[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 19
Thyroid dysfunction
Colin M. Dayan; Onyebuchi E. Okosieme; Peter Taylor
CHAPTER OUTLINE
Biological actions of thyroid hormones
Synthesis, storage and release of thyroid hormones
Iodine and thyroid hormone synthesis
Transport of thyroid hormones in blood
Entry of thyroid hormone into tissues
Thyroid hormone deiodination and regulation of extrathyroidal T3 production
Catabolism of thyroid hormones
Nuclear action of thyroid hormones
Control of thyroid hormone synthesis and secretion
Extrathyroidal factors that may affect thyroid function
THE EVALUATION OF THYROID FUNCTION
Clinical evaluation of thyroid status
In vitro tests of thyroid activity and pituitary–thyroid status
Measurement of thyroid stimulating hormone
Free T4 and free T3 measurements
Methods for measuring free thyroid hormones
Validity of commercial methods for free hormone analysis
Nomenclature of free thyroid hormone assays
Selective use of thyroid function tests
Interpreting results of thyroid function tests
Common situations in which TSH results may be misleading
Reference ranges and significant changes
Autoantibodies to thyroidal antigens
Hyperthyroidism or non-thyroidal illness?
Thyroiditis producing hyperthyroidism
Hypothyroidism resulting from Hashimoto thyroiditis
Hypothyroidism and the postpartum period
INTRODUCTION
Thyroid hormones are essential for normal growth, development and metabolism, and their production is tightly regulated through the hypothalamic–pituitary–thyroid axis. Thyroid disease is common, particularly in women, with a prevalence in the community of 3–5%. With the exception of iodine deficiency, which affects millions of people worldwide, diseases that directly affect the thyroid gland are the most common causes of thyroid dysfunction. Pituitary disease and the use of certain drugs that alter thyroid hormone synthesis or metabolism can also give rise to thyroid dysfunction. Any severe illness can produce abnormalities in the results of thyroid function tests that resolve as the patient’s illness improves. Once diagnosed, thyroid disease is usually easily treated, with an excellent long-term outcome for most patients. This chapter outlines thyroid physiology and the pathways of thyroid hormone synthesis and metabolism. The investigations used in diagnosis and management of thyroid disorders are described, together with guidance on their interpretation. The various thyroid disorders are described together with current views on their treatment.
NORMAL THYROID PHYSIOLOGY
The thyroid gland
The thyroid gland is brownish-red in colour and consists of left and right lobes connected by a midline isthmus, typically forming an ‘H’ or ‘U’. It is located anteriorly in the neck deep to the platysma, sternothyroid and sternohyoid muscles. The isthmus lies just below the cricoid cartilage and the lobes extend upward over the lower half of the thyroid cartilage. The pretracheal fascia encloses the thyroid gland and attaches it to the trachea and larynx. This explains why the thyroid gland is seen to move upwards with swallowing.
The thyroid is a highly vascular organ with blood flow ~ 5 mL/min/g of tissue (nearly twice that of the kidneys). It consists of thousands of follicles, each a spheroidal sac of epithelial cells (thyrocytes) surrounding a lumen containing colloid, mainly comprising thyroglobulin (Fig. 19.1). It also contains parafollicular C cells, which secrete calcitonin; calcitonin is discussed in Chapter 6.
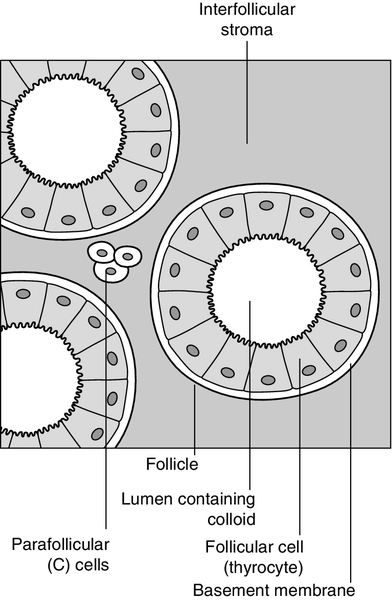
FIGURE 19.1 Structure of the thyroid follicle.
Embryologically, the thyroid is derived from the floor of the pharyngeal cavity and migrates from the base of the tongue to its final position in the neck along the thyroglossal duct. Abnormalities of this migration give rise to ectopic glands that may not function normally. Occasionally, the gland is completely absent.
The primary function of the thyroid is to synthesize and secrete thyroid hormones (Fig. 19.2). Under normal circumstances the gland synthesizes approximately 80 μg (110 nmol) of thyroxine (T4) per day and 6 μg (10 nmol) of 3,5,3′-tri-iodothyronine (T3).
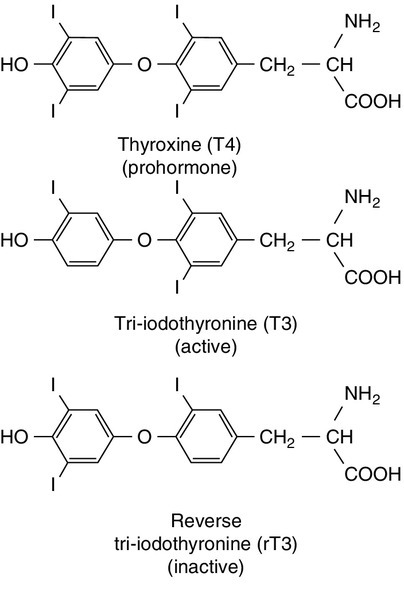
FIGURE 19.2 Structures of the iodothyronines.
Biological actions of thyroid hormones
Thyroid hormones produce enhancement of many intracellular events and promote differentiation and growth; they are essential for normal fetal and neonatal development.
Thyroid hormones increase mitochondrial oxidative phosphorylation and maintain amino acid and electrolyte transport into cells. They increase calorigenesis and oxygen consumption in most tissues, though not in brain. Thyroid hormones stimulate the synthesis of proteins, including structural proteins and enzymes. They regulate all aspects of carbohydrate metabolism, including increasing gluconeogenesis and accelerating insulin degradation. Thyrotoxicosis can, therefore, lead to a deterioration of glycaemic control in diabetes mellitus.
Stimulation of lipid metabolism leads to a fall in plasma cholesterol concentration, as degradation is increased to a greater extent than synthesis. Bone turnover is stimulated, resorption more so than mineralization, raising plasma calcium concentration in some patients. In general, the effect of thyroid hormones on the turnover of any substance will depend on the relative effects on synthesis and degradation. The actions of thyroid hormones increase demand for coenzymes and vitamins.
Complex interactions occur with the sympathetic nervous system and many of the signs and symptoms of hyperthyroidism, including palpitations, tremor and sweating may be attributed to these. For example, thyroid hormones amplify the adrenergic system through the α1 and β3 adrenergic receptors and are also the main determinant of basal metabolic rate (BMR). The increase in metabolic rate involves inhibition of AMP-activated protein kinase (AMPK) in the hypothalamus. However, the mechanisms responsible for many interactions between thyroid hormone and the sympathetic nervous system have not been identified.
Most changes in biochemical parameters produced by changes in thyroid status are not sufficiently specific or of such magnitude to be useful as tests of thyroid function, though the association may be sufficiently close that the finding of an abnormality should provoke a request for testing for previously unsuspected thyroid disease. For example, hypothyroidism is a relatively common and easily treatable cause of raised plasma cholesterol; however, the effect on lipids in subclinical hypothyroidism is modest. Equally, it may be helpful (and avoid unnecessary investigations) to anticipate these changes in cases of known thyroid dysfunction and to expect their normalization after treatment of the thyroid disorder. Some examples are listed in Box 19.1.
Synthesis, storage and release of thyroid hormones
Synthesis of T4 and T3 occurs on thyroglobulin (Tg), a glycoprotein of molecular weight 660 000 Da that contains many tyrosyl residues. Thyroglobulin is synthesized by the thyrocytes and exported to be stored within the colloid of the follicular lumen. Incorporation of iodide into Tg requires hydrogen peroxide and thyroid peroxidase (TPO), an enzyme that is synthesized within the follicular cell and transported to the apical membrane. Thyroid hormones are synthesized at the interface between the apical membrane of the thyrocyte and the colloid of the follicular lumen.
The process of thyroid hormone synthesis, storage and secretion requires a series of highly regulated steps (Fig. 19.3).
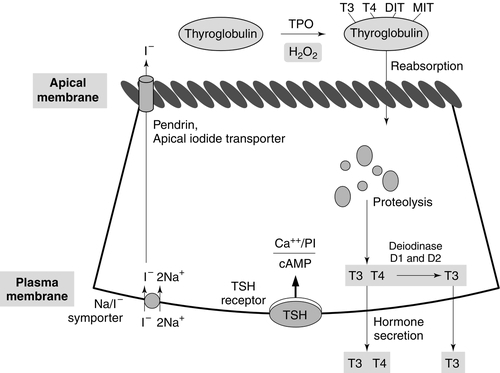
FIGURE 19.3 Sites of biochemical processes within the follicular cell (thyrocyte).
• Oxidation of iodide to iodine by thyroid peroxidase. This occurs on the luminal side of the apical membrane and requires TPO and hydrogen peroxide, which is generated by a calcium-dependent flavoprotein enzyme system situated at the apical membrane.
Recently, the dual oxidase molecules DUOX1 and DUOX2, which contain peroxidase-like and NADPH oxidase-like domains have been identified as essential for H2O2 production. They require maturation or activation factors (DUOXA1 or 2) for translocation of DUOX from the endoplasmic reticulum to the apical plasma membrane where H2O2 production occurs. Mutations in DUOX2 and DUOXA2 have both recently been identified as causes of hypothyroidism, resulting from diminished H2O2 production.
Halogenases collect the iodide removed from thyroglobulin as it is broken down in the cell.
Antithyroid drugs such as propylthiouracil and mercaptoimidazoles (methimazole and carbimazole) exert their action through inhibiting the action of TPO.
• Incorporation of iodine into tyrosyl residues on thyroglobulin. Mono-iodotyrosine and di-iodotyrosine (MIT and DIT) are formed through the actions of TPO (organification).
• Coupling of two iodotyrosyl residues in the thyroglobulin molecule. This is also catalysed by TPO and produces T3 and T4 that remain linked to Tg. When iodine supply is limited, the proportion of T3 produced on Tg increases. Iodinated Tg is stored in the follicular lumen.
• Internalization of Tg and release of T4 and T3. When there is a demand for thyroid hormone, this is signalled by an increase in plasma thyroid stimulating hormone (thyrotrophin; TSH) concentration. Thyroglobulin is then internalized by pinocytosis and appears as colloid droplets that fuse with lysosomes and undergo proteolytic degradation to release T3 and T4. Any MIT and DIT is deiodinated and the iodine conserved.
• Delivery of T4 and T3 into the circulation. This is probably achieved by thyroid hormone transporters. The large stores of T4 and T3 incorporated into thyroglobulin allow secretion of T4 and T3 more quickly when required than if it had it had to be synthesized.
Thyroid stimulating hormone appears to stimulate each of the above processes. It also stimulates the expression of many genes in thyroid tissue including thyroglobulin, sodium iodide symporter (NIS) and interleukin-8. It also causes thyroid hyperplasia and hypertrophy.
Iodine and thyroid hormone synthesis
Iodine deficiency represents a major public health issue for over two billion people who live in areas of iodine deficiency and especially for the 43 million in whom the deficiency is severe enough to cause learning difficulties. The recommended intake of iodine is 150 μg/day for adults but this may rise to 250 μg during pregnancy and lactation. Iodine is obtained from the ingestion of foods such as seafood, seaweed, kelp, dairy products, some vegetables (e.g. soybeans and potatoes) and iodized salt.
High iodine intakes can induce hypothyroidism, goitre and sometimes hyperthyroidism (see later). A large excess of iodide, when given acutely, results in acute inhibition of thyroid hormone release. It also inhibits the adenylate cyclase response to TSH (see below) and iodination of thyroglobulin; this is termed the Wolff–Chaikoff effect. After a few days of exposure to high iodide concentrations, thyroidal iodide uptake is very low, the intrathyroidal iodide concentration falls and the synthesis of iodinated thyroglobulin recommences. This effect can be used to prepare a thyrotoxic patient for thyroidectomy. However, if high iodine intake persists for more than ten days, then thyrotoxicosis can occur in individuals with a pre-existing multinodular goitre or those from iodine deficient areas; this is termed the Jod–Basedow effect.
Transport of thyroid hormones in blood
When T4 and T3 are secreted into the bloodstream, they become bound to plasma carrier proteins. Three proteins share this function: in order of decreasing affinity for iodothyronines they are thyroxine binding globulin (TBG); thyroxine binding prealbumin (TBPA, also known as transthyretin), and albumin. These binding proteins bind approximately 70% (TBG), 20% (TBPA) and 10% (albumin) of the circulating thyroid hormones. T4 is more tightly bound than T3 to each of these proteins. Approximately 0.02% of T4 and 0.2% of T3 are in the unbound or ‘free’ form in plasma in normal subjects, so that, although the total molar concentration of T4 is some 50 times that of T3, the concentration of free T4 is only about three times that of free T3. The half-life of T4 is 5–7 days; that of T3 is 1–2 days.
Free hormone hypothesis
For most of their actions, thyroid hormones need to enter the cell and bind to nuclear receptors. The free hormone hypothesis suggests that only the unbound (free) fraction of thyroid hormone is able to cross the plasma membrane and enter the nucleus. This hypothesis is supported by the observation that, in clinical practice, thyroid status correlates better with the concentration of the free hormone fraction than with the protein-bound fraction. The precise function of TBG and TBPA is unclear, but a number of roles have been suggested including acting as a reserve or buffer to prevent rapid changes in free hormone concentration in blood and also to prevent loss of thyroid hormones through filtration and excretion at the kidney. One further hypothesis is that TBG may be involved in targeting the delivery of thyroid hormone in a differential manner to different organs or facilitating the efflux of thyroid hormones from some tissues. Whatever their function, the absence or overexpression of the transport proteins is not associated with any obvious detrimental effects. There are difficulties in measuring the concentrations of free hormones and this is discussed later in this chapter.
Entry of thyroid hormone into tissues
Specific transporters appear to be required to facilitate the entry of the free thyroid hormones into cells. A number of important thyroid hormone transporters have been characterized. The organic anion transporting polypeptides (OATPs) are one group of important transporters. OATP1C1 appears to be specific for T4 and reverse tri-iodothyronine (3,3′5′-tri-iodothyronine: rT3); it is expressed exclusively in the capillaries of the brain and may be important for T4 transport across the blood–brain barrier. Two L-type amino acid transporters (LAT1 and LAT2) have been identified among the members of the heterodimeric amino acid transporter family, that transport thyroid hormone in many tissues. Human monocarboxylate transporter 8 (MCT8) is another active thyroid hormone transporter. It has a preference for T3 and is expressed in a variety of tissues including the brain, where it appears to be involved in the uptake of T3 by neurons, the primary target for thyroid hormones during brain development. The MCT8 gene is located on the X chromosome and mutations in the gene have been identified in a small number of boys with severe psychomotor retardation and elevated plasma T3 concentration (Allan–Herndon–Dudley syndrome). These mutations result in an inhibition of T3 supply to neurons leading to a defect in brain development. The characteristic pattern of thyroid function tests remains unexplained.
Entry of thyroid hormone into liver cells appears to be ATP-dependent. In rat hepatocytes, it is inhibited by a range of compounds that accumulate in plasma during illness. These compounds include free fatty acids, bilirubin and indoxyl sulphate. The uptake of thyroid hormones by the pituitary appears to occur through a different mechanism from that occurring in the liver. For example, unlike in the liver, the pituitary thyroid hormone transporters are not energy dependent; they can maintain or increase the uptake of T4 and T3 in low energy states and are not inhibited by bilirubin.
Thyroid hormone deiodination and regulation of extrathyroidal T3 production
Thyroxine is produced entirely by the thyroid gland, approximately 80 μg being produced each day. However, only 20% of T3 is produced directly from the thyroid gland; 80% is produced by the extrathyroidal deiodination of T4. Approximately 6 μg of the T3 in blood originates from thyroidal synthesis; 25 μg is formed by 5-deiodination of T4 in extrathyroidal tissues, a reaction catalysed by the D1 and D2 isoenzymes of iodothyronine deiodinase. T4 is considered to be a prohormone, T3 being responsible for the biological actions of thyroid hormones. Most tissues express one or more of a family of three iodothyronine deiodinases (D1, D2 and D3) that are responsible for providing a local source of T3 from T4 or for providing a source of T3 in plasma. In humans, it has proved difficult to determine which deiodinase and which tissues are responsible for providing most of the circulating T3. Classically it has been thought that D1 in liver and kidney provided most circulating T3, but some studies have suggested that, in humans, D2 may also provide an important source. Genetic association studies may provide more conclusive evidence. To date common genetic variants (single nucleotide polymorphisms) in DIO1 have been robustly associated with variation in the T3/T4 ratio in serum at genome wide levels of significance (p = < 1 × 10− 8), but as yet no associations have been observed with single nucleotide polymorphisms in DIO2.
Figure 19.4 summarizes the characteristics of the three deiodinases. All are selenoenzymes requiring adequate intake of dietary selenium for their expression. D1 carries out either 5′-deiodination, giving T3, or 5-deiodination, giving the inactive compound rT3. D2 appears to provide a very important local source of T3 in some tissues, including the brain, pituitary and brown adipose tissue. D3 catalyses the conversion of T4 to rT3 and is thought to be an important extrathyroidal control mechanism to regulate delivery of T3 to its receptors, particularly in the developing fetus.
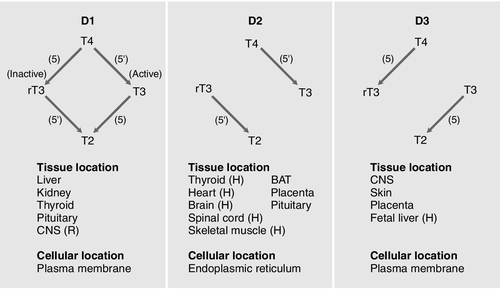
FIGURE 19.4 Characteristics of the iodothyronine deiodinases. In non-thyroidal illness, 5′-mono-deiodination is impaired leading to a decreased production of T3 and impaired breakdown of rT3. BAT, brown adipose tissue; CNS, central nervous system; (H), human not rat; (R), rat not human; T2, 3,3′-di-iodothyronine. (From Beckett G J, Arthur J R. Journal of Endocrinology 2005; 184:455–465. © Society for Endocrinology, with permission.)
Catabolism of thyroid hormones
Sulphation, glucuronidation, deamination, oxidative decarboxylation, ether cleavage and deiodination form the main routes for the inactivation and degradation of thyroid hormone. These metabolites are excreted via bile or in urine.
Nuclear action of thyroid hormones
Tri-iodothyronine exerts its effects through interaction with nuclear thyroid hormone receptors (TR) that have a high affinity and high specificity for T3. Nuclear thyroid hormone receptors are a family of ligand-regulated transcription factors that are associated with chromatin. Several α and β isoforms of TR are produced and these usually dimerize with the retinoid X receptors (RXRs). TRα1 is widely expressed with particularly high expression in cardiac and skeletal muscles, whereas TRβ1 is predominantly expressed in the brain, liver and kidney and TRβ2 is primarily limited to the hypothalamus and the pituitary.
The TR/RXR complex binds to target response elements located in the promoter region of the target genes. The formation of the T3-TR/DNA complex and subsequent recruitment of a variety of transcriptional coactivators leads to activation of the target genes, giving increased mRNA and protein production. In some circumstances, gene expression may be switched off. Mutations in the TRβ gene lead to a decreased affinity of the receptor for T3, leading to the autosomal dominant clinical syndrome of ‘thyroid hormone resistance’ (discussed later in this chapter).
Control of thyroid hormone synthesis and secretion
Classic feedback regulation
The most important regulator of thyroid homoeostasis is TSH. This dimeric peptide hormone comprises a β-subunit, required for binding to the TSH receptor, and an α-subunit. The α-subunit is the same as that of luteinizing hormone, follicle stimulating hormone and human chorionic gonadotrophin. The β-subunit is specific for TSH: both subunits are required for bioactivity. The subunits contain carbohydrate chains that are essential for the biological activity of the molecule. Modifications to these carbohydrate residues occur in different thyroid states, which alter both the bioactivity of the molecule and its plasma half-life. For example, in primary hypothyroidism, TSH has increased bioactivity and an increased plasma retention time, while in secondary hypothyroidism the bioactivity of the molecule is diminished. Most immunoassays are unable to recognize modifications to the carbohydrate chains. There is a circadian rhythm of TSH secretion, plasma concentrations being highest between midnight and 04.00 h and lowest at about midday. Thyroid stimulating hormone release also occurs in a pulsatile fashion. These changes in TSH are not reflected in the serum thyroid hormone concentrations.
