CHAPTER 43
Molecular clinical biochemistry
CHAPTER OUTLINE
Mutation, the source of diversity and disease
Genesis of an individual: the formation of gametes
Genes in families and populations
The variable expression of genetic disease
THE TECHNIQUES OF GENETIC ANALYSIS
Detection of specific sequences in DNA
THE APPLICATIONS OF DNA ANALYSIS
Inherited diseases – some examples
INTRODUCTION
If we distinguish the actual combination of genes possessed by an individual, that is the genotype, from the observable activity of those genes, the phenotype, then study of inherited disease in clinical biochemistry laboratories has traditionally been concerned exclusively with analysis of phenotypes. The last two decades have witnessed a dramatic change in this situation: molecular biological techniques are now a much more common part of the repertoire of clinical biochemistry laboratories. Initially, the identification of genes responsible for inherited diseases involved heroic efforts, requiring expensive, complex and extremely time-consuming procedures plus a few strokes of luck. Once a gene has been identified, however, modern analytical techniques make detection of mutations more straightforward than before.
Although each nucleated human cell contains about two metres of deoxyribonucleic acid (DNA) (around 3 billion bases), it is the fundamental simplicity of DNA – its building blocks comprise just four nucleotides – that favours its automated analysis. With a few exceptions, every cell in the body of an individual contains a complete copy of their DNA (or genome). For this reason, genetic analysis can be carried out on almost any nucleated cell type (such as lymphocytes or buccal mucosal cells) that can conveniently be collected. The application of DNA analysis now extends well beyond diagnosis of the classic inherited diseases to include, for example, the diagnosis and prognosis of cancer.
This chapter provides a general background to clinical laboratory applications of molecular genetic analysis. The emphasis is on diagnostic techniques with the potential for automation, utilizing polymerase chain reactions (PCRs), since classic techniques such as Southern blot analysis are not widely used in hospital biochemistry laboratories, but tend to be restricted to specialist molecular genetics departments. As far as possible, only a very basic knowledge of molecular biology has been assumed, but several excellent introductions to the topic are available (see Further reading) and a glossary is provided on page 872.
GENES AND GENE EXPRESSION
What is a gene?
A common working definition is that a gene is a sequence of nucleotide bases in DNA that codes for a single polypeptide, but the complexity of genomic organization is such that it is probably unwise to adhere rigidly to any one definition of the gene. Towards the end of the 19th century, it was already accepted that linear groups of ‘invisible self-propagating vital units’ were present in chromosomes. Mendel’s discovery (1865) that inheritance is particulate was rediscovered and publicized at the beginning of the 20th century, and the term gene was introduced to describe Mendel’s ‘particulate elements’ in 1909. By 1911, a specific gene (for colour blindness) had already been assigned to a particular chromosome (the X chromosome). With the work of Garrod, who first presented his studies on alkaptonuria in 1902, the association of specific diseases with inherited Mendelian traits became established.
Certain stains produce clearly defined bands on chromosomes, so the location of genes is described according to the number of the chromosome on which they are found, whether they are on the long (q) or short (p) arm, and the band number. For example, the location of the α1-antitrypsin gene is described as 14q31–32.3, meaning that it is found on the long arm of chromosome 14 in the region of bands 31–32.3. The locations of some of the genes that have been mapped to the X chromosome are shown in Figure 43.1.
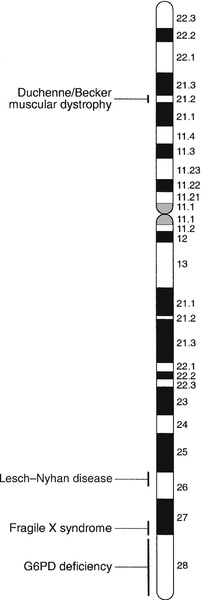
FIGURE 43.1 The mapping of genes to specific sites on the X chromosome. The regions in the X chromosome where the genes associated with Duchenne and Becker muscular dystrophy (the dystrophin gene at p21.2), Lesch–Nyhan disease (the hypoxanthine-guanine phosphoribosyl transferase or HGPRT gene at q26.1-q26.2), fragile X syndrome (the FRAXA gene at q27.3) and glucose 6-phosphate dehydrogenase (the G6PD gene at q28) deficiency are located are shown.
After the double helical structure of DNA had been discovered in 1953, the rather abstract concept of a gene became more tangibly associated with a physical structure. Nucleic acids consist of two complementary polymers of nucleotides. Each nucleotide consists of a purine or pyrimidine base, linked to a phosphorylated pentose. In DNA, the pentose is deoxyribose and the bases are adenine (A), guanine (G), cytosine (C) and thymine (T). In ribonucleic acid (RNA), the pentose is ribose and the pyrimidine uracil (U) replaces thymine. Protein coding sequences (exons) are interrupted by non-coding sequences (introns), which are variable in number (up to 50 in collagen genes, for example) and size (up to several thousand base pairs). As a consequence, although the knowledge that three bases code for an amino acid allows us to predict that the coding sequence for an average protein of 400 amino acids will be 1200 nucleotides, the complete gene could be an order of magnitude larger. The boundaries between exons and introns are critically dependent on the GT-AG rule: that is, introns almost invariably begin with GT (or GU in RNA) and end with AG. The structure of a hypothetical gene is shown in Figure 43.2. The promoter region of DNA, which precedes the coding region (‘upstream’ from the 5′ end of the gene, that is, in the opposite direction to transcription) is intimately involved in permitting and regulating expression. Some genes code for RNA (e.g. ribosomal and transfer RNA) that is not translated into protein, and modifications in the process of intron removal can result in one sequence of DNA participating in synthesis of different proteins, so that certain genes can be considered to overlap.
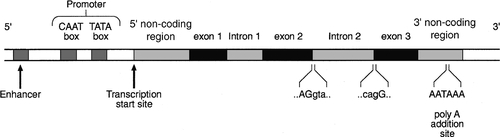
FIGURE 43.2 The structure of a hypothetical gene. Coding sequences (exons) are shown in black, introns and non-coding regions are shaded and regulatory regions are in dark shading. Bases in exon sequences are shown in upper case, while bases in intron sequences are shown in lower case, illustrating the GT-AGT rule for starts and ends of introns.
The Human Genome Project
The Human Genome Project (HGP) represents an outstanding piece of multinational cooperation to map the entire human DNA sequence. The project, started in 1990, had several aims, the first of which was to determine the entire base pair sequence. The sequence of 3 billion base pairs was announced in draft form in 2000 and the complete sequence in 2003. It was thought initially that the human genome would consist of approximately 100 000 different coding genes. As the HGP neared completion, it emerged that the actual number would be closer to 30 000.
The ‘Encode’ project
Protein coding sequences and introns account for about 20% of DNA. The function of the remainder is being elucidated but in 2012, the initial findings of the ‘Encode’ project, which had been examining what had previously been called ‘junk’ DNA, were published. It appears that the remaining 80% does have a function within the genome, with much of the non-protein coding DNA appearing to code for RNA transcripts that may have other regulatory functions alongside those of gene enhancers and promoters.
That the number of genes is much lower than expected appears to be because many genes can perform multiple functions, and it now appears that these functions may be regulated by the remainder of the genome. These discoveries have implications for the investigation and diagnosis of genetic disease, and issues concerned with gene expression will have a growing role in clinical genetics.
Gene expression
The differentiated properties of each cell are determined by the pattern of genes in the cell that are active or dormant. In any one cell, only a small percentage of genes are likely to be actively engaged in directing synthesis of RNA at any one time. Many of these are ‘housekeeping’ genes, which are expressed in virtually all cell types. Much remains to be learned concerning the factors that determine whether or not a gene is expressed, but regulation of gene expression is clearly determined by proteins that interact with DNA.
In higher (eukaryotic) organisms, DNA is found within the nucleus and the mitochondria, although the mitochondria have a very small percentage of the total DNA within the cell and a very small number of genes. Nuclear DNA is complexed with basic proteins (histones) to form chromatin. At regular intervals, DNA is wrapped around complexes of eight histones to form nucleosomes. The copying of DNA into RNA (transcription) is performed by RNA polymerase, which initiates transcription by interacting with the promoter region of a gene. Gene expression is inhibited if nucleosomes cover a promoter region and many factors that regulate transcription probably do so by competing with histones for binding to the promoter region. Within each promoter region, there are several elements that bind specific proteins capable of interacting with RNA polymerase and associated proteins. One of the key proteins in this group binds to the so-called ‘TATA box’ element (actually a TATAAAA or related sequence), which is found in most eukaryotic promoters and is usually located around 30 base pairs (bp) upstream from the transcription start site. Other conserved sequences, such as CAAT, also bind transcription factors and are found within the promoter region. Another class of regulatory sequence in DNA, the enhancer region, binds regulatory molecules, which include the steroid hormone receptors. Enhancer sequences may be some distance from the gene that they regulate, but the proteins that bind to them may nevertheless interact with the transcriptional apparatus as a result of looping in the DNA molecule.
The initial RNA transcript (pre-mRNA) is modified in several ways before it leaves the nucleus (Fig. 43.3). First, a ‘cap’ structure (7-methylguanosine) is attached to the 5′ end and a sequence of about 200 adenylic residues (poly A) is added at the 3′ end. The non-coding introns are then removed by a two-step splicing mechanism to form a mature messenger RNA (mRNA). Splicing, which takes place within ‘spliceosomes’ (complexes of RNA and proteins), requires cleavage at the 5′ and 3′ ends of the intron and ligation (joining) of the exons. The specific boundary sequences found at splice junctions (see above) act as signals for splicing. Comparison of large numbers of splice junctions reveals these ‘consensus sequences’ to be of the general form AGgta at the 5′ junction and cagG at the 3′ junction (where bases in the intron sequence are in lower case). Given that pre-mRNA molecules can contain up to 65 exons and an intron may consist of thousands of nucleotides, it is remarkable that the correct sites for splicing can be chosen.
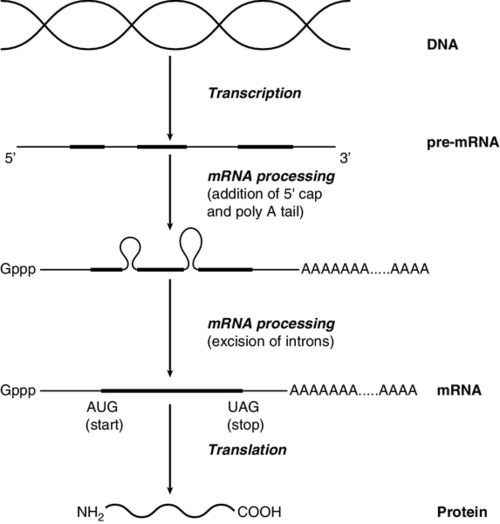
FIGURE 43.3 Transcription and mRNA processing. After transcription, processing of precursor mRNA involves capping, whereby GTP is attached to the 5′ end of the mRNA precursors via a 5′-5′ triphosphate linkage (i.e. in the reverse orientation to all other nucleotides); addition of around 200 adenylate residues to form a poly(A) tail at the 3′ end; and splicing, in which introns are excised and exons spliced together. Translation of mRNA into protein is initiated by ribosomes and transfer RNA at the AUG codon and terminated at one of the stop codons (UAG, UAA or UGA).
Finally, the process of translation involves the activity of ribosomes, transfer RNA and a variety of other molecules which synthesize a protein using the mRNA code as a template. A group of three nucleotides (a codon) specifies an amino acid and most amino acids are coded for by more than one codon (i.e. the genetic code is degenerate). In principle, each RNA sequence can be decoded in three different reading frames, depending on which triplet is chosen as the first codon. In practice, the reading frame is determined by the site of initiation, which always occurs at an AUG codon (AUG codes for methionine, but the initiating methionine is cleaved from proteins in eukaryotic cells). Translation stops at any one of three stop codons (UAA, UAG or UGA). Any subsequent modification of a protein, such as proteolytic cleavage or addition of carbohydrate, is known as post-translational modification.
Mutation, the source of diversity and disease
The accepted terminology, which referred to the ‘normal’ gene in a population as the ‘wild type’, has changed and the terms ‘normal’ and ‘mutant’ (or ‘variant’ if pathogenicity is unclear or questionable) are now preferred. However, the genetic constitution of populations is in a constant state of flux with new genes appearing as a result of mutation and deleterious genes being removed by natural selection. Mutations can be broadly divided into those that change the genetic code at a specific location (point mutations or single nucleotide polymorphisms, SNPs) and those that result in gain or loss of genetic material (deletions, duplications and insertions). Point mutations can result from incorrect insertion of a base during DNA replication by DNA polymerase or spontaneous decomposition reactions such as depurination and deamination. Mutagenic chemicals that increase this error rate include those that mimic the natural bases or distort the structure of DNA and those that chemically modify DNA. Ultraviolet light also causes point mutations, particularly by formation of pyrimidine dimers. Point mutations in which a purine is replaced by another purine (e.g. A replaced by G) or a pyrimidine is replaced by another pyrimidine are known as transitions, while replacement of a purine by pyrimidine (e.g. G replaced by C) or vice versa is known as a transversion. Gain or loss of genetic material can result from various errors, including chromosomal breakage and unequal crossing over. Insertion of viral sequences into DNA can also disrupt the genetic code and the rate of spontaneous chromosomal breakage can be markedly increased by ionizing radiation. Whenever the number of bases deleted or inserted is not a multiple of three, the reading frame of the mRNA is altered (frameshift mutation) and the RNA sequence subsequent to the mutation becomes nonsense.
Accumulated damage to DNA would rapidly overwhelm the organism, but repair mechanisms recognize and repair damaged DNA so that fewer than 1 in every 1000 accidental base changes results in a stable mutation. It is estimated that stable point mutations are acquired at a rate of about 1 in every 109 base pairs during each cell generation. Consequently, an average gene of about 103 coding base pairs is likely to acquire a mutation once in every 106 cell generations. As might be expected, individuals with inherited defects in the enzymes responsible for DNA repair are markedly more susceptible to the effects of environmental mutagens.
A significant proportion of germline point mutations are thought to be caused by modification of methylated cytosine residues. DNA methylation, restricted in eukaryotic cells to cytosine residues, usually at CpG dinucleotides (CpG denotes C-phosphate-G in a linear sequence, in distinction from a CG base pair) is not present in all such organisms but is thought to play an important role in ensuring stable inheritance of expression patterns when cells divide. Spontaneous deamination of the 5-methylcytosine creates thymidine, and problems can then arise when the normal guanine on the complementary strand becomes an adenine and the mutation cannot be detected by DNA repair mechanisms. Consequently, methylation of cytosine can produce mutation ‘hot spots’ (i.e. sequences associated with an unusually high frequency of mutations or recombination).
Some mutations may have no effect on the structure of a protein – either because the genetic code is degenerate and the new sequence codes for the same amino acid, or because some amino acids in a protein can be substituted without producing any significant effect on the function of the protein. However, some apparently ‘silent’ mutations may have an effect on the protein product, not because of the actual base change involved, but rather through an effect on splicing by activating cryptic splice sites or destroying splice enhancers. Mutations that change the three-dimensional structure of a protein and so alter its function or stability may do so by a variety of mechanisms (Fig. 43.4).
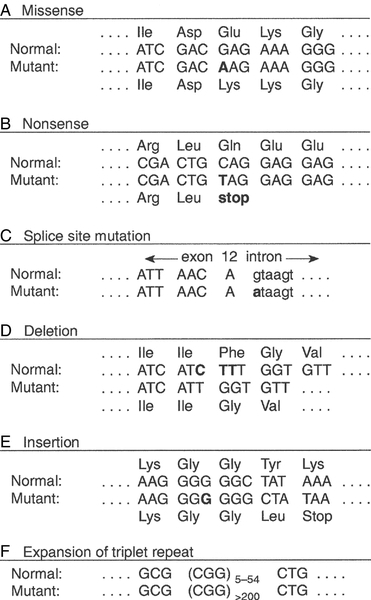
FIGURE 43.4 Examples of mutations. (A) G to A transition in the α1-antitrypsin gene, a missense mutation resulting in substitution of glutamic acid by lysine at position 342, producing the Z variant associated with α1-antitrypsin deficiency. (B) Transversion of C to T in the steroid 21-hydroxylase gene, converting the codon for glutamine to a stop codon, one of the mutations causing congenital adrenal hyperplasia. (C) G to A transition in the 5′ splice site of intron 12 in the phenylalanine hydroxylase gene, resulting in deletion of exon 12, the most frequent cause of phenylketonuria in Caucasians. Bases in intron sequences are shown in lower case. (D) Deletion of three bases in the cystic fibrosis transmembrane conductance regulator (CFTR) gene resulting in deletion of phenylalanine at position 508, the most frequent mutation causing cystic fibrosis in Caucasians. (E) Insertion of a G in the hypoxanthine-guanine phosphoribosyl transferase gene, a frameshift mutation that results in the Lesch-Nyhan syndrome. (F) Amplification of a CGG triplet repeat in the FMR-1 gene, which causes the fragile X syndrome.
