[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 20
Metabolic response to stress
CHAPTER OUTLINE
INTRODUCTION
Stress can be defined as any influence, arising either internally or externally to the body, that threatens to disrupt normal structure, function or behaviour. Not surprisingly, complex mechanisms have evolved to protect the body against such threats, whether physical or psychological. It may be surmised that during man’s evolution, the most important stresses were external, e.g. attack and lack of food, water or shelter. For many people, such factors are now of little or no relevance, but others, for example psychological stresses, have become more prevalent; stressors such as trauma and infection remain universal.
The body’s response to stress tends to be qualitatively similar whatever the stress, although the severity of the stress affects the response quantitatively, albeit with considerable intra- and interindividual variation.
It is reasonable to suppose that the development of the metabolic response to stress was subject to evolutionary pressures, and that the response is potentially beneficial. For some aspects this clearly is the case, but, as will become apparent, some aspects of the response to severe stressors, particularly sepsis, appear potentially harmful to the body as a whole. One can only speculate how this might have come about. The metabolic response to stress is complex, and includes processes that act to propagate the response as well as to curtail it and thereby limit its extent, both physiologically and anatomically.
Conventionally, stressors are divided into four categories:
1. physical, e.g. heat, cold, immobilization and pain
2. psychological, e.g. anxiety and fear
3. social, e.g. bereavement, marital breakdown
4. cardiovascular and metabolic, e.g. exercise, haemorrhage and infection.
While this system helps categorize the nature of the stimulus, multiple stressors can be applied to an individual at any one time and these, in turn, can be either acute or chronic in nature. Stress responses are intended to be short lived and self-limiting, but are proportional to the intensity of the stimulus and, consequently, can range from simple localized reactions to complex processes. Systemic responses involve vascular, endocrine and metabolic changes orchestrated by the hypothalamo–pituitary axis and sympathetic nervous systems. Centrally, there is facilitation of arousal, inhibition of vegetative functions (feeding and reproduction) and activation of counter-regulatory feedback loops. Respiratory and heart rates increase and there is an increase in vascular tone, resulting in a rise in blood pressure and hence oxygen and nutrient supply to the brain, heart and skeletal muscles.
The intensity of stress may be gauged by peak concentrations of stress hormones and neurotransmitters, by physiological changes, such as increases in heart rate and blood pressure, and by the length of time for which these changes persist during stress and following the cessation of stress. Whilst intended to confer a survival advantage, the stress response can become part of the pathological process. In severe stress, responses that appear potentially advantageous if limited in extent (e.g. mobilization of muscle protein to provide glucose as an energy source) become harmful.
THE RESPONSE TO STRESS
Initiation of the stress response
Figure 20.1 summarizes the complexity of the systemic responses to potential stimuli. Stimulation of the hypothalamus and locus coeruleus leads to the release of pituitary hormones and activation of the sympathetic nervous system. The locus coeruleus, hypothalamus and brainstem are closely linked, both anatomically and functionally. The locus coeruleus and hypothalamus mutually innervate and stimulate each other. This positive feedback system allows for activation of the systemic stress response by any stimulus that initiates either side of this loop. The locus coeruleus is located in the lateral floor of the fourth ventricle, and represents the major pool of noradrenaline (norepinephrine) secreting neurons in the brain. It receives afferents from many areas, including the hypothalamus, the cerebellum, prefrontal cortex and the hypoglossi. The cingulate gyrus and amygdala also innervate the locus coeruleus, transmitting emotional and pain stressors. Nerve fibres from this nucleus innervate the spinal cord, brainstem, thalamic relay nuclei, cerebellum and hypothalamus and are predominantly excitatory in action. Adrenaline (epinephrine) secreting nerve terminals are also found within the locus coeruleus and are thought to represent part of the medullary regulatory circuit.
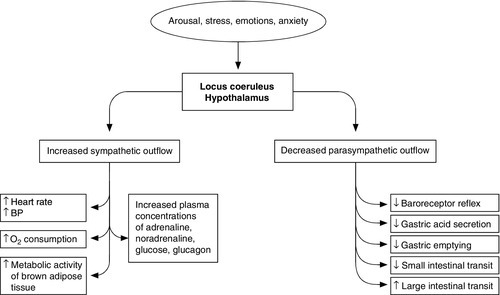
FIGURE 20.1 The systemic response to stress.
At the centre of the limbic system, the hypothalamus controls most of the vegetative (homoeostatic) and endocrine functions of the body. Combined with the locus coeruleus, it is the major effecter of the stress response.
The efferent neuronal pathways from the hypothalamus can be classified into two groups: direct monosynaptic projections to pre-ganglionic neurons in the lower brainstem and spinal cord projections to brainstem catecholaminergic neurons in the medulla that innervate neurons in the thoracic spinal cord.
Additionally, hypothalamic nuclei participate in other regulatory pathways, such as temperature control, energy homoeostasis and body fluid composition.
Hypothalamo–pituitary–adrenal axis
Stimulation of the paraventricular nucleus by afferent fibres from the limbic system and lower brainstem results in the secretion of corticotrophin-releasing hormone (CRH), into the primary capillary plexus of the hypophyseal portal system, from where it is transported to the anterior pituitary gland.
The hypothalamus also secretes the polypeptide arginine vasopressin (AVP), which is transported along nerve tracts to the posterior pituitary. Under resting conditions, CRH and AVP are secreted in a circadian and highly concordant pulsatile fashion: stressors promote a surge in AVP secretion. Vasopressin increases water retention in the kidneys, is a potent vasoconstrictor, acts as a neurotransmitter and has the ability to modulate a number of physiological processes including gluconeogenesis, platelet aggregation and inflammation.
Corticotrophin-releasing hormone and AVP act synergistically on the anterior pituitary to induce synthesis of the large preprohormone, proopiomelanocortin (POMC). The POMC molecule is processed to form a series of smaller peptides, the nature of which is dependent on the enzyme systems present within the tissue type. Anterior pituitary corticotrophin cells express prohormone convertase 1 (PC1), which cleaves POMC into ACTH, N-terminal peptide, joining peptide and β-lipotropin. In the hypothalamus the enzyme PC2 cleaves POMC to α-, β- and γ-melanocyte stimulating hormones and β-endorphin.
