26. Neurologic Disorders *
M. Terese Verklan and Suzanne M. Lopez
The developing nervous system provides ongoing challenges for researchers and clinicians. Investigations continue in a wide variety of areas, yet basic mechanisms for a pathophysiologic understanding of common events such as neonatal seizures and intraventricular hemorrhages (IVHs) remain unclear.
Improved neonatal care in recent years has not significantly reduced neurologic sequelae. How much of this is a reflection of survival of sicker and more immature infants is difficult to assess. Primary neurologic disease and secondary neurologic complications from such common conditions as cardiopulmonary disease, metabolic derangements, shock, infection, and coagulopathy still represent major problems encountered in every intensive care nursery. Serious anomalies still appear with regularity, yet in small numbers.
This chapter deals with selected topics in neonatal neurology, including congenital malformations, trauma, seizures, hypoxic-ischemic encephalopathy, and IVH.
CONGENITAL MALFORMATIONS
Physiology, Etiologic Factors, and Clinical Features
Congenital malformations of the nervous system occur when the usual sequence of maturation and development is interrupted (Table 26-1). 39 Present at birth, the etiology is multifactorial and sometimes unclear. Although strictly destructive lesions (e.g., hydranencephaly resulting from bilateral carotid artery occlusion) are separate from primary failures of morphogenesis, both may be included in the broad category of congenital malformations. The distinction between the two types lies in an understanding of the causes.
| Maturational Process | Time | Associated Defects |
|---|---|---|
| Neural tube defects (dorsal induction, neurulation) | 3-4 weeks |
Craniorhachischisis
Anencephaly
Myeloschisis
Encephalocele
Myelomeningocele
Arnold-Chiari malformation
|
| Prosencephalic development61 | 2-3 months |
Cyclopia
Holoprosencephaly
Arrhinencephaly
Septo-optic dysplasia
Agenesis of corpus callosum
Agenesis of septum pellucidum
|
| Proliferation | 2-4 months |
Microcephaly
Megalencephaly
Neurocutaneous syndromes (?)
|
| Migration61 | 3-5 months |
Schizencephaly
Lissencephaly
Pachygyria (macrogyria)
Microgyria (polymicrogyria)
Neuronal heterotopias
|
| Neuronal organization and functional organization | 6 months |
Down syndrome (?)
Mental retardation (?)
Genetic epilepsy (?)
|
| Myelination61 | 2nd trimester61 | Anoxic/ischemic damage |
Understanding congenital malformations requires an appreciation of the normal embryologic sequence. 39 The clinical and pathologic identification of normal and abnormal structures makes it possible to determine the timing of the insult or development failure. Once timing is established, an appropriate search for the cause can be made.
Neural Tube Defects
The incidence in the United States of neural tube defects (NTDs) is approximately 1 to 2 in 1000 births (see the Critical Findings box on p. 749). 41 Although the prevalence of NTDs has decreased, they are one of the most common congenital anomalies contributing to morbidity and mortality in neonates. 2 Changes in vertebral, vascular, meningeal, and dermal structures are typically found along with the defects. The more common types of NTDs include anencephaly, encephalocele, myelomeningocele, and occult spina bifida. 5 Genetic and environmental factors play a role in the development of NTDs. Familial incidence also plays a role; when one family member is affected, the risk increases by 2% to 3% in subsequent offspring and doubles if two or more family members are affected. 5 Cytogenic abnormalities are found in approximately 2% to 16% of neonates who have an isolated NTD. 35 Teratogen exposure has also been linked to NTDs. 39 A prepregnancy history of diabetes, specific drugs (especially anticonvulsants and sulfonamide drugs), and maternal hyperthermia secondary to using a hot tub or sauna have been identified as risk factors. 17 Recently, prepregnancy maternal obesity also has been linked to an increased risk. 64
The major environmental factor linked to NTDs is a dietary level of folic acid. 27 Folic acid supplements before and during pregnancy have been cited as substantially lowering the incidence of these NTDs. The U.S. Public Health Service issued a recommendation that women of childbearing years consume 400 mcg of folic acid each day to prevent NTDs. The American Academy of Pediatrics (AAP) also supports this recommendation. 9,16,27,39,41 Such an intake can be achieved by dietary supplementation of folate, adding folic acid to U.S. enriched grain products (e.g., bread, flour), and consuming foods containing folic acid (e.g., citrus fruit, beans, leafy greens). The Food and Drug Administration (FDA) required all enriched grain products to be fortified with folic acid by 1998. 6,16 In spite of this, it has been reported by the Centers for Disease Control and Prevention (CDC) that two thirds of American women fail to ingest an adequate amount of folic acid. 9 It has been argued that the amount of folic acid supplementation in grain products may be inadequate, supplying only about one fourth of daily need. 40 Noting the 26% decrease in the incidence of NTDs after the FDA required 140 mcg folic acid per 100 g of grain, the March of Dimes recommended an increase in the level of folic acid fortification. 37,62 Others, including the CDC and AAP, concur with this recommendation to increase the requirement to 350 mcg folic acid per 100 g of grain. 7
Neural Tube Defects
• As many as 50% or more of neural tube defects (NTDs) are preventable.
• Two thirds of American women fail to ingest an adequate amount of folic acid, and enriched grain products supply only one fourth of daily need.
• All women of childbearing age should consume 400 mcg of folic acid daily even when not planning to become pregnant.
• At increased risk: women with a prior NTD pregnancy. For these women, the recommended dose of folic acid is increased to 4 mg daily. It should be taken at least 1 month before conception.
• Sources: Dietary supplements, enriched grain products, and consumption of foods with folic acid content (citrus fruit, beans, leafy greens).
• The Centers for Disease Control and Prevention (CDC,) the American Academy of Pediatrics (AAP), and the March of Dimes have all recommended an increase in the amount of folic acid used to fortify grain products from 140 mcg to 350 mcg per 100 g of grain.
• Inadequate education of women continues to be a problem.
A Cochrane review concluded that supplementation of folate provides “a strong protective effect against neural tube defects.” Recommendations were made to increase availability of information about folate. Another recommendation was to advise women with a prior NTD pregnancy of the increased risk for future pregnancy and to provide them with folate supplementation. 34 For women with a prior NTD pregnancy, the recommended dosage of folic acid is increased to 4 mg daily, which should be taken for at least 1 month before conception. 6,16,20 Unfortunately, it has been reported that some health care providers in contact with women of childbearing age are not counseling them about the importance of folic acid consumption or the appropriate amount to take. 20 It should not be expected that an improved consumption of folic acid will totally prevent all NTDs because of etiologic factors such as the environment and genetics.
