inevitably complicates regular blood transfusions, and progress of their disease then depends on whether they have received regular iron chelation. If not, they begin to show signs of progressive hepatic, cardiac and endocrine disturbances including diabetes, hypoparathyroidism and delayed or absent secondary sexual development. The endocrinopathies and cardiac disease are ascribed to the labile, more toxic forms of iron that appear in cells and plasma, referred to as non-transferrin bound iron (NTBI). Throughout their teenage life these children suffer from a variety of complications due to different endocrine deficiencies. Unless iron overload is controlled by regular chelation therapy, death results in the second or third decade, from acute or intractable congestive cardiac failure. Children who are adequately transfused and fully compliant with iron chelation therapy grow and develop normally, with few or no skeletal abnormalities, and achieve sexual maturity. Even within this group there is a high frequency of growth retardation and retarded sexual maturity, with variable complications relating to iron metabolism, bone disease, endocrine abnormalities and liver disease.54
Iron chelation is usually started after 1 year of monthly blood transfusions.55,56 Currently, three iron chelating agents are available for use: desferrioxamine (DFO), deferiprone and deferasirox. DFO has been in use since the 1970s and is known to be safe and effective; side-effects seem to occur when the drug is used in high doses or when iron stores are low. The main problem with DFO is that it has to be given parenterally, and due to its short half-life, the regimen involves continuous subcutaneous infusions given over 8 hours using a syringe driver or balloon pump. Deferiprone was the first oral iron chelator to be used; it was licensed in Europe in 1999. Arthropathy and agranulocytosis (which can be fatal) are potential serious side-effects, and patients taking deferiprone are recommended to have weekly blood counts. Deferiprone seems to be particularly effective in removing cardiac iron, a therapeutic effect ascribed to its low molecular weight. Deferiprone is increasingly used in combination with DFO.57 Deferasirox is a once-daily oral iron chelator, approved in the USA and Europe since the mid-2000s.58 Large clinical trials have demonstrated its efficacy in removing liver iron in thalassemia, with increasing evidence that it also removes cardiac iron. All patients on regular blood transfusion and iron chelation therapy should be monitored regularly with a yearly review for assessment of growth, including tests for endocrine and cardiac function, iron loading (serum ferritin, MRI for liver iron concentration) and adverse effects related to therapy.56
Bone marrow transplantation (BMT) is considered the treatment of choice if there is a HLA-identical sibling. BMT should be considered only if it is clear that the child is transfusion-dependent and should be considered early as the success of BMT is reduced as the child gets older, with increasing iron overload and iron-related organ damage.59
Intermittent blood transfusions in TI are often necessary due to falls in hemoglobin caused by fever, infection and specifically human parvovirus B19 infection. It can be difficult to decide who would benefit from a short period of regular blood transfusions and when to start them. In countries with a ready supply of safe blood there is an increasing tendency to start regular transfusions, even in children maintaining hemoglobins greater than 8 g/dl, to avoid the emerging complications of skeletal deformities, pulmonary hypertension and osteopenia. There is also some evidence that this improves the quality of life, particularly with emerging options for oral iron chelation. This is not possible in much of the world and management consists of reserving transfusion for severe symptomatic anemia. The initiation of iron chelation depends on the degree of iron overload (as indicated by liver iron concentration), but as with other aspects of the management of TI, there are no clear guidelines.60
Pharmacological treatment to increase HbF and total hemoglobin levels is potentially applicable to TI, in that relatively small increases in hemoglobin levels with a corresponding reduction in ineffective erythropoiesis could help a patient thrive who would otherwise require regular transfusions.1 Hydroxyurea is the most widely used drug in this context, with encouraging results in some patients. Butyrate and other short-chain fatty acid derivatives also promote HbF synthesis, and have been used with limited clinical success in TI. A number of newer drugs are being developed which may boost HbF to a greater extent, most notably the new generation of short-chain fatty acid derivatives (SCFADs) and immunomodulatory drugs such as pomalidomide and lenalidomide.
Preventive programs in most countries now combine education, pre-conceptual, antenatal and neonatal screening, heterozygote detection and genetic counseling for a comprehensive approach in the public health management of the disease.61 Many screening programs are centered at antenatal clinics and concentrate on identifying women who are thalassemia carriers in the first trimester of pregnancy. This is done by varying combinations of blood tests and identifying women at high risk of carrying thalassemia based on their ethnic origin; this latter approach is particularly effective in areas with a low prevalence of thalassemia in the native population, such as northern Europe. If a woman is found to be a carrier, screening is then offered to her partner, and if both are carriers, they are counseled about the risk of the fetus inheriting a severe form of thalassemia and offered prenatal diagnosis, usually from 11–12 weeks’ gestation by chorionic villus sampling or amniocentesis. Parents can then make an informed choice to terminate the pregnancy if the fetus is affected. However, chorionic villus sampling and amniocentesis are invasive with an increased risk of miscarriage of about 1%. This has led to research to develop non-invasive methods of prenatal diagnosis based on maternal blood sampling. Maternal blood contains small numbers of fetal cells and also cell-free fetal DNA, both of which could potentially be used to diagnose fetal thalassemia.
Some couples at risk of having an affected child find prenatal diagnosis and selective termination unacceptable. Pre-implantation genetic diagnosis (PGD) involves the use of in vitro fertilization techniques to generate 5–15 embryos; at the eight-cell stage, one embryonic cell can be removed and tested for thalassemia alleles; it is then possible to implant only embryos without thalassemia.62 PGD, however, is currently a difficult, stressful and expensive procedure, with only 10–20% of couples taking home a baby.
β Thalassemia in association with other hemoglobin variants
HbE/β thalassemia
This is the commonest severe form of thalassemia in Southeast Asia and parts of the Indian subcontinent. Recent demographic changes, however, have resulted in HbE/β thalassemia becoming a health problem in other parts of the world, such as North America.63
The βE allele is mildly thalassemic as the mutation at β codon 26 (GAC→AAG, Glu→Lys) that gives rise to HbE, also activates a cryptic splice site, and when inherited together with β° thalassemia, results in a marked deficiency of β chain production. The clinical and hematological changes of HbE /β thalassemia are variable, ranging from severe anemia and transfusion dependency to thalassemia intermedia.10,63 There is nearly always anemia (hemoglobin values range from 4–9 g/dl) and splenomegaly.
While a large part of this phenotypic variability can be explained by the severity of the β thalassemia alleles, this cannot be the only answer since an equally broad range of clinical phenotypes has been encountered in HbE/β° thalassemic individuals who all carry null β thalassemia mutations.1,64 It can be difficult to differentiate homozygous HbE (HbE/E) from HbE/β° thalassemia on Hb electrophoresis alone since only HbE and F are observed in both cases. Genetic studies would be definitive since both parents would be HbE carriers in HbE/E, but HbE trait in one parent and β thalassemia trait in the other, in HbE/β thalassemia. Clinically, homozygotes for HbE have mild anemia and are asymptomatic, while HbE/β° thalassemia can result in transfusion-dependent thalassemia major. Variable quantities of HbA are found in HbE/β+ thalassemia cases, and the condition is milder than HbE/β° thalassemia.
HbC/β thalassemia
This is restricted to West Africans, some North Africans and southern Mediterranean populations.1 HbC/β thalassemia is largely asymptomatic and characterized by a mild hemolytic anemia and splenomegaly. A blood smear shows numerous target cells and polychromasia due to the mildly elevated reticulocyte count. Hemoglobin electrophoresis shows a preponderance of HbC and variable quantities of HbA depending on whether it is a β+ or β° thalassemia allele. The diagnosis is confirmed by demonstrating HbC trait in one parent and β thalassemia in the other.
The δβ-, γδβ-thalassemias and HPFH syndromes
This group of β-like thalassemia disorders is characterized by a reduced or absent synthesis of β- and δ-globin chains and a variable compensatory increase in γ chain production.65 They are much less common than β thalassemia. The distinction between δβ thalassemias and the HPFH syndromes is subtle and originally made on what appeared to be clear-cut clinical and hematological grounds. However, with the elucidation of the molecular basis of these conditions, it became increasingly clear that this broad classification is rather arbitrary, and that there is considerable overlap in many of the parameters that were initially used to differentiate them. It also became clear that the subtle difference between the two subgroups relates to the relatively higher compensatory increase in HbF levels in HPFH compared to the δβ thalassemias.