By convention, the DNA strand that has the same sequence as mRNA (except that it possesses T instead of U) is represented. This strand is known as the coding strand, although it is the ‘anticoding’ strand that is complementary to mRNA and therefore provides the template for mRNA synthesis.
Some amino acid changes (missense mutations), such as that which produces the Z variant of α1-antitrypsin (Fig. 43.4A), can have a profound effect on the processing or function of a protein. Some mutations (nonsense mutations) create or destroy codons for the start or stop signals of translation so that a protein of abnormal length is produced (Fig. 43.4B). Mutations at splice sites (Fig. 43.4C) frequently result in production of abnormal mRNA that is unstable. Deletion of three bases removes the codon for a single amino acid without altering the reading frame, as occurs in the most common mutation causing cystic fibrosis (Fig. 43.4D). Insertion (Fig. 43.4E) or deletion of any number of bases that is not a multiple of three will alter the reading frame so that the message becomes garbled. Amplification of triplet repeat sequences (Fig. 43.4F) has been identified as the basis of several inherited diseases. Occasionally, mutations affect regulatory regions of DNA so that the amount of protein produced is altered. Although mutations are most frequently either neutral or deleterious, rare mutations will alter the function of a protein in such a way that the fitness of an individual is improved, so contributing to evolution. Inheritance of the mutations that have accumulated in our ancestors, whether they be advantageous, neutral or deleterious, is what constitutes our individuality.
An individual inherits two copies of each chromosome (one maternal and one paternal). On each chromosome, the sequences at each site, or locus, are known as alleles. If the two alleles are identical, the individual is said to be homozygous at that locus, while if the alleles are different, the individual is said to be heterozygous for each allele. As will be seen later, genetic disease is usually heterogeneous, so that an individual said to be homozygous for a deleterious gene may be found, when studied at the molecular level, to carry a different mutation in each allele (i.e. is a compound heterozygote). When the prevalence of a mutant allele becomes more common in a population than could be maintained by new mutations alone (generally taken to be when more than 1% of the population carry the allele), there is said to be polymorphism. Many proteins in blood (e.g. haptoglobin) and on cell surfaces (e.g. human leukocyte antigen, HLA) are polymorphic.
The classic inherited diseases result from single gene defects and more than 6000 inherited diseases likely to be associated with defects in single genes have already been identified. Inherited diseases could, in theory, result from mutations in any one of the human genes – the only limitation being that the structure of some gene products is so critical that any mutation will not be compatible with life. Most of the more common diseases that afflict western society, including most cases of diabetes, atherosclerosis and hypertension, are the result of interaction between the environment and polygenic factors (i.e. they are determined by interactions between several genes). Molecular analysis of the polygenic diseases is considerably more difficult than analysis of single gene defects, but alleles that predispose individuals to development of these diseases are being identified. Both single gene defects and most multifactorial/polygenic diseases arise from mutations in the nuclear DNA but genetic diseases also arise from mutations in mitochondrial DNA and from chromosomal abnormalities. Mitochondrial DNA is extranuclear and shows almost complete maternal inheritance. Chromosomally inherited disorders include the trisomies, where faulty meiosis allows two copies of a chromosome to be present in a gamete, leading to three copies in the embryo. Trisomy of chromosome 21, for example, is responsible for Down syndrome.
If new mutations occur in germ cells, then they may give rise to an inherited disease in the next generation. The effects of mutations on non-germ cells, or somatic cells, will depend both on the gene affected and on the state of differentiation of the cell affected. The ageing process is likely to be one result of accumulated mutations in somatic cells and the central role of mutations for the development of cancer has become clearer during the last few years.
Genesis of an individual: the formation of gametes
An individual’s genotype is determined at the time of fertilization, when the chromosomes of the gametes (i.e. the male sperm and female egg) are combined. The formation of gametes (gametogenesis) is particularly relevant to an understanding of the detection of inherited disease because it is at this stage that ‘shuffling’ of genes occurs.
Normal cell division, or mitosis, involves a simple copying of each chromosome, with one identical copy being passed on to each daughter cell. To avoid a doubling in the number of chromosomes in each generation, gametogenesis involves a reduction (by half) of the chromosome number during two specialized cell divisions known as meiosis. Since the chromosome complement of parent and offspring must be equivalent, the reduction in chromosome number cannot be arbitrary: parental contributions must be equal and equivalent. This requirement can be met because each somatic cell of an individual is diploid, containing corresponding (homologous) pairs of chromosomes, one derived from the mother and the other from the father. Meiosis consists of two cell divisions: in the first, after duplication of DNA, pairing of homologous chromosomes occurs, and to ensure that each gamete receives just one member of each homologous pair, the duplicated paternal chromosome is distributed to one, the duplicated maternal chromosome to the other. The assortment between the two cells appears to be random so that each cell acquires some maternal and some paternal chromosomes. The second cell division is like ordinary mitosis, except that it is not preceded by duplication of chromosomes. As a consequence, the gametes produced are haploid, with half the normal number of chromosomes.
At first sight it might be expected that a chromosome would be transmitted from one generation to the next as an intact unit and that two genes on the same chromosome would always be inherited together. The fact that this is not so is a result of events that occur during the first meiotic division, which have important consequences for genetic analysis. As chromosomes pair prior to the first division, crossovers (or chiasmata) occur by breakage and rejoining between the chromatids of homologous chromosomes resulting in recombination (Fig. 43.5). The recombination fraction is a measure of the genetic, rather than the physical, distance between two genes (or loci). The recombination fraction for two loci can never be more than 0.5 as the resulting chromatids can only ever be recombinant or non-recombinant, no matter how many crossovers have occurred between the loci. In simple terms, linkage of two genes (i.e. a tendency to be inherited together) occurs only when the genetic distance separating them is sufficiently short to make crossover between them unlikely. The association of two genes on separate chromosomes is random but association of genes on the same chromosome is not, as it is known that crossovers do not occur at random. A process known as interference prevents a chiasma forming if one already exists nearby. When genes are associated more frequently than would be predicted by chance, they are said to be in linkage disequilibrium.
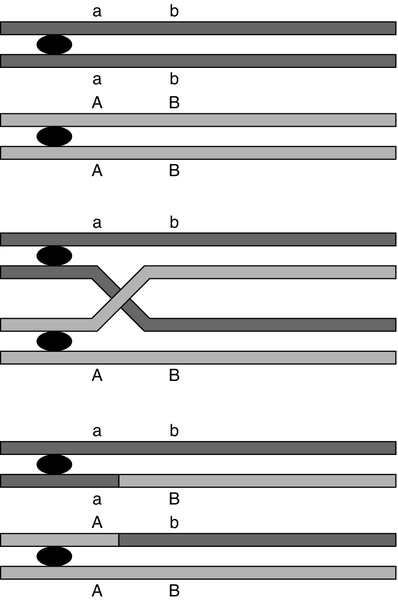
FIGURE 43.5 Crossover and linkage. Exchange of alleles as a result of crossover between homologous chromosomes. Linkage between alleles a and b (or A and B) occurs if they are sufficiently close that crossover is unlikely to occur between them.
In females, gametogenesis is initiated during fetal development and, at birth, the germ cells are in a phase of arrested maturation of the first stage of meiosis, which is not completed until ovulation. The increased risk of chromosomal abnormalities in older mothers may be explained by the fact that completion of meiosis occurs only after ovulation, when the second stage of meiosis (which is similar to ordinary mitotic division) occurs and during which fertilization can take place. This may happen up to 50 years after formation of the germ cells. In males, sperm production continues from the time of sexual maturity into old age and the large number of cell divisions involved is probably the cause for the increased number of new single gene mutations that seem to occur in the children of older men.
Genes in families and populations
Mendel introduced the concepts of dominant ‘characters’ (or traits), which are transmitted without change, and recessive traits, which become latent after cross-fertilization. In modern terms, an allele coding for a dominant trait can be said to manifest its phenotype in the heterozygous state (i.e. only one copy is required for its effects to be apparent), while an allele for a recessive trait expresses its phenotype only in homozygotes. An individual who is heterozygous for an autosomal recessive condition is described as a carrier. The types of family pedigree associated with autosomal dominant and recessive genes are illustrated in Figure 43.6A and B. Autosomal dominant conditions affect males and females equally, and affected individuals who are heterozygous for the abnormal allele will transmit it to half of their offspring. Autosomal recessive disorders occur in individuals whose parents are both carriers for the mutant gene. The risk for such patients of having affected children is 25% and the probability of any child they have being a carrier is 50%. Dominant diseases are often associated with genes coding for proteins that have structural, carrier or receptor functions, while genes coding for enzymes are often associated with recessive disorders. The explanation for this is probably that the activity of most enzymes is considerably greater than necessary for normal metabolism, so loss of up to half of the normal activity is of little consequence. Inheritance is somewhat different for alleles on X chromosomes. Dominant X-linked diseases will affect both males and females, but an X-linked recessive disease will be manifest only in males, who have only one X chromosome (Fig. 43.6C). In females, during the very early stages of embryogenesis, one or other X chromosome is inactivated in every cell; consequently, females can be carriers but will usually only suffer from an X-linked recessive disease if they are homozygous. However, sometimes, in some disorders, the inactivation is not random but occurs in such a way that only the normal chromosome is inactivated, with the result that only mutant alleles are expressed in critical tissues (‘skewed’ X-inactivation) and the individual becomes a manifesting heterozygote. This has been reported in several X-linked recessive disorders including, for example, Duchenne muscular dystrophy.
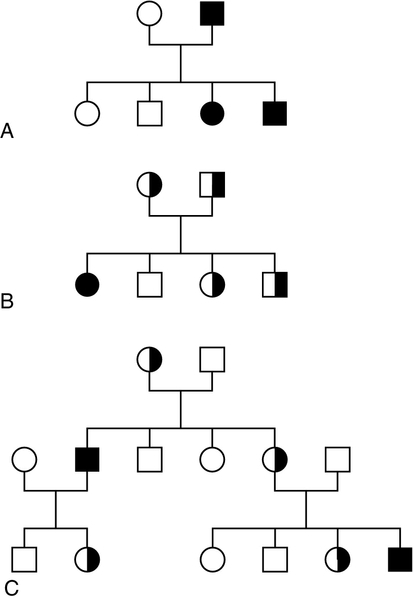
FIGURE 43.6 Patterns of inheritance in families. Inheritance of (A) a dominant condition, (B) a recessive condition and (C) an X-linked recessive condition. Squares represent males and circles represent females. Open symbols represent normal individuals, while fully shaded symbols refer to affected individuals and half-shaded symbols represent carriers.
When both alleles are expressed in a heterozygote, each producing its phenotype independently, inheritance is said to be codominant. This type of inheritance is seen most clearly when phenotypes are determined by immunological or biochemical tests – for example, in testing for blood groups and in restriction fragment length polymorphism (RFLP) analysis and, more recently, in the growing field of pharmacogenetics (see later), where it is possible to distinguish both alleles at each locus.
Of the inherited diseases currently recognized, the overwhelming majority are due to problems in nuclear DNA and are autosomal dominant, autosomal recessive or sex linked. However, it is now known that some diseases are associated with the small amount of DNA that is present in mitochondria. These diseases are maternally inherited, since the mitochondria in the fertilized egg are of maternal origin.
The relative frequency of inherited disease varies markedly between populations: for example, cystic fibrosis and α1-antitrypsin deficiency are associated primarily with northern Europeans, while red cell disorders (thalassaemia, sickle cell anaemia and glucose 6-phosphate dehydrogenase deficiency) are found primarily in people of Mediterranean, Oriental or African origin, and Tay–Sachs disease is found primarily in Ashkenazi Jews. In some populations, the prevalence of an inherited disease is due to a ‘founder effect’, as with variegate porphyria in South Africans, which can be traced to a single couple who emigrated from Holland in the 1680s. Autosomal recessive diseases that are particularly widespread in larger populations are likely to represent a balanced polymorphism in which disadvantage to homozygotes is balanced by an advantage to the larger number of heterozygotes. With some red cell disorders (e.g. HbS, the sickle cell trait), the balanced polymorphism is clearly a response to the environment, in this case with heterozygosity conferring resistance to malaria caused by Plasmodium falciparum.
In a large population, the relative frequencies of different alleles tend to remain constant and a simple mathematical formula allows calculation of the frequency of different genotypes. If two alleles, A and a, occur at a given locus and their frequencies are p and q, respectively, then:
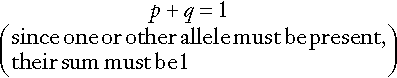
It can be shown that the genotypes AA, Aa and aa have frequencies p2, 2pq and q2, respectively (the Hardy–Weinberg law). Use of this law allows simple calculation of carrier frequencies for autosomal traits. For example, if the homozygote frequency (q2) for cystic fibrosis is about 1 in 2500, q is 1/50, p is 1 − q or 49/50 (~ 1) and the heterozygote frequency (2pq) is about 1/25.
The variable expression of genetic disease
Several factors dictate that each genetic disease is associated with symptoms of quite variable nature and severity.
It is often naively assumed that all mutations of a particular gene will have identical consequences for the organism, but this is far from being true. Different mutations in a given gene are quite likely to give rise to different phenotypes, just as a particular phenotype can result from many different mutations in the same gene or even in different genes. Since many different mutations that have deleterious effects on a particular gene are present in most populations, it is not surprising that molecular analysis often reveals that individuals who have been described as homozygous are in fact compound heterozygotes, that is, they are affected by two different deleterious alleles.
Because the haemoglobin gene has been investigated in detail, it provides a good illustration of the complexity of genetic disease. Several hundred abnormal haemoglobins have been identified. The majority have amino acid substitutions resulting from single base changes and the consequences range from complete absence of protein to variant haemoglobins with function indistinguishable from that of normal (see Chapter 29). As study of other genes progresses, similar complex arrays of mutations of every imaginable type and with differing consequences are being discovered. With the introduction of screening for phenylketonuria (PKU), it soon became apparent that the disease is heterogeneous. It has been shown that, in PKU, phenotypic heterogeneity is related to the level of phenylalanine hydroxylase activity expressed in each patient which, in turn, is determined, in classic PKU, by the particular mutations that are present in the two alleles of the phenylalanine hydroxylase (PAH) gene. However, not all cases of neonatal hyperphenyalaninaemia are due to PAH deficiency. Benign hyperphenyalaninaemia arises from a transient liver immaturity and does not lead to the disease. Two rare causes of PKU are deficiencies in the enzymes dihydropteridine reductase and dihydrobiopterin synthetase, leading to PKU with a severe phenotype. Glucose 6-phosphate dehydrogenase (G6PD) deficiency, which is an X-linked recessive condition estimated to affect as many as 500 million people worldwide, has also proved to be extremely heterogeneous at both the phenotypic and molecular levels. Some variants appear to have no clinical consequences, while at the other extreme, severe defects in G6PD cause hereditary non-spherocytic haemolytic anaemia (see Chapter 27). Intermediate defects are associated with haemolytic anaemia only in the presence of precipitating factors (e.g. infection, ingestion of fava beans or certain drugs). Analysis of the protein had indicated the existence of about 400 variants of G6PD and a similar number of mutations have now been described, but many of these appear not to cause disease. Many of the mutations causing the most severe disease are clustered near the carboxy end of the enzyme in the region of the putative NADP binding site.
A further level of heterogeneity in genetic disease occurs as a result of varying degrees of penetrance and expressivity. Penetrance refers to the degree to which the mutation causes disease. Thus, in some disorders, the presence of the mutant gene is disease-causing in some individuals but not others, demonstrating variable penetrance, whereas in diseases that are fully penetrant, the presence of the mutation will always lead to the disease phenotype. For example, the C282Y (Cys282Tyr) mutation in the HFE gene (causing substitution of the cysteine residue at position 282 of the human haemochromatosis protein by tyrosine), causes haemochromatosis in some individuals but results in a completely normal phenotype in others. Expressivity is a slightly different aspect of a gene’s effect and refers to the presence of variable phenotypes arising from the same mutation. It can be age-related or determined by the environment (as when drugs such as barbiturates precipitate attacks of acute intermittent porphyria). An example of a disorder demonstrating variable expressivity is Waardenburg syndrome, where the ‘full’ syndrome includes several phenotypic features (such as different coloured eyes, a white forelock or deafness) but where, within a single affected family, different individuals may have only one feature, which is not always the same one within that family. Variable expressivity and penetrance tend to be features of dominant, rather than recessive, conditions.