At the end of the first embryonic week, the primitive streak is present on the rostral surface of the embryo. A second streak, the notochordal process, develops alongside the primitive streak. The notochord is responsible for the induction of both the neural plate and the neurenteric canal. Cells proliferate along the lateral margin of the neural plate to form the neural folds around the central neural groove. 61
Cells at the apex of the neural folds make up the neural crest. Schwann cells, pia-arachnoid cells, sensory ganglia, melanocytes, and various secretory cells arise from the neural crest. The neural folds meet and fuse with the rostral (anterior) and caudal (posterior) ends (neuropore), closing by approximately the end of the fourth embryonic week. 61
Failure of development at this stage results in the defects of neurulation (or dorsal induction). The most severe of these defects is craniorachischisis, in which there is significant malformation of the brain (as in anencephaly), absence of the posterior skull, and an open spine along the full length of the spinal cord. Only a few affected embryos survive to early fetal stages. 61
Anencephaly is similar to craniorachischisis without the spinal defect. There is essentially no normal brain tissue above the brainstem and thalami, and parts of those structures are malformed. Onset is thought to occur before 24 days’ gestation. About one fourth of the fetuses survive into the neonatal period, but three fourths are stillborn. The majority of anencephalic infants die within the first week of life without intensive care. 61
Myeloschisis involves the failure of the posterior neural tube to close. There is no well-defined sac protruding from the defect. 8
Encephaloceles are caused by a limited failure of closure at the rostral (head) end of the neural tube. Extensions of meninges or brain tissue through the skull may occur on the ventral or rostral surface. 61
Myelomeningoceles (or even the more limited meningoceles) are a limited form of myeloschisis with failure of closure at the caudal (tail) end of the neural tube. With meningocele, the meninges protrude through the vertebrae and are contained within a sack. The spinal cord and nerve roots are generally in normal position, which improves the outcomes for these children. Unfortunately, myelomeningoceles, the more common defect, result in protrusion of both meninges and spinal cord through the opening in the spinal column. Neurologic deficits occur below the level of the protrusion. 35,47,56 The Arnold-Chiari deformities usually are included here. These malformations, often seen with myelomeningoceles, involve structures of the brainstem and cerebellum. Generally, the cerebellar tonsils are pulled down through the foramen magnum, and the brainstem is elongated in later life. Hydrocephalus is common. Dilation of ventricles often occurs without increased head circumference or clinical symptoms of increased intracranial pressure in this group of infants; therefore serial computed tomography (CT) or ultrasonographic scans should be performed. Symptoms of brainstem involvement may be present. Open myelomeningoceles and anencephaly (any defect in which the spinal or cranial contents are “open” to the outside) are associated with an elevation of alpha fetoprotein (AFP) in the amniotic fluid. This is important in prenatal diagnosis. 61
Segmentation Defects
After formation and closure of the neural tube, the development of the different regions of the brain begins to occur. Suprasegmental structures are formed. The division of the brain into hemispheres, formation of the ventricular system, and formation of the major gyral patterns are all part of this period of development. Major areas of the brain, including the cerebellum, basal ganglia, brainstem nuclei, thalamus, and hypothalamus, form at this time. 61 Defects of segmentation and cleavage occur during this phase of neural development. For unknown reasons, defects of segmentation and cleavage are far less common than defects of neurulation. Because these malformations involve abnormalities of ventral induction rather than dorsal induction (e.g., neurulation), the face, eyes, nose, mouth, and hair are also involved in the malformation. These features always should be investigated carefully for specific anomalies.
Holoprosencephaly is characterized by a single midline lateral ventricle, incomplete or absent interhemispheric fissure, absent olfactory system, midfacial clefts, and hypotelorism. The most severe form of holoprosencephaly is cyclopia (a single fused midline eye) and supraorbital nasal structure. At times, the nasal structure and eye are absent. An intermediate form is cebocephaly, which includes ocular hypotelorism (abnormally decreased space between the eyes) and a flat nose with single nostril. 61
When any of these malformations are suspected or when features suggestive of them are seen, careful examination of the hair, eyes, ears, mouth, and nose may reveal other related anomalies.
Migration and Cortical Organizational Defects
A critical aspect of brain development has yet to be described. The remaining development of the brain takes over twice as long as previously described development and includes cellular proliferation, migration, organization, and myelination. Cells that later form the cerebral cortex begin in the germinal matrix (near the caudate nucleus around the lateral ventricles). These cells then migrate in a radial fashion to their final positions near the surface of the brain. Abnormalities of cellular migration result in collections of gray matter in unusual places (heterotopias), abnormal gyri and sulci, abnormal spaces in the brain, and frequent clinical signs of gray matter dysfunction. Frequently, these clinical problems are not apparent in the newborn period.
Microcephaly means “small brain” and is manifested as a head circumference measuring greater than two standard deviations below average for infants at that gestational age. 17 Microcephaly may be (1) genetic (dominant, recessive, sex-linked) or chromosomal (translocation [see Chapter 27]); (2) caused by teratogens (cocaine, alcohol); (3) caused by infection (rubella, cytomegalovirus); or (4) of unknown cause. Occasionally there is a paucity of germinal matrix cells or they fail to adequately migrate, resulting in a brain cortex with a decreased number of neuronal cells. 61
In lissencephaly, the brain is smooth in appearance, having little or no gyri (convolutions). Although not generally present at birth, microcephaly usually occurs within the first year in type I lissencephaly. Appearance is marked by hollowing at both temples, a small jaw, and hypotonia. Neonatal seizures may be present, but seizures are more commonly present at 6 to 12 months of age. Another form of type I lissencephaly is Miller-Dieker syndrome, in which craniofacial deviations occur. In type II lissencephaly, macrocephaly is generally present at birth or develops soon afterward. Retinal, cerebellar, and muscular abnormalities always are present. 61
Clinical features of lissencephaly include seizures, microcephaly, hypotonia, feeding problems, scarcity of movement, upturned nares, temporal hollows, small jaw, protruding and long upper lip, and abnormalities on the electroencephalogram (EEG). Later, mental retardation and severe spasticity may be noted and death may occur. 61
Additional Defects
Cerebellar malformations are quite varied. Most often, at least a portion of the cerebellum is preserved, but total absence is possible. Hemispheric aplasia or vermal aplasia is seen, and familial forms have been reported. The Dandy-Walker cyst is another complex malformation involving the cerebellum in which the fourth ventricle is dilated into a cystic structure. The foramina of Magendie and Luschka are atretic, and hydrocephalus results. The cerebellum is small and displaced upward. Associated anomalies include heterotopias, agenesis of the corpus callosum, aqueductal stenosis, and syringomyelia. Causation is unknown. The differential diagnosis includes an arachnoid cyst of the posterior fossa. In the case of an arachnoid cyst, the fourth ventricle is not part of the malformation and is normal, although it may be displaced. 61
Clinical features of the Dandy-Walker cyst include frequent progressive hydrocephalus, associated malformations that cause additional specific symptoms, possible absence of symptoms in the newborn period, enlargement of the occipital shelf and posterior part of the skull, and clinical symptoms of increased intracranial pressure. 61
Craniosynostosis is the abnormally early closure (fusion) of the bones of the skull. Causation of this malformation is unknown. The premature closure of sutures may involve one or many sutures, with resulting deformity of the skull. Numerous terms are used to describe the shapes the skull assumes when craniosynostosis is present. 61
Craniosynostosis should be suspected in the presence of microcephaly or misshapen head. Appropriate evaluation requires x-ray films of the skull and a CT scan to define which, if any, of the sutures are stenosed and what problems might exist with brain structure (pressure or malformation). 61
Hydrocephalus may occur in many different situations from many separate causes. An inherited X-linked form exists. Intrauterine infection is another cause. Hydrocephalus may be associated with many of the malformations described previously. Hydrocephalus results when the normal flow of ventriculospinal fluid is obstructed. This may be the result of an atretic portion of the ventricular system, blockage from the outside, inflammation within the ventricular system causing a permanent blockage, or (very rarely) overproduction of ventriculospinal fluid. 35,61
Data Collection
The diagnosis of malformations of the central nervous system (CNS) may be quite obvious (as in anencephaly) or more subtle. Careful examination of all newborns results in the identification of most malformations. At times, the diagnosis is suspected not on the basis of findings on examination but because of an accompanying sign, such as seizures. 5
Two very important tests have become available in recent years that allow prenatal diagnosis of certain congenital malformations of the nervous system. Ultrasonographic examination (an abdominal ultrasound scan of the mother) provides an opportunity to identify certain malformations by viewing the fetus during development. Hydrocephalus, encephaloceles, myelomeningoceles, and anencephaly may be identified prenatally. Determination of AFP in the amniotic fluid and maternal serum allows the identification of anencephaly and open myelomeningoceles. A non-enclosed nervous system is associated with a significant rise in AFP in the amniotic fluid. Amniocentesis provides the amniotic fluid necessary for this determination. Testing of maternal serum for AFP also is an option and at some centers may be used along with ultrasonography for diagnosis, allowing amniocentesis to be omitted. 61 Clinical signs and symptoms have been described for each individual nervous system malformation presented earlier in this chapter.
