CHAPTER 16
The clinical management of diabetes mellitus
Ian W. Seetho; John P.H. Wilding
CHAPTER OUTLINE
Pharmacological management of cardiovascular risk
GLUCOSE-LOWERING THERAPY IN DIABETES
Insulin use in type 1 diabetes
Glycaemic management in type 2 diabetes
Sulfonylureas (and related insulin secretagogues)
Peroxisome proliferator activator γ analogues
Glucagon-like peptide 1 analogues
Dipeptidyl peptidase IV inhibitors
Insulin use in type 2 diabetes
Immunotherapy for type 1 diabetes
OBSTACLES TO ACHIEVING GLYCAEMIC CONTROL
Intercurrent illness, ‘sick day rules’ and stress
CHRONIC COMPLICATIONS OF DIABETES
Hyperosmolar hyperglycaemic state
INTRODUCTION
The rise in prevalence of diabetes presents some of the greatest medical and economic challenges of the 21st century. Despite advances in our understanding and management of diabetes mellitus, it continues to exact a huge toll both in terms of morbidity and mortality.
In this chapter, we consider dietary, lifestyle and macrovascular risk optimization measures for people with diabetes. Glucose-lowering treatment is discussed, followed by obstacles patients face in order to achieve their glycaemic goals, particularly hypoglycaemia. Later sections discuss the management of chronic and acute complications of diabetes, and its management in particular circumstances, for example in critical illness and during pregnancy.
GENERAL ASPECTS OF MANAGEMENT
The management of patients with diabetes should involve a multidisciplinary team composed of diabetologists, diabetes specialist nurses, dietitians, podiatrists, the patient’s general practitioner and other specialists if specific complications are present.
In addition to therapies that specifically target glycaemic control, nutritional management, lifestyle modifications such as adequate physical exercise and smoking cessation, and structured patient educational programmes are key components in the management of patients with diabetes.
Nutrition
Patients with diabetes should follow a healthy diet and make appropriate nutritional choices to reduce cardiovascular risk and improve glycaemic control. A dietitian with specialist knowledge in diabetes should take the lead role in providing nutritional care. Dietary advice should be tailored specifically for each patient, considering their personal and cultural preferences, beliefs, lifestyle and the changes that the patient is willing and able to make.
The principles of diet include eating regular meals with a reduction in simple carbohydrate intake with an increase in complex carbohydrates, a high fibre content and an energy intake to achieve or maintain an ideal body weight, or in those who are overweight or obese, to achieve and sustain modest weight loss. The key aim is to balance energy input and expenditure and ensure a good quality diet.
Contrary to the general perception that a diet in diabetes needs to be unique or special, there is no special ‘diet for diabetes’. People with diabetes, except perhaps in specific circumstances such as renal failure, should eat the same healthy diet as recommended to those who do not have diabetes. Thus, no more than 30% of total energy intake should be in the form of fat, and the ratio of unsaturated to saturated fats should be increased with saturated fat intake ideally making up < 7% of total calories and trans fatty acid intake minimized. Replacement of saturated fats with mono- or polyunsaturated fats aids lowering of cholesterol.
Carbohydrate (preferably in the form of slowly absorbed complex carbohydrate with a low ‘glycaemic index’ rather than simple sugars) should provide approximately 55% of total energy intake with protein providing the remaining 15%. Foods that improve glycaemic control and might reduce cardiovascular risk include whole grains and green vegetables. Conversely fried foods, high glycaemic index diets and red and processed meats may increase cardiovascular risk.
Carbohydrate intake can significantly affect postprandial blood glucose concentrations. In type 1 diabetes, matching insulin doses to the amount of carbohydrate consumed (carbohydrate counting) is effective in improving glycaemic control. Patients who are on fixed insulin doses or biphasic insulin regimens should be advised to aim for consistency in carbohydrate and glycaemic index dietary intake.
In patients treated with diet only or diet and oral hypoglycaemics, total energy intake in terms of carbohydrate consumption should be monitored as this is important in achieving glycaemic control. Strategies employed may include carbohydrate counting, exchanges or food portions based on past experience. However, as each individual is different, there is no fixed recommended ideal macronutrient composition of carbohydrate, fat and protein in the diet. Nevertheless, some studies have shown improved body weight and glycaemic control with low carbohydrate diets through a reduction in energy intake over the short term.
Sodium intake should not generally exceed 6 g/day and plentiful fruit and vegetables (‘five portions a day’ being the current mantra) should be consumed, mindful of the fact that some fruits (e.g. grapes) may contain large amounts of sugar and should not be taken in excess. A total daily dietary fibre intake of 40 g is ideal but rarely achieved in practice. Except in specific and unusual circumstances, it is a general principle that the pharmacological treatment of diabetes should be tailored to the dietary intake and lifestyle of the individual and not vice versa. Fructose reduces postprandial glycaemia when it is used as a replacement for sucrose or starch, but it may have adverse metabolic effects including causing dyslipidaemia and insulin resistance. Non-nutritive sweeteners are safe when consumed within recommended daily limits.
It is currently recommended that patients with new onset type 2 diabetes should be managed with dietary and lifestyle modification with consideration for early introduction of metformin (discussed later). The degree of adherence to a particular diet will affect the outcome and a diet that is both palatable and acceptable is more likely to be followed.
Exercise
Patients with diabetes should be encouraged to participate in regular exercise of the kind that would benefit the rest of the population. Regular low-intensity exercise, such as brisk walking (so as to be slightly breathless while talking at the same time), swimming or cycling for 30 min three to five times a week should be a realistic goal for many individuals. Exercise improves glucose disposal (by increasing the expression of type 4 glucose transporters, GLUT4) in skeletal muscle, among other mechanisms), prevents progression from impaired glucose tolerance to type 2 diabetes (by ~ 50%), increases basal metabolic rate and has convincingly been demonstrated to reduce the risk of cardiovascular events.
Some patients with cardiovascular disease require detailed assessment (e.g. electrocardiogram, review of medications) before taking part in exercise programmes, but the few absolute contraindications to exercise in patients with diabetes (e.g. tight aortic valvular stenosis) apply equally to individuals without diabetes.
In insulin-treated patients, exercise may be associated with either an increased frequency of hypoglycaemia or hyperglycaemia as blood glucose concentrations are influenced by the timing, type and quantity of insulin, carbohydrate intake and intensity and duration of physical activity. Careful monitoring of blood glucose concentrations is necessary to guide the adjustment of insulin therapy and carbohydrate intake, and the strategy adopted may depend on whether the exercise is planned or unplanned.
Aerobic exercise improves glycaemic control and lowers low density lipoprotein (LDL) cholesterol in patients with type 2 diabetes, but has little effect on other lipid parameters. Resistance training has effects on both glycaemia and cardiovascular risk factors such as systolic blood pressure, fat mass and insulin resistance.
Studies have shown that it is safe for individuals with type 2 diabetes who are treated by diet alone or in conjunction with oral hypoglycaemic agents to exercise in both the fasting and post-meal states with the most beneficial effects on blood glucose concentrations observed postprandially. As with dietary modification, exercise programmes must be individually designed in order to encourage compliance.
Smoking cessation
Smoking is hazardous to all, but is particularly so in people with diabetes. The macrovascular risks of smoking are compounded with those of diabetes itself. Smoking is associated with increased mortality and predisposes to microvascular complications of diabetes such as retinopathy and nephropathy. Smoking is potentially diabetogenic: it is a risk factor for the development of type 2 diabetes, and can influence glycaemic control by impairing insulin sensitivity and glucose tolerance.
Smoking is at least as prevalent in patients with diabetes as in the general population and young women in particular may perceive the habit as an adjunct to weight management.
Many approaches to the supportive management of patients have been described. These include smoking-cessation clinics and nicotine replacement therapy. Such forms of therapy apply equally to people with and without diabetes.
Education about diabetes
Structured educational programmes have been developed in Europe and North America. In the UK, a five-day intensive educational course called the Diet Adjustment For Normal Eating (DAFNE) programme provides opportunities for support and learning for patients with type 1 diabetes. This course teaches those with type 1 diabetes how to adjust their insulin doses according to carbohydrate portion intake, and allows participants to learn about aspects of lifestyle and management related to their diabetes.
For patients with newly diagnosed type 2 diabetes, the Diabetes Education and Self Management for Ongoing and Newly Diagnosed (DESMOND) programme offers support to patients in identifying their health risks and enables goal-setting, developing confidence in self-management of their condition.
Pharmacological management of cardiovascular risk
Despite the fact that patients with diabetes are uniquely susceptible to microvascular complications, around 80% of patients with type 2 diabetes will die of macrovascular disease. Ischaemic heart disease, stroke and peripheral vascular disease are some 2–4 times more common in patients with diabetes than in the non-diabetic population. Many available risk tables do not even include powerful diabetes-related risk factors such as microalbuminuria. Type 2 diabetes is associated with dyslipidaemia. There are elevated concentrations of small, dense LDL particles that are more prone to oxidation, which increases their ability to damage the vascular endothelium. Reduced high-density lipoprotein (HDL) concentrations result in reduced protection against atheroma: increased triglyceride-rich lipoprotein concentrations predispose to atheroma formation.
The risk of mortality from cardiovascular disease is higher in patients with diabetes and increases further with additional cardiovascular risk factors such as hypercholesterolaemia, hypertension and smoking. Diabetes is so powerful a risk factor that patients should, in most cases, be treated as if they had already suffered a cardiovascular event, rather than have their treatment assessed on the basis of primary prevention studies.
Apart from recommendations regarding the dietary and other lifestyle measures detailed above, there is a degree of uncertainty about which patients should receive preventative treatment with agents such as aspirin, lipid-lowering therapy or angiotensin-converting-enzyme (ACE) inhibitors, and what targets to pursue at any specific stage of the disease. Guidelines have been prepared by various organizations in countries with populations of differing genetic background and socioeconomic structures. None can obviate the need for individualized risk–benefit assessment, and it is important to involve patients in setting their own treatment goals, and management plans.
It is likely that future guidelines will recommend a lifetime risk calculator that uses a range of risk factors to calculate the absolute risk of an event over the rest of the patient’s lifetime. This has the effect of promoting more aggressive intervention to modify risk factors from an early age. Risk calculators can also be used to help patients to understand their personal risk and how changes can affect it, in order to improve compliance.
Aspirin
Aspirin (75 mg daily) is recommended for secondary prevention of cardiovascular disease in patients with diabetes. It is effective in reducing cardiovascular morbidity and mortality in high-risk patients with previous cardiovascular disease. The risk of adverse effects associated with this dose of aspirin depends on many factors, including the age of the patient, intake of other drugs (corticosteroids and non-steroidal anti-inflammatory drugs), presence of other disease (e.g. untreated proliferative retinopathy, uncontrolled hypertension, asthma) and medical treatment (e.g. forthcoming surgery). The risks of major gastrointestinal haemorrhage appear to be increased by a factor of 1.5–2 with approximately one additional event per 500 patient years under trial conditions (approximately 0.2% per patient per year). Intracranial haemorrhages are increased by one event per 3–5000 patient years.
The use of aspirin for secondary prevention of macrovascular disease in all patients in whom it is not contraindicated is well established. However, in primary prevention in patients with diabetes with no prior cardiovascular events, the use of aspirin has been more contentious.
The current Joint British Societies guidance (JBS2) recommends aspirin 75 mg daily in patients with diabetes aged > 50 years, those who are younger but have had the disease for more than 10 years, and those who are already receiving treatment for hypertension, with controlled blood pressure < 150 mmHg systolic and < 90 mmHg diastolic. The UK National Institute for Health and Care Excellence (NICE) guidelines have stated that low-dose aspirin should be offered to patients aged > 50 years if blood pressure is below 145/90 mmHg and to those aged < 50 years who have significant cardiovascular risk (features of the metabolic syndrome, strong early family history of cardiovascular disease, smoking, hypertension, existing cardiovascular disease or microalbuminuria).
However, a more recent meta-analysis found no significant beneficial effects of using aspirin for primary prevention of cardiovascular disease in diabetes, and the Prevention of Progression of Arterial Disease and Diabetes (POPADAD) study demonstrated that aspirin did not significantly reduce the risk of major cardiovascular events even in patients with asymptomatic peripheral arterial disease. At present, there is little evidence to support the use of aspirin in patients aged < 30 years and, in those < 16 years, aspirin should be avoided because of an increased risk of Reye syndrome.
Lipid-lowering agents
Effective management of dyslipidaemia reduces the risk of cardiovascular disease; plasma lipid concentrations should be monitored at least annually in patients with diabetes. Most people in the developed world, with or without diabetes, would benefit from a less atherogenic lipid profile. Lifestyle modifications such as dietary restriction of saturated fat, cholesterol and trans-unsaturated fat, increased intake of ω-3 fatty acids, viscous fibre and plant sterols, achievement and maintenance of an ideal body weight and taking adequate exercise are recommended, whether or not further treatment is required. Secondary causes of dyslipidaemia such as alcohol excess, hypothyroidism or liver disease should be considered in all patients prior to initiating any lipid-lowering therapy. For patients with diabetes, poor glycaemic control (denoting inadequate insulin action) may also cause excess very low density lipoprotein (VLDL) production and hypertriglyceridaemia. Treatment with metformin, pioglitazone and insulin have all been shown to improve this, either via increasing insulin action or by other specific mechanisms (e.g. by reducing the flux of non-esterified fatty acids to the liver in the case of pioglitazone).
Many primary and secondary prevention trials have convincingly demonstrated the benefits of lipid-lowering therapy in reducing macrovascular risk in subjects with and without diabetes. Many of these studies have involved the use of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase inhibitors (statins), and have shown that lowering LDL-cholesterol is approximately linearly related to risk reduction down to concentrations of 2.2 mmol/L or even below.
The American Diabetes Association (ADA) currently recommends statin therapy, in addition to lifestyle factors, as secondary prevention for patients with diabetes who have cardiovascular disease, with a goal for LDL-cholesterol of < 1.8 mmol/L. Treatment is recommended for primary prevention in all patients with diabetes over the age of 40, and in younger people who have additional cardiovascular risk factors, with an LDL-cholesterol target of < 2.6 mmol/L. Statins should be used with caution, and only on specialist advice, in children and are contraindicated in pregnant women.
The typical diabetic dyslipidaemia comprises raised triglycerides, intermediate density lipoprotein (IDL) and small, dense LDL concentrations together with low HDL concentrations. These abnormalities are particularly atherogenic, with the result that the significance of dyslipidaemia in diabetes is underestimated by routine measurements of total lipid subfractions. Fibric acid derivatives have a role in the management of the raised triglycerides in diabetic dyslipidaemia. The ADA currently recommends targets for triglycerides of < 1.7 mmol/L, and for HDL-cholesterol of > 1.0 mmol/L in men and > 1.3 mmol/L in women. Both the JBS2 and NICE guidelines (NICE Clinical Guideline 66) recommend a target total cholesterol < 4.0 mmol/L and LDL < 2.0 mmol/L. Alternatively, JBS2 recommends a 25% reduction in total cholesterol with a 30% reduction in LDL-cholesterol, if this achieves lower absolute concentrations. Both guidelines recommend statins for those with diabetes who are aged > 40 years and for those aged 18–39 years with diabetes complications (retinopathy and nephropathy), poor glycaemic control, hypertension, features of the metabolic syndrome or family history of premature CVD in a first degree relative.
Combination lipid-lowering therapy, to achieve additional lowering of LDL, may be considered in order to reach desired targets if these are not reached with maximum tolerated statin doses. To date, however, there is little evidence to show that combination therapy to lower LDL produces significant cardiovascular risk reduction over and above statin therapy alone. We would advocate a strategy of: (1) assessing overall risk and choosing targets for LDL, triglyceride and HDL; (2) excluding secondary causes of hyperlipidaemia and optimizing glycaemic control; (3) optimizing lifestyle options, and (4) targeting LDL-cholesterol with a statin. These drugs may in addition produce minor favourable effects on HDL-cholesterol and triglycerides. It should be noted that therapy targeting HDL or triglycerides does not have the evidence base of statin treatment.
