[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 32
Biochemistry of articular disorders
CHAPTER OUTLINE
DISORDERS OF THE ARTICULAR SYSTEM
The connective tissue diseases
ARTICULAR INVOLVEMENT IN ENDOCRINE AND METABOLIC DISEASES
LABORATORY TESTING IN ARTICULAR DISEASE
INTRODUCTION
While biochemical abnormalities themselves are responsible for few articular disorders, clinical biochemistry plays an important part in the diagnosis, treatment and monitoring of many of these conditions.
This chapter starts with a description of the articular system and its diseases, which will put the role of biochemistry in these conditions into perspective.
Crystal synovitis, as seen in gout and pseudogout, is the only common articular condition caused by an underlying biochemical abnormality. This is described in some detail along with examination of synovial fluid, the one biochemical investigation that is primarily the province of the rheumatologist.
Some diseases with a primary biochemical abnormality like diabetes, alkaptonuria, endocrine conditions and haemochromatosis have articular manifestations and also warrant discussion.
Immunological abnormalities are at the root of an important group of rheumatic diseases, the connective tissue diseases. Autoantibody formation is a major disease manifestation and, in some hospitals, tests to identify and measure autoantibodies are performed in the biochemistry laboratory. These tests are described briefly.
THE ARTICULAR SYSTEM
The joint is the pivotal structure of the articular system. Joints can be considered as discontinuities in the skeleton that permit controlled mobility. They have different structures depending on their functional requirements. When no movement is needed, the joint is bound together by tough fibrous tissue (e.g. the skull ‘sutures’). In cartilaginous joints, the bone ends are joined by compressible fibrocartilage and reinforced by a surrounding tough fibrous tissue (as in the symphysis pubis and the manubriosternal joint) and permit only a limited amount of movement. This type of joint tends to be located centrally. If a moderate or wide range of movement is required, a space must exist between the bone ends, forming a discontinuous ‘synovial’ joint. Most of the peripheral joints fall into this category.
The most important joints from a clinician’s point of view are the peripheral ones, which enable our arms and legs to move through such a wide range, our hands to be endowed with such dexterity and strength and our feet to carry us so uncomplainingly when we stand, walk, run and jump.
While synovial joints have the same basic structure and physiology, individual joints have evolved differently, depending on their situation and what is expected of them. For example, the knee, ankle and finger joints, which move mainly in one plane, are ‘hinge’ joints, while the hip and shoulder, which move in all directions, are ball and socket joints. Of course, to carry out even the simplest of tasks, like opening a door or walking up a step, we have to coordinate the movement of several joints, not to mention contracting and relaxing the muscles on either side of the joints.
The archetypical synovial joint is an enclosed space with a negative pressure (Fig. 32.1). The bone ends that move against each other (the articular surfaces) are covered with articular cartilage. This is made up of proteoglycans and collagen, combined in such a way as to allow it to absorb huge forces of pressure like a shock absorber, while providing a shiny surface for smooth, low-friction movement. A healthy joint is lubricated by a small amount of synovial fluid (0–4 mL). This is an ultrafiltrate of plasma with additional components secreted by the synovium. The most important of these is hyaluronate, a linear repeating disaccharide with a molecular weight of some 107 Da. This provides the fluid with its viscoelastic properties without which the cartilage would fail and smooth movement would be impossible. Cartilage and synovial fluid together maintain coefficients of friction of < 0.02.
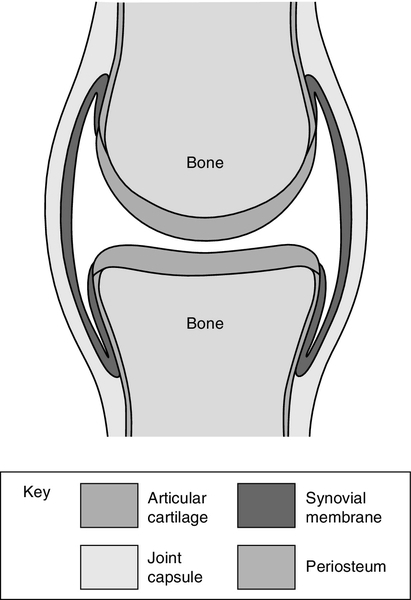
FIGURE 32.1 A typical synovial joint.
The bone ends are enclosed by the tough joint capsule which is lined by the synovial membrane, a structure only a cell or two thick (Fig. 32.1). Synovial cells phagocytose intra-articular debris, secrete many components of synovial fluid and possess immunological functions.
While we talk of ‘articular disorders’, it is not really possible to consider the joint in isolation. Each joint depends on other structures like ligaments, tendons, muscles and bone for its function and stability. Bone and muscle disease are dealt with in separate chapters.
DISORDERS OF THE ARTICULAR SYSTEM
There are over 100 rheumatic disorders with different underlying causes, different treatments and different outcomes. These conditions can be broadly divided into non-inflammatory diseases, of which osteoarthritis is the most important, and inflammatory conditions such as rheumatoid arthritis (RA), ankylosing spondylitis and systemic lupus erythematosus (SLE or lupus).
Osteoarthritis (OA)
The most common form of articular disease is osteoarthritis, in which the articular cartilage becomes fissured and gradually wears away. The joint tries to heal itself by forming bony out-growths on the sides of the joints (osteophytes). Trauma to a joint predisposes to OA, as do some rare biochemical disorders like alkaptonuria. However, in most cases, apart from an inherited tendency and increasing frequency with age, the cause cannot be identified. For many years, OA was thought to be due to degeneration or ‘wear and tear’. Recently, we have appreciated that a degree of inflammation is often present if sought and that calcium pyrophosphate and basic calcium phosphate crystals are frequently found in osteoarthritic joints (see below).
At present, we have no treatment to influence this process. The knee and hip are often affected and, if the pain and limitation to mobility are severe enough, the orthopaedic surgeon will remove the joint and replace it with an artificial one, usually with very gratifying results.
Inflammatory arthritis
The other major class of articular disease is inflammatory arthritis. This consists of a group of systemic immunological diseases that focus their attention on the synovium of the joint. Rheumatoid arthritis (RA) is the archetypical form of inflammatory arthritis. In RA, there is an imbalance between the proinflammatory and anti-inflammatory cytokines that favours the induction of autoimmunity (an immune reaction against one’s own tissues), chronic inflammation and joint damage. The synovium becomes inflamed, resulting in pain, tenderness, heat and stiffness in the joint as well as a systemic reaction consisting of malaise and fatigue. An acute phase reaction ensues, which can be detected and monitored by measurement of plasma C-reactive protein (CRP) concentration or the erythrocyte sedimentation rate (ESR). In what may well be a separate process, in time the synovial cells increase in size and number, the synovium becomes thickened and develops the changes of chronic inflammation with infiltration by macrophages and lymphocytes. There are also alterations in the small blood vessels. In addition, the amount of synovial fluid in the joint increases in quantity, which adds to the swelling and dysfunction of the affected joint. A number of different cytokines associated with the rheumatoid process can be found in the synovial tissue and fluid; these include tumour necrosis factor alpha (TNFα), interleukins one (IL-1) and six (IL-6). There are also abnormalities of the T and B cell lymphocytes. If left to its own devices, the thickened synovium will spread over the cartilage and erode it and the neighbouring bone. This results in joint damage, deformity and disability.
