Chapter 44 Abnormalities of Cone and Rod Function
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
Congenital color vision deficits as well as retinitis pigmentosa and its related disorders are covered in separate chapters (respectively, Chapter 10, Color vision and night vision, and Chapter 40, Retinitis pigmentosa and related disorders).
Disorders of the cone system
Achromatopsia
Congenital achromatopsia is a rare disorder, with an incidence of roughly 1 in 30 000.1 Individuals with this disorder have poor vision from birth and complain of poor color discrimination and photosensitivity, which relates to their visual acuity decreasing in bright light rather than actual discomfort. Patients with complete achromatopsia, also known as rod monochromatism, are generally considered to lack cones and have vision worse than 20/200. On the other hand, patients with incomplete achromatopsia, also known as atypical achromatopsia, may have slightly better visual acuity in the range of 20/80–20/200. Complete achromats have no color vision whereas incomplete achromats may have some residual color vision. Interestingly, because their color vision loss is congenital, even complete achromats may be able to identify colors. They generally perceive colors as lighter or darker shades of gray and may have learned that red is a darker shade of gray and yellow is a lighter shade of gray and so forth. Consequently, specific color vision tests (discussed below and in Chapter 10,) are designed to uncover this characteristic. Pendular nystagmus may be present, which can improve with age. Family history can be useful as achromatopsia displays an autosomal recessive inheritance pattern.
Diagnosis
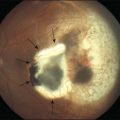
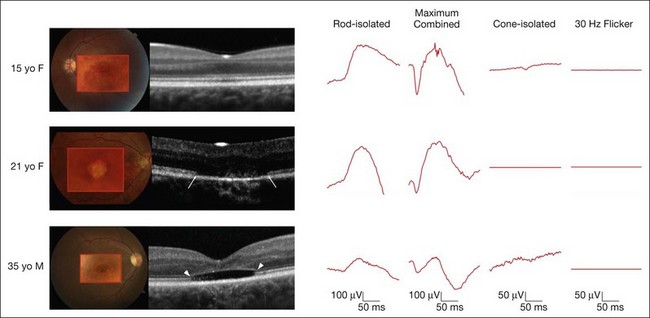
On clinical examination, patients may have a normal fundus, or have subtle granularity or atrophy of the macula. The nerve may be normal or show some temporal pallor. Electrophysiologial testing is crucial to making the diagnosis in achromatopsia. Photopic and 30 Hz-flicker electroretinograms will show nearly or completely nonrecordable cone responses in the face of normal or near-normal rod responses (Fig. 44.1). Multifocal electroretinograms will be severely diminished. Full-field electroretinography (ERG) is critical to diagnosis as it helps differentiate achromatopsia from other entities that share a normal fundus exam and associated symptoms. For instance, an infant with Leber congenital amaurosis will also have nystagmus, extremely poor vision, and photosensitivity but will have extinguished photopic and scotopic ERGs. A blue cone monochromat will have poor vision, but will have recordable cone responses when stimulated with blue light on a yellow background. A patient with optic neuropathy would have optic nerve pallor with otherwise unremarkable fundus findings as well as decreased vision and color perception, but would show normal photopic and scotopic ERGs.
Other ancillary tests are also helpful in the diagnosis. Congenital achromats may be able to identify several of the pseudochromatic (Ishihara) color plates because they may have learned to distinguish colors as different shades of gray. Farnsworth D-15 testing may reveal a scotopic axis between the deutan and tritan axes. The Sloan achromatopsia test uses an achromat’s correlation of different shades of gray to various colors in order to distinguish them from normal individuals.2 Dark-adaptation curves will show a lack of a cone–rod break, reflecting the lack of cone function (Fig. 44.2). Visual field testing may reveal a small central scotoma but the peripheral visual fields should be either mildly constricted or normal, and, most importantly, they remain stable over time. Progression on serial visual field testing should raise suspicion for other disorders such as a cone–rod dystrophy or retinitis pigmentosa with a cone–rod pattern.
The ERG changes precede the less specific and sometimes more subtle findings seen on optical coherence tomography (OCT) (Fig. 44.1). A recent report calculated the frequency of these findings in a large series of 77 eyes of achromatopsia patients.3 Among the most obvious and common is disruption of the inner/outer-segment (IS–OS) junction occurring in 70% of these eyes. A hyporeflective, optically empty cavity may be seen in the cone layer of the foveola (60%), preferentially weighted to the nasal side of the foveal center, where there is higher density of cone photoreceptors. Also present in the majority of patients is foveal hypoplasia (83%). Disruption of the RPE and outer retinal atrophy can be seen in a minority of patients (18%). Although achromatopsia has traditionally been thought to be stationary, the imaging studies suggest that this view should be reconsidered.4 Progressive cone death may occur in achromatopsia patients with age as suggested by comparing the patient’s OCT to age-matched controls as well as a preferential age-dependence of features such as the hyporeflective zone, thinning of the outer nuclear layer, and RPE atrophy.3,5 Given the overlap in genetic and clinical findings, it is also plausible that some patients diagnosed with complete achromatopsia were incomplete achromats who later progressed.
Although the technique is not readily available in most clinics, adaptive optics scanning laser ophthalmoscopic imaging of the macular photoreceptor mosaic in a patient with complete achromatopsia has helped shed light on the pathophysiology.6 This case has revealed that the macular area has significant gaps within the photoreceptor mosaic, with a total absence of photoreceptors bearing the characteristics of normal healthy cones. These remaining photoreceptors appeared to have morphologic characteristics typical of rods.
