CHAPTER 14
Acute and chronic liver disease
Adrian Bomford; Roy A. Sherwood
CHAPTER OUTLINE
CLASSIFICATION OF LIVER DISEASE
ACUTE HEPATITIS AND ITS SEQUELAE
Laboratory criteria for liver transplantation
Differential diagnosis of chronic hepatitis
PRIMARY SCLEROSING CHOLANGITIS
Liver pathology in alcoholic liver disease
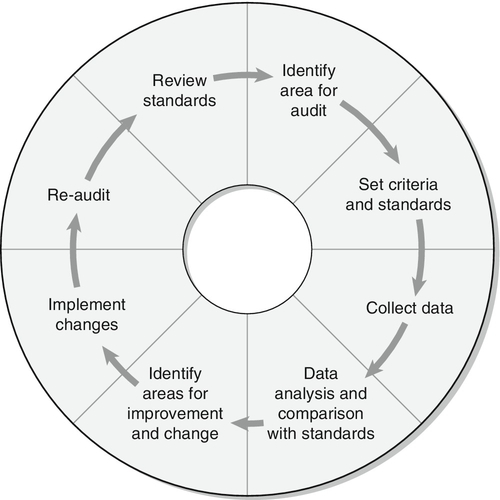
Use of laboratory tests in clinical practice
Non-alcoholic fatty liver disease
Vascular disturbances in cirrhosis
Sex hormones and their binding proteins
NEOPLASTIC DISEASE OF THE LIVER AND BILIARY TRACT
Hepatocellular carcinoma and α-fetoprotein
INHERITED METABOLIC DISORDERS INVOLVING THE LIVER
Iron overload and hereditary haemochromatosis
Other inherited metabolic diseases
CLASSIFICATION OF LIVER DISEASE
Liver disease is best classified according to its presumed aetiology (Box 14.1). This is then qualified by the pathological state of the liver where known or inferred: usually cirrhosis, inflammation (i.e. hepatitis) or cholestasis. Thus, terms such as alcoholic cirrhosis, viral hepatitis or cholestasis of pregnancy all conform to this classification. Some idea of the severity of the disease is given by the terms ‘compensated’ and ‘decompensated’. The term ‘compensated’ implies that, although the liver has been damaged it can, because of its large functional reserve, still function adequately. On the other hand, the term ‘decompensated’ implies that the liver is failing in some vital function. The terms are not precisely defined, but the development of ascites with or without peripheral oedema, jaundice or hepatic encephalopathy, in a patient known to have liver disease, would all be regarded as signs of decompensation. Finally, the presumed evolution of the disease in time is given by the terms fulminant, acute, subacute or chronic. In chronic liver disease, additional terminology often employed to provide an indication of severity, especially when assessing histological changes in the liver, includes the terms active or inactive and mild, moderate or severe.
ACUTE HEPATITIS AND ITS SEQUELAE
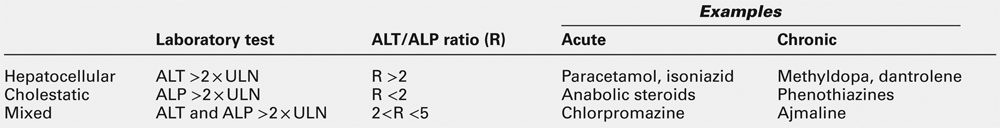
The term ‘hepatitis’ indicates that there is hepatic inflammation, for which there are many causes: viral infections, drugs and toxins (including alcohol) are common, while autoimmune causes are infrequently encountered in the primary care setting, but in a tertiary centre make up a substantial proportion of patients. The standard ‘liver function tests’ (LFTs) are useful to determine that hepatitis is present, how severe it is, to document the progression of the disease and to assess the response to therapy. However, their role in identifying the aetiology of the inflammation is limited.
When the patient presents with an acute hepatitis, the liver function tests characteristically show the so-called ‘hepatitic’ picture:
• a modest (less than twice the upper limit of the reference range) increase in plasma alkaline phosphatase (ALP) activity
• bilirubinuria and, in more severe cases, hyperbilirubinaemia, which is clinically detectable as jaundice when the total plasma bilirubin rises to > 40–50 μmol/L.
As a broad generalization, plasma activities of the aminotransferases reflect the severity of the disease. Certainly, patients who progress to fulminant hepatitis usually have exceptionally high plasma activities, with elevations of 20–40 times the upper reference limit. However, the relationship is not strictly quantitative, for lower activities (2–3 times the upper reference limit) can be found in individuals with severe hepatic necroinflammatory activity on histological examination of liver biopsy material.
Differential diagnosis
There are two aspects to this part of the diagnostic process. The first is to distinguish between viral hepatitis and other, non-viral, causes of a hepatitic illness and then, within each group, to identify the causative agent. Generally speaking, the standard LFTs are not helpful in separating viral from non-viral causes, and further serological testing, radiological imaging or histological examination will be required.
Plasma aminotransferase activities are not usually grossly raised in alcoholic hepatitis; in this condition, a ratio of AST:ALT of > 2 is characteristic, while in hepatitis due to other agents the ratio is usually < 2. In fulminant hepatitis, aminotransferase activities may be close to the reference ranges by the time the patient reaches hospital, emphasizing that results of LFTs can change rapidly with time and depend on the particular clinical course that ensues. Generally speaking, acute infection with hepatitis B virus is more severe than that caused by hepatitis A, and biochemical abnormalities are more protracted. Raised plasma IgM concentration and atypical lymphocytes are more characteristic of type A, but the standard LFTs do not permit distinction between the different types of viral hepatitis.
Acute viral hepatitis
The specific diagnosis is established by serological tests for the hepatitis viruses (Table 14.1). The extent to which individual patients require biochemical and serological investigation depends on the clinical situation. For example, in children, a clinical diagnosis can be established with reasonable certainty if the clinical features are compatible with hepatitis A (infectious hepatitis), particularly if the physician knows of local occurrence. In adults, particularly where there has been no contact with infectious hepatitis, LFTs should be performed together with appropriate serology.
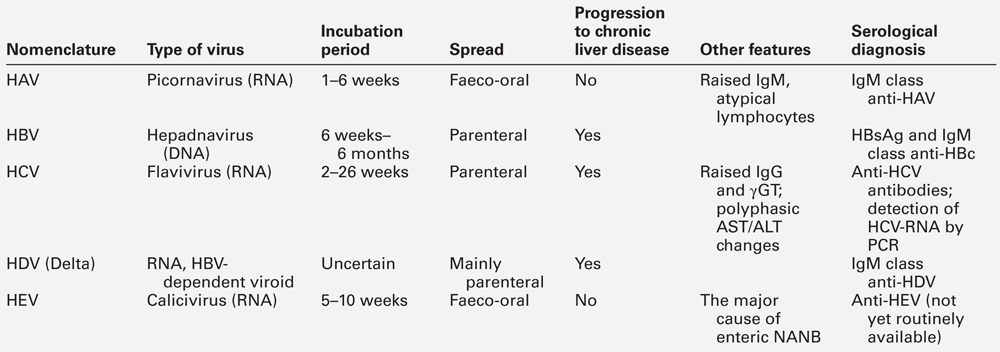
After a variable incubation period (the duration of which depends on the type of virus and the viral load), there is usually a rise in plasma aminotransferase activities, although some infants can be infected without any disturbance of LFTs. In many cases, particularly among young children, an elevated AST or ALT is the only indication of the hepatitic process and the patient remains asymptomatic. When symptoms do develop, they are coincident with the maximal aminotransferase activities. Taking infection with the hepatitis A virus as an example, the patient begins to feel unwell and lethargic and may become anorexic, with transient diarrhoea being frequent. Pyrexia can develop and the liver is found to be enlarged and tender. The urine may become dark (owing to bilirubinuria), the stools pale, and in more severe cases, jaundice becomes evident a few days later. Symptoms tend to resolve with the onset of jaundice, which usually subsides over a few days. Following infection with this virus, the patient may, despite being asymptomatic, enter a cholestatic phase. This is heralded by rising plasma activities of ALP and γ-glutamyltransferase (γGT) that can persist for many weeks and may cause considerable diagnostic confusion. Particularly in the older patient, it is an indication for ultrasound examination to rule out obstruction of the biliary tract. The jaundice usually begins to resolve before aminotransferase activities return to the reference range, but it has been a frequent observation that bilirubin may disappear from the urine while the patient remains clinically jaundiced. The explanation for this situation lies in the development of ‘bili-albumin’ (see p. 239).
Outcome of acute viral hepatitis
There are three main possible outcomes of acute viral hepatitis, each with its own characteristic progression of clinical features and pattern of LFTs.
Complete resolution
As noted above, the plasma aminotransferase activities start to rise before the onset of jaundice and often before hepatomegaly is clinically detectable. Activities begin to fall coincidentally with the onset of jaundice and symptoms, and return to normal (AST before ALT) at the same time as, or shortly before, the plasma bilirubin concentration. This is the natural history in the great majority of patients and, unless jaundice develops, many patients will be unaware that they have had an acute hepatitis.
Progression to chronic liver disease
This mode of progression is limited to viral hepatitis types B and C, but a relapsing course for hepatitis A is well described and there are occasional reports of type A triggering autoimmune hepatitis in susceptible individuals. Persistence of symptoms, signs and/or abnormal liver tests (particularly aminotransferase activity in the range of 2–10 times the upper reference limit) for more than six months constitutes, by definition, chronic hepatitis (see below). The accompanying changes in viral serology are complex and will not be considered in the present discussion.
Progression to acute liver failure
Very rarely (in < 1% of patients), any type of viral hepatitis may pursue a fulminant course. This course of events is characterized by grossly elevated plasma aminotransferase activities, an increasingly prolonged prothrombin time (PT) or international normalized ratio (INR) and the development of hepatic encephalopathy, which may occasionally develop before jaundice becomes a prominent feature. The prognosis is poor. The syndrome of acute liver failure (ALF), one cause of which is acute viral hepatitis, is described in more detail below.
ACUTE LIVER FAILURE
Acute liver failure implies the development of severe hepatic dysfunction within six months of the first onset of liver disease and in the absence of any pre-existing liver disease. Hepatic encephalopathy and a prolonged and persistent increase in PT are the characteristic features, and where these occur within eight weeks of the first symptom, the condition is known as fulminant hepatic failure (FHF). In those who survive, there are no long-term hepatic sequelae. The commonest cause in the UK is paracetamol overdose (Chapter 40) followed by viral hepatitis types B and E; ALF due to hepatitis C virus is extremely rare. Rarer causes include adverse drug reactions, other viruses, Amanita mushroom poisoning, Wilson disease, secondary hepatic malignancy, lymphoma and recreational drugs.
Laboratory features
Laboratory investigation is aimed at determining the cause of the ALF as well as assessing its consequences and complications (which are very similar irrespective of the aetiology). Measurement of the blood concentration of paracetamol is important, particularly in view of the beneficial effect of treatment by N-acetylcysteine infusion. The serology of viral causes is complicated because the hepatitis B surface antigen (HBsAg) may become undetectable before the patient reaches hospital. In general, with ALF due to causes other than paracetamol overdose, it is usually too late for specific treatment for the condition (e.g. D-penicillamine for Wilson disease) to be effective and management is by general supportive measures and, if feasible, liver transplantation.
The standard LFTs show a grossly hepatitic picture at the time of onset of the encephalopathy. Jaundice is deep and progressive and plasma aminotransferase activities of several thousand U/L are usually found, although these may have fallen considerably by the time the patient is sent to a referral centre. A rare cause of FHF is massive metastatic tumour infiltration. This is characterized by hepatomegaly (in contrast to the reduced-size liver found with most other causes of FHF) and a much more cholestatic picture – such that the AST:ALP ratio is < 4 rather than the converse, as in most other cases.
Coagulation defects are a consistent finding. Plasma concentrations of fibrinogen are decreased as are those of factors II, V, VII, IX and X. These are reflected in a prolonged PT or INR which, in the UK, is used as the main measure of severity of disease and is the most widely used parameter for following progress. Hypoglycaemia, owing to a combination of impaired gluconeogenesis and glycogen breakdown and synthesis, is such a consistent finding that glucose infusion is a routine part of management, particularly during transfer to a referral centre. Where paracetamol is involved, it may interfere with blood glucose estimation, resulting in erroneously high values. Acute kidney injury is a frequent and ominous complication that occurs in at least 50% of patients with FHF, particularly with paracetamol poisoning. In about one-half of patients, the renal lesion is acute tubular necrosis, and in the remainder it is ‘functional’ renal injury (see p. 261).