Heterozygotes for δβ thalassemia have a red cell picture similar to β thalassemia, with hypochromic microcytic red cells, but normal levels of HbA2 (<3.0%). In addition, however, there is an increased level of HbF (5–15%) that was unevenly distributed (heterocellular) among the erythrocytes. Homozygotes for δβ thalassemia or compound heterozygotes with β thalassemias are not common but have been reported to have clinical phenotypes ranging from mild anemia to thalassemia major.66 In contrast, HPFH heterozygotes have essentially normal red cell indices, normal levels of HbA2 and higher levels of HbF (15–30%) with a homogeneous pancellular distribution of the HbF. HPFH homozygotes are clinically normal, their hemoglobin levels may be increased with mildly hyochromic microcytic red cells. Individuals with HPFH/β thalassemia may have a mild anemia but are clinically asymptomatic.
A non-deletion form of δβ thalassemia due to a point mutation inactivating the β gene in cis with a point mutation in the γ gene promoter up-regulating HbF production has also been described.67 δβ Thalassemia and HPFH are clearly defined by a significant increase in HbF levels in carriers; the molecular defects are inherited in a Mendelian fashion as alleles of the β-globin complex.
About 10–15% of the healthy adult population have modest increases of HbF (1–5%) in which the inheritance patterns are less clear-cut and traditionally referred to as heterocellular HPFH because the HbF is unevenly distributed among the erythrocytes. Heterocellular HPFH was previously known as Swiss-type HPFH, after the original report in which some members of a group of Swiss soldiers were found to have slight increases in HbF levels.68,69 It is now clear that heterocellular HPFH represents the high values of HbF and F cells at the upper tail of the common HbF trait distribution.
Although of no consequence to healthy adults, when co-inherited with β thalassemia or sickle cell disease (SCD), heterocellular HPFH leads to increases in HbF that have a major ameliorating effect on the severity of disease.34,70 Heterocellular HPFH is inherited as a complex trait; multiple genes contribute to the increase in HbF levels. Recent genome wide association studies (GWAS) have mapped three major quantitative trait loci (QTLs) – Xmn1-HBG2, HBS1L-MYB intergenic region (HMIP) on chromosome 6q and BCL11A on chromosome 2p – that account for 50% of the F cell variance in healthy Europeans.12,13,71 These loci have been shown to impact HbF levels and disease severity in patients from diverse ethnic groups with SCD and β thalassemia.37–40,72,73
εγδβ Thalassemia
These are rare conditions.10 Affected newborns may present with severe hemolysis and anemia, which is self-limited. In some cases, blood transfusions are necessary during the neonatal period. It is recognized in adults by the hematological phenotypes of β thalassemia trait with normal levels of HbA2 and HbF. Only heterozygotes have been identified; presumably the homozygous state would not survive early gestation. The molecular defects result from large deletions of the β-globin gene cluster which involve the β-LCR.65,74–76 The deletions fall into two categories: group I removes all, or a greater part of the complex including the β-globin gene and the β-LCR; group II removes extensive remote upstream regions including the β-LCR but leaving the β-globin gene itself intact, despite which, its expression is silenced because of the absence of the upstream β-LCR. There is no output from the globin genes of the affected cluster. The associated phenotypes of the two groups of deletions are similar.
α Thalassemias
α thalassemia can be regarded as α+ or α°, reflecting either a reduction or complete absence of α-globin synthesis from the affected chromosome.1,77 The geographical distribution of α thalassemia is very similar to that of β thalassemia; in some parts of the tropical and subtropical regions where it is prevalent, carrier frequency for the mild form (α+) reaches 90%. The more severe defect, α° thalassemia, is prevalent in the Mediterranean region and southeast Asia where carrier frequency can reach 10%. Although the α thalassemias are more common than β thalassemias, they pose less of a public health problem since the severe homozygous states cause death in utero and the milder forms that survive into adulthood do not cause a major disability.
Genetic basis of disease
Normal individuals have four α-globin genes arranged as linked pairs, α2 and α1, at the tip of each chromosome 16, the normal α genotype being written as αα/αα.1 α Thalassemia most commonly results from deletion of one (−α/) or both (−−/) α genes of the linked pair from chromosome 16, causing a reduction (α+) or absence (α°) of α-globin from the affected chromosome, respectively. The homozygous and heterozygous states for α° thalassemia are represented as −−/−− and αα/−−, respectively; while those for α+ thalassemia are −α/−α and αα/−α, respectively. As for the β-like genes, full expression of the α-like globin genes is critically dependent on the presence of the major upstream regulatory element (MCS-R2 or HS-40) which lies 40 kb upstream of the cluster. Although more than 50 deletions removing one (−α/) or both (−−/) genes have been characterized, the majority of α thalassemia is caused by six deletions (/−α3.7, /−α4.2, /−−SEA, /−−MED,/ − ( α)20.5, and /−−FIL).1
The common α° thalassemias are caused by complete or partial deletion of both the structural α-globin genes. These deletions vary in size and are geographically isolated, with two particularly common ones, one in southeast Asia (/−−SEA) and the other in the Mediterranean region ( /−−MED). Rarely, α° thalassemias arise from deletions of the upstream α-globin regulatory elements (MCS-R) with the downstream α-globin genes intact but completely inactivated. These deletions are highly variable in size (up to 150 kb have been reported); the smallest of these remove MCS-R1 (HS-48) and MCS-R2 (HS-40), demonstrating the importance of these elements in expression of the α-globin genes.78
The molecular basis of α+ thalassemia is more complicated; the commonest forms result from deletion of one of the linked pairs of globin genes (/−α3.7or /−α4.2).1 Less commonly, both the α-globin genes are intact, and α+ thalassemia results from point mutations (T) that inactivate one of the pair. These point mutations involve the critical sequences that control the various stages of gene expression as encountered in the β thalassemias. To date, 69 causes of non-deletion α thalassemia have been described; 46 of these occur in the dominant α2 gene (/αTα), 17 in the α1 gene (/ααT), and the others on a /−α chromosome (/−αT). In general, the non-deletional α thalassemia determinants have a more severe effect on α-globin output and hematological phenotype than simple deletions that remove one or the other α-globin gene. This may be explained by the majority of mutations affecting the dominant α2 gene, where expression predominates over the α1 gene. Another explanation is that, unlike deletional α thalassemia, there appears to be no compensatory increase in expression of the linked functional α gene when the other is inactivated by a point mutation.
Pathophysiology
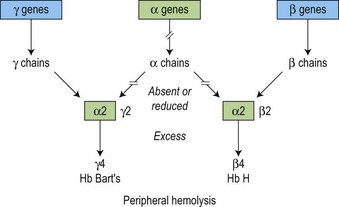
There is a fundamental difference in the pathophysiology of the α and β thalassemias (Fig. 9.7). Because γ4 and β4 tetramers are soluble, they do not precipitate to a significant degree in the bone marrow, i.e. erythropoiesis is more effective than in β thalassemia. However, these β4 tetramers do precipitate as the red cells age, forming inclusion bodies in the mature erythrocytes, resulting in peripheral hemolysis due to the red cell membrane damage and obstruction in the spleen (Figs 9.7 and 9.8). The degree of anemia and the amount of the tetramers (HbH and Bart’s) produced reflects the severity of the reduction in the output of the α-globin chain.
Genotype-phenotype correlation
HbH disease lies between the two ends of the clinical spectrum, the asymptomatic α thalassemia trait and Hb Bart’s hydrops fetalis. As in β thalassemia intermedia, HbH disease spans a wide range of clinical and hematological phenotypes,79,80 the diagnostic feature being the presence of HbH inclusions in the peripheral red blood cells (Figs 9.8 and 9.9). The most severe forms can be lethal late in gestation or in the perinatal period (HbH hydrops fetalis).1 The molecular basis of this order is equally heterogeneous, varying with the geographic distribution of the different α thalassemia variants.79,80 HbH disease most commonly results from the interaction of α° and deletional α+ thalassemia (−−/−α). Less often it can result from the interaction of α° thalassemia with non-deletional forms of α thalassemia (−−/αTα) or from homozygous non-deletional α thalassemia (αTα/αTα). A less severe form of HbH disease in southeast Asia commonly arises from homozygosity or compound heterozygosity for Hb Constant Spring (αCSα/α CSα) or (αCSα/−−). Very low levels of this elongated α-globin chain (5–8% of the total hemoglobin in homozygotes) are found; the defective αCS chain production is a consequence of the instability of the αCS mRNA.81
Diagnosis
HbH disease
Patients with HbH disease run an hemoglobin level of 7–10 g/dl with moderate reticulocytosis. Again, typical thalassemic changes are seen in the blood film. On incubation of the red cells with brilliant cresyl blue, numerous inclusion bodies are generated by precipitation of the HbH which are tetramers of β chain (β4), forming typical ‘golf balls’ (Fig. 9.8). Hemoglobin analysis shows 5–40% HbH, with the major component being HbA and a normal or reduced level of HbA2. Sometimes, there is also a small amount of Hb Bart’s.