Treatment
Limited treatment is available for congenital malformations of the nervous system. A variety of strategies are available for reducing secondary complications or providing earlier management to handle these complications more efficiently.
The greatest efforts and accomplishments have been made for infants with congenital malformations who might be expected to have productive lives. When secondary complications are managed appropriately, the majority of children with myelomeningoceles are ambulatory (total or partial) and continent of urine. 61
Myelomeningocele generally is surgically repaired as soon as possible (within 24 to 48 hours).5,35,47,61 Prevention of infection is paramount. In addition to sterile technique, prophylactic antibiotics have been shown to be beneficial. 61Trauma to the area should be avoided by keeping the infant in the prone position and maintaining sterile gauze moistened with warm, sterile normal saline. Tape should be avoided. 5,35,44 Preventing fecal contamination is vital. Several authors recommend the use of a sterile, plastic drape fastened above the anus but below the lesion to keep fecal material isolated from the site. 35,44Latex precautions also should be initiated, because these infants have an increased propensity for developing sensitivity to latex.5
In addition, spina bifida has been repaired in utero at early gestational age. Such repairs have risks for both mother and fetus but have resulted in reported significant drops in the number of infants developing hydrocephalus that would require a postnatal ventriculoperitoneal shunt. 15 Because of a lack of improved neurologic outcomes overall, a multicenter randomized controlled trial (RCT) is underway to compare intrauterine therapy to conventional postnatal care to better identify procedure-related benefits and risks. 15
Some of the malformations are lethal very soon after birth (anencephaly), limiting management options for comfort measures and family support. When appropriate, genetic counseling should be requested. For other malformations, treatment requires management of symptoms such as seizures, signs of increased intracranial pressure, and infection. A consult to neurosurgery is indicated. Other helpful consults may include physical therapy, infectious diseases, urology, and orthopedics. 35 For hydrocephalus, shunting may become necessary. See Box 26-1, the Parent Teaching box on pp. 754-755 and Figure 26-1). 61
BOX 26-1
• Positioning:
• Place infant on unaffected side (may position on shunt side with “doughnut” over operative site once incision has healed). Keep head of bed flat (15 to 30 degrees) to prevent too-rapid fluid loss.
• Support head carefully when moving infant.
• Turn q 2 hr from unaffected side of head to back.
• Shunt site:
• Use strict aseptic technique when changing dressing.
• Pump shunt if and only as directed by neurosurgeon.
• Observe for fluid leakage around pump.
• Observe and document all intake and output. Watch for symptoms of excessive drainage of CSF:
• Sunken fontanel
• Increased urine output
• Increased sodium loss
• Observe, document, and report any seizure activity or paresis.
• Observe for signs of ileus:
• Abdominal distention (serially measure abdominal girth)
• Absence of bowel sounds
• Loss of gastric content by emesis or through orogastric tube
• Perform range-of-motion exercises on all extremities.
• Observe and assess for symptoms of increased intracranial pressure (shunt failure):
• Increasing head circumference (measure head daily)
• Full or tense fontanel
• Sutures palpably more separated
• High-pitched, shrill cry
• Irritability and/or sleeplessness
• Vomiting
• Poor feeding
• Nystagmus
• Sunset sign of eyes
• Shiny scalp with distended vessels
• Hypotonia and/or hypertonia
• Observe and assess for signs of infection:
• Redness or drainage at shunt site
• Hypothermia and/or hyperthermia
• Lethargy and/or irritability
• Poor feeding and/or poor weight gain
• Pallor
• Parent teaching:
• Demonstrate and receive return demonstration of drug administration.
• Teach parents side effects of medications.
• Document on NICU’s routine discharge teaching checklist with routine care.
 |
| FIGURE 26-1
(From Harrison H, Kositsky A: The premature baby book, New York, 1983, St Martin’s Press.)
|
The one situation in which microcephaly could be considered surgically treatable is total craniosynostosis. Generally, skull deformity is present in infants with craniosynostosis, and it is always wise to consider the presence of craniosynostosis in any infant with a small head. If present, total craniosynostosis should be treated surgically. 35
The management of congenital hydrocephalus consists primarily of early shunting as soon after birth as possible. Fetal surgery for placement of a ventriculoamniotic shunt has been proposed, but an improvement in outcomes compared with surgery after birth is uncertain. In addition, hydrocephalus in a fetus is often associated with serious developmental abnormalities that may increase morbidity and mortality. 61
In Volpe’s series, outcome was variable and the procedures were not as reliable as hoped. It was not always possible to distinguish true hydrocephalus from ventriculomegaly without increased pressure. Shunting soon after birth often produces a far better outcome than would be assumed, with minimal motor deficit and only a mild to moderate deficit in intellect. 61
Wolfson Children’s Hospital Parent Handout: Newborn Ventriculoperitoneal Shunt (For Use With Ventriculoperitoneal Shunt Teaching Checklist)
Purpose of Ventriculoperitoneal Shunt
• Ventricles are compartment-like spaces that are located in the normal brain. Spinal fluid forms daily in these ventricles. This clear fluid flows out over the brain and down around the spinal cord. Spinal fluid helps cushion the brain from injury, keeps the brain moist, and carries away waste products.
• Hydrocephalus is a condition in which an abnormally large amount of spinal fluid builds up in your baby’s ventricles and usually is caused by a blockage in the spinal fluid path. Because the ventricles continue to make spinal fluid daily, a buildup of fluid occurs when it cannot escape. This excess fluid can cause pressure on the brain and result in permanent damage to the brain unless it is properly treated.
• The purpose and function of your baby’s VP shunt is to allow the excess spinal fluid to drain through a tube from the ventricle into the abdomen, where it is absorbed.
Pathway of the Ventriculoperitoneal Shunt
• A small incision is made on the scalp, and the tube is passed through the skull and into the ventricle. Located under the skin, the tube passes behind the ear, down the side of the neck, and continues to the abdomen, where a second incision is made to put the end of the tube into the abdominal cavity. A third incision is sometimes needed in the neck area with some babies.
• The scalp incision will be hidden as your baby’s hair grows. You will see and feel the shunt tubing (like a large vein under the skin), but it is barely noticeable after the baby gains weight.