Laboratory criteria for liver transplantation
Currently, with the best supportive care, 30–50% of patients with fulminant hepatic failure will survive; the figures are rather better for those with paracetamol overdose and rather poorer for those with viral hepatitis, particularly those classified as non-A–E, or drug reactions. This poor prognosis has led to the introduction of liver transplantation as a therapeutic option. It is clearly important to reserve this option for those with the worst prognosis and who are most unlikely to recover with supportive treatment. With this in mind, a number of laboratory-based criteria have been identified to ascertain the likelihood of death. The major adverse factors are an arterial hydrogen ion concentration > 50 nmol/L (pH < 7.3), and progression to grade III coma (see p. 259) with an INR of > 10 and plasma creatinine concentration > 300 μmol/L. Recently, there has been considerable interest in the measurement of plasma factor V concentration. If the ratio of factor V to factor VIII is very low, the chance of survival without transplantation is very small.
CHRONIC HEPATITIS
Chronic hepatitis is usually defined as a condition in which clinical or biochemical features of liver disease persist for more than six months. However, it has become clear that many patients can have the condition for much longer periods before it manifests itself in this way. Indeed, occasionally, such individuals may present with what at first appears to be an acute hepatitis that, on further investigation, proves to be an acute exacerbation of a previously asymptomatic chronic process (see below). On histological grounds, the condition was previously classified into two categories: chronic persistent hepatitis (CPH), defined as inflammation confined to the portal tracts with no necrosis of hepatocytes, and chronic active hepatitis (CAH) characterized by inflammatory cells spilling out into the hepatic parenchyma and damaging periportal hepatocytes in a hallmark pattern described as ‘piecemeal necrosis’. Chronic persistent hepatitis was believed to be a benign condition of little clinical significance, with a good prognosis. Chronic active hepatitis, in contrast, was envisaged as a disorder with a more serious prognosis, which frequently progressed to cirrhosis, with the inflammatory activity often continuing even after cirrhosis developed (so-called ‘active cirrhosis’).
Although CPH and CAH were purely histological descriptions of the morphological changes seen in patients with chronic liver disease, they were embraced by hepatologists and gastroenterologists as clinical entities. However, during the 1970s and 1980s, it was increasingly recognized that the associated changes could be found in patients with several quite distinct causes of chronic hepatitis (Table 14.2). Somewhat surprisingly, rather than questioning the validity of CPH and CAH as distinct syndromes, this led to expansion of the list of diseases considered to be capable of causing them. By the 1990s, the previously accepted view that CPH was associated with a good prognosis was being challenged, because it became apparent that CPH and CAH are changes at either end of a spectrum of morphological changes seen in chronic hepatitis, and that transitions between them can and do occur during flares of disease activity and periods of remission, regardless of the aetiology of the process.
TABLE 14.2
Differential diagnosis of chronic hepatitis
| Type | Laboratory tests | Comments |
| Viral | ||
| type B | HBsAg | |
| type C | Anti-HCV; HCV-RNA by PCR | Has characteristic histological features |
| Alcoholic | Blood alcohol, γGT, MCV, desialylated transferrin | Laboratory tests are supplementary to clinical history |
| Drugs | May develop autoantibodies | Oxyphenisatin, methyldopa, isoniazid, dantrolene etc. |
| α1-Antitrypsin (AT) deficiency | Low α1-AT concentration; typical phenotype (PiZZa) | Characteristic eosinophilic globules on histology |
| Wilson disease | Low caeruloplasmin, high tissue copper and urinary copper concentration | Low ALP activity; low AST/ALT activity for degree of inflammation |
| Autoimmune | ||
| type 1 | Anti-SMA and/or ANA | Markedly raised γ-globulin and IgG |
| type 2 | Anti-LKM antibodies |
a For explanation, see α1-Antitrypsin deficiency on p. 268. ANA, antinuclear antibody; LKM, liver-kidney microsomal antibody; MCV, mean corpuscular volume; SMA, smooth muscle antibody.
Other criteria for CPH and CAH were also found to be untenable. A requirement for markedly elevated plasma aminotransferase activities for diagnosis of CAH could not be upheld because it was recognized that these enzymes are poor correlates of histologically assessed activity in chronic liver disease, and that mild elevations of aminotransferases do not exclude severe disease. The temporal criterion for duration of disease of at least six months (to distinguish chronic from acute liver disease) also proved difficult to apply because it is often not possible to define the time of onset and, as noted above, patients presenting with acute hepatitis who had clear evidence of chronic liver disease were being identified.
Current recommendations for defining and describing chronic hepatitis are that the terms CAH and CPH should be abandoned in favour of precise morphological descriptions, graded for necroinflammatory activity and staged for degree of fibrosis. While the term ‘piecemeal necrosis’ may be retained, the terms ‘periportal hepatitis’ or (preferably) ‘interface hepatitis’ should be used to describe the changes previously associated with CAH. Description of the changes previously associated with CPH should employ terms such as mild or moderate portal or periportal hepatitis (as appropriate) without significant necrosis. All of these terms should be qualified by aetiological designations (e.g. autoimmune hepatitis, chronic hepatitis B, C, D etc.), wherever possible and practicable.
Laboratory investigation plays a crucial role in the differential diagnosis and management of chronic hepatitis (see Table 14.2), although histological examination of liver biopsy material is usually required to assist in assessing severity and providing additional aetiological information. Typically, chronicity is defined as persistence of a hepatitic pattern of abnormal LFTs over several months. The plasma aminotransferase activities are usually elevated to 2–10 times the upper reference limits. Plasma alkaline phosphatase is often normal or only slightly raised, although higher values may be seen in patients where cirrhosis has developed with associated distortion of the hepatic architecture. Plasma bilirubin concentration is often also normal or mildly elevated but, in severe cases, there can be a profound hyperbilirubinaemia with jaundice. The PT/INR and other markers of coagulation are often mildly abnormal, and the plasma albumin concentration may be at the lower end of the normal range, reflecting decreased hepatic synthetic function. Marked hypoalbuminaemia is, however, a late feature associated with advanced cirrhosis. Depending on the aetiology of the chronic hepatitis, plasma immunoglobulin concentrations may also be elevated, sometimes quite markedly (see below).
The above abnormalities may be a sequel to an acute hepatitis, in which case the aminotransferase activities may have risen to 10–20 times the upper reference limits before falling back to the somewhat lower values more characteristic of chronic progression. However, a clinically evident acute phase may not have been apparent especially if the patient did not develop jaundice. Indeed, chronic hepatitis is not infrequently diagnosed incidentally during routine health screening or investigation of some other condition. Notably, profound fatigue is a common feature of all forms of chronic hepatitis, and the latter should, therefore, be considered in the differential diagnosis of chronic fatigue syndromes.
Differential diagnosis of chronic hepatitis
Viral hepatitis types B and C
The specific diagnosis is based on virological tests. The detection in serum of the hepatitis B surface antigen (HBsAg) indicates that the patient is infected with the hepatitis B virus (HBV). Clinical information and the results of LFTs can be useful in deciding whether the infection is acute, but this can be confirmed by the appearance of IgM antibodies against the HBV core antigen (HBc). The gradual disappearance of HBsAg, with sequential detection of IgG antibodies to HBc and antibodies to surface antigen (HBsAb) indicates clearance of the virus, an event that occurs in the majority of adults (90%) acutely infected with it. Other markers of viraemia are derived from different components of the virus, including hepatitis B ‘e’ antigen (HBeAg) and HBV DNA, and these disappear with viral clearance. The decline in HBe antigenaemia is accompanied by the appearance of antibodies to the ‘e’ antigen (HBeAb) and is known as seroconversion. In contrast to infection acquired in adulthood, perinatal infection becomes chronic in 95% of patients, with the virus persisting into adult life. Chronic infection is associated with a number of outcomes. Many individuals are asymptomatic, with the virus detected incidentally and a serological profile in which HBsAg and HBeAb are detected and HbeAg is negative. Although such people frequently have normal liver function tests, and have long been termed ‘healthy carriers’ and been considered to be of low infectivity, it has become clear that a significant proportion have marked histological damage on liver biopsy and detectable circulating HBV DNA. Others have clear evidence of cirrhosis and, in such individuals, there is a 5% annual risk of developing hepatocellular carcinoma.
Hepatitis C virus (HCV) infections are identified initially by detection of antibodies (anti-HCV) to the virus and chronic infections confirmed by seropositivity for the viral RNA (HCV-RNA).
Antiviral therapy for chronic hepatitis B and C, using interferons, with or without other antiviral agents to inhibit viral replication, is now well established, and comprehensive guidelines for treatment exist. Thus, tests for genomic material of both viruses (HBV-DNA and HCV-RNA, respectively) are usually performed, initially to assess the viral load before treatment and subsequently to monitor the patient’s response to treatment. Plasma aminotransferase activities are also usually monitored as an adjunct to the above, since they are cheap to perform on a frequent basis and a return to normal values suggests at least some response to treatment. In the case of HBV, successful treatment is heralded by a change from HBeAg to HBeAb positivity and this event is accompanied by an acute hepatitic illness, the so-called ‘hepatitic flare’ (Fig. 14.1). Hypothyroidism is a side effect of interferon therapy, particularly in patients with chronic hepatitis C, and thyroid function tests should be monitored before treatment and at 3-monthly intervals.
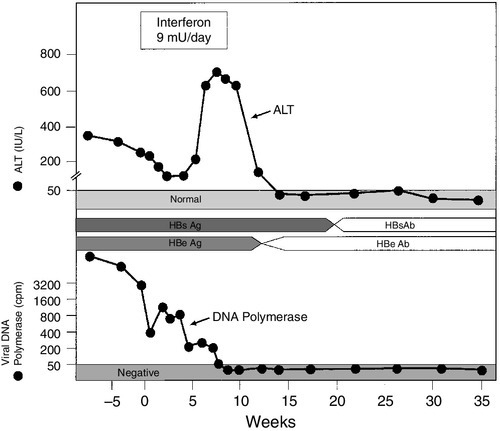
FIGURE 14.1 Changes in ALT activity in a patient with chronic hepatitis B virus infection successfully treated with interferon. Note the hepatitis response heralding clearance of HBsAg and appearance of the antibody. Clearance of the virus is also indicated by a decrease in viral DNA polymerase activity.
Procedures for the handling of potentially dangerous and infectious samples are not the concern of this chapter, but it is worth noting that as many as 1–2% of inner city populations may be carriers of either HBV or HCV. The majority of these will have no symptoms of liver disease and their blood samples may be referred to the laboratory for causes unrelated to liver disease. Caution should therefore be exercised in the handling of all blood products in view of the possibility of occult hepatitis viral infections.
Alcohol
In perhaps 10% of patients with alcoholic liver disease, the histological features of interface hepatitis, similar to those in other forms of moderate/severe chronic hepatitis can be observed. There is evidence that some such cases may be related to coexistent hepatitis C virus infection or other underlying causes. The problem that this presents for differential diagnosis is discussed in detail under the heading of alcoholic liver disease.
Wilson disease
Chronic hepatitis is a rare presentation of this rare disease (see p. 267), but is of considerable importance because treatment can be instituted and further liver damage avoided. Generally, the plasma activities of aminotransferases are low in comparison with the extent of histological activity. The diagnostic work-up is considered in detail below, but the initial screening procedures include measurement of plasma copper and caeruloplasmin concentrations and ocular slit-lamp examination for Kayser–Fleischer rings. The diagnosis must be considered in all patients with chronic hepatitis, particularly those in the younger age groups.
α1-Antitrypsin deficiency
This is another rare cause of chronic hepatitis (see p. 266). Plasma α1-antitrypsin concentrations are usually low but, occasionally, because of active inflammation, they may be within the reference range. It is therefore essential that the α1-antitrypsin phenotype is determined, particularly in younger patients.
Autoimmune hepatitis (AIH)
This is a rare cause of chronic hepatitis. However, it is one of the few causes of chronic liver disease in which drug treatment is highly effective in the great majority of patients, and accurate diagnosis is essential. This is established partly by careful exclusion of the other causes listed above and partly by the finding of a suggestive pattern of biochemical, immunological and histological abnormalities. The biochemical liver tests show a hepatitic pattern (see above), but plasma aminotransferase activities are often only moderately elevated and bilirubin concentrations are frequently normal. Typically, there is a marked hypergammaglobulinaemia with selective elevation of the IgG concentration (which can be as high as 100 g/L). Approximately 80% of AIH patients will also have significant titres (> 1:40) of organ non-specific autoantibodies (smooth muscle antibody (SMA), antinuclear antibody (ANA) or anti-liver-kidney microsomal (LKM) antibodies). Standard treatment involves immunosuppressive therapy with prednisolone and the steroid-sparing agent azathioprine.
