[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 40
Poisoning
CHAPTER OUTLINE
INTRODUCTION
Poisoning can be defined as an interaction between a foreign chemical (toxin) and a biological system that results in damage to a living organism. In general, the medical profession has been more concerned with the acute effects of toxins and the clinical management of toxicity, but chronic effects of toxins have much more importance on a global scale. For example, acute ingestion of ethanol may result in intoxication, which can cause death directly from its acute depressant effects and also through intoxication-related accidents and violence. However, alcohol-related liver disease, myopathy, hypertriglyceridaemia and cancers are all effects of chronic alcohol abuse and together, cause far more deaths than the acute effects of alcohol. Other types of chronic or delayed effects of toxins include mutagenicity, carcinogenicity and teratogenicity. For example, many cases of bronchogenic carcinoma are a direct result of exposure to cigarette smoke. Increasing concerns about environmental contamination by chemicals has added a further dimension to toxicology, especially as many chemicals, such as pesticide residues, may be found in small amounts in the general population, who do not appear to be suffering any ill effects.
The interaction between a toxin and the biological system is highly complex. Once the biological system has been exposed to the toxin, the toxin is subject to various processes. In the case of a drug, this is usually termed pharmacokinetics, but in the case of a poison, the term toxicokinetics may be used. This includes absorption, for which the route of administration is of great importance, as are the dose of the toxin and the duration of exposure. Once a toxin has entered the bloodstream, its distribution depends to a large extent, on its physicochemical characteristics, including its degree of lipid or water solubility and its degree of ionization. Metabolism is a further important process, which usually renders toxins inactive, but which may also convert some substances of low toxicity into highly toxic metabolites. Classic examples of this include paracetamol (acetaminophen), methanol and ethylene glycol. Finally, toxins are excreted, although some remain within the body for many years. All of these processes determine the amount of toxin that interacts with tissues to produce a toxic effect.
The study of the interaction between toxins and tissues is called toxicodynamics. An understanding of the nature of this interaction is essential for an intelligent approach to the diagnosis and management of poisoning. This interaction may cause functional and metabolic disturbances that produce the characteristic clinical features of poisoning and the laboratory changes that may be essential for diagnosis or management. These changes are summarized in Figure 40.1.
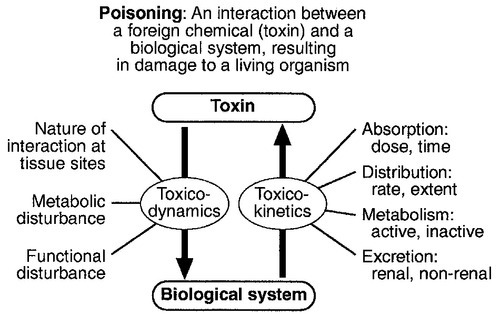
FIGURE 40.1 The factors involved in poisoning.
This chapter covers several of the more common types of acute poisoning and also some aspects of chronic poisoning. Clinical features are given in some detail, as these frequently integrate with the metabolic and biochemical changes.
AETIOLOGY OF POISONING
Intrauterine
A number of drugs, such as tretinoin, should not be taken by women intending to become pregnant because of their known teratogenicity. Other drugs, such as phenytoin and several other anticonvulsants, are teratogenic but their continued use may be considered necessary during pregnancy. As far as poisoning and overdose during pregnancy are concerned, the fetus generally only suffers damage secondary to the mother’s illness, and the management is no different from that of overdose in non-pregnant women. One exception is carbon monoxide, which has a higher affinity for fetal haemoglobin than adult haemoglobin; the mother should be treated aggressively even in apparently mild poisoning. Antidotes such as acetylcysteine or desferrioxamine should not be withheld because the patient is pregnant.
Neonates
During the neonatal period, particularly in premature infants, there is a risk of iatrogenic overdose because of the poor metabolic and excretory capacity relative to body weight. Particular care should be taken with drugs such as theophylline, digoxin, chloramphenicol and morphine.
Infants
The commonest cause of poisoning between the ages of one and four is termed accidental poisoning because of the natural exploratory activities of children at this stage of life. Each year, this results in 20 000 admissions to hospital in Britain, but only about ten fatalities. The commonest agents involved are paracetamol and oral contraceptives, but most fatalities occur from tricyclic antidepressants, salicylates, iron, methadone and quinine. Another form of poisoning in this age group is non-accidental injury, which is usually inflicted by a carer such as the mother, and the child may present with repeated episodes of floppiness and collapse, depending on the substance responsible. This is most commonly with a drug prescribed for the mother, though salt and other readily available chemicals have been used (see Chapter 44).
Childhood
Later in childhood, substance abuse, particularly volatile substance abuse (‘glue sniffing’), can cause serious problems. There are about 30 deaths each year in Britain from this cause. Acute and chronic alcohol toxicity are often underestimated in this age group. Intentional drug overdose is common, but suicide is rare.
Adult life
In early adult life, particularly in females, drug overdose in the form of parasuicidal gestures is common, though rarely fatal. In later adult life, suicidal intent is commoner and parasuicidal gestures are less frequent. The commonest causes of suicidal poisoning in England are tricyclic antidepressants, analgesic drugs and carbon monoxide. Male suicides are approximately three times as frequent as female suicides. Accidental poisoning in adults is usually domestic, the commonest agent being carbon monoxide, or industrial, in which a wide range of chemicals may be involved. Homicidal poisoning, though rare, may go undetected if it is not suspected.
TYPES OF LESION IN POISONING
Many types of poisoning involve a highly specific interaction between the toxin and one type of tissue. This has led to a ‘target organ’ oriented approach in toxicology, which is of great help in understanding poisoning, where damage to a single organ or tissue produces a characteristic clinical and pathological picture. Examples include poisoning with paracetamol, salicylates, cholinesterase inhibitors and cardiac glycosides. In other instances, the type of toxicity may be less clearly definable. The acute effects of ethanol are mainly due to its central nervous depressant effect. Cocaine, tricyclic antidepressants, many volatile substances, some β-adrenergic blocking drugs and dextropropoxyphene can all produce cardiotoxicity due to a quinidine-like sodium channel blocking effect. However, the clinical features are definable as a toxic effect on the heart.
An important way in which many poisons can affect the organism is by interference with the oxygen pathway from the inspired air to cellular respiration. Thus, a reduction in the oxygen content of the inspired air may cause acute hypoxaemia, rapidly leading to collapse and coma. Interference with the mechanics of respiration by poisons may result in hypoxaemia and hypercapnia (type II respiratory failure), while disturbances of oxygen transfer produce hypoxaemia alone (type I respiratory failure). Even if the oxygen content of the air is not diminished and respiration is functioning adequately, disturbances to other processes may prevent oxygen reaching its site of action (see Chapter 5). The oxygen-carrying capacity of the blood may be reduced by the presence of carboxyhaemoglobin or methaemoglobin or, more rarely, by acute haemolysis. Cardiac output may be reduced by a number of poisons that cause cardiac arrhythmias, depress the contractility of the heart or cause extreme vasodilatation. The final step where poisons may interfere with the oxygen pathway is blockage of the cytochrome enzyme chain as occurs with toxins such as cyanide and hydrogen sulphide. These effects and the main poisons involved are summarized and outlined in Table 40.1.
TABLE 40.1
Some typical causes of blockade of the oxygen pathway due to poisoning, with mechanisms and typical values
Note that whatever the initial effect of a poison, there may be subsequent progression down the list of effects towards cell death. The effects of two or more poisons may be additive
| Cause | Mechanism | Effect |
| Asphyxiant gases (e.g. butane, methane, carbon dioxide, nitrogen) | Hypoxic gas mixture | Reduced inspired oxygen fraction (FiO2) (normal ~ 0.21) |
| Respiratory depression (e.g. opioids, barbiturates, other sedatives and hypnotics). Respiratory muscle disorders: paralysis (e.g. organophosphates, botulinum toxin) or spasm (e.g. strychnine, phencyclidine) | Failure of ventilation (type II respiratory failure) | Reduced alveolar oxygen tension (PAO2) (normal ~ 13.3 kPa) |
| Aspiration pneumonitis Adult respiratory distress syndrome (e.g. paraquat) |
Failure of oxygen transfer (type I respiratory failure) | Reduced arterial oxygen tension (PaO2) (normal 10–13.3 kPa) |
| Carboxyhaemoglobin (carbon monoxide); methaemoglobin (e.g. nitrites); haemolysis (e.g. arsine, stibine) | Loss of functioning haemoglobin | Reduced arterial oxygen content (CaO2) (normal 18–21 volumes %) |
| Myocardial depressants (e.g. β-blockers, calcium antagonists, tricyclic antidepressants, dextropropoxyphene, cocaine) | Reduced cardiac output | Reduced tissue oxygen delivery (QO2) (normal 12–16 mL/kg per min) |
| Chemical asphyxiants (cyanide, hydrogen sulphide) | Block of cytochrome enzyme chain | Reduced tissue oxygen consumption (VO2) causing failure of oxidative metabolism (normal 3–4 mL/kg per min). Cell death |
DIAGNOSIS AND MANAGEMENT OF POISONING: GENERAL PRINCIPLES
Diagnosis
In most cases of poisoning, the diagnosis is provided by the history and clinical examination. In some patients, however, there may be no history, though poisoning may be suspected because of the circumstances or the clinical features. In rare cases, the clinical presentation may mimic other medical conditions and it is necessary to consider the diagnosis of poisoning, especially when there are unusual features of the illness. Examples of these conditions include carbon monoxide, lead and paraquat poisoning, and drug and substance abuse.
The clinical features of poisoning depend most of all on the type of agent involved, but also on the route of exposure (oral, intravenous, percutaneous or inhaled), the duration of exposure and time elapsed since exposure, and also the patient’s age and medical background (conditions such as diabetes, asthma, chronic renal failure and epilepsy). Clinical examination may reveal signs of injection marks, cutaneous burns, buccal corrosion, tablet residues in the mouth or cutaneous blisters. These blisters (originally called ‘barbiturate blisters’) may be found following overdose with barbiturates, benzodiazepines, tricyclic antidepressants or chloral hydrate, or exposure to carbon monoxide. There may be evidence of pulmonary aspiration or secondary hypostatic pneumonia in comatose patients.
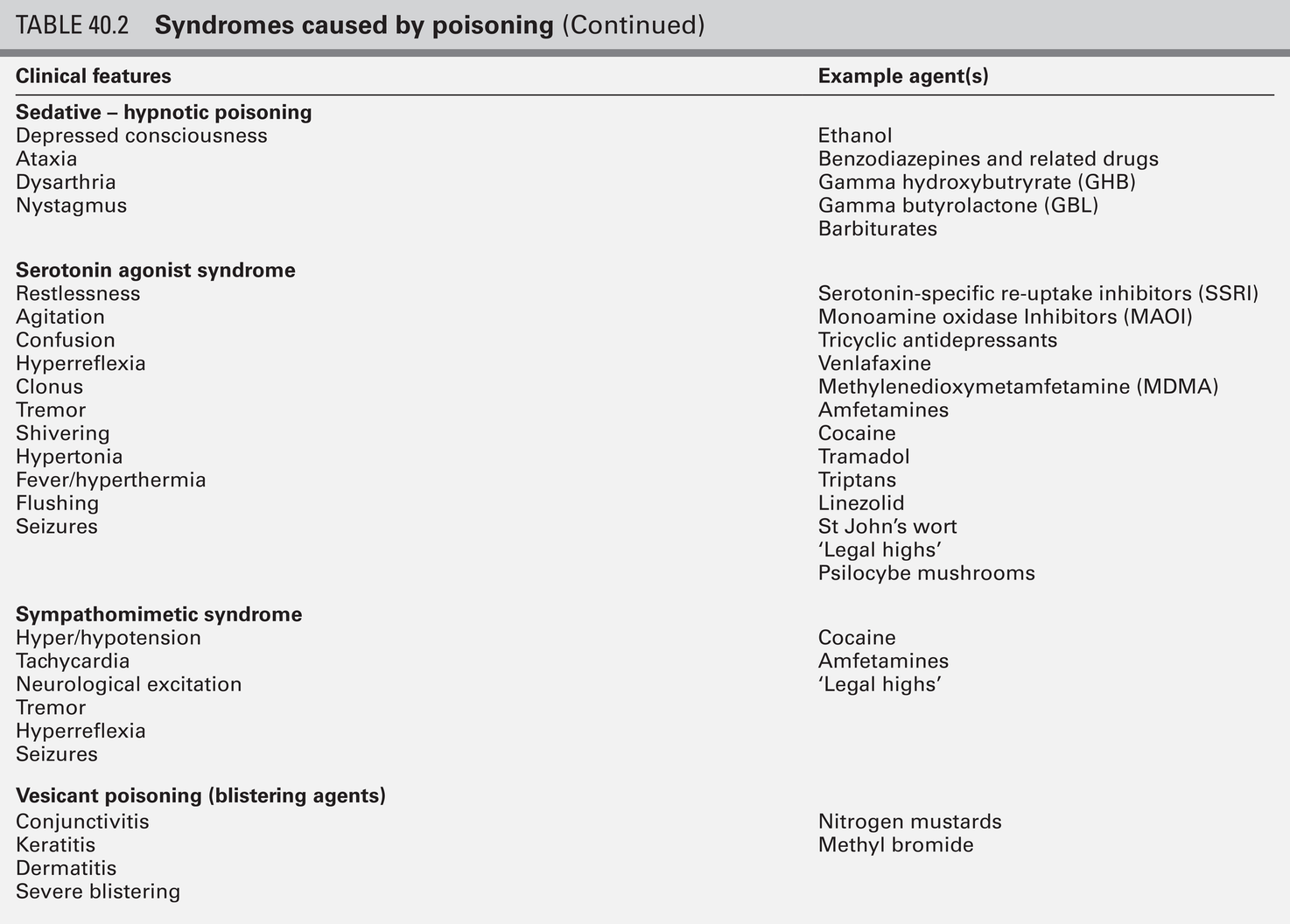
Clinical examination may indicate that a specific agonist/receptor pathway has been either over-stimulated or blocked by a poison. These syndromes are termed toxidromes and provide a useful initial guide to poison identification when the agent involved is unknown (Table 40.2).

