[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 36
Biochemical aspects of neurological disease
Paul Hart; Clare M. Galtrey; Dominic C. Paviour; Min Htut
CHAPTER OUTLINE
Toxic and metabolic encephalopathy
Vitamin B12 deficiency (subacute combined degeneration of the spinal cord)
Small fibre painful axonal neuropathy
Acute inflammatory neuropathies and variants
Chronic kidney disease and established renal failure
Nutritional peripheral neuropathies
Neuropathy associated with bariatric surgery
Ataxia with isolated vitamin E deficiency
Early onset ataxia with oculomotor apraxia and hypoalbuminemia
Fragile X-associated tremor/ataxia syndrome
Hexosaminidase deficiency (GM2 gangliosidoses)
Cerebrotendinous xanthomatosis (cholestanolosis)
Neuronal ceroid lipofuscinosis
INTRODUCTION
Despite the significant advances seen in laboratory and imaging diagnostics, many of the conditions seen by neurologists are still diagnosed on purely clinical grounds. In neurology, there remains no substitute for a meticulous clinical history and careful examination.
Neurological investigations include all imaging modalities, neurophysiological tests of both the central and peripheral nervous system, and laboratory based investigations, including haematology, biochemistry, immunology, histopathology and cytology. This chapter will focus on the neurological conditions for which biochemical abnormalities are pertinent. Separate chapters cover the biochemistry of cerebrospinal fluid and muscle disease and other conditions that overlap with general neurological practice.
ENCEPHALOPATHY
‘Encephalopathy’ is a term for any diffuse disease of the brain that alters brain function or structure. It is a common condition that encompasses coma, acute confusional state, delirium and dementia. The hallmark of encephalopathy is an altered mental state.
Encephalopathy is caused either by processes that directly affect the structure of the brain or by systemic and metabolic factors (see Box 36.1). Encephalopathy can occur acutely (over hours to days); subacutely (over weeks to months) or chronically (over years) (Table 36.1). Although the mechanism of many causes of encephalopathy is well understood, the pathophysiology of metabolic encephalopathy is less clear but is likely to involve changes in amino acids and neurotransmitter profile.

For patients presenting with altered consciousness, a detailed history often has to be taken from family, friends and other healthcare professionals, to determine pre-existing medical conditions and time of onset of altered mental state. For example, patients presenting with altered consciousness following apnoea or cardiac arrest are likely to be suffering from a hypoxic–ischaemic encephalopathy but alternative causes need to be excluded. Access to medication, previous psychiatric problems or use of illicit drugs may suggest intoxication. If the patient is known to have diabetes, hypo- or hyperglycaemia is likely. Patients on diuretics are at risk of hyponatraemia.
Both a general systemic examination and detailed neurological examination are required to determine the cause of encephalopathy. Specific breath smells suggest certain causes, e.g. ammonia-like (uraemia); fruity-smelling (ketoacidosis); musty or fishy (acute hepatic failure); onion (paraldehyde), and garlic (organophosphates). Hypertension may indicate amfetamine or cocaine intoxication or a hypertensive encephalopathy. Examination of the skin may show signs of chronic liver disease, haemodialysis fistula, previous sternotomy scars from cardiac surgery or injection marks from intravenous drug use.
Neurological examination should be divided into focal (localizing) and global (non-localizing) signs. Global signs suggest a process affecting the whole brain and include altered consciousness (acute confusional state, delirium and coma). The level of coma can be quantified with the Glasgow Coma Score, for which the minimum score is 3/15 (Table 36.2).
TABLE 36.2
Glasgow coma scale (GCS)
The total is obtained by adding together the scores for each column
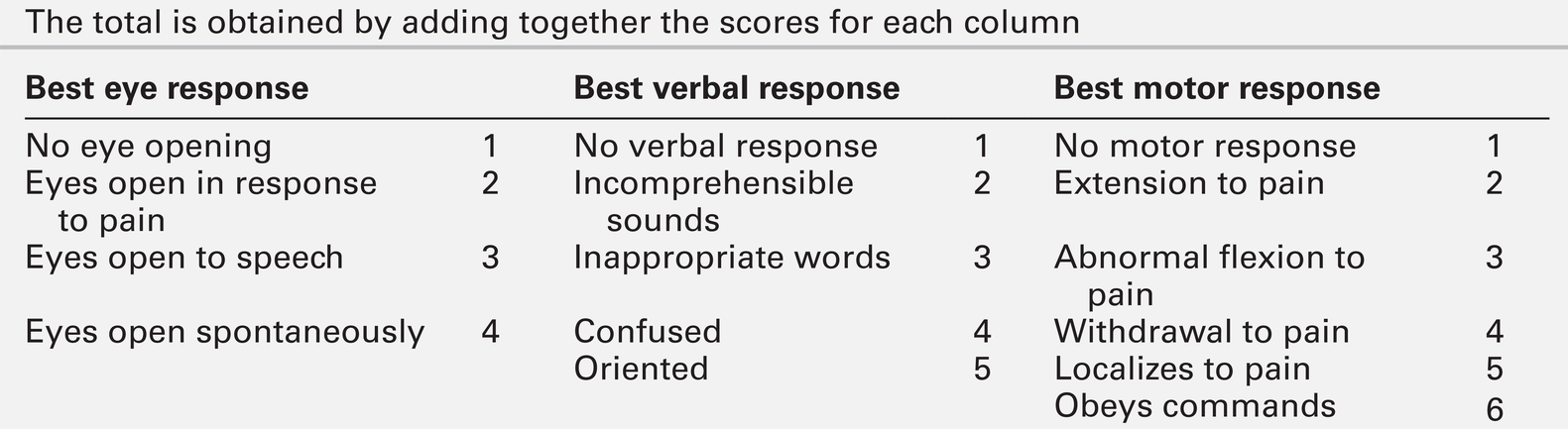
Other global neurological signs include generalized seizures, tremor, asterixis (abnormal jerking tremor in hands seen in liver flap) and myoclonus (involuntary brief jerk-like twitching, often multifocal). Focal signs indicate a localized problem within one part of the brain, for example visual or language disturbance from cerebral cortex pathology or eye movement disturbance, slurred speech or swallowing problems from brainstem pathology. Focal neurological signs usually suggest a primary neurological cause of encephalopathy.
Two-thirds of encephalopathies are secondary to systemic metabolic factors. It is therefore important that these are excluded before assuming there is a neurological cause. Many metabolic encephalopathies are reversible if corrected promptly. Investigations for a patient with encephalopathy should include blood tests, urinalysis, imaging studies, and, when indicated, cerebrospinal fluid examination and an electroencephalogram. Increasingly, laboratory investigations can be used to make a definite diagnosis of neurological causes of encephalopathy owing to the improved recognition of antibodies for autoimmune encephalitis syndromes and biomarkers in neurodegenerative disease (Table 36.3).
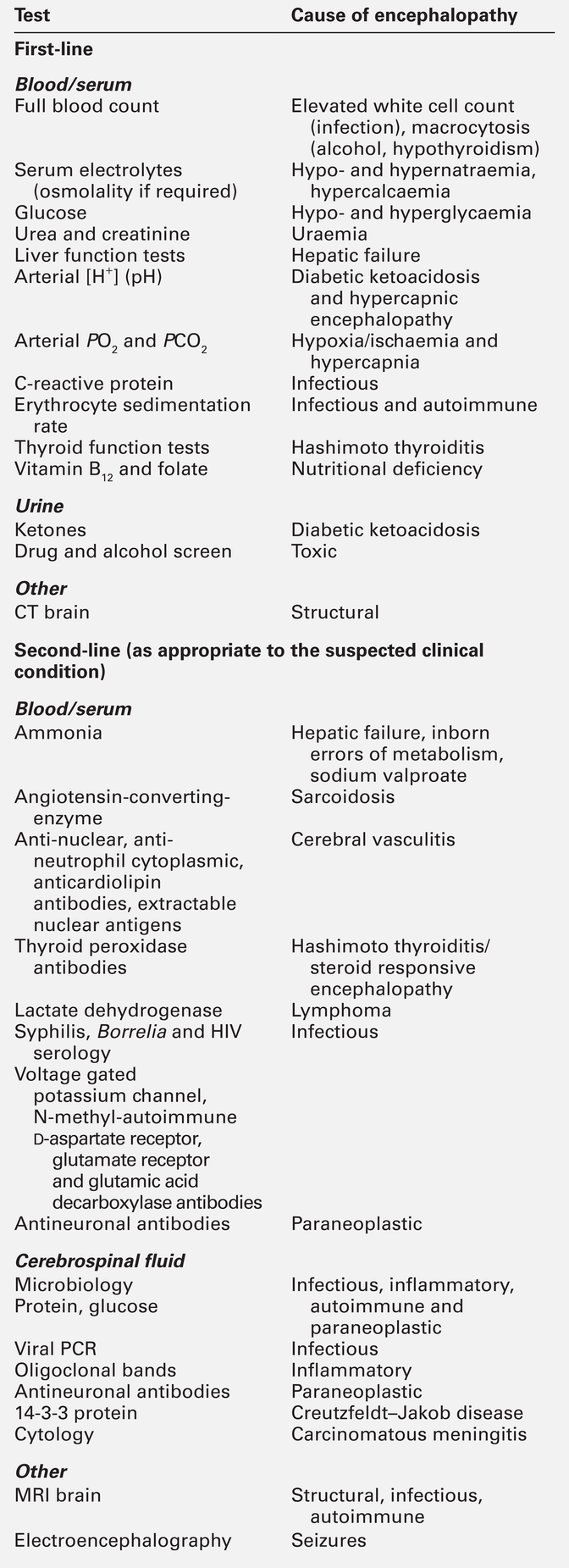
Toxic and metabolic encephalopathy
Toxic encephalopathy is caused by exogenous substances including solvents, drugs, radiation, paints, industrial chemicals, and certain metals. Some of the more common causes are discussed below. More details can be found in Chapter 40.
Carbon monoxide
This colourless and odourless gas is the most common cause of accidental poisoning in Europe and North America, as well as being a major cause of suicidal deaths. When 20–30% of total haemoglobin is bound to carbon monoxide, it causes headache, nausea and shortness of breath; when 50–60% is bound, coma results. Carbon monoxide exposure is detected by measuring the carboxyhaemoglobin concentration in blood. Carbon monoxide poisoning is treated with oxygen, using hyperbaric facilities in severe cases, if available.
Alcohol
Alcohols, including ethanol, produce an altered mental state by central nervous system depression. Methanol and ethylene glycol similarly cause direct central nervous system depression but are also toxic via metabolism in the liver to formic acid (methanol) and glycolic and oxalic acids (ethylene glycol).
Methanol and ethylene glycol concentrations can be measured in the blood but usually the suspicion is raised by other biochemical abnormalities. Poisoning with either causes a high anion gap metabolic acidosis. Treatment is by blocking the action of alcohol dehydrodenase with ethanol or fomepizole to decrease the production of toxic metabolites.
Opioids
Overdose causes coma, pinpoint pupils and respiratory depression. This can be from iatrogenic, accidental or suicidal overdose (e.g. morphine) or from use of illicit drugs (e.g. heroin). Diagnosis is usually suspected from the clinical triad but a rapid urine screening test can confirm opioid exposure. Treatment is with naloxone, given as a bolus dose followed by an infusion if the patient responds, in addition to supportive care.
Thiamin (vitamin B1) deficiency
Wernicke–Korsakoff encephalopathy is a clinical triad of confusion, ataxia and ophthalmoplegia (eye movement disorder). It comprises two syndromes. First, Wernicke encephalopathy, an acute or subacute confusional state with ataxia and ophthalmoplegia and second, Korsakoff dementia, characterized by severe amnesia and confabulation. The former is often reversible but the latter is irreversible and persistent. Wernicke–Korsakoff encephalopathy is caused by thiamin deficiency and is most commonly seen in alcoholics but can also occur in patients with anorexia, hyperemesis gravidarum, those receiving total parenteral nutrition, or in the context of malabsorption and refeeding syndrome. The role of measurement of plasma thiamin concentrations in diagnosis and monitoring remains controversial and functional assessment by assay of red cell transketolase is no longer used by most laboratories for diagnosis. Therefore, laboratory tests are aimed at excluding other conditions and providing evidence of risk factors (e.g. macrocytosis and deranged liver function tests in alcoholism). Brain magnetic resonance imaging (MRI) can show signal change in the thalami, mammillary bodies, tectal plate and periaqueductal grey matter. Treatment is with parenteral thiamin. In the UK, normal practice is to give thiamin in a vitamin complex of riboflavin, nicotinamide, pyridoxine and ascorbic acid. Early treatment can reverse the symptoms rapidly and prevent dementia. Administration of intravenous glucose (including as part of parenteral nutrition) to patients who are severely malnourished, can exhaust their (already depleted) supply of thiamin and precipitate Wernicke–Korsakoff encephalopathy. Thiamin should therefore be administered before starting a glucose infusion in patients at high risk.
Vitamin B12 deficiency
This causes a typical pattern of degeneration of the white matter producing encephalopathy, myelopathy (subacute combined degeneration of the spinal cord), peripheral neuropathy and optic neuropathy. Vitamin B12 concentrations are easily measured and, if deficiency is corrected early, further damage can be prevented (See p. 688 and p. 693).
Liver failure
Hepatic encephalopathy occurs as a consequence of liver function disturbance and its severity is graded from 0 to 4 (Table 36.4).