Monitoring response to therapy
Plasma aminotransferase activities do not correlate well with histologically assessed disease activity in AIH. However, such tests, together with plasma IgG concentrations, are cheap and can be repeated more easily than liver biopsy, so are used routinely to monitor response to immunosuppressive therapy. Some 80–95% of AIH patients respond to the standard therapy of prednisolone and azathioprine, with aminotransferase activities and IgG concentrations falling to at least 50% below their initial values within three months. Dosages are titrated against the aminotransferase and IgG values, being progressively reduced to maintenance doses as these parameters fall towards the normal range that, in most patients, is achieved within one year of starting treatment. However, this ‘biochemical remission’ does not represent a complete response. It usually requires at least a further year of treatment before complete histological remission is achieved. Current recommendations are that treatment should be continued for at least two years (preferably four), with regular monitoring of aminotransferase activities and IgG concentrations; before any attempt is made to withdraw treatment, histological remission should be confirmed. Withdrawal of all immunosuppression is rarely achieved, possibly because it is not routinely attempted, but gradual reduction in the dose of prednisolone to very low levels, or complete withdrawal, is feasible so that immunosuppression can be maintained with azathioprine alone. If complete withdrawal of immunosuppressive treatment is achieved, the patient should be monitored indefinitely, because even a complete histological response does not represent a true ‘cure’ and there is a lifelong risk of relapse of the disease. The latter is heralded by a rise in aminotransferase activities and IgG concentrations. Importantly, interpretation of biochemical test results should not focus solely on the upper reference limits for the various parameters but should take account of what may be normal for the individual patient. By definition, half of the normal population will have values below the geometric means of the reference ranges and, for such individuals, a rise in a parameter to (or just above) the upper reference limit may represent a doubling of their normal value.
PRIMARY BILIARY CIRRHOSIS (PBC)
This is a chronic cholestatic condition of unknown cause in which there is destruction of septal and interlobular bile ducts. Females are affected far more frequently than males. Most patients present with pruritus, jaundice or non-specific symptoms including tiredness and hepatic pain. However, an increasing number are detected incidentally when abnormal liver tests, particularly an increased activity of alkaline phosphatase, are detected during opportunistic screening. Although PBC is considered to be an autoimmune liver disease, and twin and family studies suggest that there is a significant genetic component, it is clearly a complex disease and little progress has been made in defining the genes responsible.
Biochemical liver function tests reveal a characteristic cholestatic picture with, as the disease progresses, increasing plasma alkaline phosphatase (ALP) (of biliary origin), increasing bilirubin concentration and falling albumin. The plasma concentrations of both pentameric and monomeric IgM are frequently raised (cf. AIH). The most specific serological tests are those for antimitochondrial antibodies (AMA), which are detectable in about 95% of patients. Antimitochondrial antibodies recognize several distinct autoantigens, but the most characteristic for PBC are the M2 antibodies that react with epitopes on the E2 components of the pyruvate dehydrogenase complex (PDC-E2), the branched chain oxoacid dehydrogenase (BCOADH-E2) and the oxoglutarate dehydrogenase complexes. Laboratory tests, particularly the plasma bilirubin concentration, are used in assessing prognosis (see p. 248).
PRIMARY SCLEROSING CHOLANGITIS (PSC)
This is a progressive disease characterized by diffuse inflammation and fibrosis of the extrahepatic and/or intrahepatic biliary system, which leads to obliteration of the intrahepatic ducts and, eventually, biliary cirrhosis (see below). It is diagnosed on the basis of characteristic cholangiographic appearances together with compatible clinical, biochemical and histological features, and exclusion of several other conditions known to cause secondary sclerosing cholangitis, such as biliary calculi and previous biliary tract surgery. Men are affected more often than women and, again, although considered an autoimmune disease, the disease is genetically complex and remains poorly characterized.
With the advent of endoscopic retrograde pancreatography (ERCP), PSC began to be diagnosed earlier in its natural history, particularly following the initial finding of persistently abnormal LFTs in patients with inflammatory bowel disease, which coexists with PSC in more than 50% of cases. In the early stages, there is usually a minor elevation of plasma aminotransferase activities and moderate elevation of ALP, which progresses, over several years, to a very severe cholestatic condition with deep jaundice. By this stage, plasma ALP activity often reaches more than ten times the upper reference limit. Death from liver failure ultimately results unless a liver transplant can be performed. However, the disease has been known to recur in the grafted liver. Rapid clinical deterioration with a progressive cholestatic picture may indicate the development of cholangiocarcinoma, a complication with a lifetime risk of 10–15% in this condition. CA19-9 concentrations may be elevated when a cholangiocarcinoma has developed but this is not a specific finding as CA19-9 is also increased in other cholestatic liver diseases. Copper retention appears to be a general feature of chronic cholestatic conditions: in PSC most patients will have raised hepatic copper concentrations (of a similar order to that seen in Wilson disease and primary biliary cirrhosis) and urinary copper concentrations are typically raised. However, in contrast to Wilson disease, the plasma caeruloplasmin and copper concentrations will remain within the reference range (see p. 267).
ALCOHOLIC LIVER DISEASE
Excessive alcohol (ethanol) consumption is by far the commonest cause of liver disease in the Western world, although it often exists as part of a wider spectrum of social, psychological and pathological effects of alcohol-related damage. Most current evidence suggests that alcohol itself is the primary cause of the liver damage, although associated malnutrition may be a contributory factor. Considering that there is an enormous variation in the amount of alcohol drunk by different individuals, the time course over which it is consumed and individual susceptibility to tissue injury, it is not surprising that the adverse effects on the liver vary widely.
Ethanol metabolism
Alcohol is absorbed from the stomach and small bowel. Absorption is most efficient when there is no other food (particularly carbohydrate) in the gut and when its concentration in the ingested fluid is of the order of 20%.
The liver is exposed to the highest concentration of alcohol in blood, owing to the blood supply by the hepatic portal vein, and is responsible for more than 95% of its metabolism. The concentrations attained after ingestion of a standard amount of ethanol depend, among other things, on sex, weight, previous exposure to alcohol, the type of alcoholic beverage and rate of gastric emptying. Ethanol is oxidized to acetaldehyde mainly by the cytosolic enzyme alcohol dehydrogenase, but also, particularly at high concentrations, by the cytochrome P450 system. Acetaldehyde is particularly toxic. It is metabolized by aldehyde dehydrogenase in the mitochondria to acetate, which is, in turn, oxidized by peripheral tissues to carbon dioxide and water. Both enzyme systems can be induced by alcohol. The intoxicating and metabolic effects are mediated directly by ethanol but it is likely that acetaldehyde is an important factor in causing tissue damage through the formation of adducts that in turn lead to functional impairment of hepatic proteins. The metabolism of ethanol also predisposes to various metabolic problems including hypoglycaemia and non-respiratory acidosis (see Chapter 40).
Liver pathology in alcoholic liver disease
Three patterns of histological change in liver tissue have been described in association with ethanol ingestion, although there is considerable overlap and all three features can be present on occasions. Fatty liver (steatosis) appears to be a response to excessive alcohol consumption in all individuals. It is considered to be a reversible feature, resolving with abstinence, and does not progress to chronic liver disease provided abstinence is maintained. In alcoholic hepatitis, the appearances are of steatosis but also sometimes with features of interface hepatitis. This is a serious condition with a marked propensity to progress to fibrosis and cirrhosis – the third and final stage of chronic alcoholic liver disease. Additional morphological changes may be seen in patients who have other coexisting disorders. For example, people with hereditary haemochromatosis or chronic hepatitis C may be at increased risk of developing liver disease from alcohol, and porphyria cutanea tarda is often associated with excessive alcohol consumption.
Biochemical abnormalities
Alcoholic steatosis
Biochemical changes are minimal and consist of subclinical hyperbilirubinaemia and a very mild elevation of plasma aminotransferase activities. Very occasionally, a cholestatic syndrome develops, but associated alcoholic pancreatitis leading to biliary obstruction should be considered if jaundice is marked. Episodes of delirium tremens and alcoholic myopathy may give markedly raised plasma aminotransferase activities. Plasma γGT activity is elevated in the majority of patients (although it is not a specific finding), but usually reflects enzyme induction rather than hepatic injury.
Alcoholic hepatitis
There is a wide spectrum of severity associated with this histological diagnosis, ranging from the classic syndrome of deep prolonged jaundice, hepatic failure, fever and leukocytosis through to a complete absence of symptoms and physical signs. Alcoholic hepatitis generally occurs after heavy bouts of drinking and when subjects have been drinking heavily for several years. The laboratory tests reveal anaemia, usually with a leukocytosis and, consistently raised aminotransferase activities. However, the AST values are seldom above ten times the upper reference limit and ALT values are usually lower. This results in an AST:ALT ratio of > 2, and a value below this in subjects with hepatitis suggests that alcohol is not a major aetiological factor.
Alcoholism and haemochromatosis
Iron overload is common in alcoholic liver disease, occurring in perhaps 50% of patients (see p. 265). Occasionally, this may progress to a similar degree to that seen in hereditary haemochromatosis, and the patient may have similar abnormalities in biochemical parameters of iron status and endocrine function. More commonly, it appears that excessive alcohol consumption leads to the unmasking of haemochromatosis in patients with this genetic disease.
Porphyria cutanea tarda
As with haemochromatosis, this genetically determined condition, comprising bullous skin lesions on exposure to sunlight, liver disease and iron overload, is often brought to medical attention by excessive alcohol consumption. The underlying biochemical defect is a deficiency of uroporphyrinogen decarboxylase (see Chapter 28).
Use of laboratory tests in clinical practice
Biochemical tests of alcohol intake and its misuse fall into three categories:
• assessment of the effect of alcohol on protein metabolism, i.e. carbohydrate deficient transferrin
• assessment of the extent of liver damage associated with alcohol misuse by measurement of plasma enzymes.
Alcohol and metabolites
Saturation of the normal pathways of alcohol metabolism results in the induction of minor pathways with the formation of metabolites with high specificity for excess alcohol intake: glucuronidation leading to ethyl glucuronide (EtG) formation; sulphation to ethyl sulphate (EtS) formation, and incorporation into 5-hydroxytryptamine to form 5-hydroxytryptophol. Ethanol in blood or urine remains detectable only for 24–36 h, even after ingestion of significant amounts, so false negative results may be obtained in subjects misusing alcohol if they can abstain for 1–2 days prior to testing. The urinary 5-hydroxytryptophol:5-hydroxyindole acetic acid ratio has close to 100% specificity for excess alcohol consumption, but mass spectrometry is required for its measurement and the test has not been adopted routinely. Recently, interest has grown in the use of EtG and EtS measurements as these metabolites have high specificity for alcohol misuse and can be detected in urine up to 72–90 h after significant intake. A commercial immunoassay exists for EtG, but the combined measurement of EtG and EtS requires mass spectrometry. Many workers favour the latter approach because of reports of both false positive and negative EtG results in the presence of a urinary infection with bacteria capable of either removing the glucuronide or glucuronidating ethanol in vitro. Ethyl glucuronide and EtS do, however, offer the potential for detection of ‘binge’ drinkers that has been notoriously difficult to achieve in the past.
Effects of alcohol on protein metabolism
Carbohydrate deficient transferrin (CDT) is defined as the asialo- and disialo- forms of transferrin that usually account for < 1.6% of the total transferrin found in plasma. The formation of CDT is directly proportional to alcohol intake: ethanol (or its metabolites) appears to inhibit the enzymes responsible for addition of the carbohydrate side-chains and induce sialidase that removes the terminal sialic acid residues from the side-chains. The percentage CDT is independent of the extent of any alcohol-related liver damage except in patients with severe cholestatic liver disease, where reduced clearance may cause a falsely high result. Hepatic failure can cause analytical difficulties in CDT measurement because of low total transferrin concentrations from reduced synthesis. As the plasma half-life of CDT is 10–14 days, a raised percentage of CDT is strongly suggestive of chronic excessive alcohol consumption with sensitivity and specificity both approaching 85%. Measurement of serum immunoglobulin concentrations may also be helpful because alcoholic liver disease is often associated with a selective increase in IgA (cf. AIH and PBC).
Plasma enzymes
Alcoholic steatosis can cause an increase in the aminotransferases or γ-GT that is reversible with abstinence, although the degree of increase correlates poorly with the extent of liver damage assessed histologically. Progression to cirrhosis is usually accompanied by abnormal plasma enzyme activities often suggesting intrahepatic cholestasis, although in some patients with established cirrhosis who abstain from further alcohol intake, these may revert to normal. For a more detailed discussion, including the role of the measurement of mitochondrial AST, see Chapter 13.