Signs and Symptoms of Shunt Infection
• The shunt is at risk for infection because it is a foreign object located inside the body. You will have to watch for these signs of shunt infection and report them immediately to your doctor:
• Temperature of 101° F or higher
• Swelling, redness, or drainage along the pathway of the shunt tube
• Lethargy or irritability (change in behavior)
• Loss of appetite or poor feeding
Signs and Symptoms of Shunt Failure/Increased Intracranial Pressure
• The spinal fluid contains proteins and chemicals that may build up and block off the shunt. It is also possible for tissue within the brain or abdomen to block the shunt or for the shunt device itself to fail. This shunt failure (malfunction) means that the spinal fluid will once again build up and result in pressure on the brain and possible irreversible damage. Therefore it is very important for you to watch for the signs of increased pressure in the brain that occurs with shunt failure and report them to your doctor immediately:
• Lethargy or sleepiness
• Unusual irritability, fussiness, or excessive crying
• Repeated vomiting
• Poor feeding
• Bulging soft spot when baby is sitting up quietly
• Shrill, high-pitched cry
• Eyes that look downward
• Increase in spaces between the bones of the skull
• Seizures/posturing
Reason and Importance of Prompt Treatment of Health Problems
• Prompt treatment of your baby’s health problems (e.g., ear infections, skin infections) is important to prevent infections spreading to the shunt. It is also vital to seek medical care for signs of shunt infection or failure as noted.
Importance of Close Medical Follow-up
• Your baby will have to be followed up by a neurosurgeon and your pediatrician after being discharged. Bring the baby to every follow-up appointment so that your baby’s head can be measured and physical condition can be evaluated. Your baby will also go to the Developmental Evaluation Clinic where a specialist in baby development can examine him or her. If development problems occur, this will ensure early diagnosis and treatment.
Care of the Shunt
• You can handle, cuddle, and play with your baby like any baby. Your baby also can sleep in any position after the initial postoperative period.
 |
Monitoring of pregnancies with fetal ultrasound allows the detection of congenital hydrocephalus. Induction of lung maturation with steroids has been suggested to allow a preterm delivery (with a smaller head) without excessive pulmonary complications. In this way, a permanent shunt can be placed sooner than with term delivery. 61
Complications
Many of the expected complications were dealt with previously in the sections describing the malformations and their associated problems. It is difficult to separate true complications from problems resulting from the malformation. For example, hydrocephalus develops in many infants with myelomeningocele and may be present at birth. 5,47 Other complications or associated problems of myelomeningocele include bowel and bladder incontinence, meningitis, urinary tract infections, and paralysis. 35
Malformations carry with them altered anatomy and physiology that is reflected in abnormal function. Common general problems include seizures, mental retardation, sensorimotor abnormalities, disturbances in primary sensory function such as vision and hearing, orthopedic problems, and vegetative functions. 61
The problems encountered are ordinarily explained on the basis of the malformation. Often midline defects in the brain (particularly at the base of the brain) have clinical problems involving the hypothalamus. Diabetes insipidus may be present.
To some extent, the anatomy predicts the types of problems. Involvement of the cortex causes seizures, retardation, and sensorimotor problems. White matter damage can cause spasticity. If the brainstem is involved in the malformation, apnea, deafness, sleep disturbance, oculomotor disturbances, and problems with sucking and swallowing may be seen. Spinal cord lesions cause quadriplegia or paraplegia. Genitourinary problems and, to a lesser extent, gastrointestinal problems also are seen.
Apnea and other brainstem findings may occur when the malformation involves the brainstem, as in Arnold-Chiari deformity, Dandy-Walker cyst, occipital encephalocele, and arachnoid cyst.
Pituitary-hypothalamic dysfunction may manifest itself in impaired temperature regulation, thyroid abnormalities, diabetes insipidus, and adrenal insufficiency.
Most of the complications occur after the newborn period, although causation is present at birth and includes seizures, retardation, spasticity, genitourinary problems, and orthopedic problems. In many circumstances, the problem is already present but functional expression, such as impaired ambulation, mental retardation, or deafness, is lacking. In the infant’s follow-up examinations, careful attention must be given to problems likely to develop or intensify with age. When a specific malformation is identified, it is necessary to become familiar with the expected problems, not only to anticipate problems as they appear but also to lessen any secondary damage that might occur if they go unrecognized.
Parent Teaching
Parents of an infant born with congenital malformations are faced with a stressful event that may develop into a major life transition. Parents, especially mothers, report feelings of guilt and self-blame, although they may not initially share these feelings with hospital staff. After the birth of a malformed child, they go through stages of grief (see Chapters 29 and 30): shock or denial, anger, bargaining, depression, and acceptance. Some authors question whether full acceptance occurs for the family of the handicapped child because of return of grief and sorrow each time a developmental milestone is missed or the child experiences illness. 38,44
Social support received from hospital personnel, family, and friends can help parents feel less stressed and more able to cope with the illness of their infant. The ability of the staff to accurately anticipate and assess parental feelings and concerns can be invaluable when assisting families through this difficult time. Parents should be encouraged to verbalize their feelings and fears in a supportive environment. Reassurances, when appropriate, should be provided (e.g., parents were not responsible for the congenital malformation; it is normal for the mother to experience [or at least report] more fears than her husband). The ultimate goal of intervention is to reduce stress, assist families to confront fears, improve coping, and facilitate the bonding process.38
Infants with congenital malformations present such a complex variety of problems that parent teaching and emotional support need to begin as early as possible. Often parents know from the time of birth or earlier that a major problem exists. In other circumstances, the anomaly is detected only after appropriate studies are performed.
When the infant is not viable, care should be directed at meeting the emotional needs of the family. Every effort should be made to give family members positive experiences and memories by encouraging early parental holding of the infant and, whenever possible, participation with care (see Chapter 30). Anticipatory counseling from social services and chaplain staff can help the family during grieving and with funeral arrangements. There are also questions about etiologic factors and genetics, and these questions should be dealt with according to the family’s wishes (see Chapter 27).
If serious handicaps are anticipated and the infant is expected to survive, the parents should be encouraged to participate in the care of the infant from the beginning. Both adjustment and specific aspects of care within the circumstance will be enhanced and learning will be more effective if parents are supported. A multidisciplinary team approach to parent education and support allows individualized hospital resources for specific needs of the patient and family. In addition to medical, nursing, social service, and chaplain involvement, team members can be drawn from psychology, developmental specialists, physical therapy/occupational therapy, and other services based on specific needs and circumstances. Parent teaching and support must be individualized according to the anomaly. When available, support groups, integrative discharge planning, and specialized clinics can help with post-discharge care and parent education.
