130 Septic Shock
 Incidence
Incidence
Septic shock is the form of acute circulatory shock that occurs secondary to severe infection. The incidence of severe sepsis and septic shock is rising, partly related to medical progress that allows individuals to survive longer, resulting in increased numbers of older, debilitated, or immunocompromised patients passing through the intensive care unit (ICU). Some 10% to 15% of ICU patients develop septic shock at one time or another, and the mortality rate is 50% to 60%.1 Somewhat lower mortality rates have been reported in some trials evaluating the effects of new therapeutic interventions,2 but such studies include a number of exclusion criteria that are often associated with high mortality rates—cirrhosis, immunosuppression, and “do-not-resuscitate orders,” for example—so it is perhaps not surprising that mortality rates are lower in these therapeutic trials than in “real life.”
 Etiology of Septic Shock
Etiology of Septic Shock
The organisms involved in severe sepsis and septic shock are most often bacterial. While in the past gram-negative organisms were most commonly implicated, increasingly gram-positive organisms are isolated, such that roughly similar numbers of gram-positive and gram-negative organisms are now involved.1 Septic shock can also be caused by a fungal or parasitic infection. In a third of patients, no infectious agent is identified.1 About half of infections are nosocomial in origin. Although an infection can arise anywhere, the lung is presently the most common source of infection (40%), followed by the abdomen (20%), indwelling venous and arterial catheters and primary bacteremias (15%), and the urinary tract (10%).1
 Pathophysiology of Septic Shock
Pathophysiology of Septic Shock
The pathophysiology of septic shock is complex and covered in detail in Chapter 129 (Reinhart & Bloos). Essentially, the systemic sepsis response starts with recognition of an invading organism or its toxins. Among the bacterial factors, one of the best-known toxins is lipopolysaccharide (LPS), which is part of the outer gram-negative bacterial membrane, but other bacterial-derived factors include lipoteichoic acid and peptidoglycan. In certain cases, essentially infections involving Staphylococcus aureus or β-hemolytic group A Streptococcus, the formation of superantigens results in toxic shock syndrome.
The early humoral response involves the complement and contact (kinin-kallikrein) systems. Immune cells, principally monocytes/macrophages and polymorphonuclear neutrophils (PMN), are not only able to recognize pathogenic agents and their products so they can phagocytose and destroy them, but also release a series of mediators which can themselves activate other cells. Among the cell membrane receptors implicated in the recognition of pathogenic agents are the so-called Toll-like receptors (TLR), a family of 10 members. Of these, TLR4 is the receptor for LPS; TLR2 for a number of products from gram-positive bacteria such as peptidoglycans, mycobacteria, and yeasts; and TLR9 for bacterial DNA.3 In response to cellular stimulation, intracellular signaling is activated, resulting largely in activation of transcriptional factors, including nuclear factor kappa B (NF-κB), which in turn are responsible for initiation of proinflammatory reactions. A number of cytokines, two of the key players being tumor necrosis factor alpha (TNF-α) and interleukin (IL)-1 that interact synergistically, are released by macrophages and other cells. TNF-α and IL-1 are particularly important proinflammatory cytokines whose administration in animals can reproduce all the features of septic shock including hypotension and development of multiple organ failure. A host of secondary mediators including lipid mediators, oxygen free radicals, proteases, and arachidonic acid metabolites are also released by macrophages, PMNs, and other cells. Vasodilator substances such as nitric oxide (NO) and prostaglandins are released by endothelial cells and are responsible for the early hemodynamic changes of sepsis. NO in particular is a powerful vasodilator acting on vascular smooth muscle. Increased NO production is essentially due to induction of inducible NO synthase (iNOS) by proinflammatory cytokines. The formation of large quantities of NO can also have secondary toxic effects on cells. NO can block mitochondrial respiration, directly via inhibition of cytochrome a,a3 and by reaction with superoxide radicals, resulting in the production of peroxynitrite, which inhibits various phases of mitochondrial respiration.4 These effects result in depletion of cellular adenosine triphosphate (ATP) and potentially severe detrimental effects on cell function. It is important to note that the inflammatory response also causes release of vasoconstrictor substances including thromboxane and endothelins.