Non-alcoholic fatty liver disease (NAFLD)
This comprises a spectrum of conditions ranging from simple hepatic steatosis (excessive accumulation of fat in hepatocytes) to end-stage chronic liver disease: these conditions are clearly associated with the epidemic of obesity and diabetes recognized in Western populations. Previously thought to be a rare and relatively benign condition, developing mainly in middle-aged females in association with obesity, insulin resistance and hypertriglyceridaemia, NAFLD is emerging as one of the commonest causes of chronic liver disease. Steatosis is an adaptive response of the liver to insulin resistance. When this is accompanied by endogenous insults such as oxidative and free radical damage, mitochondrial dysfunction and endotoxaemia, themselves resulting in part from excessive amounts of fat in liver cells, an inflammatory condition termed non-alcoholic steatohepatitis (NASH) is induced in susceptible individuals. Typical histological features include micro- and macrovesicular steatosis, mild to moderate portal and lobular inflammation (often with small clusters of polymorphs), liver cell ballooning and perisinusoidal fibrosis. Mallory’s hyaline (typically associated with alcoholic liver disease) may also be present. Non-alcoholic steatohepatitis can induce fibrosis and, in some individuals, this process will result in cirrhosis with all the attendant complications including hepatocellular carcinoma. Unfortunately, it is not possible at present to predict which individuals will experience a progressive course. Liver function tests are mildly or moderately abnormal in most patients at presentation, but longitudinal studies indicate that progression of fibrosis can occur despite return to normal of aminotransferase activities in response to weight loss.
THE CONCEPT OF CIRRHOSIS
Cirrhosis is not a clinical diagnosis, but a pathological description of the liver in which there is:
• nodular regeneration
• a disturbance of the normal hepatic architecture, that is, a distortion of the normal relationship of the portal tracts to the central veins (see Chapter 13).
It is the end result of chronic liver diseases that are usually associated with recurrent episodes of necrosis, cell death and attempts by the liver to regenerate. Liver function is disturbed, not only because of the cirrhosis per se, which has little effect on standard LFTs, but because of continuing immunologically mediated liver cell damage, the capillarization of the sinusoids due to fibrosis (see Chapter 13) and, on occasions, because of the effects of the initiating agent such as alcohol. Efforts to make a diagnosis of cirrhosis on any grounds other than histological are unreliable.
A great part of clinical hepatology is taken up with the diagnosis and management of the complications of cirrhosis: hepatic encephalopathy, ascites and the hepatorenal syndrome, infections, primary hepatic malignancy and endocrine dysfunction.
Hepatic encephalopathy
This is seen in patients with advanced cirrhosis, and a precipitating factor such as administration of a sedative, a large protein intake (including blood from variceal haemorrhage), infection or electrolyte imbalance can usually be identified. It is particularly frequent in subjects who have undergone a portocaval shunt for treatment of portal hypertension. As with the encephalopathy associated with acute liver failure, the severity of the condition is graded from I–IV:
I – alert but with asterixis (‘hepatic flap’), inversion of sleep rhythm
II – confused, disorientated, slow mentation, inappropriate behaviour
III – restless, aggressive, sleepy, uncommunicative
IV – coma.
The diagnosis is essentially a clinical one and, although characteristic electroencephalographic appearances are described, these are not used diagnostically. Similarly, the blood ammonia concentration is usually raised but correlates only poorly with the degree of encephalopathy.
Vascular disturbances in cirrhosis
Cirrhosis is characterized by the development of systemic vasodilatation, which results in a decrease in effective arterial blood volume, low systemic vascular resistance and a hyperdynamic circulation with high cardiac output. The mechanism underlying these changes is not known with certainty, but may involve increased vascular synthesis of nitric oxide because of increased activity of inducible nitric oxide synthase, as well as increased production of prostacyclin and other vasodilators such as glucagon, substance P and calcitonin gene-related peptide. In response to vasodilatation, several homoeostatic systems are triggered, including an increase in renal sympathetic activity, activation of the renin–angiotensin–aldosterone system and increased production of antidiuretic hormone (ADH). In some patients, vascular resistance and effective arterial blood volume are restored, but in others, particularly when cirrhosis is complicated by severe portal hypertension, water retention and vasoconstriction are insufficient to restore circulating blood volume and these subjects present with hypotension and a hyperdynamic circulation, together with high plasma concentrations of renin, noradrenaline (norepinephrine) and ADH. These hormones have a profound effect on renal vasculature, causing progressive vasoconstriction and a reduction in plasma flow and glomerular filtration, resulting in marked salt and water retention. These changes have a central role in the development of ascites.
Ascites
Ascites, the excessive accumulation of extracellular fluid in the peritoneal cavity (over 30 L in some patients), is usually a late complication of cirrhosis. Two key factors underlie its development, namely portal hypertension operating at the level of the sinusoids, and sodium and water retention. Portal hypertension occurs as cirrhosis develops because of progressive collagen deposition within the space of Disse and nodule formation, resulting in disruption of the vascular architecture of the liver. Increased resistance to portal flow and increased hydrostatic pressure within the sinusoids favour transudation of tissue fluid into the peritoneal cavity. Portal hypertension is a critical permissive factor in the development of ascites. The second key factor in the development of ascites is, as described above, salt and water retention with enhanced sodium reabsorption occurring in both the proximal and distal tubules of the kidneys. In some patients with ascites, normal concentrations of aldosterone are found, leading to the suggestion that enhanced renal sensitivity to the hormone underlies the increased distal tubular sodium reabsorption. The role of hypoalbuminaemia in the development of ascites in patients with liver disease is controversial. Some consider this to have a permissive role in localizing the fluid collection to the peritoneal space, while others consider that plasma albumin concentration has little influence on ascites formation. Instead, they suggest that the oncotic pressure gradient across the sinusoids is low (because albumin crosses the fenestrations) so ascites develops only when trans-sinusoidal filtration exceeds the drainage capacity of the lymphatic system, as occurs in patients with portal hypertension.
Ascites usually develops in patients with advanced, decompensated disease, but the tendency to retain salt is present before ascites actually develops and a sudden increase in dietary salt intake may lead to the generation of ascites that clears spontaneously when the salt intake is restricted. Excess water retention leading to hyponatraemia is frequent and is attributable to the mechanisms described above. There are, however, several liver and non-liver diseases other than cirrhosis (Box 14.2) that can lead to the formation of ascites, and the establishment of a precise diagnosis is important for its correct management.
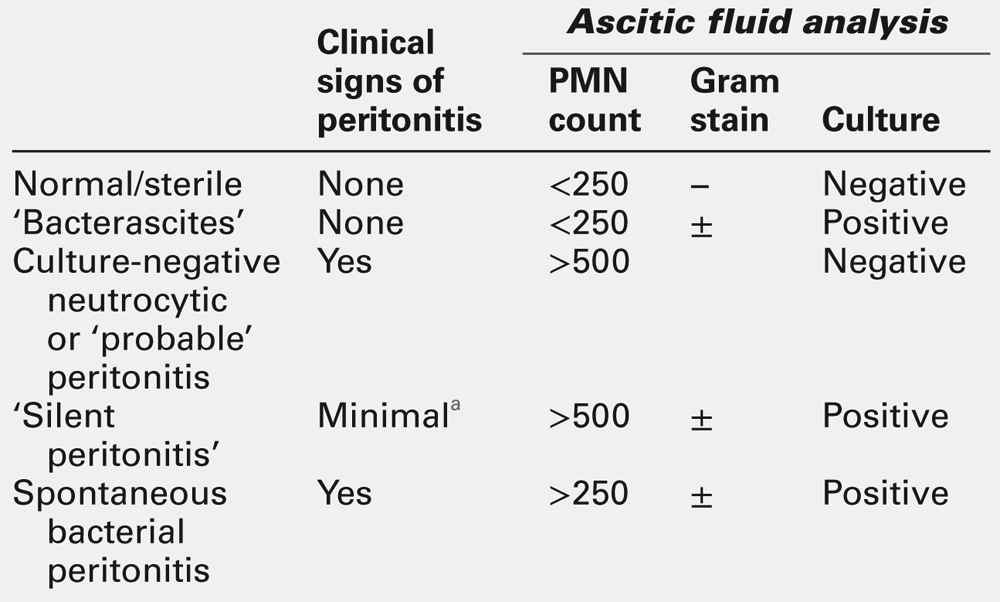
If the standard LFTs are abnormal in a patient with ascites, then it is likely that one of the liver diseases listed in Box 14.2 is responsible and it is often already known that a patient who develops ascites has cirrhosis. However, patients with liver disease can develop ascites for reasons other than cirrhosis. For example, patients with alcoholic cirrhosis may develop tuberculous ascites, pancreatic ascites or ‘malignant’ ascites if hepatocellular carcinoma supervenes. Cardiac causes can usually be diagnosed confidently on clinical grounds, but the other causes may be difficult to distinguish and laboratory investigations have an important role to play. Ascitic fluid should be aspirated using an 18-gauge needle and syringe (a so-called ‘diagnostic tap’) for analysis. The investigations and their interpretation are shown in Table 14.3. It should be recognized that sodium retention is not a diagnostic feature of cirrhotic ascites: equally intense sodium retention can occur in malignant ascites.
TABLE 14.3
Differential diagnosis of ascites based on inspection and laboratory tests

a Chylous ascites probably represents escape of splanchnic lymph into ascites.
b Concentrations may be lower when cirrhotic ascites is complicated by tuberculosis.
c The characteristic laboratory feature is a high concentration of triglycerides.
The figures for ascitic protein given in Table 14.3 are given only as guidelines. Exceptions are frequent and clinical utility is limited, but as the changes become more extreme, so their diagnostic specificity increases. Thus, a protein concentration < 10 g/L is not infrequent in uncomplicated cirrhosis, but virtually rules out malignant disease. Conversely, protein concentrations > 35 g/L are the rule in malignant ascites and are unusual in uncomplicated cirrhosis.
Several tests have been proposed to increase the specificity of total ascitic protein measurement in differentiating between cirrhotic and malignant ascites. These include calculation of the plasma to ascites albumin gradient and measurement of ascitic lactate dehydrogenase activity or ascitic cholesterol concentration (Fig. 14.2). Adenosine deaminase activity in ascites of > 60 U/L is particularly sensitive and specific for tuberculous ascites.
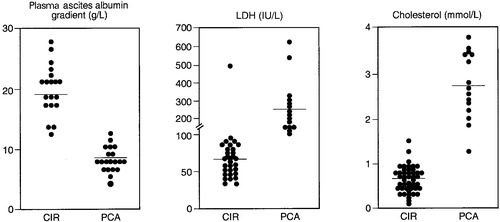
FIGURE 14.2 Plasma ascites albumin gradient and ascitic fluid concentration of lactate dehydrogenase (LDH) and cholesterol in patients with cirrhosis (CIR) and peritoneal carcinomatosis (PCA). Reproduced with permission from Arroyo V et al. Ascites, renal failure and electrolyte disorders in cirrhosis. Pathogenesis, diagnosis, and treatment. In: McIntyre N et al. (eds). Oxford Textbook of Clinical Hepatology. Oxford: Oxford University Press; 1991.
Monitoring treatment of ascites
Ascites due to cirrhosis is usually managed by a combination of dietary salt restriction and diuretic therapy, though paracentesis with albumin infusion is now being used more widely. The aim should be to achieve a net fluid loss of 500 mL/24 h until the ascites clears. Plasma urea, creatinine, sodium and potassium concentrations should be checked daily at the start of treatment. A rising plasma urea or creatinine and falling plasma sodium concentration (< 130 mmol/L) indicate impending acute kidney injury and should prompt a reduction of diuretic treatment. Diuretics should not be used when plasma sodium is < 125 mmol/L. Plasma creatinine concentration is the more useful test, as impaired urea synthesis in patients with cirrhosis makes the plasma urea a less sensitive indicator of renal function. Plasma potassium needs careful monitoring if furosemide is used because of the risk of hypokalaemia, which may precipitate encephalopathy. So, too, can hyperkalaemia, which may occur with spironolactone if renal impairment supervenes.
Acute kidney injury
The onset of acute kidney injury (AKI) is indicated by rising plasma concentrations of creatinine and urea and usually, but not always, a urinary output falling to < 300 mL/24 h. A particularly dangerous complication, as already mentioned, is hyperkalaemia. Such developments are likely to precipitate encephalopathy in patients with liver disease. The major clinical problem in such patients is an idiopathic form of AKI that is widely known as the ‘hepatorenal syndrome’ (see below) and usually associated with advanced liver disease, ascites and encephalopathy. This should not be confused with several other situations in which renal disease and liver disease coexist: polycystic disease, infections such as leptospirosis, circulatory failure and the immune complex glomerulonephritis associated with hepatitis B virus infection or the presence of cryoglobulins in hepatitis C infection. Acute kidney injury may also develop when cirrhotic patients are fluid depleted, and following surgery to relieve obstructive jaundice. The latter syndrome is caused by acute tubular necrosis, but the mechanism is unknown.