Another aspect of inheritance, known as imprinting, may help to explain processes such as variable penetrance and expressivity. Contrary to the assumptions of classic genetics, it now appears that expression of some genes depends on whether they are of maternal or paternal origin. The molecular mechanism of imprinting, which is likely to occur during meiosis, involves DNA methylation, which ‘marks’ certain genes and ensures that they are preferentially expressed in the next generation. If such a gene is imprinted in the maternal line, it will continue through a woman’s daughters but not through her sons, although both may be affected. The converse is true for paternally imprinted genes, which will be transmitted through sons but not through daughters. Consequently, if an imprinted gene contains a deletion, offspring will not show expression of the gene product even in the presence of a normal gene on the opposite chromosome, as this will be ‘switched off’.
Prader–Willi and Angelman syndromes are good examples of disorders arising due to abnormalities in a region carrying imprinted genes (chromosome 15 at 15q12,). Prader–Willi syndrome, features of which include hypotonia and hyperphagia, is produced by deletion of the paternal alleles at 15q12. Angelman syndrome, which is associated with ataxic movements and seizures, is also associated with deletion of 15q12 but, in this case, of the maternal allele. In some cases of Prader–Willi syndrome, rather than deletion of the paternal alleles, loss of the paternal chromosome occurs together with maternal isodisomy (two copies of the same allele from the mother) or heterodisomy (one copy of each maternal allele). There are several possible mechanisms by which two alleles can be inherited from one parent. Trisomies, for instance, usually result in spontaneous abortion, but if one chromosome is then lost there is a one in three chance that resulting cells will have a normal complement of chromosomes but with one pair of chromosomes derived from a single parent. The interesting possibility that a recessive disease can be inherited from one carrier parent then arises. This unusual mode of inheritance has been demonstrated in some patients with cystic fibrosis, but it is not yet clear how frequently it occurs in this or other diseases.
THE TECHNIQUES OF GENETIC ANALYSIS
Detection of specific sequences in DNA
Analysis of DNA is heavily dependent on the availability of techniques to identify specific nucleotide sequences. Fortunately, the function of DNA has resulted in the evolution of proteins capable of recognizing specific DNA sequences and it is inherent in the structure of DNA that one strand should recognize and bind (hybridize) specifically to its complementary strand. Most of the techniques currently used in DNA technology exploit one or other of these properties.
Use of proteins that recognize DNA sequences: restriction endonucleases
Without restriction enzymes, much of the molecular biological analysis carried out in the last 30 years would not have been possible. These enzymes are widespread in bacteria: over 3000 having been recognized so far, of which around 600 are available for commercial/analytical use. Each enzyme is named after the species of bacteria in which it was found (e.g. EcoRI from Escherichia coli), and in which it probably fulfils a defensive function, cleaving molecules of foreign DNA. The usefulness of these enzymes derives from the fact that they do not cleave DNA at random, but recognize and cut specific nucleotide sequences. The most commonly used restriction enzymes recognize sequences of 4–6 nucleotides that have a two-fold axis of symmetry and are therefore said to be palindromes (i.e. the sequence reads the same on the complementary strand) (Fig. 43.7A). Digestion of DNA by a particular enzyme provides reproducible fragments whose size will depend on the frequency with which the enzyme recognition site occurs. On average, a 4-bp site occurs every 256 bp and a 6-bp site every 4096 bp. While some enzymes (e.g. HaeIII) cut in such a way that ‘blunt ends’ are produced, others (e.g. EcoRI) cut asymmetrically so that ‘sticky ends’ are left, which are extremely useful for reannealing fragments to produce recombinant DNA (Fig. 43.7B).
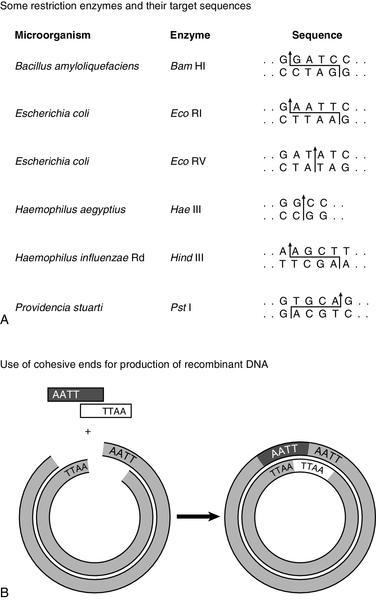
FIGURE 43.7 Restriction enzymes and recombination. (A) Arrows indicate how specific DNA sequences are cleaved by restriction enzymes. (B) Cohesive ends produced by restriction enzymes can be used for annealing of DNA sequences, which can then be joined by a ligase to form a recombinant DNA molecule.
In addition to allowing reproducible cleavage of DNA to a manageable size, restriction enzymes are also valuable tools for analysing molecular diversity and identifying the individuality of DNA sequences. Differences in DNA sequences between individuals may create or destroy sites for restriction enzymes (i.e. there is polymorphism of restriction sites). Thus, the distance between restriction sites will often differ between individuals and between the maternal and paternal strands of DNA. The pattern of restriction sites can therefore provide a ‘signature’ for each individual strand of DNA. The different populations of DNA fragments produced on digestion by an enzyme are known as restriction fragment length polymorphisms or RFLPs.
Restriction enzymes have more recently acquired a new use in preparing genomic DNA for the various techniques employed in ‘next generation’ sequencing.
Hybridization: probes and the polymerase chain reaction (PCR)
A probe is a sequence of DNA (or RNA) that has been labelled in order to identify complementary base sequences by molecular hybridization. The two strands of DNA can be dissociated (‘denatured’ or ‘melted’) in various ways, such as by heating or addition of alkali. Denaturation for a given fragment of DNA occurs at a specific temperature, and the temperature at which 50% of the duplex is dissociated is known as the Tm. When the temperature is lowered to just below the Tm, hydrogen bonds begin to reform between complementary bases, a process known as annealing or renaturation. If the probe and target DNA are mixed before reannealing is allowed to occur, the probe can be used to ‘find’ its complementary sequence. The conditions under which reannealing occurs (in particular salt concentration and temperature) determine the degree of stringency of the hybridization.
Probes can be used to detect their complementary sequences, traditionally after DNA (or RNA) fragments have been separated by electrophoresis. Digestion of genomic DNA with a restriction enzyme will produce a million or so fragments of different sizes and electrophoresis has the great advantage of allowing simple determination of the size of fragments detected by probes. The now classic technique of Southern blotting involves the transfer of electrophoretically separated bands of DNA to a sheet of nitrocellulose or nylon. Complementary DNA sequences are then detected by hybridization with labelled probes. The technique was originated by Dr (later Professor) E M Southern. Subsequently, the terms Northern and Western blotting have been used for processes in which RNA or proteins, respectively, are transferred.
In the past, all DNA analysis required some technique of visualizing the products of the reaction, and with probe hybridization this involves labelling the probe in some way. 32Phosphorous radioactive labelling, although still in use for blotting protocols, was superseded by the use of fluorescent dyes, particularly for gene sequencing, a system which allowed the development of large, high-throughput, DNA analysers, as well as techniques such as real-time PCR. However, some of the technologies employed in ‘next generation’ sequencing systems require neither gels nor dyes, as will be discussed later. Some of the newer systems allow for an electrical signal to be generated when hybridization of probe to target occurs, which may eventually lead to the development of point-of-care systems.
Probes, generally of a few thousand bases, may be sequences cut from genomic DNA or they may have been produced by making complementary DNA (cDNA) to an mRNA species. The latter procedure uses the enzyme reverse transcriptase, which transmits genetic information in the ‘reverse’ direction, that is, from RNA to DNA. Genomic and cDNA sequences differ, particularly in the absence of intron sequences from the latter. The tolerance of probes for mismatching of base sequences will depend on their size and on the stringency of hybridization. Shorter oligonucleotide probes are often more useful for direct identification of point mutations since conditions can be chosen such that hybridization occurs only when there is complete complementarity between probe and target. Because hybridization of a relatively smaller proportion of bases in the probe is required, larger genomic or cDNA probes will usually recognize corresponding sequences of DNA from different individuals in a population or even different but related genes.
Cloning, where the sequence of interest was ‘grown’ in bacteria such as E. coli after being inserted into the bacterial genome using bacteriophage viruses, has been largely superseded as a method of producing probes by the ability to create synthetic oligonucleotide sequences, although cloning as a technique is still employed in areas of research.
Without doubt, the commonest use for synthetic oligonucleotides has been as ‘primers’ in the polymerase chain reaction (PCR). After its introduction in 1985, PCR supplanted many of the more tedious techniques of molecular biology and opened up completely new possibilities. Essentially, a means of cloning DNA without the need for vectors or bacteria, PCR uses the enzyme DNA polymerase to copy DNA. To do this, the enzyme needs two oligonucleotide primers that are complementary to sequences flanking the region of interest in the target DNA, with one on each strand (Fig. 43.8). Computer and web-based programs are available for designing primers (usually 20 or more bases) in order to choose sequences likely to be most suitable for the PCR reaction and to maximize specificity by ensuring that the complementary sequence is virtually unique in the genome. The target sequence is amplified exponentially by repeated cycles of enzymatic copying. In the first cycle, double-stranded DNA is denatured by heating to between 92°C and 96°C for 5 min, and then cooled to (usually) between 55°C and 60°C, so that the oligonucleotide primers can anneal to their complementary sequences in the target DNA. For optimal specificity, the highest annealing temperature possible is used to minimize extension of primers bound non-specifically. Extension of the primers by DNA polymerase, using added nucleotides and the DNA target as a template, is then allowed to take place. Use of a heat-stable DNA polymerase, which can withstand the heating cycles, avoids the necessity of adding fresh enzyme at each cycle and allows extension to take place at a high temperature (72°C). After heat denaturation, excess primers can then anneal to the newly synthesized DNA as well as the original DNA strands and the process is repeated. In 30 cycles, amplification of over one million-fold can be achieved. Originally, the only enzyme available for PCR was that from Thermus aquaticus (Taq polymerase), but more have now been identified and developed, making amplification of much longer sections of genomic DNA possible.
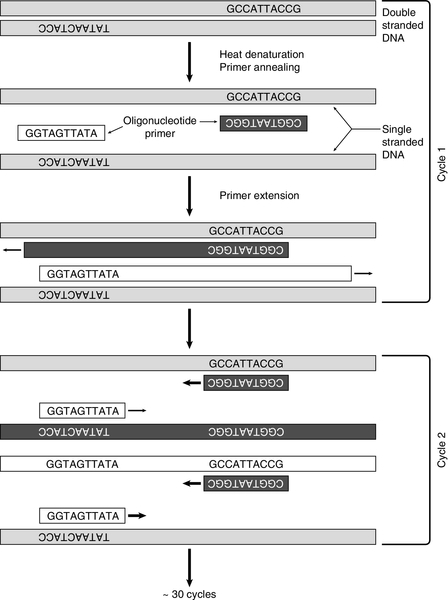
FIGURE 43.8 The polymerase chain reaction. After denaturation of DNA, primers anneal to complementary sequences. During the first cycle, a heat-stable DNA polymerase (usually Taq) initiates synthesis of two new strands. After a further denaturation cycle, primers anneal to the newly synthesized DNA as well as the original sequences and four new strands are synthesized (cycle 2). With an exponential increase in the number of DNA strands, after around 30 cycles the sequence will have been amplified about one million times.
Stringent control of conditions, such as the concentrations of magnesium and nucleotides in the reaction mixture, have usually been needed to maintain the specificity of a PCR although conditions that maximize polymerase fidelity may reduce PCR efficiency. Where such manipulation was necessary, it was predominantly dependent on the sequences involved, especially that of the primers. However, the ability to amplify longer sections of DNA means that primers can be ‘picked’ to suit the conditions required, allowing the use of universal ‘master mixes’ and thus a greater standardization of PCR assays. PCR is so sensitive that it has been used to amplify the DNA from a single cell and it can be used with samples obtained from materials as diverse as ancient mummies, fossils, hair follicles, preimplantation embryos and fixed pathological specimens. The large quantities of amplified DNA produced by PCR, which can be detected using a variety of visual labels, have eliminated the need for radiolabelling. The extreme sensitivity of the technique is also the source of one of its disadvantages – contamination by extraneous DNA (e.g. from the operator) can create havoc, so that strict precautions must be taken to avoid artefacts.
Non-isotopic fluorescent labels are employed in the widely used technique of real-time PCR. There are several versions of this technique but the basic principle is that a PCR reaction is followed, in real time, by monitoring the signals produced by dye-labelled probes that bind to the accumulating products. Various modifications to the technique allow real-time PCR to be used for quantitation of a target sequence (hence, its other, abbreviated name of qPCR) and determining dosage (the number of copies of a gene in a cell) and gene duplication as well as for mutation detection. Quantitative analysis has become increasingly important as a tool for microbiologists for determining such things as viral load in samples from patients suffering from diseases such as human immunodeficiency virus infection.
Not to be confused with real-time PCR, reverse transcriptase PCR (rtPCR) uses purified RNA as the start point and uses the enzyme reverse transcriptase to produce cDNA, which can then be amplified using conventional PCR and used to examine the protein expressed by the gene of interest. Potentially, this allows investigation of both the effect of any sequence variants in a gene on its protein product, and the possible phenotypic consequences.
Detection of mutations
The search for mutations causing disease involves either the detection of a previously identified, known mutation or the screening of the gene to search for an unknown mutation. In clinical samples, the process will usually begin with the former and proceed to the latter if none of the former is found. There are now a variety of methods available for mutation detection and these can be divided into manual, low-throughput techniques and automated, high-throughput ones. The basic principles of the manual methods often form the basis of the automated ones and it is for this reason that several of the manual techniques are described here as they demonstrate principles of analysis and the properties of DNA that are exploited, however it should be acknowledged that in most large genetics laboratories these have largely been superseded by automated high-throughput systems.
Detecting known mutations
The principles of several manual techniques for detecting known mutations are shown in Fig. 43.9. The allele-specific oligonucleotide (ASO) technique utilizes synthetic oligonucleotide probes (about 19 residues) – one that is complementary to the normal and one to the mutant allele, with the site of mutation in the middle of the complementary region (Fig. 43.9A). Under appropriate conditions of hybridization, the oligonucleotides will bind only when there is complete complementarity. This technique was first used for detection of the βs allele of the β globin gene that is responsible for sickle cell anaemia. Although ASO testing can be performed directly on genomic DNA, it is more easily carried out after prior amplification of the region of interest using PCR. If a mutation (or the corresponding normal sequence) happens to be a target site for one of the many restriction enzymes, then testing for the presence or absence of a restriction site by restriction endonuclease allele recognition can be used to analyse genotypes (Fig. 43.9B). This technique was also first used for detection of the βs allele. Again, initial PCR amplification simplifies the procedure.
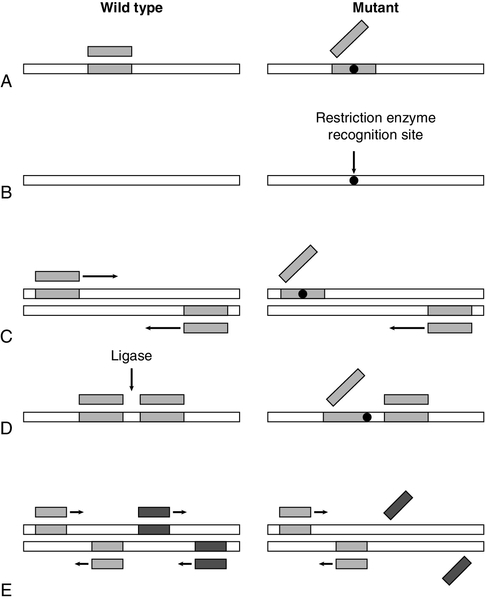
FIGURE 43.9 Some techniques for detecting known mutations. (A) Allele-specific oligonucleotides. A synthetic oligonucleotide complementary to the normal sequence will bind to wild-type, but not mutant, DNA (while an oligonucleotide complementary to the mutated sequence will bind to mutant DNA but not wild-type DNA). (B) Restriction endonuclease allele recognition. A mutation may create a recognition site for a restriction enzyme which is absent in the wild-type sequence. Consequently, digestion with the enzyme would cleave mutant DNA into two fragments, but would have no effect on wild-type DNA (similarly, a mutation may destroy a recognition site for a restriction enzyme so that wild-type DNA is cleaved, but not mutant). (C) Amplification refractory mutation system (ARMS). Primers, one of which spans the site of mutation, are designed for a PCR reaction. Primers complementary to wild-type sequence amplify normal DNA, but not mutant DNA (for confirmation, a second set of primers which are complementary to the mutated sequence is also designed so that a positive reaction is obtained with mutant DNA). (D) Ligase-mediated allele detection. Oligonucleotides flanking the site of mutation are joined by a ligase only if they are complementary to the DNA sequence. One of the oligonucleotides corresponds to a constant region, while the other is complementary to either wild-type or mutant DNA. (E) Multiplex PCR for detection of deletions. Simultaneous PCR reactions for amplification of two or more sequences detect any deletions which remove recognition sites for PCR primers.
