CHAPTER 9
Renal tubular disorders and renal stone disease
David Makanjuola; Marta Lapsley
CHAPTER OUTLINE
Isolated abnormalities of tubular function
Generalized tubular defects (Fanconi syndrome)
INTRODUCTION
Most patients with renal disease have some element of renal tubular involvement, but the other manifestations of the disease tend to be clinically more obvious and important. However, in a small number of patients, the clinical picture results primarily from a disorder of renal tubular function. These disorders can be inherited or acquired, and can affect tubular handling of a limited number of specific substances or encompass more generalized defects.
Renal tubular defects are conveniently considered with renal stone formation, since calculi sometimes form as a result of one of these conditions.
RENAL TUBULAR DISORDERS
Introduction
Hereditary renal tubular disease includes certain developmental disorders of the tubules, for example polycystic renal disease and medullary cystic disease. While these can result in disorders of renal function, including renal tubular function, they will not be considered in detail here. Renal tubular physiology will be discussed briefly, followed by a discussion of some well-recognized functional disorders of the renal tubules.
Physiology
Renal function is discussed in detail in Chapter 7, but in essence the process involves filtration at the glomeruli, followed by modification of this glomerular filtrate by both tubular reabsorption and tubular secretion. Since 170 L of filtrate are formed each 24 h, but only about one-hundredth this amount of urine is produced, reabsorption is quantitatively the more significant (Table 9.1). This is largely an active, energy-requiring process and explains why the kidneys account for some 6–8% of the resting oxygen consumption of the body, while representing < 1% of body mass.
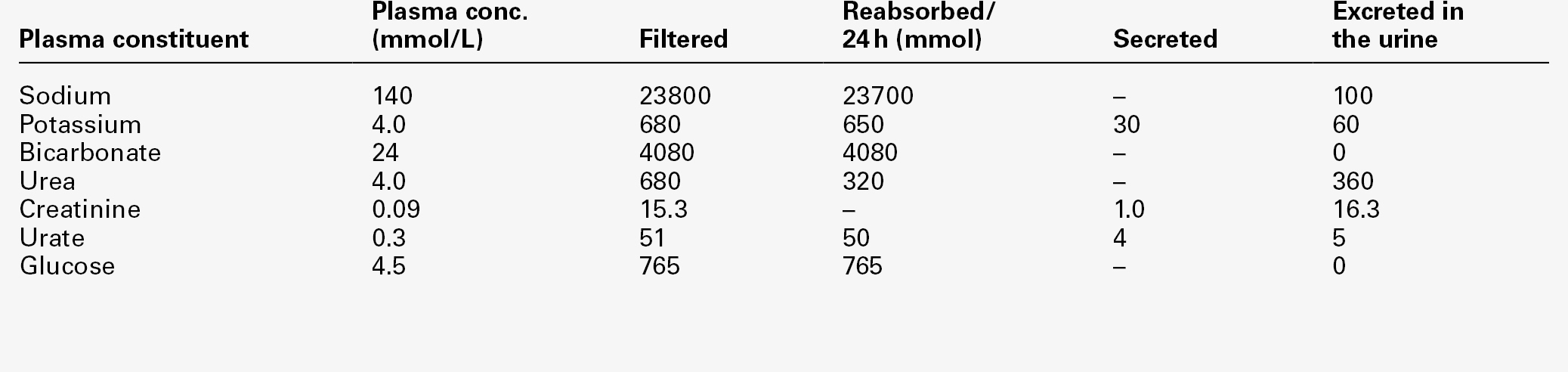
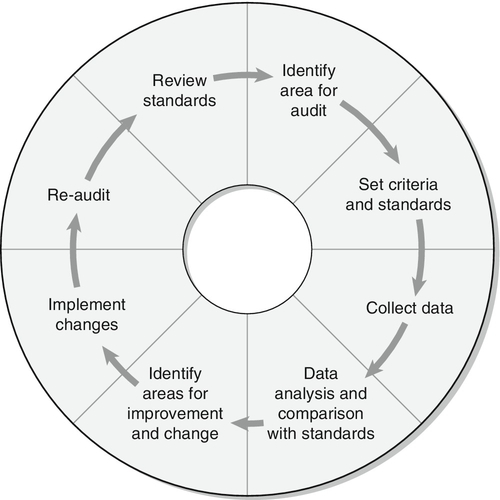
Some of the mechanisms by which active transport in the renal tubules occurs are shown in Figure 9.1. The control of renal tubular handling of certain substances is covered in detail in other chapters, for example sodium and water in Chapter 4. Only the renal tubular handling of substances that are important in disorders of renal tubular function will be considered further here.
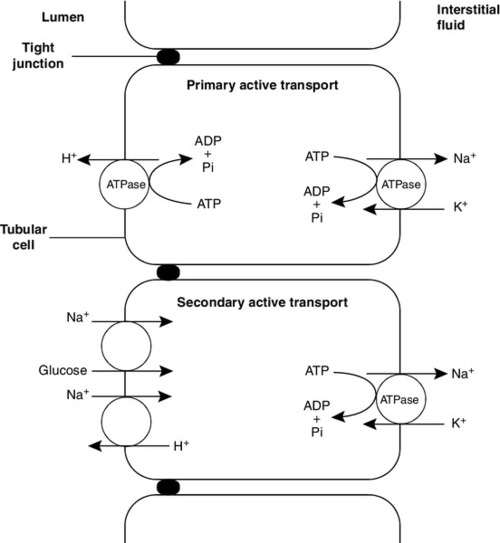
FIGURE 9.1 Active transport mechanisms in the renal tubule.
Glucose is absorbed with sodium ions in the early part of the proximal tubules, in a secondary active transport process. Glucose and sodium bind to a common carrier protein (SGLT 2, see later) in the luminal membrane and sodium moves down its electrochemical gradient, carrying glucose into the cell. Na+,K+-ATPase in the non-luminal (basolateral) membrane of the tubular cells pumps the sodium ions out into the interstitial fluid, while glucose is transported in the same direction by the glucose transporter GLUT 2.
Amino acids are also reabsorbed in the early part of the proximal renal tubules, again by a secondary active transport system linked to sodium reabsorption. There appear to be separate cotransporter proteins for certain groups of amino acids, although some of these probably have overlapping specificities. The process is driven by the Na+,K+-ATPase in the basolateral membrane pumping sodium out of the cell, with amino acids leaving by passive or facilitated diffusion.
Phosphate reabsorption in the renal tubules is influenced by the dietary intake of phosphate, certain hormones and a variety of other factors, and these are described in Chapter 6. In summary, about 90% of the inorganic phosphate in the plasma is freely filtered at the glomeruli, and then about 75% is reabsorbed in the proximal tubules. A small, variable amount is also absorbed in the distal tubules, but overall reabsorption is incomplete and up to 40 mmol/24 h appears in normal adult urine. The rate-limiting step in reabsorption appears to be a secondary active transport system linked to sodium reabsorption, with a phosphate/sodium cotransporter located in the luminal membrane of the tubular cells.