The hepatorenal syndrome (HRS)
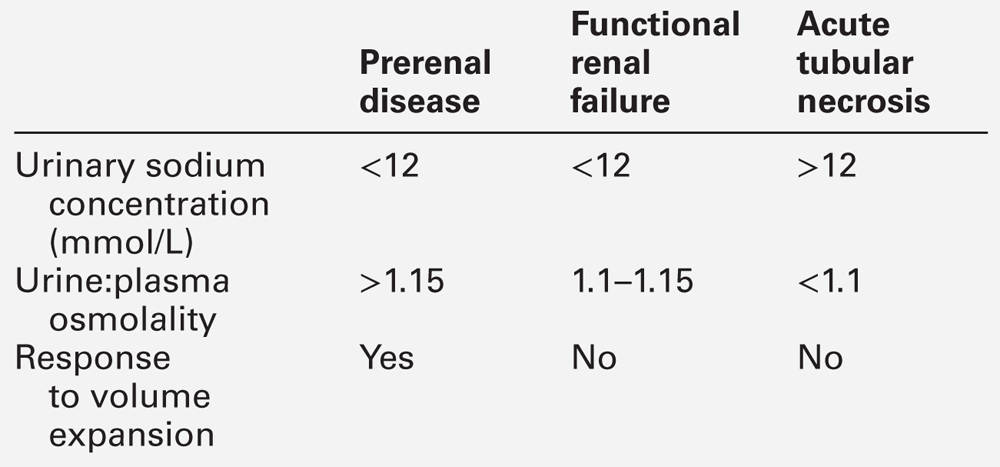
This condition is also known as functional renal failure (FRF). The characteristic feature is that the kidneys are normal when examined histologically, that is, there is disturbance of function but not of structure; specifically, the usual appearances of acute tubular necrosis are not present. Furthermore, the impairment is progressive and not reversed by fluid repletion, suggesting that this is not simply a pre-renal disorder. The precise pathogenesis is unknown, but underlying factors include those detailed in the earlier section on vascular abnormalities in cirrhosis, which lead to intense renal vasoconstriction resulting in decreased renal blood flow and a reduced glomerular filtration rate. Functional renal failure occurs classically in patients with advanced chronic liver disease, but may also occur in severe acute liver disease.
In patients known to have advanced cirrhosis, the condition can be diagnosed by simple laboratory tests and clinical observation. Reduced urine flow and rising plasma concentrations of urea and creatinine are common to all forms of AKI, but the characteristic feature of FRF, which distinguishes it from acute tubular necrosis, is the dramatic degree of sodium retention (Table 14.4). The urinary sodium concentration is usually < 12 mmol/L and often < 5 mmol/L. The urine:plasma osmolality is usually > 1, and this contrasts with patients with acute tubular necrosis, in whom the urinary sodium concentration is invariably > 12 mmol/L and often > 20 mmol/L, and the urine/plasma osmolality is about 1. Infusion of terlipressin, a non-selective agonist of V1 vasopressin receptors, together with albumin, reverses FRF in ~ 75% of patients.
Sex hormones and their binding proteins
Men with cirrhosis are frequently impotent, infertile and feminized (i.e. they may have gynaecomastia, a female distribution of body hair and testicular atrophy). Workers in the USA, where alcohol is responsible for a very high percentage of all chronic liver disease, have tended to attribute these symptoms to the alcohol rather than the chronic liver disease. In the UK, where at least 50% of patients with cirrhosis have non-alcohol related disease, sexual dysfunction is still frequently seen and it seems most likely that alcohol may act both on its own and in concert with chronic liver disease to cause this disorder. The biochemical and endocrine changes that accompany chronic liver disease are now well recognized but they correlate poorly with the clinical symptoms.
This section describes the changes in endocrine tests that are found in patients with liver disease of various causes and severity, but it is important to note that the changes described are not necessarily the cause of symptoms. The discussion below refers mainly to men; the effect of liver disease on sexual function in women has been less well studied.
Physiology and biochemistry
This topic is discussed in detail in Chapter 23 but a summary is presented here for convenience. The major circulating androgen is testosterone secreted by the testicular Leydig cells (0.17–0.35 mmol/24 h) under the control of luteinizing hormone (LH). The androgenic effect of androstenedione, and other adrenal androgens such as dehydroepiandrosterone, is probably attributable to their peripheral conversion to testosterone. Testosterone can be metabolized to its more active metabolite dihydrotestosterone (DHT) by 5α-reduction, or to oestrogens (by aromatization) or can be degraded in the liver, which is responsible for clearing about 50% of testosterone during the first pass in males. Oestrogens also circulate in men, the most potent being oestradiol (strictly 17β-oestradiol, but often abbreviated to E2), which is derived from aromatization of testosterone. Testosterone and 17β-oestradiol circulate bound to albumin (low affinity, unsaturable binding) and sex hormone-binding globulin (SHBG, high affinity, saturable binding). Only a small amount of each hormone exists in the unbound state and this is presumed to be the biologically active fraction. Spermatogenesis is under the control of follicle stimulating hormone (FSH), although LH is also required, acting indirectly on the seminiferous tubules by stimulating testosterone secretion by the adjacent Leydig cells.
Changes in men with cirrhosis
The plasma total testosterone concentration is at the lower limit of the reference range in most men with well-compensated liver disease and falls as the disease progresses. The oestradiol concentration, on the other hand, is usually at the upper limit of the reference range and rises as the disease progresses, and the ratio of oestradiol to testosterone is thus elevated. Because SHBG concentrations are invariably also raised and the affinity of SHBG for testosterone is considerably greater than that for oestradiol, the ratio of free oestrogen to free testosterone is even greater. The low testosterone concentrations are attributable to reduced testicular production and occur despite a decrease in metabolic clearance rate caused by the high SHBG concentrations. The elevated oestrogen concentrations are not due to impaired hepatic clearance as originally thought; increased production, probably due to enhanced peripheral aromatization, appears to be the most likely mechanism.
In the presence of low testosterone concentrations, a compensatory increase in pituitary secretion of luteinizing hormone (LH) would be expected, but this does not always occur, implying that a primary testicular defect is often complicated by hypothalamic–pituitary dysfunction. As the disease progresses, LH concentrations tend to fall. Thus, the characteristic abnormalities may be represented as a spectrum (Table 14.5). Similar, but less pronounced changes occur in postmenopausal women with chronic liver disease. Three-quarters of men with cirrhosis have oligospermia and this is associated with normal plasma concentrations of FSH. The absence of a compensatory rise in FSH has been attributed to a hypothalamic defect that is characteristic of patients with cirrhosis. Absence of testicular atrophy, a normal LH or a normal LH or FSH response to clomifene or gonadotrophin releasing hormone predict recovery of sexual function in alcoholic men who abstain from further drinking.
TABLE 14.5
Changes in plasma sex hormone concentrations in men with chronic liver disease in relation to its severity
| Well compensated cirrhosis | Poorly compensated cirrhosis | |
| Testosterone (T) | Low/normal | Low |
| 17β-oestradiol (E2) | High/normal | High |
| SHBG | High | High |
| Total E2/T ratio | High | Very high |
| LH | Normal | Low |
Hypogonadism is a prominent feature in men with hereditary haemochromatosis. Unlike the situation with other types of cirrhosis, such as that due to alcohol, impotence may occur very early in the disease (and even before the cirrhosis develops). It is probably due to a combination of hypogonadotrophic hypogonadism, concurrent hepatic cirrhosis and diabetic autonomic neuropathy. The pituitary dysfunction, which is probably the most important factor, is caused by selective iron deposition in the gonadotrophin-secreting cells. Plasma concentrations of LH (and prolactin) are subnormal in the majority of men with clinical hypogonadism associated with liver disease.
Changes in liver function during pregnancy
Abnormalities (occasionally marked) in the LFTs may occur during normal pregnancy. These have been attributed, not unreasonably, to changes in the hormonal environment and there is good experimental evidence that oestrogens are involved. Thus, similar abnormalities are also seen in women taking the contraceptive pill or other oestrogenic preparations.
The most pronounced change is a tendency towards cholestasis in the last trimester – intrahepatic cholestasis of pregnancy. The plasma activities of γGT and ALP rise in late pregnancy but the latter is attributable mainly to placental ALP. In a small percentage of women, however, this tendency is exaggerated and symptoms of jaundice and/or pruritus develop. Although different names are given depending on the predominant symptom, they probably form part of a spectrum. Signs and symptoms disappear within a few days, or even hours, of delivery but recur with varying degrees of severity in subsequent pregnancies or on exposure to oestrogen containing preparations. Female relatives of patients with intrahepatic cholestasis of pregnancy are at an increased risk of developing the same syndrome. Plasma total bile acid concentration is typically increased 5–10-fold and this may sometimes be the only abnormality.
Severe liver disease leading to acute liver failure is a rare complication of pregnancy. Although these conditions probably represent a clinicopathological continuum, the clinical features and timing during pregnancy, results of laboratory tests and maternal and fetal outcomes allow differentiation between acute fatty liver of pregnancy, the ‘haemolysis, elevated liver enzymes and low platelets’ (HELLP) syndrome and liver impairment in eclampsia and hyperemesis gravidarum. Liver rupture is a rare complication of fatty liver and the HELLP syndrome, with high mortality. With the widespread introduction of testing for viral hepatitis in pregnancy and biochemical screening, it is now clear that abnormal LFTs can occur in up to 3% of pregnancies. Details of appropriate investigations and management of liver disease in pregnancy are available in specialized texts.
Not surprisingly, the excretory defect in patients with the Dubin–Johnson syndrome (Chapter 13) is worsened by oestrogens, and such patients often first present with, or suffer from, increasing jaundice during pregnancy or on first exposure to the contraceptive pill. Pregnancy and the contraceptive pill are also associated with the development of benign liver tumours and increased blood coagulability leading to hepatic vein thrombosis, but both of these complications are very rare.
Changes in hormonal profiles during or after pregnancy can also precipitate, or exacerbate pre-existing AIH in women. Occasionally, this rare condition presents for the first time during the second or third trimesters of pregnancy or in the immediate postpartum period. More frequently, patients with pre-existing AIH, who have been in treatment-induced remission, experience flares of their disease during or after pregnancy. These flares are unpredictable and AIH patients who become pregnant, therefore, require more frequent monitoring of their serum aminotransferase activities during pregnancy and for up to six months after delivery, so that their immunosuppressive therapy can be increased (or reintroduced) at an early stage to avoid the development of subacute liver failure, which carries a high risk for both mother and fetus. The mechanisms underlying these effects of hormonal changes on the activity of this disease are not well understood, but it is known that hormones have profound effects on the immune system. Thus, high oestrogen and prolactin concentrations promote mainly a cell-mediated response, while low oestrogen and high progesterone concentrations favour an antibody-mediated pathway.
Glucose intolerance
Most patients with hepatic cirrhosis exhibit intolerance to glucose. This is probably due to insulin resistance at the level of peripheral glucose uptake and glycogen synthesis by muscle. The fasting plasma concentration of insulin is increased because of a combination of decreased hepatic extraction, enhanced secretion and portosystemic shunting.
Hypoglycaemia, presumably because of the vast functional hepatic reserve, is rare in liver disease, being virtually confined to patients with acute liver failure, galactosaemia, fructosaemia or, even more rarely, primary hepatic malignancy. Hypoglycaemia can occur in acute liver failure and can be severe and persistent; for this reason such patients should be routinely maintained on a glucose infusion, particularly if they are being transferred between hospitals.
In patients with type 2 diabetes mellitus and liver disease who require treatment with oral hypoglycaemics, biguanides are best avoided because of the danger of lactic acidosis. Most sulfonylureas are metabolized in the liver, and those with a long plasma half-life should be avoided in patients with liver disease because of the risk of hypoglycaemia owing to accumulation of the drug.
DRUGS AND THE LIVER
Biochemical tests play a crucial role in the recognition and monitoring of adverse effects of therapeutic drugs. An International Consensus Meeting has clarified the terminology best used for reporting and recognizing such reactions, and for inferring causality by the temporal relationship between drug administration and putative hepatic dysfunction. It should be emphasized here that this discussion is not about patients with liver disease, but patients with other conditions who develop some form of liver dysfunction during or after treatment. Table 14.6 lists the definitions and gives some classic examples. In the absence of histological evidence, the term ‘drug-induced liver injury’ is preferred to ‘hepatitis’ or ‘cirrhosis’. Almost all the pathological conditions described in this chapter can be caused by drugs, chemicals or toxins. The reader is referred to standard texts for a detailed list of possible associations between liver disease and xenobiotics.
NEOPLASTIC DISEASE OF THE LIVER AND BILIARY TRACT
As with those in other organs, liver tumours are classified as benign or malignant and the latter are subclassified as primary (relatively rare) or secondary (relatively common). Benign liver tumours are of little clinical significance other than as a very occasional association with the use of oral contraceptive preparations and their increasing incidental detection during ultrasound or computerized tomography (CT) imaging. The extent to which incidentally detected space-occupying lesions of the liver should be further investigated is difficult and controversial: most will be benign and of no clinical significance. They should be investigated, however, because of the devastating consequences of missing the occasional malignant tumour that, when detected early, may be amenable to surgical resection. In general, LFTs are normal in subjects with benign lesions, so there is little doubt that further investigation should be undertaken if LFTs are abnormal in any respect.