TABLE 36.4
Grading of hepatic encephalopathy (West Haven classification system)
| Grading | Symptoms |
| 0 | Minimal hepatic encephalopathy. Lack of detectable changes in personality or behaviour. Minimal changes in memory, concentration, intellectual function, and coordination. Asterixis absent |
| 1 | Trivial lack of awareness. Shortened attention span. Impaired addition or subtraction. Hypersomnia, insomnia, or inversion of sleep pattern. Euphoria, depression, or irritability. Mild confusion. Slowing of ability to perform mental tasks. Asterixis can be detected |
| 2 | Lethargy or apathy. Slurred speech. Obvious asterixis. Drowsiness, lethargy, gross deficits in ability to perform mental tasks. Obvious personality changes, inappropriate behaviour and intermittent disorientation, usually regarding time |
| 3 | Somnolent but can be aroused. Unable to perform mental tasks. Disorientation about time and place, marked confusion, amnesia. Occasional fits of rage, present but incomprehensible speech |
| 4 | Coma with or without response to painful stimuli |
Two situations can lead to severe hepatic encephalopathy – acute fulminant hepatic failure and decompensation of chronic liver disease. In the UK, acute liver failure is most commonly from paracetamol overdose or viral hepatitis, and chronic liver disease from alcohol abuse or chronic hepatitis B or C infection. Encephalopathy is thought to develop because hepatocellular dysfunction produces neurotoxins (e.g. ammonia), false neurotransmitters and benzodiazepine-like substances. In chronic liver disease, there can be the additional effect of porto–systemic shunting. This allows significant quantities of ammonia, formed in the bowel from protein, to reach the systemic circulation. The severity of the neurological disorder correlates poorly with ammonia concentrations so measurement rarely contributes to management. ‘Liver function tests’ will confirm that the concentration of bilirubin is high, often with abnormal liver enzyme activities and, more importantly, poor synthetic function with abnormal clotting. Plasma paracetamol concentration can be measured if overdose is a possibility. Patients with acute liver failure may require liver transplantation for survival but the encephalopathy of chronic disease may be reversed by conservative measures (see Chapter 14 for further details).
Chronic kidney disease and established renal failure
Uraemic encephalopathy presents with apathy, fatigue, inattentiveness and irritability, followed by confusion, hallucinations, slurred speech, tremor and asterixis. The mechanism is unclear but correction of uraemia reverses the encephalopathy. Suggested mechanisms include retention of organic acids, elevation of phosphate concentration in the cerebrospinal fluid (CSF), increased calcium content of the cerebral cortex owing to the action of PTH, and alteration of concentrations of neurotransmitters or proinflammatory cytokines. Although the degree of the rise in urea correlates with the severity of encephalopathy, it is not thought to be causative. The investigation, monitoring and treatment of uraemia are discussed in Chapter 7.
Respiratory failure
Hypercapnia occurs in respiratory failure either secondary to lung disease (e.g. chronic obstructive pulmonary disease) or to mechanical problems such as neurological disease (e.g. myasthenia gravis). Clinically, hypercapnia presents with headache, papilloedema, mental slowing, drowsiness, confusion, coma and asterixis. The mechanism is unclear but thought to be due to a direct effect of carbon dioxide possibly on the hydrogen ion concentration (pH) of the CSF. Hypercapnia can be confirmed by measurement of PCO2 on an arterial blood sample. Coma can occur with PCO2 > 9 kPa. Treatment is of the underlying cause and, once corrected, there is no prolonged cerebral damage (see Chapter 5 for further details).
Cardiorespiratory failure
Hypoxic–ischaemic encephalopathy is caused by hypoxaemia and/or reduced blood flow to the brain from failure of the cardiac system, the respiratory system or both (e.g. myocardial infarction, drowning). When cerebral perfusion pressure falls, there is a failure of autoregulation leading to ischaemia and hypoxia and to cell death by both necrosis and apoptosis. The diagnosis is usually evident from the history. Myoclonus is common (Lance–Adams syndrome). There is a wide spectrum of clinical outcomes depending on the degree and duration of the insult from full recovery to severe prolonged disability and death.
Disorders of glucose metabolism
Hypoglycaemia causes confusion, seizures and coma and can cause permanent neurological damage if not reversed quickly. Up to 15% of patients with diabetes will have at least one episode of hypoglycaemic coma in their lifetime. Recurrent hypoglycaemia, such as that caused by an insulinoma, can be mistaken for epilepsy. The brain contains 1–2 g of glucose stored as glycogen, which is utilized within approximately 30 min if blood glucose concentration remains low. Ketones can also be used as an energy source but this is not sufficient in prolonged hypoglycaemia, nor are they available when hypoglycaemia is caused by high insulin concentrations. The most common causes of hypoglycaemia are accidental or deliberate overdose of insulin, insulinoma, and depletion of liver glycogen (e.g. acute liver failure, alcohol binge, severe starvation). Hypoglycaemia is easily detected by blood glucose measurement at the bedside (although it should be confirmed by laboratory measurements) (see Chapter 17 for further details).
Hyperglycaemic coma occurs in two forms – diabetic ketoacidosis and the hyperosmolar hyperglycaemic state. Coma is a late feature of diabetic ketoacidosis and develops when hyperglycaemia, dehydration, acidosis and shock are severe. Cerebral oedema is a rare but often fatal complication both of the condition and its treatment. The coma of the hyperosmolar hyperglycaemic state usually develops more insidiously and occurs in elderly patients, often with previously undiagnosed diabetes mellitus. Extreme hyperglycaemia with greatly increased plasma osmolality, electrolyte losses and dehydration exacerbated by impaired thirst awareness, are all thought to contribute to the development of the encephalopathy (see Chapter 16 for further details).
Hyponatremia
Hyponatremia has many causes, including liver and cardiac failure, syndrome of inappropriate antidiuresis (neurological causes of which include head trauma, bacterial meningitis, encephalitis, cerebral infarction, subdural and subarachnoid haemorrhage), vomiting and diuretic use. The severity of the symptoms is related to the rapidity of decline in plasma sodium. This is because hyponatremia causes extracellular hyposmolarity and a tendency for free water to shift from the vascular space to the intracellular space. When plasma sodium concentration falls slowly, over a period of several days or weeks, the brain is capable of compensating by extrusion of solutes to the extracellular space. This reduces the flow of free water into the intracellular space and symptoms are much milder for a given degree of hyponatraemia. When the plasma sodium concentration falls rapidly, this compensatory mechanism is overwhelmed and severe cerebral oedema may ensue. For example, a sodium concentration falling from normal to < 110 mmol/L over 24–48 h will often cause coma. It is necessary to correct the plasma sodium concentration in a controlled manner because of the risk of central pontine myelinolysis if the increase is too rapid (see Chapter 4).
Central pontine myelinolysis is characterized by quadriparesis, dysphagia, dysarthria, diplopia and altered consciousness. Areas of the brain other than the pons are also often involved. Magnetic resonance imaging reveals signal change within these areas but the appearances may be normal early in the course of the disease. Central pontine myelinolysis may prove fatal, and incomplete recovery is common in survivors.
Hypernatraemia
Common causes of hypernatraemia include diabetes insipidus, hyperglycaemia, diarrhoea and fluid deprivation. Again, it is the rate of change of sodium concentration that is important rather than the actual values. A sodium concentration of > 160 mmol/L can cause coma due to cerebral cellular dehydration if it develops rapidly. Treatment depends on the cause of the disorder and the total body fluid volume (see Chapter 4).
Hypercalaemia
Severe hypercalcaemia (> 3.4 mmol/L) can be lead to coma, preceded by fatigue, headache, muscle weakness, irritability and confusion. High concentrations of calcium ions decrease neuronal excitability, which together with dehydration is thought to cause the neurological problems (see Chapter 6).
Septic encephalopathy
Severe extracranial infection can be associated with an impaired mental state. The pathophysiological mechanism involves reduced cerebral blood flow and oxygen extraction by the brain, cerebral oedema, disruption of the blood–brain barrier owing to inflammatory mediators acting on the cerebrovascular endothelium, and abnormal neurotransmitter compositions. Diagnosis of sepsis can be aided by the finding of a raised white blood cell count and raised inflammatory markers, and by blood cultures. Treatment is of the underlying infection with supportive care.
Autoimmune encephalopathy
The last decade has seen significant advances in our understanding of antibody-mediated encephalopathies. There are two types of antibodies detectable in blood in these conditions (Table 36.5): paraneoplastic antibodies to intracellular targets that are associated with cancer but are not pathogenetic, and antibodies to the extracellular domain of neuronal cell-surface proteins that directly cause encephalitis and are also often associated with cancer. Autoimmune encephalitis typically presents with subacute memory loss, psychiatric and behavioural disturbance and seizures. The detection of one of the specific antibodies supports a diagnosis of paraneoplastic encephalitis. Treatment is of the underlying cancer and with immunotherapy.
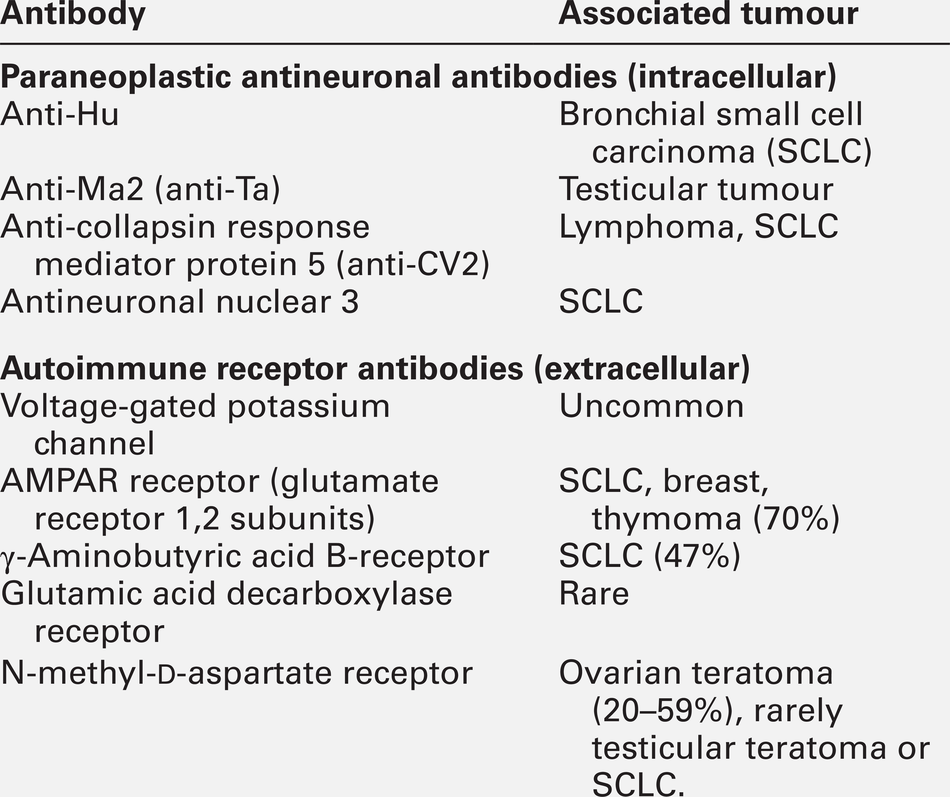
Hashimoto encephalopathy presents with seizures, behavioural and psychiatric manifestations, movement disorders and coma and is associated with the presence of high titres of anti-thyroglobulin or anti-thyroid peroxidase (TPO, antimicrosomal) antibodies. The condition can present without any features of thyroid disease. It is unclear whether the anti-thyroid antibodies are pathogenetic or simply represent an immune epiphenomenon but it is important not to miss this diagnosis as the condition responds to treatment with steroids.
Dementia
Dementia is a syndrome characterized by progressive deterioration of cognitive function without alteration of consciousness. Cognitive deficits most commonly affect memory, but other cognitive domains such as language, praxis, visual perception and most notably executive function are also often affected. Diagnosis of dementia is generally by clinical criteria during life, although many of its causes are defined on the basis of histopathological criteria with the consequence that definitive diagnosis can only be made at post-mortem. However, over the last few years a number of laboratory CSF biomarkers have emerged and have an increasing role in diagnosis. Cerebrospinal fluid Aβ-40, Aβ-42, total tau and phosphorylated tau proteins are the most sensitive biomarkers for the diagnosis of Alzheimer disease and 14-3-3 protein for the diagnosis of Creutzfeldt–Jakob disease (see Chapter 34).
SPINAL CORD DISORDERS
Spinal cord disease usually presents with motor and/or sensory symptoms in either the upper and lower or just the lower limbs, depending on the site of the insult. Clinical signs include increased tone in the limbs, brisk reflexes and, typically, well-preserved muscle bulk. Bowel and bladder function can be compromised. The list of possible causes of spinal cord disease is extensive and includes vascular, degenerative, compressive, inflammatory, malignant, metabolic and infectious conditions. Metabolic causes include vitamin B12 deficiency (Box 36.2) and adrenomyeloneuropathy. A normal MRI scan of the spinal axis will exclude a significant number of differential diagnoses.
Vitamin B12 deficiency (subacute combined degeneration of the spinal cord)
Deficiency of vitamin B12 can produce haematological and neurological abnormalities: features of gastrointestinal disease may also be apparent (see Chapter 12). From an early stage, it is the spinal cord that is predominantly affected. The onset of symptoms is insidious, with symmetrical, uncomfortable, tingling paraesthesiae, initially in the feet but later involving the hands. As the condition progresses, the gait becomes ataxic (sensory ataxia owing to loss of proprioception) and the limbs become weaker and increasingly spastic. Additional problems include peripheral polyneuropathy, optic neuropathy, psychiatric disturbance and dementia.
The exact pathophysiological process leading to neuronal damage in vitamin B12 deficiency is unknown. Vitamin B12 is involved as a cofactor in the conversion of homocysteine to methionine and of methylmalonyl- CoA to succinyl-CoA. It is possible that the impaired synthesis of methionine leads to a depletion of S-adenosylmethionine, which is required for myelin synthesis.