The production of TSH is controlled by a stimulatory effect of the hypothalamic tripeptide, thyrotrophin releasing hormone (TRH, thyroliberin), mediated by a negative feedback from circulating free T3 and free T4 (Fig. 19.5). Free T3 and free T4 inhibit TSH synthesis and release directly, by inhibiting transcription of the TSH subunit genes and indirectly by inhibiting TRH release. Free T3 and free T4 also decrease TSH glycosylation and therefore its biological activity. It is thought that the hypothalamus, via TRH, sets the level of thyroid hormone production required physiologically, and that the pituitary acts as a ‘thyroid-stat’ to maintain the level of thyroid hormone production that has been determined by the hypothalamus. Twin studies have shown that in individuals without thyroid disease, TSH and thyroid hormone concentrations are largely genetically determined. The setpoint in an individual is relatively narrow with intraindividual variation being less than half the interindividual variation.
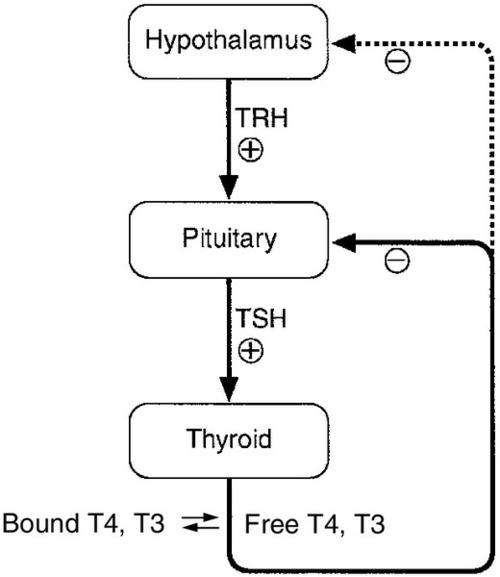
FIGURE 19.5 The ‘classic’ hypothalamo–pituitary–thyroid axis. The production of thyroid stimulating hormone (TSH) is controlled by a stimulatory effect of the hypothalamic tripeptide, thyrotrophin releasing hormone (TRH, thyroliberin), mediated by a negative feedback from circulating free T3 and free T4. Thyroid stimulating hormone may also exert autoregulatory fine control on its own production. Dopamine, somatostatin, glucocorticoids, leptin and catecholamines all may have an influence on the axis.
Other mechanisms
The classic feedback regulation of TSH release described above is an oversimplification. Dopamine, somatostatin and glucocorticoids inhibit TSH release, and these agents, together with cytokines, may be important modifiers of plasma TSH, particularly in patients with non-thyroidal illness (NTI) (Fig. 19.6). Leptin and catecholamines regulate TSH indirectly by stimulating TRH production. There is accumulating evidence that TSH can provide autoregulation via a direct action of TSH on the hypothalamus and at the pituitary. In the pituitary, folliculo-stellate cells have been identified that express TSH receptors: interaction of TSH with these cells may produce fine control on TSH release through the release of cytokines, which in turn inhibit TSH release from thyrotrophs. It has been argued that interaction of TSH receptor antibodies with the folliculo-stellate cells explains why patients with Graves disease may continue to have suppressed TSH weeks or months after normal thyroid hormone concentrations have been achieved through carbimazole therapy.
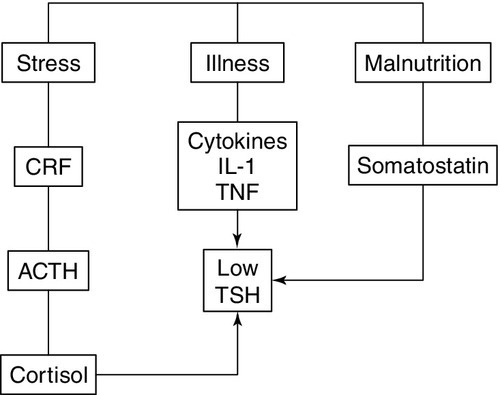
FIGURE 19.6 The hypothalamo–pituitary–thyroid axis in non-thyroidal illness. Dopamine, somatostatin, and glucocorticoids inhibit TSH release, and these agents together with cytokines may be important modifiers of serum TSH, in particular in patients with non-thyroidal illness. ACTH, adrenocorticotrophic hormone; CRF, corticotrophin releasing factor; IL-1, interleukin-1; TNF, tumour necrosis factor.
Thyroid stimulating hormone modifies thyroid hormone synthesis by binding to a specific receptor on the surface of the thyrocyte. The TSH receptor is a single protein with a large extracellular amino-terminal domain involved in the binding of TSH, seven transmembrane domains and a short intracellular carboxy-terminal domain involved in the activation of G-protein modulators of the adenylate cyclase-protein kinase-A system. These characteristics are shared with receptors for gonadotrophins: their extracellular domains show some 40% homology, the remainder being more highly conserved. This may explain the weak thyroid-stimulating activity of human chorionic gonadotrophin (hCG). Binding of TSH results in activation of adenylate cyclase and accumulation of cyclic AMP. The calcium and phosphoinositol signalling pathways may also be activated by TSH. After about an hour, an increase in release of thyroid hormones is seen. In the longer term, TSH increases the synthesis of iodinated thyroglobulin and also causes a general increase in the metabolism, size and activity of the follicular cells.
Extrathyroidal factors that may affect thyroid function
Age
Fetus
Thyroxine and TSH are detectable in fetal plasma at 10–12 weeks of gestation. Plasma concentrations of total and free T4, total and free T3 and TBG increase during gestation, total and free T4 reaching adult concentrations at about 36 weeks, but total and free T3 reaching only the lower limit of the adult range at this time. The concentration of TSH also increases with gestational age but is within or above the adult reference range throughout gestation. Marked time- and organ-specific changes occur throughout gestation in the expression of the deiodinases, which are thought to coordinate the orderly maturation of enzyme systems responsible for development of the brain and the other organ systems in the fetus.
Neonate
During the first 24 h after birth, there is a rapid, transient increase in the release of TSH, T3 and T4 that is considered to be an adaptive response to birth. Thyroid stimulating hormone peaks during the first 30 min, which then stimulates T3 and T4 production during the first 24–36 h of postnatal life. This effect may be attenuated in infants delivered prematurely. Screening for congenital hypothyroidism should be carried out after at least three days to avoid spurious results.
Infancy and childhood
Plasma concentrations of TSH are within the adult range but free T3 is higher than in adults. Free T4 concentrations tend to be at the lower end of the adult range. This hormone pattern suggests it may arise as a result of increased D1 activity in children. After puberty, no major changes in thyroid function occur, except in the pregnant woman.
Elderly
In old age, there is little change in thyroid function tests and age-related reference ranges are not required. There is a modest decrease in T4 secretion but without an accompanying change in circulating T4. A slight fall in T3 may occur in those over 80 years of age. In patients on T4 replacement, the dose of T4 may have to be decreased with age. There is also increasing evidence that the normal TSH distribution curves appear to be shifted towards higher value ranges in individuals aged over 80. Age-specific analysis of TSH concentrations and anti-thyroid antibody titres measured in the National Health and Nutrition Examination Survey (NHANES) demonstrated that 12% of subjects aged 80 and older without any evidence of underlying autoimmune thyroiditis had TSH concentrations > 4.5 mU/L. What is currently unclear is whether higher TSH concentrations confer a survival advantage or simply represent a degree of non-autoimmune age-related thyroid failure.
Pregnancy
In normal pregnancy there is a large increase in plasma TBG concentration that arises from both an oestrogen stimulated increase in synthesis and diminished clearance of the protein. There is also an increase in the pool size of extrathyroidal T4 distribution and an increase in the deiodination of thyroid hormones in the developing placenta. The result of these changes is that, during pregnancy, there is a marked increase in the requirement for iodine and an approximately 50% increase in T4 production occurs if the supply of iodine is adequate.
In order to maintain homoeostasis, an increase in total T4 and total T3 concentrations occurs, which reach a new steady state around mid-gestation. This requirement for increased thyroid hormone production requires an ideal iodine supply during pregnancy of approximately 250 μg/day. Similarly, women with hypothyroidism taking T4 replacement will need an increase in the dose of T4 of approximately 25–50 μg/day during pregnancy within the first six weeks of gestation, aiming for a TSH no higher than 2.5 mU/L. Some centres recommend that women double their pre-pregnancy levothyroxine dose on two days a week as soon as pregnancy is confirmed to reduce the risk of maternal hypothyroidism. Thyroxine replacement should be monitored carefully using both TSH and free T4, since even modest degrees of hypothyroxinaemia in early pregnancy have been associated with an impaired IQ in the infant.
In pregnancy, free T4 and free T3 concentrations may initially show a slight rise, thought to be due to the weak thyroid stimulating action of hCG, present in very high concentration in early pregnancy. This increase in free hormones can lead to suppression of TSH, such that low or sometimes undetectable concentrations may be found in up to 20% of patients during the first trimester. It is important that these changes in early pregnancy are not construed as suggesting that the patient has thyrotoxicosis. As pregnancy progresses, the concentrations of free thyroid hormones fall and TSH begins to rise, although rarely increasing above the reference range seen in non-pregnant subjects. Free thyroid hormone concentration may be lower than those found in non-pregnant women. It is important that trimester-related reference ranges are used for both TSH and free thyroid hormones. Patients can usually return to their pre-pregnancy levothyroxine replacement regimen immediately post-delivery; however, they should be monitored to ensure that this is still the optimal dose.
Hyperemesis gravidarum (severe vomiting in the first trimester of pregnancy) is associated with high plasma total and free thyroid hormone and low TSH concentrations, making it difficult to distinguish this condition from severe true thyrotoxicosis (thyroid crisis). It is believed that hCG is responsible for the thyroid stimulation; the condition usually resolves by the second trimester. Thyroid stimulating hormone receptor antibodies are negative in patients with hyperemesis but positive if the patient has Graves disease.
Thyrotrophin releasing hormone can cross the placenta from mother to fetus, but TSH does not. Thyroid hormones cross the placenta, but this transplacental flux is regulated in part by changes in expression of placental deiodinase as pregnancy progresses. A maternal supply of thyroid hormones (particularly T4) to the fetus is particularly import in the first trimester until a functional fetal thyroid has developed. However, since a fetus with congenital thyroid deficiency develops relatively normally in utero, this suggests that some maternal-fetal transfer of thyroid hormone must be maintained throughout pregnancy. Iodide and antithyroid drugs also pass to the fetus. Indeed, antithyroid drugs are transferred more readily than thyroid hormone, and for this reason treatment of thyrotoxicosis in pregnancy with a ‘block and replace’ regimen (high dose antithyroid drugs ‘rescued’ by simultaneous thyroid hormone replacement) is contraindicated in pregnancy as it would result in a relatively hypothyroid fetus. The treatment of hyperthyroidism in pregnancy requires careful consideration. Radioactive iodine is contraindicated and the choice of antithyroid drug is currently the subject of debate. Propylthiouracil has been traditionally preferred to carbimazole/methimazole owing to fears of scalp and gastro-oesphageal defects in fetuses exposed to carbimazole/methimazole in the first trimester. However, recent concerns have arisen owing to the occurrence of propylthiouracil induced hepatitis, which has occasionally required liver transplantation and rarely has resulted in maternal death. Autoantibodies may cause additional problems in managing patients with hyperthyroidism in pregnancy. In patients with Graves disease, the transplacental passage of thyroid stimulating immunoglobulins may cause transient neonatal thyrotoxicosis, while the presence of anti-TPO antibodies is associated with an increased risk of maternal hypothyroidism and miscarriage.
Non-thyroidal illness
Patients attending or admitted to hospital suffering from any of a wide range of chronic or acute non-thyroidal illnesses (NTI), often have abnormalities in thyroid function tests. A low T3 may often be found even though the patients are clinically euthyroid; this has been termed the sick euthyroid syndrome.
Several mechanisms are involved, including:
• alterations in the hypothalamic–pituitary–thyroid axis leading to decreased hypothalamic stores of TRH and suppression of TSH release due to increased concentrations of dopamine, cytokines, cortisol and somatostatin (see Fig. 19.6). While the degree of TSH suppression in NTI is often not as great as that found in hyperthyroidism, there is considerable overlap in TSH concentrations found in the two conditions
• changes in the affinity characteristics and in the plasma concentrations of the thyroid hormone-binding proteins. These changes give rise to alterations in the plasma concentrations of both the free and total thyroid hormones
• impaired uptake of thyroid hormones by the tissues
• decreased production of T3 in the peripheral tissues
• changes in the T3 occupancy and function of the T3 receptors.
The contribution of each of the above mechanisms may vary with the severity and stage of the illness, and thus the pattern of thyroid function tests may be extremely variable and may mimic the profile seen in either primary or secondary thyroid disease. Interpretation of thyroid function tests is complicated further by the effects of drugs and methodological problems associated with free hormone measurements.
Thyroid hormone metabolism is markedly affected by fasting and illness, with the magnitude of these changes tending to be proportional to the severity of the illness (Fig. 19.7). Extrathyroidal conversion of T4–T3 is reduced, leading to a marked decline in plasma total and free T3, which may fall to undetectable concentrations. Reverse T3 concentration increases, primarily owing to impaired catabolism rather than increased synthesis. These changes are often considered to be an adaptive response for energy conservation, as rT3 is not metabolically active. Concentrations of free fatty acids and other substances that can compete with thyroid hormones for binding to plasma proteins may rise. This can produce a transient increase in free T4. Drugs that compete for T4 binding sites will have a similar effect. Uptake of thyroid hormone into cells may be impaired, either directly by endogenous inhibitors or indirectly as a result of impairment of the active transport systems of the cells. Finally, administration of corticosteroids or dopamine may suppress TSH release.
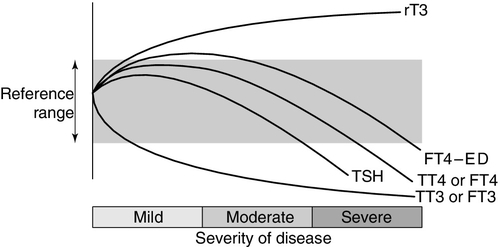
FIGURE 19.7 The effects of illness on the concentration of thyroid hormones and thyrotrophin. The profile for free T4 obtained using equilibrium dialysis is shown (FT4-ED). The concentration of free T4 found using routine assays is method dependent, with some assays showing the profile of FT4-ED, while others follow the profile of total T4 (TT4). Free T3 (FT3) shows a similar profile to total T3 (TT3).
Methodological shortcomings render the results of free hormone analysis liable to serious error. Equilibrium dialysis methods usually show a normal or slightly raised free T4 in patients with illness of mild to moderate severity, but most commercial assays tend to show normal or low free T4 results in sick patients. Normalization or transient rebound changes of thyroid parameters may be seen during recovery from NTI or refeeding after starvation. In particular, TSH may rise transiently into the hypothyroid range. It should be noted that, although TSH is in general the most reliable test of thyroid function, in hospitalized patients a TSH of < 0.1 mU/L is twice as likely to be due to NTI as to hyperthyroidism, and an increased TSH is as likely to be due to recovery from illness as to hypothyroidism. Because of the poor predictive value of thyroid function tests in hospitalized patients, these tests should only be requested if the reason for hospital admission is considered, on clinical grounds, to be related to a thyroid problem. Also, abnormal thyroid function tests during illness may need to be rechecked after the acute illness has resolved.
While the changes in thyroid hormone metabolism associated with illness might theoretically lead to tissue hypothyroidism, most trials of T4 and T3 administration in severe non-thyroidal illness have not shown improved outcome.
Drugs
Drugs may interfere with TSH secretion or the production, secretion, transport and metabolism of thyroid hormones (Table 19.1). Some drugs modify thyroid status while others produce abnormal thyroid function test results in clinically euthyroid subjects. Certain agents (in particular iron preparations) impair the absorption of T4 from the gut, and patients on thyroxine therapy should be advised to take their T4 at least 4 h apart from these medications. Patients should also be advised to avoid caffeine within 45 min of taking levothyroxine as caffeine can also impair absorption. Patients taking levothyroxine are likely to require an increase in replacement dose if drugs such as phenytoin or carbamazepine are also prescribed as they increase hepatic metabolism of T4. Phenytoin, carbamazepine, furosemide and salicylate compete with thyroid hormone binding to plasma binding proteins and may increase plasma free T4 concentrations. The influence of drugs on modifying free thyroid hormone concentrations may be method specific. For example, depending on the method, free T4 may be measured as being normal, high or low in patients given heparin or taking phenytoin or carbamazepine. Amiodarone, lithium and interferon can induce thyroid dysfunction and are covered in detail later in this chapter.
TABLE 19.1
Drugs affecting thyroid function
| Mechanism | Example of drug |
| Decrease in TSH secretion | Dopamine, glucocorticoids, octreotide, cytokines |
| Decrease in thyroid hormone secretion | Lithium, amiodarone, iodide |
| Increase in thyroid hormone secretion | Lithium, amiodarone (rare), iodide |
| Decrease in thyroidal synthesisa | Carbimazole, propylthiouracil, lithium |
| Increase in TBG | Oestrogens, tamoxifen, heroin, methadone, clofibrate, raloxifene |
| Decrease in TBG | Androgens, glucocorticoids, anabolic steroids |
| Displacement of thyroid hormones from plasma proteins | Furosemide, fenclofenac, salicylates, mefenamic acid, carbamazepine |
| Increased hepatic metabolism | Phenytoin, carbamazepine, rifampicin, barbiturates |
| Impaired T4 and T3 conversion | β-antagonists, amiodaronea, radiocontrast dyes, e.g. iopanoic acid |
| Impaired absorption of thyroxineb | Cholestyramine, aluminium hydroxide, ferrous sulphate, calcium salts, sucralfate, soya protein |
| Altered immunityc | Interleukin-1, interferons, tumour necrosis factor-α, interleukin-2 |
Other drugs listed produce abnormal thyroid function tests but patients remain euthyroid. Amiodarone is the exception (see text).
a Cause decrease in thyroid hormone synthesis or secretion and alter thyroid status.
b Interfere with absorption from GI tract. Patients on T4 therapy should be advised to take their T4 at least 4 h apart from these medications.
c These cytokines can cause transient hypothyroidism or thyrotoxicosis, the mechanism of which is unclear.
THE EVALUATION OF THYROID FUNCTION
This section describes the methods that have been developed in order to assess the functional state of the thyroid gland. In most cases, the presenting clinical features will lead to thyroid function tests being performed for confirmation of the provisional diagnosis.