Clinical features
The clinical disorders resulting from α thalassemia range from death in utero (Hb Bart’s hydrops syndrome) to a completely silent carrier state.1 Hemoglobin Bart’s hydrops, caused by homozygosity for α° thalassemia, occurs only in populations where α° thalassemia is common, notably in the Mediterranean and in Southeast Asia. Affected infants are usually stillborn with gross pallor, generalized edema and massive hepatosplenomegaly. The placenta is enlarged and friable and frequently causes obstetric difficulties. All these findings are caused by severe intrauterine anemia. There is no production of α chain, and hence neither fetal nor adult hemoglobin. The hemoglobin consists of approximately 80% Hb Bart’s (γ4 tetramers) and 20% Hb Portland (ζ2γ2). Presumably affected infants survive to term because they continue to produce embryonic hemoglobin. Apart from fetal death, there is a high incidence of toxemia of pregnancy and obstetric complications due to the large placenta.
α Thalassemia with mental retardation (ATR) syndromes
These are rare forms of α thalassemias found in association with a variety of developmental abnormalities, in particular with mental retardation, and hence they are often referred to as α thalassemia with mental retardation (ATR) syndromes.1 One group, ATR-16, results from extensive deletions and rearrangements of 1–2 Mb removing many genes including the α globin genes, from the tip of chromosome 16.1,82 The other group, ATR-X, describes a syndrome that includes α thalassemia and multiple developmental abnormalities, such as urogenital anomalies and mental retardation due to mutations in the ATRX gene, located on the X chromosome (Xq13.1–q21.1).
ATR-16 shows a remarkable variation in severity of α thalassemia, mental retardation and associated developmental abnormalities. The sub-telomeric deletion/rearrangement is de novo in all cases, and in many cases, neither parent is a carrier for α thalassemia, and the patient has a phenotype of severe α thalassemia trait (αα/−−). However, less commonly, one parent may be a carrier for mild α thalassemia (αα/−α) that has been transmitted to the child resulting in HbH disease (−−/−α) with mental retardation. Analysis of the length and characteristics of the rearrangements suggest that sub-telomeric deletions of up to ~900 kb appear to be associated with a normal phenotype. The region between 900 and 1700 kb from the 16p telomere contains 16 genes and is deleted in all families with the characteristic features of ATR-16. Deletions beyond 2000 kb result in a contiguous gene syndrome characterized by α thalassemia with tuberous sclerosis and polycystic kidney disease.1 However, the critical genes responsible for the mental retardation and other developmental abnormalities associated with ATR-16 have yet to be identified.
Acquired α thalassemia
Very rarely, α thalassemia can be acquired in patients with a variety of hematological disorders within the myelodysplastic syndromes (MDS).1 These individuals are predominantly elderly males of northern European origin. They have a normal complement of α globin genes (αα/αα) and in all cases where data is available, there is no evidence of pre-existing α thalassemia and thus the α thalassemia and abnormal erythropoeisis is presumably acquired as a clonal genetic abnormality of the MDS. The blood films of such patients show dimorphic features with populations of red blood cells containing HbH inclusions. The marked hypochromic microcytic anemia is associated with an almost absent α-globin chain synthesis. Acquired mutations in the ATR-X have been found83 and thought to have been acquired as passenger mutations as part of the pre-leukemic disorder.
Structural hemoglobin variants
More than 1000 structurally different hemoglobin variants have been described (http://globin.cse.psu.edu/globin/hbvar/), but only three, sickle hemoglobin (HbS), HbC and HbE, occur at a high frequency in different populations.1 Many of the hemoglobin variants are harmless and have been discovered in population surveys using electrophoretic analyses of human hemoglobin. Since only variants which alter the charge of the hemoglobin molecule are detectable in routine electrophoresis, this number is probably an underestimate. Diseases resulting from structural abnormalities of hemoglobin are shown in Table 9.2. In this section only those abnormal hemoglobins of clinical importance are described.
Table 9.2 Clinical disorders due to structural hemoglobin variants
| 1. | Sickle syndromes causing hemolysis and vaso-occlusion: |
| HbSS | |
| HbS and interaction of HbS with other Hb variants | |
| (Hbs S/C, S/O-Arab and S/D-Punjab) | |
| Compound heterozygosity of HbS with β thalassemia | |
| HbS/β thalassemia) | |
| 2. | Chronic hemolysis – unstable hemoglobin variants (congenital Heinz body anemia, CHBA), |
| e.g. Hb Köln, Hb Bristol | |
| 3. | Congenital polycythemia – high oxygen affinity Hb variants |
| 4. | Congenital cyanosis – low oxygen affinity Hb variants |
| M hemoglobins | |
| 5. | Hypochromic microcytic anemia (thalassemic hemoglobinopathy), |
| e.g. HbE – β structural variant | |
| Hb Constant Spring – α structural variant | |
| δβ fusion variants – e.g. Hb Lepore | |
| 6. | Drug induced hemolysis, e.g. Hb Zurich |
Sickle cell disease
Background
Sickle cell disease (SCD) was first described by James Herrick from Chicago in 1910 in a student from Grenada.84 Presence of peculiar elongated and sickle-shaped red blood cells in the peripheral blood films suggested the term sickle cell anemia. In 1949 Pauling et al demonstrated that this sickling phenomenon was related to an abnormal hemoglobin present in all patients with sickle cell anemia.85 Subsequently, in 1956 Ingram showed that the sickle hemoglobin differed from normal adult hemoglobin (HbA, α2β2) by the single substitution of glutamic acid to valine at position 6 in the β subunit.86
The sickling disorders occur predominantly in black African populations but they are also prevalent throughout the Mediterranean, Middle East and parts of India. Carriers for the βS gene are protected from Plasmodium falciparum, thus explaining the high gene frequencies in those malarious regions, although the mechanisms underlying such protection are still not clear. The βS gene in these diverse population groups is caused by the same molecular defect (β codon 6 GAG to GTG), found on four different β haplotypes in Africa, known as the Senegal, Benin, Central African Republic (or Bantu) and the Cameroon types. In addition, it is associated with a different β haplotype (Arab-India) in Saudi Arabian and Asian Indian sickle patients.1 The evidence suggests multiple independent origins of the βS mutation although gene conversion on regionally specific β haplotypes cannot be excluded.
Genetic basis and pathophysiology
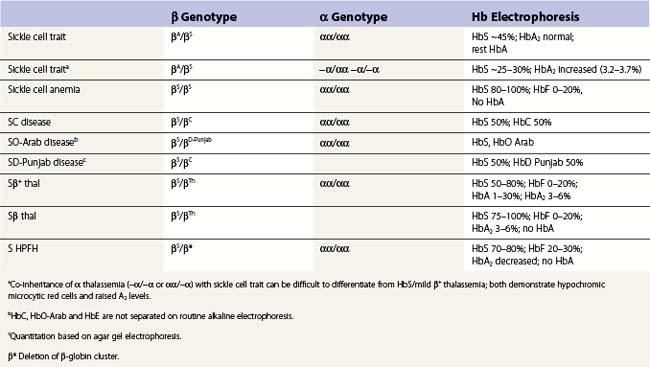
Apart from the homozygous state for the βS gene, the syndrome of sickle cell disease (SCD), can also arise from the compound heterozygous state for HbS and β thalassemias (HbS/β thalassemia) and other structural variants such as HbS C (HbSC disease) and D (HbSD) (Table 9.3).