Clinical molecular genetic testing for the four known genes responsible for the majority of achromatopsia cases is available from multiple laboratories (http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/clinical_disease_id/211350?db=genetests; accessed February 2012). A brief discussion of our molecular understanding of achromatopsia is available online.
Molecular basis of achromatopsia
Most cases of congenital achromatopsia are associated with four genes, which are primarily inherited in an autosomal recessive fashion. All mutations affect proteins involved in cone phototransduction. Two major mutations affect proteins that code for the alpha- and beta-subunit of cone cyclic-GMP gated (CNG) cation channels. CNGB3 codes for the beta-subunit and is responsible for approximately 45% of complete achromatopsia. It was originally identified in the Pingelapese people in the western Pacific nation of Micronesia.7 The second most-common mutation (25%) is in the CNGA3 gene, which codes for the alpha-subunit.8,9 The third gene that is associated with complete achromatopsia (<2%) is GNAT2, which codes for the alpha-subunit of cone transducin. Transducin is involved in the signal transduction cascade, and acts to close the cGMP channels.10,11 Finally, most recently, mutations in PDE6C have also been identified as a cause of achromatopsia (<2%).12 This gene, which has also been implicated in retinitis pigmentosa and congenital stationary night blindness, codes for the catalytic subunit of cone photoreceptor phosphodiesterase.
The vast majority of incomplete achromatopsia has been linked to missense mutations in CNGA3, which are inherited in an autosomal recessive fashion. At one time, it was felt that this was because mutations in CNGB3 and GNAT2 were primarily those leading to nonfunctional truncated products and thus only yielding the complete achromatopsia phenotype. However, the discovery of a frameshift mutation in GNAT1 that results in a truncated protein associated with an incomplete loss of color vision argues against this.13 As only a few of the mutations identified to date in CNGA3 are exclusive to incomplete achromatopsia while most mutations can also be associated with complete achromatopsia, additional epigenetic modifiers are being sought that may attenuate the color vision and visual acuity defects in incomplete compared to complete achromatopsia.
Treatment
There is currently no treatment for the underlying defects in achromatopsia. Photophobia can be reduced with tinted lenses, particularly orange or red lenses since rod photoreceptors are less sensitive to orange and red lights. Red-tinted soft contact lenses that transmit between 400 and 480 nm can reduce the stigma of having to wear dark glasses even indoors.14 Low vision aids may help to enhance visual acuity. Great strides have been made in research whereby adenoviral-mediated gene transfer has been used to correct the defect in all three genetic forms of human achromatopsia in corresponding animal models.15–17 As these gene-based therapies eventually move to human trials, identification of mutations in patients with achromatopsia will become crucial.
Cone monochromatism and blue cone monochromatism
Cone monochromatism is a congenital disorder in which two of the three cone systems (long- [L], middle- [M], and short-wavelength [S]) are absent or nearly absent. Monochromatism involving preservation of either the red or the green cone system is extremely rare, with an incidence estimated at 1 in 100 million.18 These patients have a normal visual acuity, no nystagmus and a normal ERG, residual red and green sensitive pigments by fundus densitometry, and spectral sensitivity curves similar to normal patients.19–21 Given its rarity, the mechanism remains poorly understood and is mentioned here mainly for completeness.
Diagnosis
Fundus examination may be normal, or may reveal progressive pigment irregularities and macular atrophy on ophthalmoscopy and autofluorescence.22–24 By history, symptoms, and visual acuity, these individuals resemble achromatopsia and thus there are several tests that may help the clinician distinguish between the two. Because they have functional S cones, these patients will have rudimentary dichromatic color discrimination and they can be distinguished from achromats based on the presence of residual tritan discrimination on color testing. One can also use Berson plates that have been specifically developed for this purpose.25–27 Although the photopic and 30 Hz cone flicker will be absent under normal testing conditions, an S-cone signal can be recorded with a blue light-flash on a yellow background.28 Scotopic and bright-flash ERGs can be normal or attenuated.22,28,29 The disorder has been reported to be either stable or progressive, and it is unclear whether this variability is due to the specific mutation, epigenetic influences, and/or environmental factors. Clinical molecular genetic testing is currently available for some of the mutations involved with blue cone monochromatism (see online for a discussion of molecular mechanisms).
Molecular basis of blue cone monochromatism
The genetic basis of blue cone monochromatism can be explained in light of the high degree of variability in the expression of L and M opsins. Humans have one L opsin gene and one or more M opsin genes, which sit in a tandem array on the X chromosome. The S opsin gene resides separately, on chromosome 7.30,31 To date, the primary mechanisms by which blue cone monochromatism occurs involve either a one-step or two-step mechanism.30,32,33 Approximately 40% of patients will have a one-step deletion that affects the locus control region, an upstream conserved sequence which regulates transcription such that only one opsin gene is expressed per cone photoreceptor.23,24,32 The other 60% have a two-step mutation process. The first step involves a nonhomologous recombination event between the L and M opsin genes, which reduces the number of genes in the opsin array on the X chromosome to one. A second mutation then occurs in the remaining opsin gene, rendering it ineffective.34 Not all cases are explained by mutations in the structure of the opsin array and it is likely that additional mechanisms will continue to be identified.32,33
Progressive cone dystrophies
These are inherited retinal dystrophies that usually present within the first three decades of life, although there are rare reports of patients presenting after the fifth decade.36–38 Decreased visual acuity and central scotomas, some degree of color vision loss, and photophobia are the usual symptoms. The visual defects become worse over time. Some are purely cone dystrophies in which rod function remains normal. In these individuals, night blindness is not a major complaint, nor is peripheral field loss. However, there are many patients who, sooner or later, will have diminished rod function and will manifest the accompanying symptoms at that time. Cone–rod dystrophies are primarily nonsyndromic, but can also be associated with syndromes including Bardet–Biedl and spinocerebellar ataxia, so attention must be paid to a review of systems and the medical history. All mechanisms of inheritance are possible with cone–rod dystrophies, and a careful collection of family history will assist with narrowing down the myriad of mutations responsible for this phenotype, as well as with genetic counseling.