Non-steroidal anti-inflammatory drugs, e.g. diclofenac, ibuprofen and naproxen are used less frequently than they once were. Although they often provide symptomatic relief, they increase the risk of cardiovascular events, can cause chronic kidney disease and have no influence on the long-term outcome of the disease.
Of more concern in the long term, is the chronic inflammation that results in joint damage and disability. Until about 20 years ago, there was little that we could do to slow or even stop the inexorable progress of this destructive process, meaning that many people with RA became very disabled with joint deformity and pain. However, there have been dramatic improvements in our drug treatment of this most unpleasant condition. We have learnt that we need to diagnose the condition as early as possible. This enables treatment with one or more disease modifying anti-rheumatic drugs (DMARDs) (e.g. methotrexate, leflunomide, sulfasalazine), to be started as soon as possible. As a rule, these drugs rapidly bring the arthritis under control, but different drugs suit different people so sometimes a degree of trial and error is required. DMARDs are taken for long periods and can cause side-effects such as liver damage or bone marrow suppression, and regular blood tests (full blood count, ESR, CRP and liver function tests) are used to monitor their safety as well as the activity of the disease.
More recently, the treatment of RA has been transformed by the introduction of so-called ‘biologics’ (monoclonal antibodies designed to counteract the effects of cytokines or alter the function of T or B cell lymphocytes). They are expensive and need to be taken long term, so at present they are used in the cases where DMARDs have been unsuccessful. However, in time, they may become the first-line in treatment of RA. Biologics in common use include agents against TNFα (e.g. etanercept) and anti IL-6 (tocilizumab), as well as B cell depleters (rituximab) and T cell blockers (abatacept). This is a field of active research with the potential to transform the treatment not only of RA but of many other immunological diseases.
Other examples of chronic inflammatory arthritis include ankylosing spondylitis, which mainly affects the small joints of the spine, the arthritis associated with psoriasis and the ‘reactive’ arthritis one sees in response to certain intestinal or genitourinary infections.
The connective tissue diseases
The connective tissue diseases (CTDs) are a fascinating group of conditions in which, for reasons we do not understand, the normal fine balance of the immune system gets disturbed, causing the body to react against itself. In health, the immune system consists of series of counterbalancing mechanisms that marshal defences against alien material like bacteria, viruses or foreign tissue, while not reacting in this way against itself. The identification of foreign tissue (the antigen) is the first step in the process and is followed by a series of defensive reactions including manufacturing antibodies. In CTDs, the person’s ability to recognize their own tissue goes awry and they form antibodies against their own tissue (autoantibodies). In some cases, these are responsible for the clinical manifestations of the disease; in others they are a peripheral effect of the underlying disease process. Theories of the pathogenesis of autoimmune diseases are discussed in Chapter 30.
Systemic lupus erythematosus (SLE) mainly affects younger women and can involve one or more of the bodily systems. It is characterized by the formation of autoantibodies against the cell’s nucleus, or part of the nucleus, which are believed to be responsible for many of the manifestations of the disease.
Joint pain is common, although it is unusual to see joint damage such as occurs in RA. SLE can result in a variety of manifestations in other systems, for example various rashes, haemolytic anaemia and glomerulonephritis leading to renal impairment, as well as cerebral, lung and heart problems. Some of these can be life threatening.
Autoantibodies against cell nuclei (antinuclear factor, ANF) are present in the plasma in almost every patient with SLE. However, they are also frequently present in other CTDs and in a proportion of healthy people. Antibodies to specific components of the nucleus are more specific. For example, antibodies against double-stranded DNA are strongly suggestive of SLE.
Management depends on the systems affected. The joint and skin manifestations can often be controlled with hydrochloroquine and/or low doses of prednisolone. More severe cases require the use of high doses of corticosteroids with immunosuppression using cyclophosphamide, azathioprine or mycophenolate mofetil.
Other CTDs are associated with autoantibody formation, although in these cases the disease is not believed to be caused by specific antibody-antigen complexes as in SLE. Examples of these conditions include Sjögren syndrome, polymyositis, scleroderma and primary biliary cirrhosis.
For details and associated immunological abnormalities, see Table 32.1. Table 32.1 is, of necessity, an oversimplified guide to some of the more important signs and symptoms seen in the CTDs, with their associated antibodies. There is often a crossover of antibodies in the various diseases and the antibodies are by no means seen in every case.
TABLE 32.1
The connective tissue diseases, their common clinical features and frequently associated autoantibodies
| Disease | Clinical features | Autoantibodies |
| SLE | Rashes, joint pains, glomerulonephritis, haemolytic anaemia, leukopenia, neuropsychiatric lupus, pericarditis, pleurisy, vasculitis, fatigue | Anti-nuclear factor Anti-double-stranded DNA Anti-Sm Anti-Ro Anti-La |
| Scleroderma | Thickened skin, Raynaud phenomenon, pulmonary hypertension, calcinosis, pulmonary fibrosis | Anti-nuclear factor Anti-centromere |
| Sjögren syndrome | Dry eyes, dry mouth, joint pains, fatigue, Raynaud phenomenon | Anti-nuclear factor Anti-Ro Anti-La Rheumatoid factor |
| Polymyositis | Muscle weakness and wasting, joint pains, pulmonary fibrosis, fatigue | Anti-nuclear factor Anti-Jo-1 Anti-synthetase |
| Dermatomyositis | Proximal muscle weakness and wasting, rashes, joint pains, pulmonary fibrosis | Anti-nuclear factor Anti-Mi2 |
| Wegener granulomatosis | Vasculitis, sinusitis, pulmonary infiltration, glomerulonephritis, rashes | cANCA |
| Microscopic polyangiitis | Glomerulonephritis, joint/muscle pains, rashes, lung infiltration | pANCA |
| Primary biliary cirrhosis | Jaundice, itching, joint pains, Raynaud phenomenon | Antimitochondrial |
cANCA, cytoplasmic anti-neutrophil cytoplasmic antibody; pANCA, perinuclear anti-neutrophil cytoplasmic antibody; SLE, systemic lupus erythematosus.
Aches and pains
If we move from the laboratory to the clinic or surgery, ‘articular disorders’ take on another perspective. About one-fifth of the patients in a GP’s surgery will complain of musculoskeletal symptoms, such as aches, pain, stiffness or disability. Only a proportion of these patients will have a demonstrable disease or significant anatomical abnormality. Some will have abnormalities associated with the way they move their musculoskeletal system (‘mechanical’ pain), while in other cases, no abnormality can be identified. This does not mean the symptoms are imagined, but in the absence of an abnormality, attempts to diagnose and ‘cure’ a disease will, of course, be futile. Doctors are trained to diagnose diseases and then treat them. Too many doctors are uncomfortable when they are not able to make a diagnosis. This may provoke them to order unnecessary investigations in these often anxious patients, thus exposing both parties to the dangers of false positives, laboratory or clerical error and misinterpretation of the results.