Clinical evaluation of thyroid status
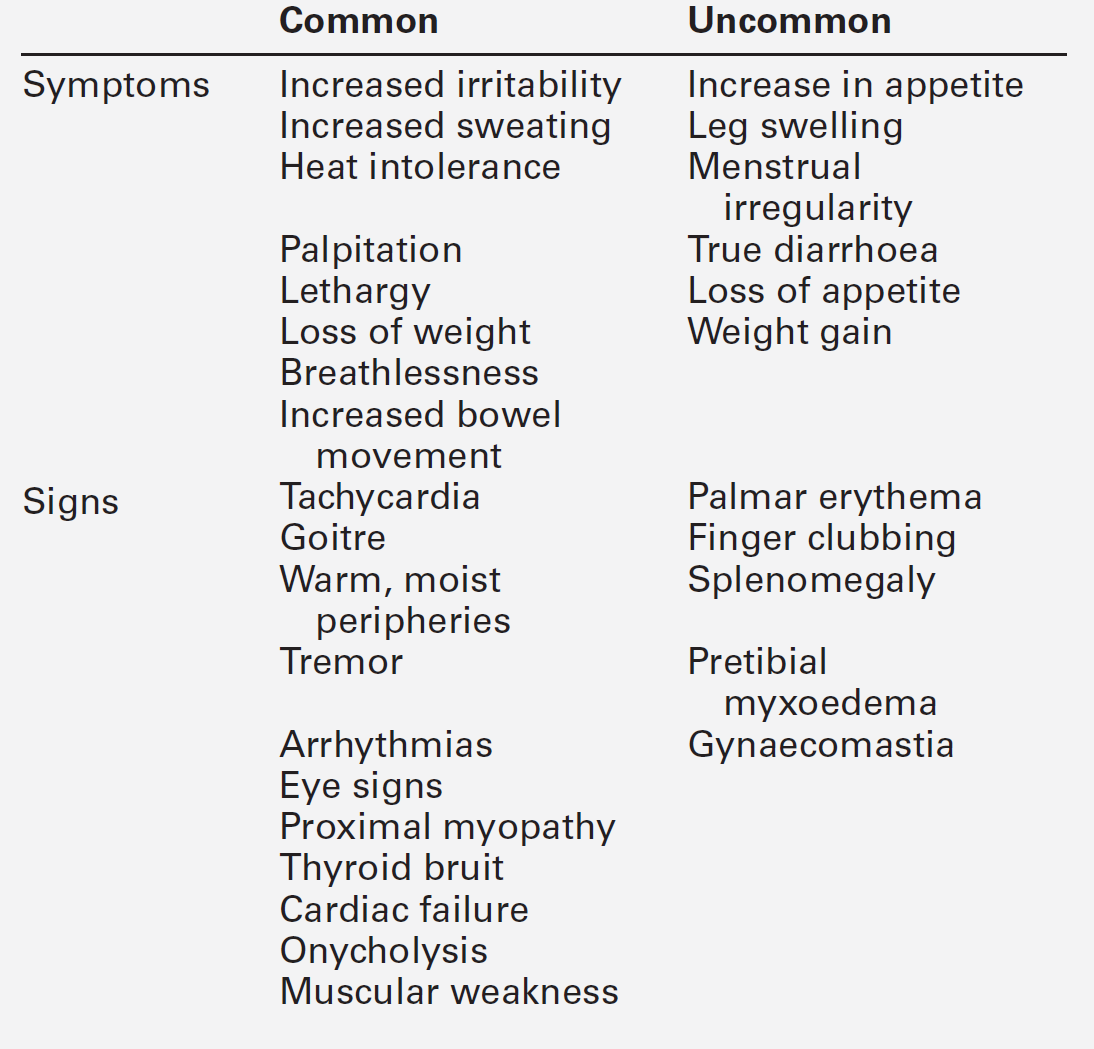
Disordered thyroid function may be detected clinically by demonstrating both changes in the thyroid gland itself and the systemic effects of under- or overproduction of thyroid hormones. In some instances, the clinical presentation may be virtually diagnostic of a specific condition and investigations are required only for confirmation. In others, the clinical features may be less specific and more thorough investigation is required. The signs and symptoms of thyroid dysfunction are listed in Tables 19.3 and 19.6. The normal thyroid can often be palpated and may also be apparent on inspection of the neck, particularly in young, thin women. Abnormal thyroid enlargements may be diffuse or asymmetrical. As a result of connective tissue attachments to the trachea, thyroid swellings move upwards on swallowing and a similar phenomenon is found with tongue protrusion in the case of thyroglossal cysts. The normal gland has a ‘rubbery’ feel; in Graves disease and diffuse colloid goitre, the consistency is softer. By contrast, the thyroid in Hashimoto disease is often rather firm, often with a palpable pyramidal lobe. Thyroid carcinoma (anaplastic) and Riedel thyroiditis result in a rock-hard gland that may, in the case of carcinoma, be irregular in outline. Increased blood flow through a hyperactive gland is often associated with a bruit. This may be maximal over the superior thyroidal artery rather than the gland itself and, in some cases, may also produce a palpable thrill.
In vitro tests of thyroid activity and pituitary–thyroid status
The tests used to investigate thyroid dysfunction can be grouped into:
• tests to elucidate the cause of thyroid dysfunction, e.g. thyroid autoantibody and serum thyroglobulin measurements, thyroid enzyme activities, biopsy of the thyroid, ultrasound and isotopic thyroid scanning
• thyroglobulin measurements, which are used to monitor treatment and detect recurrence in patients with follicular or papillary carcinoma.
Measurement of TSH and thyroid hormones should be performed to determine the patient’s thyroid status before requesting the additional tests that seek to determine the cause of any thyroid dysfunction.
Measurement of thyroid stimulating hormone
The measurement of TSH in a basal serum sample by a sensitive immunometric assay provides the single most sensitive, specific and reliable test of thyroid status in both overt and subclinical primary thyroid disorders. In primary hypothyroidism, TSH is increased, while in primary hyperthyroidism TSH is usually < 0.02 mU/L. However, TSH alone is not a reliable test for detecting thyroid dysfunction arising from hypothalamic–pituitary dysfunction and in other specific instances as outlined below.
Thyroid stimulating hormone is now measured by immunometric assays (IMAs) that utilize non-competitive labelled antibody methods with non-isotopic labels. These IMAs use highly specific monoclonal antibodies raised to epitopes on α- and β-subunits of the TSH molecule. The IMA technique uses a sandwich- assay configuration using two antibodies. The ‘functional sensitivity’ of a TSH assay defines the minimum concentration of TSH that can be quantified in routine use. To obtain the functional sensitivity of a routine assay, a precision profile is constructed using data obtained over many assay runs with multiple batches of reagent and different operators. The concentration of TSH that has a coefficient of variation of 20% taken from the precision profile defines ‘functional sensitivity’. It is essential that TSH assays are sufficiently sensitive for clinical diagnostic purposes and they must have a functional sensitivity of < 0.02 mU/L.
Free T4 and free T3 measurements
Theoretical considerations
The plasma concentrations of free thyroid hormones are extremely low and their quantification in the presence of large concentrations of bound hormones has proved challenging. Poor assay design of many free hormone methods has produced a mass of conflicting literature regarding the typical ranges of free thyroid hormone concentrations in patients with NTI and taking a variety of drugs.
The free hormone concentration results from the equilibrium between the bound (BP) and free hormone.
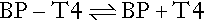
The proportion of T4 that binds to the binding protein is dictated through the law of mass action by the affinity of the binding protein (Keq) multiplied by its concentration. This is known as the ‘relative binding capacity’. Thus, if the affinity or concentration of the binding protein decreases, the proportion of T4 bound will also diminish and the free hormone concentration will increase. In humans, the free T4 fraction is mostly affected by changes in the affinity and concentration of TBG; changes in the affinity and concentration of albumin or transthyretin have little effect on free T4.
Free hormone measurements involve sampling the free hormone fraction. This can be done using physical separation of the free from bound hormone using a semipermeable membrane (equilibrium dialysis or ultrafiltration) or, alternatively, by adding an antibody that ‘captures’ a proportion of the free hormone pool. The removal of free hormone from the original equilibrium will result in further hormone dissociating from the binding protein to create a new equilibrium. A crucial requirement of any valid method for free hormone measurement is that during the assay process there should be minimal disruption of the original equilibrium, and this can be achieved by minimizing the dilution of the specimen and sampling only a small proportion of the hormone pool. The pH and the ionic composition of any buffer also need to be controlled. The inadequacies of many commercial assays have led to the suggestion that the term free hormone estimate is used for all free hormone assays used in clinical practice.
Methods for measuring free thyroid hormones
Equilibrium dialysis and ultrafiltration methods
These are widely regarded as reference methods, although neither is completely satisfactory. The long dialysis period (~ 12–18 h) used for equilibrium dialysis can, in certain samples (e.g. patients given heparin), lead to the production of non-esterified fatty acids (NEFAs) from triglyceride, which may displace thyroxine from its binding proteins and thus increase the free T4 concentration. A similar increase in free T4 will occur if such patient samples are stored unfrozen prior to analysis.
Validity of commercial methods for free hormone analysis
Routine assays are given a variety of names depending on the methodology used, but all usually involve: (1) adding an antibody to diluted serum to ‘capture’ a proportion of the free T4 pool and (2) estimating the unoccupied T4 binding sites on the antibody by adding labelled T4. These commercial assays have additional problems over those of equilibrium dialysis. First, the antibody used will tend to disrupt the original equilibrium, an effect that is dependent on the overall thyroxine binding capacity of the sample and the affinity and concentration of the antibody. Second, the binding capacity of the patient sample may be quite different to that used in the assay calibrators. The overall effect of this is that samples with low binding capacity (NTI, low TBG etc.) tend to have a negative bias introduced, while samples with high binding capacity (high TBG, e.g. pregnancy) may have a positive bias introduced. Many commercial assays include albumin, added in an attempt to reduce non-specific binding and nullify the effects of NEFA on free T4 concentrations. Unfortunately, albumin binds T4 and, if present in sufficient concentrations, will cause significant disruption of the equilibrium. Again, samples with low serum binding capacity will be more prone to this effect than samples with normal or high binding capacities. These various effects explain why free T4 assays may appear to produce low, normal or raised free T4 concentrations in the same sample taken from patients with NTI.
In a true free hormone assay the calculated and measured free hormone concentrations in a set of serially diluted serum samples should be closely approximate. Assays that show a significant reduction in free T4 at these dilutions are likely to produce heavily negatively biased results in patients with low serum binding capacity. Many current commercial assays for free T4 fail these validation tests. The development of free T3 assays has been even more problematic than for free T4.
Nomenclature of free thyroid hormone assays
The names given to free hormone assays include one-step analogue, two-step back-titration and labelled antibody assays. The principles for each of these assays are widely reviewed in the literature but the name given to an assay does not guarantee its validity or performance.
The free T4 and free T3 index methods, that estimate the free hormone concentration in the presence of protein-bound hormone, have now largely been replaced by the direct measurement of free T4 and free T3.
Total T4 and total T3
Plasma concentrations of total T4 and total T3 are measured by immunoassay using diluents that incorporate a chemical agent that releases the thyroid hormones from their binding proteins. In clinical practice, most problems associated with total hormone measurements arise in patients with modified hormone binding capacities that, in turn, may increase or decrease the total hormone concentration. Pregnancy and oestrogen medication give rise to increased concentrations of TBG and thus increases in total T3 and total T4. Some causes of abnormal TBG concentration (and thus, abnormal total thyroid hormones) are given in Box 19.2. Genetic variants of TBG, transthyretin and albumin have been described that have altered hormone-binding characteristics, which in turn modify the total concentration of thyroid hormones in the circulation. Non-thyroidal illness results in the production of modified TBG with diminished affinity for thyroid hormone. Many drugs may also lower the hormone binding capacity of plasma. Some patients produce autoantibodies to the thyroid hormones that increase the hormone binding capacity of plasma and, in addition, produce marked assay interference.
The use of total hormone measurements is now less popular following the introduction of more reliable methods for free hormone measurements. However, total hormones are still considered to have a role in clarifying thyroid status in some patients with NTI where the results of free thyroid hormone and TSH measurements do not concur.
Selective use of thyroid function tests
Many laboratories measure random TSH as the initial test of thyroid function. This approach is not infallible, but is least likely to produce an abnormal result in NTI and in patients on various forms of drug treatment. It can detect both overt and subclinical disease. A normal TSH concentration effectively excludes primary thyroid dysfunction. If an abnormal result is obtained, thyroid hormone measurements are then made to confirm that thyroid dysfunction is present, and to determine the severity of the disease.
Initial measurement of both TSH and free T4 together can provide a more satisfactory method of assessing thyroid status, since in some situations a single TSH result may be misleading. If this strategy is followed, a significant number of cases will arise in which one test is abnormal while the complementary test is normal. It is thus essential to understand and appreciate the factors that can affect the results of thyroid function tests.
Interpreting results of thyroid function tests
Table 19.2 provides a guide to the interpretation of thyroid function tests.

Situations in which TSH usually provides the correct estimate of thyroid status
Overt primary hyperthyroidism
Thyroid stimulating hormone is nearly always < 0.1 mU/L and often < 0.02 mU/L, owing to feedback inhibition on the pituitary. Free and total T4 and T3 concentrations are nearly always increased, but in a very small percentage of hyperthyroid patients, total T4 and free T4 are both normal, whereas both total T3 and free T3 are increased: this condition is known as T3 hyperthyroidism or T3 thyrotoxicosis.
Overt primary hypothyroidism
Thyroid stimulating hormone is invariably increased, often to > 20 mU/L, as feedback inhibition of the pituitary is diminished, and free T4 and total T4 are low. Free T3 and total T3 measurements are of no value here, since normal concentrations are often observed because of increased peripheral deiodination of T4–T3.
Subclinical thyroid disease
Thyroid disease presents as a spectrum of clinical and biochemical features of varying severity. The clinical diagnosis of mild thyroid disorders is often difficult, and the only biochemical abnormality may be an abnormal serum TSH concentration. For example, many clinically euthyroid patients with multinodular goitre or with exophthalmic Graves disease have a TSH < 0.1 mU/L, but normal free and total thyroid hormone concentrations. The combination of a low TSH together with normal thyroid hormone concentrations in a patient is known as ‘subclinical hyperthyroidism’, but this description is unsatisfactory, since it rests solely on the results of chemical investigations. Similarly, a diagnosis of ‘subclinical hypothyroidism’ is given to those patients with an increased TSH but normal thyroid hormone concentrations.
Before the diagnosis of subclinical thyroid disease can be made, causes of an abnormal plasma TSH other than thyroid disease must be excluded. These include pregnancy, NTI, drug treatment and assay interference.
Common situations in which TSH results may be misleading
Assay interference from endogenous heterophilic antibodies
Some individuals have antibodies in their plasma that react with a range of animal immunoglobulins (heterophilic antibodies). These antibodies interfere with a wide range of immunoassays. For example, a normal or even elevated TSH result may be found in some thyrotoxic patients owing to this type of assay interference.
Pregnancy
Thyroid stimulating hormone is a reliable indicator of thyroid status in the second and third trimester of pregnancy, but in the first trimester, a low TSH may be found in up to 20% of women. Thyroid function testing in pregnancy is covered earlier in this chapter.
Secondary thyroid disorders
Thyroid stimulating hormone concentration is normal in about half of patients with central (pituitary) hypothyroidism, and in a few, TSH may be slightly elevated. Circulating TSH has been shown to have reduced bioactivity in central hypothyroidism and thus free T4 and total T4 concentrations are usually low. When pituitary disease is known to be present, TSH concentrations should not be used to assess the thyroid status.
Very rarely, hyperthyroidism is due to a TSH-secreting tumour. Persistent hyperthyroid symptoms associated with elevated free T4 and free T3 but raised or normal TSH are consistent with this diagnosis, once the common problems of assay interference or NTI have been eliminated. Serum α-subunit concentrations are often increased in these patients.
Abnormal pituitary T3 receptor function, due generally to mutations in the thyroid hormone β receptor, can result in disordered thyroid function not reflected by changes in TSH. In this disorder, the thyrotroph is resistant to the normal negative feedback regulation, so that plasma thyroid hormone concentrations are raised in the presence of a normal or raised TSH. Depending on the balance of effects between receptor function in the pituitary and the rest of the body, the patient may be clinically euthyroid, hyperthyroid or even mildly hypothyroid. This condition is termed ‘thyroid hormone resistance’.
Reference ranges and significant changes
By convention, a reference range usually only comprises 95% of values from a reference population; therefore 2.5% of the population will have values above the reference range and 2.5% below. Thyroid hormone concentrations show a normal distribution in reference populations. For TSH, the reference population shows a log normal distribution even when subjects who are thyroid peroxidase (TPO) antibody positive are excluded. Thyroid stimulating hormone reference ranges should be established using specimens collected between 08.00 h and 18.00 h and using 95% confidence limits from log transformed data. The reference population should have no personal or family history of thyroid dysfunction, be on no medication known to alter TSH secretion and have no thyroid antibodies detectable by a sensitive assay. Method-related reference ranges need to be used for all thyroid function tests. Typical adult reference ranges are: TSH 0.3–4.0 mU/L, total T4 60–160 nmol/L, total T3 1.2–2.6 nmol/L, free T4 9–25 pmol/L and free T3 3–8 pmol/L.
In neonates, infants and children, age-related reference ranges are recommended. Trimester-related reference ranges should be available for use in pregnancy.
Knowledge of the analytical variability and estimates of the intraindividual and interindividual variability allows a calculation of differences in the results of thyroid tests that can be considered as clinically significant when monitoring serially a patient’s response to therapy. These changes may be method dependent but are in the order of:
Miscellaneous tests
Thyrotrophin releasing hormone test
In this dynamic test of pituitary-thyroid function, the TSH response to an intravenous dose of thyrotrophin releasing hormone (TRH) is measured. Formerly, it was used as a test of hypothyroidism when TSH estimates were insensitive (exaggerated TSH response) and as a test of pituitary function. Nowadays it is only occasionally used in the diagnosis of thyroid hormone resistance or TSH-secreting adenoma.
Thyroglobulin
Thyroglobulin (Tg) is normally present in the circulation in very small amounts and, although concentrations may be raised in many thyroid disorders, its measurement (by immunoassay) is essentially reserved for follow-up of patients with thyroid cancer (post total thyroidectomy and radioiodine ablation), in whom elevation of previously suppressed concentrations may indicate tumour recurrence. The measurement of Tg is challenging, as up to 30% of patients with thyroid cancer have antithyroglobulin antibodies in their serum that may interfere with the assay. Rarely, demonstration of suppressed Tg concentration in the presence of raised T4 and T3 is used to diagnose factitious hyperthyroidism due to illicit abuse of thyroid hormone.
The measurement of serum thyroglobulin is also useful in identifying the cause of congenital hypothyroidism as all patients with organification disorders, and those with severe thyroid-stimulating hormone receptor defects, will have raised concentrations when their circulating TSH is increased, except for those with thyroglobulin synthesis defects where the thyroglobulin concentration usually remains low. Thyroglobulin measurements can also provide useful information about the presence of thyroid tissue, for example, if serum thyroglobulin is measurable where there is apparent athyreosis on imaging, this implies that radiologically undetectable hypoplastic thyroid tissue is present.
α-Subunit
The pituitary glycoprotein hormones TSH, follicle stimulating hormone (FSH) and luteinizing hormone (LH) are composed of two subunits, the α-subunit being common to all three hormones and the β-subunit specific to each hormone. The plasma concentration of the α-subunit can be measured by immunoassay, without cross-reaction from complete hormone molecules, and it may be raised in patients with pituitary tumours. Raised concentrations are also found in some postmenopausal women.
Autoantibodies to thyroidal antigens
Antibodies to thyroid peroxidase (TPOAb)
Originally known as thyroid microsomal antibodies, these antibodies are found in almost all (95%) patients with autoimmune hypothyroidism secondary to Hashimoto thyroiditis early in the course of the disease, and in some patients with other autoimmune thyroid diseases. The antibodies are polyclonal and their target antigen has been identified as the thyroid peroxidase enzyme, which plays critical roles in thyroid hormone synthesis. These antibodies are frequently present in individuals with subclinical hypothyroidism as well as in 2–5% of biochemically euthyroid women (rising to over 10% over the age of 70). The prevalence of these antibodies is lower in men, but also increases with age, being present in 2.5% of the male population aged over 75. They may play a pathogenetic role in some patients with destructive autoimmune thyroid disease, as they have been shown both directly to fix complement on the thyrocyte and to direct cell-mediated cytotoxic reactions against thyrocytes, but in the majority of cases the damage to the thyroid is believed to be T cell rather than antibody mediated.
Knowledge of the presence of this group of antibodies can be of value in several circumstances. The presence of a high titre of TPOAb in the presence of goitrous hypothyroidism is characteristic of Hashimoto disease; they may also be seen in association with the early, transient hyperthyroidism sometimes associated with this condition. In pregnancy, the presence of antithyroid peroxidase antibodies is a significant risk factor for miscarriage, maternal hypothyroidism and the future development of postpartum thyroiditis with disturbed thyroid function. In more general terms, the finding of TPOAb in high titre is indicative of thyroid autoimmunity and may hence be associated with present or future occurrence of other organ-specific autoimmune disease. Patients with ophthalmic Graves disease (eye signs typical of Graves ophthalmopathy but without a present or past history of hyperthyroidism) are often found to have these antibodies, confirming the association with autoimmune thyroid disease despite normal thyroid function. In patients with subclinical hypothyroidism, the presence of TPOAb is an indicator to consider commencing T4 replacement, as such patients are at significant risk of developing overt hypothyroidism in the future (50% will progress to biochemical hypothyroidism over the next 20 years).
Antibodies to thyroglobulin (TgAb)
Antibodies to thyroglobulin are also found in patients with thyroid autoimmunity, but at lower frequency than the TPOAb. These antibodies do not fix complement and are not known to play a direct pathogenetic role in the aetiology of autoimmune thyroid disease in man. The only reason to measure these antibodies is in patients being treated for follicular or papillary cancer, to indicate possible assay interference in Tg assays. Antibodies to thyroglobulin can interfere with assays for Tg, giving falsely elevated Tg concentrations in radioimmunoassays and falsely decreased concentrations in IMAs. In patients being monitored for differentiated thyroid cancer, a rising titre of TgAb is associated with a poor prognosis.