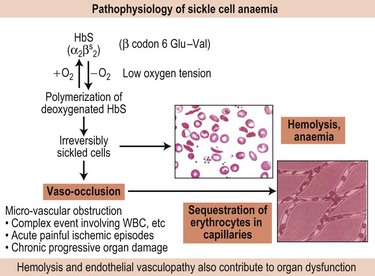
Fundamental to the pathophysiology of sickle cell disease is the polymerization of the sickle hemoglobin (HbS) which is dependent on several factors – concentration of the HbS itself, oxygen saturation, pH, temperature and other factors such as the 2,3-diphosphoglycerate concentration (2,3-DPG) (Fig. 9.10). Fully oxygenated HbS cannot enter the polymer phase whereas partially oxygenated or deoxygenated HbS can.87 Mixed hybrids of HbS with non-S hemoglobins, e.g. HbA (α2ββS) and HbC (α2βCβS), have a 0.5 probability of entering the polymer phase while those with HbF, i.e. (α2γβS) and HbA2 (α2δβS), cannot enter the polymer phase.
Despite the identical sickle mutation, SCD has long been appreciated to exhibit extraordinary phenotypic variability which reflects the genetic background of the individual and the co-inheritance of different modifying genes interacting with the different environmental backgrounds.88 Two case reports and a pilot twin study89 suggest that environmental factors may be of greater importance in determining clinical expression and complications of SCD. These reports showed that, despite identical β- and α-globin genotypes with similarities in growth, hematological and biochemical parameters, the identical twins had quite different acute pain rate and sickle-related complications.
Nonetheless, genetic factors that influence the primary event of HbS polymerization, such as the causative genotype (HbSS vs. HbSC, HbSβ thalassemia), co-inheritance of α thalassemia and genetic determinants for producing HbF are major predictors of SCD severity. HbF levels are a major predictor of survival and complications in SCD; the risk of early death is inversely associated with HbF levels.70 The beneficial effect of HbF is twofold: i) HbF inhibits HbS polymerization, and ii) its presence in the red blood cells reduces the concentration of intracellular HbS. The three recently identified HbF loci have also been shown to impact sickle phenotype.13,37,73 α Thalassemia which is present in about 30% of SCD patients is associated with less hemolysis, higher hematocrit and hemoglobin levels, lower mean corpuscular volume (MCV) of the red blood cells, and lower reticulocyte counts. The resulting increased hematocrit and blood viscosity may increase certain vaso-occlusive complications such as acute painful episodes, acute chest syndrome and avascular necrosis.
At the secondary level, it is likely that genetic variants controlling differences in vascular hemodynamics, endothelial adhesion, red cell membrane proteins and red cell hydration account for much of the individual variability in vaso-occlusive complications and hemolysis. Genetic studies based on candidate genes have associated several genes with some complications of SCD including stroke, leg ulcers, priapism and avascular necrosis.90 However, candidate genes were selected based on our understanding of the sickle pathophysiology. Genome wide association studies (GWAS) that permit a hypothesis-free and unbiased assessment of all genes should enlighten us on the key pathways and molecular mechanisms underlying the sickle pathophysiology. For such a complex disease, GWAS will require large numbers (thousands) of well characterized patients.
Diagnosis
Diagnosis is established by a combination of several tests for the detection and quantification of HbS plus the other Hbs for the elucidation of the different genetic interactions: hemoglobin electrophoresis on cellulose acetate (pH 8.6) and agar (pH 6.2) and positive sickling or solubility tests.91 In sickle cell anemia (HbSS), it is usual to see sickled cells, Howell–Jolly bodies and changes typical of hyposplenism. In HbSC disease target cells are prominent and HbC crystals may be present and in HbSβ thalassemia (or HbS with α thalassemia) hypochromic microcytic target cells are present.
Because the gene for HbS (and the structural hemoglobin variants, HbC and HbE) and the thalassemias (both α and β) occur together at high frequency in many populations, it is not uncommon for an individual to inherit genes for both types of condition, posing difficulties in diagnosis. Although these diagnostic difficulties can normally be resolved by family studies, often family members may not be accessible and one has to rely on a careful assessment of the hemoglobin electrophoresis and the red cell indices. In sickle cell trait (Table 9.3) uncomplicated by α or β thalassemia the level of HbS varies between 40% and 45% and is always less than 50%. This is because the βS chain is positively charged and is less able to compete with the negatively charged βA chains for the positively charged α-globin subunits.92,93 Carriers for α thalassemia and HbS have lower HbS levels, varying from ~25% (in −α/−α) to ~35% (in αα/−α). Electrophoretically there may be little difference between HbSS with high levels of HbF and HbS/β° thalassemia but the latter is accompanied by hypochromic microcytic red cells and an elevated HbA2 level. These features are also present in HbS/β+ thalassemia, but in this case, there is some HbA and the HbS is, of course, more than 50%. However, increased levels of HbA2 do not always reflect the co-existence of β thalassemia. In terms of electrostatic attraction for the α globin subunit, the δ globin subunit lies between that of βA and βS. Since the δ globin chain has a greater affinity for the α subunit compared to βS, co-inheritance of α thalassemia with HbS can mimic HbS/β thalassemia. In both scenarios, the HbS level is lower than expected, the red cells hypochromic microcytic and the level of HbA2 increased.
Clinical features
Sickle cell trait (carrier status) is a benign condition and generally does not cause any clinical disability. However, under certain extreme conditions, such as severe pneumonia, flying in unpressurized aircraft or rigorous physical exercise with ensuing dehydration, vaso-occlusive episodes can occur.94,95
Acute chest syndrome is characterized by acute dyspnea, pleuritic pain and fever, accompanied by a new pulmonary infiltrate on chest X-ray. It is often accompanied by a significant fall in the hemocrit which may reflect sequestration of the sickled cells in the pulmonary vessels. The distinction from pulmonary infection is often very difficult, particularly as infection and infarction usually co-exist. Acute chest syndrome is a frequent cause of admission but can be triggered by acute pain; it is the commonest cause of death in young patients.70 The brain is the major site of morbidity in children; acute vaso-occlusion in the CNS usually presents in childhood, either as fits, transient neurological symptoms resembling ischemia attacks, or with a fully developed stroke.96 Overt stroke affects about 6% of 10-year-olds with a peak between the ages of 2 and 5 years (incidence of 1.02 per 100 patient years), being rare in infants less than 1 year of age. Recurrent attacks are common; 70% of patients experience a recurrence within 3 years and many are left with permanent motor and intellectual disabilities. Long-term/lifetime blood transfusion is recommended for secondary prevention of stroke. Children with HbSS are at highest risk followed by HbS/β° thalassemia and HbSC disease. Priapism, defined as a sustained, painful and unwanted erection, is a well recognized complication in young men with SCD. It results from vaso-occlusion of the outflow vessels from the corpora cavernosa by sickled erythrocytes. This complication may present as multiple short-lived episodes (’stuttering’ priapism) which may progress to ‘severe prolonged’ priapism lasting several days and leading to permanent penile dysfunction.
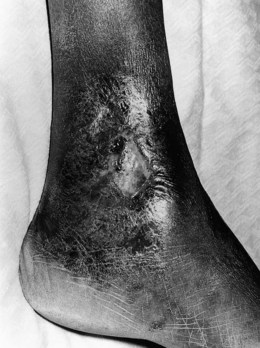
Repeated vaso-occlusive events and vasculopathy related to the chronic hemolysis ultimately result in end-organ damage and almost any organ can be affected. An observational study noted that by the fifth decade, ~50% of patients would have one or more of sickle-related end-organ damage.97 Avascular necrosis of the femoral head may lead to total disability, frequently requiring a total hip replacement. Virtually every patient with sickle cell anemia has some form of renal impairment. Sickling of the erythrocytes is enhanced in the hypertonic, hypoxic and acidotic environment of the renal medulla leading to progressive infarction of the medullary papillae. There is progressive inability to concentrate urine, polyuria, nocturia and enuresis, which is common in children. Eventually the glomerular damage causes end-stage renal failure, particularly in patients over 40 years of age. Due to the chronic hemolysis, pigment gallstones are very common and are seen in one third of SS patients by the age of 10 years. However, it is difficult to assess its clinical significance as not all patients develop clear-cut cholecystitis. Gilbert’s disease, which is present in about 30% of Africans, is a predisposing factor. Recurrent chronic leg ulceration is common and can be a major handicap (Fig. 9.11). The lesions normally occur just above the medial or lateral malleoli and seem to be more common in those patients with severe anemia. Proliferative retinopathy leading to progressive visual loss is an important ocular complication although this is more common in HbSC disease.