Other effects of the inflammatory reaction that accompanies septic shock include expression of adhesion molecules on vascular endothelium and circulating cells (platelets, PMNs, and monocytes), allowing adhesion of activated leukocytes and their migration into subendothelial tissues. Alterations in intercellular endothelial junctions result in increased capillary permeability and generalized edema. Alterations of coagulation and fibrinolysis complete the picture, with proinflammatory mediators creating a procoagulant state. Briefly, activation of tissue factor on the surface of various cells, particularly monocytes and endothelial cells, initiates the coagulation system.5 In addition, sepsis causes a significant reduction in plasma levels of natural anticoagulants such as protein C, protein S, and antithrombin by reducing their synthesis, increasing their consumption, and increasing their clearance. Thrombolysis is also stimulated with an increase in levels of plasminogen activator inhibitor (PAI-1). The net result is a balance in favor of procoagulant processes, often leading to disseminated intravascular coagulation (DIC) and participating in the microcirculatory disorder that leads to multiple organ failure and death in many patients with severe sepsis.
During the sepsis response, antiinflammatory mediators including IL-4 and IL-10 are also released, which limit the effects of the proinflammatory mediators and can lead to a state of relative immunosuppression, sometimes called immunoparalysis.6
 Classification
Classification
Following recommendations from the Sepsis Conference,7 patients with septic shock may be classified according to the letters PIRO:
P = Predisposing Factors
Each patient has specific characteristics. For example, an individual receiving long-term immunosuppressant therapy requires a different approach than someone who was previously healthy. Factors associated with lifestyle, such as alcoholism, may influence the course of septic shock.8 Patient age and gender may also be important. Increasingly, genetics are being considered, and studies are discovering which genetic factors can influence the development of and survival from severe sepsis. In particular, a polymorphism of the TNF-α promoter gene has been associated with increased risk of sepsis.9 Multiple other polymorphisms that may influence the response of the host to pathogenic organisms have been described, including for IL-1 receptor antagonist (IL-1ra), TLR2, and IL-6.10 Improved understanding of these aspects should help better direct therapeutic strategies.
I = Infectious Insult
This refers to the specific characteristics of the infection, that is, the agent or pathogen involved (e.g., gram-positive versus gram-negative, bacteria versus fungus),11 the source of the sepsis (e.g., urinary tract versus respiratory tract),12 and the degree of extension of the infection (e.g., pneumonia confined to one lobe of one lung versus generalized bilateral lung involvement, appendicitis versus generalized peritonitis). All these factors can influence the severity of the sepsis response and the patient’s likely response to therapy.
R = Host Response
This refers to the factors involved in the inflammatory response of the host to the infection, assessed largely by the presence or absence of the signs and symptoms of sepsis (e.g., degree of elevation of white blood cell count, CRP, procalcitonin). Each patient mounts a different response dependent on various factors including those previously discussed, and a patient’s response will vary with their clinical course and treatment.13
O = Organ Dysfunction
This refers to the degree of organ dysfunction related to sepsis and can be evaluated using various scoring systems, including the SOFA (sequential organ failure assessment) score,14 which uses objective, readily available measures to quantify the dysfunction of six organ systems (Table 130-1). Dysfunction of each organ is rated according to a scale (0 [normal function] to 4 [organ failure]), and individual scores can then be summed to provide a total. Individual organ function as well as a composite score can thus be followed during the course of disease and treatment.
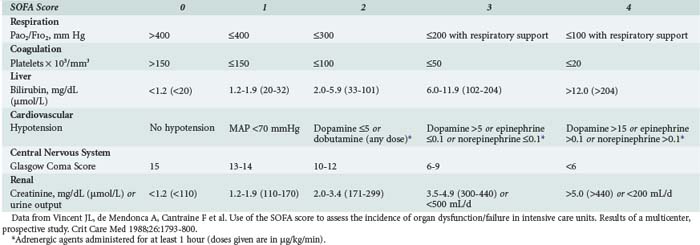
 Clinical Presentation
Clinical Presentation
One may anticipate that patients with septic shock will have fever, hyperleukocytosis, and other typical features of sepsis, but unfortunately this is not always true. Fever may be an important clue, but moderate fever can be found in other types of shock. More importantly, fever is often absent in septic shock; in fact, hypothermia may be present in 15% to 20% of cases, and this symptom is associated with higher mortality rates.15 Hyperleukocytosis is also nonspecific and can be found in other types of circulatory failure. Likewise, lactic acidosis, a hallmark of all types of circulatory failure, is usually compensated by hyperventilation, so tachypnea is not specific for septic shock. Similarly, tachycardia can be the result of the circulatory alterations associated with any type of shock.