The diagnosis is confirmed by measuring vitamin B12 concentration in the serum. In some patients, the concentration may be only ‘low-normal’. In borderline cases, measurement of homocysteine and methylmalonate concentrations can be useful. It is important that the diagnosis is made as early as possible as this is a potentially reversible condition. Magnetic resonance imaging of the spine often shows the spinal cord to be atrophic and can show signal change in the dorsal columns.
Folate deficiency
Folate deficiency is usually associated with other nutritional deficiencies. Folate antagonists (e.g. methotrexate, trimethoprim and pyrimethamine) are also potential causes. Neurological manifestations are rare and the same as with vitamin B12 deficiency. Treatment is by folate replacement, although any vitamin B12 deficiency should be treated first to prevent exacerbation of spinal cord degeneration.
Copper deficiency
Copper deficiency is rare and causes both haematological and neurological disease. Neurological manifestations include myelopathy, peripheral neuropathy and optic neuropathy. Copper deficiency was recognized in ruminants (swayback disease) long before it was identified in humans. The presentation can be similar to that of subacute combined degeneration of the spinal cord. It is usually secondary to gastric surgery, gastrointestinal diseases or total parenteral nutrition, or to excess intake of zinc or iron, which reduce copper absorption in the gut. Copper deficiency myelopathy has been attributed to ingestion of zinc contained in denture cream in some patients. Treatment is with oral or intravenous copper replacement.
Vitamin E deficiency
Vitamin E deficiency is rare, and causes a spinocerebellar ataxia, retinopathy, myopathy and anaemia. Poor diet is not usually sufficient cause unless associated with malabsorption. Abetalipoproteinemia is a rare inherited disorder of fat metabolism that results in poor absorption of dietary fat and vitamin E. Vitamin E deficiency is discussed further below (see ataxia).
Hepatic myelopathy
Hepatic myelopathy is a rare complication of chronic liver disease. It has been suggested that it is caused by the neurotoxicity of ammonia or other metabolites bypassing normal liver metabolism. It is characterized by spastic paraparesis with minimal sensory and sphincteric involvement. Diagnosis is a process of exclusion requiring normal CSF analysis and brain imaging. Liver transplantation may result in improvement, especially if performed early in the course of the disease.
Hexosaminidase A deficiency
This is a progressive neurodegenerative disease with variable age of onset and rate of progression and hence variable prognosis. The clinical presentation of the chronic form of hexosaminidase A deficiency may mimic spinocerebellar degeneration, Friedreich ataxia, or amyotrophic lateral sclerosis (see below).
Adrenomyeloneuropathy
This is the commonest clinical variant of adrenoleukodystrophy, a disorder of peroxisomal fatty acid oxidation causing the accumulation of very long chain fatty acids in myelin, adrenal cortex and Leydig cells of the testes. Patients with an adrenomyeloneuropathy presentation of this X-linked recessive disorder present in their third decade with a progressive spastic paraparesis and sphincter dysfunction. A sensorimotor peripheral neuropathy may be a feature and ataxia and dementia are seen occasionally. Adrenal insufficiency will often have been present since childhood and there may be hypogonadism. Around 50% of heterozygous females will show some symptoms later in life. The clinical findings and magnetic resonance imaging point to the diagnosis, which is confirmed by demonstrating hypoadrenalism and elevated concentrations of very long chain fatty acids (VLCFAs) in plasma and cultured skin fibroblasts. Genetic testing is available. The only gene in which mutations are known to cause adrenoleukodystrophy is ABCD1, which codes for a peroxisomal membrane transporter protein, although molecular genetic testing is rarely required to confirm the disease, especially in a male. Treatment is with steroid replacement, symptomatic management and supportive care.
PERIPHERAL NEUROPATHY
Peripheral nerve disorders are the most common neurological diseases and their prevalence has been estimated to be 2–8% of the adult population. There are many causes of peripheral neuropathy and the range of clinical presentation is wide. Peripheral neuropathy can be categorized by the clinical pattern: mononeuropathy (e.g. carpal tunnel syndrome); multiple mononeuropathy (e.g. mononeuritis multiplex in vasculitis), or distal symmetrical polyneuropathy (e.g. neuropathy in diabetes mellitus). With the help of electrodiagnostic testing (nerve conduction studies and electromyography), neuropathies can be further categorized as demyelinating, axonal or mixed type of neuropathy. Appropriate laboratory tests may help to identify a specific diagnosis. Neuropathies with a specific metabolic basis are listed in Box 36.3. Peripheral neuropathies of toxic or metabolic aetiologies are usually symmetrical, distal, sensorimotor and axonal. The American Academy of Neurology has published practice parameters to guide laboratory and genetic testing in distal symmetric polyneuropathy (see Further reading).
Neuropathy can present with a variety of signs and symptoms. Symptoms can be classified as either positive or negative. Positive symptoms reflect inappropriate nerve activities, whereas negative symptoms reflect reduced nerve activity. Positive sensory symptoms are burning or lancinating pain and paraesthesiae, whilst negative sensory symptoms are numbness, lost of proprioception and difficulty differentiating cold and hot sensations. Weakness and fatigue can be considered as negative motor symptoms, while cramps, muscle twitching and myokymia are positive motor symptoms. The findings on clinical examination of the sensory systems include sensory loss, trophic skin changes, sensory ataxia and Charcot joints. Features of motor dysfunction are dominated by lower motor neuron wasting, normal tone, absent reflexes and flexor plantar responses. Peripheral nerves should also be palpated to check for thickening, which occurs in leprosy, amyloidosis and acromegaly. Gastrointestinal and cardiovascular symptoms can be prominent features in autonomic dysfunction, causing bloating, constipation, diarrhoea, light headedness, palpitations and excessive sweating. Blood pressure should be checked with the patient lying and standing to document a postural drop caused by autonomic dysfunction. Neuropathies with a metabolic cause tend to be symmetrical, other than for some types related to diabetes, and with variable onset.
The investigations should be guided by the clinical picture. Polyneuropathy can arise in the course of many illnesses, particularly diabetes (Table 36.6). Initial investigations would reasonably include the tests listed in Box 36.4 and should yield a diagnosis in 60–90% of patients. The majority of patients will have diabetes or alcohol exposure. There are no diagnostically useful antibody assays. More invasive tests (Box 36.5) are best undertaken after consultation with a physician or neurologist with an interest in peripheral neuropathy. The nerve conduction tests will reveal whether the neuropathy is axonal or demyelinating.
TABLE 36.6
Most common causes of a symmetrical neuropathy
| Condition | Proportion of symmetrical neuropathies (%) |
| Diabetes | 11–41 |
| Chronic idiopathic axonal neuropathy | 10–40 |
| Paraproteinaemia | 9–10 |
| Alcohol misuse | 7 |
| Chronic kidney disease | 4 |
Despite all efforts, about one-third of patients with peripheral neuropathies remain without a diagnosis. About 40% of these will be found to have impaired glucose tolerance, although how much this contributes directly to the development of neuropathy is still debated. Treatment with diet and exercise reduces the rate of progression to diabetes and may also improve the neuropathy, at least in the short term.
Patients with non-painful chronic idiopathic neuropathies have been found to have higher blood triglyceride concentrations and more exposure to environmental toxins compared with controls. Currently, it is unclear whether these observations will translate into any therapeutic benefit.
Small fibre painful axonal neuropathy
There is a small subset of patients in whom small nerve fibres alone are affected and in whom pain and temperature sensation but not large fibre modalities (vibration and proprioception) are impaired. Conventional clinical neurophysiological tests are normal because they do not assess small nerve fibre function. Most patients with a small fibre neuropathy have diabetes mellitus or at least impaired glucose tolerance but a few have Sjögren syndrome and other rare causes.
Diabetic neuropathies
Peripheral neuropathy is rarely encountered in young patients with newly diagnosed diabetes mellitus but diabetes presenting in older patients is frequently associated with a neuropathy and this may be the presenting problem. Studies have suggested that after 25 years of diabetes, about 50% of patients will have evidence of a neuropathy, most commonly chronic diabetic peripheral neuropathy. Poor glycaemic control, duration of diabetes, hyperlipidaemia (particularly hypertriglyceridaemia), increased albumin excretion and obesity are risk factors for the development of neuropathy.
Although distal symmetrical peripheral neuropathy is the commonest presentation, the manifestations of diabetic neuropathy are varied and can be subdivided clinically into the symmetric polyneuropathies and the focal or multifocal neuropathies (Box 36.6).
Symmetrical polyneuropathies
The onset of distal sensory and sensorimotor polyneuropathy is insidious, with mild symptoms of numbness and paraesthesiae affecting the toes and feet. A burning pain, worse at night, may be a prominent symptom. The typical findings on examination are sensory loss to pinprick, light touch and vibration sensation in a stocking distribution, with absent ankle reflexes. Occasionally, there may be mild distal weakness. Foot ulceration and neuropathic arthropathy are potentially serious complications related to loss of pain sensation. Ulcers tend to be situated at pressure points – over the toes, heels, malleoli and metatarsal heads. In some patients, neuropathy may be precipitated by the institution of therapy with either insulin or oral hypoglycaemics and even dietary control. The neuropathy then becomes evident 4–6 weeks after starting treatment, with painful paraesthesiae affecting the lower half of the legs.
Acute painful neuropathy is a distinct entity characterized by marked weight loss over a short period of time followed by severe burning pains in the feet with contact discomfort. By maintaining good glycaemic control, patients slowly improve.
Proximal symmetrical motor neuropathy and asymmetrical proximal neuropathy (see below) are now termed lumbosacral radiculoplexus neuropathy (diabetic amyotrophy). There is often diffuse lower back pain at the onset, followed by progressive proximal weakness and wasting of the muscles. The knee reflexes are absent but sensory loss is minimal. Good glycaemic control leads to slow recovery over 12–30 months.
Focal and multifocal neuropathies
An isolated third nerve lesion is the commonest of the cranial nerve palsies, usually with sparing of pupillary function; in about half of patients, there is aching retro-orbital pain. The lesion is thought to be an infarct in the nerve trunk (i.e. it has microvascular aetiology).
Diabetic patients are more prone to isolated peripheral nerve palsies (mononeuropathy), suggesting increased susceptibility to compressive injury as the nerves are damaged at sites of pressure (e.g. the ulnar nerve at the elbow, the median nerve at the wrist).
Lower limb asymmetrical proximal motor neuropathy, also called lumbosacral radiculoplexus neuropathy, is probably caused by an underlying microvasculitis. The onset is usually subacute, with thigh pain, followed by asymmetric weakness and wasting of the lower limb muscles, most often affecting hip and knee flexors. Sensory loss is minimal. The weakness can be very severe. Recovery is slow, occurring over 2–4 years and is often incomplete.
Pathophysiology of diabetic neuropathy
The pathophysiology of diabetic neuropathy is only partially understood. A combination of direct axonal injury from hyperglycaemia, insulin resistance, central adiposity (visceral fat produces toxic adipokines), endothelial injury and microvascular dysfunction is possibly responsible. Hyperglycaemia is known to increase flux through the polyol pathway, increase nonenzymatic glycation of proteins and enhance oxidative stress.
Polyol pathway
Aldose reductase converts glucose to sorbitol, which is then converted to fructose by sorbitol dehydrogenase. In nerves from diabetic patients, accumulation of sorbitol leads to a compensatory reduction in other osmolytes such as myoinositol and taurine. The loss of myoinositol may lead to a reduction in nerve Na+,K+-ATPase, which then causes nerve conduction defects. Aldose reductase inhibitors showed early promise by reversing the biochemical changes in experimental diabetic nerves but, in clinical trials, these compounds have had no significant impact on human diabetic neuropathy.
Non-enzymatic glycation
The formation of irreversible advanced glycation end products of various components in the nerves of patients with diabetes is believed to contribute to the structural changes that are seen in diabetic neuropathy. Myelin components and axonal cytoskeletal proteins are glycated, leading to impaired cytoskeletal assembly with consequently abnormal axonal transport and axonal atrophy. Aminoguanidine, developed as a potential inhibitor of glycation, causes improvement in Na+,K+-ATPase activity and nerve conduction velocity when administered to experimental diabetic rats. It has not been tested in humans.
Oxidative stress
Diabetes is associated with the increased production of free radicals, which have the potential to cause damage to arteries. Endothelial cell function is altered and this may impair nerve blood flow and nerve oxygenation. Thioctic acid (α-lipoic acid) reduces oxidative stress, and is available in some countries for the treatment of diabetic patients with neuropathic pain.
Immune mediated neuropathies
Immune mediated neuropathies are heterogeneous disorders, with a wide range of presentations. The pathological process affects the nervous system either directly (e.g. demyelination) or indirectly (e.g. secondary to vasculitis).
Acute inflammatory neuropathies and variants
Guillain–Barré syndrome is an acute immune mediated polyradiculoneuropathy, usually with demyelination. In two thirds of patients it is preceded by an infection. The presentation is acute and evolves over a few weeks, typically with an ascending flaccid paralysis, early loss of deep tendon reflexes and minimal sensory signs. Sphincteric functions are usually spared. Significant autonomic features are common and neuropathic pain may be problematic. The salient feature of CSF analysis is elevated protein with a normal cell count. The finding of an increased cell count warrants further investigation for conditions such as human immunodeficiency virus (HIV) and Lyme disease. Antibodies associated with Guillain–Barré syndrome are shown in Table 36.7.

A variety of infections can precede the onset of Guillain–Barré syndrome and these can be associated with various anti-ganglioside antibodies. Campylobacter jejuni is associated with GM1, GM1b, GD1a, GalNAc-GD1a, GD3, GT1a and GQ1b. Haemophilus influenzae is associated with GM1 and GT1a. Mycoplasma pneumoniae and cytomegalovirus are associated with galactocerebroside GM2 antibody.