α-Melanocyte stimulating hormone acts on melanocortin receptors in the paraventricular nucleus of the hypothalamus, stimulating the sympathetic nervous system. Adrenocorticotrophic hormone and the other peptide cleavage products of POMC have a negative feedback effect on the hypothalamus to limit CRH release.
While CRH and AVP act synergistically on the anterior pituitary, only small amounts of ACTH are secreted in the absence of CRH, which is pivotal to an integrated stress response.
Cortisol
Following stimulation by ACTH, cortisol is the principle glucocorticoid produced by the adrenal cortex, accounting for 95% of glucocorticoid activity. Approximately 90% of circulating cortisol is protein bound; some is attached non-specifically to albumin, but the majority binds with high affinity to cortisol-binding globulin (CBG); only free cortisol is biologically active. Cortisol-binding globulin is an α2-globulin, produced in the liver, which behaves as a negative acute phase protein. The concentration of CBG falls in response to some inflammatory stimuli, increasing the bioavailability of cortisol.
Cortisol is a major effector of the metabolic response to stress. It is lipid soluble and readily crosses cell membranes, its activity being mediated via the glucocorticoid receptor (GR). Two isoforms of GR have been isolated, GRα, which binds cortisol and the GRβ, which does not bind cortisol and may act to inhibit the effect of endogenous glucocorticoid action. The hormone receptor complex is translocated to the nucleus where it interacts with glucocorticoid response elements and, following recruitment of co-activators, leads to increased gene transcription. Other actions are DNA independent and involve direct interactions with transcription factors, such as nuclear factor-κβ (NF-κβ), This is important in the modulation of the stress response, since NF-κβ activates the expression of interleukin-1(IL-1), IL-6 and tumour necrosis factor α (TNFα), all of which are key components of the inflammatory response and interact with the hypothalamo–pituitary–adrenal axis (HPAA).
Cortisol exerts a negative feedback effect on CRH and ACTH release, affects carbohydrate, protein and fat metabolism, and modulates the immune response. Cortisol limits glucose utilization in cellular respiration, perhaps by reducing the oxidation of NADH to NAD+ in the tricarboxylic acid cycle. High concentrations of cortisol reduce the sensitivity of peripheral tissues to insulin. This anti-insulin effect is particularly marked in skeletal muscle and adipose tissue, reducing glucose uptake and metabolism.
In extra-hepatic tissues, there is increased protein catabolism and decreased protein synthesis through reduced RNA formation and amino acid uptake. Within the liver, however, this metabolic picture is reversed, with increased amino acid uptake and the synthesis of acute phase proteins. Additionally, cortisol enhances the mobilization of fatty acids from adipose tissue, shifting oxidative metabolism to favour energy generation from glucose to fatty acids.
Thyroid hormones
Major disturbances of thyroid hormone metabolism and plasma concentration occur during the metabolic response to stress, typically with falls in the concentrations of tri-iodothyronine and often of thyroxine and, sometimes, in the critically ill, a fall in the concentration of thyroid stimulating hormone. (These changes are discussed further in Chapter 19.)
Sex hormones
The effects of trauma and sepsis on sex hormones are complex and dependent on the type of injury. Plasma concentrations of free testosterone may fall without any consistent change in the concentration of luteinizing hormone (LH), suggesting a change in the sensitivity of Leydig cells to LH or a disturbance of normal feedback regulation. The effects tend to be more marked in women, with reduced activity of the entire hypothalamo–pituitary–gonadal axis. Corticotrophin releasing hormone is known to have inhibitory effects at various levels in this axis, including antagonizing the effects of LH on Leydig cells. Chronic activation of the hypothalamo–pituitary–gonadal axis, such as has been demonstrated in long distance runners and ballet dancers, causes suppression of gonadal function in males and females.
Growth hormone
The anterior pituitary secretes growth hormone, stimulated by growth hormone releasing hormone, produced by the ventral medial nucleus of the hypothalamus. Growth hormone is a small protein that is active in most cells of the body, where it enhances amino acid uptake and protein synthesis. Under the influence of growth hormone, free fatty acids are used as an energy source in preference to carbohydrates and proteins. Growth hormone reduces glucose uptake by skeletal muscle and fat cells, promotes gluconeogenesis by the liver and consequently increases insulin secretion by the pancreas.
Adrenal medulla
Activation of the locus coeruleus by stressors causes the release of noradrenaline and increased sympathetic discharge via the brainstem and spinal cord. Pre-ganglionic sympathetic nerves pass through the sympathetic chain to innervate modified neuronal cells in the adrenal medulla. These cells secrete noradrenaline and adrenaline into the circulation. Catecholamine synthesis is an energy-dependent process with many mitochondria packing the terminal varicosities containing the secretory vesicles. Tyrosine is converted to L-DOPA and then to dopamine, which is transported into the secretory vesicles where a hydroxylation reaction converts it into noradrenaline. The majority of noradrenaline is then metabolized to adrenaline. The sympathetic nerve impulse to the adrenal medulla results in the influx of intracellular calcium and the discharge of the catecholamine secretory vesicles (in the ratio of 80:20, adrenaline:noradrenaline) directly into the circulation. The combined effect of this circulating adrenaline and noradrenaline is identical to that caused by direct sympathetic nervous system stimulation, but has a longer duration and can reach all cells of the body irrespective of their autonomic innervation.
STRESS AND THE KIDNEYS
Fluid conservation occurs as part of the stress response, often resulting in low urine outputs despite euvolaemia. Increased sympathetic nervous system activity, combined with raised concentrations of circulating catecholamines, reduces renal blood flow. This initiates the metabolism of prorenin to renin in the juxtaglomerular cells of the kidneys. Renin is released into the blood where it acts on angiotensinogen to form angiotensin I. Under the influence of angiotensin-converting-enzyme, predominantly in the lung, angiotensin I is converted to angiotensin II, a potent vasoconstrictor. Angiotensin II stimulates AVP release from the hypothalamus, as well as having a direct affect on the kidney to increase salt and water retention. Furthermore, under the influence of angiotensin II, the mineralocorticoid aldosterone is synthesized in the zona glomerulosa of the adrenal cortex, by metabolism of corticosterone by the enzyme aldosterone synthase. Whilst ACTH has a permissive role in aldosterone synthesis, it does not control the rate of release. Aldosterone acts on the cortical collecting tubules where it stimulates a Na+,K+-ATPase, leading to the retention of sodium and water. (Salt and water balance is considered further in Chapter 4 and renal function in Chapter 7.)