As described in Chapter 5, the renal tubular secretion of hydrogen ions is linked to the net effective reabsorption of bicarbonate (see Figs 5.2 and 5.3). Around 4000 mmol of bicarbonate is filtered every 24 h, but normal urine contains virtually no bicarbonate so the tubules must secrete 4000 mmol of hydrogen ions to achieve this. Although this mechanism prevents the loss of alkali into the urine, it does not result in the net excretion of acid. The tubules must also secrete the hydrogen ions produced each day by normal metabolism (see Chapter 5), a further 40–80 mmol/24 h.
There are two distinct mechanisms by which hydrogen ions are secreted into the tubular lumen (Fig. 9.1). A secondary active transport system linked to sodium operates in the epithelial cells of the early tubular segments, so that the Na+,K+-ATPase on the basolateral membrane produces an electrochemical gradient for sodium to enter the cell from the luminal surface, but in contrast to glucose and amino acids, a hydrogen ion is simultaneously secreted into the lumen. Although a very high hydrogen ion gradient cannot be achieved, this mechanism is responsible for the bulk of hydrogen ion secretion, so that most bicarbonate reabsorption occurs in the proximal tubules. The sodium–bicarbonate cotransporter, located on the basolateral membrane of these tubular cells, mediates the transport of the generated bicarbonate into the systemic circulation. There may be other hydrogen ion secretory mechanisms in the proximal tubules, but they do not appear to be quantitatively important.
In the late tubular segments (the late distal tubules and the collecting ducts), a completely different mechanism for hydrogen ion secretion exists. This is relatively independent of tubular sodium content and occurs through primary active transport. The intercalated cells (I cells) in this part of the nephron have a hydrogen ion-transporting ATPase on their luminal surfaces and, although this accounts for < 5% of the total hydrogen ions secreted, it is important because it can generate a hydrogen ion gradient of almost 1000 to 1. It is this that is responsible for the final acidification of urine and dictates the minimum achievable urinary pH of about 4.5 (or maximum hydrogen ion concentration of ~ 32 μmol/L). A chloride–bicarbonate exchanger at the basolateral membrane of the intercalated tubular cells is responsible for transporting the bicarbonate generated during this process to the systemic circulation.
Isolated abnormalities of tubular function
Glycosuria
Glucose is freely filtered at the glomeruli, but is normally then reabsorbed in the proximal tubules so that it is undetectable in urine. If plasma glucose concentrations rise to greater than ~ 10 mmol/L or if the glomerular filtration rate increases (as in pregnancy), then the capacity of the proximal tubules to reabsorb filtered glucose is exceeded and glycosuria occurs.
Generalized defects in renal tubular function may also result in glycosuria (see later), but a small group of people appear to have an isolated defect of tubular glucose reabsorption.
Hereditary renal glycosuria
Patients with this condition excrete a variable amount of glucose in their urine at normal plasma glucose concentrations. Other aspects of carbohydrate metabolism are not affected: glucose tolerance and plasma insulin concentrations are normal. The condition is inherited in an autosomal recessive manner, and two major phenotypes have been identified (types A and B), based on the exact changes in the kinetics of glucose reabsorption. Hereditary renal glycosuria is rare, and is generally recognized to be a benign condition with no clinical sequelae.
The reabsorption of glucose in the proximal renal tubules is similar to the absorption of glucose in the intestine. There is a Na+,D-glucose cotransporter on the luminal cell wall to transport glucose into the cell, with a facilitated glucose transporter (GLUT 2) on the basolateral membrane to enable the glucose to exit. The intestinal form of the Na+,D-glucose cotransporter (SGLT 1) and its gene have been well characterized, but, while the corresponding renal cotransporter (SGLT 2) is known to differ from SGLT 1, its identity in humans is less well established. Nevertheless, it is assumed that mutations in the gene for SGLT 2 cause hereditary renal glycosuria by interfering with the uptake of glucose in the proximal tubules.
There is a corresponding condition affecting SGLT 1 in the gut. This cotransporter is involved in both glucose and galactose absorption, and the classic presentation is with life-threatening diarrhoea in early infancy due to glucose and galactose malabsorption (familial glucose–galactose malabsorption). There is often an associated mild renal glycosuria, although in hereditary renal glycosuria there is no corresponding effect on the gut.
Mutations affecting GLUT 2, which facilitates transport of glucose, galactose and fructose across the basolateral membrane, are another rare cause of renal glycosuria (Fanconi–Bickel syndrome).
Amino acidurias
Amino acids are normally freely filtered at the glomeruli and then almost entirely reabsorbed in the proximal convoluted tubules. There is a maximal capacity to each reabsorptive mechanism and, in most patients with amino aciduria, some extrarenal disorder leads to accumulation of amino acid(s) in the plasma, that exceeds the reabsorptive capacity of the tubules, with consequent ‘overflow’ amino aciduria. However, as with glycosuria, generalized defects in renal tubular function can result in amino aciduria, and there are also some isolated defects in the reabsorption of particular groups of amino acids.
Cystinuria
Cystinuria is the classic example of an amino aciduria due to a defect in renal tubular function, in that the amino aciduria occurs at normal or even low plasma concentrations of the amino acids involved.
In most patients with cystinuria, there is renal loss not only of cystine, but also of the dibasic amino acids ornithine, arginine and lysine. There is also an associated failure of intestinal absorption of the same amino acids. Inspection of their molecular structures (Fig. 9.2) shows that each has two amino groups separated by 5–7 bonds, which suggests that malfunction of a single membrane carrier protein might explain the disorder. However, the true explanation is not this simple, since the clearance of cystine may exceed the creatinine clearance, suggesting secretion of cystine into the tubules and, furthermore, since dibasic amino aciduria (e.g. lysinuric protein intolerance) or cystinuria can each occasionally occur alone.
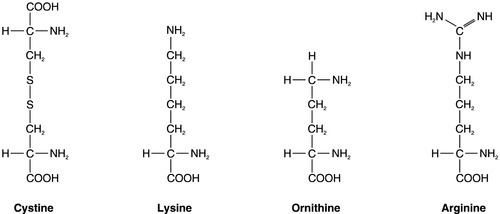
FIGURE 9.2 Chemical structures of the dibasic amino acids involved in cystinuria.
The only known clinical manifestation of cystinuria is recurrent urinary tract stone formation, the name ‘cystine’ coming from the original (erroneous) assumption that the source of these stones was the bladder. Cystine stones form readily in acidic urine. They are yellow-brown in colour and are radio-opaque because of their sulphur content, although they are less radio-dense than calcium-containing stones. There may also be some calcium deposition if there is infection secondary to the calculi. They tend to occur as staghorn or multiple recurrent stones and often require some form of surgical intervention or lithotripsy (fragmentation of stones by external shock waves). Patients with cystinuria also have a higher incidence of calcium oxalate stones than the general population. All stone formers should, therefore, be screened for cystinuria, preferably by formal amino acid measurement in the urine, as qualitative screening tests are not sufficiently sensitive. The intestinal defect appears to cause no clinical problems.