Management
In general, management requires a multidisciplinary team effort and consists of continuous general medical care, attention to good nutrition, immunization, avoidance of extremes of temperature and dehydration, and treatment of complications as they arise.98 In untreated patients, mortality is highest in the first year of life. In high risk populations, neonatal screening programs should be established to identify affected babies which should be referred to a specialist center and a comprehensive care plan initiated as soon as possible. These babies should be started on prophylactic penicillin at 3–4 months of age followed by polyvalent pneumococcal vaccine at the age of 6–12 months. There should be a systematic approach to education of parents (and later on, the child) on recognition of the different acute clinical events and signs of illness that play a critical role in successful management of children with SCD.
Most patients with SCD generally tolerate the relatively low Hb levels and blood transfusion is usually not required. Over the last decade, however, blood transfusion therapy is increasingly used in SCD with an ever expanding list of indications. Transfusion of normal blood provides benefit by correcting the low oxygen-carrying capacity as well as improving microvascular circulation by decreasing the population of sickled red cells. Regular blood transfusion also suppresses endogenous erythropoiesis and production of HbS. The choice of transfusion therapy – simple top-up, automated exchange (erythrocytapheresis), and partial exchange transfusion – depends on the needs of the patient. Except for severe anemia, exchange transfusion offers many advantages, and is increasingly used. Top-up transfusion is indicated when there is a sharp fall in the hemoglobin level due to bone marrow failure (aplastic crisis) or increased hemolysis (see Table 9.4 for indications for transfusion therapy in SCD).
Table 9.4 Indications for blood transfusion in sickle cell disease
| Acute/episodic | Long-term management | |
|---|---|---|
| •Anemia | ||
Ischemic stroke is one of the most devastating complications in young children with SCD. Early cerebrovascular disease can be detected by transcranial Doppler (TCD) scanning demonstrating increased blood flow velocity in the middle cerebral artery, and studies have shown that starting blood transfusions at this stage can prevent progressive vasculopathy and overt stroke (STOP 1 study).99 It is therefore recommended that children over 2 years of age should undergo TCD screening annually and those children at risk (abnormal blood flow velocities) started on prophylactic blood transfusion. A follow-up study (STOPI 2)100 provided evidence that discontinuing blood transfusion after reversion to normal TCD velocity led to new CNS complications. Thus, it is recommended that prophylactic blood transfusion for primary prevention of stroke is indefinite. Adolescents and adults should be screened for pulmonary hypertension by transthoracic echocardiogram. Regular screening for proteinuria and institution of ACE inhibitors in those with significant proteinuria (>1 g in 24 hours) may slow the progress of sickle nephropathy.
Anti-sickling therapy. Therapeutic approaches to reducing vaso-occlusion and hemolysis in SCD are constantly evolving. The major therapies to date have been designed to inhibit the primary event of HbS polymerization, including anti-sickling agents and induction of HbF synthesis, and rehydration of the red blood cell.1
Induction of HbF as a form of therapy was initially based on clinical and epidemiological observations which showed that even slight elevations of fetal Hb have an ameliorating effect on the clinical severity.70,101 An increase in intracellular HbF effectively reduces the concentration of HbS and mixed hybrids of HbS and HbF (α2γβS) do not form polymers. Pharmacological agents used include 5-azacytidine, a potent inhibitor of DNA methylation, arabinosylcytosine (Ara-C), hydroxyurea, erythropoietin and the butyrate analogues.102 Hydroxyurea (HU) is the agent of choice and the only agent currently approved for treatment of SCD by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMeA). Hydroxyurea has now been in use for patients with SCD for 25 years and has proven efficacy for reducing acute pain and acute chest syndrome.103 An observational follow-up study also suggested that HU therapy was associated with reduced mortality in SCD patients.104 However, only 60–80% of SCD patients respond to hydroxyurea.105 The clinical benefits of hydroxyurea are not solely dependent on HbF increases but likely to be associated with the increased nitric oxide (promoting vasodilation), the accompanying reduction in the levels of neutrophils, monocytes and reticulocytes, and improved red cellular hydration, all of which are mitigating factors of vaso-occlusion. Genetic factors may also play a role in the individual response to HU.106 Clinical indications for initiating HU include frequent acute pain and acute chest syndrome. However, potential expanding indications may include prevention of renal damage (indication significant proteinuria) and CNS damage (indication elevated TCD velocity). HU is relatively non-toxic, its myelosuppressive effects are reversible and limited evidence suggests that HU treatment in adults does not increase secondary malignancy. Despite the large body of evidence for its efficacy, HU remains under-prescribed in patients with SCD. Three reviews of the use of HU in SCD have recently been published.107–109
Bone marrow transplantation offers potential cure for sickle cell disease and may have a role in certain cases of sickle cell disease.110 Allogeneic, cord blood and chimeric marrow transplantation are theoretically attractive ways to cure SCD but until more is known about the natural history of the disease and the ability to predict the disease severity, bone marrow transplant as a form of curative treatment for all cases is not recommended.
Hemosiderosis in SCD is a growing problem, and inevitable with simple top-up blood transfusions. The pathophysiology of transfusional loading in SCD appears to be different to that in thalassemia major but a major factor may be related to the total duration of transfusion.1,111 Nonetheless, current recommendations are that patients should receive chelation therapy after twenty top-up transfusions or if liver iron concentration is more than 7 mg/g DW. (NIH publication, http://www.nhlbi.nih.gov/health/prof/blood/sickle/sc_mngt.pdf; Sickle Cell Society, UK, 2008, at http://www.sicklecellsociety.org/pdf/SCSPackageCareProp.pdf.)
Other sickling disorders
These include mainly the compound heterozygous states for HbS together with Hbs C, O-Arab, and D-Punjab as well as the inheritance of the βS gene with the different forms of thalassemia (see Table 9.3).91
Other hemoglobin variants
The other hemoglobin variants that are encountered commonly are Hbs C and E (see Table 9.5 and section on β thalassemias and sickle cell disease). HbC is the second most common variant among individuals of African ancestry. HbC is less soluble than HbA and tends to crystallize within the red cells leading to their reduced deformability. The important interactions of HbC are with HbS (to produce HbSC disease)1 and β thalassemia.
Table 9.5 Molecular abnormalities observed in thalassemic hemoglobinopathies
| Abnormality | Example |
|---|---|
| Unstable variants | Hb Suan Dok (α2109 Leu→Arg) |
| Hyperunstable variants | Hb Quong Sze (α2125 Leu→Pro) Hb Showa Yakushiji (β110 Leu→Pro) |
| Variants that cause abnormal splicing | HbE (β26 Leu→Lys) Hb Knossos (β27 Ala→Ser) |
| Variants with mutations in the termination codon | Hb Constant Spring (α2142 Term→Gln)* |
| Variants linked to thalassemic mutation | HbG-Philadelphia (/−αG-Phil) |
| δ/β fusion globin subunits | Hb Lepore |
* Hb Constant Spring is a particularly common form of non-deletion α thalassemia in Thailand.
After HbE/β thalassemia, other symptomatic forms of HbE-associated conditions include its interaction with the different genotypic forms of HbH disease.1,64 However, inclusions and HbH (β4) are present only in HbAE but not HbE/E or HbE/β° thalassemia; presumably the abnormal βE-subunits do not form tetramers. Interaction of HbE with HbS produces sickle cell disease (HbSE).
The diagnosis of HbE is based on detection or quantitation of the variant by electrophoretic or chromatographic separation of the hemoglobin in peripheral blood. Separation of hemoglobins using routine techniques, including HPLC columns, cannot differentiate HbE from HbA2. In uncomplicated heterozygous HbE, the variant forms ~35% of the total hemoglobin. As with βS, the βE globin variant is positively charged and is less able to compete with the negatively charged β-globin for the positively charged α subunits.93 Hence, in the presence of limiting amounts of α-globin when α thalassemia is co-inherited with heterozygous HbE, the proportion of HbE is reduced: ~10% (in −−/−α), 20–22% (in −−/αα or −α/−α) and 27–30% (in αα/−α).