In the severely ill patient, baseline haematological and biochemical investigations will be necessary and arterial blood gas estimations will also be required. A chest radiograph is usually indicated. The results of haematological and biochemical investigations may be of great assistance in making the diagnosis and monitoring the response to treatment. The tests to be carried out and their frequency depend very much on the poison involved. Many of these are given later in this chapter, and a general summary is given in Table 40.3. Toxicological investigations may also be required: in some cases, these may only be available in specialized laboratories. The usual samples required are blood (note that whole blood is required for measurement of carboxyhaemoglobin, methaemoglobin, cyanide and several metals) and urine (50 mL in a universal container with no preservative). Urine should always be provided where possible, since many types of analysis, such as drugs of abuse, are more efficiently carried out on urine samples. Some of these assays are qualitative, but may be necessary in order to confirm the presence of compounds, which are not easily detectable in blood.


Poisons centres now exist in most countries, with specialized facilities for provision of information on the toxicity of substances and on the management of poisoning. Most hold stocks of specialized antidotes, such as snake antivenoms. Many also have an analytical laboratory and some have patient care facilities. They are usually accessible by telephone or e-mail. In the UK, there are several poisons centres that provide information on poisoning: see Appendix 40.1.
Management
When an acutely ill patient presents, the first priority, regardless of the cause of the illness, is to establish the airway and provide immediate supportive treatment. Cardiopulmonary resuscitation or urgent ventilatory support may be needed. Depending on the patient’s state, questioning about the possible agent involved, obtaining an antidote or taking diagnostic samples may waste valuable time. In view of the frequency of opioid abuse, naloxone (an opioid antagonist) may have both a diagnostic and a therapeutic role. Once any immediate life-threatening problems have been dealt with, full supportive care must be provided while attempts are made to establish the diagnosis and treat the poisoning.
Respiratory support
In comatose patients, the most important measure is to maintain the airway and support ventilation as required, since respiratory complications are the commonest causes of death in unconscious poisoned patients. The unconscious patient should be placed in the left lateral (recovery) position in order to keep the airway patent and to minimize the risk of aspiration of gastric contents. Regular observation is essential as mechanical ventilation is not infrequently required.
Cardiovascular support
The circulation must be supported in order to maintain tissue perfusion and the electrocardiogram should be monitored, since cardiac dysrhythmias are common in poisoning. Antiarrhythmic agents and inotropic agents may be necessary.
Central nervous system complications
A brief convulsion due to cerebral hypoxia or to a toxic effect of the poison is not an indication for anticonvulsant drug therapy. However, if convulsions are repeated or prolonged, diazepam is the first-line drug. Drugs such as phenytoin, clomethiazole and thiopental may be required if there is no response to diazepam. In tricyclic antidepressant poisoning, administration of sodium bicarbonate may relieve convulsions. Phenytoin is contraindicated in tricyclic poisoning because both tricyclics and phenytoin are sodium channel blockers and these increase the risk of cardiac arrhythmias.
Body temperature
The core temperature should be recorded with a low-reading thermometer, since hypothermia may complicate poisoning with sedative and antidepressant drugs. The patient should be wrapped in a space blanket, but active rewarming is not usually required. The prognosis is better than in accidental hypothermia from other causes, and temperatures as low as 22 °C are compatible with full recovery. In hyperthermia (rectal temperature > 39 °C), reduction of body temperature is a priority. Depending on the cause of the problem, different treatments may be required. Hyperthermic patients poisoned with agents that produce severe muscle rigidity may require elective paralysis with a muscle relaxant and mechanical ventilation. Malignant hyperthermia or neuroleptic malignant syndrome can be treated with intravenous dantrolene.
Renal complications
Renal function should be monitored (bladder catheterization may be necessary if there is oliguria or severe hypovolaemia). Acute kidney injury may occur as a result of a direct toxic effect or secondary to acute haemolysis with haemoglobinuria or rhabdomyolysis with myoglobinuria. There is controversy as to whether myoglobin and haemoglobin are directly toxic to the kidneys. Most probably they cause an obstructive lesion; alkalinization of the urine increases myoglobin excretion and may prevent acute kidney injury.
General supportive care
In the severely ill patient, general supportive care may be life saving. Regular turning will be needed to prevent pressure necrosis of tissues, and care of the eyes and mouth will also be required.
Intestinal decontamination
Decontamination of the gut following ingestion of poisons is controversial. Syrup of ipecacuanha to induce emesis is now obsolete; there is little evidence that it is effective in emptying the stomach. In the comatose patient, gastric lavage is still occasionally used. However, this procedure may wash some of the poison further down the gut, and it is now rarely recommended.
The most widely used method of gut decontamination is to administer activated charcoal, which adsorbs almost all drugs and poisons. The main exceptions are iron and lithium, which are poorly adsorbed, and alcohols and glycols, whose molar load usually exceeds the adsorptive capacity of the charcoal. Repeated doses of activated charcoal may be used for sustained-release preparations that have delayed absorption, and may also be used to eliminate certain poisons from the body if there is significant enterohepatic recycling (e.g. carbamazepine and theophylline).
Antidotes
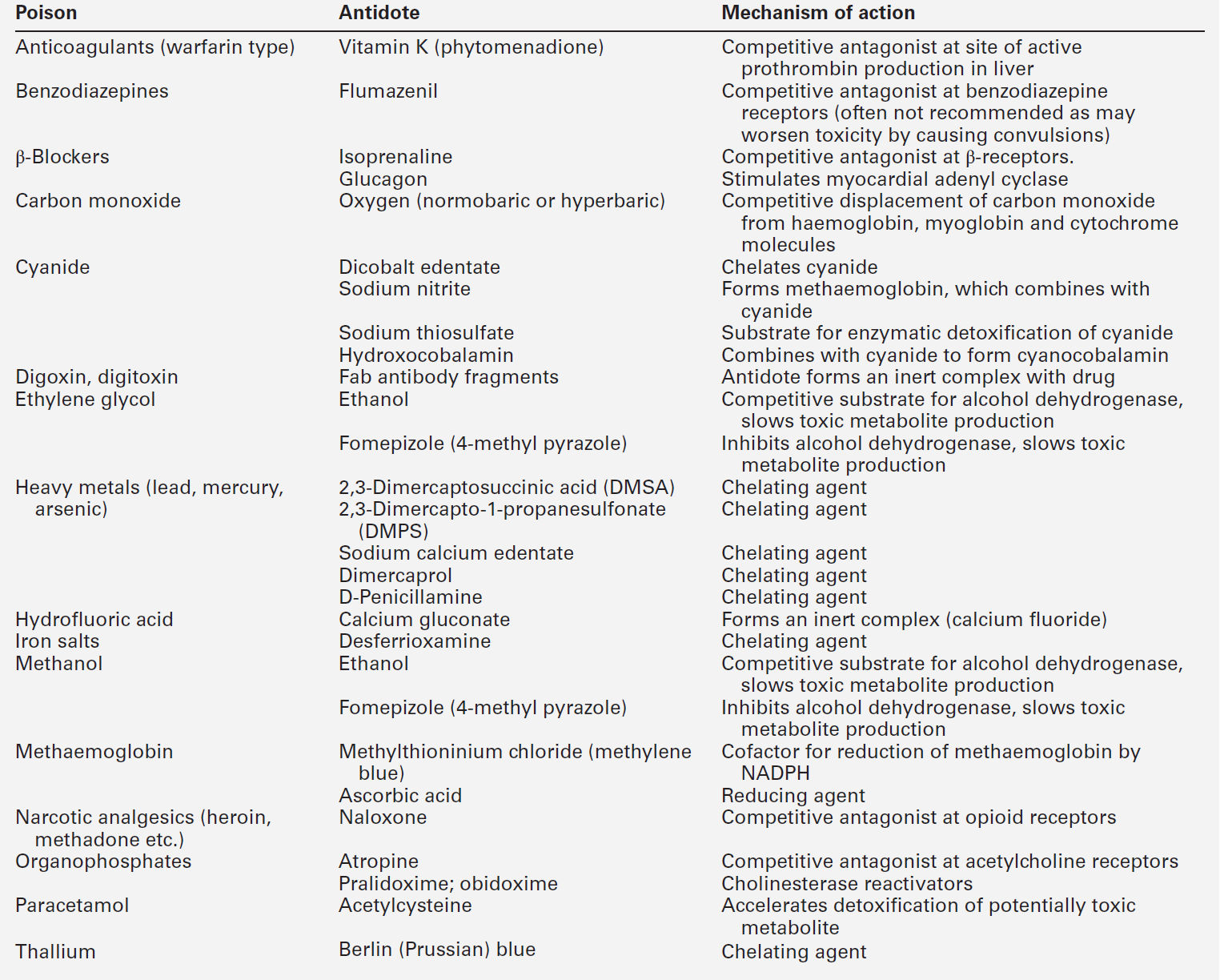
Antidotes form an important part of the management of certain poisons. In some cases (as with paracetamol, opioids, cardiac glycosides, organophosphates and snake bites), they may be life-saving. However, it should be noted that antidotal treatment is only required in a minority of poisonings. The most debated areas with regard to antidotes concern their effectiveness in ‘late’ paracetamol poisoning (when the patient presents more than 12 h after ingestion) and their effectiveness and use in the management of poisonings by metals. A list of commonly used antidotes is given in Table 40.4.
Elimination techniques
In some types of poisoning, active elimination techniques are indicated. This approach is most widely used in poisoning by salicylates, theophylline, ethylene glycol and methanol. The techniques include repeated oral doses of activated charcoal, enhanced renal elimination (by alkalinization of the urine or diuresis), haemoperfusion and haemodialysis. Haemodialysis may be required for the elimination of the poison (most notably in the case of salicylates, ethylene glycol and methanol) or for the support of renal function. Exchange transfusion may be used in infants. Peritoneal dialysis, plasmapheresis and continuous arteriovenous haemofiltration are less useful in removing poisons, although the latter is an effective technique to support renal function.
SPECIFIC POISONS
Paracetamol (acetaminophen)
Although paracetamol is safe when taken in the recommended dose, it is potentially very toxic in overdose and, since it is widely available over the counter, it is currently the commonest cause of admissions to hospital for poisoning and also of acute hepatic necrosis. It causes approximately 150 deaths each year in Britain.