Antibodies to the thyroid stimulating hormone receptor
The hyperthyroidism of Graves disease is mediated via the effect of IgG class autoantibodies that bind to the TSH receptor and produce an increase in intracellular cyclic AMP concentration. Serum concentrations of such antibodies can be correlated with thyroid gland hyperfunction. Two types of assay system are in use: the first measures the capacity of the autoantibody to inhibit the binding of labelled TSH to its receptor on human thyroid membrane preparations (TSH-binding inhibiting immunoglobulin or TBII), and the second measures the increase in cyclic AMP concentrations in thyroid preparations following stimulation by the patient’s IgG (thyroid stimulating antibody or TsAb). During periods of thyroid overactivity, these assays will be positive in 90–95% of patients.
Knowledge of TSH receptor antibody status in patients with Graves disease during pregnancy is of prognostic value: the infants of patients with a high antibody titre during the third trimester have an increased risk of neonatal hyperthyroidism secondary to transplacental passage of maternal autoantibody. Such neonatal hyperthyroidism is transient, settling as maternal antibody disappears from the child’s circulation.
Other TSH receptor antibodies (‘blocking’ antibody), although infrequently demonstrated, can inhibit gland function and lead to hypothyroidism; the predominant effect (stimulation vs blocking) may vary in a single patient with time. However, stimulation is far more commonly found.
Antibodies and ophthalmopathy of Graves disease
The aetiology of the ophthalmopathy of Graves disease, which is present in about a third of patients, is still poorly understood, but current evidence suggests an autoimmune pathogenesis and an association with smoking. Orbital muscle, connective tissue and adipose tissue become infiltrated with lymphocytes and macrophages. The extracellular compartment of the extraocular muscles and orbital fibro-adipose tissue becomes oedematous owing to water deposition caused by production of glycosaminoglycans by orbital fibroblasts and pre-adipocytes. Evidence of autoimmune responses to thyroid antigens is often present in patients with Graves ophthalmopathy. The most promising candidate autoantigen is the TSH receptor that is expressed in orbital connective tissue and in particular orbital fat (pre-adipocytes). It seems most likely that TSH receptor binding antibodies activate orbital fat cells by signaling through this receptor in a way that is subtly different from activation of thyroid cells, explaining why Graves ophthalmopathy is often, but not always, associated with stimulation of the thyroid gland. A role for activation of the insulin-like growth factor (IGF-1) receptor in orbital fat has also been proposed.
Imaging the thyroid
Standard radiography gives only limited information in the case of thyroid enlargement and is not used routinely. X-ray of the chest may show a superior mediastinal mass in patients who have retrosternal extension of a goitre, and the presence of tracheal deviation with or without narrowing of the lumen may also be noted.
Thyroid ultrasound can demonstrate the type of thyroid enlargement (diffuse or localized) and nature (solid or cystic, single or multiple) of thyroid nodules. It is, however, not possible definitively to exclude malignancy using ultrasound alone and biopsy of the nodule may be required. There is increasing use of ultrasonography to improve the quality of the content of fine needle aspirations of thyroid nodules. Colour-flow Doppler sonography is also increasingly being used to distinguish between type 1 amiodarone-induced thyrotoxicosis (increased uptake) and type 2 amiodarone-induced thyrotoxicosis (decreased/absent uptake); these conditions are covered later in this chapter.
The ability of the thyroid to concentrate radioisotopes of iodine and other substances has been utilized for many years to identify areas within the gland that are or are not functionally active. These imaging systems are of particular value in the evaluation of thyroid nodules, where the presence of nonfunctioning solid tissue is associated with a much higher risk of malignancy. Although subject to important limitations, the level of overall gland activity can also be assessed.
Thyroid scintiscanning
Using intravenous administration of radioisotopes and a scintillation camera, qualitative images of functioning thyroid tissue can be obtained and the proportion of the total dose taken up by the gland can be quantified. Of the available isotopes, 99mTc, given intravenously as pertechnetate, is concentrated within the gland but not organified into thyroid hormones and therefore diffuses out of the gland with time. This fact, together with its short half-life, means that large doses can be administered without delivering a high radiation dose to the thyroid. The gland is imaged 20 min after injection.
Disadvantages of use of this isotope are that it cannot reliably be used to identify retrosternal glands (as confusion may arise with intravascular radioactivity) and the lack of information regarding iodine organification. The use of radioisotopes of iodine circumvents this latter problem, 123I perhaps being the best isotope to use when imaging thyroid tissue in its normal site as, by comparison with other isotopes of iodine, it delivers a much lower total radiation dose to the gland. Iodine-131, with higher energy and a longer half-life, is useful where deep thyroid tissue is sought or where there is a need to detect, and treat by irradiation, functionally active metastatic thyroid carcinoma.
By quantifying the proportion of the total administered dose of isotope concentrated within the thyroid gland during a given time period, it is possible to estimate the activity of the gland, uptake increasing with increased gland activity and vice versa. However, gland uptake of isotopes of iodine may be critically influenced by many factors other than gland activity (such as dietary iodine content, coincidental administration of iodine-containing drugs such as amiodarone and previous administration of iodine-containing contrast media for radiological investigations) and the results of such uptake studies should be interpreted with caution. Biochemical tests of thyroid function have therefore superseded uptake studies in the measurement of gland activity. However, uptake studies are useful in identifying those patients with hyperthyroidism and a negligible uptake (painless thyroiditis, previous treatment with amiodarone) for whom 131I therapy would be inappropriate.
Perchlorate discharge test
Rarely, hypothyroidism and/or goitre result from defects in enzymes responsible for the organification of iodine into thyroid hormone. In such cases, iodine will be trapped within the thyrocyte but is not organified. Perchlorate, given orally as potassium perchlorate, will cause inhibition of the active transport of iodine across the thyrocyte membrane and hence lead to a loss of non-organified iodine from the gland.
HYPERTHYROIDISM
Clinical features
The clinical symptoms and signs which may result from the hyperthyroid state are summarized in Table 19.3. The clinical picture may be modified by the presence of coexisting systemic diseases and may depend on the age of the patient. Some of the thyroid diseases themselves also produce characteristic physical signs, for example the orbital and cutaneous manifestations of Graves disease. The changes seen in the major organ systems are described below.
Cardiovascular system
Many of the manifestations of hyperthyroidism relate to the increased demands placed upon the cardiovascular system. High circulating concentrations of thyroid hormones have a direct stimulatory effect on cardiac muscle. Heart rate and stroke volume are both increased at rest (including during sleep) and peripheral vascular resistance is reduced, leading to a marked rise in cardiac output in patients who have no pre-existing cardiac disease. Overt cardiac failure may result from this high output state but can also be triggered by the occurrence of arrhythmias. Atrial flutter/fibrillation is seen in over 10% of patients with hyperthyroidism and may be the presenting feature in some cases. Patients who suffer from ischaemic chest pain typically find that angina worsens as a result of the increased metabolic demands placed on the myocardium.
Thyroid crisis
Florid hyperthyroidism may result in a life-threatening illness (thyroid ‘crisis’ or ‘storm’). In these – fortunately uncommon – cases, cardiovascular symptoms and signs predominate. Circulatory collapse is characteristic and results from arrhythmia and high output cardiac failure. High output cardiac failure usually occurs in younger individuals with longstanding thyrotoxicosis; despite doubling of the cardiac output, the increase in preload and blood volume in thyrotoxicosis causes a rise in ventricular filling pressure leading to pulmonary and peripheral congestion. Pharmacological attempts to correct abnormal cardiac rhythms are usually unsuccessful in the face of continuing hyperthyroidism. Management aims to control the hyperthyroidism rapidly (using large doses of antithyroid drugs, including iodine) while treating the patient symptomatically. β-Blocking drugs such as propranolol are used, and measures to cool the patient are often needed. Use of steroids under these circumstances may be of additional value.
Gastrointestinal system
Weight loss is a classic feature of hyperthyroidism, often occurring despite an increased appetite. Increases in gastrointestinal motility lead to increased frequency of bowel movement, though true diarrhoea does not usually result. Nausea and vomiting are also uncommon, but may precede the onset of a thyrotoxic crisis. In severe hyperthyroid states, liver function can be markedly deranged, with hypoalbuminaemia and elevation of plasma aminotransferase and alkaline phosphatase activities. Paradoxically, alkaline phosphatase concentrations (of mixed bone and liver origin) can rise further initially after treatment for thyrotoxicosis, settling after 2–3 months.
Central and peripheral nervous system
Generalized hyperkinesia is very often seen in hyperthyroid individuals, frequently being accompanied by emotional lability. A fine tremor of the outstretched fingers is also characteristic. Patients complain of inability to sleep, despite often profound feelings of tiredness.
Locomotor system
Muscular weakness particularly affects proximal muscles. The aetiology of the weakness may be related to impaired phosphorylation of creatine. Muscle biopsy shows loss of type IIb muscle fibres, with replacement by fat and occasional lymphocytic infiltration. These changes reverse when euthyroidism is restored, but muscle power can take several weeks or months to return fully to normal. Periodic paralysis, associated with hypokalaemia during attacks of weakness, is also seen in association with hyperthyroidism, particularly in people of oriental descent. Myasthenia gravis is found in approximately 1% of patients with Graves disease and, similarly, myasthenia sufferers have an increased incidence of Graves disease.
Respiratory system
As part of the generalized myopathy, respiratory muscle function may be impaired and pulmonary compliance may also be reduced. Dyspnoea may result from these changes and breathlessness secondary to cardiac dysfunction may also be exacerbated. All these changes are reversible with appropriate antithyroid therapy.
Skin and hair
With prolonged elevation of thyroid hormone concentrations, diffuse hair loss is common; the nails are brittle and may become elevated from the nail bed (onycholysis). Palmar erythema may be found; the skin itself feels warm and moist due to increased cutaneous blood flow and sweating.
The skeleton
Prolonged hyperthyroidism may result in significant loss of mineral from the skeleton, which results from increased bone turnover, with resorption of bone exceeding accretion. Hypercalciuria and hyperphosphaturia are also found and urinary hydroxyproline excretion is increased, reflecting the increase in collagen turnover. Although hypercalcaemia is commonly found in hyperthyroid individuals, it is usually mild and resolves with effective antithyroid therapy.
The kidneys: mineral and water balance
Hyperthyroid individuals often complain of increased thirst and mild polyuria, even in the absence of hypercalcaemia or hyperglycaemia, and osmoregulation may be disturbed while biochemical hyperthyroidism persists, though this is rarely clinically significant. Plasma sodium and potassium concentrations do not usually alter, but urinary magnesium excretion increases and plasma magnesium concentrations may be low.
Other endocrine systems
In the female hyperthyroid patient, menstrual irregularities occur sometimes, with scanty menstrual loss and/or irregular cycle length. Although cycles usually remain ovulatory, fertility is significantly reduced. In general, plasma concentrations of free sex steroids are reduced as a consequence of increases in binding proteins, but total sex steroid concentrations may be high. Preferential metabolism of androgens to oestrogens may be responsible for the gynaecomastia seen in a small proportion of hyperthyroid males. Hypothalamo–pituitary responsiveness to exogenous gonadotrophins and basal concentrations of LH and FSH are usually normal.
The turnover of cortisol is increased in hyperthyroidism, but basal concentrations remain normal and the response to physiological stress is preserved in the absence of coincidental adrenal or pituitary disease.
Hyperthyroidism in the elderly
Although weight loss is variable, there may be anorexia leading to a clinical suspicion of malignancy. Patients are often tired and withdrawn with reduced mobility due to proximal myopathy, and the term ‘apathetic thyrotoxicosis’ has been used to describe this presentation. Atrial fibrillation, which may be associated with cardiac failure, is a common presentation in patients over 70 years, especially in those with multinodular goitre, in whom mild hyperthyroidism may have been present for some years. Any coexisting osteoporosis will be accelerated. Elderly patients often present with a single feature of hyperthyroidism (e.g. weight loss, atrial fibrillation) without the other systemic features more commonly seen in younger patients.
Causes of hyperthyroidism
Table 19.4 lists the causes of hyperthyroidism and summarizes their pathogenesis.
TABLE 19.4
The major causes of hyperthyroidism
| Cause | Pathogenesis |
| Graves disease | Hyperthyroidism due to TSH receptor stimulating antibody |
| Toxic multinodular goitre | Autonomous function in areas of a nodular gland |
| Toxic nodule | Autonomously functioning nodule |
| Chronic iodine excess | High thyroidal iodine ± areas of autonomously functioning gland or thyroid stimulating antibody |
| Thyroiditis | Transient phenomenon resulting from release of stored thyroid hormone from damaged gland |
| Pituitary tumour secreting TSH | Inappropriate release of TSH stimulating the thyroid |
| Amiodarone | High iodine content; alteration of thyroid hormone metabolism |
| Thyrotoxicosis factitia | Exogenous administration of thyroid hormone |
| Ectopic thyroid tissue | Functionally active and autonomous thyroid tissue present in extrathyroidal tumours |
| Trophoblastic tumours | Secretion of thyroid stimulator |
Thyroid stimulating hormone measurement demonstrates suppression of TSH concentrations in all cases of hyperthyroidism (except those due to inappropriate pituitary secretion of TSH, as described later). Free T4 is usually elevated in association with raised free T3 concentrations, though true T3 toxicosis (elevation of T3 alone) may be present. Specific diagnostic biochemical and immunological tests are outlined below.
When following the response of hyperthyroidism to treatment, TSH concentrations should be measured, but as it is clear that TSH suppression may persist for a variable period of time after restoration of normal free T4 and T3 concentrations, the measurement of TSH should not be used unsupported to follow response to therapy. The authors recommend that, ideally, concentrations of free T4 and T3 be measured with TSH until TSH secretion returns to normal.
Graves disease
Graves disease, an autoimmune condition of unknown aetiology, is characterized by the presence of diffuse thyroid enlargement, orbital tissue involvement (Graves ophthalmopathy) and, less commonly, skin involvement (pretibial myxoedema and thyroid acropachy) in addition to thyroid dysfunction. The disease predominantly affects females, with a female:male ratio of approximately 6:1, and peak incidence in the fourth and fifth decades. As with other autoimmune diseases, there is clearly an inherited predisposition to develop the disease, although other family members may have hypo- rather than hyperthyroidism. Excess iodine may induce hyperthyroidism in susceptible individuals, and observational studies indicate a relationship between major stress, such as loss of a close relative, and the onset of hyperthyroidism some months later. It is now well established that stress may modify the immune response.
Graves disease is also seen as the immune system recovers after a period of immunosuppression. This occurs in around 3% of patients with HIV disease after initiation of retroviral therapy and up to 15% of patients with multiple sclerosis 9–12 months after treatment with Campath® monoclonal antibody therapy as the T cell compartment repopulates.
Thyroid involvement
In Graves disease, thyroid enlargement (goitre) is typically moderate and diffuse, and the gland has a soft consistency. Because of increased blood flow through the hyperactive gland, a vascular bruit may be heard over the thyroid.
Eyes
Hyperthyroidism produces a characteristic staring expression, with the sclera being seen above and below the iris (lid retraction) and a tendency for the lid movement to lag behind that of the globe as patients look downward from a position of maximum upward gaze (lid lag). These signs may be present in any patient with hyperthyroidism. Additional and specific orbital signs and symptoms may be found in patients with Graves disease (Fig. 19.8). Similar changes may rarely occur with other autoimmune thyroid diseases such as Hashimoto thyroiditis and are listed in Table 19.5. Patients with Graves ophthalmopathy should be managed in a multidisciplinary eye clinic with the joint input of ophthalmologists and endocrinologists.
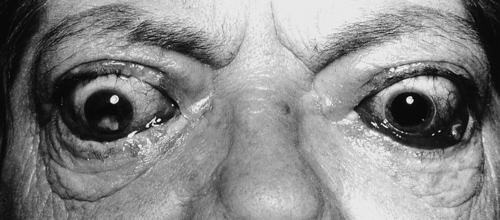
FIGURE 19.8 The appearance of a patient with severe Graves ophthalmopathy. Significant proptosis, periorbital swelling, chemosis and conjunctival infection are all present. The dilated left pupil is a consequence of the use of mydriatic eyedrops and not the disease process. (© University Hospital of Wales, Heath Park, Cardiff Medical Illustration Dept).
TABLE 19.5
The eye signs found in thyrotoxicosis
| Sign | Examination finding |
| Proptosisa | Forward protrusion of the globe |
| Lid retraction | Sclera visible above/below iris |
| Lid lag | Lid movement lags behind that of globe |
| Ophthalmoplegiaa | Failure of full ocular movement often producing diplopia |
| Chemosisa | Oedema of conjunctival sac |
| Periorbital oedemaa | Oedema and prolapsed orbital fat present around eye |
| Conjunctival infectiona | Inflammatory response |
| Visible ocular musclea | Enlarged extraocular muscle visible anteriorly |
| Loss of visiona | Compressive optic neuropathy or corneal exposure with scarring |
| Ptosis (rarely) | Drooping of upper eyelid |
a Eye signs specific to Graves disease rather than thyrotoxicosis.
Skin
Patients with Graves disease may sometimes develop indurated purple skin lesions, classically over the anterior tibia. These areas (pretibial myxoedema) contain large amounts of glycosaminoglycans and tend to be seen in patients with high concentrations of thyroid stimulating antibody and ophthalmopathy. Acropachy (similar to clubbing) is a recognized but rare feature of Graves disease.
Diagnosis
The diagnosis of Graves disease is not difficult when the typical triad of hyperthyroidism, goitre and extrathyroidal involvement is present. The diagnosis may be made in other circumstances by demonstrating the presence of thyroid stimulating antibody and is supported by the finding of diffuse, increased uptake of radioisotope on thyroid scintiscanning (see Figure 19.9A).
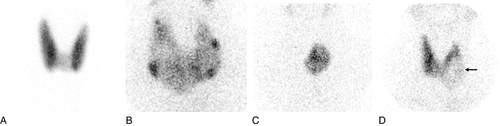
FIGURE 19.9 Isotope scans of patients with various thyroid disorders. The figure shows 99mTc scan images of (A) Graves disease showing characteristic diffuse uptake of isotope; (B) multinodular goitre with patchy uptake of isotope and inadequate activity in relation to the autonomously functioning nodules; (C) toxic adenoma causing ‘T3 thyrotoxicosis’. The suppressed TSH secretion leads to atrophy of the normal thyroid tissue, which recovers following removal of the adenoma by surgery or 131I ablation; (D) a ‘cold nodule’ with reduced uptake in relation to the rest of the gland. Fine needle aspiration revealed papillary carcinoma confirmed histologically following thyroidectomy.
Natural history
The natural history of the hyperthyroidism of Graves disease is that of successive relapse and remission over many years in the majority of patients. In about 30% there is a single episode of hyperthyroidism, lasting several months, followed by prolonged remission and even the eventual development of hypothyroidism 10–20 years later. Unfortunately, it is not possible at first presentation to identify which patients are destined to have a single episode. The antithyroid drugs, which do not influence the natural history of Graves disease, are given for an empirical period of 9–18 months, thereby identifying the ‘single episode’ patient and avoiding the use of destructive therapy.
Treatment
There are three forms of treatment of the hyperthyroidism of Graves disease: antithyroid drugs, surgery and 131I. Each is effective but none is perfect. Medical therapy with antithyroid drugs offers the only chance of lasting remission without resulting in permanent hypothyroidism. However, medical treatment is required for at least 9–18 months in most instances and overall success rates are < 50%. Surgery or radioactive iodine therapy provide rapid control of hyperthyroidism, but incur the need for lifelong thyroid hormone replacement. Radioiodine treatment with 131I may be associated with a rise in thyroid stimulating antibody concentrations and a worsening of ophthalmopathy and is absolutely contraindicated in pregnancy or the puerperium. Surgery is generally safe and effective, but carries the risks associated with anaesthesia and can be complicated by vocal cord damage or transient or permanent hypoparathyroidism. These complications are, however, minimized in the hands of an experienced surgeon.
The antithyroid drugs carbimazole (and its active form methimazole) and propylthiouracil exert their action principally by inhibiting the action of thyroid peroxidase and, therefore, thyroid hormone synthesis. Treatment is given for 9–18 months. Shorter treatment periods are associated with higher relapse rates, but treatment periods extending beyond 18 months carry no advantage. The antithyroid drugs can be given either in titrated doses or as part of a ‘block and replace’ regimen using both carbimazole and thyroxine. Both approaches are equally effective but the block and replace regimen, although generally resulting in more stable thyroid function and a reduced frequency of follow-up visits for dose titration, may be associated with slightly more side-effects than the titration. Lasting remission is only achieved in 30–40% of cases: adverse prognostic indicators include younger age, male gender, high pretreatment concentrations of T3, the presence of a large goitre and persistence of TSH receptor antibody at the end of the planned treatment period. As the drugs do not affect release of thyroid hormone from the gland, there is typically a delay of 4–8 weeks before euthyroidism is restored. Although persistently raised TSH receptor antibody concentrations at the end of therapy are associated with an increased risk of relapse, they are not sufficiently predictive to be of value in guiding clinical decision-making and testing is currently recommended only for diagnostic and not for prognostic purposes.