Severe hypertriglyceridaemia requires lifestyle and pharmacological therapy. A fibrate or ω-3 fatty acid-rich marine oils may be used to reduce the risk of pancreatitis associated with high triglycerides. Any statin–fibrate combination requires care because of the potentially increased risk of rhabdomyolysis, and monitoring of plasma creatine kinase activity may be appropriate.
Hypertension
Assiduous control of blood pressure (BP) has been shown to reduce major cardiovascular events and minimizes progression of diabetes complications. In the UK Prospective Diabetes Study (UKPDS), intensive treatment of hypertension to a median of 144/82 mmHg was compared with conventional BP treatment (median BP in the control group was 154/87 mmHg). Intensive treatment resulted in reductions in multiple diabetes-related endpoints, including death. The majority of subjects in the tight control arm of the study required three or more agents in order to achieve this. The ADA has adopted an even more aggressive target of 130/80 mmHg on the basis of epidemiological risk studies showing an inflexion in the relationship between BP in patients with diabetes and cardiovascular risk at 115/75 mmHg.
Apart from more rigorous treatment goals, management of hypertension in patients with diabetes is similar to that in patients without the condition. Initially, lifestyle changes are recommended including weight loss, exercise and dietary modifications, such as a reduction in sodium and alcohol intake and increased consumption of fruit and vegetables. However, if blood pressure remains inadequately controlled despite lifestyle modifications, pharmacological therapy may be commenced.
Some patients will have comorbid conditions that will direct the choice of drug therapy. The presence of microalbuminuria, proteinuria, mild to moderate renal impairment, diabetic retinopathy, ischaemic heart disease, stroke and left ventricular systolic dysfunction would, for example, indicate a specific role for an angiotensin-converting-enzyme (ACE) inhibitor or angiotensin-II receptor antagonist (blocker) (ARB). Given the potentially beneficial effects of ACE inhibitors and ARBs on glycaemia and in the prevention of diabetic complications such as diabetic nephropathy (see below), they are generally considered as first-line agents in patients with diabetes if lifestyle changes do not improve blood pressure.
β-Blockers, diuretics, and calcium channel blockers have all been shown to reduce cardiovascular events in patients with diabetes, and all may be used in combination with an ACE inhibitor or ARB. Most patients will require multiple agents to manage their hypertension. Generally initial treatment will be with either an ACE inhibitor or ARB, and if required, a calcium channel blocker, then a thiazide diuretic, β-blocker, α-blocker or potassium-sparing diuretic. It is important to monitor renal glomerular function and plasma potassium concentration whenever ACE inhibitors, ARBs or diuretics are used. Thiazides are effective, but unwanted effects including hyperglycaemia, erectile dysfunction, gout and dyslipidaemia have reduced their use as first line agents. β-Blockers are specifically indicated in some situations, for example stable heart failure and ischaemic heart disease, and were found to be safe and effective in the UKPDS. However, they may promote weight gain and, in some patients with impaired awareness, hypoglycaemia may be a problem. In the Anglo–Scandinavian Cardiac Outcome Trial (ASCOT), treatment with an atenolol–thiazide-based regimen was associated with 30% greater incidence of new type 2 diabetes than treatment with an amlodipine–perindopril-based regimen, and was less effective at reducing BP and cardiovascular events in patients with established diabetes.
In the past, there were concerns about the safety of the dihydropyridine group of voltage activated calcium channel blockers. However, results from the Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) and ASCOT have been reassuring, and it now seems reasonable to recommend amlodipine as a second-line agent in combination with an ACE inhibitor or ARB (although amlodipine and other calcium channel blockers inhibit the metabolism of simvastatin, which must not be co-prescribed at high dose).
α-Blockers appear effective and have favourable effects on insulin sensitivity and LDL-cholesterol concentrations. However, ALLHAT showed an apparent doubling in the incidence of heart failure (a secondary outcome measure) with an α-blocker compared with a thiazide diuretic. For this reason, α-blockers are generally reserved for use where treatment with at least two other classes of hypotensives in combination has failed to achieve the target BP.
Angiotensin-converting-enzyme inhibitors and angiotensin-II receptor antagonists
Angiotensin II (ATII) increases hepatic glucose production and decreases insulin sensitivity. The use of ACE inhibitors or ARBs consistently increases insulin sensitivity by some 15–20% in pharmacological studies. The Heart Outcomes Prevention Evaluation (HOPE) study was terminated early after interim analysis demonstrated that 4.5 years of treatment with the ACE inhibitor ramipril, 10 mg daily, reduced major vascular events and death in subjects with preserved left ventricular function, and either pre-existing vascular disease or those with diabetes and an additional vascular risk factor. The latter group also had a reduced risk (relative risk 0.84) for the development of diabetic complications. In an extension to the study to a total of 7.2 years follow-up, there was a 31% relative risk reduction for new diagnosis of type 2 diabetes.
Results of other studies and meta-analyses have, in general, not shown such large benefits in patients who already have diabetes (unless with other clearly established indications), although they have generally been restricted to relatively small subgroup analyses. While it is not clear whether all (non-pregnant) patients with diabetes should be offered these agents, ACE or AT-II inhibition is clearly indicated for subjects with diabetes and hypertension, microalbuminuria, proteinuria, mild to moderate renal impairment, diabetic retinopathy, ischaemic heart disease, stroke or left ventricular systolic dysfunction. Caution must be exercised with the use of these agents when renal artery stenosis is present (e.g. in patients with impaired renal function and absent distal pulses or a renal bruit) and in subjects with hyporeninaemic hypoaldosteronism (type 4 renal tubular acidosis), in whom dangerous hyperkalaemia may result.
GLUCOSE-LOWERING THERAPY IN DIABETES
Background
Trials in both type 1 Diabetes Control and Complications Trial (DCCT) and type 2 (UKPDS) diabetes have confirmed that microvascular, and, to a lesser extent, macrovascular, complications of diabetes may be delayed or even prevented by tight glycaemic control. The UKPDS (1998) trial in type 2 diabetes found that a 10 mmol/mol (0.9%) reduction in HbA1c from 63 to 53 mmol/mol (7.9–7.0%) over a median follow-up of ten years from diagnosis led to a reduction of 12% for any diabetes-related endpoint, 25% for microvascular endpoints and 24% for cataract extraction, and of 21% for retinopathy and 33% for albuminuria at 12 years. The improvements in macrovascular disease were less convincing and of borderline statistical significance.
The DCCT (1993) trial in type 1 diabetes showed a risk reduction of 26–63% in microvascular disease with intensive insulin therapy, but at the cost of a three-fold elevation in the incidence of severe hypoglycaemia (fits, unconsciousness) and some additional weight gain compared with the control arm of the study. However, the use of more modern insulin analogues has been associated with lower incidence of hypoglycaemia.
Insulin use in type 1 diabetes
Patients who have been diagnosed with type 1 diabetes will require insulin therapy along with general measures such as dietary and lifestyle changes. Although it is often considered that patients with type 1 diabetes have no useful islet cell function, this is not entirely true. In fact, β-Cell function is approximately 10% of normal at disease presentation, and may double after the initiation of insulin therapy, resulting in a reduced requirement for exogenous insulin and stabilization of glucose metabolism (‘honeymoon effect’). This effect does not last and, in the majority of patients, insulin secretion declines again over the next 2–5 years.
The ‘honeymoon effect’ may reflect, in part, the amelioration of glucotoxicity or lipotoxicity (see Chapter 15) on the reduced numbers of metabolically stressed β-Cells. After initiation of insulin therapy, the toxic effects of hyperglycaemia are removed, thereby relieving this metabolic stress. In some studies, as many as 10% of patients with newly diagnosed type 1 diabetes have been maintained off insulin altogether for the first year, but this is clearly not recommended in routine clinical practice because of the risks of ketoacidosis.
In a subset (~ 15%) of patients, there is retention of some detectable islet function, as assessed by C-peptide secretion, for at least 40 years. These individuals also appear to have reduced susceptibility to severe diabetic ketoacidosis (DKA), less risk of retinopathy, a more robust glucagon response to hypoglycaemia and lower average insulin requirements.
A number of trials have attempted to augment residual insulin secretion with immunosuppressive drugs (e.g. steroids, azathioprine, antithymocyte globulin and ciclosporin) or by plasmapheresis, and these measures have been met with some success but, on current evidence, their benefits have not been clearly outweighed by their risks. Studies using strict metabolic control in order to mitigate any effects of lipotoxicity or glucotoxicity on the β-Cell (e.g. with continuous subcutaneous insulin infusion, CSII) have been able to demonstrate transiently preserved insulin secretion, but only for a period of a few months, with rapid regression on discontinuation of strict metabolic control.
Regular insulin
Regular human insulin (e.g. Actrapid®, Humulin S®) has an onset of activity within 30–60 min after subcutaneous injection, a variable peak concentration at around 2–4 h, and a duration of action of 5–8 h, contrasting with its duration of action of only a few minutes when administered intravenously. This difference may be explained by the tendency for hexamerization in solution.
The hexameric form of regular insulin has to dissociate into dimers or monomers in order to be absorbed into the bloodstream. This is rate-limiting for its absorption and influences the pharmacokinetic profile. As such, in order to treat prandial glucose peaks, regular insulins have to be injected about 30 min before each meal so that the onset of action coincides with the rise in prandial glucose concentrations.
Insulin analogues
More recent developments in insulin technology have focused on the design and production of insulin analogues whose pharmacokinetic profiles are determined by modifications to non-receptor-binding regions of the insulin molecule.
Insulin aspart (Novorapid®) is homologous with regular human insulin with the exception of a single substitution of aspartate for proline in position B28 of the B chain: it is produced by recombinant DNA technology in Saccharomyces cerevisiae (baker’s yeast). Insulin lispro (Humalog®) is identical to normal human insulin except that the positions of the amino acids proline and lysine at B28 and B29 are reversed. In insulin glulisine (Apidra®), asparagine is replaced by lysine at position B3, with glutamic acid replacing lysine at B29. As a result of these changes in amino acid sequence, the rapid insulin analogues (aspart, lispro and glulisine) have a lower tendency to form hexamers, permitting more rapid absorption and a faster onset of action (< 15 min), enabling injection immediately before, or just after, eating. There is a sharper peak, more closely resembling first-phase insulin secretion in the non-diabetes state, and a shorter duration of action (3–5 h), which may reduce the incidence of hypoglycaemia before the next meal is ingested.
Intermediate-acting insulin
Isophane (NPH, Neutral protamine Hagedorn) is an intermediate-acting insulin that is a combination of soluble insulin and protamine zinc. Protamine delays the onset of action and prolongs the effects of the soluble insulin. The onset of action is within two hours, with a duration of action of 10–20 h. Most patients require two subcutaneous injections daily to provide adequate glycaemic control. Isophane has fallen out of favour because of increased risks of nocturnal hypoglycaemia and fasting hyperglycaemia.
Premixed insulin analogues
The most commonly prescribed premixed insulins are a mixture of quick acting analogue and an insulin analogue protamine complex (e.g. Novomix 30® is composed of insulin aspart-protamine complex and insulin aspart in a ratio of 70:30). These are usually given as a twice-daily regimen.
Long-acting insulin analogues
Longer-acting insulin analogues (insulin glargine and insulin detemir) are produced by genetic engineering. The onset of action is within two hours and they have a longer duration of action of up to 24 h. These insulins provide a steady basal insulin profile with minimal peak action and are injected subcutaneously once daily.
In insulin glargine, asparagine is replaced by glycine at position 21 of the α chain of the insulin molecule and the carboxyl terminal of the β chain is changed with the addition of arginine. This insulin is slowly released into the circulation because of its acidic pH, which causes it to precipitate when injected subcutaneously. In insulin detemir, tyrosine at position B30 is lost with acylation at B29 that results in a fatty acid side chain (tetradecanoic acid) becoming linked with lysine. With these alterations, insulin detemir forms hexamers with slower dissociation and a longer duration of action.
Insulin regimens
Most patients with type 1 diabetes use an insulin replacement regimen that comprises multiple-dose subcutaneous injections (three to four injections per day) consisting of basal and prandial insulin. Insulin is injected subcutaneously. Short-acting insulin (regular soluble or analogue) acts to control prandial blood glucose concentrations; long-acting insulin provides a constant background basal insulin concentration. This combination of short-acting and long-acting basal insulin is called the basal bolus regimen or multiple daily injection therapy. Most people take their long-acting insulin at bedtime in order to provide optimal glycaemic control overnight. Some patients learn how to adjust insulin doses according to carbohydrate intake (in the UK often through attendance at a DAFNE (dose adjustment for normal eating) course).
An alternative is a twice daily insulin regimen consisting of a premixed or biphasic mixture of longer- and shorter-acting insulins in ratios of 75:25, 70:30 or 50:50. Such regimens are commonly used in type 2 diabetes and in patients with type 1 diabetes before they progress to a basal bolus regimen. Some patients with type 1 diabetes prefer twice daily insulin to the basal bolus regimen, particularly those who dislike frequent injections, but it is not suited to those whose diet and activities tend to differ on a day-to-day basis, as there is less scope for dose adjustment in relation to carbohydrate intake, with attendant risks of hypoglycaemia. The longer-acting component may not provide effective glycaemic control overnight because of a waning of the effect of the previous evening’s dose, a decrease in insulin sensitivity in the early hours of the morning, and increases in growth hormone and cortisol concentrations, all of which will increase the risk of early morning hyperglycaemia (the ‘dawn’ phenomenon). Premixed insulin preparations are, therefore, not recommended for long-term treatment in type 1 diabetes and most patients are eventually recommended to convert to the basal bolus regimen for optimal glycaemic control. The relative merits and disadvantages of these two regimens, and a comparison with continuous subcutaneous insulin infusion, are outlined in Table 16.1.
Continuous subcutaneous insulin infusion
Continuous subcutaneous insulin infusion pumps (‘insulin pumps’) are portable devices that deliver a continuous subcutaneous infusion of short-acting insulin at a set rate. The pump is battery-driven and comprises a reservoir containing about 3 mL of insulin in a syringe. A syringe driver delivers insulin via a cannula that is inserted subcutaneously into the abdominal wall. The cannula is re-sited every 1–2 days to avoid skin infections. In addition to the continuous basal infusion, the patient triggers the delivery of an additional dose at mealtimes to provide prandial cover.
The advantages of a pump include having a basal insulin rate that is personalized to each individual, allowing more flexibility with regards to mealtimes as the basal rate can be supplemented with a corrective mealtime insulin dose according to the carbohydrate consumed. Studies have shown improved glycaemic control in pump users. However, in the event of pump failure, there is the risk of diabetes ketoacidosis occurring.
Clinical indications for pump therapy include poor glycaemic control despite high levels of care, disabling hypoglycaemia on multiple daily injection regimens, recurrent unpredictable hypoglycaemia, especially in patients with severe dawn phenomenon and pregnant women without acceptable glycaemic control with subcutaneous insulin. Before each patient is commenced on the insulin pump, they must attend a comprehensive educational pump course. They must be sufficiently motivated to use the pump, with a commitment to regular blood glucose monitoring, attendance at pump clinics and adherence to carbohydrate counting so that they will be able to manage their pump. Close liaison with the diabetes pump team is essential for the success of this form of therapy.
It should be stressed that when deciding which regimen to use, each patient’s specific circumstances will need to be carefully considered in light of different needs, goals and capabilities and the level of support available. An important factor is the risk of hypoglycaemia that comes with insulin therapy. There should be a balance between achieving optimal glycaemic control whilst at the same time ensuring each patient’s safety, in terms of hypoglycaemia risk, when deciding on which insulin and regimen is to be used.
Insulin administration
The recommended subcutaneous injection sites include the abdomen, upper outer thighs, upper outer arms and buttocks. It is important to avoid injecting insulin intramuscularly. This commonly occurs in the limbs of slim individuals or children and can influence insulin absorption and hence blood glucose profiles.
As a result of the trophic action of insulin, an accumulation of fat, called lipohypertrophy, may occur at sites of repeated subcutaneous insulin injections. Lipohypertrophy can affect insulin absorption, causing problems such as hypoglycaemia, but can be avoided by rotating injection sites. Insulin injection sites should be inspected when patients attend for their diabetes review.