Cystinuria occurs with equal frequency in both sexes, although males tend to be more severely affected. It may present at any time from the first year of life up to the ninth decade, with a peak incidence in the second and third decades. The prevalence of cystinuria varies between racial groups and according to whether the figures are taken from neonatal amino acid screening programmes or from known cystinuric stone formers (immaturity of the renal tubules in the first few months of life may lead to some heterozygous infants having urinary cystine outputs in the homozygous cystinuria range, leading to misclassification). However, the worldwide prevalence is estimated to be 1 in 7000, making it a relatively common inherited metabolic disorder.
The mode of inheritance is autosomal recessive, although in some families it appears to be incompletely recessive, with heterozygotes excreting more urinary cystine, ornithine, arginine and lysine than normal, although less than in the homozygous state. In the past, this has led to attempts to classify cystinuria into type I (homozygous for the fully recessive form) and types II and III (variants of the incompletely recessive form), based on combinations of the relative amounts of the amino acids excreted in the urine and/or the intestinal uptake of cystine and dibasic amino acids. The discovery of the genes involved in the recessive and the incompletely recessive forms of cystinuria has made this classification clearer, although the situation is not yet completely resolved.
The gene involved in type I cystinuria codes for a protein known as rBAT (a loose acronym for ‘related to the b0,+ amino acid transporter’, with the term b0,+ signifying broad specificity for neutral (0) and dibasic (+) amino acids). rBAT is a membrane glycoprotein that is one of the activating components of a heteromultimeric transporter for cystine and dibasic amino acids. The gene for rBAT (SLC3A1) is located at 2p16.3-p21, and, to date, more than 120 different mutations have been identified in it in patients with type I cystinuria.
A second cystinuria locus, accounting for non-type I forms of cystinuria, has been identified by linkage analysis at chromosome 19q13.1–13.2. This gene (SLC7A9) encodes a protein that acts as the amino acid transporting subunit of the membrane complex. More than 95 gene mutations have been identified. Most, but not all cases of cystinuria can be explained by mutations in SLC3A1 and SLC7A9.
Healthy individuals excrete < 10 μmol cystine/mmol of creatinine (< 130 μmol/24 h). Patients with homozygous cystinuria usually excrete > 1700 μmol/24 h and may excrete up to 5000 μmol/24 h. Heterozygotes may have an entirely normal amino acid excretion pattern in the urine or may excrete up to 1700 μmol cystine/24 h, in which case they may produce stones.
Medical treatment of cystinuria begins with maintenance of a high fluid intake throughout both day and night and alkalinization of the urine, both being aimed at decreasing the chance of precipitation of cystine in the renal tract. The solubility of cystine is pH dependent with a solubility limit of ~ 700 μmol/L at pH 7, rising to ~ 1500 μmol/L at pH 7.5. Regular monitoring is required to ensure that cystine remains well below its solubility limit throughout the day and, critically, the night. If these measures fail, then it may be possible to convert the cystine to a more soluble compound, most commonly by the use of D-penicillamine. This can form the mixed disulphide cysteine-penicillamine (Fig. 9.3), which is significantly more soluble than cystine. The aim is to keep the daily excretion of free cystine below 2000 μmol. Unfortunately, D-penicillamine frequently causes an allergic reaction and can also cause nephrotic syndrome and pancytopenia, so careful monitoring is essential. Second generation chelating agents include tiopronin (α-mercaptopropionylglycine) and captopril, although studies on the efficacy of the latter have been inconclusive. Small stones may dissolve with careful medical management, although frequent surgical intervention is required for some patients.
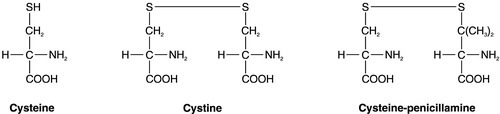
FIGURE 9.3 Chemical structures of cysteine, cystine and cysteine-penicillamine.
Occasionally, in spite of both medical and surgical treatment for stone formation, cystinuria causes sufficient renal damage to result in established renal failure. In this case, renal transplantation may be effective, since the donor kidney should not be affected by the amino acid transport defect and the patient should, therefore, remain disease free.
Hartnup disorder
A second example of an amino aciduria due to a true defect in renal tubular function, rather than an ‘overflow’ effect, is found in Hartnup disorder. This is named after the family in which it was first described and is again a defect of both renal and intestinal amino acid transport. The constant feature is a failure to reabsorb the neutral amino acids (Box 9.1) in the renal tubules, with their consequent appearance in the urine. The failure of reabsorption is not absolute, as renal clearances of the affected amino acids are generally lower than the creatinine clearance. Most affected individuals also have increased amounts of indoles (e.g. indican) in the urine, which originate from the bacterial breakdown of unabsorbed tryptophan in the gut. Rarely, the renal or intestinal lesions may occur alone.
The original description of Hartnup disease included a pellagra-like skin rash, transient cerebellar ataxia and constant renal amino aciduria. Some affected individuals have also shown psychotic behaviour, while others have learning disabilities. The pellagra-like rash and its response to nicotinamide suggest that the clinical features of the disease may be due to failure to absorb tryptophan in the intestine and reabsorb it in the renal tubules, leading to a deficiency of nicotinamide. However, investigation of siblings of individuals with Hartnup disease and the results of neonatal urine amino acid screening programmes both suggest that the typical amino aciduria (Hartnup disorder) may exist without the features of Hartnup disease. It would appear that Hartnup disorder is an autosomal recessive inherited condition, but that this is not expressed clinically without the presence of certain other genetic or environmental influences, such as poor nutrition. The causative gene, SLC6A19, is located on chromosome 5p15.33 and codes for a sodium-dependent neutral amino acid transporter.
Familial renal iminoglycinuria
A further example of a specific abnormality in renal tubular reabsorption causing a distinct pattern of amino/imino aciduria occurs in familial renal iminoglycinuria. The primary genetic defect is a mutation in either the SLC36A2 gene, which codes for a high affinity glycine, proline and hydroxyproline transporter, or the SLC36A1 gene, which codes for a low affinity transporter. However, the phenotype is variable as it may be modified by mutations in at least three other genes. Some reabsorption of these amino acids can still occur via the transport system that is not affected in any individual patient.
The condition, which is benign, is autosomal recessive, although some heterozygotes are ‘incomplete’ and have hyperglycinuria. In some but not all homozygotes, impaired intestinal transport of proline can be demonstrated.
Neonatal screening for amino acidurias suggests that familial renal iminoglycinuria occurs in 1 in 15 000 live births in a Caucasian population. However, it should be appreciated that in healthy neonates, the renal tubular reabsorption of proline, hydroxyproline and glycine is less efficient than in adults; imino aciduria normally disappears by three months and hyperglycinuria by six months of age.