CYTOKINES
Cytokines are peptides secreted by many cell types including mast cells, macrophages and endothelial cells. They are released into the extracellular fluid and can function as autocrine, paracrine or endocrine hormones and they trigger the acute phase response.
Major changes in the plasma concentrations of cytokines occur during trauma and sepsis: they are also released during psychological stress. Cytokines are produced by activated cells of the immune system and mediate communication between the immune, endocrine and central nervous systems. Cytokines are classified into three groups dependent on their function. Group 1 (IL-2, IL-3, IL-4, IL-7, IL-10, IL-11, IL-12 and granulocyte–macrophage colony stimulating factor) act on a variety of cell types as positive or negative growth factors, whilst group 2 (TNFα/β, IL-1α/β, IL-6, IFN-α/γ, IL-8 and macrophage inhibitory protein-1) have proinflammatory properties. Group 2 cytokines are responsible for the induction of fever, muscle catabolism, activation of white blood cell precursors and growth of inflammatory fibroblasts and macrophages. Group 3 contains cytokines with anti-inflammatory activity (IL-1 receptor antagonists, soluble IL-1 receptors, TNFα binding protein and IL-1 binding protein). Endothelial cells and macrophages are major producers of IL-1, IL-6 and TNFα, but virtually all nucleated cells have the ability to synthesize them.
Physical damage to cells results in the release of cytokines, some of which are chemotactic (e.g. IL-8), attracting large numbers of macrophages and neutrophils to the site of injury, where they are activated by TNFα, IL-1 and IL-6 to phagocytose dead tissue and bacteria.
Whether cytokines have localized (paracrine) or generalized (endocrine) effects is an important determinant of the extent of the inflammatory response. Cytokines having local actions are also released into the circulation where they initiate some of the systemic features of the metabolic response, such as fever (IL-1) and stimulation of hepatic protein synthesis to produce acute phase proteins (IL-6): IL-6 is also a potent stimulator of ACTH (and hence cortisol) secretion, which amplifies the stress response.
Interleukin-1, IL-6 and TNFα are particularly important cytokines. Interleukin-1 is produced by activated macrophages and mediates the inflammatory response, stimulates the HPAA and potentiates amino acid flux in skeletal muscle. Tumour necrosis factor-α, produced by macrophages and lymphocytes has similar properties to IL-1, and acts synergistically with it. Tumour necrosis factor-α stimulates inducible nitric oxide synthase, causing vasodilatation whilst also stimulating the production of cyclooxygenase, which in turn facilitates prostaglandin and thromboxane synthesis. Prostaglandin E2 acts on the hypothalamic thermoregulatory centre to cause fever and is also immunosuppressive; other prostaglandins excite A and C pain fibres. Thromboxanes stimulate platelet function and are procoagulant. Interleukin-6 may also contribute to the activation of the HPAA and to insulin resistance. Tumour necrosis factor-α, IL-1β and IFN-γ are crucial for the induction of other cytokines (e.g. IL-6 and IL-8), platelet activating factor, prostaglandins, leukotrienes and nitric oxide.
STRESS AND INFLAMMATION
Stress and glucocorticoid hormones can either enhance or suppress immune function, depending on the following factors:
• changes in leukocyte distribution and the compartments involved
• the concentration, duration and nature of the glucocorticoid exposure
• the timing of stress or stress hormone exposure relative to the stage of an immune response.
The stress response promotes a physiological synchronization of cardiovascular, musculoskeletal and neuroendocrine systems to confer a survival advantage. Despite the known anti-inflammatory effects of glucocorticoids, the body is unlikely to suppress immune function when the host is liable to wounding and infection. Indeed it is more likely that acute stress may serve to enhance the immune response.
Tissue injury (trauma, infection, hypoxia) causes the release of proinflammatory mediators such as heat shock proteins, adenosine and high mobility group box protein 1 in a characteristic damage-associated molecular pattern, and they are known collectively as ‘alarmins’. Effective immunoprotection requires rapid recruitment of leukocytes to the site of damage, which is facilitated by the binding of alarmins to endogenous receptors on neutrophils and macrophages. Subsequently, the activation of NK-κβ leads to the transcription of proinflammatory proteins, including the cytokines IL-1, IL-6 and TNFα. Following release, these cytokines further activate inflammatory cells and the vascular system leading to a systemic response.
Cortisol and adrenaline act synergistically to induce rapid changes in leukocyte distribution and to enhance cell mediated and contact hypersensitivity immunity. Stress-induced changes include a reduction in the numbers of lymphocytes and monocytes but an increase in the number of neutrophils. These changes are rapidly reversed upon cessation of the stress.
Glucocorticoids act synergistically with cytokines to enhance specific immune reactions, by inducing their release and potentiating their actions. Synergistic interactions between glucocorticoids and cytokines may be mediated by glucocorticoid induced upregulation of cytokine receptors on target cells as determined by increased cytokine binding or cytokine receptor mRNA expression. For example, glucocorticoid-induced TNF receptors have been shown to promote survival and serve as a stimulatory receptor for T cells. Furthermore, glucocorticoids increase IL-1 binding to human peripheral blood B cells.
Acute psychological stress is known to increase circulating TNFα, IL-1β and IL-10, with glucocorticoids also enhancing the biological activity of IL-2, interferon γ (IFN-γ) and granulocyte colony stimulating factor. Acute stress responses may induce a biphasic shift in leukocyte numbers. Initially, catecholamine hormones and neurotransmitters induce leukocytes to exit the spleen and bone marrow and to enter blood vessels and lymphatics. Consequently, there is an increase in blood leukocytes, predominantly natural killer (NK) cells and granulocytes. As the stress response evolves, glucocorticoid hormones initiate migration of lymphocytes to sites of infection. This redistribution leads to a reduction in the numbers of circulating lymphocytes, but is not an anti-inflammatory effect.
In contrast to acute stress, chronic stress has been shown to dysregulate immune responses by altering the cytokine balance from type 1 to type 2. Immunosenescence is accelerated and immunity decreased owing to a reduced number of circulating protective cells and increased populations of suppressor T lymphocytes.
To limit potential tissue damage after activation of the immune system, a counter-regulatory response is simultaneously triggered by inflammation and hypoxia. An important physiological role of endogenous glucocorticoids might be to suppress an ongoing immune response. Studies have demonstrated the existence of a negative feedback loop between the immune system and HPAA such that proinflammatory mediators arising from ongoing immune reactions stimulate the axis, which in turn results in the secretion of cortisol, which suppresses the immune response and prevents it from potentially damaging the host.