As a rheumatologist, each week the author sees one or two patients referred as a consequence of an inappropriately performed blood test. These patients usually have mechanical or unexplained musculoskeletal symptoms and their doctors have performed a batch of tests with vague rheumatological connotations, presumably to reassure themselves or to be seen to be doing something for the patient. The problem is that these tests are often positive in healthy people, especially at low levels. For example, low titres of ANF are widespread, and it is common for a number of anxious young women to be referred with aches and pains, ‘weakly positive ANF’ and a sheaf of internet misinformation about how fatal SLE is. Informed of their ‘positive’ test, they have often had to wait several months for their referral appointment, in fear and trepidation, and come prepared to hear the worst. False positive rheumatoid factor tests, high plasma urate concentrations and slightly raised ESRs produce a steady stream of worried well people with fears they have rheumatoid arthritis, gout or inflammatory disease, respectively. Inappropriate tests are not only of concern to the biochemist and accountant. They cause a mass of unnecessary work for the clinician, but the real victims are the unfortunate patients who have to endure long periods of unnecessary strife, stress and uncertainty.
Crystal arthritis
Just as crystals form in vitro when the concentration of a solute is sufficiently high to allow nucleation and growth, so do they form in vivo. Crystals are sometimes found in joint fluid. These include monosodium urate (MSU), calcium pyrophosphate (CPP), basic calcium phosphates, cholesterol and oxalate crystals. The body’s response to crystals varies. In some cases, the presence of crystals in a joint provokes a severe inflammatory reaction, but researchers have found crystals in joint fluid from asymptomatic patients. Different crystals tend to favour different sites. Monosodium urate is preferentially deposited in cartilage and synovium, CPP in articular fibrocartilage and basic calcium phosphates in tendons and hyaline cartilage. The formation of crystals is influenced by physical factors such as temperature and hydrogen ion concentration (pH), but it is a complex process and what follows is a practical simplification.
Hyperuricaemia and gout
Gout is the best known form of crystal arthritis and is caused by MSU crystal deposition in articular tissues. The term hyperuricaemia is used to describe excessive uric acid or urate in the blood as defined by solubility in plasma at 37 °C; that is, > 0.42 mmol/L in males and > 0.36 mmol/L in females. While there is a relationship between MSU crystal deposition and hyperuricaemia, it is by no means absolute. Hyperuricaemia without attacks of gouty arthritis is common, and sometimes, particularly during an acute attack, plasma uric acid concentrations are normal.
Uric acid is the end-product of purine metabolism (Fig. 32.2). Purines are components of nucleic acids and of nucleotides that are involved in energy transformation and phosphorylation reactions and act as intracellular messengers. They are derived from diet, the breakdown of nucleotides and from de novo synthesis. Purines are metabolized to urate via hypoxanthine and xanthine, the final step being catalysed by xanthine oxidase. The kidneys excrete two-thirds of the urate, the remainder being removed via the bowel, where it is broken down into carbon dioxide and ammonia by bacterial action. The amount of urate pooled in the body depends on the relationship between the input from diet, the breakdown of nucleotides and de novo synthesis versus the output via kidney and bowel. Hyperuricaemia results from excess production of urate or reduced excretion, or sometimes both mechanisms. The average uric acid pool size is 7.2 mmol in men and 3.6 mmol in women. Two-thirds is turned over each day. The uric acid pool of people with gout is increased to 12–24 mmol; those with deposits of urate (tophi) may have a pool as large as 180 mmol.
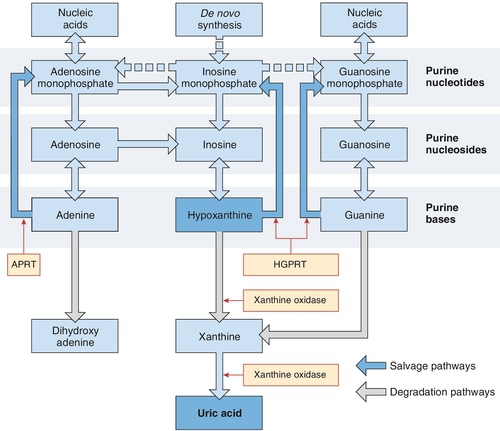
FIGURE 32.2 A simplified diagram of the pathways of purine nucleotide metabolism and uric acid synthesis in humans. APRT, adenine phosphoribosyl transferase; HGPRT, hypoxanthine-guanine phosphoribosyl transferase. (From Marshall W J, Bangert S K, Lapsley M 2008 Clinical Chemistry. 7th ed. Edinburgh: Elsevier, with permission.)
Gout tends to run in families and is frequent in certain races, such as the New Zealand Maori. This points to an inherited abnormality in urate metabolism, but while there are well-defined enzyme defects resulting in hyperuricaemia, for example hypoxanthine-guanine phosphoribosyl transferase deficiency, the cause of the Lesch–Nyhan syndrome (see p. 640), these are rare. In most cases, we can only divide people with gout into those who undersecrete uric acid from the kidney (85%) and those who synthesize an increased amount.
Gout commonly affects middle-aged males; it is most unusual in premenopausal women. Hyperuricaemia, family history, obesity, hypertension, age, alcohol consumption and renal insufficiency are risk factors for gout. Several drugs, particularly diuretics, low-dose aspirin and ciclosporin, and lead poisoning, may provoke gout by causing hyperuricaemia. Conditions with high purine turnover, such as leukaemia and lymphoma and their treatment with cytotoxic drugs, can cause gout and sometimes acute urate nephropathy by increasing the urate load.
Asymptomatic hyperuricaemia
Asymptomatic hyperuricaemia is the first stage of gout. This is seen on at least one occasion in as many as 10% of adults, the majority of whom never experience an attack of gout. In the past, we have ignored asymptomatic hyperuricaemia, but it is looking increasingly likely that it is a risk factor for acute coronary syndromes and it is also linked with obesity, type 2 diabetes, hypertension and hyperlipidaemia.
Acute gout
Acute gout usually strikes the first metatarsophalangeal joint (the bunion joint in the big toe) at night. Crystal formation is more likely at colder temperatures and this joint is cooler than more proximal ones of a similar size. The joint rapidly becomes severely painful, swollen, red and exquisitely tender to touch. The weight of the bedclothes is unbearable. There is often a systemic reaction with fever, an acute phase reaction and a leukocytosis. While acute gout most commonly attacks the great toe (70% of cases), the instep, the ankle or knee can be affected. Examination of synovial fluid drawn from an affected joint will show many MSU crystals. Left to itself, acute gout usually subsides over ten days or so.
While some patients never have another attack, there are usually further sporadic attacks. These may become more frequent and may involve several areas at a time (polyarticular gout) as the urate load increases and the disease progresses. In time, upper limb joints such as the fingers, wrists and elbows become affected.