Parent teaching for the mothers and fathers of infants with congenital anomalies should (1) be started early, (2) involve the parents in the care of the infant, (3) use the resources of the hospital for specialized help, and (4) continue after the infant has gone home from the hospital. 38
BIRTH INJURIES
Physiology and Etiology
Birth injuries (birth traumas) are the direct result of difficulties encountered during the delivery process. These may be minor injuries without expected sequelae or the direct cause of death in the neonatal period. Classification of birth injuries usually is etiologic (predisposing factors or mechanisms of injury) or anatomic. An anatomic classification is used in this discussion to illustrate commonly encountered problems (Table 26-2).
| Site of Injury | Type of Injury |
|---|---|
| Scalp |
Caput succedaneum
Subgaleal hemorrhage
Cephalhematoma
|
| Skull |
Linea fracture
Depressed fracture
Occipital osteodiastasis
|
| Intracranial |
Epidural hematoma
Subdural hematoma (laceration of flax, tentorium, or superficial veins)
Subarachnoid hemorrhage
Cerebral contusion
Cerebellar contusion
Intracerebellar hematoma
|
| Spinal cord (cervical) |
Vertebral artery injury
Intraspinal hemorrhage
Spinal cord transection or injury
|
| Plexus injuries |
Erb’s palsy
Klumpke’s paralysis
Total (mixed) brachial plexus injury
Horner syndrome
Diaphragmatic paralysis
Lumbosacral plexus injury
|
| Cranial and peripheral nerve injuries |
Radial nerve palsy/nerve injuries
Medial nerve palsy
Sciatic nerve palsy
Laryngeal nerve palsy
Diaphragmatic paralysis
Facial nerve palsy
|
The timing of birth injuries can be used to identify and describe causes. Etiologic classification of birth injuries includes uterine injury (antenatal), fetal monitoring procedures, abnormal or difficult presentations or methods of delivery, and multifactorial injuries. It should be recognized that the same injury might have multiple causations. Thus a cephalhematoma could be the result of forceps delivery, vacuum extraction, or routine vaginal delivery. A variety of specific predisposing factors increase the risk for birth injury, as follows:
• Macrosomia
• Cephalopelvic disproportion
• Dystocia
• Prematurity
• Prolonged or precipitous labor
• Breech presentation
• Forceps usage
• Rotation of fetus
• Version and extraction
• Handling after delivery
Multiple factors often are present. When multiple predisposing factors are present, a single underlying maternal disease often links them. A common example is that of a premature, macrosomic fetus with a diabetic mother in whom labor is not progressing properly.
The common factors that are present in deliveries complicated by birth injuries are as follows:
• Unusual progress of labor
• Unusual size or shape of the fetus (large for gestational age or hydrocephalus)
• Problems encountered during delivery (dystocia or forceps application)
• Unusual or unexpected presentations (breech or unexpected twin)
The maternal history must always be explored for the underlying disease process or conditions that might increase the risk for a birth injury.
Prevention
Most birth injuries may be preventable, at least in theory. Careful attention to risk factors and the appropriate planning of delivery should reduce the incidence of birth injuries to a minimum. Transabdominal ultrasonography facilitates predelivery awareness of macrosomia, hydrocephalus, and unusual presentations. Particular pregnancies then may be delivered by controlled elective cesarean section to avoid significant birth injury. Care must be taken to avoid substituting a procedure of greater risk. Often a small percentage of significant birth injuries cannot be anticipated until the specific circumstances are encountered during delivery. Emergency cesarean section may provide last-minute salvage, but in these circumstances, the injury truly may be unavoidable.
SPECIFIC BIRTH INJURIES
Injuries to the Scalp
The three commonly encountered forms of extracranial hemorrhage are caput succedaneum, subgaleal hemorrhage, and cephalhematoma and are distinguished not only in clinical manifestations but also in pathophysiology (see the Critical Findings box at right. 61 These three extracranial scalp injuries are included with neurologic birth injuries, not because they have associated neurologic problems but because the family or health care providers often raise the question of possible neurologic involvement.
Extracranial Hemorrhage
There are three common forms of extracranial hemorrhage but with different etiology and clinical assessment findings, as follows.
1. Caput succedaneum
a. Etiology: Trauma to scalp (usually vertex vaginal delivery) results in hemorrhagic edema superficial to the aponeurosis of the scalp.
b. Findings: Soft, pitting edema that crosses suture lines.
2. Cephalhematoma
a. Etiology: Mechanical trauma; most common in primiparous women, with delivery using forceps or in vacuum-assisted deliveries.
b. Findings: Firm, tense collection of blood confined by the sutures. Area often increases in size after delivery. No significant blood loss. Blood collects beneath the periosteum (subperiosteal).
c. Warning: Associated with linear skull fracture in up to 25% of the cases.
3. Subgaleal hemorrhage
a. Etiology: Forces that compress and then drag head through pelvic outlet.
b. Findings: Firm swelling that crosses suture lines and is fluctuant to palpation. Blood collection is under the aponeurosis (connective tissue connecting the occipital and frontal muscles). Bleeding (swelling) may continue after birth and dissect along tissue planes into the neck.
c. Warning: Acute blood loss may occur. Presenting symptom may be shock.
Monitor VS for signs of shock:
Elevated HR
Decreasing BP
Monitor baby for signs of shock:
Pallor
Delayed capillary refill time
Diminished tone
Respiratory distress
Transfusion may be necessary: type and crossmatch.
Serial Hct should be followed.
Elevated bilirubin is a common complication as a by-product of broken-down red blood cells.
PHYSIOLOGY AND ETIOLOGY
Caput succedaneum is caused by trauma to the scalp, usually during a routine vertex vaginal delivery. The caput is the result of hemorrhagic edema superficial to the periosteum of the scalp. Therefore spread of the edema is not restricted to suture lines and is soft and pitting because of its superficial location. 35,61
Forces that compress and drag the head through the pelvic outlet are associated with subgaleal hemorrhage. Significant acute blood loss can occur with shock as the presenting symptom. Bleeding may continue after birth with enlargement of the accumulated blood and dissection of the blood along tissue planes into the neck. Such a hemorrhage carries the greatest potential for complications, but fortunately it is the least common form of birth injury to the scalp. 32,35,61
Cephalhematoma is a subperiosteal collection of blood that is confined by the skull sutures. The incidence is 1% to 2% of all live births. The cause is nearly always mechanical trauma, and its occurrence is more common in primiparous women and in forceps or vacuum-assisted delivery. Males are generally more likely to be affected than females. It is associated with an underlying linear skull fracture in up to 25% of cases. The firm, tense collection of blood frequently increases in size after birth, but significant blood loss does not occur. 5,35,61
DATA COLLECTION
With caput succedaneum, physical examination reveals soft, pitting edema that is diffuse and crosses suture lines. Laboratory tests are not needed. 35,61
Because the subgaleal collection of blood is under the aponeurosis (connective tissue connecting the occipital and frontal muscles) and superficial to the periosteum, subgaleal hemorrhage crosses suture lines. It is firm but fluctuant to palpation. Vital signs should be carefully monitored for symptoms of shock. Pallor, delayed capillary refill time, diminished tone, respiratory distress, elevated heart rate, or decreasing blood pressure should be observed for and treated promptly. Transfusion may be necessary. The hematocrit should be serially followed, and bilirubin levels should be determined during recovery.35,61
Cephalhematomas may occur anywhere but are most commonly found in the parietal area on one side. Because the location of the blood is subperiosteal, the blood is confined by suture lines. Symptoms are normally absent. A skull fracture underlying the cephalhematoma is present in 10% to 25% of affected infants. X-ray examination of the skull defines the fracture. Rare complications include infection, osteomyelitis, hyperbilirubinemia, meningitis, and late-onset anemia. 35,61
TREATMENT
Usually, no treatment is necessary for any of these three lesions. In subgaleal hemorrhage, treatment of blood loss and shock may be necessary. During resolution, the breakdown of the blood may cause hyperbilirubinemia requiring treatment (see Chapter 21). 35,61
PARENT TEACHING
Parents of an infant with caput succedaneum should understand that the swelling is outside of the cavity of the brain and will usually reabsorb within 48 hours. 35Careful preparation of the parents for the acute side effects of subgaleal hemorrhage is important. Parents should be warned of the possibility of swelling and discoloration of the face, head, and neck. The purpose of serial hematocrit and bilirubin checks should be explained. Parents can expect 2 to 3 weeks for the swelling to resolve. 35 Parents of an infant with a cephalhematoma should be instructed that the cephalohematoma may enlarge but that they should not be concerned unless localized changes occur, suggesting secondary infection (erythema, induration, or drainage). This lesion should not be drained and may be evident for 6 to 8 weeks and leave a small calcification after reabsorption of hemorrhage. The hemorrhage can be significant enough to cause hyperbilirubinemia or anemia. Outpatient evaluation of bilirubin levels and hematocrit may be needed in some cases.