Cortisol exerts its anti-inflammatory effects through a number of mechanisms. By stabilizing lysosomal membranes, cortisol reduces the release of proinflammatory proteolytic enzymes and reduces damage to the vascular endothelium, thereby decreasing capillary leak. Cortisol is thought to have an anti-inflammatory action through the non-genomic binding of the GR-cortisol complex to the transcriptase NK-κβ, reducing the formation of prostaglandins and cytokines that would act as positive chemotactic agents for phagocytic white blood cells. Cortisol inhibits the production of IL-2, which stimulates the proliferation of T cells. It also induces the synthesis of lipocortin, an inhibitor of phospholipase A2, which is required for the synthesis of prostaglandins and leukotrienes. Concurrently, there is secretion of IL-6 an anti-inflammatory cytokine that inhibits the production of IL-1 and TNFα and thus provides negative feedback for the inflammatory cascade.
Cortisol can promote increased transcription of anti-inflammatory genes. Mitogen activated protein kinases (MAPKs) form intracellular signalling pathways that result in the production and post-transcriptional modification of proinflammatory mediators. MAPK phosphatases dephosphorylate MAPKs rendering them inactive; glucocorticoids increase the expression of MAPK phosphatases and thus can down regulate the inflammatory response.
Cells that surround the inflamed tissues express specific tissue protective receptors (TPR) that bind the type 1 cytokine erythropoietin. Binding of this ligand inhibits proinflammatory cytokine production, inhibits macrophage activity and delimits the volume of injury by countering apoptosis. Activation of TPRs also acts to recruit vascular and tissue specific stem cells and enhances tissue repair.
Catecholamines
Noradrenaline is released mainly from the postganglionic nerve endings, while the adrenal medulla is the major source of adrenaline in the circulation.
Stimulation of the sympathetic nerves to the adrenal medulla results in large quantities of adrenaline and noradrenaline being released into the circulation. The action of these catecholamines is mediated via cell surface adrenergic receptors, which are themselves bound to protein molecules that traverse the cell membrane. Binding to these receptors causes a conformational change in the protein, resulting in the opening of an ion channel or the stimulation of a second messenger enzyme system. There are two major groups of adrenergic receptors, α and β, which are further divided into subgroups (α1–2, β1–3). Noradrenaline preferentially excites α receptors in addition to having some β activity, whilst adrenaline is equally effective across both receptor groups. Sympathetic stimulation is excitatory in some tissues and inhibitory in others and similarly α and β receptors may be excitatory or inhibitory.
Circulating catecholamines enhance the sympathetic nervous system’s stimulation of the cardiovascular system. This results in arteriolar and venous constriction and increased heart rate and force of myocardial contraction. The net result is raised ventricular blood volume causing increased muscle fibre stretching, improving cardiac output and raising blood pressure.
During stress, noradrenaline and adrenaline both cause hyperpolarization of gut smooth muscle and decreased bowel motility. Circulating adrenaline stimulates the phosphorylase enzyme via cAMP, causing rapid glycogenolysis especially in the liver and skeletal muscle. Combined with the anti-insulin effects of cortisol, adrenaline and noradrenaline activate triglyceride lipase in fat cells, causing the rapid mobilization of fatty acids, shifting energy metabolism away from glucose.
Brain catecholaminergic systems utilize noradrenaline, adrenaline, dopamine and L-DOPA. Stressful stimuli accelerate both the synthesis and release of brain catecholamines, with adrenergic and noradrenergic neurons involved in the central processing of the stress response. The effect of these neurotransmitters is dependent on which receptors and transporters are expressed on target cells. Adrenaline binds to the same receptors as noradrenaline and adrenergic neurons may use noradrenaline transporters for reuptake from synaptic clefts.
Acute stress leads to transient alterations in brain catecholamine systems with transient activation of the HPAA. After a short period of time and depending on the nature of the stressor, the activity of these catecholaminergic neurons can return to baseline. Chronic, long-term or repeated stressors may induce permanent changes and keep catecholamine producing neurons chronically active.
In response to the majority of stressors, brainstem noradrenaline producing neurons stimulate the HPAA non-specifically via adrenergic receptors. In contrast, stimulation of the paraventricular nucleus of the hypothalamus is stressor specific.
Acute phase proteins
Hepatic protein synthesis shifts from constitutive proteins (e.g. albumin and transferrin), to acute phase proteins (e.g. C-reactive protein (CRP) and fibrinogen), following activation of the systemic stress response. This causes increased plasma concentrations of CRP, coagulation proteins, protease inhibitors and complement proteins. These acute phase proteins are arbitrarily divided into type I, e.g. haptoglobin and α1-acid glycoprotein that are mediated by IL-1 and TNF-like cytokines, and type II acute phase proteins mediated by IL-6-like cytokines. Since the synthesis of this group of proteins increases under stress, they are referred to as positive acute phase proteins. In contrast, constitutive proteins such as albumin and the hormone binding proteins are downregulated and classified as negative acute phase proteins. Under stress conditions, the amino acids required for this protein synthesis are derived from extrahepatic tissues, where proteolysis and reduced amino acid uptake occur, secondary to the influence of cortisol and the catecholamines.
Positive acute phase proteins have diverse functions in opsonization and trapping of microorganisms, activating complement, binding cellular remnants, neutralizing enzymes, scavenging free radicals and in modulating the host’s immune response.
C-reactive protein is a ring-shaped protein that binds to phosphocholine on the surface of dead cells and some bacteria where it acts as an opsonin, activating complement by the classical pathway. Haptoglobin binds free haemoglobin, inhibiting its oxidative activity and has anti-inflammatory capabilities, binding to major receptors on the cell membranes of leukocytes. It is removed from the circulation, along with any bound molecules, by the reticuloendothelial system. Fibrinogen (factor 1) is a high molecular weight glycoprotein involved in the clotting cascade and inflammation. It is converted by the enzyme thrombin into fibrin monomers that automatically polymerize to form fibrin fibres as part of clot formation. Fibrinogen and fibrin contribute to inflammation by inducing leukocyte migration and by modulating the inflammatory response of leukocytes and endothelial cells via an increased responsiveness to cytokines and chemokines.