Mechanisms
Paracetamol is rapidly absorbed from the upper gastrointestinal tract and the majority is metabolized by conjugation with sulfate or glucuronate to non-toxic derivatives. However, a small proportion (approximately 8–10%) is metabolized by a specific cytochrome P450 enzyme, CYP2E1, to produce a highly reactive intermediate, N-acetyl-p-benzoquinoneimine (NAPQI). This reactive metabolite can be metabolized by conjugation to form non-toxic mercapturic acid conjugates, provided glutathione is present in the liver cell. When an overdose of paracetamol is taken, the rate of production of NAPQI may exhaust existing glutathione stores and the capacity of the liver to synthesize glutathione (Fig. 40.2). In this case, NAPQI covalently binds to sulfhydryl groups in hepatocytes, forming an irreversible complex, which can result in acute centrilobular necrosis of the liver. Since paracetamol is also metabolized in the cells of the renal tubules, a similar process can also occur in the kidneys, leading to acute kidney injury. There is usually a small amount of renal damage in the presence of hepatic damage, but occasionally renal damage predominates and acute kidney injury may rarely be the presenting feature of paracetamol poisoning.
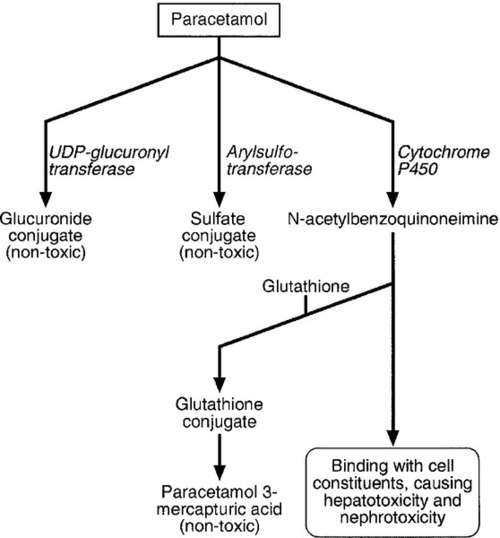
FIGURE 40.2 Metabolism of paracetamol. Induction of the cytochrome P450 pathway increases the production of the toxic metabolite, N-acetyl-p-benzoquinoneimine, causing depletion of glutathione stores; a protein-depleted diet leading to deficiency of amino acids may make less glutathione available. In both cases, increased toxicity may result following an overdose.
Toxic dose
The upper therapeutic limit for an adult of 4 g daily in divided doses is generally accepted as being safe, but doses above this can cause severe hepatotoxicity and possibly death. However, owing to the wide variation in the metabolic handling of paracetamol by the body, much larger overdoses (> 50 g) may have little effect in some individuals, producing nothing more than a minor rise in plasma aminotransferase activities.
Clinical features
In the first hours after ingestion of an overdose of paracetamol, symptoms may be minimal, unless it has been taken with some other drug, for example in a compound formulation with dihydrocodeine. There may be malaise, nausea and vomiting. A very large overdose may cause depression of consciousness or metabolic acidosis. By 24–36 h, there may be pain in the right hypochondrium. Plasma aminotransferase activities will rise at this time, but the peak values are a poor indicator of prognosis. The prothrombin time (or international normalized ration, INR) is a reliable prognostic indicator of severe hepatotoxicity. Hypoglycaemia and coagulation defects may complicate the hepatic failure. By about 48 h, the early signs of hepatic encephalopathy may appear. Death usually occurs after five or six days.
Management
Giving an antidote is more important than emptying the stomach or giving activated charcoal. After a single acute overdose, an antidote should be given if the patient’s plasma paracetamol concentration is on or above the line on the treatment nomogram (Fig. 40.3). The need for treatment following repeated supratherapeutic dosing (staggered overdose) cannot be assessed using the nomogram and treatment decisions are based on the dose ingested and the patient’s body weight. Repeated plasma paracetamol estimations should not be performed routinely, but may be helpful in doubtful cases.
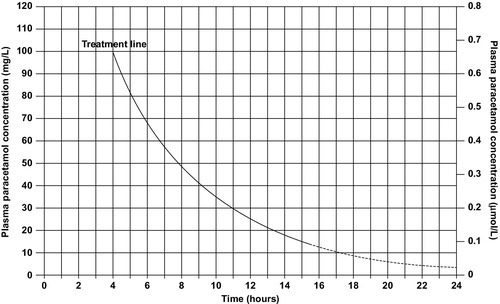
FIGURE 40.3 UK nomogram for treatment of paracetamol poisoning.
Acetylcysteine is the antidote of choice and the intravenous route is the only reliable method of treatment in a patient who is comatose or vomiting. Although the effectiveness of acetylcysteine diminishes considerably with time once 12 h have passed since ingestion, later administration improves outcome even in patients with established liver failure. Acetylcysteine should also be given if the time of the overdose is in doubt or not known. Its main adverse effect is anaphylactoid reactions, which respond to administration of antihistamines and discontinuation or slowing of the infusion.
Salicylate
Although the use of aspirin (acetylsalicylic acid) as an analgesic is decreasing, the management of salicylate poisoning remains a major challenge. The potentially lethal dose in adults is between 24 and 30 g, but death can occur in children under 18 months, from as little as 300 mg. Most fatal poisonings occur in elderly people, because they lack the metabolic reserves to cope with salicylate poisoning and also because attempted suicide with aspirin is commoner in old age. The use of aspirin preparations in children under 16 years of age is largely restricted to rheumatological indications because of the risk of Reye syndrome in children given aspirin for viral illnesses. However, this risk is very low.
Mechanisms
The mechanisms involved in salicylate poisoning are multiple and complex. The clinical features are mainly due to gastrointestinal irritation, stimulation of the respiratory centre causing respiratory alkalosis and uncoupling of oxidative phosphorylation, leading to heat production and metabolic acidosis. Many other mechanisms are also involved in salicylate poisoning. These are summarized and correlated with the clinical features in Figure 40.4.
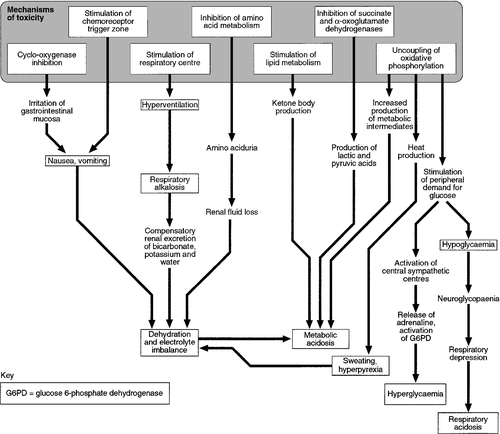
FIGURE 40.4 Pharmacological and metabolic effects of salicylates, leading to biochemical disturbances and clinical effects.
Clinical features
Salicylate poisoning usually presents with nausea and vomiting, increased rate and depth of respiration, sweating, tinnitus and sometimes deafness. Consciousness is preserved initially, but confusion, disorientation and loss of consciousness may occur, and usually indicate severe poisoning with a poor prognosis. The patient is often severely volume depleted because of vomiting, hyperventilation and sweating. Excess heat production usually causes compensatory sweating in adults but there may be hyperpyrexia in children. There is usually a combined compensated metabolic acidosis and respiratory alkalosis, with an arterial [H+] of 32–40 nmol/L (pH 7.4–7.5). Later, the [H+] rises (pH falls) as the alkali reserve diminishes, and this is a serious sign. In infants, metabolic acidosis may predominate from the start.
Salicylate overdose increases pulmonary capillary permeability, which, in severe poisoning, may cause non-cardiogenic pulmonary oedema. This presents initially as lowering of the arterial oxygen tension and subsequently becomes apparent on chest radiographs. Acute kidney injury is rare and is more likely to occur in children than in adults.
Laboratory measurements
Plasma salicylate concentration should be measured on presentation and at intervals of 4–6 h thereafter, until it has fallen below the toxic range. This is especially important, since salicylates are precipitated in an acidic environment and may therefore be deposited in the stomach, resulting in delayed absorption. Plasma concentrations may continue to rise for many hours, particularly in serious overdoses. Experience has shown that in most cases with a fatal outcome, plasma salicylate concentrations have risen progressively following admission to hospital.
Management
Activated charcoal should be given to patients presenting to hospital soon after overdose (within 1 h) and a second dose may be given if plasma salicylate concentration continues to rise. Gastric lavage is rarely, if ever, indicated but may be considered if a patient has presented early to hospital after taking a massive overdose. All symptomatic patients should have central venous pressure monitored, and fluid replacement and correction of electrolyte imbalances, especially hypokalaemia, are priorities. Hypoglycaemia may need to be corrected. Elimination of salicylate can be promoted by administration of sodium bicarbonate to produce a urine pH of > 7.5 ([H+] < 32 nmol/L). This has been clearly demonstrated, but the concept of ‘forced diuresis’ is now no longer used, since the excretion of salicylate is not influenced by urine volume. A urinary alkalinization regimen is recommended if the plasma salicylate concentration is greater than 500 mg/L (3.6 mmol/L). Haemodialysis may be necessary in severe poisoning with a plasma salicylate concentration > 900 mg/L (6.4 mmol/L), or lower than this if complications are present. As well as removing salicylate, haemodialysis can also correct acid–base and electrolyte imbalances and is therefore preferred to charcoal haemoperfusion. The prothrombin time should be measured; it is rarely prolonged, but if it is, phytomenadione (vitamin K) should be administered intravenously.
Chloroquine
In overdose, this drug can produce hypokalaemia ([K+] often < 2 mmol/L), coma, convulsions and sudden cardiovascular collapse. An acute overdose of > 5 g, systolic blood pressure < 85 mmHg, a QRS complex duration > 0.12 s and a blood chloroquine concentration > 25 μmol/L are all regarded as predictors of a fatal outcome. Active removal methods such as haemoperfusion and haemodialysis are ineffective. Aggressive resuscitation with the use of intravenous noradrenaline (norepinephrine) and diazepam may be life-saving. After treatment, severe hyperkalaemia may ensue, particularly if potassium salts have been given to correct hypokalaemia.
Digoxin
Clinical features
Digoxin and other cardiac glycosides, including plant toxins from oleander (Nerium oleander) and yellow oleander (Thevetia peruviana), increase the force of contraction of the myocardium. Overdose increases the irritability of ventricular muscle, resulting in extrasystoles, ventricular tachycardia and fibrillation. Conduction is depressed and sinus bradycardia and various degrees of block may also occur. Since digoxin inhibits Na+,K+-ATPase, hyperkalaemia is a feature of digoxin poisoning. A plasma potassium concentration > 5.3 mmol/L (in the absence of any other cause of hyperkalaemia) suggests severe poisoning with an increased likelihood of cardiac toxicity. Occasionally, the plasma potassium concentration itself may rise rapidly to life-threatening levels and require urgent treatment.
Other clinical features of acute poisoning include headache, nausea, vomiting, abdominal discomfort, confusion, disorientation and visual blurring or distorted colour vision.
Management
Activated charcoal effectively adsorbs digoxin and should be used if the patient is not vomiting. Atropine may reverse bradycardia. Marked hyperkalaemia should be reversed with intravenous insulin and glucose. There is a concern that administration of intravenous calcium to counteract the effects of hyperkalaemia could worsen digoxin toxicity: however, observational studies do not demonstrate any adverse effects. Whenever there is evidence of severe poisoning such as cardiovascular collapse or refractory hyperkalaemia, digoxin-specific Fab antibody fragments should be obtained urgently. Given intravenously in an appropriate dose, this treatment should produce reversal of signs of toxicity within 20–30 min. These Fab antibodies interfere with commonly available assays for digoxin, so measurements of serum digoxin concentration after treatment will not be reliable.
Iron
Toxicity
Iron absorption is normally regulated by the intestinal mucosa. In overdose, iron-containing medication is corrosive and damages the mucosa, so that iron ions are absorbed in toxic amounts. When the plasma iron exceeds the iron binding capacity of transferrin, unbound iron circulates freely and may damage the liver, kidneys, cardiovascular system and central nervous system, leading to multi-organ failure.
Toxic doses in terms of elemental iron content are:
• mild-to-moderate poisoning: > 20 mg/kg
• severe poisoning: > 75 mg/kg
• lethal dose: > 150 mg/kg.