Chronic tophaceous gout
In the later stage of untreated disease, the patient may have continuous inflammation of many joints with associated pain, tenderness and immobility. In addition, crystals of urate are deposited in masses of chalky material called tophi, which sometimes ulcerate and discharge through the skin. These are often seen over joints and bony prominences such as the points of the elbows. They can occur in the joint and inside bone causing permanent damage, pain, deformity and disability. Urate crystals are often the focus for renal stone formation. At this stage, gout is a miserable business.
Diagnosis
When gout presents in the classic manner, the diagnosis is obvious. However, infection in a joint also makes it hot, red and painful and sometimes it can be difficult to differentiate between crystal arthritis and infection. Because both conditions cause fever, leukocytosis and an acute phase reaction, the only reliable way to make the diagnosis is to aspirate fluid from the joint and examine it for crystals (using polarizing light microscopy, see Fig. 32.3) and for bacteria (Gram stain and culture). Rarely, the two conditions coexist, so both tests should be performed.
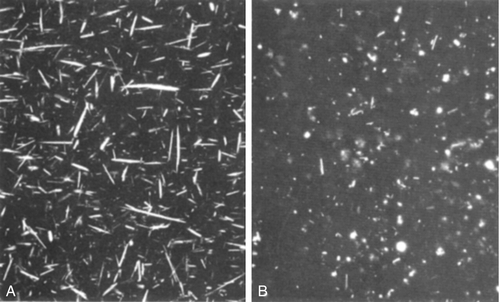
FIGURE 32.3 Sodium urate (needle shaped, A) and calcium pyrophosphate (rhomboid, B) crystals viewed microscopically. Under polarized light, the former are negatively, the latter positively, birefringent.
For the reasons mentioned above, plasma urate concentrations are not helpful in diagnosis, although very high values are convincing and sometimes influence decisions about drug treatment.
Treatment
There are three strands to the treatment of gout. Immediate action is required to counteract the severe inflammation that is producing such pain and disability. In straightforward cases, anti-inflammatory drugs, such as diclofenac or naproxen, can rapidly achieve this. Colchicine, which stabilizes lysosomal membranes, can be used when anti-inflammatory drugs are inadvisable, for example in patients with kidney or stomach conditions. These drugs settle the inflammation but have no effect on crystal formation or the underlying problem of the increased urate load. The second strand to treatment is to try to persuade the patient to modify his lifestyle. More exercise, less alcohol, improved diet and weight loss are the goals. They are more easily achieved in some than in others.
The third strand of treatment is to reduce the urate load. This will have built up over years and so will take time to reduce. Reduction is usually achieved by the use of allopurinol, a potent purine xanthine oxidase inhibitor, which blocks the conversion of xanthine to urate. Xanthine is soluble and is excreted renally. The decision to start allopurinol should be influenced by the frequency of the attacks of acute gout, the presence of tophi and/or chronic polyarticular gout and very high urate concentrations. Allopurinol gradually leaches away the high urate load and, in time, urate concentrations become normal and tophi are absorbed. There is a danger of allopurinol provoking an acute flare of gout when it is first used, so it should not be started until the acute attack has been controlled. Once treatment with allopurinol has been established, urate concentrations should be checked. The target is a value of < 0.3 mmol/L and the dose of allopurinol should be titrated accordingly.
The treatment of uncomplicated gout is simple and rewarding if patients can be persuaded to take their tablets regularly. Male patients often seem to find it difficult to continue their medication when they feel well and this is by far the commonest cause of failure of treatment. However, treating complicated gout can be challenging. These patients often have chronic kidney disease, cardiac disease, peptic ulcers and are taking warfarin, all of which are relative contraindications to the use of anti-inflammatory drugs. They often require diuretics (which may provoke gout) to control heart failure and gout is frequently a problem in transplant patients because of the use of the hyperuricaemia-inducing anti-rejection drug, ciclosporin. In these cases, injecting corticosteroid into the affected joint controls the acute inflammation. If several joints are affected, systemic adrenocorticotrophic hormone (ACTH) or corticosteroids are necessary.
Lowering urate concentrations with allopurinol has been the mainstay of the treatment of gout for the past 40 years. It is inexpensive and in most cases is very effective. However, drug reactions do occur and there has been a need for other urate lowering agents. Recently, febuxostat, a non-purine selective xanthine oxidase inhibitor, has been introduced and is showing great promise. It does not inhibit enzymes involved in purine and pyrimidine metabolism, as does allopurinol and may in time become the drug of first choice.
All mammals apart from humans and Dalmatian dogs possess the enzyme urate oxidase (uricase) which oxidizes urate to allantoin. This is 5–10 times more soluble than uric acid and so is more effectively excreted by the kidney. However, urate oxidase was lost in early human evolution. The reasons for this are not clear but it has been proposed that its loss was an advantage because uric acid is a powerful antioxidant and scavenger of free radicals that might protect against oxidative damage and so prolong life and decrease the risks of cancer. As a result, humans are prone to gout. The selective inbreeding of Dalmatian dogs has produced a striking white animal with black spots but also one without urate oxidase. Hence, the Dalmatian’s tendency to the development of gout and the formation of urate renal stones, which can be treated with allopurinol.
Until recently, this interesting information was of peripheral interest. However, researchers have investigated the use of urate oxidase to treat hyperuricaemia in humans. There are two preparations. Rasburicase, a recombinant form from the fungus Aspergillus flavus, is used to prevent acute urate nephropathy, which can occur when large tumour loads are treated with chemotherapy (tumour lysis syndrome, see p. 114). However, its very short circulating life and the fact patients sometimes develop antibodies to the drug limits its use. These disadvantages led to the development of PEG-uricase, a recombinant porcine urate oxidase, to which multiple strands of polyethylene glycol (PEG) of average molecular weight 10 000 Da have been attached. This greatly prolongs its circulating life and it is non-immunogenic. However, it has to be given intravenously at two-weekly intervals and can cause flares of gout and infusion reactions. At present, its use is reserved for patients with gout that is difficult to control who cannot take standard treatment. It may be noted that accurate measurement of urate in patients treated with this drug is difficult, as it continues to act in vitro.
Calcium pyrophosphate deposition (CPPD)
This is the other common form of crystal arthritis. It is not as frequent as gout and is less well understood. Calcium pyrophosphate crystals are preferentially deposited in articular cartilage where calcification can be seen on X-rays (chondrocalcinosis). Affected joints can become acutely inflamed in a way that resembles gout (hence CPPD’s other name of pseudogout). The knee and the wrist are most frequently affected. An acute phase reaction is common and, again, finding CPP crystals in the joint fluid makes the diagnosis. In some cases, there is chronic involvement of several joints, which can cause diagnostic confusion with other inflammatory articular diseases.
Calcium pyrophosphate deposition becomes more common with age. Symptomless chondrocalcinosis is often seen on X-rays of the elderly. Attacks are sometimes triggered by trauma unrelated to the joint (of which surgery is an excellent example), and rheumatologists are often asked to see elderly postoperative patients in the surgical wards with a hot, swollen, ‘infected’ wrist or knee. Almost invariably, pseudogout is responsible.