Unstable hemoglobin disorders
Structural changes in the globin subunits can lead to instability of the hemoglobin molecule causing it to precipitate intracellularly, detectable by supravital staining as globular aggregates known as Heinz bodies. These inclusions reduce the life span of the erythrocytes and cause variable hemolysis, an integral part of the syndrome of congenital Heinz body hemolytic anemia (CHBA).91 The true incidence of CHBA is not known; according to the Globin Gene Server (http://globin.cse.psu.edu), more than 25% of the 1000 hemoglobin variants were designated ‘unstable’ and associated with varying degrees of reticulocytosis and hemolytic anemia. Many of the unstable variants are also associated with a thalassemic phenotype.17,26 A basis for the phenotypic variation of these disorders lies in the variable instability of the abnormal globin subunits. Those at the severe end of the spectrum are not able to form functional tetramers and precipitate in the red cell precursors causing a functional deficiency and a thalassemic phenotype, while the less unstable globin variants are able to form a hemoglobin tetramer that survives the different stages of red cell maturation only to precipitate in the mature red cell causing hemolysis in the peripheral circulation. Hemolysis is highly variable in intensity and often precipitated by infections and exposure to chemical oxidants.
The majority of CHBA follows an autosomal dominant pattern of inheritance; affected individuals are almost exclusively heterozygotes.91 Unstable hemoglobins are not common, generally limited to single families, and only the proband is affected, suggesting that the mutation has arisen by a de novo mutation. There are two exceptions: Hb Köln (β98 Val→Met) has been reported in various ethnic groups and often observed as a de novo mutation.20 Hb Hasharon (α47 Asp→His) is found in Italian families and Ashkenazi Jews, and causes hemolytic anemia in newborns.20
Hemoglobin variants with abnormal oxygen binding
The molecular basis underlying the abnormal oxygen binding of these structural hemoglobin variants lies in the perturbance of the transitional state between hemoglobin conformations – R or ‘relaxed’ in which it has a high affinity for oxygen, and low affinity for effectors such as 2,3-DPG, and T or ‘tense’ when it has a relatively low affinity for oxygen and high affinity for the allosteric affectors.112
Mutations for high oxygen affinity variants have been described only in the heterozygous form with the exception of one α chain variant, Hb Tarrent (α126 Asp→Asn) in which, on the basis of the Hb electrophoresis quantitation, two of the four α-globin genes were affected.113 Since most of the affected individuals are likely to have four α-globin genes (αα/αα), α-globin variants cause a less clinically significant condition compared to those of the β subunit.
The increased oxygen affinity causes a functional anemia and tissue hypoxia, which in turn leads to increased output of erythropoietin and an elevated red cell mass. High affinity hemoglobin variants follow an autosomal dominant inheritance pattern; all affected individuals are heterozygotes.91 A positive family history is useful but occasionally the variants arise from de novo mutations. Most affected individuals are completely healthy and identified through a routine blood count which shows increased hemoglobin or hematocrit. The individual may also have a ruddy complexion. There is no splenomegaly and, apart from an increased hemoglobin concentration and hematocrit, the white cell and platelet counts are within normal limits. Diagnosis is made by excluding other causes for erythrocytosis, and by demonstrating a left-shifted oxygen dissociation curve with a reduced P50 value, and a normal 2,3-diphosphoglycerate (2,3-DPG) value. Diagnosis is confirmed by hemoglobin analysis, using either mass spectrometry or DNA sequence analysis. A normal hemoglobin electrophoresis or HPLC does not exclude a diagnosis of high affinity hemoglobin.
Diagnosis of a low oxygen affinity hemoglobin is made by excluding other causes of cyanosis, especially cardiopulmonary, and differentiation of the cyanosis from that of methemoglobinemia.1 A simple test is to expose the individual’s blood to pure oxygen when it will turn bright red in cases of low oxygen affinity hemoglobin and cardiopulmonary disease. In contrast, in the blood of patients with methemoglobinemia, M hemoglobins and sulfhemoglobinemia retain their abnormal color despite exposure to pure oxygen. Most of the low oxygen affinity hemoglobin variants are unstable, and determination of the whole blood P50 is not always reliable. Hemoglobin electrophoresis, mass spectrometry of hemoglobin and DNA sequence analysis are useful tools for diagnosis.
Hemoglobins M
A striking clinical feature of the M hemoglobins is the dusky blue appearance of the skin and mucous membranes.91 In the vast majority of cases, the cyanosis is due to an excess of deoxyhemoglobin in the blood from a congenital cardiopulmonary defect. Rarely, it can be caused by a low oxygen affinity hemoglobin variant. Next in the differential diagnosis are the methemoglobinemias or sulfhemoglobinemias, usually caused by a deficiency of the enzyme cytochrome b5 reductase.114 The enzyme enables red cell hemoglobin to be maintained in a reduced form and deficiency of the enzyme results in oxidized hemoglobin and a cyanotic appearance. A much rarer cause of methemoglobinemia is the presence of one of the M hemoglobins. The M hemoglobins are inherited in an autosomal dominant pattern. Affected individuals present with cyanosis that varies from brownish to slate grey but are otherwise asymptomatic. If cyanosis is present from birth, it is normally due to an α chain variant, whereas β chain HbM produces cyanosis after the first few months of life when adult hemoglobin (α2β2) synthesis becomes established.
Thalassemic hemoglobinopathies
This group of hemoglobin disorders includes structural hemoglobin mutants associated with a thalassemia phenotype, that is, hypochromia and microcytosis with or without anemia.10 The phenotype ranges from the mild forms of heterozygous thalassemia to that resembling thalassemia intermedia. In some cases, chronic severe hemolytic anemia with splenomegaly is a feature. The molecular abnormalities are remarkably heterogeneous and includes single amino acid mutations causing hemoglobin variants such as Hbs E and Knossos with abnormal splicing, amino acid deletions, elongated or truncated globin chains and fusion globins such as Hb Lepore (Table 9.5).
Some of the α-globin mutants are invariably linked to a tandem α-globin gene deletion and hence the concomitant deficiency, for example HbG-Philadelphia (/-αG-Phil). Thalassemic hemoglobinopathies also include an increasing number of α and β structural variants in which the amino acid substitutions confer such instability that they are readily degraded in the bone marrow erythroid precursors resulting in an ineffective erythropoiesis and a thalassemic phenotype.17
1 Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. 2nd ed. Cambridge, UK: Cambridge University Press; 2009:826.
2 Higgs DR, Wood WG. Long-range regulation of alpha globin gene expression during erythropoiesis. Curr Opin Hematol. 2008;15(3):176-183.
3 Cantor AB, Orkin SH. Hematopoietic development: a balancing act. Curr Opin Genet Dev. 2001;11(5):513-519.
4 Blobel GA, Weiss MJ. Nuclear factors that regulate erythropoeisis. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge, UK: Cambridge University Press; 2009:62-85.
5 Tolhuis B, Palstra RJ, Splinter E, et al. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell. 2002;10(6):1453-1465.
6 Stamatoyannopoulos G. Control of globin gene expression during development and erythroid differentiation. Exp Hematol. 2005;33(3):259-271.
7 Wijgerde M, Grosveld F, Fraser P. Transcription complex stability and chromatin dynamics in vivo. Nature. 1995;377:209-213.
8 Sankaran VG, Xu J, Orkin SH. Advances in the understanding of haemoglobin switching. Br J Haematol. 2010;149(2):181-194.
9 Feng WC, Southwood CM, Bieker JJ. Analyses of β-thalassemia mutant DNA interactions with erythroid Krüppel-like factor (EKLF), an erythroid cell-specific transcription factor. Journal of. Biological Chemistry. 1994;269:1493-1500.
10 Weatherall DJ, Clegg JB. The Thalassaemia Syndromes, 4th ed. Oxford: Blackwell Science; 2001.
11 Liu P, Keller JR, Ortiz M, et al. Bcl11a is essential for normal lymphoid development. Nat Immunol. 2003;4(6):525-532.
12 Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39(10):1197-1199.
13 Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci USA. 2008;105(5):1620-1625.
14 Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322(5909):1839-1842.
15 Sankaran VG, Xu J, Ragoczy T, et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009;460(7259):1093-1097.
16 Weatherall DJ, Clegg JB. Thalassemia – a global public health problem. Nature Medicine. 1996;2(8):847-849.
17 Thein SL. Structural variants with a beta-thalassemia phenotype. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge, UK: Cambridge University Press; 2001:342-355.