Clinical features
The clinical course of iron poisoning can conveniently be divided into four phases. During the first 6 h there may be vomiting, abdominal pain and diarrhoea, resulting from direct irritation of the gastrointestinal mucosa. The vomitus or stools may be dark or blood stained and may smell metallic. Severe fluid loss can lead to lethargy, convulsions, coma, metabolic acidosis and shock. Leukocytosis (> 15 × 109/L) and hyperglycaemia (> 8.3 mmol/L) indicate severe poisoning. This phase usually resolves after 6 h and the patient is often relatively well for the next 18 h. By about 24 h, the patient’s condition may worsen: this indicates severe poisoning. Features in this third phase include lethargy, coma, convulsions, cardiovascular collapse, metabolic acidosis, hypoglycaemia and renal and hepatic failure with coagulation abnormalities. Finally, a fourth phase of poisoning may follow the acute episode; it consists of stricture formation that may lead to small bowel obstruction 2–5 weeks after poisoning.
Analysis
Blood should be taken 2–6 h after ingestion. Measurement of serum iron concentration after 6 h may underestimate the amount of free iron because of distribution into tissues. Serum iron concentrations in excess of total iron binding capacity indicate severe poisoning, but the estimation of iron binding capacity may be unreliable in overdose because of the shortcomings of the methods used. The serum iron concentration at 4 h after ingestion is the most reliable guide:
• < 3 mg/L (55 μmol/L): mild toxicity
• 3–5 mg/L (55–90 μmol/L): moderate toxicity
• > 5 mg/L (> 90 μmol/L): severe toxicity.
Treatment
Chelation therapy
Desferrioxamine binds free iron and the non-toxic complex is excreted by the kidneys. The decision to use it should be based upon the patient’s clinical condition and on laboratory analysis. Once parenteral desferrioxamine has been given, colorimetric assay methods for iron are misleading because they measure both free and chelated iron.
Other metals
Poisoning with lead, mercury, bismuth, thallium or arsenic is uncommon, but may arise from deliberate, accidental or occupational exposure. Lead poisoning in children may be due to pica or follow the use of surma eye make-up in Asian children; in adults it is usually the result of occupational exposure.
The characteristic features of lead toxicity are anaemia with punctate basophilia of the red cells, peripheral muscle weakness, abdominal pain, colic and constipation, and encephalopathy if concentrations are high. Chronic poisoning may produce learning difficulties in children. Chelating agents such as 2,3-dimercaptosuccinic acid (DMSA) and 2,3-dimercaptopropane sulfonate (DMPS) are more effective than the traditional sodium calcium edetate and other chelating agents. DMSA and DMPS are water-soluble analogues of dimercaprol. DMSA is more effective in chelating lead, while DMPS can be used for mercury. Important advantages of DMSA and DMPS over the older chelating agents are that they can be given orally as an outpatient treatment and do not interfere with zinc and copper metabolism.
Orthopaedic implants, such as hip replacements, may contain cobalt and chromium metal. If the implant is functioning well, blood metal concentrations will be very low. However, elevated concentrations > 119 nmol/L (cobalt) or > 134.5 nmol/L (chromium) are associated with a failing implant and may lead to local tissue reactions, with malignant tumours being described in case reports. In the UK, the Medicines and Healthcare Products Regulatory Agency has issued guidance about the follow-up of patients who have specific types of hip implants. They recommend at least annual assessment of patients during the life of the implant, with more frequent assessment if whole blood concentrations of either chromium or cobalt are above the defined limits. It is unclear if elevated blood cobalt or chromium concentrations produce systemic toxicity in the setting of a failing hip replacement. However, hip revision should be considered if they are persistently elevated.
Organophosphates
Organophosphate poisoning is a major public health issue worldwide resulting in 200 000 deaths each year. Most of these occur through ingestion with suicidal intent. In developed countries, however, the number of poisonings is low and deaths are very rare. The most serious poisoning occurs by ingestion; cutaneous absorption and inhalation of sprays rarely cause serious toxicity.
Toxicity
The acute toxicity of these compounds is due to the inhibition of the enzyme acetylcholinesterase by phosphorylation, resulting in an accumulation of acetylcholine at postganglionic parasympathetic nerve endings (muscarinic receptors), parasympathetic ganglia (nicotinic receptors) and neuromuscular junctions (nicotinic receptors). All the organophosphates inhibit both red cell acetylcholinesterase and plasma cholinesterase (pseudocholinesterase) and this provides the basis for biological monitoring of toxicity (red cell measurements being preferred for acute toxicity). For monitoring of occupational exposure, regular measurement of plasma cholinesterase activity should be carried out: a reduction of the pre-employment value by 30% indicates excessive exposure and the worker should be removed from exposure pending recovery of enzyme activity.
Phosphorylated acetylcholinesterase is relatively stable, which means that the features of poisoning may persist longer than the presence of the organophosphate in the bloodstream. Spontaneous reactivation of the enzyme depends mainly on the chemical structure of the organophosphate. Exposure to compounds such as dimethylphosphates and dimethylphosphorothioates leads to dealkylation of the enzyme (a process referred to as ‘ageing’), which makes the enzyme inaccessible to reactivation, either spontaneously or by administration of reactivating agents such as pralidoxime. In poisoning by carbamates, the affected acetylcholinesterase undergoes rapid spontaneous reactivation.
Clinical features and management
Early symptoms of acute exposure to organophosphates are non-specific but lead on to more characteristic features. In mild-to-moderate poisoning there may be headache, blurred vision, miosis, excessive salivation, lacrimation, sweating, wheezing and lethargy. The patient should be kept under observation for at least 24 h. Severe poisoning may cause coma, convulsions, respiratory muscle paralysis, bradycardia and hypotension.
The first step is to maintain a clear airway and ensure adequate ventilation, after which atropine should be given until atropinization is achieved, that is, the heart rate is > 80/min, secretions are inhibited or pupils are enlarged.
Pralidoxime (a specific cholinesterase reactivator) should ideally be started within 4 h of exposure. In patients who present late to hospital, enzyme inactivation becomes less reversible and pralidoxime is unlikely to have any effect if given after 24–36 h.
The patient may relapse after apparent recovery, as a result of an acute myopathy that is distinct from the acute toxicity or the delayed neuropathy that may occur after several weeks. This has been called the ‘intermediate syndrome’.
Alcohols and glycols
One of the important features about alcohols from the clinical chemistry point of view is that they all cause a raised osmolar gap; comparison of measured and calculated osmolality can be used for the early confirmation of the potential severity of poisoning by alcohols and glycols.
Ethanol (ethyl alcohol)
Ethanol is probably the best known of all toxins. There are 100 deaths per year in England and Wales from acute alcohol toxicity, but the numbers of deaths from other causes, including road accidents and accidents in the workplace and also from chronic hepatic damage and cancers, is considerably greater than this, with the total toll being over 25 000 deaths each year. When alcohol is taken acutely, effects on behaviour can be demonstrated at blood concentrations as low as 200 mg/L (4.3 mmol/L). Intellectual performance, judgement and coordination are progressively impaired at increasing blood concentrations. The legal limit in the UK for being in charge of a motor vehicle is 800 mg/L (17.4 mmol/L). By 1600 mg/L (35 mmol/L), most people have marked impairment of coordination and are obviously drunk. However, tolerant individuals may have a blood alcohol concentration of > 5000 mg/L (110 mmol/L), without apparent behavioural impairment. By contrast, death from acute toxicity may occur in non-tolerant individuals with blood ethanol concentrations in the range 2000–3000 mg/L (43–65 mmol/L). A fatal outcome from acute alcohol toxicity may result from cardiac or respiratory depression or from aspiration of vomit.
The effects of ethanol are increased by sedative and hypnotic agents, and deep coma may occur in a patient who has taken a combination of a relatively small amount of alcohol plus a small overdose of a benzodiazepine. The clinical assessment of a patient with a severe head injury may be confused by alcohol intoxication. In children, convulsions and hypoglycaemia may result from acute alcohol intoxication. Alcohol potentiates rebound hypoglycaemia and may also cause marked hypoglycaemia in adults. Alcoholic ketoacidosis may follow bouts of heavy drinking (see Chapter 5).
Dilute ethanol enhances gastric emptying, but high concentrations (e.g. at the strength found in spirits) may cause gastric irritation and can delay stomach emptying. In rare cases, haemodialysis may be indicated to remove ethanol from the bloodstream, but supportive care is usually sufficient.
Heavy ingestion of alcohol may lead to the clinical picture of alcoholic hepatitis, with fever, leukocytosis and hepatomegaly. More chronic ingestion can lead to hypertriglyceridaemia; hyperuricaemia is also common in chronic heavy alcohol drinkers. A pseudo-Cushing syndrome occurs in a proportion of alcoholics and may be accompanied by an alcoholic myopathy. The commonest pathological sequel of long-term alcohol abuse is alcoholic cirrhosis, which is accompanied by typical physical signs and the biochemical changes of hepatic impairment. Acute bleeding may occur from ruptured oesophageal varices. The most important tests for detecting chronic abuse, apart from the detection of alcohol in a morning blood or urine sample, are plasma γ-glutamyltransferase activity and carbohydrate deficient transferrin concentration (see Chapter 13).
The alcohol-dependent patient who discontinues drinking because of an accident or a hospital admission may develop the alcohol withdrawal syndrome, in which there is a risk of convulsions and death. Initial irritability may develop into frank hallucinations and infusion of a sedative drug such as clomethiazole may be needed.
Methanol (methyl alcohol)
Methanol is a common constituent of car engine antifreezes. Model aircraft fuel also contains methanol, as do some screenwashes, varnishes and thinners. As little as 10 mL of pure methanol in a child and 50 mL in an adult can be fatal, causing profound metabolic acidosis, coma, convulsions and blindness.
Plasma osmolality, acid–base status and renal function should be checked in all patients with methanol ingestion. Direct measurement of blood methanol concentration is preferable to measurement of plasma osmolality (which is a surrogate indicator), but may not be available in a clinically useful timeframe. If plasma osmolality is normal and there is no acidosis (normal osmolal and anion gap), treatment is not required. Enzymatic metabolism of methanol to toxic metabolites can be prevented by either fomepizole (4-methyl pyrazole) or ethyl alcohol administration. Fluid administration may be required to maintain an adequate urine output for elimination of methanol. Acidosis should be corrected with intravenous sodium bicarbonate. Haemodialysis is indicated if visual disturbance is present, there are features of CNS toxicity or severe metabolic acidosis or if the patient develops acute kidney injury.
Ethylene glycol
Ethylene glycol is a common constituent of automobile antifreezes. If ingested, it tends initially to cause signs of alcoholic intoxication followed later by tachycardia, pulmonary oedema, convulsions and then acute kidney injury (see Fig. 40.5). Metabolic acidosis, leukocytosis, hypocalcaemia and crystalluria are characteristic, owing mainly to the metabolic production of oxalate, which then combines with calcium. Oxalate deposition may lead to meningism and acute tubular injury. The presence of calcium oxalate crystals in the urine is typical and is a useful diagnostic feature.
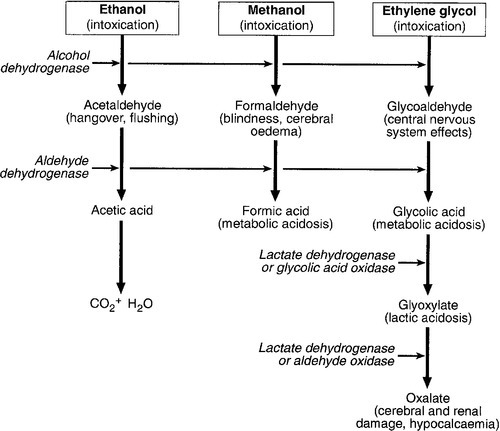
FIGURE 40.5 Metabolism and clinical effects of ethanol, methanol and ethylene glycol. Clinical effects are given in brackets; enzymes are in italics. The higher affinity of ethanol for alcohol dehydrogenase is the rationale for its use as an antidote in poisoning with other alcohols. Fomepizole also inhibits metabolism of alcohols by its high affinity for alcohol dehydrogenase.
The main principles of treatment are as for methanol poisoning: to delay the metabolism of ethylene glycol to toxic metabolites by administering fomepizole or ethyl alcohol, to correct the metabolic acidosis and to hasten the elimination of ethylene glycol by increasing urine output or, in severe cases, by using haemodialysis. In addition, plasma calcium concentration should be monitored (hypocalcaemia should be corrected if it occurs), as should renal function (acute kidney injury may require haemodialysis).