Diagnosis
OCT may show thinning of the outer retinal layers, limited mostly to the central retina.39 Fundus autofluorescence can show central or parafoveal areas of increased or decreased autofluorescence with variable involvement of the periphery. However, the OCT and autofluorescence changes are nonspecific and may be similar in a number of other retinal degenerations as well. It is therefore important to remember that it is the electrophysiological evaluation coupled with these imaging studies which will help make the definitive diagnosis.40
Although not readily available in most clinics, adaptive optics imaging has been used to investigate eyes with cone dystrophy. With this technique the photoreceptor mosaic shows increased cone spacing in cone dystrophy compared to normal subjects or retinitis pigmentosa patients.41
The diversity of mutations responsible for cone and cone–rod dystrophies currently precludes widespread screening for mutations. However, this may change as more efficient screening mechanisms such as microarrays become more readily available.42 A brief discussion of the molecular mechanisms of cone–rod dystrophies is given online.
Molecular basis of cone–rod dystrophies
The progressive cone–rod dystrophies are extremely genetically heterogeneous and may be associated with autosomal dominant, autosomal recessive, and X-linked inheritance patterns. At the time of publication, RETNET (http://www.sph.uth.tmc.edu/Retnet/) listed 23 genes with mutations that have been associated with a cone–rod dystrophy. For some of these genes, known mutations predominantly result in cone–rod dystrophy phenotypes.43 The principal gene in this category is CRX, a homeobox gene controlling rod and cone photoreceptor development, which may be associated with up to 10% of autosomal dominant cone–rod dystrophy. Mutations in CRX can also result in a retinitis pigmentosa or Leber congenital amaurosis phenotype, but as of yet the disease phenotype cannot be accurately predicted based on the mutation.44 Furthermore, some CRX mutations have been associated with extensive phenotypic heterogeneity within the same family.45 This suggests that there may be modifiers of CRX gene function that remain to be elucidated to help explain the wide range of clinical and genetic heterogeneity in this disorder.
Cone–rod dystrophy phenotype may also be seen with specific mutations in genes that are typically associated with other retinal diseases. For example, ABCA4 is most commonly associated with Stargardt disease, but is also responsible for 30–60% of cases of autosomal recessive cone–rod dystrophies.46 PRPH2 and RPGR are usually associated with autosomal dominant and X-linked retinitis pigmentosa, respectively, but can also cause cone–rod phenotypes of the same inheritance pattern.47,48 Many of the genes associated with Leber congenital amaurosis, including GUCY2D, can also cause a cone–rod dystrophy phenotype.49 Since less than half of cone–rod dystrophy cases are associated with known genetic mutations, much remains to be uncovered about the molecular mechanisms of this disease.
Congenital stationary night blindness
Congenital stationary night blindness (CSNB) is generally associated with nonprogressive defects in scotopic vision and/or dark adaptation with otherwise normal visual function. The initial description of the Nougaret family by Belgian ophthalmologist Cunier,50 which was later expanded by Nettleship,51 ultimately encompassed nine generations and 2121 patients, and established that this was indeed a nonprogressive “stationary” night blindness. In fact, this classic description still bears the name Nougaret and represents one of the largest pedigrees in ophthalmology and one of the first documentations of a dominantly inherited disorder.52
Since these initial reports, CSNB is now recognized to have many variants, which are associated with various inheritance patterns (autosomal dominant, recessive, or X-linked). Although most are truly stationary, some mutations lead to progressive visual loss. Myopia is linked to some forms, but not all. Finally, while the original family had a normal fundus, CSNB variants with fundus abnormalities are well characterized. We now know that the heterogeneity of CSNB is secondary to the variety of mutations that cause this condition. Clinical testing of some of the CSNB genes is currently available (for a list of genes and laboratories offering testing, see the GeneReviews website http://www.ncbi.nlm.nih.gov/sites/GeneTests/?db=GeneTests; accessed February 2012). At the time of publication, a new gene array is being developed in order to facilitate rapid and economical screening of all known mutations for CSNB.53 As emerging therapies become available, there is a continued need for clinicians to recognize the clinical characteristics to determine whether their patients may benefit from emerging treatments.54
Clinical suspicion, careful clinical examination and testing, in conjunction with appropriate genetic testing will ultimately help clinicians make the diagnosis of the particular variant of CSNB. A molecular classification that parallels and supplements the conventional clinical classification was elegantly laid out by Dryja in his Jackson Memorial Lecture from 2000.55
CSNB with normal fundi
CSNB patients with normal fundus examination may have normal visual acuity and may not complain of night blindness. Only some forms (X-linked variants) are associated with myopia and generally have subnormal vision (~20/50). On electrophysiologic testing, patients with CSNB and normal fundi generally have severely abnormal rod-mediated ERGs, and some forms have modestly reduced cone amplitudes. Even when reduced in amplitude, cone responses are expected to have a normal implicit time in these patients. A delay in the rod or cone peak implicit time with relatively high amplitudes is more likely a harbinger of progressive retinal degeneration.56 Other ancillary tests are of less value in these patients, who have normal visual fields. Color vision as measured by conventional psychophysical testing is also typically normal.