Symptomatic relief can be obtained in nearly all patients by also using β-blocking drugs for these first few weeks of therapy in order to reduce their tremor and tachycardia. In pregnant thyrotoxic patients, careful monitoring is required in order to keep the dose of antithyroid drugs to a minimum, especially during the last trimester, as these compounds cross the placenta and may render the fetus hypothyroid. ‘Block and replace’ treatment should not be used as T4 crosses the placenta only poorly. It is generally recommended to maintain free T4 and free T3 concentrations in the high–normal range to ensure sufficient transfer of thyroid hormones to the fetus to counter the effects of transplacental passage of the antithyroid drugs.
Iodine-131 can be used therapeutically to treat hyperthyroidism due to Graves and other thyroid disorders. Radioiodine works initially by interfering with organification of iodine, but later predominantly inhibits replication of thyrocytes, by inducing radiation damage in the gland and hence controlling thyroid overactivity, after a delay of weeks or months. The major side-effect of this form of therapy is long-term hypothyroidism, with up to 80% of patients becoming hypothyroid within the first year, if a standard dose of 400 MBq is used. In some individuals, there is a transient rise in thyroid hormone concentrations in the first few weeks after radioiodine before they begin to fall. In patients with Graves disease, the appearance or worsening of thyroid eye disease may occur, and in patients with active eye disease it is recommended that radioiodine is avoided and/or steroid prophylaxis for 6–8 weeks is given (e.g. prednisolone 20 mg reducing by 5 mg every two weeks). Graves ophthalmopathy is associated with smoking and cessation should be advised, particularly prior to radioiodine treatment. There is no evidence of an increase in the risk of malignancy following therapeutic doses of radioiodine. The dose should be repeated if hyperthyroidism persists after six months. Note that transient hypothyroidism at three months is occasionally seen with return of thyrotoxicosis by six months following radioiodine requiring repeat therapy.
Thyroidectomy is a highly effective treatment for Graves disease. Patients should be rendered euthyroid prior to operation using antithyroid drugs. In some centres, the drug is stopped for two weeks prior to surgery and potassium iodide substituted; this reduces the size and vascularity of the gland and makes surgery technically easier. To avoid the risk of relapse and the requirement for repeat surgery, near-total thyroidectomy (rather than subtotal thyroidectomy) is now routinely performed for thyrotoxicosis, rendering all patients hypothyroid postoperatively, and treatment with thyroid hormone is routinely administered immediately post surgery.
Treatment of Graves ophthalmopathy
A third of patients with Graves disease have thyroid eye involvement; sight loss is rare but may occur if untreated in 3–5% of patients. In addition, most patients have impaired quality of life and suffer significant psychological distress due to the disfiguring appearance of the eyes. In its classic form thyroid eye disease is easily recognizable. However, some patients suffer with eye disease for months to years before a diagnosis, which may be missed in patients without eye symptoms, patients with unilateral eye disease or patients who are euthyroid at presentation since orbitopathy may precede or post-date the development of hyperthyroidism. General management measures include smoking cessation and maintenance of normal thyroid status. Radioiodine therapy is best avoided in patients with moderate to severe eye disease (see above) but may be administered with glucocorticoid cover in mild cases. Local measures, e.g. lubricants, ointments and corrective prisms, will be adequate for mild disease. Glucocorticoids are first-line therapy in active moderate to severe disease. Other treatments that have been tried include ciclosporin, methotrexate, intravenous immunoglobulins, orbital radiotherapy, and the B cell depleting agent, rituximab. Selenium is an antioxidant with a non-specific anti-inflammatory effect that has been shown to reduce symptoms in mild Graves ophthalmopathy.
Toxic multinodular goitre
Hyperthyroidism arising in a previously multinodular goitre occurs in an older population than that affected by Graves disease; patients are typically over the age of 50, with females being affected more than males.
Clinical features
The cardiovascular features of hyperthyroidism tend to be prominent in this, often elderly, group of patients, though all the features of hyperthyroidism mentioned earlier may occur. The goitre itself is classically nodular and may be large, often having been present for many years prior to the onset of thyroid dysfunction.
Diagnosis
The biochemical diagnosis of hyperthyroidism in this situation is fairly straightforward, with suppression of TSH, though thyroid hormone concentrations may not be grossly elevated, in some cases lying at the upper limit of normal. Thyroid autoantibodies are not usually present. Thyroid scintiscanning shows a patchy uptake of isotope, with multiple hot and cold areas being seen throughout the gland (Fig. 19.9B). Radioiodine uptake values may lie at or above the upper limit of normal, but are not usually grossly elevated, and thyroid ultrasound can be used to confirm the multinodular nature of the gland.
Treatment
In general, radioiodine is the treatment of choice for the patient with toxic multinodular goitre as relapse invariably occurs after withdrawal of antithyroid drug therapy. Antithyroid drugs can be used until radioiodine becomes effective. Where compressive symptoms result from gland enlargement, surgery should be used in place of radioiodine.
Toxic adenoma
Isolated, autonomously functioning adenomas within the thyroid account for 5% of cases of hyperthyroidism. Half of these patients have ‘T3-thyrotoxicosis’ and the resulting clinical hyperthyroidism is usually mild.
Diagnosis
Scintiscans of the gland in cases of toxic adenoma show an area of high uptake in the region of the nodule, with suppression of uptake in the remaining, normally responsive, areas of the gland (Fig. 19.9C).
Treatment
Patients with proven toxic adenoma may be treated successfully either with radioiodine, which is concentrated by the hyper-functioning adenoma, or by surgery. Radioiodine is usually the treatment of choice and post 131I hypothyroidism is less common than with Graves disease.
Thyroid stimulating hormone-secreting pituitary tumour
Very rarely, adenomas of the pituitary gland secreting TSH (TSHomas) may produce hyperthyroidism.
Diagnosis
The biochemical key to making this diagnosis is the persistence of TSH secretion despite definite overproduction of thyroid hormones. The differential diagnoses are thyroid hormone resistance (see p. 400), drugs (amiodarone/amfetamine), and acute psychotic illness. Interference with one or other of the thyroid function assays should always be excluded, for example by repeating the tests on another assay platform. In TSHomas, high circulating concentrations of the α-subunit of TSH may be found in the serum, TSH secretion is not increased by the administration of TRH and a goitre is present in the absence of immunological evidence of thyroid autoimmunity. Pituitary MRI or CT imaging should be performed, but not before the biochemical diagnosis is established, as the lesion may be too small to visualize and because incidental non-functioning microadenomas of the pituitary are common. If large, the pituitary lesion may produce local damage, with reduction or loss of secretion of other anterior pituitary hormone and impairment of the visual fields. TSH secreting pituitary adenomas may co-secrete other anterior pituitary products such as prolactin and growth hormone.
Treatment
Treatment consists of surgical removal of the tumour or, alternatively, pharmacological reduction of TSH secretion. The long-acting somatostatin analogue octreotide has been reported to reduce TSH secretion and tumour size in individual cases and dopaminergic agonists such as bromocriptine and cabergoline may also be effective. Ablative antithyroid therapy will lead to control of hyperthyroidism, but will not deal with the primary problem and is, therefore, not generally indicated.
Other causes of hyperthyroidism
In some of the areas discussed below, the term ‘hyperthyroidism’ is strictly not applicable, as the elevation of thyroid hormones derives from extrathyroidal sources. However, for continuity, this term will be used to represent states of clinical and biochemical thyroid hormone excess.
Iodine
In individuals with goitre due to previous iodine deficiency, chronic administration of excess iodine in the diet can induce a hyperthyroid state. This phenomenon (sometimes called the Jod–Basedow phenomenon) is particularly likely to occur in patients who already have a degree of pre-existing thyroid autonomy, expression of which may have been masked by the lack of iodine. In iodine-replete areas, a similar phenomenon may be seen, especially in individuals with a multinodular goitre and in some cases the hyperthyroidism reverses completely when the source of additional iodine is withdrawn from the diet. The precise relationship between iodine dose and thyroid response is clearly complex, since it is dependent not only on the individual’s prior exposure to iodine and the degree of gland autonomy, but also on the time course of iodine administration; use of large doses of iodine as a therapeutic means of rapidly treating hyperthyroidism has quite the opposite of the effect seen with chronic administration of more moderate doses. In iodine-induced hyperthyroidism, radioiodine uptake is characteristically reduced and urinary iodine excretion increased, with coincident TSH suppression and evidence of elevated T4 and elevated or high normal T3 concentrations.
Amiodarone
This anti-arrhythmic drug contains iodine and, at therapeutic dosage, generates 6–12 mg of free iodine per day compared with the recommended iodine intake of 0.2 mg/day. The drug can produce abnormalities in thyroid function tests in up to 50% of patients, but may also induce true thyroid dysfunction. Amiodarone has a structure similar to that of thyroid hormones and inhibits the peripheral conversion of T4 to T3 catalysed by iodothyronine deiodinase D1. Resulting concentrations of T4 may, therefore, be high and T3 low, with an increase in rT3. Thyroid stimulating hormone may rise transiently during the first few weeks of treatment, but by four months, most patients who are euthyroid will have normal or sometimes suppressed concentrations of TSH. The drug inhibits both iodine uptake by the thyroid and entry of T4 into cells and can also cause both iodine-induced hypothyroidism and hyperthyroidism. The frequency of these two conditions depends on the ambient iodine intake. In areas of high iodine intake (e.g. Worcester, USA), amiodarone produces hypothyroidism in up to 20% of patients receiving the drug, while hyperthyroidism only occurs in about 2% of patients. Conversely, in areas of Italy, where iodine intake is low, hypothyroidism occurs in 5% of patients and hyperthyroidism in 10%. In patients with an underlying thyroid abnormality (e.g. multinodular goitre or subclinical Graves disease), the high iodine content can induce thyrotoxicosis (amiodarone-induced thyrotoxicosis type I) that is treatable with antithyroid drugs), but the drug may also induce a destructive thyroiditis (amiodarone-induced thyrotoxicosis type II) that is not readily responsive to antithyroid drugs, but is often responsive to steroids. A mixed picture is also seen, and occasionally urgent thyroidectomy is required for thyrotoxicosis refractory to both forms of therapy. It is therefore important to evaluate patients before they commence therapy with amiodarone. This should include clinical examination and a basal measurement of TSH, free T4, free T3 and TPOAb. After starting treatment, thyroid function tests should be repeated at six months and thereafter every six months, including the year after the drug is stopped. The long half-life of the drug means that changes in thyroid test results may persist for some time after ceasing therapy.
Thyrotoxicosis factitia
Cases of hyperthyroidism occur due to self-administration of thyroid hormone, taken either as T3 or T4. Radioiodine uptake is low, goitre is absent and thyroglobulin concentrations are low. Unless there is a prior history of thyroid autoimmunity, thyroid-directed antibodies will be absent. Thyroid stimulating hormone will be undetectable; total and free T4 concentrations will be high if the patient is taking T4 and suppressed if the patient is taking T3, and total and free T3 concentrations will be high in both cases. Careful supervision of patients with serial measurement of thyroid function in hospital may be necessary, and specific psychological assessment and counselling may be required after the diagnosis has been made.
Ectopic thyroid tissue
Metastatic thyroid follicular carcinoma may rarely produce sufficient thyroid hormone to result in hyperthyroidism. Other tumours, such as ovarian teratomata, may contain functional thyroid tissue in sufficient quantity to produce symptoms and signs of thyrotoxicosis (struma ovarii): endogenous thyroid radioisotope uptake will be suppressed and functioning tissue demonstrable in the tumour under these most uncommon circumstances.
Other thyroid stimulators
Trophoblastic tumours such as choriocarcinoma, hydatidiform mole and metastatic embryonal testicular carcinoma may secrete human chorionic gonadotrophin (hCG) and produce hyperthyroidism.
Subclinical hyperthyroidism
This term is used to describe patients with the biochemical picture of suppressed serum TSH but normal concentrations of T3 and T4. Subclinical hyperthyroidism is present in about 0.5–3.0% of the population and rises in prevalence with age. Most patients are asymptomatic and revert to normal thyroid status over time. However, a small proportion of patients (1–5% per annum) will progress to overt hyperthyroidism (suppressed TSH and elevated T3 and T4). Patients with a fully suppressed TSH concentration (< 0.01 mU/L) are more likely to progress to overt hyperthyroidism than those with partial TSH suppression (TSH 0.1–0.4 mU/L). Patients with subclinical hyperthyroidism are at increased risk of atrial fibrillation and osteoporosis and, once persistent abnormal thyroid function over a period of six months is confirmed, treatment may be indicated to prevent these complications. Other indications for treatment are listed in Box 19.3. Radioiodine is generally used as the underlying aetiology is almost always nodular thyroid disease, although this form of treatment is contraindicated in pregnancy.
Hyperthyroidism or non-thyroidal illness?
It may be difficult to determine whether the finding of suppressed serum TSH and raised free T4 are due to non-thyroidal illness (NTI) or hyperthyroidism, especially if the serum T3 concentration is in the upper part of its reference range. A raised total T4 is uncommon in NTI. Furthermore, features such as ophthalmopathy, goitre or unexplained atrial fibrillation will favour hyperthyroidism. In the absence of these clinical signs, measurement of the TSH receptor antibody and isotope imaging may be helpful in detecting Graves disease and nodular goitre. If there still remains doubt, repeat thyroid function testing after 3–6 months is indicated.
HYPOTHYROIDISM
Clinical features
The clinical features seen as a consequence of low circulating concentrations of thyroid hormone are summarized in Table 19.6.
Cardiovascular system
A reduction in resting cardiac output occurs. Cool peripheries are characteristic, owing to a reduction in cutaneous blood flow, which contributes to the intolerance to cold that is so often present. In the thorax, cardiac dilatation may be seen on chest X-ray and occurs in association with pericardial effusion, which occasionally leads to compromised myocardial function. Ischaemic chest pain is uncommon, more frequently being seen in hypothyroid patients with coincidental ischaemic heart disease when they first receive thyroid hormone replacement therapy. The cardiac changes reverse with appropriate thyroid replacement therapy.
Gastrointestinal system
Most individuals show a moderate weight gain despite reduced appetite, due primarily to fluid retention. Intestinal absorption of nutrients may be affected both by reduced absorption rates and increased intestinal transit time, but net absorption may actually be reduced, normal or even increased. Results of biochemical liver function tests are usually normal.
Central and peripheral nervous system
Deficiency of thyroid hormones in fetal or early neonatal life, if not promptly treated, results in irreversible damage to the CNS, with structural abnormalities evident on histological examination. The classic picture of congenital hypothyroidism is fortunately only rarely seen now, due largely to the introduction of neonatal screening for hypothyroidism and the efficacy of therapy, if introduced sufficiently early in life.
In adult life, neurological defects resulting from hypothyroidism are usually reversible. The characteristic features are of generalized slowing in intellectual function, with inanition, slow mentation, somnolence, and occasionally, a frankly psychotic state. Speech becomes slow and the voice coarse and gruff in nature, the latter in part due to oedema within the vocal apparatus. Cerebellar ataxia may be seen with prolonged hypothyroidism and may become irreversible with delay in treatment. Seizures may also occur in severe cases. Peripheral nervous system manifestations are also common, with compression of the median nerve at the wrist being perhaps the best known (carpal tunnel syndrome). Relaxation of the tendon jerks is characteristically delayed.
Locomotor system
Muscular stiffness is a particularly common complaint in hypothyroidism and relates to reduced relaxation rate. The muscles show abnormal structure on microscopy, with loss of striations, oedema, swelling of fibres and relative deficiency of type II fibres. Muscular weakness is often evident clinically and plasma activities of muscle enzymes such as creatine kinase may be raised.
Respiratory system
Similar changes to those described above may occur in the respiratory muscles, with abnormal muscle function leading, in patients with pre-existing lung disease, to exacerbation of any carbon dioxide retention and sleep apnoea. Chest X-ray may show pleural effusions, though these are seldom large.
Skin and hair
Increased water binding occurs as a result of deposition of mucopolysaccharides in the skin, in common with the other tissues. The indurated oedema that results gives rise to the ‘myxoedematous’ appearance of the typical hypothyroid patient. Associated anaemia and hypercarotenaemia through impaired conversion of β-carotene into retinol may render the skin pale or yellow, respectively. Body hair tends to be easily lost, though the classic description of loss of the outer-third of the eyebrows is rarely seen today.
The skeleton
Thyroid hormone deficiency in early life leads to abnormalities of the epiphyses with marked reduction in linear growth and stunted final height. Short stature may be an apparently isolated presentation of hypothyroidism in childhood. In common with all systemic illnesses, prolonged hypothyroidism in childhood leads to retardation of bone age compared with chronological age. Rates of bone turnover are reduced, leading to a reduction in the pool of exchangeable calcium. Plasma concentrations of calcium and phosphate remain normal. Alkaline phosphatase activity tends to be low in children with hypothyroidism.
The kidneys: mineral and water balance
Renal blood flow and glomerular filtration are both decreased, but total body water has been shown to increase with hypothyroidism, owing to impaired renal excretion of water that itself results from a reduction in delivery to the distal tubule and abnormal osmoregulatory function in the hypothalamus and posterior pituitary. Although exchangeable body sodium is increased, the dilutional effect typically leads to a mild hyponatraemia. Plasma creatinine and urea concentrations remain normal.
Reproductive system
In adults of both sexes, hypothyroidism leads to a reduction in libido and subfertility. Menorrhagia due to failed progesterone secretion with anovulatory cycles is common in the female, as is oligospermia in the male. These changes may be related to impaired luteinizing hormone secretion, particularly in longstanding cases. Basal gonadotrophin concentrations are, however, typically within the normal range, unless pituitary disease is responsible for the hypothyroid state. A reduction in sex hormone binding globulin concentration leads to an elevation in free sex hormone concentrations, with a reduction in total concentrations of both oestrogen and testosterone following from both this and alterations in sex steroid synthesis.
Other systems
Cortisol turnover is often reduced and, in some patients, the cortisol response to hypoglycaemia is blunted, although response to exogenously administered ACTH is normal. Hyperprolactinaemia is a common finding, resulting from increased TRH release. The hyperprolactinaemia correlates with the degree of elevation of TSH, although frank galactorrhoea is only relatively infrequently found.
A normochromic anaemia, which may be either normo- or macrocytic in nature, is often seen and is likely to reflect diminished production of erythropoietin, with low red cell mass. Macrocytosis is frequently ascribed to coincidental occurrence of vitamin B12 deficiency due to autoimmune pernicious anaemia, but a deficiency in the response to available B12 and concurrent malabsorption of folate from the gut may contribute. If menorrhagia has been prolonged or severe, microcytosis, rather than macrocytosis, may be present as part of an iron deficiency state. Plasma concentrations of clotting factors VIII and IX may be reduced in hypothyroidism and platelet adhesiveness reduced, causing a mild bleeding tendency that may exacerbate the existing risk of anaemia. The effects of thyroid hormones on lipids have been outlined earlier in this chapter. Glucose absorption from the gut and uptake by the tissues from plasma are both delayed, but in patients with established diabetes, a characteristic increase in sensitivity to exogenous insulin is seen, probably due to decreased clearance of insulin.
Causes of hypothyroidism
Causes of hypothyroidism are listed in Table 19.7.
TABLE 19.7
Causes of hypothyroidism
| Disease | Pathogenesis | |
| Primary (due to disease of the thyroid gland) | Thyroiditis: Hashimoto; postpartum; de Quervain (rarely); Riedel (rarely) |
Autoimmune destruction of the gland Prolonged inflammatory damage Gland becomes fibrosed |
| Primary myxoedema | Gland atrophy; post-autoimmune in many cases | |
| Ablative | Post-radioiodine or surgery; gland bulk reduced | |
| Athyreosis | Abnormal or absent thyroid tissue | |
| Dyshormonogenesis | Abnormal synthesis of thyroid hormones | |
| Drugs | Inhibition of thyroid hormone synthesis or induction of thyroid autoimmunity | |
| Iodine deficiency | Lowered gland iodine; impaired hormone synthesis | |
| Acute iodine excess | Transient inhibition of hormone synthesis may become permanent in the presence of coexisting thyroid destructive autoimmune activity | |
| Secondary (due to extrathyroidal disease) | Anterior pituitary failure: tumours; granulomatous deposits; Sheehan syndrome; post pituitary destructive therapy; developmental abnormalities; post-head injury |
Loss of TSH stimulation of thyroid |
| Hypothalamic dysfunction | Loss of TRH stimulation of anterior pituitary |
Measurement of plasma TSH concentrations provides the cornerstone of the biochemical evaluation of hypothyroidism. As circulating concentrations of thyroid hormone fall, TSH secretion increases and is used to monitor thyroid status. Secretion of T3 is preferentially maintained in the presence of the high TSH concentrations that accompany declining thyroid function. T3/T4 ratios, therefore, rise and plasma T4 concentrations correlate better with thyroid activity than do plasma T3 concentrations. Replacement therapy for primary hypothyroidism can be monitored using plasma TSH concentrations. The particular problems resulting from pituitary failure are described below. Hashimoto disease and other forms of thyroiditis are described in the following section on thyroiditis. It should be noted that not all raised concentrations of TSH equate to hypothyroidism: of possible sources of error clinically, the most important is hypoadrenalism, which results in a mild to moderately raised TSH (with completely normal free T4) but treatment with thyroxine (rather than steroids) will increase metabolism of any remaining cortisol and precipitate a potentially fatal hypoadrenal crisis.