Drug and substance abuse
Many drugs and chemicals are widely abused and some cause serious medical complications. There are approximately 600 deaths from opioid abuse, 150 from cocaine and 30 deaths from volatile substance abuse each year in the UK and, although other abused substances cause fewer deaths, they remain an important cause of morbidity.
Apart from opioid toxicity, the management of poisoning with this group of agents is mainly symptomatic. Confirmation of exposure is often necessary for diagnostic or medicolegal reasons and Table 40.5 gives a guide to the typical times during which urine tests remain positive. Immunologically based point-of-care urine tests are commonly used for diagnosis of poisoning by drugs such as opioids, cocaine, cannabis, amfetamine and metamfetamine (usually includes 3,4-methylenedioxymetamfetamine, MDMA, see below). Tests are also available for benzodiazepines, methadone (which is structurally different to other opioids and does not give a positive result in tests for opioids), and phencyclidine (ketamine will cross-react in this test).
TABLE 40.5
Duration of positive results in urine after a typical dose taken by a drug abuser
| Substance | Limit of detection | Time of detection |
| Amfetamine | 0.25 mg/L | 1–2 days |
| Barbiturates | 0.5 mg/L | 1–3 days (longer for phenobarbital) |
| Cannabis | 50–300 μg/L | 2 days to 3 weeks (depends on usage and limit set) |
| Cocaine | 0.3 mg/L | 2–3 days |
| Codeine | 0.25 mg/L | 1–2 days |
| Dihydrocodeine | 0.25 mg/L | 1–2 days |
| LSD | 0.5 mg/L | 2–3 days |
| MDMA (‘Ecstasy’) | 0.1 mg/L | 1–2 days |
| Methadone | 0.25 mg/L | 2–5 days |
| Morphine | 0.5 mg/L | 1–2 days |
| Propoxyphene | 0.3 mg/L | 2–4 days |
LSD, lysergic acid diethylamide; MDMA, 3,4-methylenedioxymetamfetamine.
Amfetamines
The amfetamines are frequently abused for their stimulant effects. Amfetamine sulphate may be injected, inhaled or taken orally. Methylamfetamine (‘crystal meth’ or ‘ice’) comes in the form of crystals that can be smoked and is about 20 times more potent than amfetamine sulphate, but has similar effects.
The main complications of amfetamine toxicity include agitation, convulsions, cardiac arrhythmias, hyperthermia and myoglobinuric acute kidney injury, which can be aggravated by volume depletion. There is no specific pharmacological antidote and management is generally supportive. Sedation and anticonvulsants will be needed for the patient who is agitated or fitting, and maintaining an acidic urine markedly increases the excretion of amfetamine and shortens its elimination half-life. However, if there is a suspicion of rhabdomyolysis, for example following repeated seizures, or if the plasma creatine kinase activity is raised or myoglobinuria is present, then alkalinization of the urine is recommended to prevent myoglobinuric kidney injury.
3,4-Methylenedioxymetamfetamine (MDMA)
3,4-Methylenedioxymetamfetamine, also known as ‘Ecstasy’ or ‘E’, is an amfetamine derivative with different properties from amfetamine; it typically produces euphoria, empathy and a subjective sense of increased energy, which is the reason why it has become popular as a ‘dance drug’. First synthesized in 1914, it was briefly used as a mood-modifying agent, but was banned in the UK in 1977. In the usual doses (tablets or capsules contain 30–150 mg; users take between half and five orally during the course of an evening), it has few adverse effects in the majority of people. The commonest effects are trismus, tachycardia, sweating and agitation. A small proportion of users develop muscle pain and stiffness, which may persist for a week. When the drug is taken before or during strenuous exercise such as dancing, it may lead, in rare cases, to collapse, convulsions and acute hyperthermia, rapidly followed by disseminated intravascular coagulation and rhabdomyolysis. Deaths have occurred after a single dose.
The reason for collapse is related to the combination of physical hyperactivity and inadequate fluid replacement, but it is not known why only a very small number of individuals are affected. Management initially should be directed to restoring fluid volume and reducing body temperature. Intravenous dantrolene may be effective. Ingestion of MDMA may also produce a serotonin syndrome, which is characterized by a triad of altered mental status (agitation, confusion), neuromuscular hyperactivity (clonus) and autonomic instability (hyperthermia, tachycardia). In order to prevent the hyperthermia becoming fatal, paralysis and ventilation may be required. Another complication of MDMA use is hyponatraemia, which usually occurs when excess fluid has been taken but there has been insufficient exercise to sweat off the fluid. This occurs because MDMA causes serotonin production, which in turn causes release of excess arginine vasopressin (antidiuretic hormone) from the posterior pituitary. The patient is likely to be confused and may have convulsions, but has a normal body temperature. Plasma sodium will be ≤ 125 mmol/L. The most important step in management is to avoid giving any fluids; most patients will recover spontaneously.
Although MDMA is taken because it causes the release in the nervous system of large amounts of serotonin, which is one of the neurotransmitters responsible for the ‘high’ caused by the drug, this leads to depletion of serotonin and a ‘midweek low’ is a well-known consequence of MDMA consumption. Serotonin neurotoxicity has been demonstrated in animals, and a reduction in cognitive abilities, particularly memory, has been demonstrated in humans following chronic intermittent use.
Heroin (diamorphine)
Heroin abuse is common in the UK, causing over 500 deaths per year. Many of these are from respiratory failure, which may occur after the first use of the drug. Experienced users may die from acute respiratory depression or aspiration of vomit following loss of tolerance after a period of abstinence. Tolerance is rapidly lost; the dose requirement may fall by 10-fold over a period of 2–3 days of abstinence, so that the dose which previously produced euphoria may prove fatal.
The classic signs of opioid toxicity consist of depressed consciousness, which may amount to profound coma, small or ‘pinpoint’ pupils (pethidine is an exception to this and the atropine in Lomotil®, a mixture of diphenoxylate and atropine, may cause dilated pupils) and respiratory depression. The pattern of opioid induced respiratory depression is characteristic and consists of a slowing of respiration, whereas other respiratory depressants tend to decrease the depth of respiration with less effect on the rate. An adequate dose of naloxone should reverse these symptoms; an initial dose may only produce a minimal or partial response and further doses may be required. The presence of other central nervous depressant drugs or other causes of coma may mask the response to naloxone.
Once the diagnosis of opioid toxicity has been confirmed by administration of naloxone, the choice can be made as to whether to continue reversal with an infusion of naloxone sufficient to maintain adequate spontaneous respiration or whether to ventilate the patient until the drug is metabolized. Diamorphine has a short elimination half-life of 2–4 h, so that treatment may not need to be prolonged. Other opioid drugs such as buprenorphine, dihydrocodeine, methadone and dextropropoxyphene have considerably longer elimination half-lives and a longer period of recovery should be anticipated.
Complications of heroin overdose include a chemical pneumonitis owing to aspiration of vomit, non-cardiogenic pulmonary oedema and non-traumatic rhabdomyolysis. The question of provoking heroin withdrawal symptoms in an addict by administration of naloxone is often raised; opioid withdrawal is a non-fatal condition, which is, in any case, short-lived because of the short duration of effect of a single dose of naloxone (10–30 min).
Lysergic acid diethylamide (LSD)
Lysergic acid diethylamide is a synthetic hallucinogen, which may lead to a patient being admitted to an intensive therapy unit because of behavioural disturbances, accidents or suicide attempts. There is no specific antidote.
Cocaine
The hydrochloride is typically snorted through a straw or a rolled up bank note, while the free base form, in small crystals or ‘rocks’, is drawn directly into the lungs by flaming a rock with a cigarette lighter. Cocaine produces marked vasospasm and hypertension, and patients most frequently present to emergency departments with chest pain. They may also present with confusion, convulsions, myocardial infarction, acute heart failure or cerebral haemorrhage. There is no specific antidote, but intravenous diazepam relieves the chest pain and reduces the risk of convulsions. After this, blood pressure may be controlled with agents such as intravenous nitrates. Other management is supportive.
Cannabis
Tetrahydrocannabinol, the active ingredient of the cannabis plant, combines with receptors in the brain and periphery to cause mental effects together with vasodilatation and tachycardia. Effects last a few hours, but the drug can persist in tissues for a long time. Acute overdose is unlikely to be fatal, but heavy use is cumulative and can lead to agitation, hallucinations and paranoid behaviour.
The main psychoactive component of cannabis, Δ9-tetrahydrocannabinol (THC), reaches peak concentration in the blood within minutes of smoking, but rapid absorption by fatty tissues then leads to a rapid fall. However, one of the main metabolites of cannabis, 11-nor-Δ9-tetrahydrocannabinol carboxylic acid (carboxy-THC), which is detectable in the blood shortly after use, persists in the blood for a week or more. Immunoassays do not distinguish between active THC and its inactive metabolites. For this reason, blood or urine tests may remain positive for a long time after cannabis consumption, and cannot be taken to indicate intoxication.
Solvents
Volatile substance abuse (solvent abuse, ‘glue sniffing’) is relatively common in teenagers. Most obtain a sensation of intoxication within seconds of inhalation and come to little harm. The commonest substance abused is toluene. However, if the dose is too high, coma and convulsions may ensue and inhalation of vomit is a possibility. Since volatile substances (particularly chlorinated hydrocarbons) sensitize the myocardium to catecholamines, cardiac arrhythmias may occur and cause a fatal outcome. Butane from cigarette lighter refills may be inhaled directly; the gas may cause asphyxia or its cooling effect may cause cardiac arrest through vagal inhibition.
Chronic complications of solvent abuse are unusual, but brain, liver and kidney damage have all been reported. Carcinogenesis has not been documented. Management is generally symptomatic; cardiac arrhythmias may respond to an intravenous dose of a β-blocker.
Benzodiazepines
The benzodiazepines are widely thought to be non-fatal in overdose, but about 200 deaths occur every year in the UK from respiratory depression, aspiration of vomit or hypothermia. Usually such deaths occur when benzodiazepines are combined with other agents such as ethanol. In most patients with an uncomplicated benzodiazepine overdose, the coma amounts to a deep sleep with an adequate gag reflex and preserved tendon reflexes and response to painful stimuli.
Patients rarely need management in an intensive therapy unit, but sometimes the drug has been co-ingested with ethanol, opioids, barbiturates or tricyclic antidepressants. The question may arise as to whether flumazenil should be given as a therapeutic or diagnostic test. Reversal of benzodiazepine toxicity could provoke convulsions due to simultaneously ingested tricyclic antidepressants. In general, it should only be used in carefully considered circumstances.
Theophylline
Clinical features
Most oral preparations of theophylline are sustained- release formulations. The patient may be asymptomatic on presentation. Symptoms usually take several hours to develop and include vomiting, abdominal pain, haematemesis, irritability and hyperventilation. A sinus tachycardia is usual and there may be hypotension, cardiac arrhythmias (most commonly supraventricular tachycardia), convulsions, hypokalaemia and hyperglycaemia. Coma is not common. Rhabdomyolysis and acute kidney injury are rare complications.
Management
Activated charcoal in repeated doses is the treatment of choice for severe poisoning. This is indicated if the patient is clinically severely poisoned and/or has a plasma theophylline concentration > 40 mg/L. In life-threatening poisoning, charcoal haemoperfusion can be used, if available.
Antidepressants
Tricyclic antidepressants
Acute overdose with tricyclic antidepressants is the commonest cause of poisoning admissions to intensive therapy units in the UK. The mechanisms of toxicity are complex and are due to at least three pharmacological effects:
• an anticholinergic effect, which causes delayed gastric emptying, sinus tachycardia and mydriasis
• blockade of noradrenaline uptake at adrenergic synapses, which may produce hypotension
• a quinidine-like or membrane-stabilizing effect. In low doses, this last effect is antiarrhythmic, but in overdose, it delays conduction and depresses contractility. The quinidine-like effect slows sodium flux into cells and is the mechanism underlying cardiac toxicity. It may be aggravated by hypoxaemia (Fig. 40.6).