 Hemodynamic Changes
Hemodynamic Changes Monitoring
Monitoring
Any patient with septic shock requires monitoring with an arterial catheter to enable reliable and continuous assessment of arterial pressure. Changes in systolic and pulse pressure in mechanically ventilated patients during the respiratory cycle may also indicate a greater likelihood of response to a fluid challenge.16 The arterial catheter also facilitates blood sampling, notably for blood gas analysis.
Invasive Versus Less-Invasive Monitoring
The role of the pulmonary artery catheter (PAC) in critically ill patients has been questioned.17 However, although no study has conclusively demonstrated positive effects of this type of monitoring on outcome,18–20 information obtained from the PAC may help in guiding patient management.21 The PAC is useful not only for monitoring pulmonary artery occlusion pressure (PAOP) and cardiac output but also allows assessment of mixed venous oxygen saturation (SvO2), a highly useful parameter because a fall in SvO2 is generally associated with inadequate oxygen transport. Importantly, the PAC is not necessary in all patients but is likely to be of use in complex cases, particularly in patients with concomitant cardiopulmonary disease.
Less-invasive monitoring techniques are increasingly being used. Echocardiography can provide useful additional information, largely to visualize the degree of ventricular filling and ejection volume. However, echocardiography requires an experienced operator, gives no information on the adequacy of cardiac output for the patient’s needs, and is difficult to perform continuously, so information is intermittent. Other less-invasive methods of monitoring cardiac output include PiCCO, LidCO, transesophageal Doppler techniques, and even bioimpedance or bioreactance techniques.22 However, measurement of cardiac output in isolation is not very helpful in most critically ill patients.
Blood Lactate Levels
Blood lactate level is an important biological variable in determining the adequacy of perfusion and oxygenation. Normal blood lactate level is around 1 mEq/L, and hyperlactatemia becomes pathological above 2 mEq/L. Although in other forms of circulatory shock, hyperlactatemia is due to cellular hypoxia, in septic shock additional mechanisms may play an important role in raising blood lactate levels. In sepsis, blood lactate levels may be raised by an increase in cellular metabolism, by inhibition of pyruvate dehydrogenase, and by reduced clearance. Repeated measurements enable one to assess the efficacy of treatment and have a predictive value superior to derived oxygenation parameters.23 The evolution of blood lactate levels enables a global evaluation of the state of the shock, although in view of the relatively slow rate of change, blood lactate levels cannot be used to guide resuscitation.
Peripheral Perfusion Parameters
Other techniques for monitoring peripheral perfusion have been developed. While the sublingual region is not one that would immediately seem to be of most interest, it is easily accessible, and using techniques of orthogonal polarization spectral (OPS) or sidestream darkfield (SDF) imaging, heterogeneity of microcirculatory flow and reduced perfused vessel density and proportion of perfused vessels can be observed (Figure 130-1) and quantified in patients with sepsis.24,25 Moreover, the impact of therapeutic interventions on such changes can be monitored,26,27 opening the possibility that monitoring the microcirculation could be used to guide treatment.

Near-infrared spectroscopy (NIRS) is a technique that uses the differential absorption properties of oxygenated and deoxygenated hemoglobin to evaluate tissue oxygenation (StO2). Analysis of changes in StO2 during a circulatory stress test, such as a brief episode of forearm ischemia (venous or arterial occlusion), may be more useful to quantify sepsis-induced microvascular dysfunction than an isolated StO2 value.28
Although these techniques have demonstrated clearly the presence of alterations in the microcirculation in patients with severe sepsis, which are associated with prognosis,29,30 further research is needed to fully evaluate the relevance of these values to early resuscitation and care of critically ill patients.
 Management
Management
Management of the patient with septic shock involves three inseparable components: treatment of the infection, cardiovascular resuscitation, and immunomodulation (Figure 130-2). Detailed guidelines for the management of patients with severe sepsis or septic shock have been published.31

Cardiovascular Resuscitation
The VIP ruse proposed by Weil and Shubin32 should be followed. Each patient is in fact a VIP, but the letters refer here to Ventilation, Infusion, and Pump.