The mainstay of treatment for Guillain–Barré syndrome is intravenous immunoglobulin or plasmapheresis, usually in an intensive care setting.
Chronic inflammatory demyelinating polyneuropathies and variants including paraproteinaemic neuropathies
Chronic inflammatory demyelinating polyneuropathy (CIDP) is a heterogeneous group of acquired immune-mediated neuropathies. It may present in isolation or as part of a systemic disease. The classic form of CIDP is symmetrical with motor involvement greater than sensory. Weakness is present in both proximal and distal muscles. It can be associated with various systemic diseases such as HIV infection, connective tissue disorders and paraproteinaemia (see Chapter 30). Serum protein electrophoresis is therefore one of the investigations that should be carried out routinely in all adult patients presenting with a peripheral neuropathy. Treatment of CIDP includes intravenous immunoglobulin, plasmapheresis and immunomodulating treatment including steroids.
Monoclonal gammopathy of unknown significance
This can cause a neuropathy that is slowly progressive over many years and has a symmetrical distribution, first affecting the feet with numbness and paraesthesiae but later involving the hands. Muscle weakness and wasting occurs months or years after the onset of sensory symptoms. The cause of the neuropathy is unknown. The commonest paraprotein to be detected is IgM, which can bind to peripheral nerves via the carbohydrate moiety in myelin-associated glycoprotein (MAG). Antibodies to MAG are found in 60% of patients. IgG and IgA neuropathies are less common, but are more responsive than IgM neuropathy to immunotherapy and plasmapheresis.
Multiple myeloma
This rarely causes an isolated neuropathy. When it does occur, it is sensorimotor in type and may be axonal and demyelinating. Treatment of the myeloma may stabilize the neuropathy.
Waldenström macroglobulinaemia
This is commonly associated with a sensorimotor neuropathy in addition to the more generalized symptoms of fatigue, weakness and a bleeding diathesis.
POEMS syndrome
Polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes (POEMS syndrome) has a peak incidence at age 50–60 years. It is distinct from the neuropathy associated with myeloma and its pathogenesis remains unknown. The main clinical features are of a progressive sensorimotor neuropathy associated with a monoclonal plasma cell proliferative disorder and osteosclerotic bone lesions. The light chain is invariably λ rather than κ, and when present, the heavy chain is either A or G and only occasionally M. The endocrine disturbance can appear at any stage of the illness and can involve adrenal, thyroid, pituitary, parathyroid, gonadal or pancreatic function. There is no specific diagnostic test for POEMS syndrome.
Chronic kidney disease and established renal failure
Peripheral neuropathies are not uncommon in patients with chronic kidney disease (CKD). A variety of peripheral nerve disorders may occur: carpal tunnel syndrome, ulnar and femoral neuropathy and uraemic polyneuropathy. The pathogenesis probably involves anatomical vulnerability, ischaemia, deposition of β2-microglobulin-associated amyloid and tissue compression around nerves.
The neuropathy associated with non-diabetic renal disease is of insidious onset, progressing over months and is present in 60–100% of patients on dialysis. Neuropathy generally develops only at CKD stage 5 and is characterized by paraesthesiae, tingling and burning of the feet, cramps and restlessness of the legs. Weakness is uncommon and, when present, is relatively mild. Patients may also develop autonomic features, with postural hypotension, impaired sweating, diarrhoea, constipation or impotence. Although there is a correlation between the impairment of nerve conduction and decreased glomerular filtration rate, starting dialysis or increasing its rate does not seem to improve an established neuropathy. However, improvement is seen after renal transplantation. The neuropathy has been attributed to the inability of dialysis membranes to clear ‘middle molecules’ (see Chapter 7), which are thought to be neurotoxic. The evidence to support this hypothesis is conflicting.
Liver disease
The prevalence of peripheral neuropathy in chronic liver disease is variable. Alcohol is the commonest cause of neuropathy associated with liver disease owing both to the direct neurotoxic effect of alcohol and to liver failure. Peripheral neuropathy can occur as a direct result of a primary liver disease although the underlying mechanism of neuropathy is unclear. It can also be part of a systemic disease that affects both the liver and peripheral nervous system for example, vasculitis. Most patients with chronic liver disease have evidence of an axonal neuropathy. There may be an additional autonomic neuropathy, which carries a poor prognosis, and carpal tunnel syndrome is common. Acute peripheral neuropathy, especially Guillain–Barré syndrome, can be associated with viral hepatitis. Primary biliary cirrhosis is associated with a painful sensory neuropathy with sensory ataxia.
Endocrine disturbances
Hypothyroidism
Hypothyroidism frequently causes carpal tunnel syndrome because of deposition of mucopolysaccharide complexes within the carpal tunnel. Adequate thyroxine replacement is often the only treatment necessary.
Hyperthyroidism
Hyperthyroidism can also cause carpal tunnel syndrome that typically resolves with treatment. The mechanism is thought to be the same as in hypothyroidism. Thyroid storm can be associated with acute neuropathy.
Acromegaly
Acromegaly causes swelling of the median nerve from oedema and/or hypertrophy, resulting in an increased incidence of carpal tunnel syndrome. Treatment of the acromegaly improves the symptoms. There can also be a polyneuropathy, which is often painful. The relationship between peripheral neuropathy and excess growth hormone is not clear and can be complicated by coexisting diabetes mellitus.
Nutritional peripheral neuropathies
Nutritional deficiencies and excess alcohol are associated with peripheral neuropathies as well as encephalopathy and spinal cord disorders (see previous sections).
Vitamin B12 deficiency
This is commonly associated with peripheral neuropathy, which may be precipitated by nitrous oxide anaesthesia. Measurement of plasma vitamin B12 concentration may give a low-normal value even though there is a true functional deficiency. It is therefore recommended that, for patients with a neuropathy and borderline B12 value, plasma methylmalonate and homocysteine concentrations should also be checked. In one review, patients with abnormal methylmalonate and homocysteine concentrations accounted for 15% of those previously labelled as ‘cryptogenic neuropathy’ as the plasma vitamin B12 concentrations were within reference limits. Interestingly, a megaloblastic anaemia is rarely seen in patients with a vitamin B12 deficiency state and neurological complications.
Intramuscular injection of vitamin B12 is the standard treatment for such deficiency, but the recovery of neuropathy is variable, and may not occur at all.
Thiamin (vitamin B1) deficiency
Also known as beri-beri, this is now rarely encountered in developed countries; when it does occur, it is most often associated with alcoholism and severe anorexia. Thiamin and its coenzyme thiamin pyrophosphate are required for mitochondrial function and myelin formation. A deficiency state has to be more prolonged before the peripheral nerves become affected than is the case with encephalopathy. The symptoms are numbness and pain that start in the feet and may later involve the hands. Although direct measurements of thiamin concentration are available, their use in diagnosing thiamin deficiency is controversial. If deficiency is suspected clinically, treatment should be given immediately either orally or intravenously. Reversal of the neuropathy is unpredictable and it may take 12 months for any improvement to be seen.
Vitamin B6 (pyridoxine) deficiency
This deficiency causes an axonal neuropathy that develops very slowly and typically results in distal numbness and mild weakness. In contrast, a severe ataxic painful neuropathy can result from vitamin B6 toxicity at doses of more than 200 mg/day.
Vitamin E deficiency
Vitamin E deficiency only rarely causes an isolated neuropathy; measuring plasma vitamin E concentrations should not be part of the routine screen in neuropathy. Chronic vitamin E deficiency in developed countries is usually associated with disorders of lipid absorption. Vitamin E has a role in maintaining cell membrane structure and as a free radical scavenger. After long exposure to low concentrations of vitamin E, ataxia and axonal neuropathy may develop. The neurological features can improve with oral vitamin E treatment and it is also recommended that high doses of vitamin A should be given at the same time.
Niacin (vitamin B3), pantothenic acid (vitamin B5) and folic acid deficiencies
These are also associated with peripheral neuropathy.
Chronic hypophosphataemia
This is seen most commonly in patients on long-term parenteral nutrition and may result in a Guillain–Barré like syndrome.
Copper deficiency
Copper deficiency may be associated with gastric surgery and may mimic subacute combined degeneration of the cord (see p. 689).
Neuropathy associated with bariatric surgery
Untreated, up to 62% of patients who have had bariatric surgery develop a peripheral neuropathy. The neuropathy is predominantly sensory. Some 31% develop features of encephalopathy, and 28% have Wernicke–Korsakoff syndrome. The onset varies from 3 to 20 months following surgery; outcomes are mixed. The most frequent associated deficiencies are iron, thiamin, folate, vitamin D and calcium. Delayed (> 2 years after surgery) deficiency of copper has also been reported. Current guidelines recommend trace element and vitamin replacement and careful monitoring after surgery to minimize the development of neurological and other complications.
Strachan syndrome
Previously termed Jamaican neuritis, Strachan syndrome was first identified in the West Indies during periods of famine. It is characterized by a painful, predominantly sensory, ataxic neuropathy, and may also be associated with optic neuropathy, sensorineuronal hearing loss, dorsal lateral myelopathy, glossitis and stomatitis. It is thought to be caused by multiple deficiencies of thiamin, niacin, riboflavin, pyridoxine and cobalamin, and hence treatment is administration of multivitamins, although recovery may only be partial.
Metabolic neuropathies
Refsum disease (heredopathica atactica polyneuritiformis)
This is an autosomal recessive metabolic disease, characterized by accumulation of phytanic acid in plasma and tissues. It is caused by deficiency of peroxisomal phytanoyl-CoA hydroxylase (phytanic acid oxidase), which converts phytanic acid to α-hydroxyphytanic acid; mutations in the gene for this enzyme (located on chromosome 10p13) have been identified in most patients, although other genes may also be involved. A progressive symmetrical demyelinating sensorimotor neuropathy, cerebellar ataxia, retinitis pigmentosa initially presenting with night blindness, and ichthyosis are hallmarks of the disease. The age of onset of the disease can vary from childhood to adulthood, and as the condition progresses, the patient develops an ataxic gait, distal limb weakness and numbness. Other important clinical features include a ‘salt and pepper’ type of retinal pigmentation (flecks of dark pigment with areas of whitish depigmentation), nerve deafness, cataracts and cardiomyopathy. Skeletal deformities, including kyphoscoliosis, pes cavus, and shortened metacarpals and metatarsals, are also encountered. Optic atrophy, cataract and vitreous abnormalities cause further deterioration of vision.
The neuropathy starts distally with marked wasting and weakness. Symptoms of pain and paraesthesiae occur in some but not all patients. The tendon reflexes are lost as the condition progresses and there is loss of sensation (especially vibration sensation and joint position sense) in a glove and stocking distribution. The peripheral nerves may be enlarged. Without treatment, there is gradual deterioration, but about half of those affected may enter a phase of remission that can last for several years.
The diagnosis of Refsum disease is made on the basis of clinical findings and a plasma phytanic acid concentration > 200 μmol/L. However, plasma accumulation of phytanic acid is not specific for the condition as raised concentrations may occur in other peroxisomal disorders. In addition, patients have been recorded with defective α-oxidation but normal phytanic acid concentrations. Therefore, to be certain of the diagnosis of Refsum disease, it has to be shown that there is a defect in α-oxidation. This can be done by measuring the production of 14CO2 from a cultured skin fibroblast monolayer using (1-14C) phytanic acid as substrate. Cerebrospinal fluid protein concentration is often raised. Molecular genetic testing is available.
Phytanic acid is derived exclusively from the diet, therefore dietary control is the treatment of choice. This requires the elimination, as far as possible, of all dairy products and any fatty meat or oily fish. However, such a diet is protein and energy poor so liquid nutritional supplements are often required. In those who respond with a fall in plasma phytanic acid concentrations, progression of the neurological damage is halted, and in those with neuropathy, there is improvement in nerve conduction velocities. As dietary manipulation takes time to correct plasma phytanic acid concentrations, weekly plasma exchange can be used to attain normal concentrations quickly for patients with more rapid clinical progression.
Porphyric neuropathy
Porphyria is a rare but important cause of peripheral neuropathy. Its diagnosis depends on demonstrating an abnormality in haem metabolism (see Chapter 28). Only the hepatic porphyrias (acute intermittent porphyria, AIP, hereditary coproporphyria and variegate porphyria) are associated with neuropathy, the most severe attacks being associated with AIP. Within 2–3 days of the onset of an acute attack, the neuropathy begins with back and limb pains and the early development of marked symmetrical limb weakness, which may spread to involve the cranial nerves and respiratory muscles. The patient may have paraesthesiae and the sensory loss may be either in a symmetrical glove and stocking distribution or patchy. Autonomic features can also be present – hypertension, tachycardia, constipation and bladder disturbance. Other clinical problems such as seizures or psychiatric disturbance and a normal CSF protein may help to distinguish porphyric neuropathy from the Guillain–Barré syndrome. The exact pathogenetic mechanism is not clear but evidence suggests that both direct toxic effects of 5-aminolaevulinic acid and intracellular metabolic derangements, including reduced activity of Na+,K+-ATPase, contribute to the neurological disorders.
Treatment of an acute episode includes avoiding any drugs that may exacerbate the condition, and appropriate sedation and pain relief. A glucose infusion should be commenced. The use of specific measures, for example haematin, is discussed in Chapter 28. Recovery is usually good but may be incomplete in some patients.