Glycaemic management in type 2 diabetes
When type 2 diabetes is diagnosed, lifestyle modification should be advised initially, and metformin commenced in combination with lifestyle if HbA1c does not reach target after three months. If glycaemic control remains poor after 3–6 months, further oral agents, or a glucagon-like peptide 1 (GLP-1) receptor agonist or insulin may be added. Some patients with marked hyperglycaemia may be commenced on insulin at diagnosis if it is deemed that non-insulin agents will not provide the necessary control of blood glucose.
Metformin
Metformin belongs to a class of drugs called the biguanides that act to reduce insulin resistance, therefore improving insulin sensitivity. Metformin is the only biguanide currently available. It has an established role in the treatment of type 2 diabetes in combination with diet and lifestyle measures. The UKPDS study showed that obese patients treated with metformin had reduced myocardial infarction (by 6%), stroke (by 3%) and mortality (by 7%). These effects were independent of improved glycaemic control.
Metformin has the clear advantage over many other drugs of improving glycaemic control without causing weight gain. Metformin does not cause hypoglycaemia and may even lead to weight loss by suppressing appetite. As such, it is often prescribed as first line treatment in patients who are overweight. Several of the adverse features of the metabolic syndrome, including raised circulating concentrations of non-esterified fatty acids (NEFAs), triglycerides and plasminogen activating inhibitor-1 (PAI-1), are improved by metformin. Metformin has an effect on blood glucose that is additive to, and broadly equivalent to, that of sulfonylurea monotherapy. In type 2 diabetes, metformin can be used as monotherapy, or combined with insulin or with sulfonylureas, dipeptidyl peptidase IV (DPP-4) inhibitors, GLP-1 analogues or thiazolidinediones. In type 1 diabetes it is sometimes used together with insulin for obese adults, although this usage is currently unlicensed. Metformin may also be considered in patients who are not overweight.
Metformin has a useful role in diabetes prevention; a 31% relative reduction in the progression of diabetes was observed in the Diabetes Prevention Programme (DPP) placebo-controlled study, with greatest effect in younger and more obese subjects. Metformin also has a possible role in the treatment of polycystic ovary syndrome (PCOS), alone or in combination with drugs acting on sex steroid metabolism, particularly in those patients who are also glucose intolerant.
Mechanism of action
Metformin decreases hepatic gluconeogenesis and glucose output, improves peripheral glucose uptake and utilization in insulin-sensitive tissue such as muscle and adipose tissue, and may reduce intestinal glucose transport. Its actions depend on the presence of adequate β-Cell function, and therefore insulin in the circulation. The exact mechanism of action of metformin is not clear but it may work through the activation of an intracellular AMP-activated protein kinase.
Lactic acidosis
Lactic acidosis is a rare but serious complication that can occur whilst taking metformin. Lactic acidosis was more frequently reported with phenformin, another biguanide that was subsequently withdrawn in most countries in 1977 after 306 documented cases. While phenformin excretion relies upon hepatic hydroxylation (pharmacogenetically deficient in approximately 10% of Caucasians), metformin is subject to renal tubular secretion, and its excretion depends only on renal function.
Lactic acidosis presents with non-specific symptoms such as lethargy, nausea, vomiting, altered level of consciousness and abdominal pain. Biochemical features of lactic acidosis are those of an elevated anion gap metabolic acidosis with high blood lactate concentrations. There appears to be no direct correlation between blood concentrations of metformin and lactate, and the overwhelming majority of cases of lactic acidosis have occurred in the setting of major comorbidities (Table 16.2). These include conditions associated with increased lactate formation (e.g. cardiac failure, pulmonary disease), reduced lactate metabolism (e.g. hepatic failure) and reduced metformin and lactate excretion (e.g. renal failure).
TABLE 16.2
Cautions with and contraindications to the use of metformin
| Caution or contraindication | Reason |
| Old age | Renal and other organ impairment |
| Tissue hypoxia | Increased lactate production via anaerobic glycolysis, reduced gluconeogenesis |
| Pulmonary disease | Increased lactate production via tissue hypoxaemia and anaerobic glycolysis |
| Liver failure, ethanol | Reduced lactate clearance via inhibition of gluconeogenesis |
| Iodinated radiological contrast media | Risk of acute kidney injury, competition for renal tubular secretion |
| Cardiac impairment | Increased lactate production via poor tissue oxygen delivery |
| Shock states, severe dehydration | Increased lactate production via poor tissue oxygen delivery |
| Advanced microvascular disease | Risk of chronic kidney disease |
| Severe infection | Increased lactate production |
| Drugs affecting tubular secretion of metformin | Contrast media, cimetidine, digoxin, others |
| Type 2 diabetes itself | Microcirculatory disturbances, peripheral vascular disease, increased incidence of cardiac and renal disease, reduced activity of pyruvate dehydrogenase in insulin resistance favouring anaerobic metabolism |
There is an increased risk of lactic acidosis in patients treated with metformin who receive radiological contrast media. This may be avoided by withholding metformin for a few days before and after the intended radiological procedure.
A systematic review has shown that there is no evidence from prospective comparative trials or from observational cohort studies that metformin is associated with an increased risk of lactic acidosis, or with increased plasma concentrations of lactate, compared with other antihyperglycaemic treatments, if prescribed as licensed. Patients with significant cardiac, renal or liver dysfunction should not receive metformin. As metformin is renally excreted, there is the attendant risk of accumulation in acute or chronic kidney disease, resulting in an increased risk of lactic acidosis. It is recommended by NICE that prescription of metformin should be reviewed in patients with a glomerular filtration rate (GFR) < 45 mL/min/1.73m2 and it is to be avoided in those with a GFR < 30 mL/min/1.73 m2. Some physicians recommend that metformin should not be used if the patient’s plasma creatinine concentration is raised to any extent; others argue that guidelines have been progressively tightened with no reduction in the incidence of lactic acidosis, which has anyway always been very low throughout the history of metformin usage. Although some formularies suggest avoiding metformin when plasma creatinine concentration is raised above certain thresholds, cardiac dysfunction is probably a more important, but less well recognized, contraindication to its use.
Other unwanted effects of metformin
The major side-effects of metformin include gastrointestinal problems such as nausea, abdominal discomfort, metal taste and diarrhoea. Many patients will experience these symptoms after commencing metformin but will eventually develop tolerance. These side-effects are generally dose dependent, and can be minimized by commencing at a low dose and then slowly titrating the dose upwards, or by using a slow-release preparation. Approximately 80% of the total benefit of metformin is seen with doses of around 1.5 g/day, which is close to the median tolerated dose. Metformin induces malabsorption of vitamin B12 and folate but this is rarely clinically significant.
Sulfonylureas (and related insulin secretagogues)
First generation sulfonylurea (sulphonylurea) derivatives were first used to treat diabetes in the 1950s, following the observation in 1942 that certain sulphonamide antibiotics could provoke severe hypoglycaemia in patients treated for typhoid fever. They act as insulin secretagogues, reducing blood glucose concentrations by augmenting the first-phase release of immediately releasable (reserve) insulin from β-Cells. Unlike glucose-stimulated insulin release, there is little direct effect on insulin production. First generation sulfonylureas, such as chlorpropamide, were beset with unwanted effects including the syndrome of inappropriate antidiuresis (SIADH) and flushing. These drugs have largely been replaced by more potent and shorter acting second and third generation drugs (e.g. gliclazide) that lack these unwanted effects.
Mechanism of action
Central to the release of insulin from secretory granules (but not the only mechanism involved) is the closure of an ATP-sensitive membrane-bound inwardly rectifying 140 kDa K+ channel, KATP. In β-Cells, this channel is a hetero-octamer of pore-forming units (KIR 6.2) together with regulatory ATP binding cassette (ABC) protein sulfonylurea receptor 1 (SUR-1) molecules. The regulatory subunit, SUR-1, contains 13–17 transmembrane domains with a large number of possible protein kinase A or C phosphorylation sites, together with a number of distinct binding sites for other molecules (including the A and B sites to which sulfonylureas and other exogenous compounds attach, as discussed below). The existence or nature of the endogenous ligand at sulfonylurea binding sites is not clear. Closure of KATP channels depolarizes the plasma membrane, causing rapid influx of calcium ions via voltage dependent calcium channels. The resultant increase in free ionized cytosolic calcium concentration ([Ca2 +]i) triggers cytoskeletal trafficking of secretory granules to the plasma membrane and release of insulin by a process of exocytosis. Amplification of the signal generated by KATP closure occurs via a number of incompletely understood mechanisms, including direct mobilization of calcium from intracellular stores (e.g. by the action of glucagon-like peptide (GLP)-1 analogues). The effects of KATP channel opening are terminated by extrusion of K+ via a voltage gated K+ channel.
The principal mechanism of action of the sulfonylurea drugs is to close the KATP channel by binding to the A and B sites on SUR-1. Conversely, diazoxide, used in the treatment of certain hyperinsulinaemic conditions, is a potent opener of KATP channels, and thiazide diuretics also open β-Cell KATP channels (causing impairment of glucose tolerance).
Adverse effects of sulfonylureas
Concern has existed for many years that sulfonylurea drugs may have harmful cardiovascular effects (mainly arrhythmias). This concern was first raised in relation to tolbutamide (the University Group Diabetes Programme, UGDP). The action of sulfonylureas on cardiac KATP channels suggests a plausible mechanism for adverse cardiac effects.
In contrast to these predominantly theoretical reservations, analysis of UKPDS data does not support the suggestion of adverse cardiovascular effects from sulfonylureas. At the present time, and in the light of the Diabetes Insulin–Glucose in Acute Myocardial Infarction (DIGAMI) study results, many physicians would agree that discontinuation of sulfonylurea drugs and substitution with insulin infusion is probably indicated in the setting of acute coronary syndromes. Some would also advocate the use of selective agents (e.g. nateglinide or glimepiride) in other cardiac patients, pending definitive data.
Other unwanted effects of sulfonylureas
Unwanted effects of the sulfonylureas include hypoglycaemia (particularly in elderly patients treated with longer-acting agents and in renal/hepatic failure), serious immunological reactions (Stevens–Johnson syndrome and other rashes), marrow dyscrasias, hepatitis, weight gain (typically 1–3 kg) and precipitation of acute porphyria. Contraindications to the use of these drugs include type 1 diabetes, pregnancy, lactation and significant hepatic and renal insufficiency.
Hypoglycaemia owing to deliberate or inadvertent overdose of sulfonylureas requires prompt and aggressive management. The first-line treatment is with a bolus of 50% glucose followed by continuous infusion of 10% or 20% glucose. If blood glucose cannot be maintained at safe levels with this treatment alone, hydrocortisone, glucagon or octreotide may be useful.
Indications and clinical usage
Sulfonylurea and derivative drugs are used to treat patients with type 2 diabetes who have adequate insulin reserve, but in whom lifestyle and dietetic improvements have failed to control hyperglycaemia. Doses are titrated to blood glucose and HbA1c responses; primary failure of either to improve may indicate advanced β-Cell failure. Because of their tendency to promote weight gain, sulfonylureas are best used as first-line agents in non-obese patients, but may be added to metformin, thiazolidinediones or both in obese patients with secondary failure of these agents. Insulin therapy can be added if and when necessary. Weight gain with repaglinide and nateglinide (see below) may be less than with conventional sulfonylureas, and severe hypoglycaemia may be less common.
The extent to which postprandial hyperglycaemia rather than fasting hyperglycaemia contributes to diabetic complications remains the subject of considerable debate. Nevertheless, because of its ability to augment glucose-stimulated insulin release (as opposed to basal insulin release), meglitinides may be useful when postprandial hyperglycaemia in a patient with type 2 diabetes is the dominant clinical problem.
Meglitinides
Repaglinide and nateglinide are prandial glucose regulators that form the meglitinide class of drug. They are indicated in the treatment of patients with type 2 diabetes who have poor glycaemic control on diet control or metformin therapy. These agents are short-acting and act to increase insulin secretion by closing KATP channels of the β-Cell membrane via binding to a receptor distinct from that of sulfonylureas. Repaglinide binds to the B site of SUR-1 and nateglinide binds to the A site. The meglitinides are used in patients as monotherapy (repaglinide only) or in combination with metformin (repaglinide and nateglinide). Given the rapid onset and short duration of action, these agents are taken between 15 and 30 min before each meal to provide postprandial glycaemic control. As repaglinide and nateglinide are rapidly metabolized in the liver and excreted in bile, these agents should not be used in patients with hepatic impairment.
Adverse effects of meglitinides
Hypoglycaemia is a common adverse effect. Other reactions include gastrointestinal effects such as abdominal pain, diarrhoea, constipation, nausea and vomiting; hypersensitivity responses such as pruritus, rashes and urticaria may occur.
Peroxisome proliferator activator γ analogues
Thiazolidinediones were originally developed as ligands for the orphan nuclear peroxisome proliferator activator receptor family (PPARα, PPARγ and PPARδ). The endogenous activators of the nuclear PPAR receptors are fatty acids and fatty acid-derived eicosanoids, and it is now known that the actions of the fibrate group of lipid-lowering agents are mediated via PPARα receptors. Activation of PPARs leads to the formation of heterodimers with the retinoid X receptor (RXR), bound to its own endogenous ligand, retinoic acid. These PPAR–RXR dimers bind to specific gene regulatory sequences (peroxisome proliferator response elements, PPREs), modulating the transcription of multiple (> 40) target genes.
Thiazolidinediones are insulin sensitizers that act as ligands at the PPARγ receptors, which regulate gene expression and transcription of glucose transporter molecules GLUT-4, lipoprotein lipase, fatty acid transporter protein and fatty acyl CoA synthase. The PPARγ nuclear receptors are found in adipose tissue and also in liver and muscle. Their activation leads to improved insulin responsiveness by increasing glucose uptake, and adipose lipogenesis by regulating expression of adipose tissue adipokines.
Troglitazone, the first of these compounds to be introduced, was withdrawn following cases of idiosyncratic hepatotoxicity leading to acute liver failure. At present, the only thiazolidinedione in clinical use is pioglitazone. The other drug in this class, rosiglitazone has been linked to an increased risk of cardiovascular disorders such as myocardial infarction and heart failure, and to bone fractures. It is no longer recommended for use.
Mechanisms of action
It has been suggested that the insulin-sensitizing effects of PPARγ agonists are almost exclusively a result of a ‘fatty acid steal’ mechanism, owing to their effect on adipose tissue, rather than direct effects on the pancreas, liver or peripheral insulin-sensitive tissues. This is because PPARγ receptors are expressed chiefly in adipose tissue, with only low levels of expression in other insulin-sensitive tissues. Thiazolidinediones, acting via PPARγ, increase free fatty acid uptake into adipose tissue (by ~ 60%) and also increase fatty acid oxidation in the liver, heart, kidneys and skeletal muscle. These effects on adipose tissue are brought about by altering adipocyte gene expression, pre-adipocyte differentiation and fat distribution (favouring redistribution from central to subcutaneous depots). By such actions, hepatic NEFA uptake is reduced (by ~ 40%), rendering the liver more insulin-sensitive and giving these agents a potential role in the treatment of hepatic steatosis. However, thiazolidinediones have multiple actions, and it is by no means established that ‘fatty acid steal’ is their dominant mechanism of action.
Peroxisome proliferator activator receptor α is expressed in tissues that metabolize fatty acids extensively such as liver, kidneys, heart, and muscle. Its activation increases plasma HDL-cholesterol concentrations by transcriptional induction of the HDL apolipoproteins, apoA-I and apoA-II (see Chapter 37). Triglyceride concentrations are reduced by decreased hepatic apoC-III production and increased lipoprotein lipase expression. Low density lipoprotein subclass distribution is shifted toward larger particle species by changes in lipoprotein composition and interaction. Combination PPARα/γ agonists are being developed in the hope that a single tablet may be able to benefit both the adverse lipid and glycaemic profiles of type 2 diabetes.
Pioglitazone may be used alone or in combination with metformin or a sulfonylurea, or with both as ‘triple therapy’. While this may delay the need for insulin in some patients, the usual scenario is that pancreatic reserve is failing and that further delays in introducing insulin treatment are potentially hazardous. Pioglitazone is also licensed in the UK for prescription with insulin when metformin cannot be used. The maximum effects of pioglitazone take some 4–12 weeks to develop, and patients may experience a deterioration in glycaemic control during this period if switching from another agent. Pioglitazone is also sometimes used in the treatment of hepatic steatosis (an unlicensed indication). This use, however, should be considered carefully in each patient because of the associated risks.