I = Infusion
Septic shock is accompanied by absolute and relative hypovolemia, the result of various mechanisms:
Assessment of an adequate volume state is essentially clinical: restoration of arterial pressure, improvement of cutaneous perfusion, improved urine output, and improved mental state. The central venous pressure (CVP) can be a useful guide, but it is not possible to define in advance the CVP level that should be reached in any individual patient. However, monitoring CVP or PAOP is essential to limit the risk of pulmonary edema. In fluid replacement, it is preferable to use a fluid challenge technique in which filling pressures are measured at regular intervals during fluid administration (Table 130-2).33 If a PAC is in place, it is recommended that fluid replacement be given until cardiac output reaches a plateau and further fluid causes no further increase in cardiac output.
| Define | Example |
|---|---|
| The type of fluid | Ringer’s lactate |
| The rate of infusion | 500 mL in 20 min |
| The goal | Mean arterial pressure >75 mm Hg |
| The limits | Central venous pressure 16 mm Hg |
There has been considerable debate as to which fluid should be used in sepsis, but it is the quantity of fluid rather than the type of fluid per se that is of greatest importance. Because of their propensity for leakage into the extravascular space, greater volumes of crystalloids are needed to achieve the same effect as colloid,34 thus potentially increasing the risk of edema, but colloids are more expensive and carry their own risks. In particular, there has been considerable controversy about the use of albumin in critically ill patients, but a large multicenter study performed in Australasia (the SAFE study)35 showed that albumin administration was not associated with worse outcomes.
P = Pump (Vasoactive Agents)
Dopamine was often recommended as the first-line drug for its mixed β- and α-adrenergic effects. However, a recent randomized controlled study showed no differences in mortality rates in patients with shock treated with dopamine or norepinephrine as first-line vasopressor, but dopamine use was associated with increased adverse effects, notably arrhythmias.36 Norepinephrine is, therefore, the preferred first-line vasopressor in patients with septic shock. Epinephrine should not be used as a first-line vasopressor in septic shock; it can have deleterious effects on the splanchnic circulation.37 Dobutamine is often added to vasopressor therapy, particularly when using norepinephrine, to increase cardiac output by its positive inotropic effects.
Immunomodulation
The link between coagulation and inflammation led to the suggestion that some of the key coagulation proteins may have beneficial effects in sepsis. Administration of activated protein C (drotrecogin alfa [activated]) early in severe sepsis or septic shock reduced mortality2 and morbidity.38 In addition to its anticoagulation effects, activated protein C has important antiinflammatory effects, can influence cell signaling, and has antiapoptotic effects,39 which may help explain why it has been shown to have beneficial effects in sepsis while other anticoagulants (antithrombin, tissue factor pathway inhibitor) have not. Drotrecogin alfa (activated) is indicated in patients with severe sepsis (sepsis associated with organ dysfunction) at a dose of 24 µg/kg/h via a continuous intravenous (IV) perfusion for 96 hours. It has not been shown to be effective in patients with less severe sepsis40 or in children41 and should not be used in patients who have recently undergone a surgical procedure.40 Drotrecogin alfa (activated) administration is associated with an increased risk of hemorrhage,42 such that it is contraindicated in patients with a high risk of bleeding. Infusions should be stopped 2 hours prior to any surgical intervention but may be restarted 12 hours after major interventions or sooner for more minor procedures if hemostasis is assured. The high costs of the drug may also limit its use, although its cost-effective profile is similar to many other accepted ICU therapies.43
The administration of steroids for patients with sepsis was proposed many years ago, but at the large doses studied (about 30 mg/kg of methylprednisolone) was never shown to have a beneficial effect on survival. More recently, the concept of relative adrenal insufficiency, based on the response to an ACTH test, reawakened interest in steroids, and moderate doses of corticosteroids (50 mg hydrocortisone IV every 6 hours) in patients with septic shock were shown to restore the activity of vascular adrenergic receptors, without excessive immunosuppressive effects, thus improving hemodynamic status and reducing mortality.44 Although initially it was recommended that this treatment strategy be guided by an ACTH test, this has now been abandoned because of difficulties with the interpretation of such tests.31 Moreover, a recent multicenter study failed to confirm the beneficial effects of moderate dose corticosteroids in patients with less severe sepsis.45 Interestingly, a recent post hoc analysis of a large multicenter trial suggested that administration of low-dose vasopressin in combination with corticosteroids was associated with improved mortality rates and reduced organ dysfunction compared to the combination of norepinephrine and vasopressin.46