Fabry disease (angiokeratoma corporis diffusum; α-galactosidase deficiency)
This is an inherited X-linked recessive disorder of glycosphingolipid metabolism resulting from a deficiency of the enzyme ceramide trihexosidase (α-galactosidase). The gene for this enzyme (GLA) lies on the long arm of the X chromosome between Xq21.33 and Xq22. However, females heterozygous for the condition have low enzyme activity and can be mildly affected clinically. The accumulating ceramide trihexoside consists of sphingosine (a long chain amino alcohol) linked to a long chain fatty acid. The trihexose (glucose–galactose–galactose) is linked to the primary alcohol group on C1 of ceramide. Because of the low ceramide trihexosidase activity, cleavage of the terminal galactose from the ceramide trihexoside does not occur.
The clinical effects are produced by the deposition of ceramide trihexoside (globotriaosylceramide) in the vascular endothelium. The characteristic manifestations are renal impairment, neuropathic pain and cutaneous angoikeratomas in a bathing trunk distribution. The onset of symptoms is in late childhood or early adolescence, with episodes of severe shooting and burning pains and paraesthesiae in the feet and hands (Fabry crisis), which may be severe enough to confine the patient to a wheelchair. Exercise, fatigue and emotional stress are recognized precipitants of acute painful episodes. The symptoms subside as the patient gets older. In contrast, the characteristic purple skin lesions, corneal dystrophy and renal involvement causing hypertension and CKD become more evident with age. Involvement of the myocardium and mitral valve disease (incompetence and prolapse) are very common, but established renal failure is the usual cause of death. Clinical examination reveals only minimal abnormalities suggesting a peripheral neuropathy. Anhidrosis may suggest autonomic involvement and nerve conduction studies show a mild axonal neuropathy with a selective loss of small myelinated fibres and unmyelinated axons.
Diagnosis in males is reliably made by measuring ceramide trihexosidase activity in plasma, leukocytes or cultured skin fibroblasts. Blood spots can be used for screening at-risk populations. Enzyme activity can also be measured in duodenal or jejunal biopsy material. Glycolipid can be measured in urine sediment. Molecular genetic testing is the most reliable method for the diagnosis of carrier females. Predictive testing is available for at-risk asymptomatic adults and for high risk pregnancies. Standard treatment is enzyme replacement therapy. There have been no human trials of gene replacement to date.
Cerebrotendinous xanthomatosis (cholestanolosis)
Cerebrotendinous xanthomatosis is a rare, autosomal recessive lipid storage disease with multiorgan involvement. The clinical manifestations usually start at infancy and develop during the first and second decades of life. Peripheral neuropathy, especially the subtype of axonal sensorimotor neuropathy, is common. Patients may also develop a progressive cerebellar ataxia, juvenile cataracts, chronic diarrhoea and tendon xanthomas. A deficiency of hepatic mitochondrial 27-hydroxylase has been identified. Biochemical findings include high plasma and tissue cholestanol concentration, normal to low plasma cholesterol concentration, decreased chenodeoxycholic acid, increased concentration of bile alcohols and their glycoconjugates and increased concentrations of cholestanol and apolipoprotein B in cerebrospinal fluid. Molecular genetic testing for CYP27A1, the only gene in which mutations are known to cause cerebrotendinous xanthomatosis, is available.
Treatment with chenodeoxycholic acid inhibits abnormal bile acid synthesis and is most effective in reducing elevated plasma cholestanol concentrations and reducing bile alcohols. The use of hydroxymethylglutaryl-CoA (HMG-CoA) reductase inhibitors (statins) may offer some help in slowing the neurological progression.
Tangier disease
This is a rare autosomal recessive disorder of lipid transport, characterized by a deficiency of plasma high density lipoprotein (HDL) and tissue storage of cholesteryl esters, especially in the tonsils, giving them a distinct orange–grey colour (see also Chapter 37). The cause of the neuropathy is not clear, but it seems unlikely that low HDL alone is responsible. Neuropathy occurs in about two-thirds of patients and may be the initial symptom in one half of cases, although in others it is evident only on clinical examination. The low plasma concentrations of HDL are thought to be from increased breakdown. There is hypocholesterolaemia with normal or increased concentrations of triglycerides. There is no specific treatment.
Amyloidosis
There are several types of amyloidosis, including primary, secondary, familial and dialysis-related. The associated constitutional symptoms of weight loss and other organ involvement (e.g. heart or kidneys) should provide the clue to consider amyloid as a cause of neuropathy.
Amyloid neuropathy occurs in approximately 15% of patients with primary amyloidosis, and the median duration of symptoms before diagnosis is 29 months. It can be symmetrical or focal, and can have variable degrees of sensory and/or motor involvement. The neuropathy is chronic, debilitating and relentlessly progressive. Autonomic disturbances also occur. Associated biochemical abnormalities are summarized in Box 36.7.
Secondary amyloidosis occurs in conditions such as chronic infection and rheumatoid arthritis and will not be discussed in detail here.
Familial amyloid polyneuropathy (FAP) is usually dominantly inherited and caused by a mutation in the gene coding for transthyretin (TTR, prealbumin), a protein that is produced in the liver and normally functions as a carrier molecule for thyroxine and retinol. As the same mutation is found in Swedish, Portuguese and Japanese patients, it is likely that the original mutation was in the Viking population that travelled to Europe and the Far East.
The diagnoses of primary and familial amyloidosis can only be made by demonstrating amyloid deposits in appropriately selected tissue. Initial samples are obtained from low-risk sites such as abdominal wall subcutaneous fat, bone marrow or rectum, and only if these are normal should an affected organ be biopsied. If TTR amyloid is detected, the gene should be sequenced to confirm the mutation, which will allow appropriate genetic counselling.
In FAP, younger patients with aggressive disease may be considered for liver transplantation, which seems to have a favourable effect on the course of neuropathy but not on cardiac or ocular lesions. Oral administration of tafamidis meglumine, which prevents misfolding and deposition of mutated TTR, is under evaluation in patients with FAP. High dose melphalan and stem cell transplantation can be effective. Supportive treatment such as management of neuropathic pain and gastroparesis is also very important. Treatment with melphalan and prednisone gives a survival benefit for patients with primary amyloidosis.
Mitochondrial disorders
The term ‘mitochondrial encephalomyopathy’ reflects the major neurological features – central nervous system and muscle – associated with mitochondrial disorders. Muscle disorders and the relevant mitochondrial enzyme defects are discussed in Chapter 33. It is not uncommon to find some minor peripheral nerve involvement in mitochondrial diseases (approximately 30% of patients), but it is rare for it to be the presenting or dominant clinical feature. Table 36.8 summarizes the main associations.
TABLE 36.8
Neuropathy in mitochondrial disease
| Type | Mitochondrial disease |
| Axonal | MELAS (A3243G mutation). Sensorimotor neuropathy can be acute or subacute |
| Demyelinating | MNGIE. Symmetrical sensorimotor neuropathy (occasionally axonal change predominates) MELAS |
MELAS, mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes; MNGIE, mitochondrial neurogastrointestinal encephalopathy.
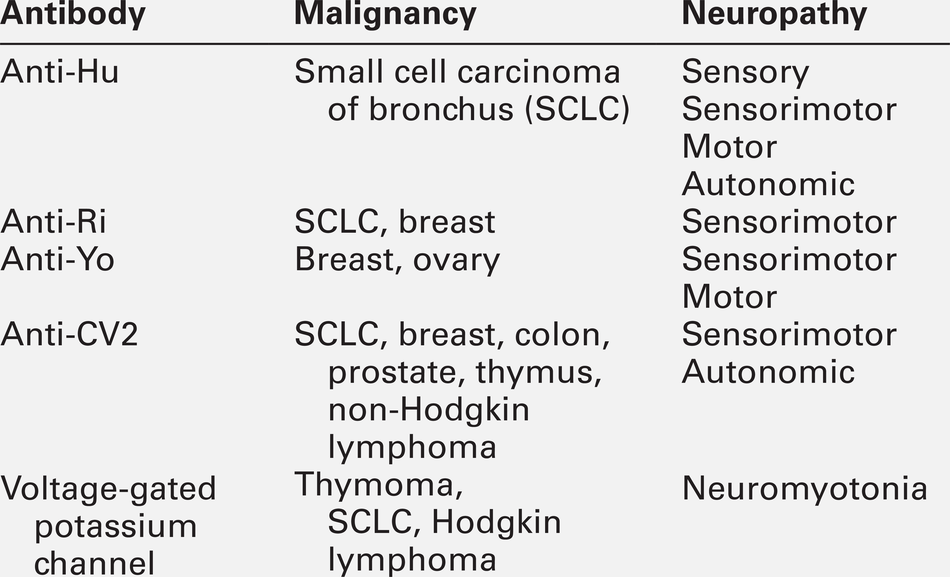
Paraneoplastic neuropathies
The paraneoplastic neurological syndromes occur in < 1% of malignancies and can present months or years before a cancer diagnosis. More than 25% of paraneoplastic neurological syndromes present as diseases of the peripheral nervous system, including sensory and autonomic neuropathies and Lambert–Eaton myasthenic syndrome.
The most common underlying malignancy in paraneoplastic neuropathies is a small cell carcinoma of bronchus, occurring in 50–75% of patients, presenting classically with a subacute sensory neuropathy and associated with anti-Hu antibodies (also known as antineuronal nuclear antigen antibodies or ANNA-1) in the plasma or CSF. In the majority of patients, the neurological involvement extends beyond the peripheral nervous system, and includes brain stem or limbic encephalitis and cerebellar degeneration.
The other nervous system-specific antibodies are outlined in Table 36.9. Although useful to confirm a clinical diagnosis of paraneoplastic syndrome, most of these markers are not specific enough to determine the origin of the cancer. Examination of CSF often shows an elevated protein with oligoclonal bands and an increased lymphocyte count. The tumour remains undetectable in 15% of patients.
The response to immunomodulatory therapies (steroids, immunosuppressants, intravenous immunoglobulin and plasma exchange) is variable.
MOVEMENT DISORDERS
The term ‘movement disorders’ refers to the neurological conditions that have as their predominant feature an excess or a paucity of movement, which may be voluntary or automatic. Disorders in which there is a predominant excess of movement are often referred to as hyperkinesias. Dyskinesia is also often used as a term referring to hyperkinetic involuntary movements. The five major categories of dyskinesias, in alphabetical order, are chorea, dystonia, myoclonus, tics and tremor.
Disorders with a paucity of movement are referred to as the hypokinesias. The characteristic hypokinetic disorder is Parkinson disease, in which there is bradykinesia (a slowness of movement), reduced amplitude of movements, rigidity and loss of postural reflexes. Tremor (a dyskinesia) is also a common feature of Parkinson disease.
Many of the movement disorders are associated with alterations in the basal ganglia or their connections. They can also arise as a consequence of cerebellar disease, which typically results in impairment of coordination (asynergy, ataxia), problems with judgement of distance (dysmetria) and a kinetic limb tremor.
Myoclonus and many forms of tremors do not appear to be related primarily to basal ganglia pathology, and often arise as a consequence of abnormal neural activity elsewhere in the central nervous system. Myoclonus can also be present in diseases that involve the cerebellum, such as mitochondrial diseases and storage disorders (e.g. sialidosis and ceroid lipofuscinosis).
In the late onset sporadic movement disorders, protein misfolding and aggregation, with neuronal dysfunction and cell death, is the likely underlying pathological process. In diseases with a younger age of onset, genetic abnormalities leading to cell damage (often owing to deficiencies of proteins involved in mitochondrial function) or biochemical abnormalities (such as deficiencies in enzymes involved in dopamine synthesis) as well as metabolic or storage disorders are more likely.
Parkinsonism
Parkinsonism may be caused by many conditions other than Parkinson disease (PD). However, by far the commonest cause of parkinsonism is idiopathic PD, which is the commonest condition seen in adult movement disorder clinics. Whilst a large number of other movement disorders are genetic in aetiology, patients with a monogenic form of PD are rare. Abnormalities in ten genetic loci have so far been identified in PD. Mutations of the PARK2 gene, which encodes the protein parkin (a component of a ligase complex that has a role in protein degradation) are the commonest autosomal recessive mutations in young onset patients. Mutations of PARK8, which result in abnormalities of LRRK2 (leucine-rich repeat kinase 2, a protein kinase) are the commonest dominant mutations identified. There are likely to be a large number of susceptibility genes for the development of PD, the most recently recognized significant one being a glucocerebrosidase gene mutation, which in homozygous individuals causes Gaucher disease.
The clinical syndrome of PD arises predominantly as a consequence of loss of nigrostriatal dopaminergic neurons but other neuronal populations including serotonergic, noradrenergic and cholinergic neuronal transmission are also important and are increasingly recognized as having a likely role in the development of the non-motor features of PD, such as autonomic dysfunction (postural hypotension, abnormal sweating, bladder, bowel and sexual dysfunction), mood disorders, sleep disorders and dementia.
The phenotype of monogenic PD is almost identical to that of sporadic disease; individuals are very responsive to medication but dystonia in the limbs may be present at onset and patients often develop early motor complications. The so-called ‘atypical parkinsonian syndromes are much less common than idiopathic PD and are generally not responsive to the usual first line treatment, levodopa, progress more rapidly and lead to a greater loss of independence with a mean survival of 3–9 years.
There are other rarer causes of parkinsonism, including mitochondrial disorders and neurometabolic disorders. Drug-induced parkinsonism from dopamine blocking drugs is an important differential diagnosis in anyone presenting with a relatively acute disorder.
Tremor
Tremor is a rhythmic oscillation of one or more body parts and is probably the commonest type of movement disorder presenting to neurologists. Tremor can be classified according to its phenomenology (rest, action, kinetic, task specific and body part involved), frequency and amplitude or, more specifically, by aetiology (e.g. Parkinson disease, essential tremor or secondary tremor as in thyrotoxicosis). The parkinsonian syndromes and other degenerative disorders are the commonest causes of rest tremor, along with secondary causes, such as toxins and drug therapies. The commonest causes of action or kinetic tremors are enhanced physiological tremor (stress, anxiety and exercise), essential tremor and cerebellar disease.