Another method for identifying point mutations exploits the specificity of the primers used for PCR: the amplification refractory mutation system (ARMS) (Fig. 43.9C), also known as allele-specific amplification, is based on the observation that oligonucleotides that are not exactly complementary to the target DNA sequence frequently do not function as primers in PCR. The method uses two forward primers, one which is complementary to the normal sequence and one which is complementary to the mutant sequence, in combination with a common reverse primer. In yet another technique – ligase-mediated allele detection (Fig. 43.9D) – DNA ligase is used to couple two oligonucleotides at the site of a mutation. If one of the pair of nucleotides corresponds to either the normal or the mutant sequence, the ligase will only link the two oligonucleotides if there is a perfect match between the oligonucleotides and the target DNA, which has usually been amplified by PCR.
Because of the heterogeneity of genetic disease, one of the biggest obstacles to the application of DNA analysis is the requirement for simultaneous detection of multiple mutations. Since it is possible to perform several PCR reactions simultaneously (‘multiplex PCR’, Fig. 43.9E), several of the systems for the detection of mutations can be adapted to identify more than one mutation in a single assay. Alternatively, a reverse ASO method (in which oligonucleotides are immobilized and the test material is used as a probe) allows simultaneous detection of several alleles. Such a principle underlies one form of the increasingly available ‘microarray’ or DNA chip, systems of DNA analysis. There are basically two forms of microarray. In the first, target DNA samples are immobilized on the chip and are then interrogated using labelled probes. The technique requires great precision in locating the sample sequences precisely on the chip, which needs to be done using robotic delivery systems. The second form is a reverse hybridization method in which the chip production procedure is highly complex but involves synthesizing thousands of different oligonucleotide sequences in situ on glass or silicon slides; these are the probes. The target DNA is amplified in a reaction that labels it with fluorophore, and allowed to hybridize with the immobilized probes. Bound label is detected using a laser scanner, with perfect sequence matches between sample and probe demonstrating stronger signals, and the signals analysed using digital imaging software. The second method may be more appropriate to clinical analysis but the complexity of production means that chips tend to be produced by specialist manufacturers. At the moment, the instrumentation, and the chips themselves, remain costly but, as with all advances in molecular analysis, costs are likely to fall as the technology becomes more widespread.
In some inherited diseases, the gene responsible is particularly prone to deletions, copy number changes or duplications. Screening a gene by simultaneous amplification of several regions of DNA by multiplex PCR can be used effectively to screen such genes for deletions. Sequences that are absent or of decreased molecular weight can be detected simply by electrophoretic separation of the amplified sequences, although more precise methods have now been developed, for example multiplex ligation-dependent probe amplification (MLPA) (see Muscular dystrophy, p. 865).
Scanning or screening methods
To detect mutations when the precise site of mutation in a given individual is unknown, mutation screening methods were developed (Fig. 43.10). The sequence to be screened (e.g. one complete exon in a gene) is usually amplified first by PCR. One of several techniques that are capable of detecting the presence of single base differences in the sequence can then be used. In cleavage mismatch detection (Fig. 43.10A), hybridization of mutant and normal DNA strands produces a heteroduplex, and chemical or enzymatic techniques are used to cleave the strands at the site of mismatched base pairs. The size of fragments produced then allows localization of mutations within the sequence. Denaturing gradient gel electrophoresis and temperature gradient gel electrophoresis are methods that take advantage of the sudden decrease in electrophoretic mobility that occurs when a double-stranded molecule of DNA begins to dissociate (Fig. 43.10B). A homoduplex molecule (i.e. a duplex of complementary strands) will begin to dissociate and decrease its mobility at a characteristic point in a gradient of either denaturing agent or temperature. If denatured normal DNA is allowed to re-anneal in the presence of mutant DNA, heteroduplex molecules will form with a mismatch in almost complementary strands, which will begin to denature early so that the electrophoretic profile is altered. The technique of single-stranded conformational polymorphism (SSCP) is based on the fact that sequences of single-stranded DNA fold into specific conformations (Fig. 43.10C), so that normal and mutant DNA sequences can be separated by acrylamide gel electrophoresis. All of these, essentially manual, techniques are still used and remain valid techniques, but with rising workloads and the need for rapid results, they too are being increasingly overtaken by automated methods using large analytical platforms. Conformational sensitive capillary electrophoresis (CSCE) and denaturing high performance liquid chromatography (DHPLC) both utilize the principles of heteroduplex formation, with detection based on the different mobilities produced by conformational changes. Conformational sensitive capillary electrophoresis uses fluorescence detection to screen for the presence of mutations in a DNA sequence, while DHPLC uses altered retention times in an HPLC system.
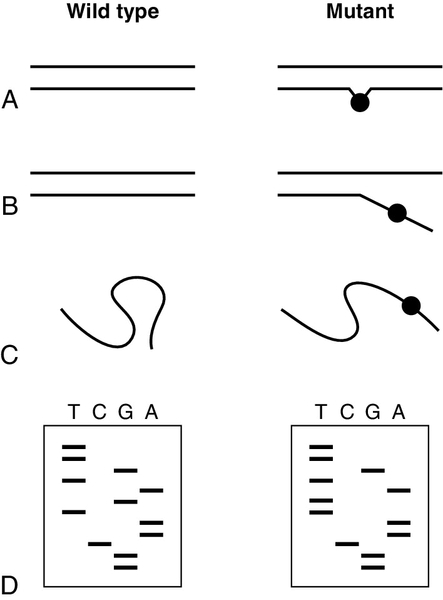
FIGURE 43.10 Detection of unknown mutations. (A) Cleavage mismatch. If a heteroduplex is formed between wild-type and mutant DNA, unpaired bases at mutation sites are susceptible to chemical modification and subsequent cleavage. (B) Denaturing or temperature gradient electrophoresis. As DNA migrates through a gel with a gradient of temperature or denaturant, the rate of migration changes suddenly when the strands begin to separate. The presence of a mutation will alter the point at which this event occurs. (C) Single-strand conformational polymorphism. Molecules of single-stranded DNA form three-dimensional structures determined by their sequence, so that in non-denaturing gels, mobility is determined by sequence as well as length. The presence of a mutation may alter the three-dimensional structure formed and hence mobility. (D) DNA sequencing (dideoxy method of Sanger). Sequencing gels are read starting from the smallest fragment. Thus, the wild-type sequence is GGCAATGATGTT and the mutant sequence is GGCAATTATGTT.
If one of the above scanning techniques reveals the presence of a mutation, DNA sequencing can then be used to identify, or confirm, the precise mutation. Two techniques for sequencing of DNA were introduced in 1977, one by Sanger and the other by Maxam and Gilbert. Both techniques share the same basic principle in that a series of single-stranded DNA molecules, each one base longer than the last, is generated. These molecules can be separated by electrophoresis to generate a ‘ladder’ from which the sequence can be read. The dideoxy terminator method of Sanger (Fig. 43.10D) uses DNA polymerase to synthesize a complementary copy of the target DNA starting from a primer annealed close to the region of interest. The enzyme can incorporate dideoxynucleosides, but chain elongation then ceases immediately because these nucleoside analogues lack a 3′ hydroxy group. DNA synthesis is carried out in four separate incubation mixtures, each containing the four deoxynucleoside triphosphate substrates (one of which is labelled) in addition to a low concentration of just one of the four dideoxynucleoside analogues. When incubation is terminated, a population of labelled DNA molecules of varying lengths will have been produced. All molecules will have the same 5′ end, but will vary in length to a base-specific 3′ end (e.g. all terminating in A if dideoxy ATP was used). Originally, it was necessary to clone a fragment of DNA before sequencing could be carried out, but sequencing now uses PCR products obtained from either genomic DNA or cDNA. The use of radioactive (32P) labelling followed by polyacrylamide electrophoresis (PAGE) gel and autoradiography has been superseded by DNA analysers which use four different dyes for the four different nucleosides, and automated read outs. The availability of these instruments, plus automated software for mutation and variant ‘calling’, makes sequencing more attractive for detecting mutations, and some laboratories use automated DNA analysers and sequencers as their front-line mutation detection system for some genes.
Tracking of mutant genes
When the gene causing a disease has not been identified, or it is not feasible to identify the precise mutation causing a defect, it may nevertheless be possible to predict, by gene tracking, whether a particular individual is affected provided a closely linked marker is available. Even when a disease gene has been identified and cloned, because the gene screening methods described above are not always 100% sensitive, gene tracking methods may still be useful to identify affected individuals. To be useful, a marker must exhibit a degree of polymorphism so that it is possible to distinguish between alleles associated with normal and mutant genes. Thus for a marker to be informative, an individual who is heterozygous for the disease locus must also be heterozygous for the marker. For genetic analysis, information on phase is also required, that is, it is necessary to determine which allele is linked to (tracks with) the mutation in an individual who is heterozygous. Some of the situations that may occur with a recessive disease are illustrated in Figure 43.11. Family 1 is fully informative and the disease gene is associated with allele a, so that the fetus would be affected only if it were also homozygous for allele a. In family 2, the analysis is informative only for the mother (in whom the mutant gene is linked to allele b), so that there is a 50% probability that the fetus can be predicted to be unaffected and a 50% chance that the fetus will be predicted to be at 50% risk. In family 3, the analysis is completely uninformative, since both parents are homozygous for the marker. In family 4, a marker with greater polymorphism is used, which is entirely informative.
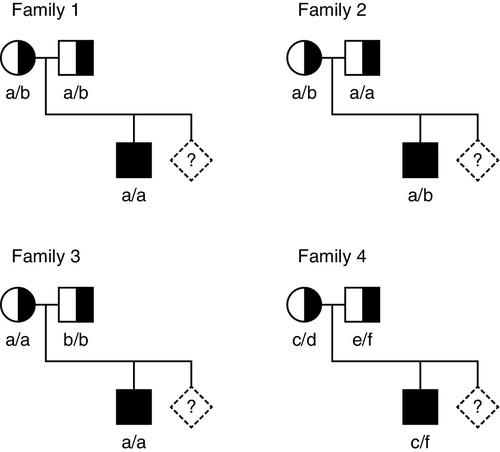
FIGURE 43.11 Linkage analysis for a recessive condition. Linkage of alleles a, b, c, d, e and f is shown in four families. Half-shaded symbols represent heterozygotes for the condition. Fully shaded symbols refer to affected individuals and diamonds refer to a fetus for which diagnosis is requested.
Often a polymorphic marker can be found within the gene of interest, but if a linked marker outside the gene is used, there is an increased possibility of errors being made as a result of crossover. Even with intragenic markers, in large genes such as the dystrophin gene, there is a distinct possibility of crossover occurring between a marker and a mutation site. Similarly, there can be a degree of uncertainty as to whether an affected member of a pedigree has a new disease-causing mutation; again this may be particularly true in cases of muscular dystrophy. The risk of incorrect diagnosis can be minimized by the use of multiple markers, but diagnosis will nevertheless be based on an estimate of probability. Other disadvantages of linkage studies are the expense, time and need to test other family members, so these techniques are used mainly in specialist laboratories. The developments of ‘next generation’ sequencing described below are likely to allow great improvements in identifying, and tracking, pathogenic mutations in such diseases and pedigrees.
There are several types of polymorphic marker that are suitable for tracking inheritance of particular alleles within families. Polymorphism at restriction sites (Fig. 43.12A) has been used extensively for linkage analysis, both for identification of the genes causing disease and for genetic analysis in families. Throughout the human genome, about one base in 1000 is polymorphic and around one in six random base changes creates or abolishes a restriction site. The majority of these polymorphisms are of no consequence to the organism, but their detection with restriction enzymes has provided what was, until recently, the most important technique for genetic mapping and linkage studies. Restriction fragment length polymorphisms (RFLPs) can be identified either by digestion of genomic DNA with the enzyme and subsequent use of a probe to identify fragments separated by electrophoresis or by PCR amplification of the region surrounding the restriction site, followed by restriction digestion and direct visualization of fragments after electrophoresis. A limitation of RFLP analysis is that the maximum polymorphism at any one site, can be no more than 50% (i.e. presence or absence of the site), and strategies that use relatively small numbers of SNPs have now been largely superseded.
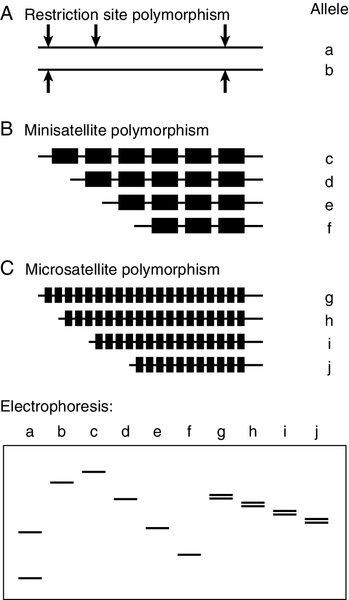
FIGURE 43.12 Polymorphic markers used for linkage studies. (A) Alleles a and b are produced by polymorphism at a restriction site; (B) c, d, e and f, by polymorphism in the number of repeats in a minisatellite; (C) g, h, i and j, by polymorphism in the number of repeats in a microsatellite. Electrophoresis of a produces two small fragments after digestion, while b is not cleaved and remains as one large fragment. Electrophoresis of alleles c–j detects different sizes of mini- and microsatellites, but the latter may be complicated by separation of the two DNA strands (see text).
However, the use of SNPs for gene tracking has now come full circle as a consequence of the Human Genome Project and the production of a dense SNP map of the human genome. This knowledge, combined with the development of high-throughput microarray technology, means that it is possible to obtain SNP chips linked to large SNP databases; the use of large numbers of SNPs makes these chips more informative.
The discovery of hypervariable sequences in DNA consisting of a variable number of tandem repeats (VNTRs), with considerable polymorphism in the number of repeats, has found widespread application. Such sequences consist of short (10–60 base pairs) oligonucleotide sequences (‘minisatellites’) repeated 20–100 times, so that PCR amplification spanning the region (or excision of the region with a restriction enzyme) produces fragments which vary considerably in size in different individuals (Fig. 43.12B). The high degree of heterozygosity makes these VNTR loci particularly informative and, since they are inherited in a simple Mendelian fashion, they can be used in just the same way as RFLP analysis. Detection of these repeat sequences can be used, for example for determining zygosity in twins.
Similar repetitive sequences, with around 10–60 copies of motifs consisting of 1–4 bases (short tandem repeats or ‘microsatellites’) have been identified more recently (Fig. 43.12C). Unlike minisatellites, which are frequently located near the ends of chromosomes, microsatellites are found scattered more evenly. One of the most common microsatellites, the (CA)n repeat (where n is roughly 15–30), occurs on average every 30 000 bases. Polymorphism in the length of these sequences can be detected by PCR amplification and used to track mutant genes in families. Dinucleotide repeats such as AC may separate into two bands on electrophoresis because the AC strand migrates faster than TG. Because fainter bands, which are probably generated during amplification, may also be apparent, there are advantages to using microsatellites with tri- and tetranucleotide repeats that are less susceptible to these problems, though the latter occur considerably less frequently in the genome.
Using a multilocus probe capable of hybridizing under low-stringency conditions to VNTR sites at several chromosomal sites, digestion with restriction enzymes gives a characteristic profile for each individual. The technique of ‘DNA fingerprinting’ has found most widespread use in forensic studies (e.g. the identification or elimination of crime suspects using samples of blood or semen) and in proving or disproving family relationships in civil cases (e.g. confirmation of maternity or paternity).
Analysis of these repeat sequences can now be performed quite straightforwardly by determining fragment length using capillary electrophoresis, a technique with greater accuracy than traditional gel based electrophoresis.