Dent disease
Also known as X-linked nephrolithiasis, Dent disease is most often caused by a mutation in the gene coding for a voltage gated chloride channel (CLC-5) expressed predominantly in the proximal renal tubules. However, a significant number of patients have a mutation in the gene usually associated with the oculocerebrorenal syndrome of Lowe, OCRL. Both defects are thought to cause failure of the endosomal acidification that is required to allow recycling of the proximal tubular membrane receptor, megalin, which mediates protein reabsorption. The syndrome is characterized by hypercalciuria, nephrocalcinosis, recurrent calcium-containing renal stones, low molecular weight proteinuria and chronic kidney disease eventually leading to established renal failure. Glycosuria, phosphaturia, amino aciduria and rickets may also occur in affected males. Carrier females rarely present clinically, but often have increased urinary excretion of low molecular weight proteins. Mutations of the gene encoding another voltage gated chloride channel (CLC-Kb) are responsible for some cases of Bartter syndrome (see p. 57).
Phosphate transport defects
Specific disorders of renal tubular phosphate handling can be inherited or acquired. There is reduced proximal tubular reabsorption of phosphate (in particular, the TmP/GFR is reduced, as explained in Chapter 6) and hypophosphataemia. The normal response to this would be an increase in the 1α-hydroxylation of vitamin D, and if this is adequate, the end result would be hypercalciuria (with an increased risk of renal stones), but no bone disease. If the vitamin D response is inadequate, then the result may be bone disease with hypercalciuria (e.g. as in hereditary hypophosphataemic rickets with hypercalciuria) or bone disease alone (e.g. X-linked hypophosphataemic rickets and oncogenic hypophosphataemic osteomalacia). Inherited and acquired forms of hypophosphataemic bone disease are discussed further in Chapter 31.
The phosphate loss, and other changes in more generalized forms of renal tubular disorder, may also be severe enough to cause bone disease (see p. 174).
Renal tubular acidosis
The renal tubular acidoses (RTAs) are a group of hyperchloraemic metabolic acidoses that occur secondarily to an abnormality in urinary acidification. The plasma bicarbonate concentration is low, the plasma chloride raised and the anion gap normal, in contrast to the acidosis seen with reduction in the glomerular filtration rate in which the plasma bicarbonate is low, chloride normal and anion gap increased.
Renal tubular acidoses are classified according to the presumed site of the failure of hydrogen ion secretion. In proximal (type 2) RTA, there is a failure in bicarbonate reabsorption, while in distal RTA, there is a failure in net acid excretion. If failure of hydrogen ion secretion is the sole defect in the distal nephron, it is known as classic distal (type 1) RTA, whereas if the defect is more generalized, causing hyperkalaemia as well as acidosis, it is known as generalized distal (type 4) RTA. These conditions are described in Chapter 5, but the underlying mechanisms are briefly summarized again here, and details of diagnostic tests are given in Appendix 9.1. Type 3 RTA is a mixture of types 1 and 2 and is very rare. The term was originally used to describe a transient condition occurring in infants with immature tubules. However, it is now usually applied to a rare inherited deficiency of carbonic dehydratase type II, in which infants also have cerebral calcification, mental retardation and osteopetrosis (see Chapter 31).
Proximal (type 2) renal tubular acidosis
Isolated proximal RTA may be autosomal recessive or dominant. A mutation in the sodium–bicarbonate cotransporter gene (SLC4A4) is responsible for the recessive form; the molecular basis for the dominant form is yet to be elucidated. The net effect is that bicarbonate reabsorption is impaired, leading to metabolic acidosis and bicarbonaturia, with a decrease in the plasma bicarbonate concentration until the filtered load of bicarbonate falls to a point at which hydrogen ion secretion is sufficient to allow reabsorption of all filtered bicarbonate. Thus, a new steady state is established, in which there is a metabolic acidosis with a low plasma bicarbonate concentration (15–20 mmol/L) but no bicarbonaturia.
Administration of bicarbonate to such patients will establish a steady state closer to normal, but quite large doses of bicarbonate are required (e.g. 5–15 mmol/kg body weight/24 h), since as the plasma bicarbonate rises, so does the magnitude of renal bicarbonate loss. Hydrogen ion secretion in the distal tubules is normal and the urine can be acidified in response to an acid load.
Proximal RTA may occur as an isolated defect, but is more usually part of a generalized defect of proximal tubular function, the Fanconi syndrome (see p. 174).
Distal (type 1) renal tubular acidosis
Decreased activity of the apical H+-ATPase (proton pump) is probably the commonest defect in distal RTA and may be the end result of a number of different pathological processes. The most common of these is Sjögren syndrome, which should be excluded in any patient presenting in adulthood. Other dysproteinaemic and autoimmune conditions may also cause acquired distal RTA, as may chronic hypercalciuria with nephrocalcinosis or treatment with drugs such as amphotericin, lithium and analgesics. Inherited forms (which may be autosomal dominant or recessive) include mutations in the genes encoding the H+-ATPase pump and the chloride–bicarbonate exchanger. There is a complex relationship between distal RTA, nephrocalcinosis and kidney stone formation. In some patients, hypercalciuria appears to be the primary abnormality, causing interstitial and tubular damage with calcium deposition and secondary RTA. In others, RTA is the primary condition causing increased calcium phosphate resorption from bone in an attempt to buffer the retained acid; the subsequent hypercalciuria is proportional to the severity of the acidosis.
While other distal tubular functions remain unimpaired, hydrogen ion secretion is reduced so that urinary pH cannot be lowered below 5.5. A resultant increase in the exchange of potassium for sodium ions in the distal tubules causes significant renal potassium loss and the condition is usually accompanied by hypokalaemia.
Diagnosis depends on the demonstration of an inappropriately high urinary pH (> 5.5) during either spontaneous acidosis or an acid loading test (see Appendix 9.1). Treatment involves potassium supplementation together with bicarbonate. Unlike proximal RTA, the dose of bicarbonate required is small (e.g. a little over 1 mmol/kg body weight/24 h in adults), since there is only a minor degree of bicarbonaturia and this does not increase as the plasma bicarbonate concentration rises.
Patients with incomplete distal RTA have low urinary citrate excretion and urine pH persistently > 5.5 but are able to maintain their plasma bicarbonate concentrations within the reference range, possibly by increasing proximal tubular ammonium production. They may also have hypercalciuria and are at risk of developing osteoporosis, nephrocalcinosis and calcium phosphate kidney stones. Hypercalciuria may be reversed and the risk of developing osteoporosis and kidney stones reduced by alkali therapy, which decreases calcium phosphate resorption from bone.