Primary myxoedema
In a large proportion of patients with primary hypothyroidism, no goitre, or history of goitre, will be present. These individuals will have high plasma TSH concentrations with low total and free T4 concentrations, and the majority will have anti-peroxidase and/or anti-thyroglobulin antibodies. Thyroid growth blocking antibodies have also been described in these patients. While the majority of patients may represent end-stage Hashimoto thyroiditis with lack of clinical recognition of the early phases of the disease, clearly other immunological processes may be responsible for some of them.
Post-surgery or post-radioiodine
Although the hypothyroidism developing after surgery or 131iodine therapy for Graves disease is usually permanent, it may be temporary, especially in the first 3–4 months. If symptomatic at this early stage, patients should be treated with a relatively small dose of thyroxine (50–75 μg daily) and the need for continued treatment assessed at six months. If serum TSH remains elevated, thyroxine therapy will be required lifelong and the dose needs to be increased. If at that stage serum TSH is normal or undetectable, stopping the thyroxine for four weeks and retesting thyroid function would be the appropriate action.
Congenital hypothyroidism
Causes of congenital hypothyroidism are listed in Box 19.4. Clinical detection of hypothyroidism in the neonate may be difficult. In many developed countries, screening programmes measuring TSH or T4 on dried bloodspots from heel-prick blood samples (taken at six days when phenylketonuria screening is performed) have been operating for some years with considerable success. Abnormal thyroid development is responsible for most congenital hypothyroidism. Scintiscanning may be used in order to help to clarify the diagnosis.
Inability to organify trapped thyroidal iodide can be recognized by an abnormal perchlorate discharge test and is usually associated with goitre. Patients with mild forms of this type of defect may also have deafness (Pendred syndrome), but are in this case not often hypothyroid. Ineffective transport of iodide into the thyroid gland can produce both goitre and hypothyroidism and will be found in individuals with abnormally low radioiodine uptake. The block to transport can, in some cases, be overcome by treating patients with high doses of oral iodide, reducing goitre and restoring normal thyroid function.
Deficiency of iodotyrosine dehalogenase produces impaired deiodination of iodotyrosine, such that large amounts of mono-and di-iodotyrosines appear in the blood, and iodine deficiency can result from the urinary loss of large quantities of deaminated metabolites of these molecules. Again, large doses of iodine may reduce the severity of the hypothyroidism and the accompanying goitre. Biochemically, parenteral administration of mono- and di-iodotyrosines will be followed by their rapid urinary excretion and this can be used as a diagnostic test for the condition.
Diversion of thyroidal iodine into metabolically inactive or less active iodoproteins, rather than thyroid hormones, can lead to hypothyroidism. Radioactive iodine uptake will be high, due to high TSH drive to synthetic activity within the gland, and the abnormal quantities of iodinated products of the gland can be measured in serum. Thyroid hormone replacement therapy is required.
Maternal TSH receptor-blocking antibodies can cross the placenta to produce transient hypothyroidism in the fetus, which resolves within a few weeks after birth. In these patients, bloodspot TSH values are usually only modestly raised. Testing for TSH receptor antibodies would distinguish these infants from those requiring T4 replacement therapy.
Lithium treatment
Lithium therapy may lead to reduced thyroid hormone synthesis in a similar manner to the acute effects of iodine, but more commonly aggravates any underlying autoimmune disease. Female patients are more often affected and thyroid autoantibodies are often present. Lithium can, therefore, cause hypothyroidism (in approximately 14% of women and 5% of men taking lithium) but also, less commonly, induces hyperthyroidism. Patients taking lithium should have their thyroid function tests measured every six months. Patients on lithium who have positive TPO antibodies are at increased risk of developing thyroid dysfunction. Lithium itself has been used as a treatment for hyperthyroidism and as an adjunct to radioiodine treatment (as it enhances radioiodine uptake) but because of the potential for toxicity, this approach has not entered routine practice.
Cytokine therapy
Treatment with a variety of cytokines (see Table 19.1) can result in hypothyroidism that is more common in patients with underlying autoimmune thyroid disease. This is most commonly observed in patients being treated with interferon therapy for viral hepatitis. It is usually self-limiting once cytokine therapy ceases, but may need temporary treatment with thyroxine.
Iodine
Both excess and deficiency of dietary iodine may result in hypothyroidism.
Although oral use of large doses of iodine would normally lead to transient suppression of thyroid hormone synthesis, iodine can rarely produce more lasting hypothyroidism, particularly in individuals with Hashimoto disease.
Environmental iodine deficiency remains a major cause, worldwide, of goitre and hypothyroidism. Clinical euthyroidism is often maintained by preferential production of T3 and compensatory thyroid hyperfunction, leading to large goitre formation.
Secondary hypothyroidism
Loss of TSH drive to thyroid function may result from any of the causes of pituitary gland hypofunction. Recognition of the aetiology of this type of hypothyroidism is critical and is usually made by observing a low T4 accompanied by an inappropriately ‘normal’ TSH concentration. In some cases of secondary hypothyroidism, TSH may be slightly increased. Increased T3:T4 ratios are not seen in this type of hypothyroidism as TSH drive is absent.
Treatment of hypothyroidism
Myxoedema coma
Profound hypothyroidism may produce the state known as myxoedema coma. This is a relatively uncommon endocrine emergency that may be triggered by factors such as infection, cold exposure, the use of sedative medications or non-compliance with treatment. The classic presentation is that of decompensated hypothyroidism with altered sensorium, marked bradycardia, hypothermia and circulatory collapse. Patients should be admitted to an intensive care unit where possible. Therapy is supportive with thyroid hormone replacement using nasogastric T4 or parenteral T3 therapy. Replacement doses of glucocorticoids should be given until coincidental adrenal insufficiency has been excluded, infections should be treated aggressively and further heat loss prevented to allow progressive rewarming to occur. Despite appropriate therapy, the mortality from this condition remains high, often with arrhythmias in the first 24 h after commencing therapy, and early recognition and careful management in a critical care setting offers the best chance of recovery.
Thyroid hormone replacement therapy
Treatment of primary or secondary hypothyroidism is with oral synthetic levothyroxine. The usual replacement dose is 1.6 μg/kg per day, which equates to about 100–125 μg daily. Patients are customarily advised to take levothyroxine 30–60 min before breakfast since food and caffeine interfere with its absorption. Bed time administration is equally effective and may be suited to patients who take a number of other medications. Treatment can usually be initiated at full replacement doses except in elderly individuals and patients with cardiac disease who will require more cautious starting doses. Serum T4 and TSH should be checked 6–8 weeks after initiation, making any minor adjustment to the dose necessary to restore TSH to normal. In the elderly and in patients with established ischaemic heart disease, levothyroxine replacement should begin with 25 μg daily, increasing by 25 μg increments every 4–6 weeks. In patients with hypopituitarism, with both secondary adrenal and thyroid failure, it is important that levothyroxine is not started before hydrocortisone replacement, as this might precipitate adrenal crisis by increasing the metabolic rate in a patient unable to secrete cortisol.
The objective of treatment is to achieve clinical well-being and restore biochemical euthyroidism, i.e. a serum TSH within the laboratory reference range. Patients who remain symptomatic despite full replacement doses may benefit from additional dose increases to keep TSH concentration in the lower half of the reference range. Lowering TSH to below the reference range is not advisable as there is evidence that such individuals with exogenous hyperthyroidism have an increased risk of atrial fibrillation and osteoporosis. The response to treatment in patients with secondary hypothyroidism should be based on FT4 measurements since TSH secretion is deficient in pituitary disease. The therapeutic goal in such cases would be a FT4 in the upper-third of the reference range.
Although thyroid hormone replacement with synthetic levothyroxine is effective in most cases, a significant proportion of patients report that they remain symptomatic. The management of such patients can be challenging and factors that may predispose to inadequate thyroid hormone replacement include poor patient compliance, inadequate dosage and administration, concurrent use of medications which interfere with levothyroxine availability and the presence of comorbidities which impair levothyroxine absorption. The combination of a raised serum T4 and TSH is suggestive of poor compliance, the patient having been over zealous in the few days prior to thyroid function testing in an attempt to compensate for missing doses earlier. Some individuals may remain symptomatic even after normalization of thyroid function. Although some of such patients may benefit from further dose titrations to attain a low normal TSH, it is important to consider alternative causes of persistent symptoms such as diabetes mellitus, obesity, Addison disease, anaemia, depression, coeliac disease, obstructive sleep apnoea and post-viral syndromes. In practice, such investigations are frequently negative and whether some individuals with persistent symptoms will benefit from combined treatment with T4 and T3 remains contentious. The use of T3 in replacement is much harder to titrate and monitor, is not recommended by professional bodies and should not be conducted away from specialist centres with programmes of long-term follow-up for these individuals. Recent delineations of common genetic variations in the deiodinase and thyroid hormone transport proteins suggest that such polymorphisms may account for some of the variability in the response to thyroid hormone treatment.
Subclinical hypothyroidism
This is the term used to describe patients who are clinically euthyroid with normal serum T4 concentration but raised TSH, usually < 10 mU/L. There may be mild non-specific symptoms, such as tiredness, weight gain and depression. Now viewed as the mildest form of thyroid failure, it is common practice to treat with thyroxine if antibodies are positive and/or serum TSH is > 10 mU/L in order to ‘nip things in the bud’ before the progression to overt thyroid failure (5% per year), and the possibility of loss to follow-up and later presentation with severe hypothyroidism. Other potential indications for treatment in patients with subclinical hypothyroidism are shown in Box 19.5. Recovery from non-thyroidal illness should be excluded before a diagnosis of subclinical hypothyroidism is made, emphasizing that in all cases thyroid function tests should be repeated and confirmed as remaining abnormal before initiating therapy.
THYROIDITIS
Inflammation of the thyroid gland (thyroiditis) can result in both overactivity and underactivity of the gland.
Thyroiditis producing hyperthyroidism
Autoimmune thyroiditis (typified by Hashimoto disease) is a common cause of hypothyroidism and, as such, will be reviewed in detail below. However, in the early phase of autoimmune damage to the gland, release of stored thyroid hormone may lead to transient hyperthyroidism in some patients. This usually lasts about 6–8 weeks, followed by mild and equally transient hypothyroidism and then recovery, although, in the long term, permanent hypothyroidism is common. Known as silent thyroiditis, the biochemical pattern is similar to that observed in postpartum thyroiditis and in de Quervain thyroiditis.
Diagnosis and treatment
Depending on the phase of the illness, thyroid function may show hyperthyroidism, euthyroidism or hypothyroidism. Radioactive iodine scans in the hyperthyroid phase of the illness will typically show reduced isotope uptake since the hyperthyroidism is not due to excessive synthesis of thyroid hormones but to the release of pre-formed hormones. For the same reason, treatment with antithyroid drugs is ineffective, although a β-blocker may help to alleviate symptoms of hyperthyroidism.
Hypothyroidism resulting from Hashimoto thyroiditis
In Hashimoto disease, the commonest form of autoimmune hypothyroidism, destructive thyroiditis results from both a cell-mediated and humoral attack on the thyroid tissue. Females are affected with greater frequency than males. The gland is typically enlarged but small and firm in early cases, with a palpable pyramidal lobe. At this stage, hyperthyroidism may transiently occur, but further gland damage rapidly leads to permanent hypothyroidism, at which stage goitre regresses and the gland remnant is composed of fibrous tissue.
Diagnosis
Thyroid-directed autoantibodies (described earlier) are almost invariably present in the early phase of the disease, though they may ultimately become undetectable over a period of years. In about half of patients with Hashimoto thyroiditis, serum TSH is elevated and T4 normal (subclinical hypothyroidism), the other patients having overt thyroid failure.
Immunological and biochemical evidence of other organ-specific autoimmune diseases such as diabetes, pernicious anaemia, Addison disease and hypoparathyroidism may also be found.
Other forms of thyroiditis
De Quervain thyroiditis is an unusual condition, which often follows a viral illness. Patients typically present with pain in the thyroid and neck region with fever and malaise. Thyroid function tests may transiently become hyperthyroid, then hypothyroid, before returning to normal. Rarely, permanent hypothyroidism may follow repeated episodes. Riedel thyroiditis is a condition of unknown aetiology. Extensive replacement of the thyroid with fibrous tissue causes the gland to become stony hard. Hypothyroidism is a rare complication.
Hypothyroidism and the postpartum period
Postpartum thyroiditis, due to autoimmune thyroid disease, may develop during the early postpartum period, and is typically histologically and biochemically identical with Hashimoto disease. The classic presentation is of a transient hyperthyroid phase which is followed by a hypothyroid phase. Published series suggest that the overall incidence of the condition is between 5% and 9% and that 40% of patients have failed to recover euthyroid function by one year post-delivery. Of those who do recover normal thyroid function, a further 20–30% will have become hypothyroid within 3–4 years. After the initial episode, the future risk of developing the syndrome increases in successive pregnancies.
Although the presence of antithyroid antibodies during pregnancy is a risk factor for postpartum thyroid disease, at most only half of these women will go on to develop frank thyroid dysfunction. Swings in thyroid function may contribute to the aetiology of postnatal depression and thyroid function should be tested in all such patients.
NEOPLASIA
Thyroid neoplasia forms a small but clearly important proportion of all thyroid disease. Thyroid cancer is the most common endocrine malignancy and accounts for more mortality than all the other endocrine cancers combined. The types of primary and secondary thyroid neoplasm are summarized in Box 19.6. The differentiated thyroid cancers (DTC), papillary and follicular thyroid cancer, are the most common histological types. Papillary thyroid cancer is most frequent in iodine sufficient parts of the world and is associated with a favourable prognosis. Papillary microcarcinomas (diameter < 1 cm), are now increasingly detected owing to the growing use of imaging techniques such as ultrasound, computerized tomography or positron emission tomography scans. Anaplastic thyroid cancer is relatively uncommon and typically follows an aggressive course with rapid thyroid enlargement and poor prognosis. Primary thyroid lymphomas almost always arise on a background of Hashimoto thyroiditis and also present with rapid gland enlargement.
Diagnosis
The usual presentation is with a solitary nodule in the thyroid. Although thyroid nodules are common in the general population, only about 5% of such nodules are malignant. Malignancy is suspected in patients with thyroid nodules if there is a history of rapid enlargement, a family history of thyroid cancer or a past medical history of head and neck irradiation as therapy for another disease (Box 19.7). Clinical suspicion of the diagnosis is further raised by the finding of a nonfunctioning solitary nodule on thyroid scintiscanning (Fig. 19.9D) that is shown on ultrasound examination to be solid (although cystic lesions may also be malignant). The diagnosis of thyroid carcinoma ultimately depends on histological examination of the gland following an excision biopsy or a fine needle aspirate of the nodule. Biochemical assessment (other than establishing the usually euthyroid status of the patient and excluding evidence of autoimmune processes) plays little part in making the diagnosis. Measurement of plasma thyroglobulin is of no diagnostic value, but is essential for long-term monitoring for recurrence.
Treatment
The management of thyroid cancer should be undertaken in a multidisciplinary care setting comprising health professionals with expertise in the field including oncologists, surgeons, endocrinologists, pathologists and radiologists. For papillary and follicular carcinoma, treatment is by total thyroidectomy and, unless the carcinoma is small (< 1 cm in diameter), in which case the prognosis is excellent, adjunctive treatment is given with ablative doses of 131I. In order to maximize the effect of the radioiodine, the patient should stop any thyroxine for four weeks beforehand to achieve a serum TSH of > 25 mU/L. Alternatively, symptomatic hypothyroidism can be avoided by increasing the serum TSH with the use of recombinant human TSH (rhTSH), thus allowing levothyroxine therapy to continue uninterrupted. Whole body scanning, taking advantage of the therapeutic 131I, will identify any metastatic disease at this stage. Thereafter, thyroxine should be given either in suppressive or replacement doses depending on individual patient risk (Box 19.8). The differentiated thyroid carcinomas are TSH dependent and suppression of TSH has been shown to improve outcomes in patients with high risk disease. Thus, full suppression (TSH < 0.1 mU/L) is recommended for patients with persistent disease, while partial suppression (TSH 0.1–0.5 mU/L) is advised in disease-free high-risk patients. TSH suppression is not required in patients with low or very low risk (Box 19.8). Patients on suppressive therapy should undergo periodic cardiovascular and bone mineral density evaluation because of the risk of osteoporosis and atrial fibrillation. Further evaluation should be undertaken six months after initial treatment and should comprise a neck examination, cervical ultrasound and rhTSH-stimulated Tg measurement. Patients without clinical or ultrasound evidence of disease who have normal stimulated Tg concentrations (< 1.0 ng/mL) are considered to be in remission and will then require long-term surveillance with annual neck examinations and basal Tg measurements.
Tumour markers
Following total thyroidectomy and 131I ablation, there should be no normal thyroid tissue remaining, and any thyroglobulin detected in the serum must arise from the current tumour or from metastases. The sensitivity of the current thyroglobulin assays is poor and the most meaningful results are obtained following TSH stimulation, either as a result of thyroxine withdrawal or by rhTSH stimulation. An undetectable serum thyroglobulin, which fails to increase following TSH stimulation, is indicative of cure and a reason for less intensive follow-up. Response to treatment of recurrent disease or metastases can be monitored by measuring the serum thyroglobulin concentration. The utility of thyroglobulin measurements is limited in the presence of thyroglobulin antibodies, as these antibodies interfere with the thyroglobulin assays. Tests of thyroglobulin recovery have been used in a bid to overcome this problem, but these tests are not entirely reliable and the clinician must be aware of the limitations of thyroglobulin assays in monitoring patients with differentiated thyroid cancer.
Medullary carcinoma of the thyroid secretes calcitonin, which may therefore, be used as a tumour marker. The tumour occurs in both sporadic and familial forms, and because of the association with syndromes of multiple endocrine neoplastic (MEN) disease, patients should be screened for the other manifestations of MEN (e.g. hyperparathyroidism and phaeochromocytoma). If the diagnosis of MEN is confirmed, first-degree relatives of the index patient should also be screened for these diseases. Multiple endocrine neoplasia is discussed in detail in Chapter 41.
SYNDROMES OF RESISTANCE TO THYROID HORMONES
These occur most commonly due to a mutation in the nuclear T3 receptor (TRβ). The mutation may be inherited (usually autosomal dominant) or occur de novo. Thyroid hormone resistance has been detected in approximately in 1 in 50 000 live births and affects males and females equally. The condition is characterized by elevated concentrations of total and free thyroid hormones, normal or slightly increased plasma TSH, normal or exaggerated response to TRH and absence of the usual symptoms and metabolic consequences of thyroid hormone excess. Goitre is often present. It is important to distinguish these patients from those with a pituitary TSHoma (Table 19.8). The SHBG concentration is often normal in patients with thyroid hormone resistance, but is increased in TSHoma. Suppression of TSH secretion by high doses of T3 is suggestive of thyroid hormone resistance rather than a TSHoma. A useful pointer to thyroid hormone resistance is to obtain thyroid function test results in first-degree relatives. DNA analysis to demonstrate the mutation in the TRβ gene should be performed to confirm the diagnosis. Some patients appear to have a relatively selective pituitary resistance and present with hyperthyroid-type symptoms. Management is complex and should be reserved for specialist centres. In many cases no treatment is required.