The treatment of fever is controversial. Increased body temperature increases oxygen requirements, but the increased cellular metabolism may form part of the body’s natural defense. Animal studies have suggested that control of fever may be detrimental,47 and the release of heat shock proteins in fever may have important protective effects.48 A multicenter study in patients with severe sepsis reported that ibuprofen, a cyclooxygenase inhibitor, was well tolerated but did not reduce mortality.49
High-flow hemofiltration techniques can remove a range of bacterial products and mediators but are not without risk, notably because this process can remove beneficial products such as hormones and medications, including antibiotics, as well as potentially harmful substances.50 Clinical studies have provided conflicting data regarding the effects of these techniques on outcomes.51,52
Nutritional Support
Organ Support
Organ dysfunction can involve any organ and can be quantified using the SOFA score (see Table 130-1). Techniques for individual organ support are covered in separate chapters, but an overview is given here.
Respiratory Alterations
Respiratory failure is a common complication of sepsis and is characterized by hypoxemia associated with the presence of bilateral infiltrates on chest radiograph, with no evidence of left hear failure (normal PAOP). The diagnosis of acute respiratory distress syndrome (ARDS) is made when the PaO2/FIO2 ratio is less than 200 mm Hg; the less severe form, acute lung injury (ALI), is defined as a PaO2/FIO2 less than 300 mm Hg.56
When starting a patient on mechanical ventilation, several factors need particular attention:
Renal Alterations
Sepsis is the leading cause of acute renal failure in the ICU.60 Renal function can worsen as a result of circulatory changes associated with vasoconstriction of the afferent arteries and reduced glomerular filtration rate. In addition, management of the patient with sepsis often involves the administration of nephrotoxic agents—for example, certain medicines or contrast agents for radiologic examinations.
Unfortunately, there is no prophylactic approach to renal failure other than to try and maintain adequate renal perfusion and overall volume state. Administration of low (renal)-dose dopamine is ineffective at preventing renal failure,61 and diuretics may be harmful.62
Cerebral Function Alterations
Circulatory shock is typically accompanied by an alteration of intellectual function, initially manifested as confusion without real coma and reversible with resolution of shock. Cerebral alterations can be prolonged, and the patient is then said to have septic encephalopathy. The exact cause of the encephalopathy is unclear, although various mediators of sepsis have been implicated. Investigations are of little use except to exclude other causes. The electroencephalogram (EEG) generally shows a slow diffuse slowing, whereas cerebral computed tomography (CT) and cerebrospinal fluid examination are normal.63
 Conclusion
ConclusionKey Points
Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699-709.
De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362:779-789.
Dellinger RP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296-327.
Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250-1256.
Important report of the Sepsis Definitions Conference and introducing the PIRO concept.
Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358:111-124.
Study demonstrating no beneficial effect of corticosteroids in patients with septic shock.
1 Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med. 2006;34:344-353.
2 Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699-709.
3 Salomao R, Martins PS, Brunialti MK, et al. TLR signaling pathway in patients with sepsis. Shock. 2008;30(Suppl 1):73-77.
4 Hollenberg SM, Cinel I. Bench-to-bedside review: nitric oxide in critical illness–update 2008. Crit Care. 2009;13:218.
5 Amaral A, Opal SM, Vincent JL. Coagulation in sepsis. Intensive Care Med. 2004;30:1032-1040.
6 Volk HD, Reinke P, Docke WD. Clinical aspects: from systemic inflammation to “immunoparalysis. Chem Immunol. 2000;74:162-177.
7 Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250-1256.
8 Moss M, Parsons PE, Steinberg KP, et al. Chronic alcohol abuse is associated with an increased incidence of acute respiratory distress syndrome and severity of multiple organ dysfunction in patients with septic shock. Crit Care Med. 2003;31:869-877.