Coagulation factors
Coagulation may be enhanced during the stress response to infection, with the inflammatory and clotting cascades being closely linked. Endothelial cell injury activates the clotting cascade through binding of factor VIIa to tissue factor, leading to the generation of thrombin. This catalyses the formation of fibrin and activates platelets, resulting in the formation of a clot. During stress, there are increased concentrations of circulating procoagulants such as fibrinogen and the cytokine IL-6, which stimulate the expression of tissue factor. Concurrently, platelets are activated by proinflammatory cytokines and adrenaline. There is downregulation of physiological anticoagulant mechanisms such as proteins C and S (negative acute phase proteins) and inhibition of fibrinolysis. Consequently, inflammation-induced coagulation is characterized by widespread intravascular fibrin deposition. Impairment of thrombolysis occurs because of high circulating concentrations of plasminogen activator inhibitor type 1.
By binding to protease activated receptors (PARs), thrombin induces leukocyte migration to the surface of the endothelium and by induction of the expression of cytokines and chemokines by leukocytes. The four isoforms of PARs (PAR 1–4) belong to a family of G-coupled receptors activated by proteolytic cleavage.
Procoagulant activity is regulated by three important anticoagulant pathways: antithrombin, the protein C system and tissue factor pathway inhibitor. During inflammation-induced coagulation, the function of all three pathways can be impaired.
The serine protease inhibitor antithrombin is the main inhibitor of thrombin. During severe inflammatory responses, antithrombin concentrations are markedly decreased owing to impaired synthesis (negative acute phase response), degradation by elastase from activated neutrophils and consumption by ongoing thrombin generation.
Antithrombin has an anti-inflammatory effect, by stimulating prostacyclin release from endothelium. The anti-inflammatory effects of cytokines are the result of blockade of both cytokine production by neutrophils and their tethering to vascular endothelium. Antithrombin also binds to specific receptors on neutrophils, monocytes and lymphocytes, blocking their interactions with the endothelium.
In severe inflammation, plasma concentrations of protein C are low due to impaired synthesis, consumption and degradation by proteolytic enzymes such as neutrophil elastase. There is also a significant downregulation of thrombomodulin, a protein cofactor expressed on the surface of endothelial cells. Under normal conditions, the thrombin–thrombomodulin complex activates protein C, but in severe inflammation the expression of this protein is inhibited by the inflammatory cytokines TNFα and IL-6. Low plasma concentrations of free protein S may compromise the function of the protein C system. In plasma, 60% of the cofactor protein S is bound to complement regulatory protein C4b binding protein (C4bBP). Increased plasma concentrations of C4bBP, as a consequence of the acute phase response, result in a relative protein S deficiency. In sepsis, the endothelial protein C receptor is downregulated, further impeding the function of this anticoagulant.
SHOCK
Shock can be classified according to its aetiology: cardiac dysfunction (either primary or secondary); loss of circulating volume (fluid loss or redistribution), or failure to maintain systemic vascular resistance. These may occur in combination, particularly when entering the final common pathway of refractory shock. If untreated, the end result, whatever the cause, is tissue hypoperfusion and impairment of cellular metabolism.
The amount of oxygen delivered to tissues is a function of the cardiac output and arterial oxygen content. In haemorrhagic shock, cardiac output falls because of reductions in preload, force of contraction and stroke volume. These changes promote compensatory sympathetic responses inducing tachycardia, peripheral vasoconstriction, tachypnoea and the activation of neurohumoral mechanisms. The adrenal glands secrete adrenaline, noradrenaline and cortisol, whilst renal hypoperfusion activates the renin–angiotensin system. Angiotensin II, a vasoconstrictor, stimulates the release of vasopressin and aldosterone. Vasopressin, a pressor agent, increases the permeability of the renal collecting ducts to water, resulting in water retention, whilst aldosterone acts at the distal renal tubule to promote sodium retention.
If the stress response is inadequate for the metabolic challenge imposed upon the body, endothelial cells switch to anaerobic metabolism, absorb interstitial fluid and swell. This causes narrowing of the lumen of arterioles, further restricting the oxygen supply. Ischaemic cells accumulate lactic acid and free radical species, which are directly cytotoxic, prompting the release of inflammatory mediators and the adhesion of neutrophil leukocytes cells. As the inflammatory cascade is propagated, the cumulative anaerobic metabolites become negatively inotropic, and energy-dependent potassium channels fail, causing arterioles to dilate, heralding the onset of refractory shock.
CARE OF THE SHOCKED PATIENT
The response to stress is typically well regulated but homoeostatic control can be lost in the presence of powerful stimuli or if a second insult occurs, for example an infection or a need for surgery after trauma.
Definitions
Patients are admitted to critical care units because of various underlying disorders, yet may display similar clinical manifestations regardless of the clinical insult. This observation led to the introduction of the term ‘systemic inflammatory response syndrome’ (SIRS) and to attempts to define shock in order to establish a consistent basis to assist diagnosis and treatment (Box 20.1). Such a classification also facilitates design of clinical trials and incorporation of their results into routine clinical practice. The term multi-organ dysfunction syndrome (MODS) is now preferred to the previously used term ‘multi-organ failure’. Multi-organ dysfunction syndrome is defined by organ specific criteria, involving clinical and investigation findings. It is now preferred as a concept as it is acknowledged that it may be appropriate to offer organ support before the function of individual organs has deteriorated to a life-threatening extent. Signs of organ system dysfunction are shown in Box 20.2.
Management
The mortality and morbidity of critically ill patients has been much improved by the Surviving Sepsis Campaign (SSC), an international forum launched in 2004 to improve the management of sepsis. Since the majority of patients with SIRS, shock and MODS have infection as a contributory factor, appropriate management of sepsis is essential.
Immediate care
Immediate measures include removing the source of infection (if practicable), collection of tissues and fluids for microbiology and the commencement of antimicrobials. Targets in this phase of management include a heart rate < 90 bpm, systolic BP > 90 mmHg (MAP > 65 mmHg), a urine output > 0.5 ml/kg/min and normalisation of serum lactate concentration (if previously raised).
Initial antimicrobial therapy must be empirical, on the basis of the likely causative organisms until the results of culture and sensitivity are known. Factors to consider include the site of the infection, knowledge of the patient’s pre-morbid condition, previous antibiotic exposure and local prescribing guidelines. In the case of antibiotics, broad-spectrum agents can often be replaced with a tailored regimen once sensitivities are known. This approach reduces the risk of promoting multi-drug resistance in pathogenic bacteria. Gram staining of specimens may reveal bacteria and recently the application of PCR (polymerase chain reaction) technology has led to rapid diagnostics in some infections.