Adverse effects
As pioglitazone may cause fluid retention, it is contraindicated in patients with heart failure; this risk may be increased if it is used in combination with insulin. The use of pioglitazone is associated with a small increased risk of bladder cancer it is therefore not recommended in patients with a history of bladder cancer or uninvestigated haematuria and should be used with caution in the elderly, given that the risk of bladder cancer rises with age.
Contraindications to the use of pioglitazone also include severe renal insufficiency, pregnancy, breastfeeding and hepatic dysfunction, with the possible exception of that arising from steatohepatitis. Fears over the potential to cause liver damage requires that plasma aminotransferase activity should be monitored two-monthly over the first year after starting treatment. Weight gain of 1–4 kg is predictable from the mechanism of action, but the potential for this to interfere with insulin sensitivity is more than offset by the favourable redistribution of adipose tissue. Fluid retention may cause a mild dilutional anaemia (haemoglobin typically falls by 10–20 g/L) and ankle oedema (in 5–10% of individuals, more when used in combination with insulin).
Glucagon-like peptide 1 analogues
Proglucagon is a product of the glucagon gene, which undergoes post-translational cleavage in pancreatic α-cells to form glucagon and in the L cells of the small bowel, to form glucagon-like peptide 1 (GLP-1), which is produced in response to food in the gastrointestinal tract. Glucose-dependent insulinotropic polypeptide (GIP) is secreted by the K cells in the duodenum. The incretin effect occurs when oral glycaemic stimuli, in the form of food intake, stimulate insulin secretion. It is mediated by the gastrointestinal incretins GLP-1 and GIP, which stimulate insulin synthesis and secretion. In addition, GLP-1 delays gastric emptying and intestinal transit and reduces food intake by promoting satiety.
The incretin response is reduced in type 2 diabetes and the incretin analogues (called incretin mimetics) have been developed for use in its treatment. Human GLP-1 has a short half-life because of the degradation of the N terminal by dipeptidyl peptidase IV (DPP-4) and therefore synthetic GLP-1 analogues (exenatide and liraglutide) have been developed that share the effects of GLP-1 but are resistant to cleavage by DPP-4.
Mechanisms of action
The actions of GLP-1 include increasing the insulin secretion resulting from any given glycaemic stimulus, slowing the rate of gastric emptying and intestinal transit, increasing satiety and reducing the secretion of glucagon. Glucagon-like peptide 1 also has trophic effects on β-Cells, stimulating differentiation and proliferation of progenitor cells in the pancreatic ductal epithelium and inhibiting β-Cell apoptosis. Glucagon and GLP-1 both stimulate insulin secretion by raising cytosolic calcium concentrations by mechanisms distinct from those involving the KATP channel of β-Cells. Receptor binding increases cyclic AMP generation, which impedes sequestration of calcium into intracellular organelles following glucose-stimulated KATP channel closure. Endogenous GLP-1 has a very short half-life (1–2 min) and is therefore not a practical therapy.
Exenatide is a synthetic analogue of a 39 amino acid exendin-4 peptide that was found in the saliva of the Heloderma suspectum (Gila monster) lizard. It requires twice daily subcutaneous injection. More recently, a once weekly preparation of exenatide has been approved. Liraglutide has a more prolonged duration of action compared to exenatide because of greater resistance to inhibition by DPP-4.
These agents are used in patients with type 2 diabetes who have inadequate glycaemic control on one or two oral agents who are intolerant of other oral agents, or in whom use of other oral agents is contraindicated. Their use should preferably be reserved for patients with a body mass index (BMI) > 35 kg/m2 or when BMI is < 35 kg/m2 and insulin therapy is not appropriate for work-related reasons because of the risk of hypoglycaemia, or when weight loss is desirable. Recently both liraglutide and exenatide have also been shown to be of benefit when used in combination with insulin, as they can help reduce insulin doses and promote weight loss.
Adverse effects
The main side-effects that limit the tolerability of GLP-1 analogues are gastrointestinal problems such as nausea, diarrhoea, vomiting and abdominal distension, and injection site reactions. Hypoglycaemia is possible when used in combination with a sulfonylurea or insulin. Renal failure has been reported with exenatide use. Pancreatitis has been reported, and should be considered in patients taking GLP-1 analogues who report severe abdominal pain. Liraglutide should not be used in patients with a history of medullary thyroid carcinoma or multiple endocrine neoplasia type 2 because of potential stimulation of thyroid C-cells, observed in rodent studies.
Dipeptidyl peptidase IV inhibitors
The GLP-1 and GIP incretins are metabolized by cleavage of the N-terminal by dipeptidyl peptidase IV (DPP-4), an enzyme that is ubiquitously expressed. Inhibiting DPP-4 activity prolongs the half-life and increases circulating concentrations of the incretins, thereby stimulating insulin secretion and inhibiting glucagon release. Members of the DPP-4 inhibitor class of drugs include: sitagliptin, saxagliptin, linagliptin and vildagliptin. These may be used as monotherapy or in combination with metformin, a sulfonylurea or pioglitazone in patients who fail to reach glycaemic targets with monotherapy; some can also be used in combination with insulin.
Adverse effects of DPP-4 inhibitors
Inhibitors of DPP-4 do not cause weight gain or weight loss. Hypoglycaemia is uncommon but may occur when they are taken with a sulfonylurea or insulin. Other adverse effects include upper respiratory tract infections, headache and nasopharyngitis. Cardiovascular outcome studies are currently ongoing but thus far, have not shown any significant reduction in cardiovascular risk.
Alpha-glucosidase inhibitors
This class of drugs (acarbose, miglitol) inhibit the α-glucosidase enzymes that break down carbohydrates into monosaccharides at the intestinal border. By delaying the breakdown of carbohydrates in the small bowel, prandial absorption of glucose is reduced, resulting in improved control of postprandial blood glucose concentrations. The use of these drugs is limited by intolerance to the gastrointestinal effects of undigested carbohydrate in the bowel, causing, in particular, excessive flatulence and diarrhoea. The gastrointestinal symptoms do, however, discourage excessive carbohydrate consumption. Hypoglycaemia may occur in association with use of insulin or sulfonylureas.
Sodium-glucose co-transporter 2 (SGLT2) inhibitors
This new class of drugs includes depagliflozin and canagliflozin. These agents block the reabsorption of glucose by the SGLT2 transporter in the proximal renal tubules, resulting in 70-80 g of glycosuria per day. They have been shown to reduce HbA1c by between 7 and 11 mmo1/mo1 when used as monotherapy or added to other agents such as metformin and sulfonylureas in dual or triple combinations, or with insulin. They also result in modest weight loss (about 2-3 kg) and reductions in blood pressure. Because of their mode of action, they are less effective in patients with renal impairment.
Side effects include an increased risk of fungal genital infections and a slightly increased risk of urinary tract infections. The intrinsic risk of hypoglycaemia is low. They should be avoided in patients taking loop diuretics because of the small risk of volume depletion.
Insulin use in type 2 diabetes
Insulin may be initiated in patients with type 2 diabetes when glycaemic control is poor or at presentation if symptoms and signs are such that type 1 diabetes cannot be excluded. Patients with type 2 diabetes require insulin treatment after a median of seven years from diagnosis. The decision to start insulin treatment is often a difficult one, and will depend on many factors relating to individual patient preference and circumstances as well as glycaemic status. It is apparent from the UKPDS that type 2 diabetes is a progressive condition, and that once fasting hyperglycaemia occurs, drug treatment, even when intensive, will improve glycaemic control only transiently, with the subsequent deterioration of β-Cell function and glycaemic control paralleling that of less intensively treated patients, albeit after some initial improvement. Many patients are reluctant to start insulin but are willing to take several oral hypoglycaemic agents. In some cases, the addition of a third oral hypoglycaemic (e.g. metformin, DPP-4 inhibitor plus thiazolidinedione or metformin, sulfonylurea plus DPP-4 inhibitor) may be effective, at least for a while. Responses to the addition of a thiazolidinedione are variable, but in some cases worthwhile. However, treatment failures are also common, and failure to reduce HbA1c values by a predetermined amount (e.g. > 10 mmol/mol (1%)) after six months should lead to reconsideration of insulin treatment, as adequate insulin reserve is required for thiazolidinedione action.
Initial insulin treatment in type 2 diabetes is usually with either a once daily intermediate acting insulin (isophane) or long-acting insulin (glargine or detemir). Some patients are commenced on a twice daily premixed insulin regimen (e.g. 30% short-acting insulin and 70% intermediate acting insulin). Insulin treatment in individuals who are overweight may lead to further weight gain. This is because of: the anabolic effects of insulin; reduced energy loss through glycosuria; possibly increased oral intake because of need to avoid or treat hypoglycaemia, and altered lifestyle, exercise and diet with insulin therapy. With increased weight, these patients face problems of increased insulin resistance, and consequently, the required insulin doses needed to maintain glycaemic control rise, leading to further weight gain.
Bariatric surgery
Weight loss is an important component in the management of overweight and obese subjects with type 2 diabetes. Bariatric surgery may be performed in selected obese patients with diabetes, preferably before the development of irreversible complications and after the exhaustion of conservative measures including diet, exercise and drug therapy. Surgery should be considered in patients with type 2 diabetes who have a BMI of > 35 kg/m2 (as recommended by NICE and the ADA). It is likely that this will be an increasingly considered option for patients with suitable psychological and medical reserves (see Chapter 11 for further details).
Bariatric surgery improves glycaemic control, depending on the surgical procedure, with ‘remission’ rates of diabetes (defined as resolution of clinical and laboratory manifestations of type 2 diabetes) reported to be as high as 78% in a meta-analysis. Surgery is associated with restoration of first-phase insulin response in up to 80% of subjects. The Swedish Obesity Study compared the effects of bariatric surgery in 2010 obese subjects with 2037 obese controls and found that bariatric surgery was associated with a decreased incidence of diabetes and, in those with type 2 diabetes at baseline, increased diabetes remission rates at two years and at ten years. Not only have improvements in glycaemia been reported well before weight loss becomes apparent, but they appear also to exceed those expected on the basis of the amount of weight lost. It has been proposed that increased secretion of a number of gut peptides with insulinotropic actions such as GLP-1 and GIP, and decreased secretion of orexigenic peptides such as ghrelin, may be responsible for the success of bypass techniques, raising the intriguing possibility that operations of this kind have a partly endocrine mechanism of action. As energy intake following surgery is changed, it may be necessary to adjust glucose lowering therapy to avoid severe hypoglycaemia.
Pancreatic transplantation
Simultaneous pancreatic and kidney transplantation has been performed in patients with established renal failure, and is a reasonable option to consider at this stage in patients with glycaemic instability, who are physically fit and psychologically well prepared. Pancreas-alone transplants have also been carried out and may sometimes be considered in patients with recurrent hypoglycaemia. Although complete independence from insulin medications is possible, this form of treatment requires immunosuppressive therapy and is limited by the availability of organs for donation.
Islet cell transplantation
Islet cell transplantation may be used in patients with severe hypoglycaemia unawareness. This uses cadaveric pancreatic islets cells that are infused though a catheter placed into the hepatic portal vein. However, it is limited by donor availability and low rates of independence from insulin medications because of graft attrition. The Clinical Islet Transplantation (CIT) consortium, a network of centres in North America and Europe, is conducting studies on islet transplantation in type 1 diabetes, but the clinical outcomes are not yet known.
Immunotherapy for type 1 diabetes
Immunomodulatory therapies for type 1 diabetes are the subject of phase II and phase III clinical trials. These treatments target specific antigens or modulate the immune system with the aim of establishing immune tolerance and preventing the destruction of pancreatic β-Cells. The concept of tolerance is important in that in autoimmune disease, the immune system loses its tolerance to a particular antigen, which may be a mechanism underlying the autoimmune nature of type 1 diabetes. Different immune-targeted treatments that target pathogenetic cells and activate regulatory cells have been developed. Nutritional studies have investigated how exposure to different dietary components may influence disease initiation.
Glutamate decarboxylase (GAD65) immunization of young non-obese diabetic (NOD) mice has prevented development of type 1 diabetes. However, in humans, there was no effect on β-Cell destruction. Inoculation with human heat shock protein 60 (Hsp60) prevented disease progression in NOD mice, whilst in humans, it produced increased C-peptide concentrations. Studies in children have not shown an improvement in β-Cell function, however. Trial evidence for anti-CD3 and anti-CD20 monoclonal antibodies therapy in humans has shown improved C-peptide concentrations and reduced insulin demand. Monthly injections with CTLA4-Ig (a T cell receptor that inhibits T cell activation) over two years in human trials also increased C-peptide concentrations. Trials of interleukin-2 showed favourable results in NOD mice. Trials with insulin therapy have not shown effects on cell function.
Nutritional studies in children showed that lower concentrations of islet cell antibodies were present in those who had been weaned in infancy onto a hydrolysed casein formula compared with those weaned to cows’ milk formula. A trial of the effects of ω-3 fatty acids showed reduced risk of islet autoimmunity in children with a genetic risk of type 1 diabetes. In another trial, vitamin D did not protect β-Cell function in patients with recent onset of type 1 diabetes.
OBSTACLES TO ACHIEVING GLYCAEMIC CONTROL
Glycaemic management of diabetes is rarely as simple as making incremental additions to a patient’s therapy until normoglycaemia is achieved. Hypoglycaemia is the main barrier to tight glycaemic control. For the majority of patients, achieving good glycaemic control involves a complex trade-off between the potential benefits (most of which are long term) and risks or obstacles (many of which are only too readily and immediately apparent).
The patterns of endogenous insulin secretion, which vary rapidly in response to changes in blood glucose concentration, are not easily mimicked by the injection of exogenous insulin into a subcutaneous depot, from which there is prolonged release into the systemic rather than portal circulation, thus partially bypassing hepatic action and first-pass clearance. This leads to an inherently increased risk of hypoglycaemia, which may be exacerbated by a number of factors related to the treatment, the individual and his or her circumstances.
Intensive control
The ADA recommends an HbA1c concentration of < 53 mmol/mol (< 7%) as a treatment goal whereas the International Diabetes Federation (IDF) recommends an HbA1c of < 48 mmol/mol (< 6.5%).
The DCCT demonstrated that in type 1 diabetes, intensive insulin treatment (multiple daily injections or CSII therapy) improved glycaemic control and reduced retinopathy, nephropathy and neuropathy complication rates. Subsequently, the Epidemiology of Diabetes Interventions and Complications (EDIC) study showed that this effect persisted for at least ten years in those subjects who had been in the intensive glycaemic control group of the DCCT.
In type 2 diabetes, the UKPDS showed reduced microvascular complications with intensive glucose control. At long-term follow-up at ten years, early intensive glycaemic control continued to reduce the risk of microvascular complications. However, a recent meta-analysis demonstrated that intensive glycaemic control in type 2 diabetes is associated with a 30% increase in the relative risk of severe hypoglycaemia. Several large trials have compared the effects of intensive versus standard glycaemic control on cardiovascular outcomes in type 2 diabetes. The Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation (ADVANCE) trial showed a significant reduction in microvascular outcomes (nephropathy and retinopathy) with tightly controlled diabetes but no significant reduction in cardiovascular outcomes. The Veterans Affairs Diabetes Trial (VADT) trial found that intensive glycaemic control did not significantly lower the incidence of cardiovascular events compared with standard glycaemic control. The Action to Control Cardiovascular risk in Diabetes (ACCORD) trial found increased mortality associated with an intensive glycaemic control to a target HbA1c of < 42 mmol/mol (< 6%), leading to the early termination of the trial.
In both type 1 and type 2 diabetes, the effect of intensive control of blood glucose on microvascular complications is therefore well established. Although the ACCORD, ADVANCE and VADT trials did not show any improvement in cardiovascular outcomes with intensive control, glycaemic goals should still be set because of the adverse microvascular consequences associated with poor glycaemic control. Both the ADA and the International Diabetes Federation recommend individualized targets for each patient and the need to balance the risks and benefits of tight control of HbA1c. Goals for patients with a history of frequent or severe hypoglycaemia, long standing diabetes, the elderly or those with limited life expectancy and patients with multiple comorbidities will need to be carefully considered. The established cardiovascular risk reduction measures such as blood pressure control, smoking cessation, lipid lowering therapy and healthy lifestyle should continue to be recommended as part of an individualized care plan.