Distal renal tubular acidosis with hyperkalaemia (type 4)
In type 4 RTA, there is a generalized impairment of distal tubular function associated with aldosterone deficiency or resistance, with decreased tubular secretion of both hydrogen and potassium ions. A relatively severe hyperkalaemia is usually the primary abnormality and the accompanying hyperchloraemic metabolic acidosis is often mild with a plasma bicarbonate concentration > 16 mmol/L.
The condition may arise because of a lack of mineralocorticoid activity (e.g. in adrenal failure), disease of the kidney resulting in impaired production of renin (hyporeninaemic hypoaldosteronism, e.g. in diabetic nephropathy) or resistance to the action of mineralocorticoids on the distal tubules (e.g. in patients treated with spironolactone). Treatment of the condition depends on the underlying cause.
Hereditary renal hypouricaemia
This is a rare disorder of the renal tubular handling of urate, in which there is a net decrease in reabsorption of urate, resulting in hypouricaemia and an increased renal urate clearance. Plasma urate concentrations are < 150 μmol/L in men and < 126 μmol/L in women. The condition is generally harmless, although some patients also have hypercalciuria, and about 25% have a tendency to stone formation. It is transmitted in an autosomal recessive manner and heterozygotes tend to have intermediate plasma urate concentrations. Mutations have been demonstrated in the gene for a urate–anion exchanger, which is found on the luminal membrane of proximal tubular cells and which accounts for the majority of urate reabsorption in normal kidneys.
Generalized tubular defects (Fanconi syndrome)
Generalized renal tubular defects tend to occur together in a distinct syndrome known as the renal Fanconi syndrome (which should not be confused with Fanconi anaemia, a form of congenital aplastic anaemia or Fanconi–Bickel syndrome, a rare disorder of carbohydrate metabolism). There is a failure in net proximal tubular reabsorption of glucose, amino acids, phosphate and bicarbonate, with consequent glycosuria, amino aciduria, phosphaturia and acidosis, together with the development of vitamin D-resistant metabolic bone disease. There may also be increased urinary losses of water and other substances, for example sodium, potassium, calcium, magnesium, urate and low molecular weight proteins. The exact mechanism by which the Fanconi syndrome occurs is not clear and there may be distal, as well as proximal, tubular dysfunction.
Causes of the renal Fanconi syndrome may broadly be divided into inherited and acquired and are listed in Box 9.2. The most common cause of inherited Fanconi syndrome in children is cystinosis, an autosomal recessive disorder of cystine transport across lysosomal membranes. The gene responsible (CTNS) is located on chromosome 17p13 and encodes a lysosomal membrane protein, cystinosin. There is accumulation of cystine in the lysosomes of most tissues, including the kidneys, and in the infantile form of the disease, the Fanconi syndrome progresses to cause glomerular damage and renal failure during childhood. Cystinosis is usually treated with cysteamine, which is concentrated inside the lysosomes and reacts with cystine to form both cysteine and a cysteine–cysteamine complex. These are able to cross the lysosomal membrane via different transporters. When administered regularly, cysteamine decreases the cystine load in lysosomes, conserves renal function and improves growth. The adequacy of treatment can be assessed by measuring the cystine content of white blood cells. If renal function becomes significantly impaired, transplantation is particularly useful, since the transplanted kidney does not have the genetic defect.
There is also an adult onset form of cystinosis in which there is no Fanconi syndrome or glomerular impairment, and an intermediate form that presents during adolescence and does eventually lead to renal failure.
Other inherited metabolic diseases that are associated with the Fanconi syndrome are listed in Box 9.2. There is also an idiopathic inherited form, but this is a diagnosis of exclusion. It is interesting that in inherited metabolic diseases for which there is a specific treatment (e.g. avoidance of lactose and galactose in galactosaemia), the Fanconi syndrome resolves on treatment, presumably because of the fall in the concentration of a toxic metabolite.
While the inherited diseases associated with the Fanconi syndrome tend to present in childhood, the acquired forms tend to occur in adults. These may also be reversible if exposure to the causative agent can be stopped.
The clinical features of the Fanconi syndrome tend to be rather non-specific and include polyuria, polydipsia, dehydration, hypokalaemia and acidosis, with impaired growth and rickets in children and osteomalacia in adults.
Treatment is primarily directed at the underlying cause, but inappropriate renal losses need to be replaced (e.g. with fluids, bicarbonate and potassium) and the bone disease treated with phosphate replacement and vitamin D.
RENAL CALCULI
Introduction
Stone formation in the urinary tract (urolithiasis) has been described since ancient times, but in the past, lower urinary tract stones, particularly those arising in the bladder, appear to have been more frequent, engendering a surgical enthusiasm for ‘cutting for stone’. In industrialized, relatively affluent populations, renal stone formation (nephrolithiasis) has increased in frequency, while bladder stone formation has almost disappeared as the prevalence of malnutrition and infection have decreased.
However, while the frequency of nephrolithiasis is acknowledged to be increasing in the developed world, precise incidence and prevalence figures have not been well established. Nevertheless, it would appear that as many as 5% of the population (possibly even more) will have a clinical stone event at some time during their lives. In previous decades, stone formation was more frequent in men than in women, but in recent years the incidence has become almost equal. The ten-year risk of further stone recurrence can be as high as 70% after a first episode, so investigation and treatment, which can reduce this figure to 25%, is worthwhile.
The clinical effects of stone formation tend to be similar whatever the type, but it is apparent that there are a number of distinct metabolic derangements that can each give rise to stones of a characteristic composition. The main types of renal stones are listed in Table 9.2 in approximate order of frequency of occurrence, and aetiological factors in the formation of each are described in detail below.
TABLE 9.2
The main types of renal stone, in approximate order of frequency of occurrence
| Type of stone | Frequency (%) |
| Predominantly calcium oxalate | 60 |
| Uric acid | 17 |
| Magnesium ammonium phosphate and calcium phosphate (struvite) | 12 |
| Predominantly calcium phosphate | 10 |
| Cystine | 1 |
The classic clinical manifestation of nephrolithiasis is renal or ureteric colic. The pain is characteristically severe, requiring opiates for analgesia, and is usually accompanied by haematuria. Some stones may be discovered incidentally when abdominal X-rays are taken for an unrelated reason, and passage of tiny stones may cause only minimal discomfort. In general, permanent intrinsic renal damage rarely occurs after an acute event unless there is superadded infection in an obstructed kidney. However, there is an increased incidence of chronic kidney disease in patients with a history of kidney stones and current UK guidelines recommend screening such patients annually with measurement of plasma creatinine and calculation of eGFR (see Chapter 7).
Pathogenesis of renal stones
The role of the kidneys in water conservation means that it is often necessary to excrete concentrated urine. Since some constituents of urine are relatively insoluble in water, to the extent that supersaturated solutions form, it is not surprising that these constituents sometimes crystallize out to form renal calculi.