Skull Fractures
Three forms of skull fracture should be identified and differentiated: linear fractures, depressed fractures, and occipital osteodiastasis. 35,61
PHYSIOLOGY AND ETIOLOGY
Linear skull fracture (a nondepressed fracture) is the most common type of skull fracture. The result of compression of the skull during delivery, a linear skull fracture most often has no associated injuries and causes no symptoms. Bleeding may be seen extracranially (common) or intracranially (rare). Intracranial bleeding causes symptoms referable to the bleeding rather than to the fracture itself. 35,61
The typical depressed skull fracture is of the “ping-pong” type, an indentation without loss of bony continuity. When forceps are used during delivery, the direct cause of injury may result but is often without complications or sequelae. When neurologic signs are present, direct cerebral injury, intracranial bleeding, or free bone fragments should be suspected. 35,61
DATA COLLECTION
A linear skull fracture usually produces no signs or symptoms unless intracranial bleeding has occurred. Skull x-ray films most frequently demonstrate a parietal fracture. A depressed skull fracture may be noted by presence of a visible depression or a palpable “ping-pong” fracture in the parietal or temporal area. No other signs and symptoms are present unless intracranial bleeding or focal irritation of the cortex causes them. Evaluation with a skull x-ray examination or CT scan is necessary to delineate the fracture and to identify complications.
TREATMENT
No treatment is necessary for a linear skull fracture. Treatment of a depressed skull fracture varies and centers on the mode of treatment and the necessity of treatment for fractures that create no neurologic symptoms. If free bone fragments or clots are identified, neurosurgical intervention is necessary. More conservative approaches are indicated when no complications are present. Noninvasive treatments such as vacuum extractors and breast pumps have been used with success to raise the depressed bone segment. 61
COMPLICATIONS
With a linear skull fracture, the single complication to be aware of is a “growing” skull fracture. A dural tear may allow leptomeninges to extrude into the fracture site, setting up the possibility of a leptomeningeal cyst. As the cyst enlarges, the edges of the fracture may fail to fuse and even spread apart, giving the appearance of a “growing” fracture. Palpation and x-ray examination demonstrate the lesion. Surgical correction may be necessary to ensure healing and prevent further complications. With a depressed skull fracture, intracranial bleeding and direct cerebral injury with seizures or residual neurologic deficit are rare.
PARENT TEACHING
Parents should be instructed to have the fracture site monitored for several months to ensure that reunion of the bone has taken place. Patients will require no other aftercare unless neurosurgical intervention was necessary or complications developed.
Intracranial Birth Injuries
Three major forms of bleeding occur intracranially: epidural hematoma, subdural hemorrhage, and subarachnoid hemorrhage (see the Critical Findings box on p. 761). Added to these are cerebellar hemorrhages, cerebellar contusions, and cerebral contusions. Each has its own particular set of symptoms and signs and complications and sequelae. IVH usually is not related to trauma and is covered separately in this chapter.
PHYSIOLOGY AND ETIOLOGY
An epidural hematoma is pathophysiologically difficult to form in newborns because of a relatively thick dura. When present, it is almost always accompanied by a linear skull fracture across the middle meningeal artery.
Subdural hemorrhage is more common in term infants than in preterm infants and occurs from trauma tearing veins and venous sinuses. Although some assume its presence represents birth trauma, several authors indicate that this is not necessarily the case. 61,65 Subdural hemorrhage has been linked with maternal use of aspirin and also to maternal ingestion of phenobarbital. 61 Four major pathologic entities are defined: (1) laceration of the tentorium, (2) laceration of the falx, (3) laceration of the superficial cerebral vein, and (4) occipital osteodiastasis. Tentorial laceration causes a posterior fossa clot with compression of the brainstem. The straight sinus, Galen’s vein, lateral sinus, and infratentorial veins may be involved. Laceration of the falx is caused by rupture of the inferior sagittal sinus. The laceration usually occurs at the junction of the tentorium and the falx, and the clot appears in the longitudinal cerebral fissure over the corpus callosum. Laceration of superficial cerebral veins causes subdural bleeding over the convexity of the brain. Subarachnoid bleeding or contusion of the brain also may be present. 36,61
A subarachnoid hemorrhage is the most common type of neonatal intracranial hemorrhage. In term infants, trauma is the most common cause, whereas in preterm infants, hypoxia is more often the cause. Small hemorrhages are more common than massive ones and usually result from venous bleeding. Underlying contusion may be present. 3,36,61
Intracranial Birth Injuries
1. Epidural hematoma
a. Occurrence: Rare.
b. Location: Bleeding occurs into the epidural space. Blood is located between the inner area of skull bone and the periosteum.
c. Pathophysiology: Most (not all) with history of traumatic labor or delivery.
d. Clinical findings:
Increased intracranial pressure (swollen fontanel).
Seizures may occur.
e. Associated problems: Almost always accompanied by a linear skull fracture.
2. Subdural hemorrhage
a. Occurrence: More common in term infants than in preterm.
b. Location: Bleeding is produced from tear of cerebral vein or sinus, which is often accompanied by a tear in the dura. Exact location of the hematoma depends on the location of the bleeding source. 61
Laceration of the tentorium
Laceration of the falx
Laceration of the superficial cerebral vein
Occipital osteodiastasis
c. Pathophysiology:
Debate as to whether its presence indicates birth trauma. Volpe indicates that most cases result from trauma. 61
Linked to maternal use of aspirin and maternal ingestion of phenobarbital.
d. Clinical findings:
Neurologically abnormal at birth, if massive bleed:
Seizures
Stupor or coma
Skew deviation of eyes
Pupil changes: unequal pupils, poorly responsive pupils, fixed and dilated pupils
Nuchal rigidity
Apnea and bradycardia
Signs of increased intracranial pressure
e. Associated problems: Risk for herniation with lumbar puncture.
3. Subarachnoid hemorrhage
a. Occurrence: Most common type of neonatal intracranial hemorrhage.
b. Location: Blood is within the subarachnoid space but not because of extension from other areas. Small hemorrhages are more common than large ones. Source believed to be small vascular channels. 61
c. Pathophysiology:
Term: Usually caused by trauma
Preterm: Usually caused by hypoxia
d. Clinical findings:
Most common: Minimal or no symptoms
Seizures (especially with term infants): “Well baby with seizures”
Apnea (especially with preterm infants)
For massive bleed (rare): Sudden and marked deterioration; death
Associated problems:
Usually none for infants without significant trauma or hypoxia.
After major bleed:
Hydrocephalus (most common sequela)
Neurologic residual
Death
Cerebral contusions are uncommon as an isolated event. Focal blunt trauma is necessary to produce a contusion. Pathologically, focal areas of hemorrhage and necrosis are seen. Shearing forces may cause slitlike tears in the white matter.
Cerebellar contusion and intracerebellar hemorrhage are uncommon events usually seen in association with occipital osteodiastasis and infratentorial subdural hemorrhage. These are catastrophic events, as described, and most often result in the death of the patient.