Dystonia
Dystonic contractions of muscles have a longer duration than myoclonus or chorea, involve simultaneous contractions of agonist and antagonist muscles, result in twisting (torsion) postures of the body parts involved, and consistently involve the same pattern of muscle movements.
Dystonia can be classified based upon the age of onset (important as the younger the age of onset, the more likely it is that it will spread to involve other body parts) the body part involved (focal, multifocal, hemidystonia, segmental or generalized) and additionally whether it is:
• primary (usually genetic or undetermined)
• a ‘dystonia plus’ syndrome with associated parkinsonism or myoclonus
• a secondary dystonia (as a consequence of brain injury or drug therapy)
• heredodegenerative dystonia (associated with diseases such as Huntington chorea)
• dystonia as a feature of other neurological diseases, such as Parkinson disease.
Adult-onset primary focal dystonia is the commonest form and typically affects the neck muscles (cervical dystonia or spasmodic torticollis). Task-specific action dystonias include writer’s cramp and musician’s dystonia. A comprehensive description of all of the dystonic disorders is beyond the scope of this chapter but important and treatable causes based on the aetiologic classification are briefly described below.
DYT1 dystonia (Oppenheim dystonia)
This is an autosomal dominant condition with a penetrance of between 30% and 40%. There is a deletion of one of a pair of CAG triplets in the TOR1A gene (also known as DYT1) on chromosome 9q. This gene codes for torsin A, a heat shock protein. The mutation is commoner in the Ashkenazi Jewish population (1 in 2000). The onset of dystonia is typically before the age of 40 and it usually involves the upper or lower limbs first. It is a pure dystonic syndrome and conventional imaging (MRI) is normal. Patients with severe generalized dystonia as a consequence of this condition may respond well to deep brain stimulation.
Dopa-responsive dystonia (DRD)
Some forms of dystonia respond to levodopa therapy (e.g. dystonia associated with Parkinson disease) but the term ‘dopa-responsive dystonia’ is specifically reserved for the syndrome arising as a consequence of an autosomal dominant mutation in the GTP-cyclohydrolase 1 gene. It is a disorder of tetrahydrobiopterin synthesis leading to inhibition of phenylalanine, tyrosine and tryptophan hydroxylases and failure of production of catecholamines and indoleamines. Dopa-responsive dystonia should be considered in anyone with early onset dystonia because treatment with L-dopa can lead to dramatic resolution of symptoms. It is an important differential to consider in any patient with presumed dystonic cerebral palsy or young onset spastic paraparesis. The most important diagnostic test is a trial of L-dopa. Other helpful tests include:
• CSF analysis to detect reduced total biopterin and neopterin concentrations
• an oral phenylalanine loading test: a positive result is an elevated plasma phenylalanine:tyrosine ratio following oral phenylalanine.
The gene causing DRD has been mapped to chromosome 14q, although approximately 40–50% of patients with DRD have no identified mutation. If a patient is diagnosed with DRD, there should be an assiduous search for other possibly affected family members (misdiagnosed in some cases to have multiple sclerosis or cerebral palsy), who may benefit from L-dopa therapy.
Tetrahydrobiopterin (TH) deficiency is usually a more serious form of DRD and is transmitted as an autosomal recessive disorder, presenting with dystonia and parkinsonism in infancy. Biochemical analysis of CSF will provide some evidence, but ultimately testing for the gene mutation is required to establish the diagnosis. Decreased CSF concentrations of homovanillic acid and 3-methoxy-4-hydroxyphenylethylene glycol, with normal 5-hydroxyindoleacetic acid concentrations, are the biochemical hallmarks of TH deficiency.
Wilson disease
Wilson disease arises as a consequence of an inborn error of copper metabolism manifesting as hepatic cirrhosis and basal ganglia damage (hepatolenticular degeneration). It can present in a number of different ways and so any patient with a movement disorder under the age of 50 years should be tested. Nearly all patients with neuropsychiatric Wilson disease have Kayser–Fleischer rings (copper deposition at the edge of the corneas), hence slit-lamp examination is an effective screening tool. The average delay from symptom onset to diagnosis is close to 24 months. The initial manifestations of the illness are neurological in ~ 40% of patients (usually after the age of 12 years). The remainder present with features of liver disease (~ 40%) or a psychiatric illness.
Wilson disease is inherited as an autosomal recessive trait. The gene responsible lies on chromosome 13q14.3 encoding for a copper-transporting P-type ATPase (ATP7B). The enzyme binds copper in its large N-terminal domain and aids in intracellular processing in the hepatocyte. Intestinal absorption of copper is normal in Wilson disease. There are two pathways for copper excretion from the hepatocyte aided by ATP7B. One is by attachment to caeruloplasmin in the Golgi apparatus, and subsequent delivery of the copper–caeruloplasmin complex into the plasma. A second is promotion of copper excretion into the bile. Mutations of ATP7B lead to failure to excrete copper by both routes, with accumulation of free copper in the circulation and in the tissues, including liver, brain, eyes, kidneys and bones.
The diagnosis and treatment of Wilson disease are discussed in Chapter 14.
Chorea
The term chorea is derived from a Greek word meaning ‘dance’. Historically, it was first used to describe St Vitus dance, which may have simply described chaotic but voluntary movements occurring in the context of religious fervour. Sydenham chorea describes a childhood chorea occurring as a parainfectious component of acute rheumatic fever but the best known ‘choreic’ disorder is probably Huntington disease.
Chorea consists of involuntary, continual, abrupt, rapid, brief, unsustained, irregular movements. Its causes can be divided into inherited and acquired. Huntington disease is the commonest inherited disorder: the rest are all rare. Inherited neurometabolic disorders such as the lysosomal storage disorders, amino acid disorders and glucose transporter deficiency syndromes can also have chorea as a predominant phenotypic feature. Of the acquired causes, focal striatal pathology (strokes and space occupying lesions), drug-induced disorders (such as the chorea seen as a complication of long-term dopaminergic therapy in Parkinson disease), chorea gravidarum and thyrotoxicosis are important causes, as are systemic lupus erythematosus, primary antiphospholipid syndrome and central nervous system (CNS) infection.
Myoclonus
The literal meaning of myoclonus is a ‘quick movement of a muscle’. Myoclonus can be positive (an active muscle contraction) or negative (sudden and brief loss of muscle tone). The commonest pathological form is probably cortical myoclonus caused by metabolic or toxic encephalopathies. Brief muscle jerks as a consequence of cortical motor discharges, sometimes in response to action or sensory stimuli, are seen.
Asterixis is a form of negative myoclonus in which there is a brief loss of muscle tone, most commonly affecting gait and sustained upper limb posture. ‘Liver flap’, with an irregular loss of wrist extensor tone seen in hepatic failure and carbon dioxide retention, is a form of metabolic upper limb myoclonus.
Tics
Tics are brief and intermittent movements of muscles and groups of muscles resulting in sudden, abrupt and repetitive movements that may mimic gestures or be fragments of normal behaviour. Tics may therefore be simple or complex movements. Gilles de la Tourette syndrome is the commonest tic disorder and is characterized by motor and vocal or phonic tics beginning in childhood, often accompanied by obsessive–compulsive disorders, poor impulse control and other behavioural problems.
ATAXIA
Ataxia, meaning a ‘lack of order’, is a term which refers to the type of clumsiness produced by dysfunction of the cerebellum or cerebellar pathways. Impaired balance, gait disorder and clumsiness of the hands, together with dysarthria and jerky tremor of the eyes (nystagmus) are the typical clinical signs. The commonest causes of acquired cerebellar ataxia are acute and chronic alcohol consumption, prescribed drug intoxication (often anti-epileptic drugs), cerebrovascular disease, metastatic cancer and multiple sclerosis. The differential diagnosis in patients where these are excluded is very long and includes all types of neurological pathological processes. The most important feature in the history is the age at onset of ataxia. Ataxia presenting in childhood is more likely to be due to an inherited disorder and onset in adulthood is more likely to be a sporadic ataxia.
The autosomal dominantly inherited cerebellar ataxias (or spinocerebellar ataxias) may be caused by a daunting list of genetic abnormalities, many of which are trinucleotide repeat disorders. No single clinical feature is specific for a particular mutation and even within families there is considerable variability in the phenotype. There are a large number of recessive ataxias but the commonest by far is Friedreich ataxia, which has a prevalence of about 1 in 50 000.
Friedreich ataxia
In this condition, in addition to the ataxia and dysarthria, there may be sensory loss and corticospinal tract signs with absent reflexes. Neurophysiological testing reveals an axonal peripheral neuropathy and skeletal abnormalities (e.g. kyphoscoliosis), hypertrophic cardiomyopathy, and diabetes are common. The genetic abnormality is an expanded trinucleotide repeat of GAA in a gene on chromosome 9q coding for the protein called frataxin. Point mutations are less common. Frataxin is a mitochondrial protein encoded by nuclear DNA and is probably involved in iron transport. Excessive iron accumulation in mitochondria is a feature of the condition.
Ataxia with isolated vitamin E deficiency
This is a rare, but important, disorder that has a phenotype similar to Friedreich ataxia but with additional ophthalmoplegia and retinitis pigmentosa. It arises as a consequence of defects in the TTP1 gene coding for α-tocopherol transfer protein that incorporates α-tocopherol into lipoproteins secreted by the liver. The reason for its importance is that it can be treated with vitamin E.
Abetalipoproteinemia
This is a rare autosomal recessive deficiency of apolipoprotein B-containing lipoproteins. The abnormality arises as a consequence of microsomal triglyceride transfer protein (MTP) deficiency. The age of onset is typically in teenage years or early adulthood and the syndrome itself arises as a result of vitamin E deficiency. The main features are a cerebellar ataxia with polyneuropathy, acanthocytosis (characteristically shaped abnormal red blood cells), a coeliac syndrome, and retinal degeneration. It can be treated with high doses of vitamin E.
Ataxia telangiectasia
Ataxia telangiectasia arises as a consequence of a mutation in the ‘ataxia telangiectasia mutated’ (ATM) gene, which codes for a protein kinase that plays an important role in cell cycle control, apoptosis and DNA double-strand break repair. As well as an ataxic neurodegenerative disorder, the abnormality leads to an increased incidence of malignancy. The disorder begins early in childhood, typically at the age of 1–2 years, with truncal ataxia and dysarthria. Oculocutaneous telangiectasias, recurrent sinopulmonary infections and malignancies, particularly leukemia or lymphoma are characteristic features. Plasma α-fetoprotein concentration is elevated, there is evidence of both B cell and T cell immune deficiency and chromosome breaks can be identified.
Early onset ataxia with oculomotor apraxia and hypoalbuminemia
This is caused by a mutation in the aprataxin gene. Cerebellar ataxia and peripheral neuropathy are the commonest features along with oculomotor apraxia. Choreiform movements of the limbs and mental deterioration are also seen. Hypoalbuminemia and hypercholesterolemia are typical and brain MRI or computed tomography show marked cerebellar atrophy. Ataxia with oculomotor apraxia type 2 has also been identified with a similar clinical picture but with mutations in the senataxin gene.
Fragile X-associated tremor/ataxia syndrome
This condition is associated with a tremor that appears very similar to that of essential tremor. Other clinical features include a cerebellar ataxia and mild to moderate executive dysfunction. Both sexes can be affected. The disease is rare and is characterized pathologically by intranuclear inclusions. An increased T2 signal in the middle cerebellar peduncle is a relatively specific MRI appearance.
Hexosaminidase deficiency (GM2 gangliosidoses)
This group of conditions is characterized by accumulation of GM2 ganglioside and glycosphingolipids within neuronal cells (and other cell types) owing to a genetically determined deficiency of the enzyme hexosaminidase. Depending on subunit composition, there are three possible isoenzymes: hexosaminidase A (αβ), hexosaminidase B (β2) and hexosaminidase S (α2). The two classic subtypes are Tay–Sachs disease and Sandhoff disease.
In Tay–Sachs disease, there is absence of hexosaminidase A (αβ) and hexosaminidase S (α2). Hexosaminidase B (β2) activities are normal or slightly elevated. The abnormality lies in the α-subunit, possibly owing to a mutation at the locus on chromosome 15. In Sandhoff disease, there are low activities of both hexosaminidase A and B. Hexosaminidase S activity is increased. There is a deficiency of the β-subunit owing to a mutation at the locus on chromosome 5.
There is a spectrum of clinical presentations of these conditions:
• juvenile onset is at 1–4 years, with dementia, seizures and ataxia. A cherry-red spot at the macula may or may not be present
• an ataxic form of GM2 gangliosidosis is characterized by a slowly progressive cerebellar or spinocerebellar ataxia with onset in late childhood or the adolescent period, which can be the manifestation of either hexosaminidase A or hexosaminidase A and B deficiencies
• a spinal muscular atrophy form is recognized with slowly progressive generalized muscle wasting and weakness. Only a deficiency of hexosaminidase A has been identified in this form, which can present in childhood or early adulthood
• an adult onset type is characterized by early onset of dementia, seizures and normal pressure hydrocephalus.
In addition to measuring hexosaminidase activities in plasma, leukocytes or cultured skin fibroblasts, a rectal biopsy should be done in adult onset cases to look for the characteristic changes in autonomic nerve fibres that are apparent on electron microscopy.
Cerebrotendinous xanthomatosis (cholestanolosis)
This is a leukodystrophy characterized by severe progressive ataxia and limb spasticity by the age of 30. Characteristic appearances include tendon xanthomas and zonular cataracts. Dementia may occur at a young age before the other physical signs are evident and it can be difficult to make a clinical diagnosis at this early stage. Pathologically, there is widespread CNS demyelination with degeneration of the spinal cord. See page 695 for further details about the underlying biochemical abnormalities and potential treatment.