Diagnosis
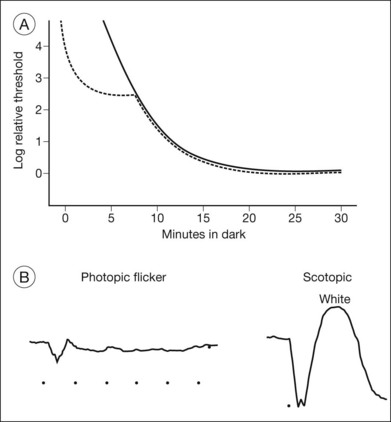
In a CSNB patient with a normal fundus, the ERG and dark-adaptation curve are very helpful in characterizing the disorder and determining the specific variant. These patients may broadly be divided into two groups depending on the presence or absence of an a-wave on ERG (Fig. 44.3). Riggs type (also known as type I) patients lack a scotopic ERG, lack both an a- and b-wave on maximum bright-field stimulation ERG, and lack a rod–cone break on their dark-adaptation curve. Without a scotopic ERG and a rod–cone break on the dark-adaptation curve, the molecular defect is predictably located at the rod photoreceptor level (Fig. 44.4).
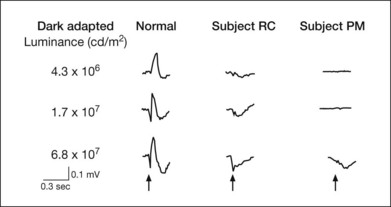
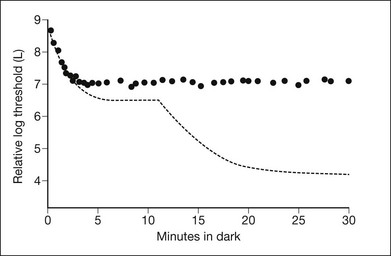
Fig. 44.4 CSNB, normal fundus, showing dark-adaptation curve of subject PM in Fig. 44.3. There is no evidence of a rod–cone break.
Schubert–Bornschein (also known as type II) patients possess an a-wave on maximum bright-field stimulation ERG but no b-wave, hence exhibiting a negative waveform. They are further classified into those who possess a scotopic ERG (incomplete form) and those who do not (complete form). In the incomplete form there is a rod–cone break on the dark-adaptation curve whereas in the complete form there is not.57 Because there is an intact a-wave (originating in the photoreceptors) but diminished b-wave (originating in the inner retina) the defect is expected to originate not in the photoreceptors but within the inner retina. Our molecular understanding of CSNB correlates with these clinical findings. More detail is available online.
Molecular basis of CSNB
For type II (Schubert–Bornschein) patients, electrophysiologic and psychophysical testing imply that the complete and incomplete forms affect both rod and cone pathways and predict that the defects may be located within the inner retinal circuitry, downstream of the photoreceptors. Not surprisingly, the mutations identified to date affect the formation and function of retinal synapses. The incomplete form has been linked to mutations in CACNA1F, a gene that encodes calcium-channels. These are believed to disrupt signaling of cones to the OFF bipolar cells or rods to the ON bipolar cells, since the calcium channels are concentrated mostly in the outer nuclear layers.58,59,60 The complete form has been linked to nyctalopin (NYX), which is an adhesion molecule involved in formation of synapses in the ON pathway of the retina.61 CACNA1F and NYX are both located on the X chromosome, causing X-linked inheritance. More recently, there have been reports of autosomal recessive forms of the Schubert–Bornschein type. These have been associated with mutations in CABP4, a calcium-binding protein located in the synaptic terminals.62 For the complete form, mutations identified include GRM6, which is a metabotropic glutamate receptor on bipolar dendrites, and TRPM1, which are transient receptor potential channels in ON bipolar cells.63–68
CSNB with abnormal fundi
CSNB with abnormal fundi includes two entities: Oguchi disease and fundus albipunctatus.
Oguchi disease
Oguchi disease refers to cases of CSNB in which there is a particular fundus finding known as the Mizuo–Nakamura phenomenon.69 In this variant, the retina appears normal following prolonged dark adaptation, but on exposure to light the retina displays a golden sheen with an unusually dark macula70 (Fig. 44.5).
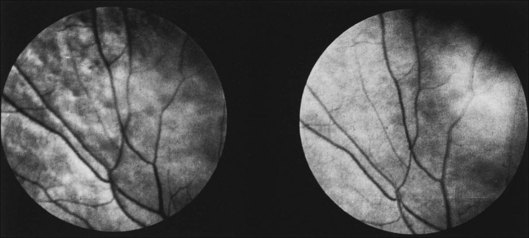
Diagnosis
Visual acuity and color vision are typically normal in this disorder. On clinical testing, in addition to the abnormal fundus appearance, patients have a dark-adaptation curve with a cone component (Fig. 44.6) but no rod–cone break, and exhibit gradual recovery of full rod sensitivity after prolonged dark adaptation of 1–2 hours. There is no scotopic ERG under normal testing conditions.71 A single-flash ERG following prolonged dark adaptation will be normal, but then subsequent flashes will not generate a response until the patient undergoes another prolonged period of dark adaptation.72
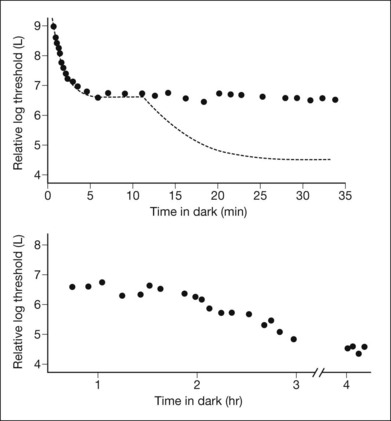
The mechanism underlying the Mizuo–Nakamura phenomenon and fundus sheen seen in Oguchi disease remains a subject of speculation. Histopathological study suggests the presence of an abnormal layer between photoreceptor outer segments and the retinal pigment epithelium (RPE).73 Recent OCT studies showed that the outer segments appeared shortened in areas with the golden sheen reflex seen in light and suggested that the sheen is related to accumulation of photoactivated rhodopsin in shortened rod outer segments.74 Other authors have shown that the IS–OS junction was better visualized on OCT following prolonged dark adaptation (and disappearance of the golden sheen), suggesting that reversible changes in the rod photoreceptors related to this mutation may explain the golden sheen reflex.75 More detail on the molecular basis is available online.