TABLE 19.8
Overview of factors that enable thyroid hormone resistance to be distinguished from TSH secreting adenomas
| Factor | Thyroid hormone resistance | TSH-secreting adenoma |
| Thyroid status | Usually clinically euthyroid | Usually clinically thyrotoxic |
| Serum α TSH subunit | Normal | Elevated |
| Serum SHBG | Often high (85%) | Normal |
| TSH response to TRH stimulation | Rise in TSH | No response |
| MRI pituitary | No tumour visualized | Primary tumour visualized (However, incidentaloma detected in 10% of normal subjects) |
| Genetic analysis of thyroid hormone receptor B gene | Mutation confirmed | No mutation |
SCREENING
Is screening for thyroid disease justified? There are undoubtedly some groups of patients in whom thyroid function testing is justified, even if there is no clinical evidence of thyroid disease. The question of screening in neonates has already been discussed. Screening is warranted in all patients with a personal history of previous thyroid disease or neck irradiation, ophthalmic Graves disease, atrial tachyarrhythmias or a history of goitre or previous thyroiditis. In view of the increased prevalence of long-term thyroid dysfunction in patients who have previously received antithyroid drugs, this group of individuals should also be offered regular thyroid function testing. Screening patients with a personal or family history of autoimmune disorders such as type 1 diabetes, pernicious anaemia, coeliac disease, Addison disease and rheumatological disorders will also yield a significant proportion of patients with undiagnosed thyroid disorders. Thyroid screening is also indicated in patients receiving medications which affect thyroid function such as amiodarone, lithium, tyrosine kinase inhibitors, α-interferon, Campath® and antiretroviral agents. Screening patients who have hyperlipidaemia (who have a higher prevalence of hypothyroidism than the normal population) is also justified. The situation is far less clear in the case of screening larger sections of the general population, such as the elderly female population or the hospital inpatient population. In the former example, the cost of such a screening programme has to be balanced against the likely pick-up rate, and in the latter, the confounding influence of non-thyroidal illness is likely to reduce the value of screening in this circumstance. Screening programmes should probably be targeted on otherwise well but ‘at-risk’ individuals in the community, rather than on the hospital population at large.
The value of thyroid screening in all pregnant women is contentious. Most international endocrine associations do not recommend universal thyroid screening but propose screening only in pregnant patients at risk of thyroid disease, e.g. women with a personal or family history of thyroid disease, co-existent autoimmune disease, goitre, thyroid antibodies, history of head and neck irradiation, or history of poor obstetric outcomes. However, such a targeted case finding approach would miss a third of pregnant women with thyroid disease. A randomized controlled trial of a thyroid screening strategy in early pregnancy showed no beneficial effect on childhood neurointellectual development at three years.
ACKNOWLEDGEMENT
We would like to thank Geoffrey J. Beckett and Anthony D. Toft who wrote the chapter in the previous edition of this book.
Further reading
British Thyroid Association. UK Guidelines for the use of thyroid function tests, http://www.british-thyroid-association.org/info-for-patients/Docs/TFT_guideline_final_version_July_2006.pdf [Accessed October 2013].
Cooper DS, Biondi B. Subclinical thyroid disease. Lancet. 2012;379:1142–1154.
Dayan CM. Interpretation of thyroid function tests. Lancet. 2001;357:619–624.
Ekins R. Measurement of free hormones in blood. Endocr Rev. 1990;11:5–46.
Franklyn JA, Boelaert K. Thyrotoxicosis. Lancet. 2012;379:1155–1166.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 19
Thyroid dysfunction
Colin M. Dayan; Onyebuchi E. Okosieme; Peter Taylor
CHAPTER OUTLINE
Biological actions of thyroid hormones
Synthesis, storage and release of thyroid hormones
Iodine and thyroid hormone synthesis
Transport of thyroid hormones in blood
Entry of thyroid hormone into tissues
Thyroid hormone deiodination and regulation of extrathyroidal T3 production
Catabolism of thyroid hormones
Nuclear action of thyroid hormones
Control of thyroid hormone synthesis and secretion
Extrathyroidal factors that may affect thyroid function
THE EVALUATION OF THYROID FUNCTION
Clinical evaluation of thyroid status
In vitro tests of thyroid activity and pituitary–thyroid status
Measurement of thyroid stimulating hormone
Free T4 and free T3 measurements
Methods for measuring free thyroid hormones
Validity of commercial methods for free hormone analysis
Nomenclature of free thyroid hormone assays
Selective use of thyroid function tests
Interpreting results of thyroid function tests
Common situations in which TSH results may be misleading
Reference ranges and significant changes
Autoantibodies to thyroidal antigens
Hyperthyroidism or non-thyroidal illness?
Thyroiditis producing hyperthyroidism
Hypothyroidism resulting from Hashimoto thyroiditis
Hypothyroidism and the postpartum period
INTRODUCTION
Thyroid hormones are essential for normal growth, development and metabolism, and their production is tightly regulated through the hypothalamic–pituitary–thyroid axis. Thyroid disease is common, particularly in women, with a prevalence in the community of 3–5%. With the exception of iodine deficiency, which affects millions of people worldwide, diseases that directly affect the thyroid gland are the most common causes of thyroid dysfunction. Pituitary disease and the use of certain drugs that alter thyroid hormone synthesis or metabolism can also give rise to thyroid dysfunction. Any severe illness can produce abnormalities in the results of thyroid function tests that resolve as the patient’s illness improves. Once diagnosed, thyroid disease is usually easily treated, with an excellent long-term outcome for most patients. This chapter outlines thyroid physiology and the pathways of thyroid hormone synthesis and metabolism. The investigations used in diagnosis and management of thyroid disorders are described, together with guidance on their interpretation. The various thyroid disorders are described together with current views on their treatment.
NORMAL THYROID PHYSIOLOGY
The thyroid gland
The thyroid gland is brownish-red in colour and consists of left and right lobes connected by a midline isthmus, typically forming an ‘H’ or ‘U’. It is located anteriorly in the neck deep to the platysma, sternothyroid and sternohyoid muscles. The isthmus lies just below the cricoid cartilage and the lobes extend upward over the lower half of the thyroid cartilage. The pretracheal fascia encloses the thyroid gland and attaches it to the trachea and larynx. This explains why the thyroid gland is seen to move upwards with swallowing.
The thyroid is a highly vascular organ with blood flow ~ 5 mL/min/g of tissue (nearly twice that of the kidneys). It consists of thousands of follicles, each a spheroidal sac of epithelial cells (thyrocytes) surrounding a lumen containing colloid, mainly comprising thyroglobulin (Fig. 19.1). It also contains parafollicular C cells, which secrete calcitonin; calcitonin is discussed in Chapter 6.
FIGURE 19.1 Structure of the thyroid follicle.
Embryologically, the thyroid is derived from the floor of the pharyngeal cavity and migrates from the base of the tongue to its final position in the neck along the thyroglossal duct. Abnormalities of this migration give rise to ectopic glands that may not function normally. Occasionally, the gland is completely absent.
The primary function of the thyroid is to synthesize and secrete thyroid hormones (Fig. 19.2). Under normal circumstances the gland synthesizes approximately 80 μg (110 nmol) of thyroxine (T4) per day and 6 μg (10 nmol) of 3,5,3′-tri-iodothyronine (T3).
FIGURE 19.2 Structures of the iodothyronines.
Biological actions of thyroid hormones
Thyroid hormones produce enhancement of many intracellular events and promote differentiation and growth; they are essential for normal fetal and neonatal development.
Thyroid hormones increase mitochondrial oxidative phosphorylation and maintain amino acid and electrolyte transport into cells. They increase calorigenesis and oxygen consumption in most tissues, though not in brain. Thyroid hormones stimulate the synthesis of proteins, including structural proteins and enzymes. They regulate all aspects of carbohydrate metabolism, including increasing gluconeogenesis and accelerating insulin degradation. Thyrotoxicosis can, therefore, lead to a deterioration of glycaemic control in diabetes mellitus.
Stimulation of lipid metabolism leads to a fall in plasma cholesterol concentration, as degradation is increased to a greater extent than synthesis. Bone turnover is stimulated, resorption more so than mineralization, raising plasma calcium concentration in some patients. In general, the effect of thyroid hormones on the turnover of any substance will depend on the relative effects on synthesis and degradation. The actions of thyroid hormones increase demand for coenzymes and vitamins.
Complex interactions occur with the sympathetic nervous system and many of the signs and symptoms of hyperthyroidism, including palpitations, tremor and sweating may be attributed to these. For example, thyroid hormones amplify the adrenergic system through the α1 and β3 adrenergic receptors and are also the main determinant of basal metabolic rate (BMR). The increase in metabolic rate involves inhibition of AMP-activated protein kinase (AMPK) in the hypothalamus. However, the mechanisms responsible for many interactions between thyroid hormone and the sympathetic nervous system have not been identified.
Most changes in biochemical parameters produced by changes in thyroid status are not sufficiently specific or of such magnitude to be useful as tests of thyroid function, though the association may be sufficiently close that the finding of an abnormality should provoke a request for testing for previously unsuspected thyroid disease. For example, hypothyroidism is a relatively common and easily treatable cause of raised plasma cholesterol; however, the effect on lipids in subclinical hypothyroidism is modest. Equally, it may be helpful (and avoid unnecessary investigations) to anticipate these changes in cases of known thyroid dysfunction and to expect their normalization after treatment of the thyroid disorder. Some examples are listed in Box 19.1.
Synthesis, storage and release of thyroid hormones
Synthesis of T4 and T3 occurs on thyroglobulin (Tg), a glycoprotein of molecular weight 660 000 Da that contains many tyrosyl residues. Thyroglobulin is synthesized by the thyrocytes and exported to be stored within the colloid of the follicular lumen. Incorporation of iodide into Tg requires hydrogen peroxide and thyroid peroxidase (TPO), an enzyme that is synthesized within the follicular cell and transported to the apical membrane. Thyroid hormones are synthesized at the interface between the apical membrane of the thyrocyte and the colloid of the follicular lumen.
The process of thyroid hormone synthesis, storage and secretion requires a series of highly regulated steps (Fig. 19.3).
FIGURE 19.3 Sites of biochemical processes within the follicular cell (thyrocyte).
• Oxidation of iodide to iodine by thyroid peroxidase. This occurs on the luminal side of the apical membrane and requires TPO and hydrogen peroxide, which is generated by a calcium-dependent flavoprotein enzyme system situated at the apical membrane.
Recently, the dual oxidase molecules DUOX1 and DUOX2, which contain peroxidase-like and NADPH oxidase-like domains have been identified as essential for H2O2 production. They require maturation or activation factors (DUOXA1 or 2) for translocation of DUOX from the endoplasmic reticulum to the apical plasma membrane where H2O2 production occurs. Mutations in DUOX2 and DUOXA2 have both recently been identified as causes of hypothyroidism, resulting from diminished H2O2 production.
Halogenases collect the iodide removed from thyroglobulin as it is broken down in the cell.
Antithyroid drugs such as propylthiouracil and mercaptoimidazoles (methimazole and carbimazole) exert their action through inhibiting the action of TPO.
• Incorporation of iodine into tyrosyl residues on thyroglobulin. Mono-iodotyrosine and di-iodotyrosine (MIT and DIT) are formed through the actions of TPO (organification).
• Coupling of two iodotyrosyl residues in the thyroglobulin molecule. This is also catalysed by TPO and produces T3 and T4 that remain linked to Tg. When iodine supply is limited, the proportion of T3 produced on Tg increases. Iodinated Tg is stored in the follicular lumen.
• Internalization of Tg and release of T4 and T3. When there is a demand for thyroid hormone, this is signalled by an increase in plasma thyroid stimulating hormone (thyrotrophin; TSH) concentration. Thyroglobulin is then internalized by pinocytosis and appears as colloid droplets that fuse with lysosomes and undergo proteolytic degradation to release T3 and T4. Any MIT and DIT is deiodinated and the iodine conserved.
• Delivery of T4 and T3 into the circulation. This is probably achieved by thyroid hormone transporters. The large stores of T4 and T3 incorporated into thyroglobulin allow secretion of T4 and T3 more quickly when required than if it had it had to be synthesized.
Thyroid stimulating hormone appears to stimulate each of the above processes. It also stimulates the expression of many genes in thyroid tissue including thyroglobulin, sodium iodide symporter (NIS) and interleukin-8. It also causes thyroid hyperplasia and hypertrophy.
Iodine and thyroid hormone synthesis
Iodine deficiency represents a major public health issue for over two billion people who live in areas of iodine deficiency and especially for the 43 million in whom the deficiency is severe enough to cause learning difficulties. The recommended intake of iodine is 150 μg/day for adults but this may rise to 250 μg during pregnancy and lactation. Iodine is obtained from the ingestion of foods such as seafood, seaweed, kelp, dairy products, some vegetables (e.g. soybeans and potatoes) and iodized salt.
High iodine intakes can induce hypothyroidism, goitre and sometimes hyperthyroidism (see later). A large excess of iodide, when given acutely, results in acute inhibition of thyroid hormone release. It also inhibits the adenylate cyclase response to TSH (see below) and iodination of thyroglobulin; this is termed the Wolff–Chaikoff effect. After a few days of exposure to high iodide concentrations, thyroidal iodide uptake is very low, the intrathyroidal iodide concentration falls and the synthesis of iodinated thyroglobulin recommences. This effect can be used to prepare a thyrotoxic patient for thyroidectomy. However, if high iodine intake persists for more than ten days, then thyrotoxicosis can occur in individuals with a pre-existing multinodular goitre or those from iodine deficient areas; this is termed the Jod–Basedow effect.
Transport of thyroid hormones in blood
When T4 and T3 are secreted into the bloodstream, they become bound to plasma carrier proteins. Three proteins share this function: in order of decreasing affinity for iodothyronines they are thyroxine binding globulin (TBG); thyroxine binding prealbumin (TBPA, also known as transthyretin), and albumin. These binding proteins bind approximately 70% (TBG), 20% (TBPA) and 10% (albumin) of the circulating thyroid hormones. T4 is more tightly bound than T3 to each of these proteins. Approximately 0.02% of T4 and 0.2% of T3 are in the unbound or ‘free’ form in plasma in normal subjects, so that, although the total molar concentration of T4 is some 50 times that of T3, the concentration of free T4 is only about three times that of free T3. The half-life of T4 is 5–7 days; that of T3 is 1–2 days.
Free hormone hypothesis
For most of their actions, thyroid hormones need to enter the cell and bind to nuclear receptors. The free hormone hypothesis suggests that only the unbound (free) fraction of thyroid hormone is able to cross the plasma membrane and enter the nucleus. This hypothesis is supported by the observation that, in clinical practice, thyroid status correlates better with the concentration of the free hormone fraction than with the protein-bound fraction. The precise function of TBG and TBPA is unclear, but a number of roles have been suggested including acting as a reserve or buffer to prevent rapid changes in free hormone concentration in blood and also to prevent loss of thyroid hormones through filtration and excretion at the kidney. One further hypothesis is that TBG may be involved in targeting the delivery of thyroid hormone in a differential manner to different organs or facilitating the efflux of thyroid hormones from some tissues. Whatever their function, the absence or overexpression of the transport proteins is not associated with any obvious detrimental effects. There are difficulties in measuring the concentrations of free hormones and this is discussed later in this chapter.
Entry of thyroid hormone into tissues
Specific transporters appear to be required to facilitate the entry of the free thyroid hormones into cells. A number of important thyroid hormone transporters have been characterized. The organic anion transporting polypeptides (OATPs) are one group of important transporters. OATP1C1 appears to be specific for T4 and reverse tri-iodothyronine (3,3′5′-tri-iodothyronine: rT3); it is expressed exclusively in the capillaries of the brain and may be important for T4 transport across the blood–brain barrier. Two L-type amino acid transporters (LAT1 and LAT2) have been identified among the members of the heterodimeric amino acid transporter family, that transport thyroid hormone in many tissues. Human monocarboxylate transporter 8 (MCT8) is another active thyroid hormone transporter. It has a preference for T3 and is expressed in a variety of tissues including the brain, where it appears to be involved in the uptake of T3 by neurons, the primary target for thyroid hormones during brain development. The MCT8 gene is located on the X chromosome and mutations in the gene have been identified in a small number of boys with severe psychomotor retardation and elevated plasma T3 concentration (Allan–Herndon–Dudley syndrome). These mutations result in an inhibition of T3 supply to neurons leading to a defect in brain development. The characteristic pattern of thyroid function tests remains unexplained.
Entry of thyroid hormone into liver cells appears to be ATP-dependent. In rat hepatocytes, it is inhibited by a range of compounds that accumulate in plasma during illness. These compounds include free fatty acids, bilirubin and indoxyl sulphate. The uptake of thyroid hormones by the pituitary appears to occur through a different mechanism from that occurring in the liver. For example, unlike in the liver, the pituitary thyroid hormone transporters are not energy dependent; they can maintain or increase the uptake of T4 and T3 in low energy states and are not inhibited by bilirubin.
Thyroid hormone deiodination and regulation of extrathyroidal T3 production
Thyroxine is produced entirely by the thyroid gland, approximately 80 μg being produced each day. However, only 20% of T3 is produced directly from the thyroid gland; 80% is produced by the extrathyroidal deiodination of T4. Approximately 6 μg of the T3 in blood originates from thyroidal synthesis; 25 μg is formed by 5-deiodination of T4 in extrathyroidal tissues, a reaction catalysed by the D1 and D2 isoenzymes of iodothyronine deiodinase. T4 is considered to be a prohormone, T3 being responsible for the biological actions of thyroid hormones. Most tissues express one or more of a family of three iodothyronine deiodinases (D1, D2 and D3) that are responsible for providing a local source of T3 from T4 or for providing a source of T3 in plasma. In humans, it has proved difficult to determine which deiodinase and which tissues are responsible for providing most of the circulating T3. Classically it has been thought that D1 in liver and kidney provided most circulating T3, but some studies have suggested that, in humans, D2 may also provide an important source. Genetic association studies may provide more conclusive evidence. To date common genetic variants (single nucleotide polymorphisms) in DIO1 have been robustly associated with variation in the T3/T4 ratio in serum at genome wide levels of significance (p = < 1 × 10− 8), but as yet no associations have been observed with single nucleotide polymorphisms in DIO2.
Figure 19.4 summarizes the characteristics of the three deiodinases. All are selenoenzymes requiring adequate intake of dietary selenium for their expression. D1 carries out either 5′-deiodination, giving T3, or 5-deiodination, giving the inactive compound rT3. D2 appears to provide a very important local source of T3 in some tissues, including the brain, pituitary and brown adipose tissue. D3 catalyses the conversion of T4 to rT3 and is thought to be an important extrathyroidal control mechanism to regulate delivery of T3 to its receptors, particularly in the developing fetus.
FIGURE 19.4 Characteristics of the iodothyronine deiodinases. In non-thyroidal illness, 5′-mono-deiodination is impaired leading to a decreased production of T3 and impaired breakdown of rT3. BAT, brown adipose tissue; CNS, central nervous system; (H), human not rat; (R), rat not human; T2, 3,3′-di-iodothyronine. (From Beckett G J, Arthur J R. Journal of Endocrinology 2005; 184:455–465. © Society for Endocrinology, with permission.)
Catabolism of thyroid hormones
Sulphation, glucuronidation, deamination, oxidative decarboxylation, ether cleavage and deiodination form the main routes for the inactivation and degradation of thyroid hormone. These metabolites are excreted via bile or in urine.
Nuclear action of thyroid hormones
Tri-iodothyronine exerts its effects through interaction with nuclear thyroid hormone receptors (TR) that have a high affinity and high specificity for T3. Nuclear thyroid hormone receptors are a family of ligand-regulated transcription factors that are associated with chromatin. Several α and β isoforms of TR are produced and these usually dimerize with the retinoid X receptors (RXRs). TRα1 is widely expressed with particularly high expression in cardiac and skeletal muscles, whereas TRβ1 is predominantly expressed in the brain, liver and kidney and TRβ2 is primarily limited to the hypothalamus and the pituitary.
The TR/RXR complex binds to target response elements located in the promoter region of the target genes. The formation of the T3-TR/DNA complex and subsequent recruitment of a variety of transcriptional coactivators leads to activation of the target genes, giving increased mRNA and protein production. In some circumstances, gene expression may be switched off. Mutations in the TRβ gene lead to a decreased affinity of the receptor for T3, leading to the autosomal dominant clinical syndrome of ‘thyroid hormone resistance’ (discussed later in this chapter).
Control of thyroid hormone synthesis and secretion
Classic feedback regulation
The most important regulator of thyroid homoeostasis is TSH. This dimeric peptide hormone comprises a β-subunit, required for binding to the TSH receptor, and an α-subunit. The α-subunit is the same as that of luteinizing hormone, follicle stimulating hormone and human chorionic gonadotrophin. The β-subunit is specific for TSH: both subunits are required for bioactivity. The subunits contain carbohydrate chains that are essential for the biological activity of the molecule. Modifications to these carbohydrate residues occur in different thyroid states, which alter both the bioactivity of the molecule and its plasma half-life. For example, in primary hypothyroidism, TSH has increased bioactivity and an increased plasma retention time, while in secondary hypothyroidism the bioactivity of the molecule is diminished. Most immunoassays are unable to recognize modifications to the carbohydrate chains. There is a circadian rhythm of TSH secretion, plasma concentrations being highest between midnight and 04.00 h and lowest at about midday. Thyroid stimulating hormone release also occurs in a pulsatile fashion. These changes in TSH are not reflected in the serum thyroid hormone concentrations.