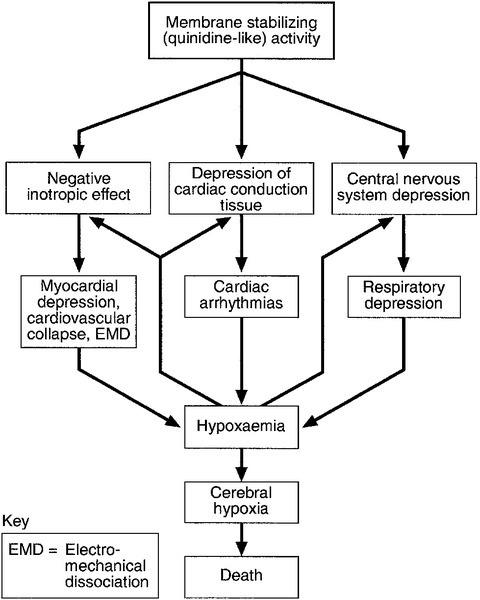
FIGURE 40.6 Mechanisms by which the membrane-stabilizing activity of drugs can cause death.
Clinical features
There may be initial nausea, vomiting, agitation and hallucinations, giving way to coma. Tendon reflexes may be equal and very brisk, with extensor plantar reflexes; as coma deepens, muscle tone becomes flaccid and reflexes are lost. The pupils are more often mid-sized than widely dilated. Convulsions are common and respiratory depression may occur.
The most serious and potentially fatal complications are cardiac arrhythmias (most commonly atrioventricular block, ventricular tachycardia or ventricular fibrillation, which may be followed by asystole) and profound hypotension, which may amount to electromechanical dissociation. Pulmonary oedema may occur. The electrocardiogram (ECG) shows a sinus tachycardia and there may be a right bundle branch block pattern. A QRS duration > 100 ms is regarded as the best indicator of risk of cardiac toxicity.
Management
Cardiopulmonary status must be assessed urgently and resuscitation commenced if necessary. Poisoning is potentially reversible, provided hypoxic cerebral damage has not occurred. In significant poisoning, the most important treatment is alkalinization with intravenous sodium bicarbonate, aiming for a blood [H+] of 32 nmol/L (pH 7.5). This is indicated even in the absence of acidosis when there is prolongation of the QRS duration on ECG, there are cardiac arrhythmias or resistant hypotension. Seizures should be treated with benzodiazepines. If recurrent, phenobarbital or thiopentone may be needed in a critical care setting. Phenytoin is contraindicated as both tricyclics and phenytoin block cardiac sodium channels. In life-threatening poisoning, lipid emulsion (e.g. intralipid) therapy may be considered; this acts as a lipid reservoir in the circulation that may ‘trap’ the drug and reduce the concentration at the receptor.
Monoamine oxidase inhibitors
Toxicity
Several drugs and foods are prohibited for patients taking monoamine oxidase inhibitors to prevent the well-known ‘cheese’ reaction, which consists of a sudden and severe rise in blood pressure owing to tyramine-provoked release of noradrenaline.
Overdose of monoamine oxidase inhibitors produces symptoms that build up over 12–24 h, with muscle twitching progressing to widespread muscle spasms, trismus and opisthotonos. The blood pressure may vary between hypotension and moderate hypertension; there is usually a sinus tachycardia and the patient is warm to the touch, sweating profusely and has fixed dilated pupils. The core temperature may rise steeply, leading to death from hyperthermia. The muscle spasms may lead to rhabdomyolysis, which can cause acute kidney injury. Disseminated intravascular coagulation may occur as a complication of hyperthermia.
Management
If the rectal temperature rises above 39 °C the patient should be electively paralysed with pancuronium and mechanically ventilated for 12–24 h both to reduce heat generation through muscle spasm (and thus correct hyperthermia) and prevent rhabdomyolysis. Hypotension is usually secondary to hypovolaemia, but dopamine may be tried if fluid replacement fails to restore blood pressure.
Other antidepressants
Lofepramine
This tricyclic drug is metabolized to desipramine, but toxicity in overdose is usually relatively mild and deaths are rare.
Trazodone
This drug is chemically unrelated to the tricyclic antidepressants. Toxicity is usually mild, with drowsiness, dizziness and occasionally coma.
Venlafaxine
This drug is less cardiotoxic than the tricyclic antidepressants, but may cause convulsions in overdose.
Fluvoxamine, fluoxetine, sertraline, paroxetine
These serotonin reuptake inhibitors rarely cause serious toxicity in overdose, though a mild serotonin syndrome may occur, and they can also interact with other drugs to produce a serotonin syndrome. Management is symptomatic.
Citalopram
This serotonin reuptake inhibitor is more cardiotoxic than others, producing QT interval prolongation.
Lithium
Toxicity
Lithium is eliminated by the kidneys and reduced renal function can lead to accumulation. The therapeutic range is narrow (plasma concentration 0.8–1.2 mmol/L in the management of hypomania or mania in bipolar affective disorder; 0.4–1.0 mmol/L for prophylaxis: note that blood should not be collected into a lithium heparin tube). Toxicity in therapeutic use can be caused by changes in fluid or electrolyte balance (particularly owing to reduction in fluid intake or increased fluid loss from diarrhoea or vomiting). Diuretic therapy or non-steroidal anti-inflammatory drugs may also cause toxicity by reducing lithium excretion.
Clinical features
Symptoms of toxicity include confusion, agitation, drowsiness, tremor, hyper-reflexia, hypertonia, ataxia, vomiting, convulsions and, rarely, electrocardiographic changes, diabetes insipidus and acute kidney injury. In acute poisoning, serum concentrations of > 5 mmol/L may be associated with minimal symptoms and the elimination half-life is relatively rapid. The development of marked neurological symptoms is an indication for active elimination. Deaths from lithium poisoning are rare, but neurological impairment may be permanent.
Management
Lithium is not adsorbed by activated charcoal but is readily excreted by the kidneys and a high urine output should be ensured. Saline diuresis enhances lithium excretion but urine alkalinization does not.
In a symptomatic patient with high serum lithium concentrations after an acute overdose, haemodialysis should be considered. Although efficiently removed by haemodialysis, lithium has a large volume of distribution leading to a ‘rebound’ rise in plasma concentrations and repeated dialyses may be necessary. Peritoneal dialysis is much less effective.
Cyanide
Cyanide can be rapidly fatal. It may be inhaled as hydrogen cyanide (either pure, as an industrial gas or as a product of combustion in cases of smoke inhalation) or enter the body by ingestion of cyanide salts or intestinal hydrolysis of cyanogenic glycosides (e.g. following massive ingestion of stone fruit kernels or apple pips) or by cutaneous absorption of cyanide salts in industrial situations.
Cyanide acts as a chemical asphyxiant, rapidly blocking cellular oxygen utilization so that cerebral function and circulation are rapidly impaired with the development of a metabolic acidosis. Early signs include hyperventilation and tachycardia, but coma, cyanosis and convulsions soon supervene. Rapid diagnosis and intervention are essential. A high inspired oxygen content (or hyperbaric oxygen, if available) is an effective treatment, though its mode of action is unclear, perhaps being due to simple displacement of cyanide. When the diagnosis is certain, dicobalt edetate can be given intravenously over 1 min, repeated as necessary, and is an effective antidote. Although relatively non-toxic when administered to a patient poisoned with cyanide, this antidote can produce severe anaphylactoid reactions with laryngeal oedema and convulsions if given to a patient who is not poisoned by cyanide. The safest course of action is not to give the antidote unless the patient’s level of consciousness is deteriorating. Another group of antidotes (amyl nitrite and sodium nitrite) act by producing methaemoglobin, each molecule of which can bind four cyanide ions. Sodium thiosulphate acts to neutralize cyanide by donating a sulphur group to produce sodium thiocyanate, in a reaction that is catalysed by rhodanese.
Subacute or chronic cyanide toxicity can also occur during prolonged nitroprusside therapy. The main clinical feature is a lactic acidosis – if none is present then cyanide toxicity can be ruled out. Cyanide measurement is not usually necessary: a rapid improvement should occur after administration of sodium thiosulphate intravenously. Other cyanide antidotes are not indicated.
Carbon monoxide
Toxicity
Carbon monoxide is a colourless, odourless gas produced by combustion of carbon-containing compounds. It has a high affinity for haemoglobin and cytochrome enzymes, and is highly toxic because it acts as a chemical asphyxiant. It accounts for around 50 deaths each year in the UK, which may be suicidal from motor vehicle exhaust fumes, or accidental, mainly from release of products of combustion (of gas, solid or liquid fuels) in the home or exposure to fire smoke.
As little as 0.1% carbon monoxide in the inspired air can be fatal over a period of several hours, while 1% can be fatal in minutes. Toxicity is due mainly to combination of carbon monoxide with haemoglobin to form carboxyhaemoglobin, but is also due to interference with the cellular uptake of oxygen as a result of its combining with cytochrome enzymes. Carboxyhaemoglobin in the blood has a half-life of about 4 h if the patient is breathing air. The half-life is markedly reduced (to 1 h) if the patient is breathing 100% oxygen and is still further reduced (to 20 min) if the patient is breathing oxygen in a hyperbaric chamber at 2.5 atmospheres, but the effectiveness of hyperbaric oxygen in diminishing the toxicity of carbon monoxide is hotly debated.
Clinical features
The symptoms of exposure are non-specific: lethargy, nausea, headache, drowsiness, hyperventilation, leading to vomiting, collapse, coma and convulsions. Elderly people may present with stroke or exacerbation of coronary heart disease precipitated by reduced oxygen-carrying capacity of the blood. Non-traumatic rhabdomyolysis may occur. If the source of exposure is not apparent, the diagnosis may also not be apparent. Once diagnosed or suspected, evidence should be sought, for example a malfunctioning flue from a fire or heating appliance, or motor exhaust fumes. A rare cause is exposure to methylene chloride, which is metabolized to carbon monoxide by hepatic smooth endoplasmic reticulum.
Specific physical signs are minimal; the ‘cherry-red’ colour of carboxyhaemoglobin is usually only noted at post-mortem. Diagnosis of exposure should be made by measuring carboxyhaemoglobin on a whole blood sample. Correlation of the percentage of carboxyhaemoglobin with clinical effects is poor and is complicated by the disappearance of carbon monoxide from the blood following exposure, while the clinical effects may remain for longer. However, normal values in non-smokers are < 1% and in smokers < 5%. Carbon monoxide is produced during the catabolism of tetrapyrroles and severe haemolysis in haemolytic anaemia can, rarely, give a carboxyhaemoglobin as high as 8%.
Management is by removal from exposure, resuscitation as necessary and immediate administration of a high inspired oxygen concentration. Blood should be taken at the earliest possible moment for carboxyhaemoglobin estimation. The role of hyperbaric oxygen therapy in carbon monoxide poisoning is controversial but should be considered in life-threatening poisoning, if the treatment is available.
Methaemoglobinaemia
Causes
The more important causes of life-threatening methaemoglobinaemia, likely to present with acute symptoms, are:
• sodium or potassium nitrite
• amyl or butyl nitrite
• sodium or potassium nitrate
• sodium or potassium chlorate.
Nitrites are more potent methaemoglobin formers than nitrates, but nitrate can be converted into nitrite by intestinal bacteria. Inhaled nitrites (amyl and butyl nitrites) produce little methaemoglobin unless the liquid is swallowed, when severe toxicity may occur. Many drugs, including sulphonamides, dapsone and local anaesthetics, can increase methaemoglobin production but are unlikely to produce concentrations exceeding 30%. It should also be noted that the cause may be iatrogenic, for example sodium nitrite given for cyanide poisoning.
Symptoms
At low concentrations, the patient may be deeply cyanosed but otherwise asymptomatic and require no treatment. At high concentrations, the patient may be dyspnoeic on exertion and have postural hypotension. At severely toxic concentrations, the patient becomes flaccid and comatose. Cardiac arrhythmias and convulsions may occur and may progress to cardiorespiratory arrest. Methaemoglobinaemia should be suspected if the skin has a blue or greyish, cyanosed appearance: the blood may be a dark or chocolate–brown colour. This colour change occurs with methaemoglobin concentrations of 15–20%, but clinical symptoms only appear at values > 20–30%, while consciousness is likely to be lost at > 50%. Death is common at > 70%.