DATA COLLECTION
For epidural hemorrhage, the signs and symptoms may be diffuse (increased intracranial pressure with a bulging fontanel) and may include focal or lateralizing seizures, eye deviation, and hemisyndromes. Laboratory tests should include x-ray examination to look for fractures and CT scanning to identify bleeding.
Infants with subdural hemorrhage are neurologically abnormal at birth. Tentorial lacerations and laceration of the falx tend to produce signs by pressure on the brainstem. These signs include skew deviation of the eyes, apnea, coma, or unequal pupils. Nuchal rigidity and opisthotonos are signs of progressive herniation. Signs and symptoms of subdural hemorrhage from laceration of the superficial cerebral veins are variable. Small clots may produce no identifiable dysfunction. Typical signs are those of focal or lateralized cerebral dysfunction, although increased intracranial pressure may occur. CT scans including views of the posterior fossa should be obtained immediately when a subdural hemorrhage is suspected. Lumbar puncture is not used as a diagnostic tool because of the risk for herniation.61
With subarachnoid hemorrhage, underlying contusions may cause focal neurologic signs. Often no significant increase in intracranial pressure is found acutely. Irritability and a depressed level of consciousness may persist. Seizures are common in term infants, whereas apnea is common in preterm infants. Diagnosis generally is made with CT scan. If a lumbar puncture is performed, it is generally done for another reason (e.g., meningitis workup) and shows elevated red blood cells (RBCs) and protein. 3,36,61 For infants without serious injury from trauma or hypoxia, the prognosis is good. 61 Focal signs predominate in cerebral contusions.
TREATMENT
Surgical evacuation of epidural and subdural clots may be necessary as emergency procedures. Subdural taps may be useful in the symptomatic infant with subdural bleeding from laceration of superficial cerebral veins. Many infants with intracranial bleeding may require treatment of seizures.36,61 In the presence of coagulation defects, prompt intervention may require platelets, vitamin K, or replacement therapy for deficient coagulation factors. 3,36,61
COMPLICATIONS
The complications of epidural hemorrhage range from none to permanent neurologic deficits with or without seizure. Sequelae of subdural hemorrhage occur in 20% to 25% of affected infants. The most common sequelae are focal neurologic signs. Seizures and hydrocephalus are seen less often. Hydrocephalus is the major potential complication of subarachnoid hemorrhage and directly alters outcome. 61
PARENT TEACHING
Because long-term outcome is variable and may be abnormal even in infants who appear normal at discharge from the nursery, parent teaching must be individualized. Emphasize the need for appropriate follow-up and intervention. Referral to available support groups is usually beneficial.
Spinal Cord Injuries
PHYSIOLOGY AND ETIOLOGY
Injuries to the spinal cord (usually the cervical portion) are seen most often in complicated breech deliveries. Before cesarean sections were routinely performed for breech delivery, fatal attempts to deliver vaginally often were associated with intraspinal hemorrhage. The breech presentation in conjunction with a hyperextended head is the most dangerous situation and is worsened by fetal depression. Traction, rotation, and torsion cause mechanical strain on the vertebral column. Cephalic deliveries are not entirely safe because of the difference in mechanical forces; a different clinical picture is seen with a higher-level lesion. 22,61
DATA COLLECTION
Clinical manifestations depend on the severity and location of the injury. Clinical syndromes include stillbirth or rapid neonatal death, respiratory failure, and spinal shock syndrome. High cervical cord injuries are more likely to cause stillbirths or rapid death of the neonate. Lower lesions cause an acute cord syndrome. Common signs of spinal shock include flaccid extremities (may involve just the lower extremities if the cervical cord is spared), asensory level, diaphragmatic breathing, paralyzed abdominal movements, atonic anal sphincter, and distended bladder. Useful laboratory tests include magnetic resonance imaging (MRI) or CT scan of the spine and somatosensory-evoked potentials to help determine the extent and site of the lesion. The differential diagnosis includes dysraphism, neuromuscular disease, and cord tumors. 22,61
COMPLICATIONS
After the acute phase, chronic lesions include cysts, vascular occlusions, adhesions, and necrosis of the spinal cord. Flaccid or spastic quadriplegia is expected. Some infants with spinal cord injuries are respirator dependent, and bowel and bladder problems continue.
PARENT TEACHING
Parents should understand fully the implications of severe injury to the spinal cord. Recovery is frequently minimal to nonexistent. Continued specialized care may be necessary, including ventilator therapy. The overwhelming implications for the family cannot be emphasized strongly enough.
An individualized multidisciplinary team approach to discharge planning is vital to parental confidence and a timely discharge. The problems of both patient and family are complex and not limited to medical concerns. A successful discharge is unlikely unless family emotional, financial, and educational concerns are addressed early in the planning process. The timely assessment of needs and involvement of supportive agencies allow resolution of problems well before the projected discharge date. Such assistance should include early family referral to available federal programs for financial aid (e.g., Supplemental Security Income [SSI]) and assistance with patient transportation to their multiple outpatient follow-up appointments. Early assessment of equipment needs and home nursing requirements is also of primary importance and should include a determination of the availability of these resources in the community, parent acceptance of their use, and whether the home can accommodate them (i.e., adequate electrical system and space).
Plexus Injuries
PHYSIOLOGY AND ETIOLOGY
Plexus injuries occur more commonly than cord injuries and result from lateral traction on the shoulder 55 (vertex deliveries) or the head (breech deliveries). 61 Risk factors include large infant, fetal depression, breech delivery, and a variety of obstetric factors. 13,35,61 Any factor resulting in a difficult vaginal delivery of the baby can increase the risk for injury (e.g., prolonged second stage of labor, placenta previa). 13,55 A study of 35,796 infants (54 with brachial plexus injury) concluded that brachial plexus injury is not predictable before delivery. 12 Some authors note the preventability of some risk factors. 61 Estimates of the incidence of brachial plexus injuries range from 0.5 to 2 per 1000 live births. 61 Extremely mild cases often have undetectable findings and may remain unidentified.
Pathologic changes range from edema and hemorrhage of the nerve sheath to actual avulsion of the nerve root from the spinal cord. Of the reported cases of plexus injuries, 90% involve the cervical nerve 5 (C5) to C7 nerve roots and are classified as Erb’s palsy. 22,61 In a small minority of cases, the C4 nerve root is also affected, causing diaphragmatic problems. The site of injury in Erb’s palsy is Erb’s point where C5 and C6 nerve roots join to form the upper trunk. Total brachial plexus palsy occurs in 8% to 9% of the cases and has findings referable to C5 to thoracic nerve 1 (T1) (and possibly C4). When T1 is involved, the sympathetic fibers become affected with an ipsilateral Horner syndrome (ptosis, anhidrosis, and miosis) and possible delay in pigmentation of the iris. Klumpke’s palsy rarely occurs in the newborn period and involves only the distal upper extremity (hand), whereas the muscles in the proximal extremity are normal. When both distal and proximal weakness occur, it should be classified as total plexus palsy. 13,61
DATA COLLECTION
Signs of brachial plexus palsies vary somewhat, most often because of the overlap of pure clinical syndromes. Shoulder and arm findings are characteristic of a true Erb’s palsy. Involvement of the hand and fingers is seen in total forms or Klumpke’s palsy. Table 26-3 lists the specific cord levels involved in various functions that might be addressed.