Molecular basis of Oguchi disease
Mutations in Oguchi disease have involved the rhodopsin kinase (GRK1) or arrestin (SAG).76,77 To date, all identified mutations are autosomal recessive requiring both copies to be affected in order to manifest a phenotype. Both rhodopsin kinase and arrestin deactivate photoactivated rhodopsin. Mutations in this gene are either less efficient or unable to inactivate rhodospin, desensitizing the rod photoreceptor. Thus, the rod photoreceptor supplies of chromophore are replenished by photoisomerized chromophore that is replaced during rhodopsin regeneration from the RPE, which takes 4–7 hours. This explains the kinetics of the dark-adaptation curve.78
Fundus albipunctatus
Fundus albipunctatus describes a subgroup of CSNB in which white or yellow dots can be seen scattered through the fundus (Fig. 44.7). Individuals may complain of night blindness early in childhood without progression, though most patients remain asymptomatic until the characteristic flecks are detected incidentally on routine fundoscopy.
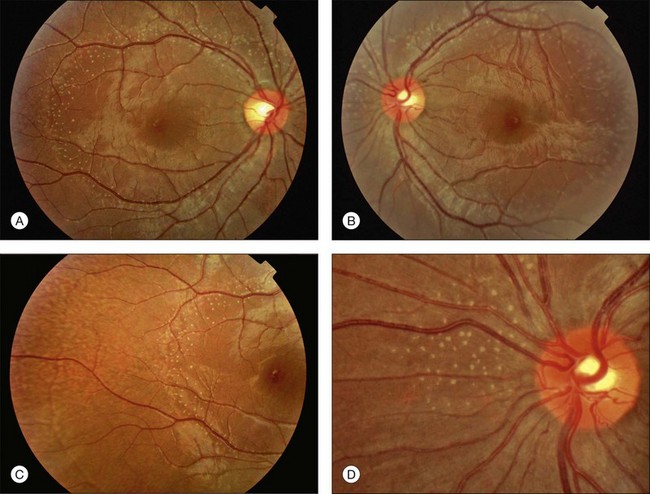
Diagnosis
As with other forms of CSNB, visual acuity and color vision are typically normal in this disorder. A scotopic ERG can be recorded but only after unusually long dark adaptation, whereas the cone ERG is usually normal. The dark-adaptation curve of fundus albipunctatus features prolongation of both rod and cone sensitivities (Fig. 44.8). Consistent with this, rod and cone visual pigment regeneration is significantly slower in patients with fundus albipunctatus (Fig. 44.9).79 Of note, there is also a form of fundus albipunctatus associated with cone dystrophy, in which there is progressive decrease in visual acuity and color vision with bull’s-eye maculopathy and significantly reduced cone ERGs.80
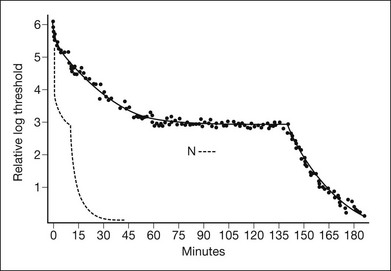
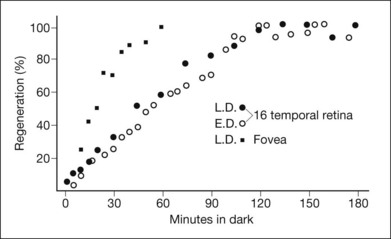
Optical coherence tomography and autofluorescence imaging have recently been used to investigate the flecks in fundus albipunctatus with and without cone dystrophy.81,82 In both reports, OCT demonstrated homogeneous dome-shaped hyperreflective deposits originating from the inner RPE that correlated with the white fundus spots and were noted to project into the outer retina, disrupting the IS–OS junction, the external limiting membrane, and the outer nuclear layer. Autofluorescence revealed areas of hyperautofluorescence corresponding to the elevated lesions on OCT. It is hypothesized that these lesions represent accumulations of retinoids secondary to the disruption of production of 11-cis retinal. The patient with cone dystrophy in conjunction with fundus albipunctatus also had a bull’s-eye lesion with corresponding loss of the IS–OS junction on OCT and abnormal circular hypoautofluoresence. More detail on the molecular basis is available online.