Certain factors predispose to this process: chronic dehydration, as may occur in hot climates, is one factor. Increased urinary excretion of certain constituents (e.g. calcium, urate or oxalate) increases the likelihood of supersaturation occurring, and alteration of urinary pH may adversely affect the solubility of some solutes. For example, urate stones are more likely to form in acidic urine, whereas alkaline conditions, as may occur with a urinary tract infection, make calcium phosphate precipitation more likely. There is evidence that specific microorganisms may precipitate early crystallization. Anatomical abnormalities of the kidneys, such as medullary sponge kidney or pelvi-ureteric junction obstruction, increase the risk of stone formation, at least in part because of urinary stasis. There is a 2–3 fold risk of stone formation in people with a family history of urolithiasis. Patients who are overweight, especially if they have insulin resistance, have an increased risk of developing both calcium oxalate and uric acid stones, partly because of excretion of excess dietary constituents in the urine and partly because of the production of persistently acidic urine.
These factors are opposed by the presence in urine of inhibitors of crystallization (e.g. magnesium, pyrophosphate, citrate and certain glycoproteins), so that crystal formation in urine is usually slower than in simple salt solutions.
Each of the types of stone listed in Table 9.2 will now be considered in more detail.
Calcium stones
Calcium-containing stones form the majority of renal calculi and may consist of calcium oxalate, calcium phosphate or a mixture of the two. Hyperoxaluria, with or without hypercalciuria, is likely to be involved in the formation of pure calcium oxalate stones, whereas pure calcium phosphate stones suggest either hypercalciuria or persistently alkaline urine.
Hypercalciuria
Hypercalciuria is present in around 30% of patients with calcium-containing stones, although the exact prevalence depends on the population studied and the upper limit accepted for the reference range (of the order of 7.5 mmol/24 h for men and 6.25 mmol/24 h for women or 0.1 mmol/kg body weight/24 h).
A minority of hypercalciuric stone formers also have hypercalcaemia, and it is then important to establish a cause for this; it is most likely to be due to primary hyperparathyroidism. The treatment is that of the underlying cause.
Normocalcaemic hypercalciuria may be divided broadly into four groups: skeletal resorption, dietary, absorptive and renal leak hypercalciuria. Skeletal resorption and hence urinary excretion of calcium may be seen in patients following prolonged immobilization, Paget disease of bone or excessive glucocorticoid activity including chronic steroid therapy. Pure dietary hypercalciuria is rare, but high consumption of dairy products, combined calcium and vitamin D supplements or salt all tend to increase urinary calcium excretion.
Increased intestinal absorption of calcium accounts for around 50% of subjects with normocalcaemic hypercalciuria. The mechanism is not well understood, but there is some evidence for increased end-organ sensitivity to vitamin D, resulting in absorption of a greater fraction of dietary calcium than in normal subjects. Plasma parathyroid hormone (PTH) concentrations may be decreased.
Renal leak hypercalciuria may be seen in renal tubular acidosis, medullary sponge kidney, chronic loop diuretic therapy and proximal tubular syndromes such as Dent disease, but is more often idiopathic, accounting for 25% of hypercalciuric subjects. Some patients may have an unrecognized bone resorption defect; indeed, it is not always possible to determine whether the primary disorder is of renal or bone origin. Patients may be differentiated from those with absorptive hypercalciuria by a calcium load test: calcium excretion is high after loading in both groups but remains > 0.23 mmol/mmol creatinine after fasting in patients with a renal leak of calcium (see Appendix 6.1). Plasma PTH is often mildly raised in response to the chronic calcium loss, although any increase is more commonly due to vitamin D deficiency. Treatment with thiazide-like diuretics reduces both calcium excretion and plasma PTH concentrations, and reduces the risk of osteoporosis.
Treatment of patients with hypercalciuria and/or calcium containing stones includes increasing fluid intake to achieve daily urine volumes of 2–3 L, correcting dietary factors such as excessive salt, protein, sugar and calorie intake, and avoiding combined calcium and vitamin D supplements. A low sodium diet enhances both sodium and calcium reabsorption in the proximal tubules and so decreases urinary excretion (calcium is reabsorbed passively along the concentration gradient produced by active sodium and water reabsorption). Restricting animal protein reduces the dietary acid load and therefore the loss of calcium from bone. Restricting sucrose and fructose consumption appears to reduce urinary calcium excretion and may help with weight loss, which reduces the risk of stone recurrence. Formal calcium restriction is no longer recommended owing to the risk of osteoporosis as well as to a paradoxical rise in stone risk from the increased absorption of free oxalate in the intestine (calcium oxalate is poorly absorbed). Subjects should be advised to consume a ‘normal’ amount of calcium per day of around 1000 mg. However, calcium supplements, especially if taken between meals, should be avoided as they are associated with an increased risk of stone formation. Thiazide-like diuretics, such as chlortalidone or indapamide, specifically reduce renal calcium excretion and are particularly successful in patients with renal leak hypercalciuria. Those with absorptive hypercalciuria may become refractory to treatment and require an occasional ‘thiazide holiday’. Response to treatment should be monitored by measuring urinary calcium excretion. Biochemical side-effects, including hypokalaemia, hyperuricaemia, hypercalcaemia and hyperglycaemia, are relatively common and should be sought routinely. Potassium citrate supplements should be added, especially if the urinary citrate excretion is below the mid range and the urine pH is regularly < 6.5, or if there is hypokalaemia. Excessive treatment with potassium citrate, resulting in the production of alkaline urine, increases the risk of calcium phosphate stone formation and should be avoided. Patients with distal renal tubular acidosis may show a sufficient fall in urinary calcium excretion in response to potassium citrate alone to avoid treatment with thiazide diuretics. This group includes patients with a partial form of the disorder in which plasma pH is normal under basal conditions, but the urine cannot be acidified when tested formally.
Hyperoxaluria
Now that oxalate can be measured reliably in the urine, it has become apparent that hyperoxaluria contributes as much, if not more, to calcium oxalate stone formation as hypercalciuria. A relatively small increase in urinary oxalate excretion above normal can have a marked effect on the risk of stone recurrence. In most instances, hyperoxaluria results from excessive dietary intake or increased intestinal absorption of oxalate; the two rare primary hyperoxalurias are described below.
The upper reference limit for urinary oxalate excretion is of the order of 400–500 μmol/24 h. A dietary excess of oxalate-containing foods can increase excretion to 700 μmol/24 h or even greater, particularly if the dietary calcium content is low.
Increased intestinal absorption of oxalate may occur in short bowel syndrome and malabsorption, whatever the cause (enteric hyperoxaluria). Unabsorbed fatty acids in the gut lumen combine with calcium, reducing the available calcium for calcium oxalate formation. This leaves larger than usual amounts of oxalate in the free form, which can be easily absorbed. In addition, exposure of the colonic mucosa to substances such as fatty acids and bile salts, which have detergent properties, may increase its permeability to oxalate and further increase the amount absorbed.