18 Cooley TB, Lee P. A series of cases of splenomegaly in children with anemia and peculiar bone changes. Trans Am Pediatr Soc. 1925;37:29.
19 Modell B, Darlison M, Birgens H, et al. Epidemiology of haemoglobin disorders in Europe: an overview. Scand J Clin Lab Invest. 2007;67(1):39-69.
20 . A Database of Human Hemoglobin Variants and Thalassemias. Online. Available from http://globin.cse.psu.edu/globin/hbvar/
21 Maragoudaki E, Kanavakis E, Traeger-Synodinos J, et al. Molecular, haematological and clinical studies of the −101 C→T substitution in the β-globin gene promoter in 25 β-thalassaemia intermedia patients and 45 heterozygotes. British Journal of Haematology. 1999;107(4):699-706.
22 Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Human Molecular Genetics. 1999;8:1893-1900.
23 Hentze MW, Kulozik AE. A perfect message: RNA surveillance and nonsense-mediated decay. Cell. 1999;96:307-310.
24 Huang S-C, Benz EJJ. Posttranscriptional factors influencing the hemoglobin content of the red cells. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge, UK: Cambridge University Press; 2001:252-276.
25 Isken O, Maquat LE. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21(15):1833-1856.
26 Thein SL. Is it dominantly inherited beta thalassaemia or just a beta-chain variant that is highly unstable? Br J Haematol. 1999;107(1):12-21.
27 Thein SL, Old JM, Wainscoat JS, et al. Population and genetic studies suggest a single origin for the Indian deletion beta zero thalassaemia. British Journal of Haematology. 1984;57:271-278.
28 Kimberland ML, Divoky V, Prchal J, et al. Full-length human L1 insertions retain the capacity for high frequency retrotransposition in cultured cells. Hum Mol Genet. 1999;8(8):1557-1560.
29 Viprakasit V, Gibbons RJ, Broughton BC, et al. Mutations in the general transcription factor TFIIH result in beta-thalassaemia in individuals with trichothiodystrophy. Hum Mol Genet. 2001;10(24):2797-2802.
30 Yu C, Niakan KK, Matsushita M, et al. X-linked thrombocytopenia with thalassemia due to a mutation affecting DNA-binding contribution of the N-finger of transcription factor GATA-1 [abstract]. Blood. 2000;96(11):495a.
31 Badens C, Mattei MG, Imbert AM, et al. A novel mechanism for thalassaemia intermedia. The Lancet. 2002;359(9301):132-133.
32 Galanello R, Perseu L, Perra C, et al. Somatic deletion of the normal beta-globin gene leading to thalassaemia intermedia in heterozygous beta-thalassaemic patients. Br J Haematol. 2004;127(5):604-606.
33 Chang JG, Tsai WC, Chong IW, et al. Beta-thalassemia major evolution from beta-thalassemia minor is associated with paternal uniparental isodisomy of chromosome 11p15. Haematologica. 2008;93(6):913-916.
34 Ho PJ, Hall GW, Luo LY, et al. Beta thalassemia intermedia: is it possible to consistently predict phenotype from genotype? British Journal of Haematology. 1998;100(1):70-78.
35 Camaschella C, Mazza U, Roetto A, et al. Genetic interactions in thalassemia intermedia: analysis of beta-mutations, alpha-genotype, gamma-promoters, and beta-LCR hypersensitive sites 2 and 4 in Italian patients. American Journal of Hematology. 1995;48:82-87.
36 Rund D, Oron-Karni V, Filon D, et al. Genetic analysis of β-thalassaemia intermedia in Israel: diversity of mechanisms and unpredictability of phenotype. Am J Hematol. 1997;54(1):16-22.
37 Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci USA. 2008;105(33):11869-11874.
38 Sedgewick AE, Timofeev N, Sebastiani P, et al. BCL11A is a major HbF quantitative trait locus in three different populations with beta-hemoglobinopathies. Blood Cells Mol Dis. 2008;41(3):255-258.
39 Galanello R, Sanna S, Perseu L, et al. Amelioration of Sardinian beta-zero thalassemia by genetic modifiers. Blood. 2009;114(18):3935-3937.
40 Nuinoon M, Makarasara W, Mushiroda T, et al. A genome-wide association identified the common genetic variants influence disease severity in beta(0)-thalassemia/hemoglobin E. Hum Genet. 2010;127(3):303-314.
41 Traeger-Synodinos J, Kanavakis E, Vrettou C, et al. The triplicated alpha-globin gene locus in beta-thalassaemia heterozygotes: clinical, haematological, biosynthetic and molecular studies. Br J Haematol. 1996;95(3):467-471.
42 Camaschella C, Kattamis AC, Petroni D, et al. Different hematological phenotypes caused by the interaction of triplicated alpha-globin genes and heterozygous beta-thalassemia. American Journal of Hematology. 1997;55(2):83-88.
43 Harteveld CL, Refaldi C, Cassinerio E, et al. Segmental duplications involving the alpha-globin gene cluster are causing beta-thalassemia intermedia phenotypes in beta-thalassemia heterozygous patients. Blood Cells Mol Dis. 2008;40(3):312-316.
44 Sollaino MC, Paglietti ME, Perseu L, et al. Association of alpha globin gene quadruplication and heterozygous beta thalassemia in patients with thalassemia intermedia. Haematologica. 2009;94(10):1445-1448.
45 Thein SL. Genetic modifiers of beta-thalassemia. Haematologica. 2005;90(5):649-660.
46 Wonke B. Bone disease in beta-thalassaemia major. British Journal of Haematology. 1998;103:897-901.
47 Galanello R, Perseu L, Melis MA, et al. Hyperbilirubinaemia in heterozygous beta-thalassaemia is related to co-inherited Gilbert’s syndrome. British Journal of Haematology. 1997;99(2):433-436.
48 Galanello R, Cipollina MD, Dessì C, et al. Co-inherited Gilbert’s syndrome: a factor determining hyperbilirubinemia in homozygous beta-thalassemia. Haematologica. 1999;84(2):103-105.
49 Piperno A, Mariani R, Arosio C, et al. Haemochromatosis in patients with beta-thalassaemia trait. Br J Haematol. 2000;111(3):908-914.
50 Andrews NC. Molecular control of iron metabolism. Best Pract Res Clin Haematol. 2005;18(2):159-169.
51 Andrews NC. Genes determining blood cell traits. Nat Genet. 2009;41(11):1161-1162.
52 Ganesh SK, Zakai NA, van Rooij FJ, et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat Genet. 2009;41(11):1191-1198.
53 Soranzo N, Spector TD, Mangino M, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet. 2009;41(11):1182-1190.
54 Borgna-Pignatti C, Rugolotto S, De Stefano P, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89(10):1187-1193.
55 Angelucci E, Barosi G, Camaschella C, et al. Italian Society of Hematology practice guidelines for the management of iron overload in thalassemia major and related disorders. Haematologica. 2008;93(5):741-752.
56 Cappellini M-D, Cohen A, Eleftheriuo A, et al, editors. Guidelines for the Clinical Management of Thalassaemia. 2nd ed. Nicosia, Cyprus: Thalassaemia International Federation; 2008:199.
57 Borgna-Pignatti C, Cappellini MD, De Stefano P, et al. Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood. 2006;107(9):3733-3737.
58 Vichinsky E. Clinical application of deferasirox: practical patient management. Am J Hematol. 2008;83(5):398-402.
59 Gaziev J, Sodani P, Polchi P, et al. Bone marrow transplantation in adults with thalassemia: treatment and long-term follow-up. Ann N Y Acad Sci. 2005;1054:196-205.
60 Taher A, Isma’eel H, Cappellini MD. Thalassemia intermedia: revisited. Blood Cells Mol Dis. 2006;37(1):12-20.
61 Cao A, Galanello R, Rosatelli MC. Prenatal diagnosis and screening of the haemoglobinopathies. In: Rodgers GP, editor. Baillière’s Clinical Haematology. London: Baillière Tindall; 1998:215-238.
62 Braude P, Pickering S, Flinter F, et al. Preimplantation genetic diagnosis. Nat Rev Genet. 2002;3(12):941-953.
63 Vichinsky E. Hemoglobin E syndromes. Hematology Am Soc Hematol Educ Program. 2007:79-83.
64 Fucharoen S. Hemoglobin E disorders. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge, UK: Cambridge University Press; 2001:1139-1154.