Next generation sequencing
One of the challenges in clinical genetics is the search for pathogenic mutations, where several genes may be implicated in a disease, or a group of disorders, for example, the peripheral neuropathies, and the clinical phenotype does not necessarily point to any one gene as the prime suspect, or where a large pedigree needs to be searched for potential disease causing mutations where one has not yet been identified in any individual of that pedigree. Traditional methods would involve testing one gene, or gene locus, after another. It would, however, potentially be more useful to screen all of the likely candidates, from all the members of a pedigree on a single sequencing run. The new high throughput technologies collectively called ‘next generation’ (or ‘massively parallel’ or ‘deep’) sequencing may well be the answer to this diagnostic problem. Several different platforms have been developed, with different sequencing technologies, but all the systems employ roughly the same essential steps.
Step 1. Genomic DNA is fragmented using restriction enzymes.
Step 2. The fragments are enriched for the regions of interest.
Step 3. All the fragments are sequenced in parallel.
Step 4. The results are aligned with a reference sequence to search for any variants (which at this stage may number in the thousands).
Step 5. The bioinformatics step, comparing these variants to a polymorphism or SNP database (which may take the number down to the hundreds).
Step 6. Variant calling, to determine if the variants detected are likely to be pathogenetic or not.
Another common feature of these new platforms is that none of them use traditional Sanger type sequencing using dNTPs. Some of the techniques include:
• use of reversible dye-terminators. Each molecule of DNA is amplified as a clone and fluorescently labelled nucleotides allow identification of each nucleotide as it is added before the dyes are removed prior to the next addition
• sequencing by ligation, whereby dye labelled oligonucleotides of known sequence are annealed and ligated to clones of amplified DNA immobilized on beads or glass slides
• ion-semiconductor sequencing, where the terminal incorporation of nucleotides uses traditional sequencing chemistry but detection of the incorporated nucleotides is achieved using a supersensitive ion sensor, essentially a form of pH meter, which detects the hydrogen ions released when each base is added, each cycle (taking only seconds) uses a different nucleotide but runs of the same nucleotide in the sequence will all be incorporated in the same cycle, giving a proportionally higher signal.
Even in the few years since the last edition of this book, both run (or ‘read’) times and cost per read have come down considerably, as has the size of the instruments, with recent ones very definitely being bench top, so why is it that these new techniques cannot automatically replace all the others currently in use? Over time, they may well do so, but at the moment the ability to sequence a whole genome or exome is not necessary for the detection of common pathogenic mutations in well characterized disorders, which tend to benefit from a targeted approach.
THE APPLICATIONS OF DNA ANALYSIS
The clinical application of DNA analysis has grown enormously since the first edition of this book, demonstrated by the increase, in both number and size, of molecular and clinical genetics departments in our hospitals.
Diagnosis of index cases
The very nature of inherited disease, with its implications for the families of affected individuals, means that most molecular testing now takes place in specialized genetics laboratories. These laboratories work closely with clinical geneticists and genetic counsellors. Consequently, any clinical biochemistry laboratory undertaking molecular testing must be aware of the consequences of a positive diagnosis. However, diagnosis of inherited metabolic diseases (e.g. amino acid and organic acid disorders) has, for many years, been undertaken in biochemistry laboratories, and the initial diagnosis of an inherited disease is still usually based on clinical history, physical signs and non-genetic laboratory tests. Confirmation of the diagnosis can then often be obtained by a specific enzyme assay or protein study, but such assays frequently require the use of cell cultures, with their accompanying problems. Consequently, in an increasing number of diseases, DNA testing is now used for the diagnosis of index cases. These are often diseases in which (a) partial or complete gene deletions, which are relatively easy to detect, are frequent (as in muscular dystrophy) or (b) the defective protein has not yet been characterized or where it is difficult to devise methods for direct analysis of the protein (as with cystic fibrosis). One other potential advantage of DNA tests for diagnosis of index cases is that mutant genes are usually present in all nucleated cells, so that in diseases in which expression of the gene is limited to organs such as liver or kidney, biopsy can be avoided.
In the use of DNA analysis for diagnosis in index cases, it is important that accurate and reliable data exist on the relationship between genotype and phenotype.
Prenatal diagnosis
Analysis of fetal DNA, often with a view to selective termination of an affected pregnancy, can be used to determine the presence of severe or potentially fatal genetic disease. Such testing is most likely to be carried out when the parents have previously had an affected child, when they have been identified as carriers, or when one of them is affected by a dominant condition.
Although fetal blood samples can be used to detect many inherited diseases – particularly those that affect proteins in red blood cells or plasma proteins – DNA analysis can be carried out without taking tissue samples from the fetus. In the earliest prenatal diagnoses of genetic disorders, amniotic fluid cells obtained by amniocentesis at 16–18 weeks of gestation were used. However, it is now more common to use chorionic villus sampling (CVS), carried out at 8–10 weeks (see Chapter 22). Potential problems with prenatal diagnosis are contamination with maternal DNA and – especially where linkage studies are being carried out – uncertainty concerning paternity. Analysis of mini- or microsatellites can help avoid both of these problems. Where the diagnosis relates to an X-linked recessive disorder, if the fetus is shown to be female, either by cytogenetic analysis or a specific molecular test for identifying the Y chromosome, no further CVS testing may be necessary, as the fetus will, at most, be a carrier and this can be determined once the child is born.
Use of PCR makes testing of individual cells possible, so that it is possible to test fertilized embryos and implant only those known to be unaffected by a particular disease. This procedure has been carried out successfully with parents who are both heterozygotes for cystic fibrosis. The implications of this procedure are considerable, not least because many parents find selective abortion an unacceptable means of avoiding affected children. The use of such techniques for in vitro fertilization (IVF) is controlled, in the UK, by law, and is overseen by the Human Fertilisation and Embryology Authority.
A recent advance has been the ability to detect cell-free, fetal DNA in the blood of the mother, bringing with it the possibility of testing the fetus for inherited disease without the need for invasive testing. Such testing does come with risks, mainly relating to avoiding contamination with the mother’s own DNA. However, where the fetus is male, looking for markers carried on the Y chromosome only overcomes this problem. The use of DNA methylation ratios to separate fetal from maternal DNA has also been described. The use of fetal DNA in this way is very much in its early stages and is currently used primarily for determining fetal gender but is also being looked at as an alternative, and early, method of screening for Down syndrome.
Screening
The word ‘screening’ is used in various ways in medicine, and in the section on detection of mutations above, it was used in the context of examining a whole gene to find disease-causing mutations in patients with a clinical diagnosis of a genetic disease. In this section, the term is used in the context of testing healthy or presymptomatic individuals for molecular evidence of disease.
Screening of individuals
Screening can be ‘targeted’ in that individuals from families with strong family histories of a disease (high risk) are included and it may involve presymptomatic or carrier (heterozygote) testing. Such screening is often applied to determining the risks of developing a particular form of cancer, for example as in breast cancer screening. For women with several close relatives with the disease, there is a targeted screening programme based on DNA analysis. Although familial breast cancer represents a small percentage of cases of the disease, screening high-risk individuals is effective and involves examining the entire BRCA1, BRCA2 and TP53 genes.
Population screening
Population screening involves making available to all, on an equitable basis, testing for certain diseases or conditions. Examples of such programmes include antenatal screening for Down syndrome and those for cervical and breast cancer screening in women (not the same as the targeted testing described above). Currently, there are no such programmes that use DNA analysis in a primary testing strategy, although proposals have been made to use detection of fetal DNA in maternal serum for screening for Down syndrome, as mentioned above.
The criteria that must be satisfied for a successful screening programme are the same, regardless of whether traditional chemical analysis or DNA techniques are used (or, indeed, imaging or cytological testing) and have changed little in recent decades. All screening programmes are fraught with ethical and organizational problems, but none of these is peculiar to the analytical technique employed. The importance of education and the provision of counselling services for the success of a screening programme have been repeatedly emphasized, but the obtaining of informed consent, the reliability of methods (i.e. numbers of false positives and false negatives), prevalence of disease in the population and the existence of a clearly recognized advantage to pre-symptomatic diagnosis (e.g. the possibility of avoiding precipitating factors in the environment) must be considered. In the UK, such screening has, since 1996, been overseen by the National Screening Committee, which also advises the government on the relevant issues.
Newborn screening to detect affected individuals with phenylketonuria and congenital hypothyroidism has been widely performed for some years using conventional biochemical tests on dried blood spots obtained by heel prick in the first two weeks of life. This scheme has now been extended to include testing for cystic fibrosis (see later section) haemoglobinopathies and medium chain acyl-CoA dehydrogenase deficiency (MCADD). While DNA diagnostic tests can also be carried out on blood spots, the programme still uses biochemical tests as the ‘frontline’ screening test. DNA testing can be used to follow-up positive screening results but currently is still employed mainly to identify the mutation(s) present in the child, rather than confirmation of the diagnosis.
At the present time, there are no plans to use DNA testing for primary screening of newborn infants. There are several reasons why this is not appropriate yet. In many genetic diseases, there can be large numbers of disease-causing mutations that produce the same biochemical (or haematological) effects. Similarly, not all mutations and variations in a gene will be pathogenic. Also, some mutations show variable penetrance or expressivity, so that whole gene screening, for example by sequencing, might create problems by identifying infants who have gene mutations but who will not necessarily develop the disease being screened for. Currently, it is still more efficient to screen by testing for the gene products. However, as DNA arrays and microchips become more accessible and cost-effective, molecular screening may become more widespread, providing there are accurate data on the genotype/phenotype correlations.
Pharmacogenetics
An area of laboratory testing in which biochemistry laboratories may find themselves involved is that of pharmacogenetics. Pharmacogenetics is concerned with identifying genetic variations that affect an individual’s response to drugs. Initially, the term was used in respect of variations in drug metabolizing enzymes, but it has now been expanded to include polymorphisms in drug receptor, drug transporter and ion channel genes. Pharmacogenomics, on the other hand, refers to the (generally commercial) application of genomic technology in drug development and therapy. The two terms are often used interchangeably and there is some difference of opinion as to whether or not this is an acceptable practice. However, even though the differences may appear to be semantic, in general, pharmacogenomic studies in drug discovery regimes mainly aim to identify suitable drug targets and are concerned with multiple genes and phenotypes, whereas pharmacogenetics aims to identify variants in individual genes with the aim of ‘personalizing’ drug dosage and thus reducing adverse drug reactions. These are responsible for high levels of therapeutic morbidity and mortality worldwide every year, with an associated cost to the health services of millions of dollars each year. Biochemical methods of identifying poor metabolizers (see below) are often cumbersome and demanding, using metabolic ratios that measure the relative concentrations of parent drug and metabolites in blood and urine.
The term ‘pharmacogenetics’, coined by Vogel in 1959, was originally brought to prominence by Kalow in descriptions of the variation in response to the muscle relaxant suxamethonium seen in patients with serum cholinesterase (butyrylcholinesterase) deficiency. Another of the earlier described pharmacogenetic effects was that observed with the enzyme N-acetyltransferase 2 (NAT2), where patients treated with the antituberculosis drug isoniazid were found to show variation in the rate at which they metabolized the drug: they were either slow, intermediate or rapid acetylators. The largest group of drug metabolizing enzymes is the cytochrome P450 (CYP P450) ‘superfamily’, which accounts for the metabolism of well in excess of 100 commonly used prescription and over-the-counter drugs. Of the large number of CYP P450 enzymes, six account for close to 90% of the drugs metabolized by this family. Of these, CYP3A4/5 account for around 50% but the most extensively characterized is CYP2D6, debrisoquine hydroxylase, accounting for 30%. It is the metabolizing enzyme for a large number and wide variety of drugs including antipsychotic, antiarrhythmic and antihypertensive drugs. Many drug metabolizing enzymes are now known to be highly polymorphic and many of them also demonstrate codominant inheritance, such that heterozygotes for normal and certain variant alleles show intermediate phenotypes. Poor metabolizers can be affected in a variety of ways, depending on whether it is the parent drug or its metabolite that is the pharmacoactive moiety. Thus, poor metabolizers may suffer the adverse effects of overdosing on a standard drug dose if the parent drug is active, or a poor therapeutic response if the metabolite is active. In the case of CYP2D6, more than 75 polymorphisms have been identified so far, and the prevalence of poor metabolizers and the frequency of different causative alleles vary significantly in different geographic and ethnic groups. Thus, in Caucasians, poor metabolizer status is found in 5–10% of individuals and is caused in nearly 90% of those cases by one of just three variant alleles, CYP2D6*3, CYP2D6*4 and CYP2D6*5. (By convention, the commonest, or normal, allele of a gene is denoted as *1.) ‘Extensive metabolizer’ is the name given to individuals with an expected or normal response to a drug, and while these are mainly *1 homozygotes, some of the other alleles, such as the *2, also produce an extensive metabolizer phenotype. Intermediate metabolizers are thought to represent heterozygotes with extensive and poor metabolizer alleles.
The molecular variation responsible for the different alleles includes not only simple changes such as SNPs, small deletions and insertions but also whole gene deletions and duplications. The phenotypic effect of these differences can be on the concentration of enzyme produced or on its function, depending on the location of the polymorphism in the translated gene product. In the case of CYP2D6, a duplication of the *2 allele leads to an ultra-rapid metabolizer phenotype.
Other clinically important drug metabolizing enzymes include thiopurine S-methyltransferase (TPMT) and uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1). Thiopurine S-methyltransferase is involved in the metabolism of the immunosuppressant azathioprine and the antileukaemia drug 6-mercaptopurine; biochemical testing for TPMT phenotype has been available for some time but the role of genotyping has yet to be firmly established. Variants of UGT1A1 are responsible for Gilbert syndrome (unconjugated hyperbilirubinaemia) and Crigler–Najjar disease but the enzyme is also involved in the metabolism of the anticancer drug, irinotecan, and prospective genotyping may be useful in the future for avoiding some of the side-effects that can occur during treatment with this drug.
With the identification of the genes and sequences for an increasing number of drug metabolizing enzymes, analysis using the new techniques for rapid, high-throughput DNA testing should allow early identification of many of the variants, thus allowing altered drug dosage prior to, or shortly after, therapy commences, and so avoiding dangerous and costly adverse drug reactions.
Inherited diseases – some examples
Single gene disorders
The diseases most often thought of as ‘genetic’ tend to be those that arise from mutations in a single gene. Some disorders may arise from mutations in any one of a collection of genes related to a particular metabolic pathway, i.e. they may have monogenic causes but are not necessarily single gene diseases.
α1-Antitrypsin deficiency
The function of α1-antitrypsin (α1AT) is to inhibit neutrophil elastase and other proteases. The rate of association between α1AT and elastase is rapid, but after cleavage at the reactive site (methionine at position 358) of α1AT by the elastase, the molecule undergoes a radical change in structure that prevents dissociation of the elastase, thus inactivating it. The gene coding for α1AT (the protease inhibitor or PI gene) spans 12 200 bases and is transcribed into a protein with a single chain of 394 amino acids.
α1-antitrypsin displays considerable polymorphism, with around 75 alleles having been detected so far. The variants are inherited in an autosomal codominant fashion, so that both alleles are expressed independently and can be detected in serum. Many of the variants, which are identified by letters of the alphabet depending on their electrophoretic mobility, function normally. The Z variant is of greatest clinical significance. In northern Europeans, around 5.3% of the population are carriers (i.e. are MZ heterozygotes) and about 1 in 2000 live births are ZZ homozygotes. Although deficiency can usually be detected by quantitation of serum α1AT, phenotyping by isoelectric focusing is more informative and reliable.
Secretion of α1AT, which is synthesized in the liver, is markedly reduced in ZZ homozygotes because the mutant protein forms insoluble polymers, which accumulate in the endoplasmic reticulum of hepatocytes. The Z variant results from a point mutation converting a lysine residue in normal (M) protein to a glutamic acid one at position 342, which is at the base of the reactive centre loop. Juvenile cirrhosis occurs in 3–10% of ZZ homozygotes as a result of damage caused by the intracellular aggregates, though a much higher percentage are affected if a sibling has liver disease. The reasons for the incomplete penetrance of this condition are not known: intrauterine infection, gut-derived proteases, autoimmunity, fever and subclinical hepatitis have been suggested as possible exacerbating factors. Additional genetic factors, possibly in chaperone proteins responsible for directing misfolded protein to the secretory pathway, or other proteins involved in α1AT clearance have also been suggested.