Primary hyperoxaluria is a rare inherited metabolic disease that must be considered if nephrolithiasis occurs during childhood (although a few affected individuals do not present until adult life). There is increased synthesis of oxalate, with a consequent increase in its urinary excretion, together with that of some other organic acids. The pattern of accompanying organic aciduria has allowed classification into two types. In type 1 primary hyperoxaluria, excessive amounts of glyoxylic and glycolic acids are excreted, whereas in the rarer type 2, excretion of these acids is normal but that of L-glyceric acid is increased. The biochemical defect in type 1 is a deficiency of the peroxisomal enzyme alanine:glyoxylate aminotransferase. In type 2, the defect is in a cytosolic enzyme, D-glycerate dehydrogenase/glyoxylate reductase. The inheritance of both types is autosomal recessive. In type 1, the 24 h excretion of oxalate is of the order of 1.5–3 mmol; it may be lower in type 2, which may have a less severe clinical course. Besides renal stone formation, there is also a tubulointerstitial nephropathy that progresses to chronic kidney disease. Plasma oxalate concentrations begin to rise as renal clearance decreases, eventually leading to widespread tissue deposition of oxalate (e.g. in myocardium, retinae and synovial membranes). However, the condition appears to exhibit marked clinical and biochemical heterogeneity.
Other causes of hyperoxaluria include ingestion of ethylene glycol, which is rapidly converted to glycolate and can lead to calcium oxalate crystals forming in the renal tubules, excessive ingestion of vitamin C, which may lead to increased oxalate formation in some individuals, and chronic Aspergillus infection. Pyridoxine is a cofactor for the alanine:glyoxylate aminotransferase enzyme, and deficiency results in mild hyperoxaluria that responds to physiological replacement doses. A syndrome of ‘mild metabolic hyperoxaluria’ has also been described in adults that responds to pharmacological doses of pyridoxine.
Treatment of calcium oxalate stone formers includes increased fluid intake, dietary restriction of oxalate (Box 9.3) and oral potassium citrate if urine citrate excretion is below mid range or the pH is consistently < 6.5. If there is no hypercalciuria, oral calcium supplements can be taken at mealtimes to decrease intestinal oxalate absorption. The starting dose is 500 mg equivalent of calcium per day, which may be increased to 10 g/day in patients with severe malabsorption and should be titrated against urine calcium and oxalate excretion. Pyridoxine reduces urinary oxalate excretion in some individuals: the starting dose should be 10 mg/day, increasing to a maximum of 500 mg. Cholestyramine can be used as a last resort to bind intestinal oxalate, but it is often poorly tolerated.
Other factors in calcium stone formation
Although uric acid can itself form renal stones (see below), hyperuricosuria may also contribute to calcium stone formation. This may be through heterogeneous nucleation following uric acid crystallization or through other mechanisms, and this remains a controversial area. Dietary restriction of proteins and purines and/or the use of allopurinol may correct hyperuricosuria and reduce the risk of recurrence.
Citrate is a recognized inhibitor of calcium stone formation, and some stone formers have low urinary citrate concentrations as the only biochemical abnormality. This is particularly so in patients with distal renal tubular acidosis, who are already at increased risk of calcium phosphate stone formation because of failure to acidify the urine adequately. Potassium citrate supplementation is effective but should be used cautiously in patients with renal impairment or hyperkalaemia. Sodium citrate should be avoided as it can increase urinary calcium excretion.
Magnesium is also a recognized stone inhibitor. Although magnesium supplementation has not been shown to reduce stone risk, it may be worth trying if definite hypomagnesuria is present.
Normal urine also contains a variety of protein-based inhibitors of stone formation, and it may well be that deficiencies or defects in these will be found to be the cause of some types of stone in the future. For example, a genetic variant of uromodulin (Tamm–Horsfall glycoprotein) has been found that has stone-promoting properties, whereas the normal type is thought to inhibit stone formation.
Infection-related stones
Infection-related (triple phosphate) stones are composed predominantly of magnesium ammonium phosphate (struvite), with variable amounts of calcium phosphate as carbonate–apatite. They form in the presence of high urinary concentrations of ammonia, bicarbonate and carbonate, which essentially means that they only form when the urine is infected with urea-splitting bacteria (e.g. Proteus, Klebsiella and Pseudomonas spp.). For this reason, and in contrast to other types of stones, they occur more frequently in women and in other people with a predisposition to urinary tract infection, such as in those with an indwelling catheter or a spinal injury. They may also occur occasionally in other stone formers. The clinical presentation is often insidious as they tend to form staghorn calculi that may cause silent deterioration of renal function. Treatment includes correcting any underlying cause if possible, and rapid access to antibiotics appropriate to the known sensitivity of previously cultured organisms. Patients can keep a supply of antibiotics at home or even take continuous prophylaxis. Complete sterilization of the urine is possible only if stones are eradicated.
Uric acid stones
Hyperuricaemia and gout are discussed in Chapter 32. In healthy people, the amount of uric acid excreted in the urine depends on, among other things, the purine content of the diet. This makes the definition of a reference range rather difficult, particularly since even at ‘normal’ excretion rates of 3.6–4.8 mmol/24 h, the urine is supersaturated with uric acid and yet most people do not form uric acid stones. Stones may form in patients with hyperuricaemia from any cause, as well as in those with renal hypouricaemia due to an isolated genetic defect or as part of a Fanconi syndrome.
Most patients with uric acid stones have normal plasma urate concentrations and urinary urate excretion, but produce concentrated urine with consistently low pH (< 5.5). Patients with an ileostomy are at particularly high risk owing to bicarbonate and water loss from the stoma. The solubility of urate decreases sharply below its isoelectric point (pI) of 5.75. This offers a useful therapeutic intervention, in that if alkali is used to maintain the urine pH at 6.5–7.0, stones can be redissolved and further formation prevented even if there is a degree of hyperuricosuria. More pronounced alkalinization of the urine should be avoided, as it increases the risk of calcium phosphate deposition. Well-motivated subjects may titrate their dose of alkali (usually potassium citrate) by testing their own urine with pH paper. Patients with severe hyperuricosuria may need allopurinol to prevent stone formation and should be advised to restrict dietary purine intake.
Cystine stones
These have already been discussed in the section on cystinuria, above.
Miscellaneous rarities
Renal stones submitted for analysis occasionally may not fall into one of the categories discussed above. Some may be entirely unrelated to the renal tract (factitious disorders), whereas others may be from the renal tract but not be true stones (e.g. blood clots, sloughed papillae, encrusted sutures). However, there are a few other inherited metabolic diseases that do result in the formation of stones of unusual composition.
In hereditary xanthinuria, there is a deficiency in xanthine oxidase, with the consequent replacement of uric acid in the urine by xanthine and hypoxanthine. In about two-thirds of affected people this remains an asymptomatic metabolic abnormality, usually detected because of very low plasma urate concentrations, but in the remaining one-third xanthine stones form in the renal tract. There may also be associated myopathy or arthritis.