Familial CPPD is well recognized, and kindred from many different countries have been described. These patients present in their twenties and usually have severe destructive joint disease. In some families, a genetic abnormality on the short arm of chromosome 5p has been identified.
Calcium pyrophosphate deposition is associated with several metabolic diseases described in other chapters. Definite associations have been established with haemochromatosis, hyperparathyroidism, hypomagnesaemia, hypophosphatasia, alkaptonuria and Wilson disease. In these cases, the onset is often at a younger age (under 55 years) and the disease is more severe than is seen in uncomplicated cases. A link with thyroid disease is now disputed.
As yet, we have little understanding of the underlying biochemical processes at play in this group of conditions. Our ‘first aid’ style of treatment, with anti-inflammatory drugs and corticosteroid injection of affected joints, reflects this.
Basic calcium phosphate deposition disease
Basic calcium phosphate (BCP) crystals include hydroxyapatite, octacalcium phosphate and tricalcium phosphate. They are often difficult to identify, which is why we still have so much to learn about these conditions. Basic calcium phosphate crystals are sometimes deposited in the tendons or in soft tissues outside the joints, where they can give rise to intense pain. This tends to occur in younger females and affects the shoulder, wrist and even the great toe, when gout may be incorrectly diagnosed. Injection of corticosteroid into the lesion often results in dramatic improvement. Mercifully, the pain usually settles down over a week or two and the calcification seen on X-ray is often resorbed within six weeks. Basic calcium phosphate crystal deposition has also been implicated in an unusual condition that causes large effusions and joint resorption in the shoulders of elderly women (Milwaukee shoulder).
There is increasing interest in these crystals because they appear to be closely involved in the process of osteoarthritis (OA). Basic calcium phosphate crystals can be found in the synovial fluid of as many as 60% of joints severely affected by OA. It is uncertain whether this is related to the cause of OA or is a downstream effect of the OA process. However, in tissue culture, BCP crystals stimulate mitogenesis and the production of metalloproteinases, which cleave cartilage matrix. This suggests BCP crystals may be involved in the cartilage breakdown seen in OA, and researchers are starting to look at the effects of agents such as phosphocitrate, which inhibit the formation of BCP crystals.
Other crystals found in synovial fluid
Other crystals can be found in synovial fluid. These include cholesterol and oxalic acid crystals. These latter occur in primary oxalosis and in patients with established renal failure on dialysis, in whom the crystals can provoke a chronic polyarthritis in the small joints of the hand.
ARTICULAR INVOLVEMENT IN ENDOCRINE AND METABOLIC DISEASES
Diabetes mellitus
Over 30% of diabetic patients have potentially disabling hand or shoulder disorders. Capsulitis of the shoulder (frozen shoulder) is particularly common in diabetes. It causes severe, disabling shoulder pain, which settles after a few months but leaves the patient with very limited mobility at the shoulder joint. The range of movement usually improves over the months. Sometimes a corticosteroid injection will help the pain, but limited movement remains a problem.
Many patients with diabetes develop changes in the connective tissues of the hands. Dupuytren contracture, a condition in which there is thickening of the palmar fascia and increasing contracture of fingers, is common in diabetes. In what seems to be a different process, called diabetic cheiroarthropathy or diabetic hand syndrome, the skin and underlying tissues become thickened, resulting in a limited range of movement of the hand and finger joints. Often the finger tendons are affected. These changes are sometimes mistaken for rheumatoid arthritis but there is no joint damage or acute phase reaction. Biopsy shows excessive fibrosis and increased deposition of dermal collagen. It has been suggested that increased tissue glycation may result in diminished collagen breakdown. Perhaps a similar sort of process is taking place in the frozen shoulders of diabetic patients.
Peripheral nerve damage is a frequent complication of diabetes. In some cases, the nerve supply to the joints is affected so that the usual sensations of joint position and joint movement are lost. Repeated trauma can take place within such a joint without the patient being aware and this can rapidly result in painless destruction and bony resorption of large areas of bone around the affected joint. This is called a Charcot joint. It occurs almost exclusively in the foot and is a major problem. However, although evidence of damage to small nerve fibres is usually present in this condition, other factors are undoubtedly involved. Charcot arthritis occurs more frequently in type 1 diabetes. The reason for this is not clear.
Other endocrine disorders
Several endocrine disorders can cause muscle and joint pains and need to be considered in the assessment of the patient with aches and pains. Only rarely will they be found to be responsible, but early diagnosis and treatment will often relieve the patient’s symptoms and can prevent further suffering and tissue damage. As ever with uncommon conditions presenting in unusual ways, the important thing is to think of the diagnosis.
Both hypo- and hyperthyroidism can result in joint pains and a variety of muscular symptoms. Hyperparathyroidism is a cause of bone pain and aching joints as well as chondrocalcinosis and pseudogout, while joint pains, degenerative arthritis, joint laxity and muscle weakness can result from effects of acromegaly on bone, joints and soft tissue. Hypercortisolism sometimes presents in the rheumatology clinic with back pain (from osteoporotic fractures) and muscle weakness.
Haemochromatosis
As described in Chapter 14, liver disease and diabetes in a male with slate grey skin is the classic presentation of haemochromatosis. However, the alert rheumatologist will discover the occasional patient with this condition presenting in the clinic with articular disease. This can happen in two ways. We have already discussed how haemochromatosis is associated with chondrocalcinosis and pseudogout. In addition, haemochromatosis itself is responsible for a characteristic form of arthritis involving the metacarpophalangeal joints of the index and middle fingers. X-rays show loss of joint space and characteristic hook osteophyte formation in these joints. There is no inflammatory reaction, but iron studies show the characteristic picture of haemochromatosis. The synovium contains much iron. Venesection unfortunately makes little difference to the joint symptoms, but does prevent further harm to the pancreas and liver. It is also important to screen the patient’s relatives so that those who have inherited the disease can be identified and treated before damage occurs.
Alkaptonuria
This rare, autosomal recessive disorder is caused by a deficiency of the enzyme homogentisic acid oxidase. This results in a build-up of homogentisic acid, an intermediary product in the metabolism of phenylalanine and tyrosine. The excess homogentisic acid is oxidized and polymerized and forms a blackish pigment, alkapton, which is deposited in cartilage and can be easily seen in the cartilage of the ears. More importantly, it is also deposited in articular cartilage, which becomes more susceptible to mechanical stress and degeneration. Arthritis of the hips, knees and shoulders develops early. Alkapton is also deposited in the intervertebral discs in the spine, which become calcified. This produces a characteristic X-ray appearance and predisposes to mechanical back problems. The diagnosis is made by identifying homogentisic acid in the urine.
LABORATORY TESTING IN ARTICULAR DISEASE
Anaemia in rheumatoid arthritis
Anaemia is common in RA. When the disease is active, there is frequently a normochromic, normocytic anaemia. This is called ‘the anaemia of chronic disease’ (see Chapter 27) and it will resolve if the activity of the arthritis can be controlled. Iron is present in the bone marrow but cannot be utilized in erythropoiesis.