Neuronal ceroid lipofuscinosis
This is another condition in which the age of presentation determines the subtype. Early development is usually normal, with the age of onset from six months to 30 years. Patients present with myoclonus, ataxia and visual loss (except in the late onset variety). If seizures are a feature, they tend to be refractory to treatment. Most cases are autosomal recessive but the adult onset can be autosomal dominant. The incidence varies throughout the world with a particular predilection for the infantile subgroup in Finland of 1:13 000 live births.
Histological examination reveals an accumulation of autofluorescent lipopigment inclusions in body tissues and the diagnosis is made from biopsy of skin, conjunctival or rectal tissue. Abnormal accumulation of S-methylated methionine has been found in the brains of affected infants and of ε-N-trimethyl lysine in those from patients with the juvenile onset form. There is a profound deficiency of endo-β-N-acetylglucosaminidase, which is required for the cleaving of chilobiosyl residues. All forms of this condition lead to learning disability, seizures, ataxia and, in the early onset types, visual loss. Life expectancy is severely limited.
Coeliac disease
This is an immunologically mediated disease (see also Chapter 12) strongly associated with certain human leukocyte antigens. It is common and in large case series, neurological complications have been reported in up to 6% of patients. Cerebellar ataxia is one of the commonest neurological associations. The occurrence of antigliadin antibodies in otherwise idiopathic ataxia requires further dedicated research studies and it remains possible that adult onset sporadic ataxia with no clear cause and associated coeliac disease is no more than a chance occurrence.
INFLAMMATORY DISORDERS OF THE CENTRAL NERVOUS SYSTEM
Inflammation is a common pathological process within the CNS, by far the commonest form being multiple sclerosis (MS). Diagnosis is made on clinical grounds together with MRI evidence. Oligoclonal bands are present in the CNS in up to 95% of patients with clinically definite MS (see Chapter 34). Other inflammatory conditions affecting the CNS include systemic lupus erythematosus, Sjögren disease, Behçet disease, polyarteritis nodosa, paraneoplastic disorders, acute disseminated encephalomyelitis and vasculitis. Mimics of MS also include granulomatous disorders (sarcoidosis, Wegener granulomatosis), diseases of myelin (metachromatic leukodystrophy, adrenomyeloleukodystrophy), vitamin B12 deficiency, and certain infectious diseases (Lyme disease, neuroborreliosis, human T-lymphotropic virus type 1, HIV, progressive multifocal leukoencephalopathy and neurosyphilis).
CONCLUSION
The conditions discussed above represent a very small fraction of all neurological disorders. There are many more for which clinical biochemistry, at our current level of understanding, has little to offer in everyday clinical practice. These include headache, epilepsy, sleep disorders, cerebrovascular disease (beyond glucose and lipid chemistry), neurological malignancies, dementia and neurological trauma. There are also many conditions, for which biochemical perturbations are an important part of diagnoses, however a full discussion is beyond the scope of this chapter. These include many other inherited metabolic diseases, a multitude of which have neurological manifestations.
ACKNOWLEDGEMENT
We would like to acknowledge the contribution of J. Gareth Llewelyn, Mark Cossburn, Alistair Church and Huw R. Morris, who wrote the chapter for the previous edition of the book.
Further reading
Albers JW, Fink JK. Porphyric neuropathy. Muscle Nerve. 2004;30:410–422.
Hughes R. Rational testing: investigation of peripheral neuropathy. Br Med J. 2010;341:c6100.
Sullivan KA, Feldman EL. New developments in diabetic neuropathy. Curr Opin Neurol. 2005;18:586–590.
Internet resources
Washington Neuromuscular Disease Center: neuromuscular.wustl.edu; [Accessed March 2013].
Website of the Washington Neuromuscular Disease Center.
American Academy of Neurology. www.aan.com/go/practice/guidelines; [Accessed March 2013].
The clinical practice guidelines page of the American Academy of Neurology.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 36
Biochemical aspects of neurological disease
Paul Hart; Clare M. Galtrey; Dominic C. Paviour; Min Htut
CHAPTER OUTLINE
Toxic and metabolic encephalopathy
Vitamin B12 deficiency (subacute combined degeneration of the spinal cord)
Small fibre painful axonal neuropathy
Acute inflammatory neuropathies and variants
Chronic kidney disease and established renal failure
Nutritional peripheral neuropathies
Neuropathy associated with bariatric surgery
Ataxia with isolated vitamin E deficiency
Early onset ataxia with oculomotor apraxia and hypoalbuminemia
Fragile X-associated tremor/ataxia syndrome
Hexosaminidase deficiency (GM2 gangliosidoses)
Cerebrotendinous xanthomatosis (cholestanolosis)
Neuronal ceroid lipofuscinosis
INTRODUCTION
Despite the significant advances seen in laboratory and imaging diagnostics, many of the conditions seen by neurologists are still diagnosed on purely clinical grounds. In neurology, there remains no substitute for a meticulous clinical history and careful examination.
Neurological investigations include all imaging modalities, neurophysiological tests of both the central and peripheral nervous system, and laboratory based investigations, including haematology, biochemistry, immunology, histopathology and cytology. This chapter will focus on the neurological conditions for which biochemical abnormalities are pertinent. Separate chapters cover the biochemistry of cerebrospinal fluid and muscle disease and other conditions that overlap with general neurological practice.
ENCEPHALOPATHY
‘Encephalopathy’ is a term for any diffuse disease of the brain that alters brain function or structure. It is a common condition that encompasses coma, acute confusional state, delirium and dementia. The hallmark of encephalopathy is an altered mental state.
Encephalopathy is caused either by processes that directly affect the structure of the brain or by systemic and metabolic factors (see Box 36.1). Encephalopathy can occur acutely (over hours to days); subacutely (over weeks to months) or chronically (over years) (Table 36.1). Although the mechanism of many causes of encephalopathy is well understood, the pathophysiology of metabolic encephalopathy is less clear but is likely to involve changes in amino acids and neurotransmitter profile.
For patients presenting with altered consciousness, a detailed history often has to be taken from family, friends and other healthcare professionals, to determine pre-existing medical conditions and time of onset of altered mental state. For example, patients presenting with altered consciousness following apnoea or cardiac arrest are likely to be suffering from a hypoxic–ischaemic encephalopathy but alternative causes need to be excluded. Access to medication, previous psychiatric problems or use of illicit drugs may suggest intoxication. If the patient is known to have diabetes, hypo- or hyperglycaemia is likely. Patients on diuretics are at risk of hyponatraemia.
Both a general systemic examination and detailed neurological examination are required to determine the cause of encephalopathy. Specific breath smells suggest certain causes, e.g. ammonia-like (uraemia); fruity-smelling (ketoacidosis); musty or fishy (acute hepatic failure); onion (paraldehyde), and garlic (organophosphates). Hypertension may indicate amfetamine or cocaine intoxication or a hypertensive encephalopathy. Examination of the skin may show signs of chronic liver disease, haemodialysis fistula, previous sternotomy scars from cardiac surgery or injection marks from intravenous drug use.
Neurological examination should be divided into focal (localizing) and global (non-localizing) signs. Global signs suggest a process affecting the whole brain and include altered consciousness (acute confusional state, delirium and coma). The level of coma can be quantified with the Glasgow Coma Score, for which the minimum score is 3/15 (Table 36.2).
TABLE 36.2
Glasgow coma scale (GCS)
The total is obtained by adding together the scores for each column
Other global neurological signs include generalized seizures, tremor, asterixis (abnormal jerking tremor in hands seen in liver flap) and myoclonus (involuntary brief jerk-like twitching, often multifocal). Focal signs indicate a localized problem within one part of the brain, for example visual or language disturbance from cerebral cortex pathology or eye movement disturbance, slurred speech or swallowing problems from brainstem pathology. Focal neurological signs usually suggest a primary neurological cause of encephalopathy.
Two-thirds of encephalopathies are secondary to systemic metabolic factors. It is therefore important that these are excluded before assuming there is a neurological cause. Many metabolic encephalopathies are reversible if corrected promptly. Investigations for a patient with encephalopathy should include blood tests, urinalysis, imaging studies, and, when indicated, cerebrospinal fluid examination and an electroencephalogram. Increasingly, laboratory investigations can be used to make a definite diagnosis of neurological causes of encephalopathy owing to the improved recognition of antibodies for autoimmune encephalitis syndromes and biomarkers in neurodegenerative disease (Table 36.3).
Toxic and metabolic encephalopathy
Toxic encephalopathy is caused by exogenous substances including solvents, drugs, radiation, paints, industrial chemicals, and certain metals. Some of the more common causes are discussed below. More details can be found in Chapter 40.
Carbon monoxide
This colourless and odourless gas is the most common cause of accidental poisoning in Europe and North America, as well as being a major cause of suicidal deaths. When 20–30% of total haemoglobin is bound to carbon monoxide, it causes headache, nausea and shortness of breath; when 50–60% is bound, coma results. Carbon monoxide exposure is detected by measuring the carboxyhaemoglobin concentration in blood. Carbon monoxide poisoning is treated with oxygen, using hyperbaric facilities in severe cases, if available.
Alcohol
Alcohols, including ethanol, produce an altered mental state by central nervous system depression. Methanol and ethylene glycol similarly cause direct central nervous system depression but are also toxic via metabolism in the liver to formic acid (methanol) and glycolic and oxalic acids (ethylene glycol).
Methanol and ethylene glycol concentrations can be measured in the blood but usually the suspicion is raised by other biochemical abnormalities. Poisoning with either causes a high anion gap metabolic acidosis. Treatment is by blocking the action of alcohol dehydrodenase with ethanol or fomepizole to decrease the production of toxic metabolites.
Opioids
Overdose causes coma, pinpoint pupils and respiratory depression. This can be from iatrogenic, accidental or suicidal overdose (e.g. morphine) or from use of illicit drugs (e.g. heroin). Diagnosis is usually suspected from the clinical triad but a rapid urine screening test can confirm opioid exposure. Treatment is with naloxone, given as a bolus dose followed by an infusion if the patient responds, in addition to supportive care.
Thiamin (vitamin B1) deficiency
Wernicke–Korsakoff encephalopathy is a clinical triad of confusion, ataxia and ophthalmoplegia (eye movement disorder). It comprises two syndromes. First, Wernicke encephalopathy, an acute or subacute confusional state with ataxia and ophthalmoplegia and second, Korsakoff dementia, characterized by severe amnesia and confabulation. The former is often reversible but the latter is irreversible and persistent. Wernicke–Korsakoff encephalopathy is caused by thiamin deficiency and is most commonly seen in alcoholics but can also occur in patients with anorexia, hyperemesis gravidarum, those receiving total parenteral nutrition, or in the context of malabsorption and refeeding syndrome. The role of measurement of plasma thiamin concentrations in diagnosis and monitoring remains controversial and functional assessment by assay of red cell transketolase is no longer used by most laboratories for diagnosis. Therefore, laboratory tests are aimed at excluding other conditions and providing evidence of risk factors (e.g. macrocytosis and deranged liver function tests in alcoholism). Brain magnetic resonance imaging (MRI) can show signal change in the thalami, mammillary bodies, tectal plate and periaqueductal grey matter. Treatment is with parenteral thiamin. In the UK, normal practice is to give thiamin in a vitamin complex of riboflavin, nicotinamide, pyridoxine and ascorbic acid. Early treatment can reverse the symptoms rapidly and prevent dementia. Administration of intravenous glucose (including as part of parenteral nutrition) to patients who are severely malnourished, can exhaust their (already depleted) supply of thiamin and precipitate Wernicke–Korsakoff encephalopathy. Thiamin should therefore be administered before starting a glucose infusion in patients at high risk.
Vitamin B12 deficiency
This causes a typical pattern of degeneration of the white matter producing encephalopathy, myelopathy (subacute combined degeneration of the spinal cord), peripheral neuropathy and optic neuropathy. Vitamin B12 concentrations are easily measured and, if deficiency is corrected early, further damage can be prevented (See p. 688 and p. 693).
Liver failure
Hepatic encephalopathy occurs as a consequence of liver function disturbance and its severity is graded from 0 to 4 (Table 36.4).
TABLE 36.4
Grading of hepatic encephalopathy (West Haven classification system)
| Grading | Symptoms |
| 0 | Minimal hepatic encephalopathy. Lack of detectable changes in personality or behaviour. Minimal changes in memory, concentration, intellectual function, and coordination. Asterixis absent |
| 1 | Trivial lack of awareness. Shortened attention span. Impaired addition or subtraction. Hypersomnia, insomnia, or inversion of sleep pattern. Euphoria, depression, or irritability. Mild confusion. Slowing of ability to perform mental tasks. Asterixis can be detected |
| 2 | Lethargy or apathy. Slurred speech. Obvious asterixis. Drowsiness, lethargy, gross deficits in ability to perform mental tasks. Obvious personality changes, inappropriate behaviour and intermittent disorientation, usually regarding time |
| 3 | Somnolent but can be aroused. Unable to perform mental tasks. Disorientation about time and place, marked confusion, amnesia. Occasional fits of rage, present but incomprehensible speech |
| 4 | Coma with or without response to painful stimuli |
Two situations can lead to severe hepatic encephalopathy – acute fulminant hepatic failure and decompensation of chronic liver disease. In the UK, acute liver failure is most commonly from paracetamol overdose or viral hepatitis, and chronic liver disease from alcohol abuse or chronic hepatitis B or C infection. Encephalopathy is thought to develop because hepatocellular dysfunction produces neurotoxins (e.g. ammonia), false neurotransmitters and benzodiazepine-like substances. In chronic liver disease, there can be the additional effect of porto–systemic shunting. This allows significant quantities of ammonia, formed in the bowel from protein, to reach the systemic circulation. The severity of the neurological disorder correlates poorly with ammonia concentrations so measurement rarely contributes to management. ‘Liver function tests’ will confirm that the concentration of bilirubin is high, often with abnormal liver enzyme activities and, more importantly, poor synthetic function with abnormal clotting. Plasma paracetamol concentration can be measured if overdose is a possibility. Patients with acute liver failure may require liver transplantation for survival but the encephalopathy of chronic disease may be reversed by conservative measures (see Chapter 14 for further details).