Molecular basis of fundus albipunctatus
Molecular studies have demonstrated that mutations in fundus albipunctatus largely affect the gene for 11-cis retinol dehydrogenase (RDH), which converts 11-cis-retinol to 11-cis retinal.83,84 This enzyme is located in the retinal pigment epithelium and is involved in production of 11-cis retinal that is then transported to photoreceptors for incorporation into the photochromophore. Recently, there has also been a report of fundus albipunctatus secondary to a mutation in RPE65, which is involved in the conversion of all-trans retinol into 11-cis retinol, a step upstream of the one catalyzed by 11-cis retinol dehydrogenase.85 Not only does the molecular basis explain the kinetics of the dark-adaptation curve and the involvement of both the rods and cones, but because the enzymes are located in the retinal pigment epithelium, this may explain the imaging findings and how the flecks of fundus albipunctatus (presumed to be retinoids based on their hyperautofluorescence) appear to arise in the RPE.81,86 Since these mutations affect regeneration of 11-cis retinal in the RPE, thus affecting both rods and cones, it is not surprising that there is an element of cone dysfunction in some forms of fundus albipunctatus. It is interesting to note that the cone system may seem more affected than the rod system in some cases of fundus albipunctatus. In these patients there is often reduced visual acuity and bull’s-eye changes within the macula. One study found that 38% of patients with fundus albipunctatus and mutations in RDH5 had extensive, progressive cone dysfunction on ERG testing and, to a lesser degree, decreased rod dysfunction.87 It is still unclear why the cones are potentially more functionally affected than rods in these patients.
Treatment for CSNB
There is currently no treatment available for most forms of CSNB. However, because the defect in fundus albipunctatus affects regeneration of 11-cis retinal, supplementation has been explored as a potential avenue for therapy. A recent nonrandomized study in which 9-cis retinal was given to such patients showed significant improvement in Humphrey visual field mean deviation, rod dark-adaptation recovery rates, and rod ERG.54 It is believed that the 9-cis retinal is transported from the liver to the retina, where it combines with opsin to generate isorhodopsin, a visual pigment with light absorption peak very close to rhodopsin. The 9-cis retinal may also reduce the activity of the retinoid cycle and decrease mislocation of opsin, all of which may potentially stabilize photoreceptors.88
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
1 Sharpe LT, Stockman A, Jagle H, et al. Opsin genes, cone photopigments, and colour blindness. In: Gegenfurtner KS, Sharpe LT. Color vision: From genes to perception. Cambridge: Cambridge University Press; 1999:3–52.
2 O’Connor PS, Tredici TJ, Ivan DJ, et al. Achromatopsia. Clinical diagnosis and treatment. J Clin Neuroophthalmol. 1982;2(4):219–226.
3 Thiadens AA, Somervuo V, van den Born LI, et al. Progressive loss of cones in achromatopsia: an imaging study using spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2010;51(11):5952–5957.
4 Thiadens AA, Slingerland NW, Roosing S, et al. Genetic etiology and clinical consequences of complete and incomplete achromatopsia. Ophthalmology. 2009;116(10):1984–1989. e1981
5 Thomas MG, Kumar A, Kohl S, et al. High-resolution in vivo imaging in achromatopsia. Ophthalmology. 2011;118(5):882–887.
6 Carroll J, Choi SS, Williams DR. In vivo imaging of the photoreceptor mosaic of a rod monochromat. Vision Res. 2008;48(26):2564–2568.
7 Sundin OH, Yang JM, Li Y, et al. Genetic basis of total colourblindness among the Pingelapese islanders. Nat Genet. 2000;25(3):289–293.
8 Kohl S, Marx T, Giddings I, et al. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet. 1998;19(3):257–259.
9 Wissinger B, Gamer D, Jagle H, et al. CNGA3 mutations in hereditary cone photoreceptor disorders. Am J Hum Genet. 2001;69(4):722–737.
10 Aligianis IA, Forshew T, Johnson S, et al. Mapping of a novel locus for achromatopsia (ACHM4) to 1p and identification of a germline mutation in the alpha subunit of cone transducin (GNAT2). J Med Genet. 2002;39(9):656–660.
11 Kohl S, Baumann B, Rosenberg T, et al. Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am J Hum Genet. 2002;71(2):422–425.
12 Chang B, Grau T, Dangel S, et al. A homologous genetic basis of the murine cpfl1 mutant and human achromatopsia linked to mutations in the PDE6C gene. Proc Natl Acad Sci U S A. 2009;106(46):19581–19586.
13 Michaelides M, Aligianis IA, Holder GE, et al. Cone dystrophy phenotype associated with a frameshift mutation (M280fsX291) in the alpha-subunit of cone specific transducin (GNAT2). Br J Ophthalmol. 2003;87(11):1317–1320.
14 Park WL, Sunness JS. Red contact lenses for alleviation of photophobia in patients with cone disorders. Am J Ophthalmol. 2004;137(4):774–775.
15 Alexander JJ, Umino Y, Everhart D, et al. Restoration of cone vision in a mouse model of achromatopsia. Nat Med. 2007;13(6):685–687.
16 Michalakis S, Muhlfriedel R, Tanimoto N, et al. Restoration of cone vision in the CNGA3-/- mouse model of congenital complete lack of cone photoreceptor function. Mol Ther. Dec 2010;18(12):2057–2063.
17 Komaromy AM, Alexander JJ, Rowlan JS, et al. Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet. Jul 1 2010;19(13):2581–2593.
18 Pitt FHG. Monochromatism. Nature. 1944;154:466–468.
19 Weale RA. Cone-monochromatism. J Physiol. 1953;121(3):548–569.
20 Weale RA. Photosensitive reactions in foveae of normal and cone-monochromatic observers. Opt Acta. 1959;6:158–174.
21 Gibson IM. Visual mechanisms in a cone-monochromat. J Physiol. 1962;161:10P–11P.
22 Kellner U, Wissinger B, Tippmann S, et al. Blue cone monochromatism: clinical findings in patients with mutations in the red/green opsin gene cluster. Graefes Arch Clin Exp Ophthalmol. 2004;242(9):729–735.
23 Ayyagari R, Kakuk LE, Bingham EL, et al. Spectrum of color gene deletions and phenotype in patients with blue cone monochromacy. Hum Genet. 2000;107(1):75–82.