Hypoglycaemia
The risk of hypoglycaemia often limits the achievement of good glycaemic control in diabetes. Although microvascular and macrovascular complications of diabetes typically evolve over a period of several years, hypoglycaemia may occur quickly, over a few minutes, and the consequences may be devastating or permanent. Some 2–4% of deaths and a much greater proportion of quality adjusted life years (QALYs) lost in type 1 diabetes are attributable to hypoglycaemia. Furthermore, the perceived danger, inconvenience, embarrassment and helplessness associated with hypoglycaemia is considerable, and the extent to which an individual patient will risk hypoglycaemia often influences how they manage their glycaemic goals and how they view care for their diabetes. For some patients, the risk of hypoglycaemia conflicts with occupational use of a motor vehicle (and hence possibly their livelihoods). In other patients (e.g. those with cancer or the very elderly), the primacy of goals such as freedom from osmotic symptoms and hyperglycaemic emergencies do not necessitate tight glycaemic control.
In addition to the factors mentioned in Table 16.3, it is clear that reduced glucoregulatory responses to hypoglycaemia in patients with type 1 diabetes and longstanding type 2 diabetes can markedly increase the frequency and severity of hypoglycaemic episodes.
TABLE 16.3
Factors that may affect the effect of insulin on blood glucose concentration
| Treatment-related factors | Comments |
| Insulin | Dose, chemical form (human, animal, analogue, lipophilia, tendency to form complexes, albumin binding), suspension (protamine zinc etc.), site and volume of injection (lipohypertrophy, exercising limbs vs abdominal wall), timing in relation to meal (30 min before, immediately before, after), time of day (insulin resistance higher in the morning), changes in insulin clearance (decreased GFR), accidental intravenous injection of insulin, warming (e.g. shower soon after injection), weather or massage of the injection site |
| Diet and absorption | Glycaemic index, carbohydrate and fat content, missed meals, gastric absorption (presence of gastroparesis, drugs causing rapid transit), changes in absorption (coeliac disease), extended interval between meals (e.g. overnight, special circumstances) and dialysis (glucose removal) |
| Gluconeogenesis and glycogenolysis | Depletion of hepatic glycogen reserves after fasting, inhibitory effects of alcohol or other drugs, liver disease |
| Insulin resistance | Time of day, associated conditions and their treatment (Addison disease, hypothyroidism), exercise, intercurrent illness, sleep disturbance, mental/emotional/social stress, weight loss, improved metabolic control (glucotoxicity), obstetric delivery or intrauterine fetal death (sudden reduction in insulin resistance), cessation of steroid therapy |
In persons without diabetes, the first response to a falling blood glucose concentration is a reduction in insulin secretion; this typically occurs at blood glucose concentrations < 4.5 mmol/L. This response is not available to subjects with type 1 diabetes or to many subjects with type 2 diabetes (either insulin or secretagogue treated). Glucagon secretion forms the next layer of defence, stimulating hepatic glycolysis and gluconeogenesis. However, most patients with either type 1 or type 2 diabetes are chronically hyperglucagonaemic and cannot respond to hypoglycaemia in this way.
The last level of defence against acute hypoglycaemia is activation of the sympathetico–adrenomedullary system, which normally occurs when blood glucose falls to < 2.8–3.0 mmol/L. This increases lipolysis and circulating NEFA production and utilization, and mobilization of substrates for gluconeogenesis further inhibits insulin secretion and acts as a stimulus to glucagon release. Importantly, activation of the sympathetico–adrenomedullary system gives rise to the first clear symptoms of hypoglycaemia. With still lower blood glucose concentrations, patients also experience symptoms of neuroglycopenia. Although neuroglycopenia usually manifests as a generalized phenomenon, focal neurological deficits may occur in the setting of relatively normal consciousness and, for this reason, capillary blood glucose testing must be performed (and any low result confirmed by formal blood glucose measurement) in all cases of apparent stroke, confusion or other acute neurological disability, and in patients presenting with unexplained seizures.
Patients with diabetes are advised to carry dextrose (glucose) tablets or quick acting carbohydrate in case of emergency. Parenteral correction of hypoglycaemia may be necessary in cases where the patient has lost consciousness or is not able to eat food. An intramuscular glucagon injection may be used to correct insulin-induced hypoglycaemia. In severe cases of hypoglycaemia, for example where the patient is unconscious, intravenous glucose may need to be administered by medical personnel. Patients who have had recurrent episodes of hypoglycaemia should be reviewed by the diabetes team to assess potential causes and ways to avoid this from happening.
Hypoglycaemia-associated autonomic failure
Inability to reduce insulin secretion and failure of the glucagon response are described above. At this point, only the sympathetico–adrenomedullary responses stand between the patient and the development of neuroglycopenia. Failure of sympathetico–adrenomedullary responses can also occur and will lead both to a reduced awareness of, and reduced conscious and unconscious response to, hypoglycaemia (hypoglycaemia unawareness). Its particular importance is that it is associated with a greatly increased risk (possibly by a factor of 25) of severe hypoglycaemia. This potentially serious situation is usually amenable to treatment (see below).
Drugs such as β-blockers may interfere both with the action of adrenaline (epinephrine) and also with the perception of sympathetico–adrenomedullary activation, and should be used with great caution in the context of hypoglycaemia unawareness. Autonomic neuropathy may lead to reduced sympathetico–adrenomedullary function and reduced hypoglycaemia awareness. It is also associated with unpredictable glycaemic excursions owing to erratic intestinal transit times. Perhaps most importantly, functional impairment of sympathetico–adrenomedullary responses may occur in type 1 or long-standing type 2 diabetes in response to preceding hypoglycaemia. Recurrent hypoglycaemic episodes lead to altered body responses to hypoglycaemia and susceptibility to further episodes.
Hypoglycaemia-associated autonomic failure (HAAF) may occur after a single episode of hypoglycaemia, although this is more frequently seen following recurrent hypoglycaemia. There is evidence to support a role for cortisol in mediating HAAF. The hypothalamo–pituitary–adrenal axis is stimulated by initial bouts of hypoglycaemia, but the resulting hypercortisolaemia may result in impaired adrenaline (epinephrine), noradrenaline (norepinephrine) and glucagon responses to subsequent hypoglycaemia. Muscle sympathetic nerve activity, an index of sympathetic nervous system outflow, is also impaired following hypoglycaemia. Infusion of pharmacological doses of cortisol in healthy subjects has a similar effect on their subsequent hormonal response to insulin-induced hypoglycaemia. However, this effect has not been replicated using lower dose infusions that achieve plasma cortisol concentrations similar to those found in hypoglycaemia, and hypoadrenal subjects without diabetes who are treated with fixed physiological cortisol replacement doses demonstrate almost complete preservation of counter-regulatory responses to a subsequent episode of hypoglycaemia, calling this hypothesis into question. Regardless of the underlying mechanism, however, management of HAAF is based upon avoidance of hypoglycaemia for several days, by slightly relaxing glycaemic control, thereby allowing autonomic responses to recover.
The principle of antecedent glycaemia affecting subsequent responses is further illustrated by the converse phenomenon of ‘clinical pseudohypoglycaemia’ This term denotes the observation that some patients with chronic hyperglycaemia experience symptoms of hypoglycaemia at blood glucose concentrations well in excess of those that would trigger responses in individuals with lower prevailing blood glucose concentrations. Management of this condition is empirical, and should involve measures to induce very gradual improvements in glycaemic control.
The Somogyi effect and the dawn phenomenon
After a counter-regulatory response to a hypoglycaemic episode (whether or not followed by the administration of (sometimes excessive) glucose to reverse the symptoms), rebound hyperglycaemia is common. Usual glycaemic control, glucose tolerance and insulin sensitivity may take several hours to be restored. A special variant of the rebound from hypoglycaemia is the Somogyi phenomenon, which occurs in patients with nocturnal hypoglycaemia. The patient may not be aware of the hypoglycaemia, even in retrospect, although awakening with malaise, headache and bedclothes damp from sweating are suggestive. The rebound from the nocturnal hypoglycaemia results in the patient waking with a blood glucose concentration higher than desirable, provoking a temptation to take at least as much (or even more) insulin the next night and thereby increasing the likelihood of a subsequent episode of hypoglycaemia. Detection of this problem requires a high level of suspicion and may involve measures such as setting the alarm clock in order to self-test, the observations of another person, or continuous blood glucose monitoring.
Subjects without diabetes show circadian changes in blood glucose concentration and glucose tolerance based upon changes in counter-regulatory hormones, insulin secretion and the availability of alternative fuels for metabolism. The most marked such circadian effect is the ‘dawn phenomenon’, which typically occurs between 04.00 h and 07.00 h and is an increase in plasma glucose concentration and decrease in insulin sensitivity consequent upon the increased secretion of counter-regulatory hormones at that time. During this period, people with diabetes usually experience modest rises (1–2 mmol/L) in plasma glucose concentrations without recent ingestion of food. A larger rise is seen in patients in whom circulating insulin concentrations are too low at this time.
Owing to both the dawn phenomenon and the Somogyi effect, there is a risk of hypoglycaemia earlier in the night whenever longer-acting insulins, particularly when administered in the early evening, are titrated to pre-breakfast blood glucose concentrations.
Exercise
A patient’s usual balance between carbohydrate intake and insulin requirements may be influenced by physical activities. The metabolic effects of exercise vary with the intensity and duration of that exercise. For example, jogging typically elicits an increase in both glucose and lipid oxidation, and plasma glucose and NEFA turnover increase. In more strenuous exercise, muscle stores of glycogen and triglycerides will become depleted. Plasma glucose concentrations may increase or decline during exercise, depending on the counter-regulatory hormonal drive to hyperglycaemia, and whether glycogen reserves are adequate.
In healthy subjects, plasma insulin concentrations decrease, permitting increased hepatic glucose output to limit the fall in blood glucose. In subjects on exogenous insulin treatment, insulin concentrations may not fall (indeed, there may be a rise if increased skin blood flow during exercise increases insulin absorption from subcutaneous injection sites) and hence hepatic glucose output may not be sufficient to prevent hypoglycaemia. After exercise, there is a phase of glycogen repletion in liver and muscle, and in subjects with diabetes, delayed hypoglycaemia may be a problem at any time up to several hours afterwards.
Strategies to control blood glucose concentrations during exercise include adjusting insulin doses that are to be taken prior to planned exercise, or ingestion of extra carbohydrate before, during or after the exercise. Because of the numerous physiological variables involved, it is often difficult to predict the best strategy in advance, and a system of trial and error with increased frequency of blood glucose monitoring may be necessary. Many patients with diabetes lead active lifestyles as regular physical exercise is important in improving insulin sensitivity and improving and maintaining cardiorespiratory fitness.
Ethanol
Ethanol (alcohol) may inhibit gluconeogenesis; in patients with diabetes, this may reduce hepatic glucose output sufficiently to provoke hypoglycaemia. Typically, this does not occur while the patient is consuming ethanol, since most beverages contain adequate carbohydrate to prevent this (or the patient eats as well as drinks): instead, hypoglycaemia occurs during the first two hours after alcohol consumption. However, the inhibition of gluconeogenesis persists for several hours and a rebound hypoglycaemic episode may occur a few hours (typically 2–4) after consuming alcohol. If the clinical features of hypoglycaemia are dismissed as the consequences of inebriation, then the failure to treat the hypoglycaemia may be serious. In addition, the effects of alcohol may impair the judgement of an individual with diabetes, who may forget to consume carbohydrate after heavy alcohol consumption or fail to adjust their insulin dose in anticipation of planned alcohol consumption. It may even be a contributory cause of ketoacidosis when insulin doses are unintentionally omitted.
Intercurrent illness, ‘sick day rules’ and stress
Intercurrent illnesses such as viral infections usually cause hyperglycaemia in people with diabetes but may also give rise to hypoglycaemia. The effect on glycaemic control may persist for up to two weeks, and insulin requirements, even during simple infections (e.g. colds), may increase by 100%, although increases of ~ 20% are perhaps more typical.
It is likely that many episodes of hyperglycaemic crisis could be prevented by appropriate self-treatment and timely medical advice during times of intercurrent illness. Treatment plans should ideally be individualized and prepared in advance. Several general principles apply to all, however. These include the concepts that:
• frequent (e.g. four-hourly) blood glucose testing is needed
• medical advice should be sought if blood glucose concentrations become and remain high despite appropriate measures (e.g. > 13–15 mmol/L)
• maintenance of good hydration is of the utmost importance.
For patients with type 2 diabetes, metformin should probably be discontinued for the duration of any severe illness, and a sulfonylurea (or, if necessary, insulin) substituted in the short term. Patients taking a sulfonylurea may require dose increments or the short-term use of insulin.
Patients with type 1 diabetes are at risk of ketoacidosis and should therefore be advised to test their urine regularly (at least twice daily) to ensure no more than light to moderate ketonuria is present. Where blood ketone testing is available, testing for blood ketones is preferred. Patients should be warned against the common misconception that if they are not eating (e.g. because of vomiting), they should stop taking insulin. Instead, essential fluid and glucose may be ingested in the form of regular small volumes of soup, rehydration fluid or proprietary sugar-containing drinks. Usual doses of basal insulin are continued and the day’s bolus (rapid-acting) insulin divided into 4-hourly quotas, which can be adjusted on the basis of self-testing. Persistently elevated or rising blood glucose concentrations, particularly in the context of increasing ketonuria, should prompt contact with an appropriate medical advisor or admission to hospital for insulin infusion.
In patients with severe intercurrent illness such as septicaemia, pancreatitis or myocardial infarction (all these are more common in diabetes), intravenous insulin is often required, even in subjects not usually requiring exogenous insulin treatment. There are now good data to support the use of intravenous insulin infusion in such circumstances and during major surgery. It is not yet clear whether the infusion of insulin per se is of benefit or whether tight glycaemic control achieved by any means confers similar advantage. These issues are discussed in greater detail in a later section.
Emotional stress of many forms will result in changes in secretion of counter-regulatory hormones and autonomic tone, which will alter glucose tolerance. Since emotional stress is often associated with alterations in sleep, eating and exercise patterns, it may be practically impossible for patients with diabetes under emotional stress to maintain usual glycaemic control.
CHRONIC COMPLICATIONS OF DIABETES
The complications of diabetes can be divided into acute and chronic complications. Acute complications include hypoglycaemia (discussed above), DKA and the hyperosmolar hyperglycaemic state (see p. 326). Chronic complications include macrovascular disease (see above), retinopathy, nephropathy and neuropathy.
Nephropathy
Diabetic nephropathy is the leading cause of established renal failure and the most common diagnosis at initiation of renal replacement therapy. Glomerular damage is pathologically described as either diffuse or nodular glomerulosclerosis (Kimmelstiel–Wilson lesions). The pathological process involves thickening of the basement membrane and of the glomerular mesangium with the accumulation of extracellular collagen matrix material. There is glomerular podocyte loss and tubulointerstitial fibrosis. Glomerular hyperfiltration is followed by microalbuminuria, proteinuria and subsequent decline in renal glomerular function.
Some 20–30% of patients with type 1 diabetes will develop renal disease (most commonly 15–25 years after diagnosis). Risk factors for its development include longer duration of disease, an earlier age at diagnosis, earlier onset of puberty, poorer glycaemic control throughout the time course of the disease, smoking and a family history of diabetic nephropathy. Nephropathy is probably less prevalent among those with type 2 diabetes (10–20% lifetime risk), but seems likely to increase substantially with the increasingly early mean age of onset of type 2 diabetes. Diabetic nephropathy is screened for by laboratory testing for urine albumin excretion (urine albumin/creatinine ratio), serum creatinine and estimated glomerular filtration rate (eGFR). Screening should be performed at least annually. (Formulae used for the calculation of eGFR are discussed further in Chapter 7.)