Some RA patients require an anti-inflammatory drug to keep their pain and stiffness at bay. These drugs have a tendency to cause erosions and ulcers in the stomach, duodenum or small bowel, resulting in blood loss, iron deficiency and a hypochromic microcytic anaemia.
Separating iron-deficiency anaemia and the anaemia of chronic disease in these patients is often tricky and is made more difficult by the behaviour of ferritin in inflammatory diseases. Ferritin is one of the several proteins that are acute phase reactants, and for this reason, its plasma concentration is usually raised when RA is active. Measurement of plasma ferritin concentration is considered to be the best test of iron deficiency (see Chapter 27), but in RA, the result should be interpreted with caution. Usually a concentration of < 12 μg/L is taken as diagnostic of iron deficiency, but in RA, rheumatologists take a concentration of < 30 μg/L to indicate iron deficiency. However, iron deficiency can exist with higher concentrations of ferritin. The presence of microcytosis favours the diagnosis of iron deficiency, but sometimes examination of bone marrow for iron stores is the only reliable way to differentiate between iron-deficiency anaemia and the anaemia of chronic disease.
The acute phase response
The acute phase response is discussed in detail in Chapter 30. Traditionally, rheumatologists have used the ESR to diagnose and then monitor an acute phase response. This is, of course, an indirect measure of a series of acute phase proteins and immunoglobulins and is particularly influenced by plasma fibrinogen concentrations. Measurement of CRP concentration is gradually superseding the ESR and is believed to correlate better with disease activity in RA. In SLE, CRP concentration is often normal; indeed, a raised value in this condition is said to suggest infection rather than increased disease activity. In truth, most rheumatologists like to use both CRP and ESR.
These simple investigations are of great help to a rheumatologist. One of our main tasks when confronted with a new patient is to differentiate between the inflammatory and non-inflammatory types of arthritis. This is not always easy and the CRP and/or ESR often provide the answer. The activity of rheumatoid arthritis waxes and wanes over time and this is reflected in the acute phase response, as is the success or otherwise of drug treatment.
Examination of synovial fluid
An acutely swollen hot joint can be a result of infection, crystals or the irritative effects of blood on the synovium. The only reliable way to determine which is responsible is to aspirate the joint fluid. Blood will be apparent to the naked eye. Gram-staining will show bacteria and polarizing light microscopy will demonstrate crystals (Fig. 32.3) and whether they are negatively (gout) or positively (CPP) birefringent. The chances of identifying crystals are increased if the specimen is centrifuged and the deposit examined at the earliest opportunity. Sometimes it is only possible to obtain a tiny amount of fluid. If expressed on to a microscope slide at the bedside and covered with a cover slip to avoid evaporation, this can yield vital information when examined in the laboratory.
Rheumatoid factor
Rheumatoid factors (RFs) are antibodies directed against the Fc portion of human IgG and are present in 75–80% of patients with RA. At present, it is believed that RF is produced as a downstream product of the rheumatoid process rather than being central to the disease process. Rheumatoid factors also occur in other connective tissue diseases, chronic infections such as tuberculosis and hepatitis C and conditions associated with hyperglobulinaemia, such as hyperglobulinaemic purpura, sarcoidosis and cryoglobulinaemia. They are found in low titres in 5% of the population, occurring more frequently with advancing age.
A positive RF is strong confirmatory evidence of RA. In a patient with polyarthritis it has a sensitivity of 70% and a specificity of 80%. However, in an unselected population its predictive value is low (20%). Very high titres of RF are associated with severe disease and are a marker of poor prognosis.
Most laboratory methods identify IgM antibodies, although IgA and IgG antibodies of uncertain clinical significance do exist. For many years, rheumatoid factor was detected by techniques using flocculation or agglutination of IgG-coated particles or cells. The Rose Waaler test, the standard method for many years, used sheep red blood cells coated with rabbit IgG, which agglutinate in the presence of RF, while the flocculation of latex beads coated with human IgG forms the basis of the RA latex test. The sheep cell tests are more specific. However, heterophile antibodies reacting directly against sheep cells sometimes give rise to false positives. Because of this, sera are usually tested on uncoated and coated sheep red cells in parallel. Laser nephelometry, enzyme linked immunosorbent assay (ELISA), immunofluorescence and radioimmunoassay can also be used to measure rheumatoid factors.
Other autoantibodies are seen in RA. In recent years, it has become standard practice to measure antibodies to cyclic citrullinated protein (anti-CCP) as well as RF. These are measured by peptide-based ELISA. The test has a sensitivity of 74% and a specificity of 94%. It may eventually supersede the time-honoured RF test but most rheumatologists use both tests at present.
Other autoantibody tests
Autoantibodies are produced in the connective tissue diseases (see above) and some cases of vasculitis. These are sometimes diagnostic, but most autoantibodies occur in several of these conditions and in healthy people. These tests are valuable in confirming a suspected diagnosis and negative tests are useful in helping to rule out diagnoses. However, because they occur in a proportion of normal people, they should not be used for screening purposes or in cases without clinical features suspicious of connective tissue disease (see p. 639). For some years, autoantibodies have been identified by indirect immunofluorescence using Hep-2 cells. In this age of automation, there is a move to streamline the process of identifying anti-nuclear antibodies. In some laboratories, automated multiplex immunoassay is used to screen for a mixture of common nuclear antigens. Positive specimens are then examined by direct immunofluorescence, which remains the gold standard. At this stage, the presence of anti-nuclear factor (ANF) is a non-specific indicator that a connective tissue disease may be present (see Table 32.1). The next step is to determine to which part of the nucleus the antibody reacts, by using highly specific tests, including enzyme linked immunoabsorbent assay (ELISA) and indirect immunofluorescence. Anti-double stranded DNA, anti-Sm, anti-Ro, anti-La, anti-centromere, anti-Jo1, anti-Mi2 and anti-mitochondrial antibodies can be identified in this way, each of which, as shown in Table 32.1, is associated with different connective tissue diseases.
CONCLUSION
In spite of the fact that biochemical abnormalities themselves are responsible for few articular diseases, we have seen how clinical biochemistry plays an important part in the diagnosis, treatment and monitoring of rheumatological conditions. Many of the chapters in this book refer to medical specialties that primarily focus on biochemical disorders, or conditions with major biochemical consequences. However, there are some, such as rheumatology, for which this is not the case. It is instructive to examine the different ways in which biochemical tests are useful in such a setting, using articular disorders as an example:
• finding urate crystals in synovial fluid makes the diagnosis of gout
• finding raised iron and ferritin concentrations in haemochromatosis arthritis and demonstrating the presence of increased iron on liver biopsy confirms the diagnosis
• finding homogentisic acid in the urine confirms the diagnosis of alkaptonuria.
2. Categorizing class of arthritis:
• the presence of ANF suggests one of the connective tissue diseases.