Although other protease inhibitors are present in the lungs, α1AT contributes over 90% of the inhibitory activity capable of blocking neutrophil elastase. In the absence of α1AT, neutrophil elastase rapidly degrades tissue matrix components in the lung so that individuals with a markedly reduced plasma concentration of α1AT are prone to develop emphysema. As a result of reduced hepatic secretion of α1AT, the plasma concentration of this protein is reduced to about 15% of normal in ZZ individuals. Not all of these individuals are affected by disease, but 60–70% of ZZ homozygotes who smoke cigarettes develop pulmonary emphysema in the third or fourth decade of life. Smoking compromises the defences of the lung further because free radicals in cigarette smoke oxidize methionine at the reactive site of α1AT, drastically lowering its ability to inhibit neutrophil elastase.
Of the other mutations affecting the α1AT gene, the S mutation (changing the glutamic acid at position 264 to valine) is more common than Z, with an allele frequency of 2–4% in northern Europeans, but the consequences are less serious. Although some degradation of the S variant occurs in the liver and the serum α1AT concentration is reduced, S homozygotes are not considered to be at increased risk of liver or lung disease, although inheritance of the S allele with an allele causing severe deficiency (such as Z) confers a mild risk of emphysema. A number of other rare mutations (including the ‘null’ mutation) are associated with a decrease in plasma α1AT activity to a level that confers a risk of emphysema, and some others are characterized by accumulation of mutant protein in hepatocytes.
DNA-based techniques for identification of mutations in the PI gene complement phenotypic studies. Prenatal diagnosis is possible in juvenile cirrhosis families, where the causative mutation can be detected in the index case. DNA sequencing is the gold standard for detecting small deletions and point mutations, and this is readily available owing to the relatively small size of the gene. Various SNP detection techniques have also been developed for common mutations (e.g. Z and S), which have the potential to be used in screening programmes. While screening for this condition has been advocated, it has not been taken up widely owing to concerns regarding the variable penetrance of the condition, and the limited efficacy of interventions such as advice on smoking cessation.
Cystic fibrosis
Cystic fibrosis is inherited in an autosomal recessive manner. The homozygous condition is associated with defective exocrine secretion and consequent malabsorption with chronic obstructive pulmonary disease. The disease has a prevalence of about 1 in 2500 live births in northern Europe and a carrier frequency of 1 in 25, though it is much less common in other populations. For many years, laboratory diagnosis relied on the demonstration of raised chloride (and, optionally, sodium) concentrations in sweat. About 77% of affected individuals can be identified using the sweat test at two years of age and about 95% at 12 years. The test is far from ideal, because it is technically difficult to perform; not all patients with compatible clinical features have a raised sweat chloride and some individuals with a raised sweat chloride have no clinical features of cystic fibrosis. In one report, up to 40% of patients referred to cystic fibrosis centres had been wrongly diagnosed because of false positive or false negative sweat tests. Serum immunoreactive trypsin concentrations tend to be higher in affected newborns, forming the basis of a UK screening programme that can detect at least 95% of cases.
The gene affected in cystic fibrosis was mapped to 7q31 using linkage analysis. The gene itself was cloned in 1989 and named the cystic fibrosis transmembrane conductance regulator (CFTR). It has a size of approximately 250 kb with 27 exons and the CFTR protein is 1480 amino acids with a molecular weight of 168 kDa. The primary role of the CFTR protein is to form a chloride channel that reduces intracellular chloride. It appears to consist of two transmembrane domains, two ATP binding regions or nucleotide binding folds, and a regulatory domain. Phosphorylation of the regulatory domain by protein kinase A results in the opening of the chloride channel.
Over 1500 pathogenic mutations have been identified in the CFTR gene, although the great majority of them are extremely rare. The mutations include frameshift, nonsense, missense and splice site mutations and deletions. However, the commonest, and the first to be identified, is a 3-bp deletion in exon 10 of the CFTR gene that results in the loss of a phenylalanine codon at position 508. This mutation, Phe508del (Phe being the three letter code for phenylalanine and ‘del’ represents a deletion) represents around 70% of cases of cystic fibrosis. The frequency of this mutation in different populations varies markedly, however, and in Europe its frequency increases along a South East to North West gradient.
Detection of several cystic fibrosis mutations by multiplex PCR can be performed simultaneously using an ARMS assay (Fig. 43.13). In this assay, the common Phe508del mutation is detected together with 28 or 32 other mutations, depending on the kit manufacturer (e.g. the mutation causing substitution of Gly-551 by aspartic acid (Gly551Asp), another that converts Gly-542 to a stop codon (Gly542X) and a splice site mutation substituting T for G immediately after the last nucleotide in exon 4 (621 + 1G > T)), but chosen to be appropriate for the local population. This strategy will, for most populations, identify the mutation responsible in more than 80% of cases. If the front-line test fails to reveal the mutation, additional mutations of progressively lower frequency are tested for and, ultimately, a full gene sequence may be the only way to identify a rare or new mutation.
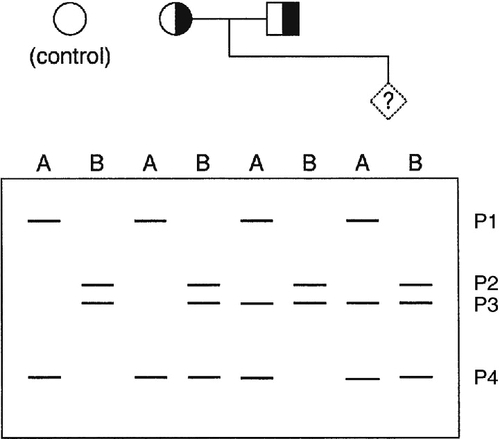
FIGURE 43.13 Simultaneous detection of the common mutations in the cystic fibrosis gene by ARMS analysis. Bands P1, P2, P3 and P4 represent the product amplified using primers complementary to the sequences spanning the 621 + 1G > T, G551D, G542X and F508del mutation sites, respectively. Two reactions are carried out on DNA from each individual. In A lanes, P1 and P4 primers are complementary to the normal sequence, while P2 and P3 are complementary to the mutant sequences. In B lanes, P1 and P4 primers are complementary to mutant sequences while P2 and P3 primers are complementary to normal sequence. Thus, the mother is a carrier for the F508del mutation and the father is a carrier for the G542X mutation. The fetus has inherited both deleterious mutations (i.e. is a compound heterozygote). Adapted from Ferrie R M, Schwartz M J, Robertson N H et al. 1992 Development, multiplexing, and application of ARMS tests for common mutations in the CFTR gene. American Journal of Human Genetics 51: 251–262, with permission.
Muscular dystrophy
Duchenne muscular dystrophy, one of the most common X-linked diseases (prevalence about 1 in 3000 live male births), results from mutations in the dystrophin gene and is characterized by progressive proximal muscle weakness in early childhood. Rarely, females can be affected, usually as a result of X-autosome translocations or skewed X inactivation. Symptoms are also apparent in 2–3% of female carriers (‘manifesting carriers’) caused by a non-random X inactivation. The diagnosis of this condition is often beyond doubt clinically, but confirmation is usually required because of its gravity. Plasma creatine kinase activity is markedly raised (often to 50–100 times normal), providing a useful confirmatory test. Becker muscular dystrophy, a less severe condition with onset in late childhood (affecting about 1 in 30 000 newborn males), is an allelic disorder, that is, it also results from mutations in the dystrophin gene.
The dystrophin gene was discovered in 1986 and turned out to be an unusually large gene, currently the largest known gene in man, spread over around 2.5 million bases. The size of the gene may be one of the reasons why it is particularly prone to new mutations: it has been calculated that a third of all cases of Becker muscular dystrophy arise from new mutations. Although at least 99% of the dystrophin gene is accounted for by introns, there are nevertheless 79 exons coding for a mRNA of 14 000 bases. The dystrophin protein has a molecular weight of about 400 000 kDa and it is expressed primarily in muscle cells. Dystrophin is normally localized to the inner surface of muscle cell sarcolemma and it is tightly linked with an oligomeric complex of glycoproteins that provides a linkage between the cytoskeleton and the extracellular matrix. When muscle biopsies of patients have been examined, it has been found that dystrophin is undetectable in patients with Duchenne muscular dystrophy, and present, altered in size or quantity, in patients with Becker muscular dystrophy.
In many patients (about 60%), the mutations causing disease are deletions or, less commonly (5%), duplications of exons. The remaining 30–35% are point mutations. Although the situation is undoubtedly more complex, in many cases it appears that Duchenne muscular dystrophy results from mutations that cause frameshifts, while the mutations causing Becker muscular dystrophy tend to be in-frame. Molecular diagnosis of muscular dystrophy has advanced considerably with the invention of PCR and its subsequent developments. Initially, diagnosis relied on RFLP linkage and cDNA probe analysis by Southern blotting. This technique was cumbersome and not always successful in identifying the disease mutation. An early use of PCR to detect deletions is shown in Figure 43.14, where ten pairs of primers were used to scan the whole dystrophin cDNA, allowing identification of some 65% of affected individuals. This was followed by the use of multiplex PCR, where deletions were detected by the failure of deleted exons to amplify in the reaction. Multiplex PCR has now been superseded as a front-line test by a technique called multiplex ligation-dependent probe amplification (MLPA). This technique involves the amplification of specifically hybridized probes and enables the rapid identification of a deletion or duplication of any of the 79 exons in the dystrophin gene. It is particularly effective in determining the carrier status of females and has been used to identify the defect in families who appeared to be normal by earlier techniques. A series of characteristic ‘printouts’ is shown in Figure 43.15. It is also possible to detect point mismatch. In families in which the defect in one allele is not due to deletion, either multiplex PCR or RFLP analysis can be used for prenatal diagnosis. Multiplex PCR can also be carried out on dried blood spots for neonatal screening. However, in the absence of any effective treatment, this will not benefit the child being screened. Any advantage conferred by the opportunity for counselling to avoid further affected children must be balanced against the effect on the parents of having to explain to their son that he has a fatal illness, and, indeed, the effect of this knowledge on the child himself.
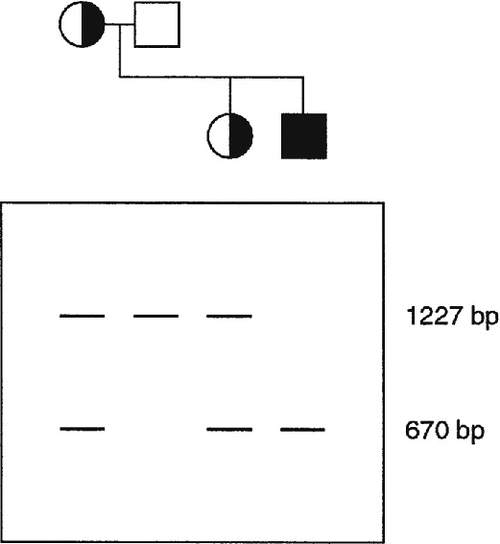
FIGURE 43.14 PCR analysis to detect deletions in the dystrophin gene. Using PCR amplification to amplify the cDNA spanning exons 43–51, the mother is shown to be a heterozygote for the normal allele (band of 1227 bp) and a mutant allele (band of 670 bp) with a deletion of exons 45–48. The father is homozygous for the normal allele but the son expresses only the mutant allele. His sister, like their mother, is a heterozygote. Adapted from Roberts R G, Bentley D R, Barby TF et al. 1990 Direct diagnosis of carriers of Duchenne and Becker muscular dystrophy by amplification of lymphocyte RNA. Lancet 336: 1523–1526, with permission.
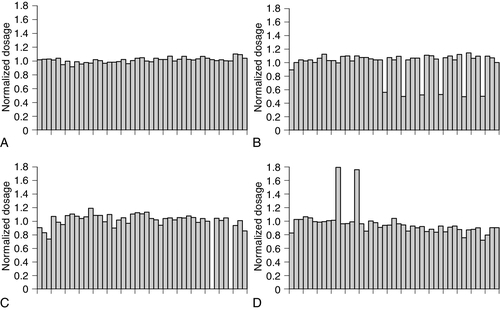
FIGURE 43.15 Results of MLPA analysis of the dystrophin gene showing normalized ratios of the hybridization signal: (A) is from a normal individual; (B) shows deleted exons in a female carrier; (C) shows deleted exons in an affected male and (D) shows duplicated exons in an affected male. In (B) (C) and (D) the affected exons are contiguous in the gene but not in the readout. Courtesy of the Bristol Genetics Laboratory, North Bristol NHS Trust.
Huntington disease
In Huntington disease, progressive chorea (irregular involuntary movements) and dementia lead to death on average 17 years after the onset of symptoms. The condition is autosomal dominant with a prevalence in the UK of 3–7 per 100 000. The trait shows complete penetrance, but age-dependent expressivity: only 10% of affected individuals have symptoms by 30 years, but this proportion reaches 95% by 70 years. Thus, most individuals who are at risk reach the age at which they may wish to have children without knowing whether or not they will develop the disease.
As a result of exhaustive research by several groups, the genetic defect causing Huntington disease was first located on chromosome 4; the gene (named ITI5) has since been pinpointed to 4p16. Inheritance of many disorders, including fragile X syndrome, myotonic dystrophy and spino-bulbar-muscular atrophy (Kennedy disease), is linked to a mechanism in which increasing expansion of triplet repeats eventually leads to disease. Huntington disease is caused by this type of mutation, with expansion of a CAG repeat in exon 1. The variable age of onset and severity of the disease correlates with the extent to which the CAG repeat has been expanded. Normal alleles contain up to 26 repeats, are stable and not associated with disease, while disease genes contain more than 39 repeats. Those with 27–35 repeats have the potential either to decrease or to expand and thus become disease alleles: they are ‘mutable’. Alleles with 36–38 repeats show reduced penetrance with some heterozygotes never developing symptoms. Disease-associated alleles containing more than 39 repeats are prone to large increases in repeats from one generation to the next, which results in a phenomenon called anticipation, where symptoms develop at a younger age.
Before the identification of the mutation, presymptomatic diagnosis using RFLP analysis and linkage studies was performed with a probability that depended on the number and informativeness of family members who could be tested. Not only did this technique leave a degree of uncertainty over the diagnosis, but diagnosis of one individual might depend on analysis of samples from other members of the family who did not wish to be tested. This technique is now used only rarely and the diagnosis is now made using PCR analysis to measure directly the size of the CAG repeat region.
The ability to identify patients with Huntington disease many years before symptoms become apparent raises some of the ethical problems of DNA testing in their most acute form. Potential psychological problems for individuals without symptoms knowing that they have a fatal disease must be considered and appropriate counselling provided. The risk of stigmatization, with difficulties in employment and obtaining insurance, makes absolute confidentiality of results crucial. The fact that the disease is transmitted in a dominant fashion also raises issues where members of one generation wish to be tested but those of another do not, as a positive result in a child almost certainly implies a positive result in the parent (depending on the repeat number). Consequently, in the UK at least, predictive testing is only undertaken using a robust protocol involving close working between clinical genetics and laboratory genetics teams.
Multifactorial and polygenic disease
While much progress has been made in understanding single gene disorders, and developing techniques for their diagnosis, such diseases are still rare in most populations. The current challenge lies in understanding those common diseases that have a polygenic or a multifactorial basis. Where a disease or disorder arises from mutations in a small number of genes, each of which contributes, it can be said to be polygenic. Diseases where the role of genetic factors is combined with environmental triggers can be thought of as multifactorial. However, in reality, conditions such as diabetes and essential hypertension, which affect millions of people worldwide, can be both and remain the focus of a lot of current investigation.
Atherosclerosis
Atherosclerosis is one such multifactorial disease and the recognition of a correlation between plasma cholesterol concentration and coronary artery disease has provided the background for genetic studies of several candidate genes. (A search for atherosclerosis on the Online Mendelian Inheritance in Man (OMIM) website gives more than 200 ‘hits’.) Environmental factors such as diet are clearly important for determining plasma cholesterol concentrations, but it has been estimated that at least 50% of the population variance of cholesterol concentration has a genetic basis, with only a small group being monogenic (requiring no, or virtually no, environmental triggers for their expression). Transport of lipids between tissues is a complex process involving formation, modification and clearance of lipoprotein particles (see Chapter 37). The plasma concentration of one of these particles, low density lipoprotein (LDL), is positively correlated with the risk of atherosclerosis. LDL particles consist predominantly of cholesteryl esters and an apolipoprotein (apoB), and their removal from plasma depends on binding of apoB to a specific cell surface receptor (the LDL receptor). Abnormalities in both the apoB and the LDL receptor genes have been shown to affect clearance of LDL, both resulting in elevated plasma cholesterol concentrations with greater elevations caused by abnormalities of the LDL receptor than of apoB.