An even rarer subtype of xanthinuria has been described, in which there is deficiency of xanthine oxidase together with sulphite oxidase, but here the main clinical concern is the neurological involvement.
Xanthine stones have also been described in patients with normal plasma urate concentrations who clearly do not have xanthine oxidase deficiency. The cause of these is unknown.
Another rare inherited disorder of purine metabolism associated with renal stone formation is adenine phosphoribosyl transferase (APRT) deficiency. This enzyme is involved in the salvage pathway for the purine base adenine, and deficiency results in increased urinary excretion of 2,8-dihydroxyadenine. This leads to stone formation in most homozygotes, although up to 15% remain clinically stone free. It is worth noting that many of the chemical tests used for uric acid also give a positive reaction with 2,8-dihydroxyadenine. In children, the finding of uric acid stones using chemical testing should, therefore, be further investigated using a more specific technique.
Very rarely, stones may be composed entirely of glycoprotein matrix or extraneous compounds such as triamterene, silicate or indinavir, all of which are excreted by the kidney.
Investigation of stone formers
All stone formers should have at least a minimum basic screen including serum creatinine, potassium, bicarbonate, urate and calcium with a 24 h urine collection for measurement of volume, calcium and oxalate. Evaluation should be at least six weeks after surgery or lithotripsy to avoid contamination of urine. Patients who have recurrent stones, a strong family history or who present before the age of 25 should have more extensive investigation as outlined in Box 9.4. The use of acid-containing bottles for urine collection is controversial because of patient safety concerns, but there is a risk of calcium oxalate and phosphate precipitation before analysis if the sample is not adequately acidified. Crystals are very difficult to redissolve, even if the urine collection is acidified on receipt in the laboratory. Accurate urate estimation requires a non-acidified collection.
There is a marked biological variation in calcium and oxalate excretion, partly owing to diet, so at least two estimations separated by a week should be requested before any treatment is started. Testing a spot urine for albumin will detect most patients with proximal tubular disorders, and can be confirmed by low molecular weight protein estimation. Urinary albumin will also be raised if there is intrinsic kidney damage or if the patient has active stone disease.
Analysis of available stones can be highly informative, although not all practitioners in the field would agree. External quality assurance surveys in the UK demonstrate that stone analysis is not generally well done, especially if the method used is qualitative. Infrared spectroscopy is the gold standard, but if this is not available, quantitative chemical techniques are adequate. Knowledge of a stone’s constituent(s) obviously directs further biochemical investigation and monitoring. It is also important to continue to analyse the stones if there is recurrence – there may be changes in the constituents that necessitate a change in management.
Treatment
Treatment of specific stone types has been covered in the relevant sections. All patients will also benefit from general advice about diet, including those with no identifiable risk factors. Adequate fluid intake is the most important, aiming to achieve daily urine volumes of at least two but preferably three litres. Periods of dehydration, such as during sporting activities, should be avoided. Most patients can be taught to increase fluid intake to maintain a pale, straw-coloured urine. Reduction of salt, refined carbohydrate and animal protein intake has been shown to have a much greater effect on stone risk than calcium restriction. In fact, the population quartile with the lowest calcium intake has a higher risk of stone formation than the quartile with the highest intake, owing to reciprocal hyperoxaluria. Calcium restriction is therefore no longer recommended. Reducing the intake of oxalate (Box 9.3) will contribute to lowering of the calcium oxalate product in the urine. However, in otherwise healthy people, over 80% of urinary oxalate is produced endogenously. Tea is particularly rich in oxalate, especially if taken without milk (calcium precipitates the oxalate), and should be avoided. Citrate excretion by the kidney may be enhanced by oral potassium citrate supplements, orange juice or bicarbonate-rich mineral water. Urinary tract infections should be treated promptly and adequately: laboratory confirmation of bacterial eradication is recommended. Long-term prophylaxis with antibiotics may be required.
CONCLUSION
Primary disorders of the renal tubules are not common, but biochemical investigations are important in both diagnosing and monitoring them.
The pathogenesis of renal stone formation is still not fully understood, but enough is known for a logical approach to be taken in the biochemical investigation of stone formers. This is something that probably has not been uniformly well done in the past.
ACKNOWLEDGEMENT
We would like to thank Stephen K. Bangert who co- authored this chapter for previous editions of the book.
Further reading
APPENDIX 9.1 DIAGNOSIS OF RENAL TUBULAR ACIDOSIS
A variety of provocative tests of urinary acidification have been used in the investigation of RTAs, but whether these can firmly diagnose the type of defect remains to be established.
In general, hyperchloraemic metabolic acidosis that is not explained by bicarbonate loss from the intestinal tract should raise the suspicion of a urinary acidification defect. The plasma potassium may give a clue as to the type (high in type 4, low in types 1 and 2). The presence of other features of the renal Fanconi syndrome suggests type 2 RTA. The amount of bicarbonate required to correct the acidosis also gives an indication of the type of RTA (type 1 responding most readily and type 2 least readily). However, it may be necessary to confirm the diagnosis by using one of the following two tests:
Urinary acidification test
This can be used to confirm the diagnosis of distal RTA. The test is not necessary if the pH of a urine specimen collected after an overnight fast is < 5.5 or if it is > 5.5 in a patient confirmed to be acidotic in the resting state. If this is not the case, the patient is given ammonium chloride at a dose of 100 mg/kg body weight. Special formulations are available to minimize gastrointestinal side-effects. Urinary pH is then measured on fresh urine samples at hourly intervals for 8 h.
In normal subjects, urinary pH should fall to < 5.5 in at least one sample. In distal renal tubular acidosis this does not occur and urinary pH usually remains > 6.5.
Ammonium chloride should not be used in patients with liver disease, in whom calcium chloride may be used as an alternative acidifying agent (1 mmol/kg body weight). Ammonium chloride is unpleasant to take and alternative tests have been sought, for example the fludrocortisone–furosemide test. The principle of this latter test is to increase sodium delivery to the distal renal tubules at a time that sodium reabsorption and, therefore, hydrogen ion excretion is maximally stimulated by a mineralocorticoid. One mg of fludrocortisone is given orally an hour before furosemide 40 mg, also orally. Urine samples are collected every 30 min for up to 6 h or until a urine pH < 5.5 is achieved. However, both false positives and false negatives can occur and ammonium chloride loading remains the definitive test.
Fractional excretion of bicarbonate
This test can be used to confirm the diagnosis of proximal RTA, as long as the patient’s plasma bicarbonate is maintained above 20 mmol/L.
Plasma and urine samples are obtained and creatinine and bicarbonate concentrations measured in each. The fractional excretion of bicarbonate is then calculated:
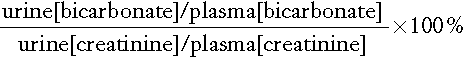
In patients with proximal RTA, the fractional excretion is > 10–15%, whereas in most patients with distal RTA it is < 10%.