24 Ayyagari R, Kakuk LE, Coats CL, et al. Bilateral macular atrophy in blue cone monochromacy (BCM) with loss of the locus control region (LCR) and part of the red pigment gene. Mol Vis. 28 1999;5:13.
25 Berson EL, Sandberg MA, Rosner B, et al. Color plates to help identify patients with blue cone monochromatism. Am J Ophthalmol. 1983;95(6):741–747.
26 Weiss AH, Biersdorf WR. Blue cone monochromatism. J Pediatr Ophthalmol Strabismus. 1989;26(5):218–223.
27 Alpern M, Lee GB, Maaseidvaag F, Miller SS. Colour vision in blue-cone ‘monochromacy. J Physiol. 1971;212(1):211–233.
28 Gouras P, MacKay CJ. Electroretinographic responses of the short-wavelength-sensitive cones. Invest Ophthalmol Vis Sci. 1990;31(7):1203–1209.
29 Moskowitz A, Hansen RM, Akula JD, et al. Rod and rod-driven function in achromatopsia and blue cone monochromatism. Invest Ophthalmol Vis Sci. 2009;50(2):950–958.
30 Nathans J, Davenport CM, Maumenee IH, et al. Molecular genetics of human blue cone monochromacy. Science. 1989;245(4920):831–838.
31 Nathans J, Thomas D, Hogness DS. Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science. 1986;232(4747):193–202.
32 Nathans J, Maumenee IH, Zrenner E, et al. Genetic heterogeneity among blue-cone monochromats. Am J Hum Genet. 1993;53(5):987–1000.
33 Gardner JC, Michaelides M, Holder GE, et al. Blue cone monochromacy: causative mutations and associated phenotypes. Mol Vis. 2009;15:876–884.
34 Michaelides M, Johnson S, Simunovic MP, et al. Blue cone monochromatism: a phenotype and genotype assessment with evidence of progressive loss of cone function in older individuals. Eye (Lond). 2005;19(1):2–10.
35 Zrenner E, Magnussen S, Lorenz B. [Blue cone monochromasia: diagnosis, genetic counseling and optical aids]. Klin Monbl Augenheilkd. 1988;193(5):510–517.
36 Krill AE, Deutman AF, Fishman M. The cone degenerations. Doc Ophthalmol. 1973;35(1):1–80.
37 Rowe SE, Trobe JD, Sieving PA. Idiopathic photoreceptor dysfunction causes unexplained visual acuity loss in later adulthood. Ophthalmology. 1990;97(12):1632–1637.
38 Ladewig M, Kraus H, Foerster MH, Kellner U. Cone dysfunction in patients with late-onset cone dystrophy and age-related macular degeneration. Arch Ophthalmol. 2003;121(11):1557–1561.
39 Sergouniotis PI, Holder GE, Robson AG, et al. High-resolution optical coherence tomography imaging in KCNV2 retinopathy. Br J Ophthalmol. May 10, 2011. E-pub ahead of print, PMID21558291
40 Wang NK, Chou CL, Lima LH, et al. Fundus autofluorescence in cone dystrophy. Doc Ophthalmol. 2009;119(2):141–144.
41 Duncan JL, Zhang Y, Gandhi J, et al. High-resolution imaging with adaptive optics in patients with inherited retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48(7):3283–3291.
42 Kitiratschky VB, Grau T, Bernd A, et al. ABCA4 gene analysis in patients with autosomal recessive cone and cone rod dystrophies. Eur J Hum Genet. 2008;16(7):812–819.
43 Hamel CP. Cone rod dystrophies. Orphanet J Rare Dis. 2007;2:7.
44 Rivolta C, Berson EL, Dryja TP. Dominant Leber congenital amaurosis, cone–rod degeneration, and retinitis pigmentosa caused by mutant versions of the transcription factor CRX. Hum Mutat. 2001;18(6):488–498.
45 Kitiratschky VB, Nagy D, Zabel T, et al. Cone and cone-rod dystrophy segregating in the same pedigree due to the same novel CRX gene mutation. Br J Ophthalmol. 2008;92(8):1086–1091.
46 Maugeri A, Klevering BJ, Rohrschneider K, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet. 2000;67(4):960–966.
47 Nakazawa M, Kikawa E, Chida Y, et al. Autosomal dominant cone–rod dystrophy associated with mutations in codon 244 (Asn244His) and codon 184 (Tyr184Ser) of the peripherin/RDS gene. Arch Ophthalmol. 1996;114(1):72–78.
48 Demirci FY, Rigatti BW, Wen G, et al. X-linked cone–rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet. 2002;70(4):1049–1053.
49 Perrault I, Rozet JM, Gerber S, et al. A retGC-1 mutation in autosomal dominant cone–rod dystrophy. Am J Hum Genet. 1998;63(2):651–654.
50 Cunier R. Historie d’une hemerolopie, hereditaire depuis siecles dans’ un famille de la commune de Vendemian, pres Montpellier. Ann Soc Med de Gand. 1838;4:385.
51 Nettleship E. A history of congenital stationary nightblindness in nine consecutive generations. Trans Ophthalmol Soc UK. 1907;27:269–293.
52 Carr RE. Congenital stationary nightblindness. Trans Am Ophthalmol Soc. 1974;72:448–487.
53 Zeitz C, Labs S, Lorenz B, et al. Genotyping microarray for CSNB-associated genes. Invest Ophthalmol Vis Sci. 2009;50(12):5919–5926.
54 Rotenstreich Y, Harats D, Shaish A, et al. Treatment of a retinal dystrophy, fundus albipunctatus, with oral 9-cis-{beta}-carotene. Br J Ophthalmol. 2010;94(5):616–621.