In the West, most malignant disease of the liver involves metastatic deposits from primary tumours of the gut, pancreas, lung or breast. Abnormal LFTs do not provide any specific information about the primary site of a tumour metastasizing to the liver. On the other hand, metastases will seldom be of sufficient size to be detectable by imaging in individuals with normal LFTs. Measurement of carcinoembryonic antigen (CEA) is of little diagnostic use, but serial measurement after resection of a primary colorectal tumour may provide early evidence of disease recurrence, particularly in the liver.
Hepatocellular carcinoma and α-fetoprotein
Primary liver cancer is relatively uncommon in the West but has been increasing due, in part, to the increased prevalence of chronic hepatitis C. However, worldwide, hepatocellular carcinoma (HCC) is one of the commonest and most rapidly progressive malignant neoplasms. In more than 75% of patients, it arises as a complication of hepatic cirrhosis. Hepatocellular carcinoma is one of the few tumours for which a serological marker, α-fetoprotein (AFP), is clinically useful and widely available, although the sensitivity of AFP is only of the order of 80% as not all tumours secrete this protein. The reference range for AFP is usually quoted as < 10 μg/L, while values of up to 107 μg/L are seen in patients with HCC. Values > 500 μg/L, in an appropriate clinical setting, are virtually diagnostic of HCC, but there is a ‘grey area’ between 10 and 500 μg/L, where similar values can be seen in patients with uncomplicated chronic liver disease associated with hepatic regeneration.
Unfortunately, many patients with small and potentially resectable tumours fall into this ‘grey area’ and, consequently, several attempts to improve the specificity of the test have been made. A steady increase in AFP concentration within the ‘grey area’ is particularly suggestive of malignant change. Also, there is good evidence that a high percentage of AFP originating from malignant cells is hyperfucosylated, and a routine test for this fraction may become available in the future. Apart from its use in diagnosis of HCC, AFP concentration is widely used to monitor response to therapy, since the concentration falls and rises in relation to the tumour mass.
A rare type of HCC, the fibrolamellar variant, is characterized by occurrence in adolescence, absence of AFP and a rather better prognosis than other types of HCC. The plasma unsaturated B12 binding protein (UBBP) and neurotensin concentrations are usually grossly elevated. The diagnosis is, however, made on histological grounds, although measurement of UBBP and neurotensin can be used to monitor treatment.
PARENTERAL NUTRITION
Abnormalities of LFTs are frequently noted in patients receiving parenteral nutrition (PN) and usually arise during or after the second week of therapy. An increase in ALP and aminotransferases to more than twice the upper limit of the reference range occurs in up to 50% of patients, but few actually develop clinical jaundice. The underlying histological lesion is usually steatosis, and there seems little doubt that over-provision of calories and infusate imbalance (particularly when glucose alone rather than glucose and fat is used as the energy source) are involved. Much anxiety has been expressed about the hepatotoxicity of fat emulsions, but this only occurs if very high concentrations are used.
The possibility that changes in LFTs might be attributable to a specific complication of the condition for which the PN is being used must be borne in mind. An additional complication of PN is the development of both gallstone disease and acalculous cholecystitis. In neonates, particularly when premature, the development of PN-related liver disease may limit its use. In such children, the hepatic lesion ranges from mild cholestasis associated with steatosis to severe cholestasis, cirrhosis and liver failure.
BACTERIAL INFECTIONS
Extrahepatic bacteraemia is frequently associated with abnormal LFTs and, particularly in the young, jaundice. The picture is usually one of cholestasis, although in certain specific instances, for example in the toxic shock syndrome (due to Staphylococcus aureus infection), the picture may be more hepatitic.
Spontaneous bacterial peritonitis may occur in subjects with ascites and liver disease, in the absence of any apparent intra-abdominal source. The condition may be clinically silent, the only manifestations being onset of encephalopathy, deterioration in renal function and worsening LFTs. Rapid detection is essential and diagnosis is based on measurement of the polymorphonuclear leukocyte count in ascites and Gram stain of a centrifuged pellet (Table 14.7). The bacterial culture of ascites should be performed by directly injecting freshly drawn ascites into blood culture bottles at the bedside, because transporting ascites to the microbiology laboratory results in a significant reduction in the success of identifying the infecting organism. The responsible organism is usually Escherichia coli or other gut-related organisms and, less frequently, pneumococci.
INHERITED METABOLIC DISORDERS INVOLVING THE LIVER
Iron overload and hereditary haemochromatosis
Iron is a toxic metal, and whole body iron balance is tightly regulated at the level of the duodenal mucosa by controlling the amount of iron absorbed from the diet. Iron excretion is limited, and in the absence of a regulated pathway of iron loss, positive iron balance leads to increased tissue concentrations. The term haemochromatosis refers to the group of genetic disorders in which, as a result of excessive absorption of dietary iron and long-term positive iron balance, iron deposition causes tissue damage, particularly to the liver, pancreas, heart, anterior pituitary and joints. Haemosiderosis implies iron overload without tissue damage, often an early stage of iron accumulation, while secondary haemochromatosis occurs in conditions requiring multiple blood transfusions and in some other haematological disorders.
Although previously considered a single gene disorder, it is now known that haemochromatosis can be caused by mutations in several genes that appear to have different functions in iron metabolism. The most common form of haemochromatosis, found almost exclusively in people of northern European descent, is caused by homozygosity for a low penetrant mutation, C282Y, in the hereditary haemochromatosis gene, HFE. This condition affects mainly men and is characterized by the insidious accumulation of iron, with the onset of symptoms and signs of iron overload delayed until the fourth or fifth decades of life. Heterozygotes for the C282Y mutation do not develop iron overload, although minor abnormalities in plasma iron and ferritin concentrations occur in approximately 15% of such individuals. Adult onset haemochromatosis is very rarely associated with mutations in TfR2, the gene encoding transferrin receptor 2, a cell surface glycoprotein involved in iron transport and uptake by cells including hepatocytes.
A contrasting group of disorders, in which the iron loading process is very rapid, presenting by the second or third decades and affecting males and females equally, is known as juvenile haemochromatosis. Although liver disease is invariably present, usually as cirrhosis, the clinical presentation in this form of genetic iron overload is with cardiac and endocrine failure. Juvenile haemochromatosis is caused by mutations in HJV and HAMP, genes that encode, respectively, hemojuvelin and hepcidin. The iron loading process resulting from the digenic inheritance of mutations in HFE and TfR2, genes normally associated with adult onset haemochromatosis, can be so rapid as to lead to the phenotype of juvenile haemochromatosis. The recent identification of the iron regulatory hormone hepcidin provides a link between HFE, TfR2 and HJV, since mutations in these genes, and in HAMP itself, cause loss of hepcidin production by the liver. This peptide acts by inhibiting dietary iron absorption and iron release from recycling and storage sites. All these forms of iron overload are characterized by hepcidin deficiency and it appears that the proteins encoded by HFE, TfR2 and HJV function as sensors of iron status on the hepatocyte membrane acting upstream of HAMP. Genotyping for mutations in HFE is now an essential part of diagnosis and screening for haemochromatosis. Mutations in the other genes are so rare that routine genetic analysis for these is not undertaken. A further variant of haemochromatosis is associated with mutations in ferroportin, a gene that encodes the hepcidin receptor.
The penetrance of the homozygous C282Y genotype in HFE-related haemochromatosis, defined in terms of severe iron overload with tissue damage manifesting as cirrhosis and type 1 diabetes, is low, probably in the order of ~ 1–2%. Biochemical penetrance, defined as an increase in transferrin saturation of > 60% and a minimally raised plasma ferritin concentration, probably occurs in 20–50% of homozygous individuals. Other undetermined genetic loci and possibly environmental factors are likely to determine penetrance, but these are currently uncharacterized.
A clinical diagnosis of haemochromatosis in a severely iron loaded subject may be made on the basis of signs of liver disease, glycosuria and a slate-grey appearance of the skin, but confirmation of the diagnosis is entirely dependent on genetic and laboratory tests. The crucial investigations are plasma iron and ferritin concentration and transferrin saturation. In a patient with full gene penetrance, the fasting plasma iron concentration is usually > 40 μmol/L (reference range 10–30 μmol/L) and transferrin is usually > 60% saturated. Plasma ferritin concentration, which is approximately proportional to the iron excess, is usually > 1000 μg/L (upper limit of reference range 200–300 μg/L) and starts to rise when hepatic iron stores exceed twice the reference range.
If these tests suggest iron overload and liver function tests are abnormal, a liver biopsy should be undertaken to assess the degree of liver damage. Histological staining for iron (using Perls stain) gives a semiquantitative assessment of the degree of iron overload, but the amount of iron in a biopsy can also be quantitated directly by inductively coupled mass spectrophotometry (ICP-MS). The reference range is up to about 20 μmol/g dry liver weight and > 40 μmol/g dry liver weight is found in patients with haemochromatosis. In less severe disease, when the patient is < 40 years of age, plasma ferritin is < 1000 μg/L, LFTs are normal and genotyping confirms the homozygous C282Y mutation, then liver biopsy is not indicated as cirrhosis is unlikely to have developed.
Treatment involves weekly venesection until the patient starts to develop iron deficiency anaemia and the plasma ferritin concentration falls to ~ 50 μg/L. Since 500 mL of blood contains about 250 mg of iron, the total body iron at the time of diagnosis can be calculated from the total volume removed. In an iron loaded individual, it is usually in the order of 10–25 g compared with less than 4 g in the normal individual. The same figures can be obtained from knowledge of the plasma ferritin concentration, which can also be used to monitor the progress of treatment (1 μg/L of ferritin is equivalent to approximately 8 mg of iron). Unless cirrhosis has already occurred, phlebotomy is successful in preventing progressive liver disease. The high risk of development of hepatocellular carcinoma remains in treated patients with cirrhosis. It is also important to screen the patient’s relatives for haemochromatosis by HFE genotyping so that treatment can be instituted if appropriate before liver damage occurs. Plasma ferritin and transferrin saturation are the most sensitive biochemical tests for presymptomatic detection in population studies.
Wilson disease
Wilson disease is an autosomal recessively inherited disease of copper metabolism with a worldwide prevalence of about 1:30 000 and a carrier rate of 1:90. It leads to excessive deposition of copper in the liver, kidneys, brain, eyes and other tissues. Like haemochromatosis, it is treatable and thus has an importance in liver disease out of proportion to its rarity. Liver disease is the most prominent aspect in about 40% of patients, the remainder exhibiting neurological, psychiatric or haematological complications. The hepatic manifestations vary widely from asymptomatic development of cirrhosis to an acute onset, often accompanied by haemolysis and evidence of renal tubular dysfunction, which may progress to fulminant hepatic failure. In such patients, the finding of a very low serum alkaline phosphatase activity may be a useful pointer to Wilson disease: the transient marked hypercupraemia is thought to lead to incorporation of copper instead of zinc into the active sites of ALP, reducing its activity. Accumulation of copper in the eyes is associated with the appearance of Kayser–Fleischer rings in the cornea (detectable by slit-lamp examination), although these may not always be present. In the brain, the copper deposition leads to neurological symptoms (see Chapter 36).
The defect in Wilson disease is attributed to mutations in the ATP7B gene on chromosome 13 (at the 13q14.3 locus), which cause failure of ATPase-dependent copper excretion. To date, more than 200 mutations have been identified, and most patients are compound heterozygotes. However, in some patients with classic Wilson disease, no mutations in the ATP7B gene have been found. Nonetheless, progress of the disease can be effectively arrested by treatment (see below) and, therefore, all first-degree relatives of patients with Wilson disease should be screened, initially by biochemical testing, to identify asymptomatic siblings who might benefit from early therapeutic intervention.
Among those presenting with chronic liver disease, the elevation of aminotransferases is said to be inappropriately low for the degree of hepatic inflammation and ALP is often within the reference range, or low in patients where zinc depletion has occurred. There may be a low-grade haemolytic anaemia leading to mild hyperbilirubinaemia, with an increased risk of pigment gallstones. Copper deposition may lead to renal tubular dysfunction with loss of potassium, glucose, amino acids and phosphate.
Diagnosis
The diagnosis, or exclusion, of Wilson disease is of crucial importance to patients and their relatives. It is seldom as straightforward as it may appear in standard texts. If there is any doubt about the diagnosis, advice should be sought from a specialist centre. The following are characteristic laboratory features (see Table 14.8).