Organ support
Balanced salt solutions (e.g. Hartmann’s, lactated Ringer’s) are the fluids of choice in the resuscitation of septic shock. Norepinephrine (noradrenaline) is typically the favoured vasopressor (because of its predominant α-adrenergic effects), often in combination with the vasodilating inotrope, dobutamine. In cases of refractory shock, vasopressin is a valuable and potent pressor agent, usually used in combination with noradrenaline and intravenous steroids. Frequently, the use of vasopressin will allow a reduction in the dose of noradrenaline, potentially limiting some of its deleterious effects. Glucocorticoids enhance vascular responsiveness to catecholamines by mechanisms that may include adrenoceptor function and prostaglandin action. Hydrocortisone is prescribed at a dose of 200 mg daily in divided aliquots or as a continuous infusion. Whilst it is not necessary to perform a tetracosactide test to identify adrenal suppression, hydrocortisone should not be commenced in septic patients who are not shocked, because of its immunosuppressive effects.
Respiratory support should be given as required. Patients receiving mechanical ventilation should undergo intermittent, planned interruptions to assess the need for ongoing assistance. If their underlying condition permits, they should also be nursed with the head of the bed elevated to 30–45° as this has been shown to limit aspiration and reduce the risk of pneumonia.
Red cell transfusion should generally be reserved for patients whose haemoglobin concentration falls to < 70 g/L, once tissue hypoperfusion has resolved, except in particular circumstances such as acute haemorrhage or myocardial ischaemia. Platelet transfusion should be administered prophylactically in severe sepsis when platelet counts are < 10 × 109/L, in the absence of severe bleeding and 50 × 109/L in the context of active bleeding, surgery or invasive procedures. Intravenous immunoglobulins are not recommended in severe sepsis or septic shock.
Continuous renal replacement therapy and intermittent haemodialysis have been shown to be of equal benefit in patients with severe sepsis. However, continuous therapies facilitate fluid balance in patients who are haemodynamically unstable.
In the first 48 h after a diagnosis of severe sepsis or septic shock, it is preferable to administer nutrition enterally, as tolerated, rather than to fast the patient or to provide only intravenous glucose. Current evidence suggests that intravenous glucose and low dose enteral feeding, increased as tolerated, is preferable to parenteral feeding in the first week of illness. Various immunomodulating agents have been used in nutritional formulations but none is currently recommended for use in patients with sepsis. The reduction in the availability of arginine in sepsis has the potential to lower nitric oxide synthesis and increase production of superoxide and peroxynitrite. Conversely, arginine supplementation has the potential to cause vasodilatation and hypotension and studies of its use in nutritional preparations have had conflicting results. Plasma glutamine concentration is reduced during severe illness. A previous meta-analysis showed a reduction in mortality associated with its use but this has not been reflected in other studies. Whilst there do not appear to be adverse effects of glutamine supplementation, a large trial (the REDOXS study) is currently in progress to investigate its use. There is some initial evidence that the administration of selenium may have a beneficial effect in SIRS and shock by providing an antioxidant defence. Further studies are required before its administration can be recommended for this specific purpose, but its use as part of standard trace element supplementation is endorsed.
There should be protocols in place for the management of blood glucose. Initially, blood glucose should be monitored every 1–2 h and insulin infusion begun if two consecutive concentrations are > 10 mmol/L. Once glucose concentrations and insulin infusion rates are stable, the frequency of monitoring may be reduced to every four hours. The upper acceptable limit for blood glucose should be 10 mmol/L in this group of patient; the stricter target of 6.1 mmol/L advocated previously was not associated with reduced mortality, but predisposed to hypoglycaemia.
Patients with severe sepsis should receive mechanical prophylaxis against venous thromboembolism using graduated compression stockings or mechanical compression devices. In addition, they should receive pharmacoprophylaxis with low molecular weight heparin unless contraindicated, e.g. thrombocytopenia, active bleeding, recent intracerebral haemorrhage.
Patients with severe sepsis and risk factors for bleeding (e.g. coagulopathy, mechanical ventilation for at least 48 h and possibly hypotension) are at risk of stress ulceration and should be given prophylaxis using proton pump inhibitors in preference to H2 receptor blockers.
Immunomodulation
The body has a non-specific, or innate, immune response provided by mechanical barriers such as the skin and gastric acid secretion, and a more specific acquired immune response. Both may be altered by treatment, either directly or as a secondary effect, e.g. proton pump inhibitors given for stress ulceration reduce gastric acidity (potentially compromising innate immunity) and nutritional support may have a beneficial effect on the immune response.
As the pathogenesis of SIRS, shock and MODS involves immune mediators, it is logical to attempt to treat the conditions by their specific modulation. Various agents have been tried for this purpose.
The use of glucocorticoids for their vasopressor effect in shock has been described above. Their effect on the immune response is via a reduction in the production of proinflammatory cytokines, particularly TNFα and IL-6, although trials of the use of glucocorticoids for immunomodulation have not shown great benefit. Similarly, trials of anti-cytokine therapies, such as monoclonal antibodies to TNFβ and recombinant IL-1 receptor antagonist, have not proven their efficacy.
Inflammatory and procoagulant responses are intimately linked and may lead to widespread injury to vascular endothelium, contributing to MODS. Protein C acts to promote fibrinolysis and has inhibitory actions on thrombosis and inflammatory responses which make it a potential therapeutic target. Activated protein C has been used in the treatment of severe sepsis but has now been withdrawn due to lack of evidence of benefit on 28-day mortality.
Some agents used routinely in the critically ill, e.g. sedatives such as propofol and benzodiazepines, and non-steroidal anti-inflammatory agents, have been shown, particularly in animal studies, to modify the immune response, although a precise clinical role is unclear. Recent evidence suggests that statins may modify the immune response but the results of clinical trials to evaluate this further are awaited.
One explanation for the lack of clinical benefit from immunomodulating therapies may be the redundancy of action that exists between the cytokines; blocking of one pathway still allows for activation and release of mediators by an overlapping route. A further reason could be that the timing of the administration of the immunomodulating agent may be of critical importance for the success of a response.