9 Teuffel O, Ethier MC, Beyene J, et al. Association between tumor necrosis factor-alpha promoter-308 A/G polymorphism and susceptibility to sepsis and sepsis mortality: a systematic review and meta-analysis. Crit Care Med. 2010;38:276-282.
10 Sutherland AM, Walley KR. Bench-to-bedside review: Association of genetic variation with sepsis. Crit Care. 2009;13:210.
11 Gao H, Evans TW, Finney SJ. Bench-to-bedside review: sepsis, severe sepsis and septic shock–does the nature of the infecting organism matter? Crit Care. 2008;12:213.
12 Volakli E, Spies C, Michalopoulos A, et al. Infections of respiratory or abdominal origin in ICU patients: what are the differences? Crit Care. 2010;14:R32.
13 Gerlach H, Dhainaut JF, Harbarth S, et al. The PIRO concept: R is for response. Crit Care. 2003;7:256-259.
14 Vincent JL, de Mendonca A, Cantraine F, et al. Use of the SOFA score to assess the incidence of organ dysfunction/failure in intensive care units: results of a multicenter, prospective study. Working group on “sepsis-related problems” of the European Society of Intensive Care Medicine. Crit Care Med. 1998;26:1793-1800.
15 Peres Bota D, Lopes Ferreira F, Melot C, et al. Body temperature alterations in the critically ill. Intensive Care Med. 2004;30:811-816.
16 Marik PE, Cavallazzi R, Vasu T, et al. Dynamic changes in arterial waveform derived variables and fluid responsiveness in mechanically ventilated patients: a systematic review of the literature. Crit Care Med. 2009;37:2642-2647.
17 Rubenfeld GD, McNamara-Aslin E, Rubinson L. The pulmonary artery catheter, 1967-2007: rest in peace? JAMA. 2007;298:458-461.
18 Harvey S, Harrison DA, Singer M, et al. Assessment of the clinical effectiveness of pulmonary artery catheters in management of patients in intensive care (PAC-Man): a randomised controlled trial. Lancet. 2005;366:472-477.
19 Wheeler AP, Bernard GR, Thompson BT, et al. Pulmonary-artery versus central venous catheter to guide treatment of acute lung injury. N Engl J Med. 2006;354:2213-2224.
20 Sakr Y, Vincent JL, Reinhart K, et al. Use of the pulmonary artery catheter is not associated with worse outcome in the ICU. Chest. 2005;128:2722-2731.
21 Mimoz O, Rauss A, Rekik N, et al. Pulmonary artery catheterization in critically ill patients: a prospective analysis of outcome changes associated with catheter-prompted changes in therapy. Crit Care Med. 1994;22:573-579.
22 de Waal EE, Wappler F, Buhre WF. Cardiac output monitoring. Curr Opin Anaesthesiol. 2009;22:71-77.
23 Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med. 2004;32:1637-1642.
24 De Backer D, Creteur J, Preiser JC, et al. Microvascular blood flow is altered in patients with sepsis. Am J Respir Crit Care Med. 2002;166:98-104.
25 De Backer D, Hollenberg S, Boerma C, et al. How to evaluate the microcirculation: report of a round table conference. Crit Care. 2007;11:R101.
26 Ospina-Tascon G, Neves AP, Occhipinti G, et al. Effects of fluids on microvascular perfusion in patients with severe sepsis. Intensive Care Med. 2010.
27 Sakr Y, Chierego M, Piagnerelli M, et al. Microvascular response to red blood cell transfusion in patients with severe sepsis. Crit Care Med. 2007;35:1639-1644.
28 Creteur J. Muscle StO2 in critically ill patients. Curr Opin Crit Care. 2008;14:361-366.
29 Sakr Y, Dubois MJ, De Backer D, et al. Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Crit Care Med. 2004;32:1825-1831.
30 Creteur J, Carollo T, Soldati G, et al. The prognostic value of muscle StO2 in septic patients. Intensive Care Med. 2007;33:1549-1556.
31 Dellinger RP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296-327.
32 Weil MH, Shubin H. The “VIP” approach to the bedside management of shock. JAMA. 1969;207:337-340.
33 Vincent JL, Weil MH. Fluid challenge revisited. Crit Care Med. 2006;34:1333-1337.
34 van der Heijden M, Verheij J, Nieuw Amerongen GP, et al. Crystalloid or colloid fluid loading and pulmonary permeability, edema, and injury in septic and nonseptic critically ill patients with hypovolemia. Crit Care Med. 2009;37:1275-1281.