TABLE 14.8
Typical laboratory findings in Wilson disease and other conditions in which Wilson disease may enter the differential diagnosis
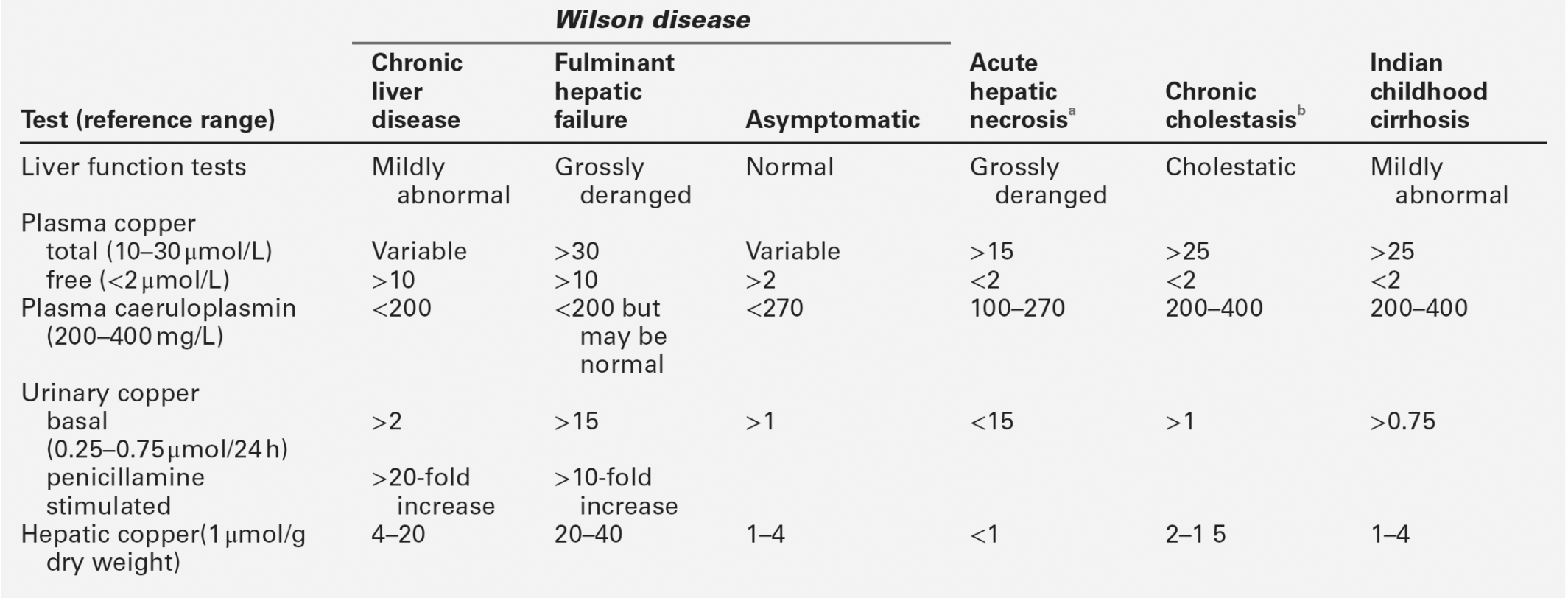
a Any severe hepatitic illness.
b Conditions such as primary biliary cirrhosis and sclerosing cholangitis.
2. A low plasma copper concentration. This is usually below the lower limit of the reference range, but similar values can be found in association with other disorders and it therefore lacks diagnostic specificity. Approximately 90% (10–25 μmol/L) of plasma copper is irreversibly bound to caeruloplasmin. The remainder (< 2 μmol/L), termed ‘free’ copper, is loosely bound to albumin and to amino acids. A ‘free’ plasma copper concentration > 7 μmol/L is virtually diagnostic of Wilson disease, but such values are found in only ~ 50% of patients. Normal total plasma copper concentrations can often be seen in patients with acute hepatocyte damage owing to release of copper trapped in the liver.
3. A high tissue copper concentration. It is clearly important that the diagnosis of Wilson disease is entertained before liver biopsy is undertaken so that an adequate sample may be taken for estimation of liver copper concentration. It is worth taking two cores to ensure that enough material is available, one for routine histology and one for tissue copper estimation, to reduce the risk of a falsely low value due to sampling error. Liver copper concentrations can, however, be raised in cholestatic liver diseases other than Wilson disease, reducing the specificity of the test.
The combination of a very low plasma copper concentration with a very high tissue copper concentration is diagnostic of Wilson disease, especially when Kayser–Fleischer rings are detected. However, not infrequently, either a liver biopsy is contraindicated because of severe coagulopathy or tissue concentrations are not high enough to be diagnostic, so other confirmatory tests may be required. The most reliable of these (and the simplest to perform) is the penicillamine challenge test. This involves collection of two 24 h urine samples for copper estimation before and after administration of D-penicillamine. This is given as two oral 500 mg doses, one at the start and one in the middle of the second 24 h period. A 10–20-fold increase in the 24 h urinary copper excretion after penicillamine administration is virtually diagnostic of Wilson disease. Advances in DNA sequencing techniques have meant that molecular diagnosis is becoming more readily available and is particularly helpful in diagnosing asymptomatic siblings once a family mutation in the ATP7B gene has been identified.
Long-term management of hepatic Wilson disease
Wilson disease is treated with penicillamine, which leads to a gradual improvement in standard LFTs. The rationale for the use of penicillamine was to increase urinary copper excretion and thereby reduce body copper. Despite the undoubted efficacy of the drug in Wilson disease, it now appears that liver copper concentrations do not change significantly and that the mechanism of action of penicillamine is considerably more complex than originally thought.
Penicillamine therapy is associated with a number of adverse drug reactions, including skin rashes, proteinuria, leukopenia and thrombocytopenia. It is particularly important to test the urine for protein during penicillamine treatment, because the drug can precipitate a number of immunologically mediated conditions including a form of nephrotic syndrome and Goodpasture syndrome. Treatment must be life-long, and even short interruptions may precipitate fatal hepatic failure. Trientine hydrochloride is an effective second-line treatment in patients who are intolerant of penicillamine. Since some may find penicillamine tablets unpleasant or inconvenient to take, and some children will never have had, or will not be able to remember having had, any symptoms of the disease, compliance may be a problem. Zinc has been used as monotherapy in children with Wilson disease; it acts as an inducer of gut mucosal metallothionein, which binds copper and leads to excretion in the stools. The best indication that an effective dose is being taken is said to be that the plasma copper concentration remains low. The use of zinc monotherapy in adults is controversial and in any event should only be introduced following treatment with either penicillamine or trientine for some years. Life-long monitoring with liver function tests and urinary copper measurements is mandatory in Wilson disease.
Indian childhood cirrhosis
This condition, which affects young children, was thought to be confined to the Indian subcontinent. The liver is grossly fibrotic and concentrations of tissue copper are in the same range as those seen in Wilson disease (see Table 14.8). Variants are now seen in other geographical areas and it appears that different mechanisms can lead to the same end-stage chronic liver disease. The Indian subcontinent variety is probably a childhood cirrhosis that requires a high environmental exposure to copper for full expression, as supplied by excess dietary copper (from milk stored in copper vessels). In the USA, a cholestatic process seems to underlie a similar form of cirrhosis with high concentrations of tissue copper. A genetically deleterious excretory defect is likely to be present. Arsenic ingestion has also been implicated. Penicillamine may be effective if given before decompensation develops.
α1-Antitrypsin deficiency
Deficiency of this glycoprotein, which is an enzyme that acts as an anti-proteinase predominantly inhibiting elastase, is associated with both pulmonary and liver disease. The condition is inherited co-dominantly and, in the classic form of the disease, histological examination of liver biopsy specimens reveals characteristic PAS-positive, diastase-resistant globules within the portal tracts. These globules represent accumulation of the defective protein within the endoplasmic reticulum. The mechanism of liver cell damage, which often progresses to cirrhosis, is ill understood.
The gene encoding α1-antitrypsin (α1-AT) is located on chromosome 14 (at 14q32.1). More than 90 genetic variants have been described, most of which are exceedingly rare and are associated with production of normal amounts of functional enzyme. The different variants (phenotypes) can be identified by their mobility on isoelectric focusing. The conventional nomenclature describes the various phenotypes by the letters ‘Pi’ (protease inhibitor) followed by an alphabetical code relating to their mobilities. The commonest (normal) phenotype is designated ‘M’ (medium) and is further subdivided into at least eight subtypes (M1, M2, M3 etc.). Of the mutations that result in α1AT deficiencies, the two commonest are designated ‘S’ (slow) and ‘Z’ (ultraslow), with homozygotes being described as PiSS or PiZZ, compound heterozygotes with defective phenotypes as PiSZ, and heterozygotes as, for example, PiMS or PiMZ. The S phenotype is mainly associated with lung disease, while the Z phenotype is strongly associated with both lung and liver disease. Other rare phenotypes are also associated with liver disease (Siiyama, MMalton, PiKing) (see also Chapter 43).
Individuals with defective phenotypes may present (or be identified incidentally) at any time from infancy to old age. The most important period is during the first few weeks of life, with about 25% of cases of the neonatal hepatitis syndrome being due to this condition. The association with liver disease is very variable. Some children, particularly those who do not develop symptoms in infancy, may never develop liver disease, while others may progress rapidly to cirrhosis and death from liver failure. The Z allele causes a Glu342Lys substitution in the protein that results in the formation of polymers that are retained in the endoplasmic reticulum of hepatocytes. These polymers are degraded via several pathways, including the ubiquitin–proteasome and the autophagy–lysosome systems. There is significant interindividual variation in the capacity of these systems, which might account for the variation in the extent of liver disease in affected individuals. The accumulation of polymers of α1-AT in the liver means that only 10–15% of the α1-AT molecules produced reach the circulation. This forms the basis for the initial screening test as plasma α1-AT concentrations are low (typically 0.1–0.2 g/L, normal 1.0–2.0 g/L). α1-Antitrypsin is a positive acute phase reactant and upregulation of α1-AT synthesis during an inflammatory response or acute hepatocellular damage can cause release of the accumulated polymers, increasing plasma concentrations to within the reference range. Strategies for investigation of possible α1-AT deficiency in these situations should include phenotyping and/or genotyping.
The hepatic porphyrias
These are so called because the major organ in which excessive production of the porphyrins or their precursors takes place is the liver (see Chapter 28). However, the four types (acute intermittent porphyria, hereditary coproporphyria, variegate porphyria and 5-aminolaevulinic acid dehydratase deficiency porphyria (Doss) porphyria) do not have any hepatic manifestations other than the occasional mild rise in plasma aminotransferase activity and an increased incidence of hepatocellular carcinoma in patients with acute intermittent porphyria. Porphyria cutanea tarda has already been described (p. 258). Asymptomatic porphyrinuria (mainly due to coproporphyrin) is common in many acute liver diseases and hepatic tumours. In erythropoietic porphyria, the liver plays a minimal role in protoporphyrin overproduction, but hepatic complications may occasionally arise: these comprise gallstones and cirrhosis associated with massive protoporphyrin deposition.
Cystic fibrosis
A wide spectrum of hepatobiliary complications of cystic fibrosis is becoming increasingly recognized as larger numbers of children survive to adulthood as a result of improvements in the treatment of the pulmonary complications of the disorder. In infancy, cystic fibrosis may present as neonatal cholestasis, often in association with meconium ileus. Older children may develop a fatty liver syndrome, but the major problem is a focal biliary cirrhosis that progresses to a generalized cirrhosis with all the complications as described previously. Extrahepatic biliary disease, with stricture of the distal common bile duct, is also frequent, although this, of itself, is probably not directly involved in the pathogenesis of the liver disease. Laboratory investigation is important in the diagnosis of this condition (see Chapter 25), but the LFTs are entirely non-specific and serve no more than to indicate that liver abnormalities are present. Liver function tests have been used to monitor response to therapy with ursodeoxycholic acid.
Other inherited metabolic diseases
Diagnosis, management and genetic counselling in children with inborn errors of metabolism form a large part of paediatric hepatology. The disorders also illustrate many aspects of applied intermediary metabolism. Most are rare and few laboratories will develop wide experience. Here, the more common types are described briefly, with more detailed accounts in the Further reading list at the end of the chapter.
Tyrosinaemia
This is an autosomal recessive condition, of which there are two types. Type I is due to deficiency of fumarylacetoacetate hydrolase (Fig. 14.3) and is the only genetically mediated disorder of amino acid metabolism that has so far been identified as a significant cause of liver disease. Type II is due to deficiency of tyrosine aminotransferase and is not associated with liver damage.
FIGURE 14.3 Intermediary metabolism of tyrosine. Classic tyrosinaemia (type I or tyrosinosis) is caused by a defect in fumarylacetoacetate hydrolase activity, but defects in other parts of the pathway are also recognized including alkaptonuria, in which there is deficiency of homogentisic acid oxidase. There are increases of all precursors (including tyrosine) in the urine, but the diagnostic test is for succinylacetone which, by reacting with sulphydryl groups, may in part be responsible for the tissue damage.
Type I tyrosinaemia usually presents at the age of 2–6 months with vomiting, diarrhoea, oedema, ascites, hepatosplenomegaly, hypoglycaemia and failure to thrive. There is a marked bleeding diathesis owing to prothrombin deficiency. Other characteristic features include systemic acidosis and renal tubular dysfunction leading to hypophosphataemic rickets. Rapid and accurate diagnosis is essential since dietary restriction of phenylalanine, tyrosine and methionine may improve the prognosis in this otherwise fatal disease. A more chronic form of the disease may present later in childhood with cirrhosis and a high incidence of hepatocellular carcinoma, although AFP concentrations may be grossly elevated even in the absence of a tumour.