CONCLUSION
The body’s response to stress tends to be qualitatively similar whatever the stress, although the severity of the stress affects the response quantitatively. In severe stress, responses that appear potentially advantageous if limited in extent become harmful. There have been improvements in the survival of critically ill patients as a result of better organ support and more effective treatment of infection, but attempts at specific immunomodulation of the inflammatory response have been disappointing.
Further reading
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 20
Metabolic response to stress
CHAPTER OUTLINE
INTRODUCTION
Stress can be defined as any influence, arising either internally or externally to the body, that threatens to disrupt normal structure, function or behaviour. Not surprisingly, complex mechanisms have evolved to protect the body against such threats, whether physical or psychological. It may be surmised that during man’s evolution, the most important stresses were external, e.g. attack and lack of food, water or shelter. For many people, such factors are now of little or no relevance, but others, for example psychological stresses, have become more prevalent; stressors such as trauma and infection remain universal.
The body’s response to stress tends to be qualitatively similar whatever the stress, although the severity of the stress affects the response quantitatively, albeit with considerable intra- and interindividual variation.
It is reasonable to suppose that the development of the metabolic response to stress was subject to evolutionary pressures, and that the response is potentially beneficial. For some aspects this clearly is the case, but, as will become apparent, some aspects of the response to severe stressors, particularly sepsis, appear potentially harmful to the body as a whole. One can only speculate how this might have come about. The metabolic response to stress is complex, and includes processes that act to propagate the response as well as to curtail it and thereby limit its extent, both physiologically and anatomically.
Conventionally, stressors are divided into four categories:
1. physical, e.g. heat, cold, immobilization and pain
2. psychological, e.g. anxiety and fear
3. social, e.g. bereavement, marital breakdown
4. cardiovascular and metabolic, e.g. exercise, haemorrhage and infection.
While this system helps categorize the nature of the stimulus, multiple stressors can be applied to an individual at any one time and these, in turn, can be either acute or chronic in nature. Stress responses are intended to be short lived and self-limiting, but are proportional to the intensity of the stimulus and, consequently, can range from simple localized reactions to complex processes. Systemic responses involve vascular, endocrine and metabolic changes orchestrated by the hypothalamo–pituitary axis and sympathetic nervous systems. Centrally, there is facilitation of arousal, inhibition of vegetative functions (feeding and reproduction) and activation of counter-regulatory feedback loops. Respiratory and heart rates increase and there is an increase in vascular tone, resulting in a rise in blood pressure and hence oxygen and nutrient supply to the brain, heart and skeletal muscles.
The intensity of stress may be gauged by peak concentrations of stress hormones and neurotransmitters, by physiological changes, such as increases in heart rate and blood pressure, and by the length of time for which these changes persist during stress and following the cessation of stress. Whilst intended to confer a survival advantage, the stress response can become part of the pathological process. In severe stress, responses that appear potentially advantageous if limited in extent (e.g. mobilization of muscle protein to provide glucose as an energy source) become harmful.
THE RESPONSE TO STRESS
Initiation of the stress response
Figure 20.1 summarizes the complexity of the systemic responses to potential stimuli. Stimulation of the hypothalamus and locus coeruleus leads to the release of pituitary hormones and activation of the sympathetic nervous system. The locus coeruleus, hypothalamus and brainstem are closely linked, both anatomically and functionally. The locus coeruleus and hypothalamus mutually innervate and stimulate each other. This positive feedback system allows for activation of the systemic stress response by any stimulus that initiates either side of this loop. The locus coeruleus is located in the lateral floor of the fourth ventricle, and represents the major pool of noradrenaline (norepinephrine) secreting neurons in the brain. It receives afferents from many areas, including the hypothalamus, the cerebellum, prefrontal cortex and the hypoglossi. The cingulate gyrus and amygdala also innervate the locus coeruleus, transmitting emotional and pain stressors. Nerve fibres from this nucleus innervate the spinal cord, brainstem, thalamic relay nuclei, cerebellum and hypothalamus and are predominantly excitatory in action. Adrenaline (epinephrine) secreting nerve terminals are also found within the locus coeruleus and are thought to represent part of the medullary regulatory circuit.
FIGURE 20.1 The systemic response to stress.
At the centre of the limbic system, the hypothalamus controls most of the vegetative (homoeostatic) and endocrine functions of the body. Combined with the locus coeruleus, it is the major effecter of the stress response.
The efferent neuronal pathways from the hypothalamus can be classified into two groups: direct monosynaptic projections to pre-ganglionic neurons in the lower brainstem and spinal cord projections to brainstem catecholaminergic neurons in the medulla that innervate neurons in the thoracic spinal cord.
Additionally, hypothalamic nuclei participate in other regulatory pathways, such as temperature control, energy homoeostasis and body fluid composition.
Hypothalamo–pituitary–adrenal axis
Stimulation of the paraventricular nucleus by afferent fibres from the limbic system and lower brainstem results in the secretion of corticotrophin-releasing hormone (CRH), into the primary capillary plexus of the hypophyseal portal system, from where it is transported to the anterior pituitary gland.
The hypothalamus also secretes the polypeptide arginine vasopressin (AVP), which is transported along nerve tracts to the posterior pituitary. Under resting conditions, CRH and AVP are secreted in a circadian and highly concordant pulsatile fashion: stressors promote a surge in AVP secretion. Vasopressin increases water retention in the kidneys, is a potent vasoconstrictor, acts as a neurotransmitter and has the ability to modulate a number of physiological processes including gluconeogenesis, platelet aggregation and inflammation.
Corticotrophin-releasing hormone and AVP act synergistically on the anterior pituitary to induce synthesis of the large preprohormone, proopiomelanocortin (POMC). The POMC molecule is processed to form a series of smaller peptides, the nature of which is dependent on the enzyme systems present within the tissue type. Anterior pituitary corticotrophin cells express prohormone convertase 1 (PC1), which cleaves POMC into ACTH, N-terminal peptide, joining peptide and β-lipotropin. In the hypothalamus the enzyme PC2 cleaves POMC to α-, β- and γ-melanocyte stimulating hormones and β-endorphin.
α-Melanocyte stimulating hormone acts on melanocortin receptors in the paraventricular nucleus of the hypothalamus, stimulating the sympathetic nervous system. Adrenocorticotrophic hormone and the other peptide cleavage products of POMC have a negative feedback effect on the hypothalamus to limit CRH release.