Microalbuminuria
The early stages of the disease are asymptomatic and include hyperfiltration (with urine albumin excretion (UAE) < 30 mg/24 h or 20 μg/min) followed by progression through microalbuminuria (UAE 30–300 mg/24 h or 20–200 μg/min) to proteinuria (UAE > 300 mg/24 h or 200 μg/min). After proteinuria has become established, the glomerular filtration rate (GFR) begins to fall and progression to established renal failure is generally inevitable, although the rate of loss of renal function may still be usefully slowed by treatment.
Differences in historical ten-year progression rates from microalbuminuria to nephropathy (~ 80%), and more recent estimates (~ 30%), lend weight to the hypothesis that aggressive treatment at the stage of microalbuminuria may be able to halt or even reverse the disease in many patients. Thus, the first and best opportunity to detect the disease clinically is at the stage of microalbuminuria. Some would even contend that risk associated with UAE is a continuous variable, with detectable increases in morbidity above the normal median value of albumin excretion of approximately 2.5 mg/24 h. Conventional dip-stick testing of urine is not usually positive at such concentrations of albumin excretion. Neither this test nor the measurement of 24 h urinary protein excretion are now recommended as screening tools. Screening should probably take place at the time of diagnosis and annually thereafter in type 2 diabetes. Although it has traditionally been thought that screening could safely be delayed for five years in type 1 diabetes, a number of studies have shown significant prevalence before five years, especially where puberty has intervened or in the presence of poor glycaemic control, and many physicians now advocate annual screening from diagnosis in this group also.
Factors giving rise to falsely elevated results include unaccustomed exercise, intercurrent illness, other renal disease, haematuria, acute deterioration in glycaemic control and urinary tract infection (which should be excluded whenever tests of microalbuminuria are positive, see Chapter 8). Furthermore, considerable day-to-day variations in urine albumin excretion rates mean that a minimum of two spot urine collections for albumin:creatinine ratio over a period of 3–6 months should be abnormal before a diagnosis of microalbuminuria can be made reliably. It should be noted that the occurrence of non-immunoreactive forms of albumin in the urine may result in underestimation of the true extent of microalbuminuria in some assays. Intermittently positive results, especially where occurring frequently without a discernible cause, do appear to increase the risk of progression to definite microalbuminuria. It is important to appreciate that microalbuminuria is not just a risk factor for the development of nephropathy, but is also an independent risk factor for coronary artery disease (indeed, one of the most potent risk factors known), being also associated with dyslipidaemia, hypertension, endothelial dysfunction and diabetic retinopathy. Patients with microalbuminuria should, therefore, undergo intensive risk factor modification, and low-dose aspirin is generally indicated for these patients. There is controversy as to whether an increase in blood pressure precedes or is a result of the development of microalbuminuria.
Management
Prevention of diabetic kidney disease is of paramount importance. It necessitates good metabolic control, adequate treatment of hypertension, avoidance of smoking, lipid-lowering therapy and avoidance of nephrotoxic drugs. Intensive glycaemic control delays onset and progression of albuminuria. However, even with these measures, data from the UKPDS and DCCT show that a significant number of patients will still develop the disease, albeit at a later stage and with slower progression. Thus, the second important feature of management remains early detection of raised UAE, which is discussed in detail above.
Good metabolic control may delay the onset of microalbuminuria and, once this is present, may halt progression or even reverse it. In general, antihypertensive therapy, maintaining systolic BP < 140 mmHg and diastolic < 80 mmHg, irrespective of the agent used, slows the development of diabetic nephropathy. The UKPDS provided evidence that blood pressure control could protect against nephropathy. Inhibitors of the renin– angiotensin system, i.e. angiotensin-converting-enzyme (ACE) inhibitors or angiotensin receptor antagonists (ARBs) confer superior long-term protection, probably because of a concomitant specific reduction of transglomerular filtration pressure, preventing increases in albumin excretion and reducing the severity of proteinuria. Angiotensin-converting-enzyme inhibitors reduce cardiovascular events and ARBs reduce progression of nephropathy. Combination ACE inhibition and ARB therapy lowers albuminuria further, but may increase the risk of hyperkalaemia and evidence for the clinical efficacy of this approach is limited. Increases in plasma creatinine concentrations of up to 20–30% from baseline are common on starting treatment and, although they do not mandate alternative treatments, monitoring of renal function is essential.
Dietary protein restriction to 0.8–1.0 g/kg body weight per day in early chronic kidney disease (CKD) or 0.8 g/kg body weight per day in advanced CKD is recommended. Blood pressure control with other agents such as diuretics, calcium channel blockers and β-blockers may be used.
End-stage disease
Despite prevention, detection and aggressive treatment of diabetic nephropathy, a significant proportion of patients will nevertheless progress towards established renal failure (ERF). Early referral to a nephrology service (usually before GFR has fallen to < 30 mL/min/1.73 m2) will usually be appropriate. Occult cardiac disease is common in this group of patients, but its angiographic investigation is hazardous owing to the risk of contrast nephropathy. Many patients with anaemia other than that caused by specific haematinic deficiencies will benefit from erythropoietin treatment at haemoglobin concentrations of < 110 g/L. Management of metabolic sequelae such as secondary hyperparathyroidism, acidosis and hyperphosphataemia are, in general, as for patients with non-diabetic kidney disease. Treatment options for ERF include haemodialysis and renal transplantation, depending on the availability of graft donors and compatibility.
Neuropathy
Neuropathy in diabetes has many different manifestations and can be focal or diffuse. The types of neuropathy include chronic sensorimotor, autonomic, diabetic amyotrophy (proximal motor neuropathy) and mononeuropathies. An acute neuropathy may also occur in the presence of hyperglycaemia (hyperglycaemic neuropathy), possibly as a result of damage to peripheral nerves.
Chronic sensorimotor neuropathy
This is most the common type of neuropathy in patients who have had diabetes for a long time. The longest nerves are usually affected, with sensory loss in a glove and stocking distribution. The sensory loss is often accompanied by loss of power and autonomic involvement. There is loss of pain and light touch, with decreased reflexes and muscle wasting in affected parts. Some patients may report painful symptoms (e.g. tingling, burning, hyperalgesia or allodynia).
Patients should be screened for diabetic neuropathy by pinprick tests, using a 10 g monofilament and a 128 Hz vibration fork applied to the distal aspects of both feet. The diagnosis is often made clinically without the need for electrophysiological studies, although these can be performed if the diagnosis is uncertain. It is important to screen for other, potentially reversible, causes of neuropathy, e.g. vitamin B12 deficiency, alcohol excess, heavy metal poisoning and neurotoxic medications, and for conditions that may require specific treatment, e.g. demyelination. Patients with sensorimotor neuropathy are at increased risk of injury to and ulceration of the feet. Charcot neuroarthropathy (Charcot foot, see below) is a common complication. All patients with peripheral neuropathy should receive regular podiatric assessment and advice regarding foot care because of the increased risk of foot ulceration.
Besides optimizing blood glucose control, the treatment for sensorimotor neuropathy includes symptomatic pain relief with agents for neuropathic pain such as duloxetine, amitriptyline, pregabalin and gabapentin.
Autonomic neuropathy
Autonomic neuropathy is seen in patients with diabetes who have longstanding poor control. Manifestations include orthostatic hypotension (a major risk factor for falls), constipation, gastroparesis (with nausea and vomiting), loss of normal Valsalva response, gustatory sweating, resting tachycardia, bladder dysfunction, erectile dysfunction and altered autonomic responses to hypoglycaemia.
Diabetic autonomic neuropathy may affect gastrointestinal motility: this condition may be occult, or manifest as constipation, diarrhoea, unexplained (or easily triggered) vomiting or malabsorption. Altered gastrointestinal motility may change the rate of absorption of nutrients from food. Rapid and unpredictable absorption is particularly unwelcome in diabetes and may completely undermine the attempts of even a well-motivated patient to maintain good glycaemic control. In some cases, diabetic ketoacidosis may be provoked as a result of repeated vomiting. If gastrointestinal motility can be improved, this renders glycaemic control somewhat easier; however, such amelioration is usually spontaneous rather than related to medical intervention. Investigations for gastroparesis include the use of isotope scintigraphy to assess solid phase gastric emptying. Prokinetic treatments such as metoclopramide, domperidone or erythromycin may be helpful in some cases to aid motility. Dietary changes may be necessary and in severe situations, parenteral nutrition may become necessary if adequate nutrition is not possible by the enteral route. A gastric pacemaker with electrodes that electrically stimulate gastric contractions may be used in severe cases.
Patients who are troubled with postural hypotension may be prescribed support stockings. In severe cases, fludrocortisone (a mineralocorticoid) or midodrine (an α-adrenergic agonist) may be used.
Autonomic neuropathy may impair hypoglycaemia awareness. This will often result in the patient (very sensibly) avoiding the possibility of severe hypoglycaemia and, as a result, glycaemic control may be more difficult to achieve. Autonomic neuropathy may be associated with increased morbidity and mortality if it affects the cardiovascular system. This is manifested by postural hypotension and resting tachycardia and is associated with an increased risk of cardiovascular events.
Erectile dysfunction and retrograde ejaculation can occur in autonomic neuropathy. If the sacral nerves innervating the bladder are affected, patients may suffer from bladder dysfunction, with bladder emptying difficulties, incontinence problems and urinary tract infections. Erectile dysfunction may be caused by multiple factors such as vascular disease, neuropathy, medication or psychological problems. Besides reviewing these factors, treatments for erectile dysfunction may include phosphodiesterase type 5 inhibitors, prostaglandins and vacuum devices (see Chapter 23).
Mononeuropathies
Mononeuropathies may result from diabetes affecting single nerves or, where multiple nerves are affected, a mononeuritis multiplex. These include third cranial nerve palsies (that may have a vascular aetiology), lateral popliteal nerve palsy and carpal tunnel syndrome.
A form of multiple mononeuropathy called diabetic amyotrophy may occur. This is caused by a reduction in the blood supply to the nerves of the lower limbs, for example the femoral nerve or the lumbar plexus, resulting in proximal weakness and wasting of muscles in the lower limbs. Amyotrophy is associated with poor glycaemic control, and optimum glycaemic control is important in attaining recovery, which is a slow process. Pain control and physiotherapy are helpful.
The feet in diabetes
Foot ulcers
Foot ulceration in patients with diabetes is a common cause of morbidity and its complications, such as sepsis, may be associated with mortality. Ulcers mainly occur in patients who have both diabetic neuropathy and peripheral vascular disease. Repeated trauma without recognition by the patient may cause infection. Foot architecture may become distorted because of callus formation, deformity and ulceration, leading to a condition called the Charcot foot (see below).
Concomitant infection of the feet does not heal well owing to repeated unrecognized foot trauma and poor vascular circulation. Infective organisms include Staphylococcus aureus, Streptococcus pyogenes and Pseudomonas aeruginosa.
Charcot foot
Charcot foot is a specific foot deformity, bilateral in approximately 20% of cases, which may arise on the background of severe sensory (and probably motor) neuropathy. Often precipitated by minor trauma, the disease may progress very rapidly over weeks or a few months, beginning with an unstable, warm, red and swollen foot (stage 0), the point at which treatment should be initiated for maximum benefit. A ‘fragmentation’ stage (stage 1) follows, with periarticular fractures, joint dislocation, instability and deformity. In the ‘coalescence’ stage (stage 2), reabsorption of bone debris occurs before finally a stable (stage 3), but deformed, foot is left at the end of the process.
The exact pathogenesis of this condition remains unclear, but abnormal protective motor reflexes may permit the development of abnormal loading forces within the foot which, in the presence of sensory neuropathy and osteopenia, may damage bone, joints and ligaments. Subluxation, or even dislocation, of the metatarsal, tarso–metatarsal or tarsal joints may occur and swelling may be caused by leakage of synovial fluid from joint capsules. Bone blood flow is usually substantially increased, perhaps as a result of abnormal sympathetic control, and this may contribute to the osteopenia. Later in the course of the disease, bones may become sclerotic. Fractures may cause the tarsal bones to collapse, resulting in an outward bowing of the arch (‘rocker-bottom foot’ deformity), further adding to the abnormal loading and risk of ulceration. The foot can be so warm and red that osteomyelitis, cellulitis or gout is suspected. Osteomyelitis may be particularly difficult to distinguish when a plantar ulcer is present and, indeed, may be very likely if there are systemic signs of infection or if bone is encountered when the ulcer is probed. Plain radiographs usually show vascular calcification and bone changes; the latter may be difficult to distinguish from osteomyelitis. Magnetic resonance imaging or bone biopsy may be required to establish the correct diagnosis in some cases.
Intravenous bisphosphonates (e.g. pamidronate), total immobilization and offloading of the foot (e.g. bed rest, pending the fitting of a total contact cast, perhaps for four months or longer) and meticulous attention to metabolic control are helpful in the acute setting. Bone markers, such as serum carboxy-terminal telopeptide of type 1 collagen, a marker of osteoclastic bone resorption, measured in venous blood taken from the dorsum of the foot, urinary deoxypyridinoline and bone-specific alkaline phosphatase, have been used in studies alongside longitudinal measurements of foot temperature to assess the response to treatment.
Eye disease
Diabetes is a common cause of visual loss, especially in the developed nations. Risk factors for the development of diabetic retinopathy include duration of diabetes, poor glycaemic control (chronic hyperglycaemia), diabetic nephropathy, hypertension, hyperlipidaemia, smoking, obesity and rapidly improving glycaemic control, for example in pregnancy or when commencing insulin pump therapy. Retinopathy is usually present after 20 years of diabetes, although the severity varies between patients. Patients with diabetic retinopathy are predisposed to cataracts, glaucoma and macular disorders.
Patients with type 1 diabetes should have an eye examination within five years of diagnosis. The prevalence of retinopathy in type 1 diabetes after 10–15 years is 25–50%. All patients with type 2 diabetes should be screened when diagnosed, reflecting the fact that the presence of type 2 diabetes may have not been recognized for some time. About 60% of patients with type 2 diabetes have non-proliferative retinopathy. All patients should be screened annually, or more frequently depending on the findings. Women with diabetes are screened more frequently during pregnancy and in the postpartum period because of an increased risk of progression of diabetic retinopathy.
Eye examinations should be carried out by an ophthalmologist or optometrist experienced at diagnosing diabetic retinopathy. In the UK, an annual retinopathy screening service is provided for all patients with diabetes. Any patient with progressive retinopathy detected at screening is referred to an ophthalmologist for follow-up investigation and, if necessary, treatment. The UK National Retinal Screening Committee classification is based on the presence or absence of both retinopathy (graded from R0 to R3) and maculopathy (graded M0 or M1) (see Box 16.1).
Initially, there is an increase in retinal perfusion. Background retinopathy denotes the formation of microaneurysms and hard exudates. Flame-shaped or dot and blot haemorrhages may follow should the microaneurysms rupture. Retinal capillaries may leak. Leakage into the macular region may cause maculopathy. Maculopathy is characterized by the formation of hard exudates in close proximity to the fovea, along with oedema and ischaemia of the macula region. It is more common in type 2 than type 1 diabetes and is the commonest cause of loss of vision.
Preproliferative retinopathy is diagnosed on the basis of the presence of cotton wool spots (soft exudates), intraretinal microvascular anomalies and venous loops with arterial occlusion. Soft exudates occur when there is infarction of the nerve fibre layer. Intraretinal microvascular anomalies result from dilated capillary beds from capillary occlusion. Continued retinal ischaemia and hypoxia resulting from small vessel occlusion triggers the release of angiogenic factors such as vascular endothelial growth factor (VEGF). Neovascularization of the retinal vasculature leads to proliferative retinopathy. These newly formed vessels are vulnerable to rupture, leading to intraocular vitreal haemorrhages and visual loss. The neovasculature is associated with fibrous tissue that may cause traction on, and detachment of, the retina or cause haemorrhage from vessel tear. New vessel formation on the iris is known as rubeosis iridis and may lead to glaucoma. Proliferative retinopathy is more common in type 1 diabetes with a prevalence of ~ 25% after 15 years.