The characteristic laboratory findings are a marked amino acidaemia, with tyrosine, phenylalanine and methionine concentrations being particularly elevated. There is also moderate derangement of LFTs, a raised INR and a markedly raised AFP. However, the diagnostic finding is the presence of succinylacetone in urine (or amniotic fluid, for prenatal diagnosis), usually with elevated urinary 5-aminolaevulinate (owing to the inhibition of porphobilinogen synthetase by the succinylacetone). Liver transplantation is being increasingly used in both forms of the disease. Treatment with nitisinone may be beneficial in slowing progression of the condition (see Chapter 24).
Galactosaemia
Galactose is generated by intestinal hydrolysis of lactose and is converted in the liver to glucose. Classic galactosaemia (see Chapter 24) is an autosomal recessive condition that is due to deficiency of the enzyme galactose 1-phosphate uridyltransferase (GALT), resulting in accumulation of galactose, galactitol (via an aldoreductase) and galactose 1-phosphate in all body tissues (Fig. 14.4). The disease usually becomes evident soon after birth when milk is introduced into the diet. It varies widely in severity but, in the majority, the LFTs are grossly deranged, with elevated plasma aminotransferases and a raised INR, indicating hepatocellular necrosis. There is a severe unconjugated jaundice due to haemolytic anaemia, accompanied by amino aciduria and proteinuria, indicating renal damage. Often, the condition rapidly progresses to liver failure, with bleeding and ascites. The mechanisms underlying the liver damage are poorly understood, but may be related to cellular ATP depletion owing to accumulation of galactose 1-phosphate in cells, which may restrict the availability of phosphate for formation of high-energy bonds. A less acute presentation may be with a chronic liver disease, cataracts and learning difficulties. Children with this condition have an increased risk of septicaemia.
FIGURE 14.4 Metabolism of galactose. The usual lesion in classic galactosaemia is a decreased activity of galactose 1-phosphate uridyltransferase, but the other enzymes listed may also be involved in some cases. Note that the epimerase activity is normal in most affected individuals so that even on a galactose-free diet, galactosaemic individuals generate enough galactose for normal development.
Galactosaemia should be suspected if sugars other than glucose are detected in urine. In this context, it should be noted that conventional urine stick tests detect only glucose and not other reducing sugars. The diagnosis requires the demonstration of low or absent GALT enzyme activity in erythrocytes and confirmation by genetic testing (see Chapter 24). Treatment is by withdrawal of lactose and galactose from the diet, which can be monitored by measurement of galactose-1-phosphate in blood samples.
Fructose intolerance
Three genetic defects in fructose intolerance have been identified: essential fructosaemia (hepatic fructokinase deficiency), hereditary fructose intolerance (fructose 1-phosphatase deficiency), and fructose 1,6-diphosphatase deficiency. All three are relatively benign conditions if fructose intake is avoided or substantially reduced. However, with high fructose intake, both hereditary fructose intolerance and fructose 1,6-diphosphatase deficiency can lead to severe liver and kidney damage. The mechanisms involved are not well understood but, by analogy with galactosaemia, the hepatic injury may be due to intracellular accumulation of phosphorylated sugars reducing availability of phosphate for ATP formation. The liver damage is reflected in grossly deranged LFTs, with markedly elevated aminotransferase activities, a conjugated hyperbilirubinaemia, hypoalbuminaemia and a raised INR, accompanied by hypokalaemia, hypophosphataemia, profound hypoglycaemia and thrombocytopenia. The renal damage leads to aminoaciduria and proteinuria.
Fructose 1,6-diphosphatase deficiency usually has less severe effects on the liver. Plasma aminotransferase activities tend to be mildly or moderately elevated and the bilirubin concentration is only mildly raised. The prominent features are hypoglycaemia, lactic acidaemia, aminoacidaemia (particularly alanine and glutamine) and a marked ketoacidosis. Diagnoses for all three conditions require demonstration of the deficiency of the particular enzyme in liver biopsy specimens, or genetic testing.
The sphingolipidoses and Niemann–Pick disease type C
The sphingolipidoses are recessively inherited lysosomal disorders in which there is a deficiency of a specific lysosomal hydrolytic enzyme and consequent deposition of complex lipids in various tissues. Type C Niemann–Pick disease is a relatively common inherited cause of liver disease in the UK. In most patients, the disease presents with a fatal neonatal hepatitis, but among those who survive, the liver disease becomes less prominent but death from progressive neurological damage is inevitable.
Glycogen storage diseases
There are 11 distinct types that comprise this group of autosomal recessive diseases, in which enzyme defects result in deficient mobilization of glycogen, deposition of abnormal forms in tissues and very variable prognosis. Types V and VII do not cause liver disease. In the remainder, which can affect the liver, clinical features are usually apparent in childhood and include gross hepatomegaly associated with hypoglycaemic attacks and growth failure. Descriptions of the biochemical lesions, specific clinical features and treatment of all the glycogen storage diseases are beyond the scope of this chapter and the reader is referred to specialist texts. Only type IV is consistently associated with cirrhosis. Liver function tests are abnormal in most types, particularly types III and IV, but jaundice is very rare. Hypoglycaemia is common to all and hyperuricaemia is prominent in types I, III and IX. Among those that affect the liver, the specific diagnoses in types II, III, IV and VI can be established by demonstrating the enzyme deficiencies in leukocytes, but liver biopsies are required for type I. Management revolves around dietary measures to maintain blood glucose concentrations. Liver transplantation is appropriate in some types, particularly types I and III, which can progress to hepatic adenomata and hepatocellular carcinoma.
LIVER TRANSPLANTATION
Liver transplantation is a rapidly expanding part of clinical hepatology. Improvement in survival figures over the last decade has largely been due to better defined indications, improvement in immunosuppressive regimens and organ preservation and improved surgical techniques. The main indication is end-stage chronic liver disease but, as already mentioned, the procedure is being increasingly used for acute liver failure and, in children, for treatment of certain inherited metabolic defects. Although the procedure is now routine in many centres, extensive preoperative assessment is still required and major complications are common. In any event, a liver transplantation programme will impose a heavy load on a clinical pathology laboratory. It is beyond the scope of this chapter to discuss all the problems that may arise, but they are summarized below.
Preoperative assessment
Selection of patients who are likely to benefit from a liver graft requires careful clinical judgement based both on the prognosis and on the quality of life with regard to the primary illness. The criteria are complex and depend on the aetiology of the liver failure and whether it is acute or is a consequence of chronic liver disease. The overall condition of the patient to withstand such a major surgical procedure needs to be carefully assessed. Thus, haemodynamic and respiratory status, patency of major blood vessels and concomitant diseases are essential considerations. The potential effects of the immunosuppressive drug therapy (to prevent graft rejection) on disease recurrence (e.g. in patients transplanted for malignant disorders or viral hepatitis), or on other complications such as pre-existing renal disease also need to be considered. ABO blood group compatibility is required but, in contrast to other solid organ grafting, pre-transplant tissue typing of recipients for human leukocyte antigen (HLA) matching is not essential, except with combined transplants (e.g. liver and kidney), because the liver appears to be an ‘immunologically privileged’ organ, and donor-recipient incompatibility leading to hyperacute rejection is a rare event (see below).
The immediate postoperative period
The quality of the functioning of the graft immediately after transplantation is a major determinant of the future clinical course and is consequently of great importance. Primary non-function of the graft, that is, a liver that does not appear to work satisfactorily from the outset, is usually due to the poor quality of the donor organ. Hyperacute rejection has been described but is a very rare cause of immediate graft failure. Its differentiation from primary non-function is difficult, but may be suspected if the graft appears to function during the first 6–24 h only to then fail.
The standard LFTs have little value during the immediate postoperative period because the patient’s blood will have been extensively diluted by multiple transfusions during the operation. Plasma aminotransferase activities on the second post-operative day are more reliable and are almost always elevated. This represents so-called ‘preservation injury’ to the donor organ acquired during its removal from the donor, and subsequent preservation and transportation prior to transplantation. Thereafter, a progressive fall in the aminotransferase activities indicates good graft function, while persistently high values suggest that the graft will not function well. Other parameters that are essential for monitoring graft function postoperatively are the blood hydrogen ion concentration (pH), glucose and coagulation factors. Hyperglycaemia is almost always present during the very early postoperative phase but, with normal graft function, the blood glucose usually normalizes over the following 24–48 h. Persistence of the hyperglycaemia, despite increasing doses of insulin, indicates poor graft function.
Acute cellular rejection occurs in 50–70% of patients during the first three weeks after transplantation, and requires rapid diagnosis and adjustment of immunosuppressive treatment to preserve the graft. It is manifest by an often abrupt increase in plasma aminotransferase activities and bilirubin concentrations. An increasing plasma bilirubin concentration seems to be the more sensitive indicator, but rising aminotransferases appear to be more specific. However, liver histology is required to confirm acute rejection. Some degree of renal impairment is often evident during the first 2–3 days, even if not present before the operation. Serious derangement of the biochemical liver tests, particularly in conjunction with evidence of a coagulopathy, is highly suggestive of interruption to the vascular supply to the graft.
Intermediate follow-up
Frequent monitoring of LFTs is required during the months after a liver transplant to detect signs of late acute or chronic rejection, along with frequent microbiological surveillance for evidence of infections. Late acute rejection (occurring > 1 month post-transplantation) is diagnosed as for early acute rejection. Chronic rejection is suspected if the LFTs were previously more or less normal but then begin to become deranged. In this situation, the aminotransferases and INR may be only mildly elevated, but there is a progressive increase in the serum bilirubin and other cholestatic indicators. These changes can also be seen in fungal, bacterial and viral infections, to which the immunosuppressed patient is particularly susceptible, but can usually be differentiated from rejection episodes on clinical and histological criteria together with microbiological investigations.
Patients are generally metabolically unstable for some months after transplantation and require careful adjustment of their immunosuppression. The principal immunosuppressive drugs currently used are ciclosporin, tacrolimus, mycophenolate mofetil and sirolimus, alone or in various combinations, sometimes with glucocorticoids and/or azathioprine. Several of these agents are nephrotoxic and potentially neurotoxic at high doses. Their metabolism is affected by a number of factors, including graft function, other drugs (e.g. antibiotics) that may be required and individual pharmacogenetic variability in their metabolism that can affect the blood concentrations at given doses. Thus, frequent monitoring (initially, two or three times per week) of the concentrations of these drugs in the blood is required during the months following transplantation in order to maintain them within the fairly narrow therapeutic ranges that provide adequate immunosuppression with minimal toxicity.
Long-term monitoring
From the laboratory standpoint, long-term monitoring of liver transplant recipients is relatively straightforward, unless complications arise. Because of the tenuous arterial supply of the common bile duct, biliary tract complications are frequent, but the laboratory features are no different from those seen in patients with other forms of biliary disease. By one year, most patients are well stabilized and require only three-monthly check-ups with routine LFTs, tests of renal function and haematological investigations. By this time also, they are usually well established on their maintenance immunosuppressive regimens and much less frequent monitoring of their blood drug concentrations is required.
Late graft loss, occurring more than one year after transplantation, is relatively rare and, today, many such patients can be rescued with a second transplant. The commonest causes of late graft failure are uncontrollable chronic rejection and recurrence of the primary disease. Thus, in patients transplanted for chronic viral hepatitis C, there is almost universal reinfection of the graft and consideration needs to be given to pre- and post-transplant antiviral therapy. In the case of patients transplanted for chronic hepatitis B, reinfection can be successfully prevented with regular immunoglobulin administration. The use of newly introduced antiviral agents is also under investigation. In those transplanted for hepatic malignancies, tumour recurrence is not infrequent and, as in other solid organ transplantation, there is an increased risk of non-hepatic malignancies, particularly lymphomas, arising either de novo or through reactivation by immunosuppression of extrahepatic tumours that have previously been apparently successfully treated. Additionally, as the numbers of long-term survivors rises, recurrence of PBC and AIH (in those transplanted for these conditions) is being seen with increasing frequency.
CONCLUSION
Liver disease is best classified according to its aetiology, qualified by the nature of the resulting pathological change. There are many aetiological factors, including viruses and other infective agents, drugs and toxins, metabolic diseases and autoimmune processes. The range of pathological responses is limited; the most frequent are hepatitis, cirrhosis and cholestasis.
Hepatitis can be acute or chronic: chronic hepatitis can lead to cirrhosis and cholestasis can be a feature of both acute and chronic liver disease. Liver disease frequently has extrahepatic manifestations, reflecting the central role of the liver in metabolism.
Liver transplantation is increasingly being employed in the management of many hitherto fatal liver diseases.