3. Supporting a diagnosis:
4. Excluding a diagnosis:
• normal iron studies virtually exclude the diagnosis of haemochromatosis.
5. Monitoring the activity of a disease:
6. Monitoring the effect of treatment:
7. Monitoring for drug toxicity:
8. Monitoring for the complications of a disease:
9. Specialty-specific idiosyncrasies:
These examples show how powerful biochemical investigations can be, when performed for a defined reason. However, we have also demonstrated that, when used inappropriately, they have the power to inflict harm and distress on the unfortunate victim. We must recall Hippocrates’ dictum ‘primum non nocere’ (first do not harm) at all times.
Further reading
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 32
Biochemistry of articular disorders
CHAPTER OUTLINE
DISORDERS OF THE ARTICULAR SYSTEM
The connective tissue diseases
ARTICULAR INVOLVEMENT IN ENDOCRINE AND METABOLIC DISEASES
LABORATORY TESTING IN ARTICULAR DISEASE
INTRODUCTION
While biochemical abnormalities themselves are responsible for few articular disorders, clinical biochemistry plays an important part in the diagnosis, treatment and monitoring of many of these conditions.
This chapter starts with a description of the articular system and its diseases, which will put the role of biochemistry in these conditions into perspective.
Crystal synovitis, as seen in gout and pseudogout, is the only common articular condition caused by an underlying biochemical abnormality. This is described in some detail along with examination of synovial fluid, the one biochemical investigation that is primarily the province of the rheumatologist.
Some diseases with a primary biochemical abnormality like diabetes, alkaptonuria, endocrine conditions and haemochromatosis have articular manifestations and also warrant discussion.
Immunological abnormalities are at the root of an important group of rheumatic diseases, the connective tissue diseases. Autoantibody formation is a major disease manifestation and, in some hospitals, tests to identify and measure autoantibodies are performed in the biochemistry laboratory. These tests are described briefly.
THE ARTICULAR SYSTEM
The joint is the pivotal structure of the articular system. Joints can be considered as discontinuities in the skeleton that permit controlled mobility. They have different structures depending on their functional requirements. When no movement is needed, the joint is bound together by tough fibrous tissue (e.g. the skull ‘sutures’). In cartilaginous joints, the bone ends are joined by compressible fibrocartilage and reinforced by a surrounding tough fibrous tissue (as in the symphysis pubis and the manubriosternal joint) and permit only a limited amount of movement. This type of joint tends to be located centrally. If a moderate or wide range of movement is required, a space must exist between the bone ends, forming a discontinuous ‘synovial’ joint. Most of the peripheral joints fall into this category.
The most important joints from a clinician’s point of view are the peripheral ones, which enable our arms and legs to move through such a wide range, our hands to be endowed with such dexterity and strength and our feet to carry us so uncomplainingly when we stand, walk, run and jump.
While synovial joints have the same basic structure and physiology, individual joints have evolved differently, depending on their situation and what is expected of them. For example, the knee, ankle and finger joints, which move mainly in one plane, are ‘hinge’ joints, while the hip and shoulder, which move in all directions, are ball and socket joints. Of course, to carry out even the simplest of tasks, like opening a door or walking up a step, we have to coordinate the movement of several joints, not to mention contracting and relaxing the muscles on either side of the joints.
The archetypical synovial joint is an enclosed space with a negative pressure (Fig. 32.1). The bone ends that move against each other (the articular surfaces) are covered with articular cartilage. This is made up of proteoglycans and collagen, combined in such a way as to allow it to absorb huge forces of pressure like a shock absorber, while providing a shiny surface for smooth, low-friction movement. A healthy joint is lubricated by a small amount of synovial fluid (0–4 mL). This is an ultrafiltrate of plasma with additional components secreted by the synovium. The most important of these is hyaluronate, a linear repeating disaccharide with a molecular weight of some 107 Da. This provides the fluid with its viscoelastic properties without which the cartilage would fail and smooth movement would be impossible. Cartilage and synovial fluid together maintain coefficients of friction of < 0.02.
FIGURE 32.1 A typical synovial joint.
The bone ends are enclosed by the tough joint capsule which is lined by the synovial membrane, a structure only a cell or two thick (Fig. 32.1). Synovial cells phagocytose intra-articular debris, secrete many components of synovial fluid and possess immunological functions.
While we talk of ‘articular disorders’, it is not really possible to consider the joint in isolation. Each joint depends on other structures like ligaments, tendons, muscles and bone for its function and stability. Bone and muscle disease are dealt with in separate chapters.
DISORDERS OF THE ARTICULAR SYSTEM
There are over 100 rheumatic disorders with different underlying causes, different treatments and different outcomes. These conditions can be broadly divided into non-inflammatory diseases, of which osteoarthritis is the most important, and inflammatory conditions such as rheumatoid arthritis (RA), ankylosing spondylitis and systemic lupus erythematosus (SLE or lupus).
Osteoarthritis (OA)
The most common form of articular disease is osteoarthritis, in which the articular cartilage becomes fissured and gradually wears away. The joint tries to heal itself by forming bony out-growths on the sides of the joints (osteophytes). Trauma to a joint predisposes to OA, as do some rare biochemical disorders like alkaptonuria. However, in most cases, apart from an inherited tendency and increasing frequency with age, the cause cannot be identified. For many years, OA was thought to be due to degeneration or ‘wear and tear’. Recently, we have appreciated that a degree of inflammation is often present if sought and that calcium pyrophosphate and basic calcium phosphate crystals are frequently found in osteoarthritic joints (see below).
At present, we have no treatment to influence this process. The knee and hip are often affected and, if the pain and limitation to mobility are severe enough, the orthopaedic surgeon will remove the joint and replace it with an artificial one, usually with very gratifying results.
Inflammatory arthritis
The other major class of articular disease is inflammatory arthritis. This consists of a group of systemic immunological diseases that focus their attention on the synovium of the joint. Rheumatoid arthritis (RA) is the archetypical form of inflammatory arthritis. In RA, there is an imbalance between the proinflammatory and anti-inflammatory cytokines that favours the induction of autoimmunity (an immune reaction against one’s own tissues), chronic inflammation and joint damage. The synovium becomes inflamed, resulting in pain, tenderness, heat and stiffness in the joint as well as a systemic reaction consisting of malaise and fatigue. An acute phase reaction ensues, which can be detected and monitored by measurement of plasma C-reactive protein (CRP) concentration or the erythrocyte sedimentation rate (ESR). In what may well be a separate process, in time the synovial cells increase in size and number, the synovium becomes thickened and develops the changes of chronic inflammation with infiltration by macrophages and lymphocytes. There are also alterations in the small blood vessels. In addition, the amount of synovial fluid in the joint increases in quantity, which adds to the swelling and dysfunction of the affected joint. A number of different cytokines associated with the rheumatoid process can be found in the synovial tissue and fluid; these include tumour necrosis factor alpha (TNFα), interleukins one (IL-1) and six (IL-6). There are also abnormalities of the T and B cell lymphocytes. If left to its own devices, the thickened synovium will spread over the cartilage and erode it and the neighbouring bone. This results in joint damage, deformity and disability.