Tight glycaemic and blood pressure control reduces the incidence and progression of diabetic retinopathy. However, improving glycaemic control should be a gradual process as a transient worsening of retinopathy has been reported with rapid improvements in glycaemic control. Smoking should be discouraged. Treatment with xenon or argon laser photocoagulation is effective in reducing visual loss in proliferative retinopathy and maculopathy. The photocoagulation aims to destroy hypoxic retinal tissue. Pan-retinal photocoagulation may be performed in patients with severe proliferative retinopathy. Focal laser photocoagulation is applied to macular oedema and leaking vessels. However, this treatment cannot reverse vision that has already been lost prior to treatment. Following therapy, some patients experience visual field constriction and night blindness. Vitrectomy may be carried out in patients with vitreous haemorrhage and this improves visual recovery. New therapies for macular oedema currently under investigation include intravitreal steroid injections and use of VEGF antagonists. Intravitreal steroid injections may be used where there is persistent loss of vision and when conventional treatment has failed. This therapy is effective but the adverse effects include an increased risk of cataracts and raised intra-ocular pressure. Concerns about chronic VEGF inhibition relate to neurotoxicity and thromboembolic events, such as myocardial infarction, stroke and thromboembolism.
Other complications
Brittle diabetes
Brittle diabetes is a term with no universally accepted definition. In 1977, Tattersall used a clinical definition, describing ‘The patient whose life is constantly being disrupted by episodes of hypo- or hyperglycaemia, whatever their cause’. This definition has been modified by various workers.
Causes of brittle diabetes include: psychological abnormalities, such as eating disorders (e.g. bulimia), personality disorders, communication disorders and manipulative behaviour. Such factors are often inferred if good glycaemic control can be achieved in hospital by the use of intravenous or nurse-administered subcutaneous insulin. If intravenous insulin therapy fails to achieve good glycaemic control, then an organic cause should be sought. These include: an unsuitable insulin regimen, intercurrent illness such as thyroid disease, Addison disease, systemic lupus erythematosus (through antibodies to insulin or its receptor), disorders of intestinal motility (erratic absorption of food) and interactions with other drugs (prescribed or non-prescribed). Problems related to insulin injections, such as faulty technique, abnormalities of local anatomy (e.g. lipohypertrophy) or subcutaneous tissue blood flow (e.g. during exercise) and changing insulin requirements in the various phases of the menstrual cycle, are well-recognized causes.
Type 4 renal tubular acidosis
Hyporeninaemic hypoaldosteronism (type 4 renal tubular acidosis, RTA) may be a manifestation of diabetic nephropathy, particularly in older patients. It presents with a hyperchloraemic, hyperkalaemic metabolic acidosis, which is out of proportion to any impairment in renal function. Type 4 RTA is frequently unmasked by inhibition of the renin–angiotensin system by ACE inhibitors or ARBs, particularly when used in combination with other agents that may increase plasma potassium concentrations (e.g. spironolactone). The precise nature of the defect in this condition is unclear. Failure of renin secretion to increase in response to posture or sodium restriction suggests an interstitial (juxtaglomerular) defect, but no anatomical correlate has been described to account for this. The failure of aldosterone release to be stimulated directly by the resulting hyperkalaemia suggests the possibility of concomitant dysfunction of the adrenal zona glomerulosa, although this may simply be a consequence of chronic hyporeninaemia.
When hyperkalaemia is modest and stable (serum potassium concentration < 6.0 mmol/L), close observation may be all that is required, but more marked elevations demand measures such as stopping offending drugs (ACE inhibitors/ARBs, β-blockers, non-steroidal anti-inflammatory drugs, potassium-sparing diuretics etc.); prescription of a low potassium diet (potassium restriction); use of potassium-wasting diuretics (thiazides, loop diuretics) and/or sodium bicarbonate (although this risks causing oedema). A differential diagnosis of autoimmune hypoadrenalism may require exclusion (especially in younger subjects with type 1 diabetes). In the occasional particularly severe and refractory case, treatment with fludrocortisone may be necessary.
EMERGENCIES IN DIABETES
General medical emergencies (e.g. stroke) in persons with diabetes are usually treated much in the same way as in subjects without diabetes, although frequent monitoring of blood glucose is necessary. Specific diabetic emergencies are discussed as specific entities, although mixed forms may occur. Diabetic emergencies are common in clinical practice and the approach to such patients requires an accurate diagnosis of the condition and appropriate management.
Diabetic ketoacidosis
Diabetic ketoacidosis (DKA) is a serious complication of type 1 diabetes and can be life-threatening. It is characterized by hyperglycaemia, acidosis and ketonaemia. Approximately 30% of patients with type 1 diabetes mellitus present with ketoacidosis. Fortunately, the onset in newly presenting patients tends to be slower than in known diabetic subjects, perhaps because of retention of some residual insulin secretion. An infection is found in ~ 35–55% of patients with ketoacidosis. Errors or omissions in treatment account for another 30%. The clinical features include dehydration, shock, vomiting, abdominal pain, acidosis (with compensatory Kussmaul respiration and, in some cases, a breath odour of ketosis) and impaired consciousness.
Four mechanisms predispose to ketoacidosis: insulin deficiency, counter-regulatory hormone excess, fasting and dehydration. Of these, the most important is insulin deficiency. Mechanisms of the development of DKA are shown in Figure 16.1. In a vicious cycle, hyperglycaemia and excess lipolysis cause dehydration and high circulating concentrations of NEFAs. The resulting acidosis and dehydration further increase the secretion of counter-regulatory hormones. The acidosis, dehydration, counter-regulatory hormones and excess lipolysis induce insulin resistance, thereby increasing hyperglycaemia and lipolysis. Protein catabolism and hypertriglyceridaemia also occur and compound the situation.
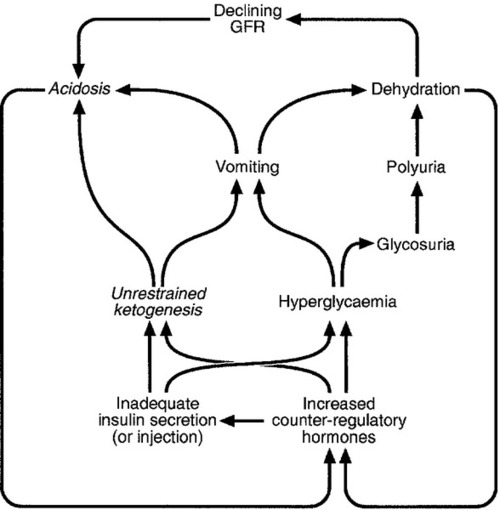
FIGURE 16.1 Vicious circles in ketoacidosis and hyperosmolar states. In both conditions, the trigger event(s) causes an initial inadequate insulin supply and hyperglycaemia. In ketoacidosis, both acidosis and dehydration cycles occur. Features in italics are restricted to ketoacidosis. GFR, glomerular filtration rate.
Rarely, patients may present with normoglycaemic ketoacidosis; typically, this occurs in situations where exercise, starvation or, occasionally, infection is the major precipitant of ketosis, but more often it is seen when a patient has taken enough insulin in a bolus to bring the blood glucose down but has not been exposed to sufficient insulin over a long enough period to suppress ketogenesis while existing ketones are cleared. The important differential diagnosis of alcoholic ketoacidosis should be considered under these circumstances (see below), particularly when a malnourished patient embarks on heavy alcohol consumption for a period of several days. Despite the unfortunate use of the term ‘diabetic coma’, it is rare for a patient with DKA to have true coma as opposed to some clouding of the sensorium. Where this occurs, alternative causes such as bacterial meningitis or a cerebrovascular event should be actively considered in the differential diagnosis.
Biochemical features
Biochemical features of DKA include hyperglycaemia, ketosis (ketonaemia and ketonuria), metabolic acidosis and uraemia. Creatinine concentrations may not accurately be measurable in the presence of heavy ketonaemia. The characteristic ketosis is a consequence of increased lipolysis and decreased fat synthesis. Excess acety1-CoA derived from β-oxidation of fatty acids is converted to the ketone bodies, acetoacetate and β-hydroxybutyrate with some decarboxylation of the former to acetone.
Bedside tests may demonstrate high blood glucose concentrations, glycosuria and ketonuria. Plasma β-hydroxybutyrate concentrations are typically three times higher than those of acetoacetate and the ratio may be even greater if there is also an element of lactic acidosis, so that the degree of ketosis may be underestimated or not recognized by urine tests for acetoacetate (see Chapter 15). This is also important in charting the recovery from ketoacidosis where ketonuria (detection of acetoacetate) may increase at a time when the patient is, by all other measures, improving and ketonaemia resolving. This effect occurs as mitochondrial redox state improves under conditions of increased tissue oxygen delivery and reduced acidosis, favouring the formation of acetoacetate from β-hydroxybutyrate. Some typical biochemical results in ketoacidosis are shown in Box 16.2.
BOX 16.2
Typical initial laboratory values in diabetic ketoacidosis
• Plasma [glucose] ~ 35 mmol/L
• Plasma [K+] > 5.3 mmol/L: whole body depletion typically 6.0 mmol/kg body weight
• Plasma [Na+] ~ 130 mmol/L: whole body depletion typically 8.0 mmol/kg body weight
• Plasma [urea] > 15 mmol/L
• Plasma [creatinine] > 150 μmol/L (if possible to measure in the presence of heavy ketonaemia)
• Plasma [ketones] (β-hydroxybutyrate and acetoacetate) > 15 mmol/L
• Plasma [Mg2 +] < 0.70 mmol/L: whole body depletion typically 0.5 mmol/kg body weight
• Plasma [phosphate] > 1.2 mmol/L: whole body depletion typically 1.0 mmol/kg body weight
• Plasma amylase 500–1000 U/L
• Plasma osmolality ~ 325 mmol/kg: whole body water depletion typically 75–100 mL/kg body weight, i.e. 7 L in typical adult
• Arterial blood gases:
• [H+] > 50 nmol/L (pH < 7.30)
• PaCO2 < 3.52 kPa
• PaO2 > 12 kPa
• 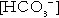 < 18 mmol/L
< 18 mmol/L
• Anion gap > 20 mmol/L
> 20 mmol/L
Management
The key to successful management of ketoacidosis is early institution of therapy and repeated clinical and biochemical assessment of the patient. The essential elements of the treatment of ketoacidosis are to replace fluid losses and to reverse the underlying metabolic disorder with continuous intravenous infusion of insulin. Ketone clearance occurs mainly through renal excretion and oxidation.
General measures
As with other medical emergencies, initial stabilization with attention to airway, breathing and circulation should precede detailed assessment. It is recommended that venous (rather than arterial) blood gases and hydrogen ion concentration (pH), and bicarbonate and potassium concentrations should be used to guide management. Appropriate investigations including blood culture and chest X-ray should be undertaken where infection is suspected, but the finding of a neutrophil leukocytosis does not, of itself, imply its presence. Although abdominal pain and a slightly elevated amylase activity do not necessarily point to the presence of pancreatitis or other intra-abdominal emergency, the possibility that DKA may have been precipitated by abdominal disease should be borne in mind.
Fluids
Fluid replacement aims to restore circulatory volume (and thereby increase the rate of ketone clearance) and correct electrolyte imbalances. There is disagreement as to the optimal rate and type of fluid replacement in DKA, partly based on fears of provoking cerebral oedema. In the UK, 0.9% saline is commonly used as the fluid of choice for initial replacement as this is available readily and comes with ready mixed potassium at the required concentrations if this is required. The disadvantage of using 0.9% saline is that of hyperchloraemic metabolic acidosis, which may lead to a slower recovery from acidosis because of renal vasoconstriction. Hartmann’s solution is often less readily available and contains a fixed, relatively low, potassium concentration.
Paediatric guidelines recommend cautious fluid replacement over 48 h. For adults, initial fluid replacement is usually rapid in the first few hours, but this should be done with caution in young adults (see below, where their greater risk of cerebral oedema is explained).
When blood glucose concentrations fall below 14 mmol/L, it is important to commence an infusion of 10% glucose to prevent hypoglycaemia while the insulin infusion is continued (normoglycaemia is often attained before the ketosis and acidosis resolves).
United States’ authorities advocate a ‘correction’ of measured serum sodium concentration for the dilution that results from osmotic movement of intracellular water to the hyperglycaemic interstitial and intravascular compartments. Various formulae have been suggested, but perhaps the most widely accepted is to add 1.6 mmol/L to measured serum sodium concentration for every 5 mmol/L that the plasma glucose concentration exceeds 5 mmol/L. If the corrected sodium starts to rise above the reference range, a switch to 0.45% saline should be considered. The possibility that the hypertriglyceridaemia sometimes encountered in DKA may give rise to pseudohyponatraemia should also be considered, particularly if the plasma is visibly lipaemic.
In general, fluid replacement of ~ 6 L over the first 24 h with attention to correcting shock very rapidly at the outset, and thereafter reducing the replacement rate, is appropriate for most patients. Polyuria, if still present, will not abate immediately after the start of therapy, so it is important to infuse enough fluid to achieve a positive fluid balance.
Insulin
The objective is to provide sufficient insulin for suppression of ketogenesis without causing precipitate falls in blood glucose or unnecessary hypokalaemia as a result of the insulin-driven intracellular movement of potassium. In practice, the insulin infusion rate required to achieve this objective will vary between patients, and over time in the same patient, as ketone clearance reduces ketone body-induced insulin resistance. However, a starting rate of 0.1 U/h per kg body weight is typical. The rate can be increased if recovery is slow. Initial bolus insulin doses are no longer recommended.
Patients who are normally on a long-acting basal insulin (detemir or glargine) should continue on their subcutaneous injections in order to provide a basal supply of insulin; this will facilitate better glycaemic control when they eventually revert back to their normal subcutaneous insulin regimens when the intravenous insulin infusion is discontinued.
Previously used variable insulin infusion rates (‘sliding scales’) are no longer used. They were designed to correct blood glucose as rapidly as possible, rather than to permit sufficient insulin to be infused to suppress ketogenesis for as long as it takes to oxidize or excrete the ketones already present in the circulation. The danger was, therefore, that large insulin doses were given in the first few hours, resulting in either too rapid a fall in blood glucose, and the need to switch to 10% glucose before sufficient saline replacement had been given or in a need to reduce the infusion rate to a level that is insufficient to suppress ketogenesis.
It should be noted that the major determinants of blood glucose reduction early in DKA are expansion of its volume of distribution and restoration of a normal glomerular filtration rate, rather than insulin-mediated glucose uptake into insulin-sensitive cells.
Potassium, magnesium and phosphate
Although whole body potassium depletion is universal in DKA, the initial serum potassium concentration may be low, normal or high. Potassium should not be given until it is clear that the patient is passing urine and renal function has been assessed. Potassium concentrations may fall precipitously with insulin treatment and frequent monitoring is necessary, especially during the early stages of therapy. In general, initial replacement rates of 5–20 mmol/h (typically 10 mmol/h), depending on the serum potassium concentration, are appropriate if the serum potassium is within the reference range. Replacement should be withheld if the potassium concentration is > 5.5 mmol/L; if it is < 3.3 mmol/L, a higher rate of infusion may be required (with cardiac monitoring), and insulin infusion may need to be suspended.
Phosphate and magnesium concentrations may also fall after the start of treatment. Although there is no hard evidence to support the routine use of phosphate replacement in DKA, there is good reason to suppose that severe hypophosphataemia (i.e. < 0.35 mmol/L) may be deleterious. Consequences of severe hypophosphataemia include respiratory and other muscle weakness, haemolytic anaemia and reduced cardiac systolic function; these probably result from reduced generation of ATP because of substrate limitation. Severe hypophosphataemia is more likely to occur when DKA is superimposed upon chronic malnutrition. It seems reasonable to suggest that severe, symptomatic, hypophosphataemia should be treated under such circumstances, e.g. with 20 mmol in 1 L intravenous fluid over several hours. Some authorities recommend that potassium replacement be given as a mixture of the chloride (2/3) and phosphate (1/3) salts throughout.
Bicarbonate
The use of bicarbonate to correct acidosis is not recommended. This is because of the risks of hypokalaemia, hypocalcaemia, paradoxical increase in cerebrospinal fluid acidosis, worsening intracellular acidosis and hypoxia (via a left shift in the oxygen dissociation curve), a possible increase in cerebral oedema risk (see below), delay in the fall in blood lactate concentration and increased ketogenesis.