Chronic kidney disease and established renal failure
Uraemic encephalopathy presents with apathy, fatigue, inattentiveness and irritability, followed by confusion, hallucinations, slurred speech, tremor and asterixis. The mechanism is unclear but correction of uraemia reverses the encephalopathy. Suggested mechanisms include retention of organic acids, elevation of phosphate concentration in the cerebrospinal fluid (CSF), increased calcium content of the cerebral cortex owing to the action of PTH, and alteration of concentrations of neurotransmitters or proinflammatory cytokines. Although the degree of the rise in urea correlates with the severity of encephalopathy, it is not thought to be causative. The investigation, monitoring and treatment of uraemia are discussed in Chapter 7.
Respiratory failure
Hypercapnia occurs in respiratory failure either secondary to lung disease (e.g. chronic obstructive pulmonary disease) or to mechanical problems such as neurological disease (e.g. myasthenia gravis). Clinically, hypercapnia presents with headache, papilloedema, mental slowing, drowsiness, confusion, coma and asterixis. The mechanism is unclear but thought to be due to a direct effect of carbon dioxide possibly on the hydrogen ion concentration (pH) of the CSF. Hypercapnia can be confirmed by measurement of PCO2 on an arterial blood sample. Coma can occur with PCO2 > 9 kPa. Treatment is of the underlying cause and, once corrected, there is no prolonged cerebral damage (see Chapter 5 for further details).
Cardiorespiratory failure
Hypoxic–ischaemic encephalopathy is caused by hypoxaemia and/or reduced blood flow to the brain from failure of the cardiac system, the respiratory system or both (e.g. myocardial infarction, drowning). When cerebral perfusion pressure falls, there is a failure of autoregulation leading to ischaemia and hypoxia and to cell death by both necrosis and apoptosis. The diagnosis is usually evident from the history. Myoclonus is common (Lance–Adams syndrome). There is a wide spectrum of clinical outcomes depending on the degree and duration of the insult from full recovery to severe prolonged disability and death.
Disorders of glucose metabolism
Hypoglycaemia causes confusion, seizures and coma and can cause permanent neurological damage if not reversed quickly. Up to 15% of patients with diabetes will have at least one episode of hypoglycaemic coma in their lifetime. Recurrent hypoglycaemia, such as that caused by an insulinoma, can be mistaken for epilepsy. The brain contains 1–2 g of glucose stored as glycogen, which is utilized within approximately 30 min if blood glucose concentration remains low. Ketones can also be used as an energy source but this is not sufficient in prolonged hypoglycaemia, nor are they available when hypoglycaemia is caused by high insulin concentrations. The most common causes of hypoglycaemia are accidental or deliberate overdose of insulin, insulinoma, and depletion of liver glycogen (e.g. acute liver failure, alcohol binge, severe starvation). Hypoglycaemia is easily detected by blood glucose measurement at the bedside (although it should be confirmed by laboratory measurements) (see Chapter 17 for further details).
Hyperglycaemic coma occurs in two forms – diabetic ketoacidosis and the hyperosmolar hyperglycaemic state. Coma is a late feature of diabetic ketoacidosis and develops when hyperglycaemia, dehydration, acidosis and shock are severe. Cerebral oedema is a rare but often fatal complication both of the condition and its treatment. The coma of the hyperosmolar hyperglycaemic state usually develops more insidiously and occurs in elderly patients, often with previously undiagnosed diabetes mellitus. Extreme hyperglycaemia with greatly increased plasma osmolality, electrolyte losses and dehydration exacerbated by impaired thirst awareness, are all thought to contribute to the development of the encephalopathy (see Chapter 16 for further details).
Hyponatremia
Hyponatremia has many causes, including liver and cardiac failure, syndrome of inappropriate antidiuresis (neurological causes of which include head trauma, bacterial meningitis, encephalitis, cerebral infarction, subdural and subarachnoid haemorrhage), vomiting and diuretic use. The severity of the symptoms is related to the rapidity of decline in plasma sodium. This is because hyponatremia causes extracellular hyposmolarity and a tendency for free water to shift from the vascular space to the intracellular space. When plasma sodium concentration falls slowly, over a period of several days or weeks, the brain is capable of compensating by extrusion of solutes to the extracellular space. This reduces the flow of free water into the intracellular space and symptoms are much milder for a given degree of hyponatraemia. When the plasma sodium concentration falls rapidly, this compensatory mechanism is overwhelmed and severe cerebral oedema may ensue. For example, a sodium concentration falling from normal to < 110 mmol/L over 24–48 h will often cause coma. It is necessary to correct the plasma sodium concentration in a controlled manner because of the risk of central pontine myelinolysis if the increase is too rapid (see Chapter 4).
Central pontine myelinolysis is characterized by quadriparesis, dysphagia, dysarthria, diplopia and altered consciousness. Areas of the brain other than the pons are also often involved. Magnetic resonance imaging reveals signal change within these areas but the appearances may be normal early in the course of the disease. Central pontine myelinolysis may prove fatal, and incomplete recovery is common in survivors.
Hypernatraemia
Common causes of hypernatraemia include diabetes insipidus, hyperglycaemia, diarrhoea and fluid deprivation. Again, it is the rate of change of sodium concentration that is important rather than the actual values. A sodium concentration of > 160 mmol/L can cause coma due to cerebral cellular dehydration if it develops rapidly. Treatment depends on the cause of the disorder and the total body fluid volume (see Chapter 4).
Hypercalaemia
Severe hypercalcaemia (> 3.4 mmol/L) can be lead to coma, preceded by fatigue, headache, muscle weakness, irritability and confusion. High concentrations of calcium ions decrease neuronal excitability, which together with dehydration is thought to cause the neurological problems (see Chapter 6).
Septic encephalopathy
Severe extracranial infection can be associated with an impaired mental state. The pathophysiological mechanism involves reduced cerebral blood flow and oxygen extraction by the brain, cerebral oedema, disruption of the blood–brain barrier owing to inflammatory mediators acting on the cerebrovascular endothelium, and abnormal neurotransmitter compositions. Diagnosis of sepsis can be aided by the finding of a raised white blood cell count and raised inflammatory markers, and by blood cultures. Treatment is of the underlying infection with supportive care.
Autoimmune encephalopathy
The last decade has seen significant advances in our understanding of antibody-mediated encephalopathies. There are two types of antibodies detectable in blood in these conditions (Table 36.5): paraneoplastic antibodies to intracellular targets that are associated with cancer but are not pathogenetic, and antibodies to the extracellular domain of neuronal cell-surface proteins that directly cause encephalitis and are also often associated with cancer. Autoimmune encephalitis typically presents with subacute memory loss, psychiatric and behavioural disturbance and seizures. The detection of one of the specific antibodies supports a diagnosis of paraneoplastic encephalitis. Treatment is of the underlying cancer and with immunotherapy.
Hashimoto encephalopathy presents with seizures, behavioural and psychiatric manifestations, movement disorders and coma and is associated with the presence of high titres of anti-thyroglobulin or anti-thyroid peroxidase (TPO, antimicrosomal) antibodies. The condition can present without any features of thyroid disease. It is unclear whether the anti-thyroid antibodies are pathogenetic or simply represent an immune epiphenomenon but it is important not to miss this diagnosis as the condition responds to treatment with steroids.
Dementia
Dementia is a syndrome characterized by progressive deterioration of cognitive function without alteration of consciousness. Cognitive deficits most commonly affect memory, but other cognitive domains such as language, praxis, visual perception and most notably executive function are also often affected. Diagnosis of dementia is generally by clinical criteria during life, although many of its causes are defined on the basis of histopathological criteria with the consequence that definitive diagnosis can only be made at post-mortem. However, over the last few years a number of laboratory CSF biomarkers have emerged and have an increasing role in diagnosis. Cerebrospinal fluid Aβ-40, Aβ-42, total tau and phosphorylated tau proteins are the most sensitive biomarkers for the diagnosis of Alzheimer disease and 14-3-3 protein for the diagnosis of Creutzfeldt–Jakob disease (see Chapter 34).
SPINAL CORD DISORDERS
Spinal cord disease usually presents with motor and/or sensory symptoms in either the upper and lower or just the lower limbs, depending on the site of the insult. Clinical signs include increased tone in the limbs, brisk reflexes and, typically, well-preserved muscle bulk. Bowel and bladder function can be compromised. The list of possible causes of spinal cord disease is extensive and includes vascular, degenerative, compressive, inflammatory, malignant, metabolic and infectious conditions. Metabolic causes include vitamin B12 deficiency (Box 36.2) and adrenomyeloneuropathy. A normal MRI scan of the spinal axis will exclude a significant number of differential diagnoses.
Vitamin B12 deficiency (subacute combined degeneration of the spinal cord)
Deficiency of vitamin B12 can produce haematological and neurological abnormalities: features of gastrointestinal disease may also be apparent (see Chapter 12). From an early stage, it is the spinal cord that is predominantly affected. The onset of symptoms is insidious, with symmetrical, uncomfortable, tingling paraesthesiae, initially in the feet but later involving the hands. As the condition progresses, the gait becomes ataxic (sensory ataxia owing to loss of proprioception) and the limbs become weaker and increasingly spastic. Additional problems include peripheral polyneuropathy, optic neuropathy, psychiatric disturbance and dementia.
The exact pathophysiological process leading to neuronal damage in vitamin B12 deficiency is unknown. Vitamin B12 is involved as a cofactor in the conversion of homocysteine to methionine and of methylmalonyl- CoA to succinyl-CoA. It is possible that the impaired synthesis of methionine leads to a depletion of S-adenosylmethionine, which is required for myelin synthesis.
The diagnosis is confirmed by measuring vitamin B12 concentration in the serum. In some patients, the concentration may be only ‘low-normal’. In borderline cases, measurement of homocysteine and methylmalonate concentrations can be useful. It is important that the diagnosis is made as early as possible as this is a potentially reversible condition. Magnetic resonance imaging of the spine often shows the spinal cord to be atrophic and can show signal change in the dorsal columns.
Folate deficiency
Folate deficiency is usually associated with other nutritional deficiencies. Folate antagonists (e.g. methotrexate, trimethoprim and pyrimethamine) are also potential causes. Neurological manifestations are rare and the same as with vitamin B12 deficiency. Treatment is by folate replacement, although any vitamin B12 deficiency should be treated first to prevent exacerbation of spinal cord degeneration.
Copper deficiency
Copper deficiency is rare and causes both haematological and neurological disease. Neurological manifestations include myelopathy, peripheral neuropathy and optic neuropathy. Copper deficiency was recognized in ruminants (swayback disease) long before it was identified in humans. The presentation can be similar to that of subacute combined degeneration of the spinal cord. It is usually secondary to gastric surgery, gastrointestinal diseases or total parenteral nutrition, or to excess intake of zinc or iron, which reduce copper absorption in the gut. Copper deficiency myelopathy has been attributed to ingestion of zinc contained in denture cream in some patients. Treatment is with oral or intravenous copper replacement.
Vitamin E deficiency
Vitamin E deficiency is rare, and causes a spinocerebellar ataxia, retinopathy, myopathy and anaemia. Poor diet is not usually sufficient cause unless associated with malabsorption. Abetalipoproteinemia is a rare inherited disorder of fat metabolism that results in poor absorption of dietary fat and vitamin E. Vitamin E deficiency is discussed further below (see ataxia).
Hepatic myelopathy
Hepatic myelopathy is a rare complication of chronic liver disease. It has been suggested that it is caused by the neurotoxicity of ammonia or other metabolites bypassing normal liver metabolism. It is characterized by spastic paraparesis with minimal sensory and sphincteric involvement. Diagnosis is a process of exclusion requiring normal CSF analysis and brain imaging. Liver transplantation may result in improvement, especially if performed early in the course of the disease.
Hexosaminidase A deficiency
This is a progressive neurodegenerative disease with variable age of onset and rate of progression and hence variable prognosis. The clinical presentation of the chronic form of hexosaminidase A deficiency may mimic spinocerebellar degeneration, Friedreich ataxia, or amyotrophic lateral sclerosis (see below).
Adrenomyeloneuropathy
This is the commonest clinical variant of adrenoleukodystrophy, a disorder of peroxisomal fatty acid oxidation causing the accumulation of very long chain fatty acids in myelin, adrenal cortex and Leydig cells of the testes. Patients with an adrenomyeloneuropathy presentation of this X-linked recessive disorder present in their third decade with a progressive spastic paraparesis and sphincter dysfunction. A sensorimotor peripheral neuropathy may be a feature and ataxia and dementia are seen occasionally. Adrenal insufficiency will often have been present since childhood and there may be hypogonadism. Around 50% of heterozygous females will show some symptoms later in life. The clinical findings and magnetic resonance imaging point to the diagnosis, which is confirmed by demonstrating hypoadrenalism and elevated concentrations of very long chain fatty acids (VLCFAs) in plasma and cultured skin fibroblasts. Genetic testing is available. The only gene in which mutations are known to cause adrenoleukodystrophy is ABCD1, which codes for a peroxisomal membrane transporter protein, although molecular genetic testing is rarely required to confirm the disease, especially in a male. Treatment is with steroid replacement, symptomatic management and supportive care.
PERIPHERAL NEUROPATHY
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