Management
In addition to supportive measures, which should include 100% oxygen, methaemoglobinaemia may be reversed by the administration of intravenous methylthioninium chloride (methylene blue). It should only be given to symptomatic patients. When the cause is chlorate poisoning, methylene blue is unlikely to reverse the methaemoglobinaemia, although ascorbic acid can be given. In extreme cases, exchange transfusion can be used, though the transfused blood may also become affected and prolong the methaemoglobinaemia. Hyperbaric oxygen, if available, may allow sufficient oxygen to be transported in solution in the plasma to maintain life.
Plant and fungal toxins
Most plants are non-toxic and most childhood ingestions of plant material do not lead to toxicity. However, many plants contain chemical agents that are potentially poisonous when accidentally ingested by children or taken as food by adults; in addition, suicidal or homicidal ingestion may occur. Two examples have already been given in this chapter: Thevetia and oleander contain cardiac glycosides, and the seeds of a number of varieties contain cyanogenic glycosides, which may cause cyanide poisoning if sufficient seeds or kernels are ingested. Other potentially serious plant poisons are included in Table 40.6.
TABLE 40.6
Plants that cause potentially serious poisoning
| Plant | Toxic principle | Possibly fatal amount (in an adult) |
| Ackee (unripe fruit) | Hypoglycin | One fruit |
| Apple or pear seeds(Malus, Pyrus spp.) | Amygdalin (cyanogenic glycoside) | 100 seeds |
| Castor oil plant(Ricinus communis) | Ricin | Two beans |
| Death cap (Amanita phalloides) | Amanitin, phalloidin (cyclopeptides) | One mushroom |
| Galerina spp. mushrooms | Amanitin-type cyclopeptide | One mushroom |
| Gyromitra spp. mushrooms | Monomethylhydrazine | One mushroom |
| Holly (Ilex aquifolium) | Ilicin | 30 berries |
| Jequirity (Abrus precatorius) | Abrin | One bean |
| Oleander (Nerium oleander) | Oleandrin (cardiac glycoside) | One leaf |
| Stone fruit kernels (Prunus spp.) | Amygdalin (cyanogenic glycoside) | 30 kernels |
| Hemlock water dropwort(Oenanthe crocata) | Oenanthetoxin | Several leaves or one root |
| Yew (Taxus baccata) | Taxine A and B | 50 needles |
Acute fungal poisoning most commonly arises from eating toxic species in mistake for edible species. Many mushrooms can cause gastrointestinal symptoms shortly after ingestion, but most of these are not associated with serious poisoning; a rule of thumb is that mushroom poisonings with symptoms starting < 6 h after ingestion are unlikely to be due to highly toxic species. A few species, especially Coprinus atramentarius, can produce an Antabuse®-like reaction if ingested with alcohol, because they contain a chemical that inhibits aldehyde dehydrogenase.
The most important group of poisonous mushrooms includes those which contain cyclopeptides known as amanitins (Amanita phalloides, A. verna, A. virosa and some Galerina and Lepiota species). After an initial period of severe diarrhoea, beginning 6–12 h after ingestion, the patient may develop fulminant hepatic failure: a single mushroom can cause death and the mortality rate from ingestion is 20%. Another important and potentially fatally poisonous mushroom species is Gyromitra esculenta, which contains gyromitrin. If this chemical is not previously removed by evaporation or boiling, severe toxicity may result from the metabolic conversion of gyromitrin to monomethylhydrazine, which is a competitive inhibitor of pyridoxal phosphate. Symptoms start suddenly 6–12 h after ingestion and consist of headache, vomiting, diarrhoea and convulsions. Liver damage may also occur. The mushroom species Cortinarius contain the toxin orellanine that causes acute kidney injury, which may require renal replacement therapy, after an initial latent, symptomless, phase of several days.
CONCLUSION
This chapter outlines the causes, mechanisms and management of the more important types of human poisoning. Poisons – drugs, chemicals and plant and animal toxins – can disrupt the physiological and biochemical functions of the body in a remarkable variety of ways. Many individual toxins produce a characteristic clinical and biochemical pattern of harm (‘toxidrome’), which sometimes provides the key to the diagnosis. Management involves supportive care, decontamination and, in some cases, the use of specific antidotes. The clinical biochemistry laboratory may become involved in the identification and quantification of poisons and also in the monitoring of biochemical responses following poisoning; close communication between clinicians and biochemists is necessary for the efficient diagnosis and management of many types of poisoning.
ACKNOWLEDGEMENT
I would like to acknowledge the contribution of the late John A. Henry, who wrote the chapter for previous editions of this book.
Further reading
APPENDIX 40.1 POISONS CENTRES
TOXBASE, the primary clinical toxicology database of the National Poisons Information Service in the UK, is available on the internet to registered users at: www.toxbase.org. It provides information on routine diagnosis, treatment and management of patients exposed to drugs, household products and industrial and agricultural chemicals.
Specialist information and advice on the treatment of poisoning is available from the UK National Poisons Information Service by telephone day and night: Tel: 0844 892 0111 (July 2013).
Help with identifying capsules or tablets may be available from a regional medicines information centre.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 40
Poisoning
CHAPTER OUTLINE
INTRODUCTION
Poisoning can be defined as an interaction between a foreign chemical (toxin) and a biological system that results in damage to a living organism. In general, the medical profession has been more concerned with the acute effects of toxins and the clinical management of toxicity, but chronic effects of toxins have much more importance on a global scale. For example, acute ingestion of ethanol may result in intoxication, which can cause death directly from its acute depressant effects and also through intoxication-related accidents and violence. However, alcohol-related liver disease, myopathy, hypertriglyceridaemia and cancers are all effects of chronic alcohol abuse and together, cause far more deaths than the acute effects of alcohol. Other types of chronic or delayed effects of toxins include mutagenicity, carcinogenicity and teratogenicity. For example, many cases of bronchogenic carcinoma are a direct result of exposure to cigarette smoke. Increasing concerns about environmental contamination by chemicals has added a further dimension to toxicology, especially as many chemicals, such as pesticide residues, may be found in small amounts in the general population, who do not appear to be suffering any ill effects.
The interaction between a toxin and the biological system is highly complex. Once the biological system has been exposed to the toxin, the toxin is subject to various processes. In the case of a drug, this is usually termed pharmacokinetics, but in the case of a poison, the term toxicokinetics may be used. This includes absorption, for which the route of administration is of great importance, as are the dose of the toxin and the duration of exposure. Once a toxin has entered the bloodstream, its distribution depends to a large extent, on its physicochemical characteristics, including its degree of lipid or water solubility and its degree of ionization. Metabolism is a further important process, which usually renders toxins inactive, but which may also convert some substances of low toxicity into highly toxic metabolites. Classic examples of this include paracetamol (acetaminophen), methanol and ethylene glycol. Finally, toxins are excreted, although some remain within the body for many years. All of these processes determine the amount of toxin that interacts with tissues to produce a toxic effect.
The study of the interaction between toxins and tissues is called toxicodynamics. An understanding of the nature of this interaction is essential for an intelligent approach to the diagnosis and management of poisoning. This interaction may cause functional and metabolic disturbances that produce the characteristic clinical features of poisoning and the laboratory changes that may be essential for diagnosis or management. These changes are summarized in Figure 40.1.
FIGURE 40.1 The factors involved in poisoning.
This chapter covers several of the more common types of acute poisoning and also some aspects of chronic poisoning. Clinical features are given in some detail, as these frequently integrate with the metabolic and biochemical changes.
AETIOLOGY OF POISONING
Intrauterine
A number of drugs, such as tretinoin, should not be taken by women intending to become pregnant because of their known teratogenicity. Other drugs, such as phenytoin and several other anticonvulsants, are teratogenic but their continued use may be considered necessary during pregnancy. As far as poisoning and overdose during pregnancy are concerned, the fetus generally only suffers damage secondary to the mother’s illness, and the management is no different from that of overdose in non-pregnant women. One exception is carbon monoxide, which has a higher affinity for fetal haemoglobin than adult haemoglobin; the mother should be treated aggressively even in apparently mild poisoning. Antidotes such as acetylcysteine or desferrioxamine should not be withheld because the patient is pregnant.
Neonates
During the neonatal period, particularly in premature infants, there is a risk of iatrogenic overdose because of the poor metabolic and excretory capacity relative to body weight. Particular care should be taken with drugs such as theophylline, digoxin, chloramphenicol and morphine.
Infants
The commonest cause of poisoning between the ages of one and four is termed accidental poisoning because of the natural exploratory activities of children at this stage of life. Each year, this results in 20 000 admissions to hospital in Britain, but only about ten fatalities. The commonest agents involved are paracetamol and oral contraceptives, but most fatalities occur from tricyclic antidepressants, salicylates, iron, methadone and quinine. Another form of poisoning in this age group is non-accidental injury, which is usually inflicted by a carer such as the mother, and the child may present with repeated episodes of floppiness and collapse, depending on the substance responsible. This is most commonly with a drug prescribed for the mother, though salt and other readily available chemicals have been used (see Chapter 44).
Childhood
Later in childhood, substance abuse, particularly volatile substance abuse (‘glue sniffing’), can cause serious problems. There are about 30 deaths each year in Britain from this cause. Acute and chronic alcohol toxicity are often underestimated in this age group. Intentional drug overdose is common, but suicide is rare.
Adult life
In early adult life, particularly in females, drug overdose in the form of parasuicidal gestures is common, though rarely fatal. In later adult life, suicidal intent is commoner and parasuicidal gestures are less frequent. The commonest causes of suicidal poisoning in England are tricyclic antidepressants, analgesic drugs and carbon monoxide. Male suicides are approximately three times as frequent as female suicides. Accidental poisoning in adults is usually domestic, the commonest agent being carbon monoxide, or industrial, in which a wide range of chemicals may be involved. Homicidal poisoning, though rare, may go undetected if it is not suspected.
TYPES OF LESION IN POISONING
Many types of poisoning involve a highly specific interaction between the toxin and one type of tissue. This has led to a ‘target organ’ oriented approach in toxicology, which is of great help in understanding poisoning, where damage to a single organ or tissue produces a characteristic clinical and pathological picture. Examples include poisoning with paracetamol, salicylates, cholinesterase inhibitors and cardiac glycosides. In other instances, the type of toxicity may be less clearly definable. The acute effects of ethanol are mainly due to its central nervous depressant effect. Cocaine, tricyclic antidepressants, many volatile substances, some β-adrenergic blocking drugs and dextropropoxyphene can all produce cardiotoxicity due to a quinidine-like sodium channel blocking effect. However, the clinical features are definable as a toxic effect on the heart.
An important way in which many poisons can affect the organism is by interference with the oxygen pathway from the inspired air to cellular respiration. Thus, a reduction in the oxygen content of the inspired air may cause acute hypoxaemia, rapidly leading to collapse and coma. Interference with the mechanics of respiration by poisons may result in hypoxaemia and hypercapnia (type II respiratory failure), while disturbances of oxygen transfer produce hypoxaemia alone (type I respiratory failure). Even if the oxygen content of the air is not diminished and respiration is functioning adequately, disturbances to other processes may prevent oxygen reaching its site of action (see Chapter 5). The oxygen-carrying capacity of the blood may be reduced by the presence of carboxyhaemoglobin or methaemoglobin or, more rarely, by acute haemolysis. Cardiac output may be reduced by a number of poisons that cause cardiac arrhythmias, depress the contractility of the heart or cause extreme vasodilatation. The final step where poisons may interfere with the oxygen pathway is blockage of the cytochrome enzyme chain as occurs with toxins such as cyanide and hydrogen sulphide. These effects and the main poisons involved are summarized and outlined in Table 40.1.