Cerebral oedema
Cerebral oedema is one of the most feared complications of ketoacidosis, occurring particularly frequently in children and adolescents in whom it is the leading cause of death in the condition. It occurs in ~ 1% of cases of DKA in children and adolescents and carries a mortality of up to 90%, despite aggressive treatment with mannitol, intubation and mechanical ventilation. The most consistent associations with the subsequent development of cerebral oedema are more severe acidosis at presentation, the larger amounts of insulin administered in the first hour and higher volumes of fluid given in the first four hours. Contrary to popular suspicion, no association with initial serum sodium concentration (whether or not corrected for glucose) has consistently been shown, although it has been suggested that those who develop cerebral oedema may have a tendency to lower corrected sodium concentrations as treatment progresses.
Resolution
Once serum bicarbonate exceeds 18–20 mmol/L, the focus of management should revert to control of blood glucose, and intravenous insulin is continued using a sliding scale. Subcutaneous insulin is recommenced once the patient is eating and does not require intravenous insulin for other reasons (e.g. sepsis). As discussed above, an apparent increase in ketosis, on the basis of urine dip-stick testing for ketones, is not necessarily significant, and need not direct therapy at this stage if all other parameters are satisfactory.
After an episode of ketoacidosis, patients should always be considered insulin dependent and treated with insulin (even if they were not originally). Steps to reduce the risk of further episodes should be taken if appropriate (e.g. reinforcement of education, change of therapy).
Hyperosmolar hyperglycaemic state
Presentation and clinical features
The hyperosmolar hyperglycaemic state (HHS) was formerly known as hyperosmolar non-ketotic hyperglycaemia. The clinical features are dehydration, severe hyperglycaemia (blood glucose usually > 30 mmol/L) and a hyperosmolar state (serum osmolality > 330 mmol/kg). By definition, ketosis is not significant with urine ketones of less than ‘+’ or blood ketones < 1.0 mmol/L. Blood hydrogen ion concentration is usually < 50 nmol/L (pH > 7.3) with a normal bicarbonate, although coexistent lactic acidosis from sepsis or myocardial infarction may lead to more severe acidosis. Patients may present with osmotic symptoms such as polyuria, polydipsia, thirst, weight loss and vomiting, and are dehydrated. Drowsiness and coma may occur as a result of the high plasma osmolality; inadequate volume replacement leads to acute kidney injury, and mortality is higher than in DKA.
The HHS occurs mainly in older subjects with type 2 diabetes and most cases occur above the age of 60 years. About half of patients have not been previously diagnosed with diabetes. Many cases arise from late presentation of diabetes with coincident cardiovascular events or infections (most commonly pneumonia or urinary tract sepsis). The cycle of hyperglycaemia, dehydration and increased counter-regulatory hormones, coincident with a history of osmotic symptoms, is similar to that which occurs in DKA, but may be more severe because of later presentation owing to the absence of significant ketoacidosis. The characteristic hypernatraemia (see Box 16.3) is caused by renal sodium resorption in response to hypovolaemia, together with osmotic diuresis and glycosuria causing persistent free water loss. The HHS usually occurs in subjects with only marginal insulin deficiency, and their insulinaemia allows sufficient glucose uptake and anti-lipolytic effect to prevent the lipolytic and ketotic problems seen in ketoacidosis.
BOX 16.3
Typical initial laboratory values in the hyperosmolar hyperglycaemic state
• Plasma [K+] ~ 4.0 mmol/L: whole body depletion typically 10.0 mmol/kg body weight
• Plasma [Na+] ~ 155 mmol/L: whole body depletion typically 8.0 mmol/kg body weight
• Plasma [urea] ~ 55 mmol/L
• Plasma [creatinine] ~ 400 μmol/L
• Whole body water depletion typically 80–120 mL/kg body weight, i.e. 8 L in typical adult
• Arterial blood gases:
• [H+] < 50 nmol/L (pH > 7.30)
• PaCO2: normal
• PaO2: normal (low if venous thromboembolism or pneumonia)
•  ~ 18 mmol/L (typically mildly elevated blood lactate)
~ 18 mmol/L (typically mildly elevated blood lactate)
• Anion gap < 20 mmol/L
Management
The management of HHS is similar to that of DKA. Fluid replacement is the mainstay of treatment but careful monitoring is necessary in order to avoid fluid overload, which may result in complications such as pulmonary oedema. Insulin requirements are usually less in HHS than in DKA. The plasma glucose concentration typically declines relatively quickly after the institution of insulin therapy, since patients are usually less acutely insulin-resistant than ketoacidotic patients. Rehydration will expand the volume of distribution of glucose, lowering its concentration, and restoration of a good urine output will permit glucose to be excreted in the urine in significant quantities (up to some 250 mmol/L). Together with insulin-independent uptake into tissues, the result is that blood glucose concentration tends to fall fairly rapidly with initial resuscitation, even if no insulin is administered. Rapid falls in plasma glucose or osmolality may predispose to cerebral oedema by encouraging the movement of water from plasma into the central nervous system, and most authorities recommend that the blood glucose concentration should if possible be reduced at a rate of no more than ~ 3 mmol/L/h. For this reason, insulin infusion is only required at very low rates (e.g. 0.5–1 U/h) in the initial stages of treatment. Controversy exists as to the ideal choice of fluid replacement. While some advocate the use of hypotonic fluids (e.g. 0.45% saline) after restoration of adequate intravascular volume, others argue that normal (0.9%) saline is, in effect, hypotonic to the patient’s plasma and is less likely than hypotonic saline to cause rapid falls in plasma osmolality that may increase the risk of cerebral oedema.
One of the major problems in the treatment of HHS is the severe hypernatraemia that often supervenes in the hours after the initiation of therapy. Commencing treatment with fluid and insulin usually induces a decrease in plasma glucose and urea concentrations, and, therefore, osmolality. However, hypernatraemia typically develops during this period, since the falling osmolality allows water to re-equilibrate into dehydrated cells while free water loss through the kidneys continues. Since there is less acidosis and more profound dehydration, plasma potassium deficits are typically less than those occurring in DKA. The intravascular volume depletion increases plasma viscosity, resulting in a hypercoagulable state and stroke, venous thrombosis and myocardial infarction are common causes of mortality. Heparin is, therefore, recommended as prophylaxis against thrombosis. Comorbidities that may have precipitated the episode should be treated; for example, infections may require antibiotic treatment.
After successful treatment of HHS, the severity of residual glucose intolerance may be very mild and the patient can often be successfully managed on diet alone, although many will be discharged on insulin initially. Patients should be educated on steps to avoid provoking factors in the future. At all stages, it is important to involve the diabetes team in the management of patients with HHS.
Other metabolic acidoses
In diabetic subjects, the vast majority of episodes of acidosis are diabetic ketoacidosis. Other metabolic acidoses are comprehensively discussed in Chapter 5. The differential diagnosis of a metabolic acidosis in a diabetic subject may occasionally be troublesome if one forgets that patients with diabetes may sometimes develop acidosis as a result of other conditions, such as severe infection, renal failure and salicylate poisoning. Diagnostic problems may also arise where there is a mixed picture (e.g. diabetic ketoacidosis and lactic acidosis secondary to sepsis, or alcoholic ketoacidosis occurring in a patient with diabetes). Metformin-associated lactic acidosis and type 4 renal tubular acidosis have been discussed earlier in this chapter.
Alcoholic ketoacidosis
Alcoholic ketoacidosis is included here, as it occasionally presents a difficult differential diagnosis from DKA (particularly given that the latter may occasionally present with an only marginally elevated blood glucose concentration) and the treatment is different in several important regards. This condition presents with ketoacidosis and low, normal or slightly elevated plasma glucose concentrations (rarely > 12 mmol/L). Patients usually have longstanding poor nutritional intake with an alcohol binge of several days’ duration, often associated with vomiting.
Ketosis is promoted by lack of insulin action in the setting of starvation, permitting mobilization of NEFAs that provide the substrate for ketone body formation as an alternative fuel. Low insulin concentrations prevent ketone body utilization by insulin-sensitive tissues and dehydration impairs ketone excretion by kidneys. Hypercortisolaemia and high growth hormone and catecholamine concentrations enhance the fatty acid release and hepatic ketogenesis consequent upon insulin deficiency.
Ketogenesis is potentiated by inhibition of hepatic metabolism of acetylcoenzyme A, owing to the action of counter-regulatory hormones such as glucagon, cortisol and catecholamines secreted in response both to the hypoglycaemia and also to extracellular fluid volume contraction (which also restricts urinary ketone elimination). In addition, alcohol metabolism depletes cellular nicotinamide adenine dinucleotide (NAD+). An increased ratio of NADH to NAD+increases the proportion of β-hydroxybutyrate relative to acetoacetate. It also restricts pyruvate formation from lactate, leading to accumulation of lactate and depletion of pyruvate, a gluconeogenic substrate. As is the case in DKA, alterations in mitochondrial redox state favour β-hydroxybutyrate over acetoacetate production. A complex acid–base disorder ensues from the combined effects of ketosis causing metabolic acidosis, and a combination of extracellular fluid contraction and vomiting causing metabolic alkalosis, with the final [H+] (pH) thus not necessarily reflecting the severity of the metabolic derangements present. Treatment differs from diabetic ketoacidosis in that insulin is not required (except if patients have coexistent diabetes), and glucose infusion (which must be preceded by thiamine repletion) forms the mainstay of therapy, together with fluid and electrolyte replenishment.
MANAGEMENT OF DIABETES IN THE HOSPITAL SETTING
The management of diabetes in the hospital setting presents a number of challenges and has the potential to affect patient outcomes. Good glycaemic control has an established role in promoting recovery from infection, improving wound healing, and, in obstetric delivery, to prevent neonatal hypoglycaemia. In the first Diabetes Mellitus Insulin–Glucose Infusion in Acute Myocardial Infarction (DIGAMI 1) Study, insulin-based compared with conventional glucose-lowering management improved survival in patients with acute myocardial infarction and type 2 diabetes. The benefit of insulin treatment was not confirmed in the second DIGAMI trial (DIGAMI 2), in which insulin was associated with an increased risk of nonfatal cardiac events, while metformin appeared to be protective against risk of death. Therefore, the type of glucose-lowering treatment used may influence the clinical outcome.
There is mixed evidence concerning mortality and the use of intensive insulin therapy in order to achieve tight glycaemic control in the hospital setting. Recent data suggest that the use of intensive insulin therapy does not reduce mortality or length of hospital stay compared with less strict control, and may increase the risk of hypoglycaemia. This may reflect the increased prevalence of ‘stress hyperglycaemia’ in hospitalized patients, or be because altered insulin action per se in the seriously ill affects the prognosis.
Many patients admitted to hospital will require changes to be made to their diabetes treatment, at least temporarily. These changes may be made necessary by the need to fast, the use of agents such as glucocorticoids or inotropes (which can directly affect blood glucose concentration), when receiving radiographic contrast material (requiring care in the use of metformin), where there is temporarily increased risk of acute kidney injury or other conditions associated with lactic acidosis (also with metformin, see Table 16.1) or during myocardial ischaemia (possibly posing a risk with some sulfonylureas). Some patients whose blood glucose concentrations are elevated during an acute illness, but who either were not previously known to have diabetes or whose blood glucose concentrations subsequently return to levels below the threshold for diabetes diagnosis may have ‘stress hyperglycaemia’.
Hospitalization presents the opportunity for a review of overall diabetes management and education: indeed, for some patients, such an opportunistic intervention may represent their only formal contact with diabetes services. It also presents an opportunity to screen for diabetes in a relatively high-risk population.
The perioperative management of patients with diabetes is to minimize changes in glycaemia brought about by the stress of surgery. Most hospitals will have formal protocols for glucose control pre- and post-surgery. Patients with type 1 diabetes will need to be on insulin at all stages to prevent diabetes ketoacidosis. Patients with type 2 diabetes will usually be able to manage on their oral agents, but may require insulin depending on the nature of the surgical procedure. Following surgery, once oral intake is adequate, most patients will be able to recommence their original treatment regimens.
PREGNANCY
For both mother and fetus, diabetes can profoundly affect the pregnancy in terms of metabolism and outcomes. Specific factors influencing the management of women who are pregnant include the presence of coexisting illness, whether they have gestational or pre-existing diabetes and whether they are likely to deliver in the relatively near future.
With pre-existing diabetes, appropriate preconceptual counselling and care is vital if a baby is planned so that glycaemic control and blood pressure are optimized and medications not recommended in pregnancy (for example statins, ACE inhibitors) are replaced. The ADA recommends a preconception HbA1c of < 53 mmol/mol (7%). Lipid control is by dietary measures. Folic acid is prescribed to reduce the risk of neural tube defects. These measures increase the chances of successful conception and lower the risk of complications such as congenital anomalies and miscarriage (Boxes 16.4 and 16.5).
In pregnancy, regular surveillance is important: glycaemic control should be monitored with regular measurement of blood glucose concentrations. Pregnancy induces a state of insulin resistance, leading to hyperglycaemia. This can lead to fetal malformation, macrosomia with attendant delivery complications, increased perinatal mortality and premature delivery. Strict glycaemic targets are set which increases the risk of hypoglycaemia. Retinal screening is important in patients because of the increased risk of progression of diabetic retinopathy. Tight preprandial and postprandial glycaemic targets are advised: NICE recommendations are < 5.9 mmol/L fasting and < 7.8 mmol/L after meals.
Insulin requirements generally rise during pregnancy because, as the pregnancy progresses, there is a reduction in insulin sensitivity, driven by human placental lactogen. This change serves to divert nutrients and the products of maternal lipolysis (fatty acids and ketones) to the fetus. It is important to recognize that a sudden fall in usual insulin requirements may be a sign of changes in placental function that herald impending delivery, and close liaison with the obstetric team is required.
During labour, it is important to monitor the mother’s blood glucose concentrations; maternal hyperglycaemia is treated by intravenous insulin and dextrose infusions. On delivery of the placenta, insulin resistance dramatically declines towards non-pregnant values. Patients with type 1 diabetes and those with type 2 diabetes who were previously on insulin pre-pregnancy revert to their pre-pregnancy insulin regimens. Those with pre-existing type 2 diabetes previously on oral glucose-lowering treatment who wish to breast feed may wish to continue on either metformin or insulin (if they were taking this during pregnancy). Patients with gestational diabetes or diet-treated pre-existing type 2 diabetes do not require any medication postpartum, unless other factors, for example sepsis or surgery, dictate otherwise. Metformin is safe whilst breastfeeding. Arrangements should be made for women with gestational diabetes to have an oral glucose tolerance test approximately six weeks postpartum if pre-discharge blood glucose values are satisfactory. Women with gestational diabetes have an approximately 50% lifetime risk of developing type 2 diabetes, and should be advised on diabetes prevention and recognition as well as being screened regularly for life.
CONCLUSION
Successful clinical management of diabetes mellitus requires an understanding of the effects of diet and exercise on glycaemic control. Type 2 diabetes is responsible for most cases of diabetes, and as ~ 80% of patients will die from macrovascular disease, it is essential to provide optimal management of all cardiovascular risk factors, including dyslipidaemia and hypertension.
The long-term management of patients with diabetes also aims to reduce the risk of the microvascular complications that affect the kidneys, nerves and eyes. Clinicians need to be skilled in strategies that reduce these risks and in the identification and management of complications when they occur.
Optimization of treatment with the growing range of hypoglycaemic agents depends in part on having a good understanding of their mechanisms of action. Advances in the formulation of insulin and production of insulin analogues also offers patients the possibility of improved glycaemic control. Effective management of blood glucose concentrations reduces risks of chronic complications and is particularly important during intercurrent severe illness and pregnancy.
The acute diabetic emergencies of diabetic ketoacidosis and the hyperosmolar hyperglycaemic state are still associated with significant morbidity and mortality. Their management depends on careful adherence of treatment protocols that are based on clear pathophysiological principals. Appropriate, frequent biochemical monitoring of patients with these emergencies is essential to a successful outcome.
ACKNOWLEDGEMENT
The authors wish to acknowledge the contributions of Dr Victor Lawrence and Dr Simon Coppack, the authors of this chapter in the second edition of this book.