65 Thein SL, Wood WG. The molecular basis of β thalassemia, δβ thalassemia, and hereditary persistence of fetal hemoglobin. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge, UK: Cambridge University Press; 2009:323-356.
66 Rochette J, Craig JE, Thein SL. Fetal hemoglobin levels in adults. Blood Reviews. 1994;8:213-224.
67 Ottolenghi S, Giglioni B, Pulazzini A, et al. Sardinian δβ-thalassemia: A further example of a C to T substitution at position -196 of the Aγ globin gene promoter. Blood. 1987;69(4):1058-1061.
68 Marti HR. Normale und anormale menschliche Haemoglobine. Berlin: Springer Verlag; 1963.
69 Marti HR, Butler R. Hämoglobin F- und Hämoglobin A2-Vermehrung bei der Schweizer Bevölkerung. Acta Haematologia (Basel). 1961;26:65-74.
70 Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease: life expectancy and risk factors for early death. New England. Journal of Medicine. 1994;330(23):1639-1644.
71 Thein SL, Menzel S, Peng X, et al. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci USA. 2007;104(27):11346-11351.
72 So CC, Song YQ, Tsang ST, et al. The HBS1L-MYB intergenic region on chromosome 6q23 is a quantitative trait locus controlling fetal haemoglobin level in carriers of beta-thalassaemia. J Med Genet. 2008;45(11):745-751.
73 Creary LE, Ulug P, Menzel S, et al. Genetic variation on chromosome 6 influences F cell levels in healthy individuals of African descent and HbF levels in sickle cell patients. PLoS ONE. 2009;4(1):e4218.
74 Game L, Bergounioux J, Close JP, et al. A novel deletion causing (εγδβ) thalassemia in a Chilean family. Br J Haematol. 2003;123(1):154-159.
75 Rooks H, Bergounioux J, Game L, et al. Heterogeneity of the epsilon gamma delta beta-thalassaemias: characterisation of three novel English deletions. Br J Haematol. 2005;128(5):722-729.
76 Harteveld CL, Voskamp A, Phylipsen M, et al. Nine unknown rearrangements in 16p13.3 and 11p15.4 causing α- and β-thalassaemia characterised by high resolution multiplex ligation-dependent probe amplification. J Med Genet.. 2005 Dec;42(12):922-931.
77 Higgs DR, Weatherall DJ. The alpha thalassaemias. Cell Mol Life Sci. 2009;66(7):1154-1162.
78 Viprakasit V, Harteveld CL, Ayyub H, et al. A novel deletion causing alpha thalassemia clarifies the importance of the major human alpha globin regulatory element. Blood. 2006;107(9):3811-3812.
79 Kanavakis E, Papassotiriou I, Karagiorga M, et al. Phenotypic and molecular diversity of haemoglobin H disease: a Greek experience. Br J Haematol. 2000;111(3):915-923.
80 Fucharoen S, Viprakasit V. Hb H disease: clinical course and disease modifiers. Hematology Am Soc Hematol Educ Program. 2009:26-34.
81 Liebhaber SA, Schrier SL. Pathophysiology of α thalassaemia. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge, UK: Cambridge University Press; 2001:391-404.
82 Wilkie AO, Buckle VJ, Harris PC, et al. Clinical features and molecular analysis of the alpha-thalassemia/mental retardation syndromes. I. Cases due to deletions involving chromosome band 16p13.3. American Journal of Human Genetics. 1990;46:1112-1126.
83 Gibbons RJ, Pellagatti A, Garrick D, et al. Identification of acquired somatic mutations in the gene encoding chromatin-remodeling factor ATRX in the alpha-thalassemia myelodysplasia syndrome (ATMDS). Nat Genet. 2003;34(4):446-449.
84 Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Arch Intern Med. 1910;6:517-521.
85 Pauling L, Itano HA, Singer SJ, et al. Sickle cell anemia: a molecular disease. Science. 1949;110:543-548.
86 Ingram VM. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature. 1956;178:792-794.
87 Noguchi C, Schechter AN, Rodgers GP. Sickle cell disease pathophysiology. In: Higgs DR, Weatherall DJ, editors. Baillière’s Clinical Haematology: The Haemoglobinopathies. London: Baillière Tindall; 1993:57-91.
88 Steinberg MH, Thein SL. Genetic modulation of sickle cell disease, in Renaissance of sickle cell disease research in the genome era, B. Pace, Editor. London, UK: Imperial College Press; 2007. p. 193–206
89 Weatherall MW, Higgs DR, Weiss H, et al. Phenotype/genotype relationships in sickle cell disease: a pilot twin study. Clin Lab Haematol. 2005;27(6):384-390.
90 Thein SL. Genetic modifiers of the beta-haemoglobinopathies. Br J Haematol. 2008;141(3):357-366.
91 Bunn HF, Forget BG. Hemoglobin: molecular, genetic and clinical aspects. Philadelphia, PA: W.B. Saunders Company; 1986.
92 Bunn HF, McDonald MJ. Electrostatic interactions in the assembly of human hemoglobin. Nature. 1983;306:498-500.
93 Bunn HF. Subunit assembly of hemoglobin: an important determinant of hematologic phenotype. Blood. 1987;69:1-6.
94 Embury SH, Hebbel RP, Mohandas N, et al, editors. Sickle Cell Disease: Basic Principles and Clinical Practice. New York, NY: Raven Press, 1994.
95 Kark JA, Ward FT. Exercise and hemoglobin S. Semin Hematol. 1994;31(3):181-225.
96 Ohene-Frempong K. Stroke in sickle cell disease: demographic, clinical, and therapeutic considerations. Seminars in Hematology. 1991;28(3):213-219.
97 Powars DR, Chan LS, Hiti A, et al. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore). 2005;84(6):363-376.
98 Steinberg MH. Management of sickle cell disease. New England. Journal of Medicine. 1999;340(13):1021-1030.
99 Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial doppler ultrasonography. New England Journal of Medicine. 1998;339(1):5-11.
100 Adams RJ, Brambilla D. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med. 2005;353(26):2769-2778.
101 Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease: Rates and risk factor. New England. Journal of Medicine. 1991;325(1):11-16.
102 Bunn HF. Induction of fetal hemoglobin in sickle cell disease. Blood. 1999;93:1787-1789.
103 Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. New England. Journal of Medicine. 1995;332(20):1317-1322.
104 Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. Jama. 2003;289(13):1645-1651.
105 Steinberg MH, Lu ZH, Barton FB, et al. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Multicenter Study of Hydroxyurea. Blood. 1997;89(3):1078-1088.
106 Ma Q, Wyszynski DF, Farrell JJ, et al. Fetal hemoglobin in sickle cell anemia: genetic determinants of response to hydroxyurea. Pharmacogenomics J. 2007;7(6):386-394.
107 Brawley OW, Cornelius LJ, Edwards LR, et al. National Institutes of Health Consensus Development Conference statement: hydroxyurea treatment for sickle cell disease. Ann Intern Med. 2008;148(12):932-938.
108 Lanzkron S, Strouse JJ, Wilson R, et al. Systematic review: hydroxyurea for the treatment of adults with sickle cell disease. Ann Intern Med. 2008;148(12):939-955.
109 Strouse JJ, Lanzkron S, Beach MC, et al. Hydroxyurea for sickle cell disease: a systematic review for efficacy and toxicity in children. Pediatrics. 2008;122(6):1332-1342.
110 Vermylen C. Bone marrow transplantation in sickle cell anemia. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge, UK: Cambridge University Press; 2001:1073-1083.
111 Vichinsky E, Butensky E, Fung E, et al. Comparison of organ dysfunction in transfused patients with SCD or beta thalassemia. Am J Hematol. 2005;80(1):70-74.
112 Perutz MF. Molecular anatomy and physiology of hemoglobin. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge, UK: Cambridge University Press; 2001:174-196.
113 Ibarra B, Vaca G, Cantú JM, et al. Heterozygosity and homozygosity for the high oxygen affinity hemoglobin Tarrant or alpha 126 (H9) Asp replaced by Asn in two Mexican families. Hemoglobin. 1981;5(4):337-348.
114 Percy MJ, Lappin TR. Recessive congenital methaemoglobinaemia: cytochrome b(5) reductase deficiency. Br J Haematol. 2008;141(3):298-308.