Familial hypercholesterolaemia
Familial hypercholesterolaemia (FH) is a dominant disorder affecting about 1 in 500 individuals and which accounts for around 5% of individuals with clinically evident atherosclerosis. In the UK, the most recent guidance from the National Institute for Care and Excellence (NICE), Clinical Guideline 71, and the subsequent Diagnostics Guidance published in 2011, recommends that a diagnosis of FH should be made based on the Simon Broome criteria, which include DNA-based as well as clinical/biochemistry criteria, and that those with a clinical diagnosis should be offered a DNA test. Of the mutations associated with FH approximately 93% are in the LDL receptor gene (LDLR). It is, however, a heterogeneous condition and mutations in the genes for apoB, proprotein convertase subtilisin/kexin type 9 (PCSK9) and LDL receptor-associated protein (LDLRAP) are now known also to cause it (see Chapter 37).
The LDL receptor gene is located on chromosome 19, spans 45 kilobases and codes for a protein of 839 amino acids. More than 400 mutations, many of which are deletions, have been identified. These mutations can be divided into five main classes:
1. ‘null’ alleles that produce no protein
2. mutations that block transport of the newly synthesized receptor from the endoplasmic reticulum to the Golgi apparatus
3. mutations that prevent the binding of LDL at the cell membrane
4. mutations producing receptors that can bind but cannot internalize LDL
5. mutations producing receptors that cannot release LDL following internalization and so are not recycled back to the cell surface – receptors must be freshly synthesized.
The apoB gene (APOB) is located at chromosome 2p24 and codes for a protein of 4536 amino acids. Apolipoprotein B-100, the full-size protein that is found in LDL, is produced by the liver while a smaller protein, apo B-48, is produced in the intestine by a unique tissue-specific process that introduces a stop codon into the mRNA. Familial defective apoB-100 is a dominant disorder, with a frequency of around 1 in 800 in the general population. Unlike deficiency in the LDL receptor, it is a much more homogeneous condition. Most cases result from one mutation, a SNP affecting codon 3500, which decreases the affinity of apo B-100 for the LDL receptor. Mutations in APOB account for around 1.5% of FH cases.
The third gene for which the NICE Diagnostics Guidance now also recommends testing is PCSK9, which encodes proprotein convertase subtilisin/kexin type 9. This protein is involved in degradation of the LDL receptor. A gain of function mutation of PCSK9 results in a reduction in the number of LDL receptors at the cell surface, thus reducing LDL uptake into cells and leading to increased circulating cholesterol. There remains some uncertainty as to the percentage of cases attributable to PCSK9 mutations.
FH, arising from mutations in the three genes described above is inherited in autosomal dominant fashion but a very small percentage (< 1%) show an autosomal recessive mode of inheritance. Mutations in the LDLRAP gene (previously referred to as the autosomal recessive hypercholesterolaemia gene, ARH) are one of the causes of this rarer form of LH. Patients with ARH have a normal LDL receptor but the receptor associated protein, encoded by LDLRAP, fails to mediate internalization of the receptor in the usual way.
Apolipoprotein E genotypes
Apolipoprotein E (apo E) mediates clearance of two other classes of lipoprotein particle (chylomicron remnants and intermediate density lipoprotein, IDL) by binding to the remnant receptor (IDL can also bind to the LDL receptor). There are three common apoE isoforms, E2, E3 and E4, characterized by different electrophoretic mobilities, with frequencies of 10, 30 and 60% respectively. The E4 variant is associated with increased cholesterol concentrations in comparison with those associated with E3, while E2 is associated with lower concentrations. Familial dysbetalipoproteinaemia (remnant, or type III, hyperlipidaemia) is associated with the E2/2 phenotype, but although almost all individuals with the condition are E2 homozygotes, the genotype shows poor penetrance so that most E2 homozygotes do not manifest the disorder, and other factors in addition to the E2 allele are clearly involved in expression of disease.
It has been estimated that about 7% of the variance in plasma cholesterol LDL concentrations found in populations can be accounted for by recognized mutations in the apo B, LDL receptor and apo E genes. Other candidate genes that may eventually prove to be involved in the complex process of atherosclerosis include those involved in cholesterol absorption, intracellular cholesterol metabolism, haemostasis and fibrinolysis. Angiotensin-converting-enzyme (ACE) regulates the concentrations of factors (angiotensin II and bradykinin) that are involved in modulation of vascular tone and proliferation of smooth muscle cells, and some recent results suggest that a polymorphism in this candidate gene is a risk factor for atherosclerosis. Two polymorphic forms of ACE have been described called the I (insertion) and D (deleted) forms, arising from the presence or absence of a 287 bp sequence in the gene. Individuals homozygous for the D form have been shown to be at increased risk of atherosclerosis, particularly those considered to be at low risk from other criteria.
Cancer genetics
Cancer can be considered to be a genetic disease. This is because it arises either from somatic mutations in cells that then become cancerous or from inherited germline mutations that lead to a predisposition to the disease, although cancers arising from a single gene defect account for fewer than 5% of cases. The genetic component is often triggered by environmental or behavioural factors (such as cigarette smoking, which increases the risk of developing lung cancer) and it is now clear that cancer is the end-point of an accumulation of somatic and germline mutations, most notably in proto-oncogenes and tumour suppressor genes.
Oncogenes and suppressor genes
The oncogenes were first identified as genes in retroviruses capable of producing tumours in birds and rodents. The first of these genes was identified in 1973, when a single gene (src) of the Rous sarcoma virus was shown to be capable of producing sarcomas in chickens. Later, it was shown that precursors to oncogenes, the proto-oncogenes or cellular oncogenes, were present in normal cells and that viral oncogenes were copies of these normal genes that had become incorporated into the retroviral genome. These viral oncogenes are able to transform infected cells once activated in some way.
Although it is now known that viruses do not cause most of the common cancers in humans and most of the oncogenes detected in this way (including src) are not of prime importance in human cancer, work with viral oncogenes made possible the rapid advances in understanding of human tumours that are now taking place. The isolation of human oncogenes was facilitated by the development of transfection techniques, in which DNA isolated from tumours can be tested for ability to transform cultured cells to a cancerous phenotype. Mechanisms for activation of proto-oncogenes include:
• amplification of a region of DNA including the oncogene
• point mutations that confer constitutive activity on the gene product.
Because the activation of only one allele is sufficient for transforming activity, oncogene mutations are generally dominant.
More than 50 cellular oncogenes have now been identified; the proteins they code for are mostly involved at some stage in the cascade of events associated with stimulation of cell division by growth factors. Thus, the sis protein is a mutated form of the platelet derived growth factor (PDGF); erbB codes for a truncated form of the epidermal growth factor (EGF) receptor; erbA codes for a mutant form of the thyroid hormone receptor; ras (a GTP-binding protein related to the hormone receptor G proteins) is a transducer of growth factor responses, and the jun and fos oncogenes mediate growth factor-induced gene expression. Oncogenes such as mos have been linked to factors that control the cell cycle.
In normal cells, the growth-promoting effects of proto-oncogenes are thought to be balanced by growth-constraining suppressor genes (tumour suppressor genes). Early evidence for the existence of such genes came from experiments in which fusion of tumour cells with normal cells resulted in hybrids with properties of normal cells. These experiments were interpreted as evidence for a tumour-suppressing gene in normal cells whose activity had been lost in tumour cells. Retinoblastoma, a rare form of eye tumour occurring in children, was later shown to result from loss of both copies of a gene (RB) located in the q14 band of chromosome 13. In familial retinoblastoma (around 40% of cases), a defect in one RB allele is inherited, so that tumours arise in cells in which the remaining (normal) allele is lost. In the remaining 60% of cases (sporadic cases), both alleles in the tumour founder cell have undergone somatic mutation. This is the two-hit hypothesis of Knudson, who proposed that the disease followed two-hit kinetics but that, in familial cases, the first ‘hit’ is inherited, that is, is a germline mutation, and the second is somatic.
Most cancers do not follow the retinoblastoma paradigm in such a straightforward manner, but one that does is neurofibromatosis type 1, associated with tumours derived from the embryonic neural crest and caused by a defect in the NF-1 gene located in the 17q11.2 chromosomal region. As with retinoblastoma, predisposition can be inherited from an affected parent as a germline mutation and tumour formation is initiated in cells that lose the activity of the remaining normal allele. The effects of suppressor gene mutations are usually only apparent when both alleles are inactivated, so that most of these genes behave in a recessive manner.
The majority of tumours that lack a functional suppressor gene (such as NF-1 or RB) are found to have two identical mutant alleles. Elimination of the normal alleles is thought to occur in these cases by mechanisms such as chromosomal non-disjunction or gene conversion, which replace the normal allele with a copy of the mutant allele at high frequency (10− 3–10− 4 per cell generation). By looking for evidence of this process, suppressor genes have been identified through demonstrating ‘loss of heterozygosity’ at specific chromosomal sites. Anonymous, highly polymorphic, DNA markers that identify heterozygous sites in normal tissue were used to demonstrate reduction to homozygosity in tumours, indicating loss of one or more alleles (Fig. 43.16), although this approach is not without pitfalls. However, the development of DNA arrays has been useful in demonstrating the possible location of tumour suppressor genes specific for different tumour types against the general background of non-specific ones. One pitfall is that simple loss of heterozygosity is not the only molecular abnormality in tumours; many demonstrate multiple structural abnormalities and some of the observed losses may be due to deficient DNA repair mechanisms or to chromosomal instability rather than the selective loss of a tumour suppressor gene. One likely mechanism for inactivation of tumour suppressor genes involves methylation of specific CpG dinucleotides in tumour suppressor gene promoters. In some cases, methylation may be an alternative to point mutation, whereas in others it appears to be the only mechanism for loss of function.
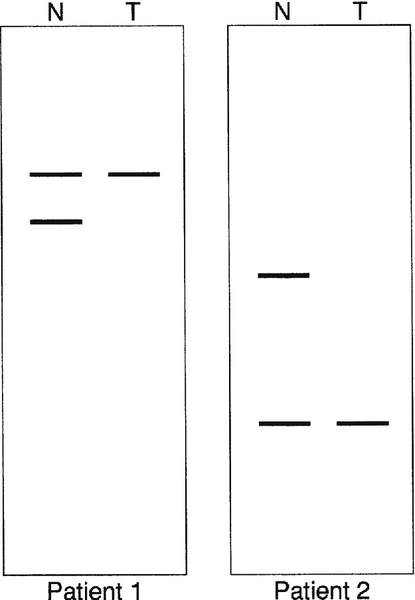
FIGURE 43.16 Loss of heterozygosity in tumours. Electrophoretic separation of digested total genomic DNA and detection of an allele on chromosome 5 with a single-locus probe in normal tissue (N) and tumour (T) of two patients with colorectal tumours. In both cases, the probe detects the alleles on DNA fragments of different size in normal tissue, demonstrating heterozygosity. Loss of heterozygosity in tumour tissue is demonstrated by disappearance of one allele. Adapted from Solomon E, Voss R, Hall V et al. 1987 Chromosome 5 allele loss in human colorectal carcinomas. Nature 328: 616–619, with permission.
One of the most studied tumour suppressor genes is TP53 (which encodes the transcription factor p53), the loss of which is a major contributor to genomic instability and is possibly the commonest single genetic change seen in cancer. The function of p53 appears to be related to apoptosis (programmed cell death) so that – unlike normal cells – any cell with a defective p53 protein is more likely to proliferate despite the acquisition of mutations. Defects in this gene, which is located at 17p12, can be from mutation or deletion, and p53 can be eliminated by inhibitive action of other gene products such as that of MDM2. Inherited mutations of TP53 are found in Li–Fraumeni syndrome, a dominantly inherited condition in which sufferers demonstrate multiple primary tumours, and are also implicated in some familial breast and colon cancers.
Another mechanism whereby suppressor genes function is illustrated by the ‘deleted in colonic carcinoma’ (DCC) gene, identified initially by loss of heterozygosity in the long arm of chromosome 18, which occurs in over 70% of colonic carcinomas. Sequencing of this gene showed that it encodes a 190 kDa protein with properties that suggest that it might be a transmembrane molecule that binds cells to the extracellular matrix or basement membranes.
The protein encoded by RB (pRB) has been shown to form a complex with the oncoproteins produced by the adenovirus SV40 and the human papillomavirus, suggesting that the ability of these viruses to form tumours results from their ability to inactivate RB. During the cell cycle, pRB switches from hyperphosphorylated to relatively unphosphorylated forms, so that pRB is likely to be involved in regulating the cell cycle.
In solid tumours, full malignancy requires acquisition of the ability to metastasize. The processes involved include changes in adhesion molecules, proteases and angiogenic factors. Little is known about the genetic changes that activate these processes, but it may be a side-effect of the overall genomic disarray seen in the cells of advanced tumours.
It has been recognized for many years that tumour cells become increasingly genetically unstable, accumulating mutations more rapidly as the tumours grow. Genetic changes, which may be acquired or inherited, can play a critical role in tumour progression by influencing mutation rates rather than growth regulation. Genes with this type of function are well illustrated by xeroderma pigmentosum, a rare autosomal recessive disease caused by defects in nucleotide excision DNA repair. Sufferers are extremely sensitive to ultraviolet light so that skin exposed to sunlight develops large numbers of freckles, which often progress to skin cancer as the damage caused by the sun cannot be repaired.
The early emphasis of research on genetic changes in cancer has been on understanding the mechanisms by which oncogenes and suppressor gene abnormalities can bring about transformation of a normal cell. The knowledge obtained thus far is now leading to progress in presymptomatic detection of tumours, and of identification of high-risk individuals, as well as in their diagnosis and treatment. This is demonstrated by the breast cancer screening programme for women at high risk of developing the familial form of disease. Sequence screening of the entire BRCA1 and BRCA2 genes in high-risk individuals (around 5% of cases) has close to 100% sensitivity, thus allowing prophylactic mastectomy when individuals are shown to have inherited the abnormal gene but, significantly, permitting reassurance for those women who have not. Testing regimens for other forms of cancer, for example bladder cancer, are being developed based on the fact that tumours shed cells. Sensitive PCR techniques can amplify the DNA of tumour cells shed into urine, blood and faeces, which can then be examined for SNPs (possibly using high-density arrays), loss of heterozygosity and microsatellite instability in characteristic tumour suppressor and oncogenes. Although not in routine use, such systems, as well as being useful for early detection of tumours, are likely to be valuable for monitoring possible disease recurrence.
The focus of clinical cancer genetics is now not only development of screening programmes capable of detecting tumours at earlier stages, but on the determination of whether or not the tumours are malignant, and how responsive they are likely to be to treatment. Studies on colorectal tumours have shown that formation of malignant tumours requires mutations in at least four or five genes, but that fewer changes are present in benign tumours. Work on glioblastoma tumours has shown that the degree of methylation in the O6-methylguanine DNA methyltransferase (MGMT) gene can predict response to chemotherapy. O6-methylguanine DNA methyltransferase is a DNA repair enzyme that removes toxic alkyl groups from the O6 position of guanine. Epigenetic silencing of MGMT by promoter CpG methylation has been associated with longer overall survival in patients with glioblastoma who, in addition to radiotherapy, received alkylating chemotherapy with an alkylating agent such as temozolomide. High levels of MGMT activity in cancer cells create a resistant phenotype by reducing the efficacy of alkylating chemotherapy.
Multiple endocrine neoplasia (MEN)
Not all ‘cancers’ or neoplasms are malignant, and some have both benign and malignant forms, a fact demonstrated by the MEN group of disorders (see Chapter 41). As the name suggests, these disorders are characterized by tumours of endocrine glands; they are familial and much is now known about their genetics. MEN1, characterized by tumours of the pituitary, parathyroids and pancreas, is inherited in a dominant fashion with a high penetrance. The gene involved is located on chromosome 11, at 11q13, and codes for the protein menin. The exact function of the protein remains unknown but at the time of writing is thought likely to be a tumour suppressor gene, as the majority of mutations found in MEN1 patients appear to result in loss of function. Pathogenesis follows Knudson’s ‘two-hit’ hypothesis as described above, whereby tumour development occurs in individuals who have inherited the first ‘hit’ as a germline mutation in the MEN1 gene when a somatic mutation occurs in a relevant endocrine cell. MEN1 is notable in that many different mutations have been described as contributing to the condition.
MEN2 also demonstrates autosomal dominant inheritance and consists of three sub-groups, all of which have medullary carcinoma of the thyroid in common. In the case of MEN2, the gene responsible is the RET proto-oncogene on chromosome 10, which codes for a receptor tyrosine kinase. However, unlike in MEN1, the germline RET mutations of MEN2 result in gain of function effects and the site of the mutation appears to be critical in determining the site of the tumour. There are also fewer loci within the gene where the small number of activating mutations are likely to be found, of great value in the molecular diagnosis of MEN2 and in contrast to the very many in the menin gene leading to MEN1.