The production of TSH is controlled by a stimulatory effect of the hypothalamic tripeptide, thyrotrophin releasing hormone (TRH, thyroliberin), mediated by a negative feedback from circulating free T3 and free T4 (Fig. 19.5). Free T3 and free T4 inhibit TSH synthesis and release directly, by inhibiting transcription of the TSH subunit genes and indirectly by inhibiting TRH release. Free T3 and free T4 also decrease TSH glycosylation and therefore its biological activity. It is thought that the hypothalamus, via TRH, sets the level of thyroid hormone production required physiologically, and that the pituitary acts as a ‘thyroid-stat’ to maintain the level of thyroid hormone production that has been determined by the hypothalamus. Twin studies have shown that in individuals without thyroid disease, TSH and thyroid hormone concentrations are largely genetically determined. The setpoint in an individual is relatively narrow with intraindividual variation being less than half the interindividual variation.
FIGURE 19.5 The ‘classic’ hypothalamo–pituitary–thyroid axis. The production of thyroid stimulating hormone (TSH) is controlled by a stimulatory effect of the hypothalamic tripeptide, thyrotrophin releasing hormone (TRH, thyroliberin), mediated by a negative feedback from circulating free T3 and free T4. Thyroid stimulating hormone may also exert autoregulatory fine control on its own production. Dopamine, somatostatin, glucocorticoids, leptin and catecholamines all may have an influence on the axis.
Other mechanisms
The classic feedback regulation of TSH release described above is an oversimplification. Dopamine, somatostatin and glucocorticoids inhibit TSH release, and these agents, together with cytokines, may be important modifiers of plasma TSH, particularly in patients with non-thyroidal illness (NTI) (Fig. 19.6). Leptin and catecholamines regulate TSH indirectly by stimulating TRH production. There is accumulating evidence that TSH can provide autoregulation via a direct action of TSH on the hypothalamus and at the pituitary. In the pituitary, folliculo-stellate cells have been identified that express TSH receptors: interaction of TSH with these cells may produce fine control on TSH release through the release of cytokines, which in turn inhibit TSH release from thyrotrophs. It has been argued that interaction of TSH receptor antibodies with the folliculo-stellate cells explains why patients with Graves disease may continue to have suppressed TSH weeks or months after normal thyroid hormone concentrations have been achieved through carbimazole therapy.
FIGURE 19.6 The hypothalamo–pituitary–thyroid axis in non-thyroidal illness. Dopamine, somatostatin, and glucocorticoids inhibit TSH release, and these agents together with cytokines may be important modifiers of serum TSH, in particular in patients with non-thyroidal illness. ACTH, adrenocorticotrophic hormone; CRF, corticotrophin releasing factor; IL-1, interleukin-1; TNF, tumour necrosis factor.
Thyroid stimulating hormone modifies thyroid hormone synthesis by binding to a specific receptor on the surface of the thyrocyte. The TSH receptor is a single protein with a large extracellular amino-terminal domain involved in the binding of TSH, seven transmembrane domains and a short intracellular carboxy-terminal domain involved in the activation of G-protein modulators of the adenylate cyclase-protein kinase-A system. These characteristics are shared with receptors for gonadotrophins: their extracellular domains show some 40% homology, the remainder being more highly conserved. This may explain the weak thyroid-stimulating activity of human chorionic gonadotrophin (hCG). Binding of TSH results in activation of adenylate cyclase and accumulation of cyclic AMP. The calcium and phosphoinositol signalling pathways may also be activated by TSH. After about an hour, an increase in release of thyroid hormones is seen. In the longer term, TSH increases the synthesis of iodinated thyroglobulin and also causes a general increase in the metabolism, size and activity of the follicular cells.
Extrathyroidal factors that may affect thyroid function
Age
Fetus
Thyroxine and TSH are detectable in fetal plasma at 10–12 weeks of gestation. Plasma concentrations of total and free T4, total and free T3 and TBG increase during gestation, total and free T4 reaching adult concentrations at about 36 weeks, but total and free T3 reaching only the lower limit of the adult range at this time. The concentration of TSH also increases with gestational age but is within or above the adult reference range throughout gestation. Marked time- and organ-specific changes occur throughout gestation in the expression of the deiodinases, which are thought to coordinate the orderly maturation of enzyme systems responsible for development of the brain and the other organ systems in the fetus.
Neonate
During the first 24 h after birth, there is a rapid, transient increase in the release of TSH, T3 and T4 that is considered to be an adaptive response to birth. Thyroid stimulating hormone peaks during the first 30 min, which then stimulates T3 and T4 production during the first 24–36 h of postnatal life. This effect may be attenuated in infants delivered prematurely. Screening for congenital hypothyroidism should be carried out after at least three days to avoid spurious results.
Infancy and childhood
Plasma concentrations of TSH are within the adult range but free T3 is higher than in adults. Free T4 concentrations tend to be at the lower end of the adult range. This hormone pattern suggests it may arise as a result of increased D1 activity in children. After puberty, no major changes in thyroid function occur, except in the pregnant woman.
Elderly
In old age, there is little change in thyroid function tests and age-related reference ranges are not required. There is a modest decrease in T4 secretion but without an accompanying change in circulating T4. A slight fall in T3 may occur in those over 80 years of age. In patients on T4 replacement, the dose of T4 may have to be decreased with age. There is also increasing evidence that the normal TSH distribution curves appear to be shifted towards higher value ranges in individuals aged over 80. Age-specific analysis of TSH concentrations and anti-thyroid antibody titres measured in the National Health and Nutrition Examination Survey (NHANES) demonstrated that 12% of subjects aged 80 and older without any evidence of underlying autoimmune thyroiditis had TSH concentrations > 4.5 mU/L. What is currently unclear is whether higher TSH concentrations confer a survival advantage or simply represent a degree of non-autoimmune age-related thyroid failure.
Pregnancy
In normal pregnancy there is a large increase in plasma TBG concentration that arises from both an oestrogen stimulated increase in synthesis and diminished clearance of the protein. There is also an increase in the pool size of extrathyroidal T4 distribution and an increase in the deiodination of thyroid hormones in the developing placenta. The result of these changes is that, during pregnancy, there is a marked increase in the requirement for iodine and an approximately 50% increase in T4 production occurs if the supply of iodine is adequate.
In order to maintain homoeostasis, an increase in total T4 and total T3 concentrations occurs, which reach a new steady state around mid-gestation. This requirement for increased thyroid hormone production requires an ideal iodine supply during pregnancy of approximately 250 μg/day. Similarly, women with hypothyroidism taking T4 replacement will need an increase in the dose of T4 of approximately 25–50 μg/day during pregnancy within the first six weeks of gestation, aiming for a TSH no higher than 2.5 mU/L. Some centres recommend that women double their pre-pregnancy levothyroxine dose on two days a week as soon as pregnancy is confirmed to reduce the risk of maternal hypothyroidism. Thyroxine replacement should be monitored carefully using both TSH and free T4, since even modest degrees of hypothyroxinaemia in early pregnancy have been associated with an impaired IQ in the infant.
In pregnancy, free T4 and free T3 concentrations may initially show a slight rise, thought to be due to the weak thyroid stimulating action of hCG, present in very high concentration in early pregnancy. This increase in free hormones can lead to suppression of TSH, such that low or sometimes undetectable concentrations may be found in up to 20% of patients during the first trimester. It is important that these changes in early pregnancy are not construed as suggesting that the patient has thyrotoxicosis. As pregnancy progresses, the concentrations of free thyroid hormones fall and TSH begins to rise, although rarely increasing above the reference range seen in non-pregnant subjects. Free thyroid hormone concentration may be lower than those found in non-pregnant women. It is important that trimester-related reference ranges are used for both TSH and free thyroid hormones. Patients can usually return to their pre-pregnancy levothyroxine replacement regimen immediately post-delivery; however, they should be monitored to ensure that this is still the optimal dose.
Hyperemesis gravidarum (severe vomiting in the first trimester of pregnancy) is associated with high plasma total and free thyroid hormone and low TSH concentrations, making it difficult to distinguish this condition from severe true thyrotoxicosis (thyroid crisis). It is believed that hCG is responsible for the thyroid stimulation; the condition usually resolves by the second trimester. Thyroid stimulating hormone receptor antibodies are negative in patients with hyperemesis but positive if the patient has Graves disease.
Thyrotrophin releasing hormone can cross the placenta from mother to fetus, but TSH does not. Thyroid hormones cross the placenta, but this transplacental flux is regulated in part by changes in expression of placental deiodinase as pregnancy progresses. A maternal supply of thyroid hormones (particularly T4) to the fetus is particularly import in the first trimester until a functional fetal thyroid has developed. However, since a fetus with congenital thyroid deficiency develops relatively normally in utero, this suggests that some maternal-fetal transfer of thyroid hormone must be maintained throughout pregnancy. Iodide and antithyroid drugs also pass to the fetus. Indeed, antithyroid drugs are transferred more readily than thyroid hormone, and for this reason treatment of thyrotoxicosis in pregnancy with a ‘block and replace’ regimen (high dose antithyroid drugs ‘rescued’ by simultaneous thyroid hormone replacement) is contraindicated in pregnancy as it would result in a relatively hypothyroid fetus. The treatment of hyperthyroidism in pregnancy requires careful consideration. Radioactive iodine is contraindicated and the choice of antithyroid drug is currently the subject of debate. Propylthiouracil has been traditionally preferred to carbimazole/methimazole owing to fears of scalp and gastro-oesphageal defects in fetuses exposed to carbimazole/methimazole in the first trimester. However, recent concerns have arisen owing to the occurrence of propylthiouracil induced hepatitis, which has occasionally required liver transplantation and rarely has resulted in maternal death. Autoantibodies may cause additional problems in managing patients with hyperthyroidism in pregnancy. In patients with Graves disease, the transplacental passage of thyroid stimulating immunoglobulins may cause transient neonatal thyrotoxicosis, while the presence of anti-TPO antibodies is associated with an increased risk of maternal hypothyroidism and miscarriage.
Non-thyroidal illness
Patients attending or admitted to hospital suffering from any of a wide range of chronic or acute non-thyroidal illnesses (NTI), often have abnormalities in thyroid function tests. A low T3 may often be found even though the patients are clinically euthyroid; this has been termed the sick euthyroid syndrome.
Several mechanisms are involved, including:
• alterations in the hypothalamic–pituitary–thyroid axis leading to decreased hypothalamic stores of TRH and suppression of TSH release due to increased concentrations of dopamine, cytokines, cortisol and somatostatin (see Fig. 19.6). While the degree of TSH suppression in NTI is often not as great as that found in hyperthyroidism, there is considerable overlap in TSH concentrations found in the two conditions
• changes in the affinity characteristics and in the plasma concentrations of the thyroid hormone-binding proteins. These changes give rise to alterations in the plasma concentrations of both the free and total thyroid hormones
• impaired uptake of thyroid hormones by the tissues
• decreased production of T3 in the peripheral tissues
• changes in the T3 occupancy and function of the T3 receptors.
The contribution of each of the above mechanisms may vary with the severity and stage of the illness, and thus the pattern of thyroid function tests may be extremely variable and may mimic the profile seen in either primary or secondary thyroid disease. Interpretation of thyroid function tests is complicated further by the effects of drugs and methodological problems associated with free hormone measurements.
Thyroid hormone metabolism is markedly affected by fasting and illness, with the magnitude of these changes tending to be proportional to the severity of the illness (Fig. 19.7). Extrathyroidal conversion of T4–T3 is reduced, leading to a marked decline in plasma total and free T3, which may fall to undetectable concentrations. Reverse T3 concentration increases, primarily owing to impaired catabolism rather than increased synthesis. These changes are often considered to be an adaptive response for energy conservation, as rT3 is not metabolically active. Concentrations of free fatty acids and other substances that can compete with thyroid hormones for binding to plasma proteins may rise. This can produce a transient increase in free T4. Drugs that compete for T4 binding sites will have a similar effect. Uptake of thyroid hormone into cells may be impaired, either directly by endogenous inhibitors or indirectly as a result of impairment of the active transport systems of the cells. Finally, administration of corticosteroids or dopamine may suppress TSH release.
FIGURE 19.7 The effects of illness on the concentration of thyroid hormones and thyrotrophin. The profile for free T4 obtained using equilibrium dialysis is shown (FT4-ED). The concentration of free T4 found using routine assays is method dependent, with some assays showing the profile of FT4-ED, while others follow the profile of total T4 (TT4). Free T3 (FT3) shows a similar profile to total T3 (TT3).
Methodological shortcomings render the results of free hormone analysis liable to serious error. Equilibrium dialysis methods usually show a normal or slightly raised free T4 in patients with illness of mild to moderate severity, but most commercial assays tend to show normal or low free T4 results in sick patients. Normalization or transient rebound changes of thyroid parameters may be seen during recovery from NTI or refeeding after starvation. In particular, TSH may rise transiently into the hypothyroid range. It should be noted that, although TSH is in general the most reliable test of thyroid function, in hospitalized patients a TSH of < 0.1 mU/L is twice as likely to be due to NTI as to hyperthyroidism, and an increased TSH is as likely to be due to recovery from illness as to hypothyroidism. Because of the poor predictive value of thyroid function tests in hospitalized patients, these tests should only be requested if the reason for hospital admission is considered, on clinical grounds, to be related to a thyroid problem. Also, abnormal thyroid function tests during illness may need to be rechecked after the acute illness has resolved.
While the changes in thyroid hormone metabolism associated with illness might theoretically lead to tissue hypothyroidism, most trials of T4 and T3 administration in severe non-thyroidal illness have not shown improved outcome.
Drugs
Drugs may interfere with TSH secretion or the production, secretion, transport and metabolism of thyroid hormones (Table 19.1). Some drugs modify thyroid status while others produce abnormal thyroid function test results in clinically euthyroid subjects. Certain agents (in particular iron preparations) impair the absorption of T4 from the gut, and patients on thyroxine therapy should be advised to take their T4 at least 4 h apart from these medications. Patients should also be advised to avoid caffeine within 45 min of taking levothyroxine as caffeine can also impair absorption. Patients taking levothyroxine are likely to require an increase in replacement dose if drugs such as phenytoin or carbamazepine are also prescribed as they increase hepatic metabolism of T4. Phenytoin, carbamazepine, furosemide and salicylate compete with thyroid hormone binding to plasma binding proteins and may increase plasma free T4 concentrations. The influence of drugs on modifying free thyroid hormone concentrations may be method specific. For example, depending on the method, free T4 may be measured as being normal, high or low in patients given heparin or taking phenytoin or carbamazepine. Amiodarone, lithium and interferon can induce thyroid dysfunction and are covered in detail later in this chapter.
TABLE 19.1
Drugs affecting thyroid function
| Mechanism | Example of drug |
| Decrease in TSH secretion | Dopamine, glucocorticoids, octreotide, cytokines |
| Decrease in thyroid hormone secretion | Lithium, amiodarone, iodide |
| Increase in thyroid hormone secretion | Lithium, amiodarone (rare), iodide |
| Decrease in thyroidal synthesisa | Carbimazole, propylthiouracil, lithium |
| Increase in TBG | Oestrogens, tamoxifen, heroin, methadone, clofibrate, raloxifene |
| Decrease in TBG | Androgens, glucocorticoids, anabolic steroids |
| Displacement of thyroid hormones from plasma proteins | Furosemide, fenclofenac, salicylates, mefenamic acid, carbamazepine |
| Increased hepatic metabolism | Phenytoin, carbamazepine, rifampicin, barbiturates |
| Impaired T4 and T3 conversion | β-antagonists, amiodaronea, radiocontrast dyes, e.g. iopanoic acid |
| Impaired absorption of thyroxineb | Cholestyramine, aluminium hydroxide, ferrous sulphate, calcium salts, sucralfate, soya protein |
| Altered immunityc | Interleukin-1, interferons, tumour necrosis factor-α, interleukin-2 |
Other drugs listed produce abnormal thyroid function tests but patients remain euthyroid. Amiodarone is the exception (see text).
a Cause decrease in thyroid hormone synthesis or secretion and alter thyroid status.
b Interfere with absorption from GI tract. Patients on T4 therapy should be advised to take their T4 at least 4 h apart from these medications.
c These cytokines can cause transient hypothyroidism or thyrotoxicosis, the mechanism of which is unclear.
THE EVALUATION OF THYROID FUNCTION
This section describes the methods that have been developed in order to assess the functional state of the thyroid gland. In most cases, the presenting clinical features will lead to thyroid function tests being performed for confirmation of the provisional diagnosis.
Clinical evaluation of thyroid status
Disordered thyroid function may be detected clinically by demonstrating both changes in the thyroid gland itself and the systemic effects of under- or overproduction of thyroid hormones. In some instances, the clinical presentation may be virtually diagnostic of a specific condition and investigations are required only for confirmation. In others, the clinical features may be less specific and more thorough investigation is required. The signs and symptoms of thyroid dysfunction are listed in Tables 19.3 and 19.6. The normal thyroid can often be palpated and may also be apparent on inspection of the neck, particularly in young, thin women. Abnormal thyroid enlargements may be diffuse or asymmetrical. As a result of connective tissue attachments to the trachea, thyroid swellings move upwards on swallowing and a similar phenomenon is found with tongue protrusion in the case of thyroglossal cysts. The normal gland has a ‘rubbery’ feel; in Graves disease and diffuse colloid goitre, the consistency is softer. By contrast, the thyroid in Hashimoto disease is often rather firm, often with a palpable pyramidal lobe. Thyroid carcinoma (anaplastic) and Riedel thyroiditis result in a rock-hard gland that may, in the case of carcinoma, be irregular in outline. Increased blood flow through a hyperactive gland is often associated with a bruit. This may be maximal over the superior thyroidal artery rather than the gland itself and, in some cases, may also produce a palpable thrill.
In vitro tests of thyroid activity and pituitary–thyroid status
The tests used to investigate thyroid dysfunction can be grouped into:
• tests to elucidate the cause of thyroid dysfunction, e.g. thyroid autoantibody and serum thyroglobulin measurements, thyroid enzyme activities, biopsy of the thyroid, ultrasound and isotopic thyroid scanning
• thyroglobulin measurements, which are used to monitor treatment and detect recurrence in patients with follicular or papillary carcinoma.
Measurement of TSH and thyroid hormones should be performed to determine the patient’s thyroid status before requesting the additional tests that seek to determine the cause of any thyroid dysfunction.
Measurement of thyroid stimulating hormone
The measurement of TSH in a basal serum sample by a sensitive immunometric assay provides the single most sensitive, specific and reliable test of thyroid status in both overt and subclinical primary thyroid disorders. In primary hypothyroidism, TSH is increased, while in primary hyperthyroidism TSH is usually < 0.02 mU/L. However, TSH alone is not a reliable test for detecting thyroid dysfunction arising from hypothalamic–pituitary dysfunction and in other specific instances as outlined below.
Thyroid stimulating hormone is now measured by immunometric assays (IMAs) that utilize non-competitive labelled antibody methods with non-isotopic labels. These IMAs use highly specific monoclonal antibodies raised to epitopes on α- and β-subunits of the TSH molecule. The IMA technique uses a sandwich- assay configuration using two antibodies. The ‘functional sensitivity’ of a TSH assay defines the minimum concentration of TSH that can be quantified in routine use. To obtain the functional sensitivity of a routine assay, a precision profile is constructed using data obtained over many assay runs with multiple batches of reagent and different operators. The concentration of TSH that has a coefficient of variation of 20% taken from the precision profile defines ‘functional sensitivity’. It is essential that TSH assays are sufficiently sensitive for clinical diagnostic purposes and they must have a functional sensitivity of < 0.02 mU/L.
Free T4 and free T3 measurements
Theoretical considerations
The plasma concentrations of free thyroid hormones are extremely low and their quantification in the presence of large concentrations of bound hormones has proved challenging. Poor assay design of many free hormone methods has produced a mass of conflicting literature regarding the typical ranges of free thyroid hormone concentrations in patients with NTI and taking a variety of drugs.
The free hormone concentration results from the equilibrium between the bound (BP) and free hormone.
The proportion of T4 that binds to the binding protein is dictated through the law of mass action by the affinity of the binding protein (Keq) multiplied by its concentration. This is known as the ‘relative binding capacity’. Thus, if the affinity or concentration of the binding protein decreases, the proportion of T4 bound will also diminish and the free hormone concentration will increase. In humans, the free T4 fraction is mostly affected by changes in the affinity and concentration of TBG; changes in the affinity and concentration of albumin or transthyretin have little effect on free T4.
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