Non-steroidal anti-inflammatory drugs, e.g. diclofenac, ibuprofen and naproxen are used less frequently than they once were. Although they often provide symptomatic relief, they increase the risk of cardiovascular events, can cause chronic kidney disease and have no influence on the long-term outcome of the disease.
Of more concern in the long term, is the chronic inflammation that results in joint damage and disability. Until about 20 years ago, there was little that we could do to slow or even stop the inexorable progress of this destructive process, meaning that many people with RA became very disabled with joint deformity and pain. However, there have been dramatic improvements in our drug treatment of this most unpleasant condition. We have learnt that we need to diagnose the condition as early as possible. This enables treatment with one or more disease modifying anti-rheumatic drugs (DMARDs) (e.g. methotrexate, leflunomide, sulfasalazine), to be started as soon as possible. As a rule, these drugs rapidly bring the arthritis under control, but different drugs suit different people so sometimes a degree of trial and error is required. DMARDs are taken for long periods and can cause side-effects such as liver damage or bone marrow suppression, and regular blood tests (full blood count, ESR, CRP and liver function tests) are used to monitor their safety as well as the activity of the disease.
More recently, the treatment of RA has been transformed by the introduction of so-called ‘biologics’ (monoclonal antibodies designed to counteract the effects of cytokines or alter the function of T or B cell lymphocytes). They are expensive and need to be taken long term, so at present they are used in the cases where DMARDs have been unsuccessful. However, in time, they may become the first-line in treatment of RA. Biologics in common use include agents against TNFα (e.g. etanercept) and anti IL-6 (tocilizumab), as well as B cell depleters (rituximab) and T cell blockers (abatacept). This is a field of active research with the potential to transform the treatment not only of RA but of many other immunological diseases.
Other examples of chronic inflammatory arthritis include ankylosing spondylitis, which mainly affects the small joints of the spine, the arthritis associated with psoriasis and the ‘reactive’ arthritis one sees in response to certain intestinal or genitourinary infections.
The connective tissue diseases
The connective tissue diseases (CTDs) are a fascinating group of conditions in which, for reasons we do not understand, the normal fine balance of the immune system gets disturbed, causing the body to react against itself. In health, the immune system consists of series of counterbalancing mechanisms that marshal defences against alien material like bacteria, viruses or foreign tissue, while not reacting in this way against itself. The identification of foreign tissue (the antigen) is the first step in the process and is followed by a series of defensive reactions including manufacturing antibodies. In CTDs, the person’s ability to recognize their own tissue goes awry and they form antibodies against their own tissue (autoantibodies). In some cases, these are responsible for the clinical manifestations of the disease; in others they are a peripheral effect of the underlying disease process. Theories of the pathogenesis of autoimmune diseases are discussed in Chapter 30.
Systemic lupus erythematosus (SLE) mainly affects younger women and can involve one or more of the bodily systems. It is characterized by the formation of autoantibodies against the cell’s nucleus, or part of the nucleus, which are believed to be responsible for many of the manifestations of the disease.
Joint pain is common, although it is unusual to see joint damage such as occurs in RA. SLE can result in a variety of manifestations in other systems, for example various rashes, haemolytic anaemia and glomerulonephritis leading to renal impairment, as well as cerebral, lung and heart problems. Some of these can be life threatening.
Autoantibodies against cell nuclei (antinuclear factor, ANF) are present in the plasma in almost every patient with SLE. However, they are also frequently present in other CTDs and in a proportion of healthy people. Antibodies to specific components of the nucleus are more specific. For example, antibodies against double-stranded DNA are strongly suggestive of SLE.
Management depends on the systems affected. The joint and skin manifestations can often be controlled with hydrochloroquine and/or low doses of prednisolone. More severe cases require the use of high doses of corticosteroids with immunosuppression using cyclophosphamide, azathioprine or mycophenolate mofetil.
Other CTDs are associated with autoantibody formation, although in these cases the disease is not believed to be caused by specific antibody-antigen complexes as in SLE. Examples of these conditions include Sjögren syndrome, polymyositis, scleroderma and primary biliary cirrhosis.
For details and associated immunological abnormalities, see Table 32.1. Table 32.1 is, of necessity, an oversimplified guide to some of the more important signs and symptoms seen in the CTDs, with their associated antibodies. There is often a crossover of antibodies in the various diseases and the antibodies are by no means seen in every case.
TABLE 32.1
The connective tissue diseases, their common clinical features and frequently associated autoantibodies
| Disease | Clinical features | Autoantibodies |
| SLE | Rashes, joint pains, glomerulonephritis, haemolytic anaemia, leukopenia, neuropsychiatric lupus, pericarditis, pleurisy, vasculitis, fatigue | Anti-nuclear factor Anti-double-stranded DNA Anti-Sm Anti-Ro Anti-La |
| Scleroderma | Thickened skin, Raynaud phenomenon, pulmonary hypertension, calcinosis, pulmonary fibrosis | Anti-nuclear factor Anti-centromere |
| Sjögren syndrome | Dry eyes, dry mouth, joint pains, fatigue, Raynaud phenomenon | Anti-nuclear factor Anti-Ro Anti-La Rheumatoid factor |
| Polymyositis | Muscle weakness and wasting, joint pains, pulmonary fibrosis, fatigue | Anti-nuclear factor Anti-Jo-1 Anti-synthetase |
| Dermatomyositis | Proximal muscle weakness and wasting, rashes, joint pains, pulmonary fibrosis | Anti-nuclear factor Anti-Mi2 |
| Wegener granulomatosis | Vasculitis, sinusitis, pulmonary infiltration, glomerulonephritis, rashes | cANCA |
| Microscopic polyangiitis | Glomerulonephritis, joint/muscle pains, rashes, lung infiltration | pANCA |
| Primary biliary cirrhosis | Jaundice, itching, joint pains, Raynaud phenomenon | Antimitochondrial |
cANCA, cytoplasmic anti-neutrophil cytoplasmic antibody; pANCA, perinuclear anti-neutrophil cytoplasmic antibody; SLE, systemic lupus erythematosus.
Aches and pains
If we move from the laboratory to the clinic or surgery, ‘articular disorders’ take on another perspective. About one-fifth of the patients in a GP’s surgery will complain of musculoskeletal symptoms, such as aches, pain, stiffness or disability. Only a proportion of these patients will have a demonstrable disease or significant anatomical abnormality. Some will have abnormalities associated with the way they move their musculoskeletal system (‘mechanical’ pain), while in other cases, no abnormality can be identified. This does not mean the symptoms are imagined, but in the absence of an abnormality, attempts to diagnose and ‘cure’ a disease will, of course, be futile. Doctors are trained to diagnose diseases and then treat them. Too many doctors are uncomfortable when they are not able to make a diagnosis. This may provoke them to order unnecessary investigations in these often anxious patients, thus exposing both parties to the dangers of false positives, laboratory or clerical error and misinterpretation of the results.
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