GENE THERAPY
Gene therapy can be characterized in several ways, but essentially, it is the use of introduced genetic material to correct disease, either to replace a defective gene product or to correct an abnormal gene. It should not be confused with the treatment of genetic disease, which currently still uses conventional therapies. Ethically, correction of genetic defects by insertion of genes into somatic cells is akin to organ transplantation. However, germline gene therapy, where introduced genes could be transmitted to future generations, is universally agreed to be unethical for use in humans, although the production of transgenic animals by introduction of genes into fertilized eggs is widely used for studying gene function and regulation. Consequently, many countries have regulatory bodies to oversee gene therapy programmes, which concentrate on somatic cell gene therapy. Such programmes target the cells, organs or tissue affected by the disease or disorder under investigation. The first replacement of a defective gene in a human (to correct immune deficiency resulting from adenosine deaminase deficiency) took place in 1990, with limited success in that while none of the ten patients in the trial was cured, no adverse effects were reported. Since that first trial, gene therapy has had a difficult history, but the picture has improved since the last edition of this book, with nearly 2000 clinical trials approved in the last five years. As the field is once again changing rapidly, this section is intended to provide an outline of the issues and principles involved.
To be a suitable candidate for potential gene therapy, it is an essential prerequisite that the gene involved has been cloned and sequenced, together with all the appropriate promoter and regulatory elements. The next step requires a mechanism for introduction of the therapeutic DNA into suitable target cells, and the affected tissue or organ must be identified and accessible. The introduction of the DNA into the target needs a vector, which may be viral (such as retroviruses or adenoviruses) or non-viral (such as liposomes) and can be ex vivo, where the patient’s own cells are cultured with the vector and then reintroduced, or in vivo, where the transformed vector is delivered directly to the affected tissue or organ. ex vivo techniques are generally to be preferred as the cells can be checked before they are returned to ensure that the desired change has been achieved.
With retrovirus vectors, the viral genome is integrated into the DNA of infected cells after reverse transcription of viral RNA into DNA. Disadvantages in the use of retroviruses are, first, the relatively small amount of DNA that can be introduced (less than ~ 7 kb), and second, that they are unable to infect non-dividing cells, which limits their use as very few cell types are continually dividing. Adenoviruses avoid these problems, since they can carry much larger segments of DNA and are also able to infect non-dividing cells, but they do have the problem of potentially being immunogenic. Indeed, a crucial part of the development of a gene therapy strategy is that the vector should produce no harmful effects. One potential danger of gene therapy using a viral vector is the initiation of cancer if the inserted gene disrupts the function of a cellular oncogene or suppressor gene. This is called insertional mutagenesis and such an effect was seen in trials for treating the immunodeficiency disorder X-linked severe combined immunodeficiency (XL-SCID), using ex vivo enrichment of patients’ lymphocytes with a retroviral gene vector. This trial was initially seen as a great success, with 9 of the 11 patients being cured, but two of them later developed leukaemia owing to an insertional activation of the oncogene LMO2. This adverse outcome led to the suspension of gene therapy trials using pools of lymphocytes. Such a problem occurs mainly with the use of retroviruses and can be avoided by using adenoviruses, but they do have the problem of immunogenicity, and are known to contain genes of their own that can be involved in the process of malignant transformation and thus may also induce malignancy but by a different mechanism. It has not been possible, thus far, to determine precisely where in the host genome the introduced gene will insert: the process is random. In the case of the XL-SCID trial, only a small number of insertions were in a position to activate the oncogene but they produced clones that outgrew the other, beneficial, clones. Consequently, until, or unless, it is possible to direct the insertion site, or avoid damaging insertions, protocols using random insertion of vectors are unlikely to be approved.
A further problem is that the introduced normal gene must be capable of expressing its normal product in sufficient amounts to be effective in correcting the host disorder. Surprisingly, it has been found that functional genes can be transfected to skeletal muscle by direct injection, but this approach does not appear to be successful for other tissues. This approach has had some success in treating haemophilia where adeno-associated virus expressing factor VIII was injected intramuscularly. The encouraging results of this strategy are thought to be because only a small increase in the amount of factor VIII has a major clinical benefit.
Non-viral methods of gene therapy include the use of ‘naked’ DNA, that is, the direct injection of DNA into target cells, and the use of liposomes, aqueous vesicles with an outer lipid bilayer, as vectors for the foreign DNA. Non-viral methods have the advantage of being non-immunogenic and so of being safer. The use of directly injected DNA is likely to be of use mainly where a small amount of product will have a significant benefit, as in the example above of factor VIII. Liposomes have the advantage of being able to carry larger quantities of DNA, but the expression of the introduced gene is short lived, meaning that repeated treatment is necessary. A potentially useful development of liposome-mediated transfer has been in the use of DNA-protein complexes designed to target cell surface receptors. The use of an appropriate protein, recognized by the cell receptor, leads to internalization to intracellular vesicles of the complex. This allows transport to lysosomes where the complex is degraded and, once the gene has escaped from the lysosome, it can be expressed.
Stem cells in gene therapy
Correction of defects in haemopoietic cells is of particular interest because stem cells in bone marrow are relatively accessible and diseases that can be corrected by bone marrow transplantation are obvious candidates for gene therapy, bearing in mind the difficulties associated with random insertion vectors described above. This is most likely to be feasible in the correction of enzyme deficiencies such as Lesch–Nyhan and Gaucher diseases, where effective therapy could be achieved with relatively low levels of gene expression.
Early stem cell work looked at the use of embryonic cells. However, such work raised ethical concerns and more recent studies have looked at inducing pluripotency in somatic cells (called iPS cells), such as fibroblasts, by introducing genes known to be involved in maintaining pluripotency. A hope for these iPS cells is that they could be used in autologous therapy in patients with genetic disease once the gene defect had been corrected in the iPS cells in vitro. A major target for gene therapy has been cystic fibrosis. In this disorder, some of the most damaging effects result from a lack of the gene protein in the lungs, and studies have indicated that restoration of as little as 5–10% of normal gene expression would be enough to produce a beneficial clinical response. An adenovirus, tropic for respiratory epithelium, was successfully exploited as a vector to insert these genes while other trials used liposomes or adeno-associated virus. However, although gene transfer was demonstrated, there were concerns about the safety of using adenoviruses that might cause an immune reaction and in both types of trial the gene expression was too short-lived. If stem cells, such as iPS cells, could be used, this could avoid some of the need for repeat administration and thus reduce the risks of an immune reaction.
Early trials using stem cells as gene therapy in this way, to correct some rare forms of inherited eye disorders, have shown some success and hold promise for the future of these techniques.
Gene therapy in cancer
A major aim of gene therapy has been the treatment of cancer, with more than 60% of approved trials targeting this area. Projects are looking at various stages of the process, including the supplementation of tumour suppressor genes and the prevention of activated oncogene expression, but also at areas such as the manipulation of tumour cells to promote apoptosis or to render them vulnerable to the host’s immune system by increasing their antigenicity. Such trials demonstrate the diverse potential of gene therapy.
So, while the work carried out in the field of gene therapy has mushroomed in the last decade, there are still many problems to be overcome before its use in clinical medicine becomes frequent or widespread. However, the knowledge gained in the process has been enormous and it may take only one significant breakthrough to produce a major leap forward.
CONCLUSION
Clinical molecular genetics has advanced tremendously in recent years, a fact demonstrated by the almost exponential rise in the establishment of genetics laboratories in hospitals in developed countries. Its perceived importance in the future of healthcare in the UK was demonstrated by the publication, in 2003, of a Department of Health White Paper, Our Inheritance, Our Future, which resulted in large injections of money into NHS genetics laboratories to fund investment in new technology and staffing and was followed by a review document in 2011.
The advances of the past ten years have perhaps not been in the direction that might have been predicted at the start but some of the most startling have been in technology. Surprisingly, some techniques, such as DNA chips and microarrays, have been slower to transfer to routine clinical use than might have been expected, almost certainly because the cost of the analytical platforms and consumables did not fall as quickly as hoped. However, other technologies such as CSCE and real-time PCR have become routine and the speed with which next generation sequencing is being applied to clinical genetics is remarkable. Consequently, predictions of high-throughput, faster turnaround analysis have become a reality – and technological development is still enabling scientific discovery.
Gene therapy is beginning once again to live up to its early promise with more successes being reported and safer solutions coming forward. Conversely, pharmacogenetics has grown much more slowly than predicted, despite evidence that shows that predictive genotyping can prevent adverse drug reactions, the reasons for this remain unclear, but unexpected complexity of the issues may play a part.
The emphasis in the coming years is likely to be ‘more and faster’; that is, increasing laboratory capacity for molecular analysis and improving turnaround times. In terms of innovation, areas such as direct analysis of cell-free DNA in plasma may well begin to make an appearance in the laboratory.
The role of molecular biology in polygenic and multifactorial diseases is receiving a lot of attention, especially as these are the conditions (diabetes, hypertension, heart disease) that affect a large proportion of the population. Linked to this is the growing area of epigenetics, an area that examines heritable characters that are not due to changes in the DNA sequence. One mechanism for such inheritance is methylation.
Other areas that are finding their way into the clinical laboratory now are expression analysis and proteomics. At its simplest, expression analysis (sometimes called ‘transcriptomics’) examines the products of genes by creating cDNA from a gene’s mRNA (usually by reverse transcriptase PCR – rtPCR) and cloning it into vectors from which the protein product can then be expressed and studied. Expression screening employs microarray technology to examine heterogeneous mixtures, usually of mRNA or cDNA, often to compare expression in different tissues. An area worthy of a book in its own right, proteomics takes expression analysis one step further, in that it is concerned with the analysis of complex mixtures of proteins, that is, with the expressed mRNAs that are actually translated. Not all mRNAs are translated into protein products and some undergo post-translational changes. Proteomics allows identification of changes in the relative abundance of expressed proteins as well as of their structure and function using mass spectroscopy techniques, in particular matrix-assisted laser desorption-time of flight mass spectrometry (MALDI-TOF MS). The technique is expected to be particularly valuable not only in the study of cancer (e.g. bladder cancer), but also, it is hoped, in its diagnosis and treatment.
Clinical molecular biology has made huge advances in recent years but there are still many more to be made.
ACKNOWLEDGEMENTS
The author wishes to acknowledge the invaluable help of the staff of the Bristol Genetics Laboratory, North Bristol NHS Trust and of David Halsall, Department of Clinical Biochemistry, Addenbrooke’s Hospital, Cambridge in the preparation of the earlier edition of this chapter. The chapter is based, with permission, on that written by Dr Michael Norman, Department of Medicine, University of Bristol, for the first edition of this book.
GLOSSARY
Allele alternative forms of a gene at the same locus
Balanced polymorphism a polymorphism that is stable in a population
Carrier an individual who is heterozygote for a recessive gene
cDNA DNA that is complementary to a mRNA molecule
Clone a cell line derived from a single cell, or gene sequences propagated by recombinant DNA techniques
Codominant pertaining to two alleles which are both expressed in a heterozygote
Compound heterozygote an individual with two different mutant alleles at the same locus
Crossover (or recombination) exchange of information between homologous chromosomes during meiosis
Diploid chromosome complement with two copies of each chromosome (as in normal human cells where the diploid number is 46)
Dominant an allele that manifests its phenotypic effect in heterozygotes
Downstream sequences further in the direction of expression (5′ → 3′)
Enhancer a regulatory DNA sequence that can function to stimulate transcription of a gene irrespective of its position and orientation relative to that gene
Epigenetic heritable traits (e.g. those caused by gene methylation) that are not dependent on DNA sequence changes
Exome all the exons in a genome
Exon any segment of an interrupted gene that is present in the mature mRNA
Expressivity variability in the severity of a genetic trait
Frameshift mutation mutation resulting from insertion or deletion of bases (but not a multiple of three) that alters the reading frame of an mRNA
Gamete haploid cell generated by meiosis (sperm or egg)
Genome the complete ensemble of genetic information of an individual
Germ cell gametes or their precursors
Haploid cell a cell containing only one copy of each chromosome
Haplotype a group of closely linked alleles that are inherited as a single unit
Heterozygote an individual with different alleles (usually one normal and one mutant) at a given locus on homologous chromosomes
Homozygote an individual having the same allele at a given locus on homologous chromosomes
Hot spot site with a high frequency of mutation or recombination
Imprinting differences in the expression of genes depending on parental origin
Intron any segment of an interrupted gene that is transcribed but removed during formation of mature mRNA
Linkage two or more loci on a single chromosome that are sufficiently close that they do not segregate independently in offspring
Linkage disequilibrium association of two loci more frequently than predicted by chance
Locus unique location on a chromosome of a gene or particular DNA sequence
Loss of heterozygosity (LOH) homozygosity (in a tumour or somatic cell) when the constitutional state is heterozygous
Meiosis a series of two modified mitoses generating haploid gametes from a diploid cell
Messenger RNA (mRNA) sequence of RNA transcribed from a gene which, after processing, codes for a protein
Microsatellite polymorphic sequences due to a variable number of tandem repeats of a di-, tri- or tetranucleotide sequence
Minisatellite polymorphic sequences due to a variable number of tandem repeats of a short sequence of ten or more nucleotides
Mitosis process of division in somatic cells
Multiplex PCR simultaneous PCR reactions performed using more than one pair of primers in the same reaction mix
Mutation a heritable change in the genetic material
Northern blot a technique for transferring RNA to a filter for subsequent detection
Oncogene a gene involved in cell development capable of causing transformation to a tumour cell
Polymerase chain reaction (PCR) a technique for amplifying a specific sequence of DNA
Penetrance the frequency with which a particular genotype is expressed
Phenotype the observable characteristics of an individual
Point mutation changes in the sequence of DNA involving single base pairs
Polygenic a trait influenced by the cumulative effects of several genes at different loci
Polymorphism the occurrence of two or more alleles at a given locus at significant frequencies in the population
Positional cloning cloning of a gene after determining its chromosomal position by linkage analysis without knowing its function.
Probe a labelled fragment of DNA used to identify complementary sequences by hybridization
Promoter region of DNA to which RNA polymerase binds before initiating transcription
Recessive an allele that produces a phenotypic effect only when present in the homozygous state
Restriction enzyme an enzyme that cleaves DNA at specific sequences
Retrovirus an RNA virus that utilizes reverse transcriptase to insert itself into the DNA of a host cell
Restriction fragment length polymorphism (RFLP) polymorphism in the size of fragments produced by digesting DNA with a restriction enzyme
Somatic cell all cells of the body except gametes
Southern blot the technique of transferring fragments of DNA after electrophoresis to a filter
Trait any phenotypically detectable character or property
Transcription production of mRNA from the DNA template
Transfection incorporation of foreign DNA into a cell
Transformation conversion of cells to a state of unrestrained growth, resembling tumour cells
Transgenic animal animal into which a foreign gene has been incorporated
Translation conversion of the mature mRNA message into a protein
Tumour suppressor gene a gene that is growth constraining and whose inactivation can lead to unrestrained growth of a cell
Upstream sequences located in the opposite direction to transcription (3′ → 5′)
Vector any plasmid, phage etc. into which foreign DNA can be inserted for cloning
Variable number of tandem repeats (VNTR) minisatellite and microsatellite sequences with polymorphism in the number of repeats
Western blot transfer of proteins to a filter after electrophoresis
Wild type the allele which is most frequent in natural populations; now referred to as ‘normal type’.
Further reading
In a subject that changes as rapidly as this one, any bibliography is likely to become outdated very quickly and so this one is short with respect to texts, listing only those likely to form a valuable ‘core’ for the subject. Readers are directed to the internet, where the most up-to-date information is to be found: several appropriate starting points are listed. Regular review of appropriate journals, many of which are available online, is also recommended, especially for technological applications.
Background
Internet resources
The British Society for Human Genetics. http://www.bshg.org.uk
An excellent place to start: it is a searchable site and has extensive links to other major websites as well as to useful online genetics journals.
OMIM database, linked through the NCBI website (PubMed). http://www.ncbi.nlm.nih.gov/omim
For information on Mendelian disorders, the OMIM site contains highly referenced entries for most disorders and genes. The NCBI website is also the starting point for links to other databases, e.g. on protein structure, as well as being the best starting point for literature searching.
Journals
Nature Genetics.
The dedicated genetics journal in the ‘Nature’ family of scientific journals.