While CRH and AVP act synergistically on the anterior pituitary, only small amounts of ACTH are secreted in the absence of CRH, which is pivotal to an integrated stress response.
Cortisol
Following stimulation by ACTH, cortisol is the principle glucocorticoid produced by the adrenal cortex, accounting for 95% of glucocorticoid activity. Approximately 90% of circulating cortisol is protein bound; some is attached non-specifically to albumin, but the majority binds with high affinity to cortisol-binding globulin (CBG); only free cortisol is biologically active. Cortisol-binding globulin is an α2-globulin, produced in the liver, which behaves as a negative acute phase protein. The concentration of CBG falls in response to some inflammatory stimuli, increasing the bioavailability of cortisol.
Cortisol is a major effector of the metabolic response to stress. It is lipid soluble and readily crosses cell membranes, its activity being mediated via the glucocorticoid receptor (GR). Two isoforms of GR have been isolated, GRα, which binds cortisol and the GRβ, which does not bind cortisol and may act to inhibit the effect of endogenous glucocorticoid action. The hormone receptor complex is translocated to the nucleus where it interacts with glucocorticoid response elements and, following recruitment of co-activators, leads to increased gene transcription. Other actions are DNA independent and involve direct interactions with transcription factors, such as nuclear factor-κβ (NF-κβ), This is important in the modulation of the stress response, since NF-κβ activates the expression of interleukin-1(IL-1), IL-6 and tumour necrosis factor α (TNFα), all of which are key components of the inflammatory response and interact with the hypothalamo–pituitary–adrenal axis (HPAA).
Cortisol exerts a negative feedback effect on CRH and ACTH release, affects carbohydrate, protein and fat metabolism, and modulates the immune response. Cortisol limits glucose utilization in cellular respiration, perhaps by reducing the oxidation of NADH to NAD+ in the tricarboxylic acid cycle. High concentrations of cortisol reduce the sensitivity of peripheral tissues to insulin. This anti-insulin effect is particularly marked in skeletal muscle and adipose tissue, reducing glucose uptake and metabolism.
In extra-hepatic tissues, there is increased protein catabolism and decreased protein synthesis through reduced RNA formation and amino acid uptake. Within the liver, however, this metabolic picture is reversed, with increased amino acid uptake and the synthesis of acute phase proteins. Additionally, cortisol enhances the mobilization of fatty acids from adipose tissue, shifting oxidative metabolism to favour energy generation from glucose to fatty acids.
Thyroid hormones
Major disturbances of thyroid hormone metabolism and plasma concentration occur during the metabolic response to stress, typically with falls in the concentrations of tri-iodothyronine and often of thyroxine and, sometimes, in the critically ill, a fall in the concentration of thyroid stimulating hormone. (These changes are discussed further in Chapter 19.)
Sex hormones
The effects of trauma and sepsis on sex hormones are complex and dependent on the type of injury. Plasma concentrations of free testosterone may fall without any consistent change in the concentration of luteinizing hormone (LH), suggesting a change in the sensitivity of Leydig cells to LH or a disturbance of normal feedback regulation. The effects tend to be more marked in women, with reduced activity of the entire hypothalamo–pituitary–gonadal axis. Corticotrophin releasing hormone is known to have inhibitory effects at various levels in this axis, including antagonizing the effects of LH on Leydig cells. Chronic activation of the hypothalamo–pituitary–gonadal axis, such as has been demonstrated in long distance runners and ballet dancers, causes suppression of gonadal function in males and females.
Growth hormone
The anterior pituitary secretes growth hormone, stimulated by growth hormone releasing hormone, produced by the ventral medial nucleus of the hypothalamus. Growth hormone is a small protein that is active in most cells of the body, where it enhances amino acid uptake and protein synthesis. Under the influence of growth hormone, free fatty acids are used as an energy source in preference to carbohydrates and proteins. Growth hormone reduces glucose uptake by skeletal muscle and fat cells, promotes gluconeogenesis by the liver and consequently increases insulin secretion by the pancreas.
Adrenal medulla
Activation of the locus coeruleus by stressors causes the release of noradrenaline and increased sympathetic discharge via the brainstem and spinal cord. Pre-ganglionic sympathetic nerves pass through the sympathetic chain to innervate modified neuronal cells in the adrenal medulla. These cells secrete noradrenaline and adrenaline into the circulation. Catecholamine synthesis is an energy-dependent process with many mitochondria packing the terminal varicosities containing the secretory vesicles. Tyrosine is converted to L-DOPA and then to dopamine, which is transported into the secretory vesicles where a hydroxylation reaction converts it into noradrenaline. The majority of noradrenaline is then metabolized to adrenaline. The sympathetic nerve impulse to the adrenal medulla results in the influx of intracellular calcium and the discharge of the catecholamine secretory vesicles (in the ratio of 80:20, adrenaline:noradrenaline) directly into the circulation. The combined effect of this circulating adrenaline and noradrenaline is identical to that caused by direct sympathetic nervous system stimulation, but has a longer duration and can reach all cells of the body irrespective of their autonomic innervation.
STRESS AND THE KIDNEYS
Fluid conservation occurs as part of the stress response, often resulting in low urine outputs despite euvolaemia. Increased sympathetic nervous system activity, combined with raised concentrations of circulating catecholamines, reduces renal blood flow. This initiates the metabolism of prorenin to renin in the juxtaglomerular cells of the kidneys. Renin is released into the blood where it acts on angiotensinogen to form angiotensin I. Under the influence of angiotensin-converting-enzyme, predominantly in the lung, angiotensin I is converted to angiotensin II, a potent vasoconstrictor. Angiotensin II stimulates AVP release from the hypothalamus, as well as having a direct affect on the kidney to increase salt and water retention. Furthermore, under the influence of angiotensin II, the mineralocorticoid aldosterone is synthesized in the zona glomerulosa of the adrenal cortex, by metabolism of corticosterone by the enzyme aldosterone synthase. Whilst ACTH has a permissive role in aldosterone synthesis, it does not control the rate of release. Aldosterone acts on the cortical collecting tubules where it stimulates a Na+,K+-ATPase, leading to the retention of sodium and water. (Salt and water balance is considered further in Chapter 4 and renal function in Chapter 7.)
CYTOKINES
Cytokines are peptides secreted by many cell types including mast cells, macrophages and endothelial cells. They are released into the extracellular fluid and can function as autocrine, paracrine or endocrine hormones and they trigger the acute phase response.
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