TABLE 40.1
Some typical causes of blockade of the oxygen pathway due to poisoning, with mechanisms and typical values
Note that whatever the initial effect of a poison, there may be subsequent progression down the list of effects towards cell death. The effects of two or more poisons may be additive
| Cause | Mechanism | Effect |
| Asphyxiant gases (e.g. butane, methane, carbon dioxide, nitrogen) | Hypoxic gas mixture | Reduced inspired oxygen fraction (FiO2) (normal ~ 0.21) |
| Respiratory depression (e.g. opioids, barbiturates, other sedatives and hypnotics). Respiratory muscle disorders: paralysis (e.g. organophosphates, botulinum toxin) or spasm (e.g. strychnine, phencyclidine) | Failure of ventilation (type II respiratory failure) | Reduced alveolar oxygen tension (PAO2) (normal ~ 13.3 kPa) |
| Aspiration pneumonitis Adult respiratory distress syndrome (e.g. paraquat) |
Failure of oxygen transfer (type I respiratory failure) | Reduced arterial oxygen tension (PaO2) (normal 10–13.3 kPa) |
| Carboxyhaemoglobin (carbon monoxide); methaemoglobin (e.g. nitrites); haemolysis (e.g. arsine, stibine) | Loss of functioning haemoglobin | Reduced arterial oxygen content (CaO2) (normal 18–21 volumes %) |
| Myocardial depressants (e.g. β-blockers, calcium antagonists, tricyclic antidepressants, dextropropoxyphene, cocaine) | Reduced cardiac output | Reduced tissue oxygen delivery (QO2) (normal 12–16 mL/kg per min) |
| Chemical asphyxiants (cyanide, hydrogen sulphide) | Block of cytochrome enzyme chain | Reduced tissue oxygen consumption (VO2) causing failure of oxidative metabolism (normal 3–4 mL/kg per min). Cell death |
DIAGNOSIS AND MANAGEMENT OF POISONING: GENERAL PRINCIPLES
Diagnosis
In most cases of poisoning, the diagnosis is provided by the history and clinical examination. In some patients, however, there may be no history, though poisoning may be suspected because of the circumstances or the clinical features. In rare cases, the clinical presentation may mimic other medical conditions and it is necessary to consider the diagnosis of poisoning, especially when there are unusual features of the illness. Examples of these conditions include carbon monoxide, lead and paraquat poisoning, and drug and substance abuse.
The clinical features of poisoning depend most of all on the type of agent involved, but also on the route of exposure (oral, intravenous, percutaneous or inhaled), the duration of exposure and time elapsed since exposure, and also the patient’s age and medical background (conditions such as diabetes, asthma, chronic renal failure and epilepsy). Clinical examination may reveal signs of injection marks, cutaneous burns, buccal corrosion, tablet residues in the mouth or cutaneous blisters. These blisters (originally called ‘barbiturate blisters’) may be found following overdose with barbiturates, benzodiazepines, tricyclic antidepressants or chloral hydrate, or exposure to carbon monoxide. There may be evidence of pulmonary aspiration or secondary hypostatic pneumonia in comatose patients.
Clinical examination may indicate that a specific agonist/receptor pathway has been either over-stimulated or blocked by a poison. These syndromes are termed toxidromes and provide a useful initial guide to poison identification when the agent involved is unknown (Table 40.2).
In the severely ill patient, baseline haematological and biochemical investigations will be necessary and arterial blood gas estimations will also be required. A chest radiograph is usually indicated. The results of haematological and biochemical investigations may be of great assistance in making the diagnosis and monitoring the response to treatment. The tests to be carried out and their frequency depend very much on the poison involved. Many of these are given later in this chapter, and a general summary is given in Table 40.3. Toxicological investigations may also be required: in some cases, these may only be available in specialized laboratories. The usual samples required are blood (note that whole blood is required for measurement of carboxyhaemoglobin, methaemoglobin, cyanide and several metals) and urine (50 mL in a universal container with no preservative). Urine should always be provided where possible, since many types of analysis, such as drugs of abuse, are more efficiently carried out on urine samples. Some of these assays are qualitative, but may be necessary in order to confirm the presence of compounds, which are not easily detectable in blood.
Poisons centres now exist in most countries, with specialized facilities for provision of information on the toxicity of substances and on the management of poisoning. Most hold stocks of specialized antidotes, such as snake antivenoms. Many also have an analytical laboratory and some have patient care facilities. They are usually accessible by telephone or e-mail. In the UK, there are several poisons centres that provide information on poisoning: see Appendix 40.1.
Management
When an acutely ill patient presents, the first priority, regardless of the cause of the illness, is to establish the airway and provide immediate supportive treatment. Cardiopulmonary resuscitation or urgent ventilatory support may be needed. Depending on the patient’s state, questioning about the possible agent involved, obtaining an antidote or taking diagnostic samples may waste valuable time. In view of the frequency of opioid abuse, naloxone (an opioid antagonist) may have both a diagnostic and a therapeutic role. Once any immediate life-threatening problems have been dealt with, full supportive care must be provided while attempts are made to establish the diagnosis and treat the poisoning.
Respiratory support
In comatose patients, the most important measure is to maintain the airway and support ventilation as required, since respiratory complications are the commonest causes of death in unconscious poisoned patients. The unconscious patient should be placed in the left lateral (recovery) position in order to keep the airway patent and to minimize the risk of aspiration of gastric contents. Regular observation is essential as mechanical ventilation is not infrequently required.
Cardiovascular support
The circulation must be supported in order to maintain tissue perfusion and the electrocardiogram should be monitored, since cardiac dysrhythmias are common in poisoning. Antiarrhythmic agents and inotropic agents may be necessary.
Central nervous system complications
A brief convulsion due to cerebral hypoxia or to a toxic effect of the poison is not an indication for anticonvulsant drug therapy. However, if convulsions are repeated or prolonged, diazepam is the first-line drug. Drugs such as phenytoin, clomethiazole and thiopental may be required if there is no response to diazepam. In tricyclic antidepressant poisoning, administration of sodium bicarbonate may relieve convulsions. Phenytoin is contraindicated in tricyclic poisoning because both tricyclics and phenytoin are sodium channel blockers and these increase the risk of cardiac arrhythmias.
Body temperature
The core temperature should be recorded with a low-reading thermometer, since hypothermia may complicate poisoning with sedative and antidepressant drugs. The patient should be wrapped in a space blanket, but active rewarming is not usually required. The prognosis is better than in accidental hypothermia from other causes, and temperatures as low as 22 °C are compatible with full recovery. In hyperthermia (rectal temperature > 39 °C), reduction of body temperature is a priority. Depending on the cause of the problem, different treatments may be required. Hyperthermic patients poisoned with agents that produce severe muscle rigidity may require elective paralysis with a muscle relaxant and mechanical ventilation. Malignant hyperthermia or neuroleptic malignant syndrome can be treated with intravenous dantrolene.
Renal complications
Renal function should be monitored (bladder catheterization may be necessary if there is oliguria or severe hypovolaemia). Acute kidney injury may occur as a result of a direct toxic effect or secondary to acute haemolysis with haemoglobinuria or rhabdomyolysis with myoglobinuria. There is controversy as to whether myoglobin and haemoglobin are directly toxic to the kidneys. Most probably they cause an obstructive lesion; alkalinization of the urine increases myoglobin excretion and may prevent acute kidney injury.
General supportive care
In the severely ill patient, general supportive care may be life saving. Regular turning will be needed to prevent pressure necrosis of tissues, and care of the eyes and mouth will also be required.
Intestinal decontamination
Decontamination of the gut following ingestion of poisons is controversial. Syrup of ipecacuanha to induce emesis is now obsolete; there is little evidence that it is effective in emptying the stomach. In the comatose patient, gastric lavage is still occasionally used. However, this procedure may wash some of the poison further down the gut, and it is now rarely recommended.
The most widely used method of gut decontamination is to administer activated charcoal, which adsorbs almost all drugs and poisons. The main exceptions are iron and lithium, which are poorly adsorbed, and alcohols and glycols, whose molar load usually exceeds the adsorptive capacity of the charcoal. Repeated doses of activated charcoal may be used for sustained-release preparations that have delayed absorption, and may also be used to eliminate certain poisons from the body if there is significant enterohepatic recycling (e.g. carbamazepine and theophylline).
Antidotes
Antidotes form an important part of the management of certain poisons. In some cases (as with paracetamol, opioids, cardiac glycosides, organophosphates and snake bites), they may be life-saving. However, it should be noted that antidotal treatment is only required in a minority of poisonings. The most debated areas with regard to antidotes concern their effectiveness in ‘late’ paracetamol poisoning (when the patient presents more than 12 h after ingestion) and their effectiveness and use in the management of poisonings by metals. A list of commonly used antidotes is given in Table 40.4.
Elimination techniques
In some types of poisoning, active elimination techniques are indicated. This approach is most widely used in poisoning by salicylates, theophylline, ethylene glycol and methanol. The techniques include repeated oral doses of activated charcoal, enhanced renal elimination (by alkalinization of the urine or diuresis), haemoperfusion and haemodialysis. Haemodialysis may be required for the elimination of the poison (most notably in the case of salicylates, ethylene glycol and methanol) or for the support of renal function. Exchange transfusion may be used in infants. Peritoneal dialysis, plasmapheresis and continuous arteriovenous haemofiltration are less useful in removing poisons, although the latter is an effective technique to support renal function.
SPECIFIC POISONS
Paracetamol (acetaminophen)
Although paracetamol is safe when taken in the recommended dose, it is potentially very toxic in overdose and, since it is widely available over the counter, it is currently the commonest cause of admissions to hospital for poisoning and also of acute hepatic necrosis. It causes approximately 150 deaths each year in Britain.
Mechanisms
Paracetamol is rapidly absorbed from the upper gastrointestinal tract and the majority is metabolized by conjugation with sulfate or glucuronate to non-toxic derivatives. However, a small proportion (approximately 8–10%) is metabolized by a specific cytochrome P450 enzyme, CYP2E1, to produce a highly reactive intermediate, N-acetyl-p-benzoquinoneimine (NAPQI). This reactive metabolite can be metabolized by conjugation to form non-toxic mercapturic acid conjugates, provided glutathione is present in the liver cell. When an overdose of paracetamol is taken, the rate of production of NAPQI may exhaust existing glutathione stores and the capacity of the liver to synthesize glutathione (Fig. 40.2). In this case, NAPQI covalently binds to sulfhydryl groups in hepatocytes, forming an irreversible complex, which can result in acute centrilobular necrosis of the liver. Since paracetamol is also metabolized in the cells of the renal tubules, a similar process can also occur in the kidneys, leading to acute kidney injury. There is usually a small amount of renal damage in the presence of hepatic damage, but occasionally renal damage predominates and acute kidney injury may rarely be the presenting feature of paracetamol poisoning.
FIGURE 40.2 Metabolism of paracetamol. Induction of the cytochrome P450 pathway increases the production of the toxic metabolite, N-acetyl-p-benzoquinoneimine, causing depletion of glutathione stores; a protein-depleted diet leading to deficiency of amino acids may make less glutathione available. In both cases, increased toxicity may result following an overdose.
Toxic dose
The upper therapeutic limit for an adult of 4 g daily in divided doses is generally accepted as being safe, but doses above this can cause severe hepatotoxicity and possibly death. However, owing to the wide variation in the metabolic handling of paracetamol by the body, much larger overdoses (> 50 g) may have little effect in some individuals, producing nothing more than a minor rise in plasma aminotransferase activities.
Clinical features
In the first hours after ingestion of an overdose of paracetamol, symptoms may be minimal, unless it has been taken with some other drug, for example in a compound formulation with dihydrocodeine. There may be malaise, nausea and vomiting. A very large overdose may cause depression of consciousness or metabolic acidosis. By 24–36 h, there may be pain in the right hypochondrium. Plasma aminotransferase activities will rise at this time, but the peak values are a poor indicator of prognosis. The prothrombin time (or international normalized ration, INR) is a reliable prognostic indicator of severe hepatotoxicity. Hypoglycaemia and coagulation defects may complicate the hepatic failure. By about 48 h, the early signs of hepatic encephalopathy may appear. Death usually occurs after five or six days.
Management
Giving an antidote is more important than emptying the stomach or giving activated charcoal. After a single acute overdose, an antidote should be given if the patient’s plasma paracetamol concentration is on or above the line on the treatment nomogram (Fig. 40.3). The need for treatment following repeated supratherapeutic dosing (staggered overdose) cannot be assessed using the nomogram and treatment decisions are based on the dose ingested and the patient’s body weight. Repeated plasma paracetamol estimations should not be performed routinely, but may be helpful in doubtful cases.
FIGURE 40.3 UK nomogram for treatment of paracetamol poisoning.
Acetylcysteine is the antidote of choice and the intravenous route is the only reliable method of treatment in a patient who is comatose or vomiting. Although the effectiveness of acetylcysteine diminishes considerably with time once 12 h have passed since ingestion, later administration improves outcome even in patients with established liver failure. Acetylcysteine should also be given if the time of the overdose is in doubt or not known. Its main adverse effect is anaphylactoid reactions, which respond to administration of antihistamines and discontinuation or slowing of the infusion.
Salicylate
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