55 Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus, and other forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. Am J Ophthalmol. 2000;130(5):547–563.
56 Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993;34(5):1659–1676.
57 Miyake Y. [Establishment of the concept of new clinical entities – complete and incomplete form of congenital stationary night blindness]. Nippon Ganka Gakkai Zasshi. 2002;106(12):737–755. discussion 756
58 Strom TM, Nyakatura G, Apfelstedt-Sylla E, et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19(3):260–263.
59 Sieving PA. Photopic ON- and OFF-pathway abnormalities in retinal dystrophies. Trans Am Ophthalmol Soc. 1993;91:701–773.
60 Nakamura M, Ito S, Terasaki H, Miyake Y. Novel CACNA1F mutations in Japanese patients with incomplete congenital stationary night blindness. Invest Ophthalmol Vis Sci. 2001;42(7):1610–1616.
61 Bech-Hansen NT, Naylor MJ, Maybaum TA, et al. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat Genet. 2000;26(3):319–323.
62 Zeitz C, Kloeckener-Gruissem B, Forster U, et al. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am J Hum Genet. 2006;79(4):657–667.
63 Dryja TP, McGee TL, Berson EL, et al. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc Natl Acad Sci U S A. Mar 29 2005;102(13):4884–4889.
64 Zeitz C, van Genderen M, Neidhardt J, et al. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest Ophthalmol Vis Sci. 2005;46(11):4328–4335.
65 Audo I, Kohl S, Leroy BP, et al. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2009;85(5):720–729.
66 Li Z, Sergouniotis PI, Michaelides M, et al. Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am J Hum Genet. 2009;85(5):711–719.
67 Nakamura M, Sanuki R, Yasuma TR, et al. TRPM1 mutations are associated with the complete form of congenital stationary night blindness. Mol Vis. 2010;16:425–437.
68 van Genderen MM, Bijveld MM, Claassen YB, et al. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am J Hum Genet. 2009;85(5):730–736.
69 Oguchi C. Ueber einen Fall von eigenartiger Hemeralopie. Nippon Ganka Gakkai Zasshi. 1907;11:123.
70 Mizuo G. On a new discovery in the dark adaptation on Oguchi’s dsease. Acta Soc Ophthalmol. 1913;17:1148.
71 Carr RE, Gouras P. Oguchi’s disease. Arch Ophthalmol. 1965;73:646–656.
72 Gouras P. Electroretinography: Some basic principles. Invest Ophthalmol. 1970;9(8):557–569.
73 Kuwakara Y, Ishihara K, Akiya S. Histopathological and electron microscopic studies of the reitna of Oguchi’s disease. Acta Soc Ophthalmol Jpn. 1963;67:1323.
74 Hashimoto H, Kishi S. Shortening of the rod outer segment in Oguchi disease. Graefes Arch Clin Exp Ophthalmol. 2009;247(11):1561–1563.
75 Yamada K, Motomura Y, Matsumoto CS, et al. Optical coherence tomographic evaluation of the outer retinal architecture in Oguchi disease. Jpn J Ophthalmol. 2009;53(5):449–451.
76 Fuchs S, Nakazawa M, Maw M, et al. A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet. 1995;10(3):360–362.
77 Yamamoto S, Sippel KC, Berson EL, et al. Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet. 1997;15(2):175–178.
78 Carr RE, Ripps H. Rhodopsin kinetics and rod adaptation in Oguchi’s disease. Invest Ophthalmol Vis Sci. 1967;6:426–436.
79 Carr RE, Ripps H, Siegel IM. Visual pigment kinetics and adaptation in fundus albipunctatus. Doc Ophthalmol. 1974:193–204.
80 Nakamura M, Lin J, Miyake Y. Young monozygotic twin sisters with fundus albipunctatus and cone dystrophy. Arch Ophthalmol. 2004;122(8):1203–1207.
81 Querques G, Carrillo P, Querques L, et al. High-definition optical coherence tomographic visualization of photoreceptor layer and retinal flecks in fundus albipunctatus associated with cone dystrophy. Arch Ophthalmol. 2009;127(5):703–706.
82 Genead MA, Fishman GA, Lindeman M. Spectral-domain optical coherence tomography and fundus autofluorescence characteristics in patients with fundus albipunctatus and retinitis punctata albescens. Ophthalmic Genet. 2010;31(2):66–72.
83 Gonzalez-Fernandez F, Kurz D, Bao Y, et al. 11-cis retinol dehydrogenase mutations as a major cause of the congenital night-blindness disorder known as fundus albipunctatus. Mol Vis. 30 1999;5:41.
84 Yamamoto H, Simon A, Eriksson U, et al. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat Genet. 1999;22(2):188–191.
85 Schatz P, Preising M, Lorenz B, et al. Fundus albipunctatus associated with compound heterozygous mutations in RPE65. Ophthalmology. 2011;118(5):888–894.
86 Voigt M, Querques G, Atmani K, et al. Analysis of retinal flecks in fundus flavimaculatus using high-definition spectral-domain optical coherence tomography. Am J Ophthalmol. 2010;150(3):330–337.
87 Niwa Y, Kondo M, Ueno S, et al. Cone and rod dysfunction in fundus albipunctatus with RDH5 mutation: an electrophysiological study. Invest Ophthalmol Vis Sci. 2005;46(4):1480–1485.
88 Maeda A, Maeda T, Palczewski K. Improvement in rod and cone function in mouse model of fundus albipunctatus after pharmacologic treatment with 9-cis-retinal. Invest Ophthalmol Vis Sci. 2006;47(10):4540–4546.