35 Finfer S, Bellomo R, Boyce N, et al. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004;350:2247-2256.
36 De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362:779-789.
37 De Backer D, Creteur J, Silva E, et al. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med. 2003;31:1659-1667.
38 Vincent JL, Angus DC, Artigas A, et al. Effects of drotrecogin alfa (activated) on organ dysfunction in the PROWESS trial. Crit Care Med. 2003;31:834-840.
39 Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161-3172.
40 Abraham E, Laterre PF, Garg R, et al. Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med. 2005;353:1332-1341.
41 Nadel S, Goldstein B, Williams MD, et al. Drotrecogin alfa (activated) in children with severe sepsis: a multicentre phase III randomised controlled trial. Lancet. 2007;369:836-843.
42 Bernard GR, Macias WL, Joyce DE, et al. Safety assessment of drotrecogin alfa (activated) in the treatment of adult patients with severe sepsis. Crit Care. 2003;7:155-163.
43 Angus DC, Linde-Zwirble WT, Clermont G, et al. Cost-effectiveness of drotrecogin alfa (activated) in the treatment of severe sepsis. Crit Care Med. 2003;31:1-11.
44 Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862-871.
45 Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358:111-124.
46 Russell JA, Walley KR, Gordon AC, et al. Interaction of vasopressin infusion, corticosteroid treatment, and mortality of septic shock. Crit Care Med. 2009;37:811-818.
47 Su F, Nguyen ND, Wang Z, et al. Fever control in septic shock: beneficial or harmful? Shock. 2005;23:516-520.
48 Wheeler DS, Wong HR. Heat shock response and acute lung injury. Free Radic Biol Med. 2007;42:1-14.
49 Bernard GR, Wheeler AP, Russell JA, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N Engl J Med. 1997;336:912-918.
50 Vincent JL. Sepsis: clearing the blood in sepsis. Nat Rev Nephrol. 2009;5:559-560.
51 Payen D, Mateo J, Cavaillon JM, et al. Impact of continuous venovenous hemofiltration on organ failure during the early phase of severe sepsis: a randomized controlled trial. Crit Care Med. 2009;37:803-810.
52 Cruz DN, Antonelli M, Fumagalli R, et al. Early use of polymyxin B hemoperfusion in abdominal septic shock: the EUPHAS randomized controlled trial. JAMA. 2009;301:2445-2452.
53 Van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359-1367.
54 Vincent JL. Blood glucose control in 2010: 110 to 150 mg/dL and minimal variability. Crit Care Med. 2010;38:993-995.
55 Hermanides J, Vriesendorp TM, Bosman RJ, et al. Glucose variability is associated with intensive care unit mortality. Crit Care Med. 2010;38:838-842.
56 Bernard GR, Artigas A, Brigham KL, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818-824.
57 The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301-1308.
58 Kress JP, Pohlman AS, O’Connor MF, et al. Daily interruption of sedative infusions in critically ill patients undergoing mechanical ventilation. N Engl J Med. 2000;342:1471-1477.
59 Strom T, Martinussen T, Toft P. A protocol of no sedation for critically ill patients receiving mechanical ventilation: a randomised trial. Lancet. 2010;375:475-480.
60 Andrikos E, Tseke P, Balafa O, et al. Epidemiology of acute renal failure in ICUs: a multi-center prospective study. Blood Purif. 2009;28:239-244.
61 Bellomo R, Chapman M, Finfer S, et al. Low-dose dopamine in patients with early renal dysfunction: a placebo-controlled randomised trial. Australian and New Zealand Intensive Care Society (ANZICS) Clinical Trials Group. Lancet. 2000;356:2139-2143.
62 Mehta RL, Pascual MT, Soroko S, et al. Diuretics, mortality, and nonrecovery of renal function in acute renal failure. JAMA. 2002;288:2547-2553.
63 Iacobone E, Bailly-Salin J, Polito A, et al. Sepsis-associated encephalopathy and its differential diagnosis. Crit Care Med. 2009;37:S331-S333.