| Part Examined | Spinal Level |
|---|---|
| Diaphragm movement (downward) | C4 (C3-5) |
| Deltoid muscle | C5 |
| Spinatus muscle | C5 |
| Biceps muscle | C5-6 |
| Brachioradialis muscle | C5-6 |
| Supinator of arm | C5-6 |
| Biceps tendon reflex | C5-6 |
| Wrist extensors | C6-7 |
| Long extensor of the digits | C6-7 |
| Triceps tendon reflex | C6-7 |
| Wrist flexor | C7-8, T1 |
| Finger flexors | C7-8, T1 |
| Dilator of iris (sympathetic chain, Horner syndrome) | T1 |
| Eyelid elevator (full elevation) (same as above) | T1 |
| Moro reflex (shoulder abduction) | C5 |
| Moro reflex (hand motion) | C8-T1 |
| Palmar grasp | C8-T1 |
Evaluation of diaphragmatic function by x-ray examination is at times necessary. CT myelography or MRI may be necessary to identify nerve root avulsion, which generally should be suspected when recovery does not occur. Electromyography often shows abnormalities early in the course of the injury, suggesting that the process actually may have begun in the last weeks of pregnancy rather than at the time of delivery. 61
Clinical syndromes of plexus injuries include Erb’s palsy, total palsy, and Klumpke’s palsy. Erb’s palsy accounts for about 90% of plexus injuries. 22,61 It involves the upper part of the plexus, C5 to C7 and occasionally C4. The shoulder and upper arm are involved, and the biceps reflex is decreased. When C4 is involved, diaphragmatic dysfunction is present.
Total palsy occurs less frequently than Erb’s palsy. Plexus involvement is diffuse (C5 to T1 and occasionally C4). The upper and lower arm and hand are involved. Horner syndrome (ptosis, anhidrosis, and miosis) exists when T1 is involved. The diaphragm is affected when C4 is involved. Biceps and triceps reflexes are decreased. 22,61
Klumpke’s palsy is rare in the neonatal period (see the preceding). 61 The lower part of the plexus, C8 to T1, is involved. The lower arm and hand also are involved. T1 involvement is associated with Horner syndrome. Triceps reflex is decreased.
TREATMENT
Treatment includes passive range-of-motion exercises followed by a gradual increase of activity to the affected limb. Treatment may include immobilization for 1 to 5 days to prevent contractures initially; finger and wrist splints also may be necessary. 61 For infants failing to achieve sufficient functional recovery by 3 months of age, surgical intervention is considered. 13,61
COMPLICATIONS
Associated trauma may occur and should be carefully investigated. Common associated injuries include clavicular fracture, shoulder dislocation, cord injury, facial nerve injury, and humeral fracture. Full recovery of plexus function was seen in 88% to 92% of cases in the first year of life during the National Collaborative Perinatal Study. 61 Children who show no signs of improvement during the first 3 months after delivery should be referred to a clinic specializing in brachial plexus injury. Rarely, nerve graft surgery of the injured nerve root is necessary.
PARENT TEACHING
Parents should be taught passive range-of-motion exercises to encourage the infant’s mobility and prevent contractures. Instructions should begin before discharge from the hospital. Usually a neonatal nurse or occupational or physical therapist gives the instructions.
Parents may equate the presence of a brachial plexus injury with poor obstetric care. This is often not the case. The awareness of early changes on electromyography should be used to help families understand that the factors causing injury to the plexus begin before the onset of labor.
Cranial and Peripheral Nerve Injuries
Median and sciatic nerve injuries usually are postnatal and result from brachial and radial artery punctures (median nerve) and inferior gluteal artery spasm (umbilical artery line drug instillation). Recovery is variable.
Median nerve palsy is manifested by decreased pincer grasp, decreased thumb strength, and the continuous fixed position of the fourth finger. Sciatic nerve palsy presents with decreased hip abduction and distal joint movement. Hip adduction, flexion, and rotation are normal, because the femoral and obturator nerves control them. Radial nerve damage usually is seen in conjunction with a humeral fracture. Prolonged labor is normally present. Congenital bands may also be causative. Recovery takes place over weeks to months. Radial nerve palsy is manifested by wrist drop (decreased finger and wrist extension) and normal grasp.
Laryngeal nerve palsy may be seen in conjunction with facial or diaphragmatic paralysis. If the paralysis is unilateral, a hoarse cry may be heard. Bilateral involvement causes breathing to be difficult and the vocal cords to remain closed in the midline. It is essential to rule out intrinsic brainstem disease. Often the presence of other brainstem-related abnormalities such as oculomotor problems, apnea, or facial palsy helps clarify this. Evoked potentials, both brainstem auditory and somatosensory, also may help rule out brainstem involvement.
Laryngeal nerve palsy is manifested by difficulty in swallowing (superior branch), difficulty in breathing (bilateral), and difficulty in vocalizing (recurrent branch). Also, the head is held high and fixed laterally with slight rotation. Severe cases may require tracheotomy and assisted feedings by gavage or gastrostomy tube. 61
Diaphragmatic paralysis is most often seen in association with plexus injuries (80% to 90% have an associated plexus injury) and has the same cause. Some series involving unilateral paralysis have a mortality rate of 10% to 20%. Most patients recover fully in 6 to 12 months. Although fewer than 10% of patients have bilateral diaphragmatic paralysis, the mortality rate for these patients is almost 50%. Treatment has consisted of using rocking beds, electric pacing of the diaphragm, continuous positive airway pressure (CPAP), respirators, or plication. Because diaphragmatic paralysis may occur in other conditions such as a myotonic dystrophy, attention to the differential diagnosis is important, particularly when an associated brachial plexus problem is not present. 61
Diaphragmatic paralysis is demonstrated by respiratory difficulty in the first few hours of life. X-ray film shows elevation of the hemidiaphragm with paradoxic movement that may disappear on positive end-expiratory pressure (PEEP) or CPAP. 22,61
Facial palsy may be part of intrinsic brainstem disease (see previous discussion of laryngeal nerve palsy) or other conditions such as Möbius syndrome, myotonic dystrophy, or facial muscle agenesis. When it is traumatic in origin, facial palsy is thought to be caused by the position of the face on the sacral promontory at the exit of the nerve from the stylomastoid foramen. 61 Normally, both the upper (temporofacial) and lower (cervicofacial) branches are involved. Known complications (from lack of total resolution) include contractures and synkinesis. Cosmetic surgical procedures occasionally are necessary but often are delayed for years.
Facial palsy is seen on the left side in 75% of cases. Features include a widened palpebral fissure, flat nasolabial fold, and decreased facial expression. Most infants completely recover within 3 weeks, although some infants continue to have deficits months later. 61
PARENT TEACHING
Infants with facial palsy may require the use of artificial tears if unable to completely close the eye on the involved side. Occasionally it may be necessary to tape the eye to prevent injury to the cornea. Parents also should be taught to expect some drooling of formula from the corner of the mouth during feedings.
Most infants with laryngeal nerve palsy recover in the first 6 to 12 months of life. Symptoms initially require supplemental parent education and support. An infant’s risk for aspiration necessitates careful feeding and appropriate response if choking occurs. Additional education for gavage feedings, a tracheotomy, or an apnea monitor may be necessary for the parents of a few infants. The teaching requirements for the infant with diaphragmatic paralysis must also be tailored to meet the individual needs and circumstances of the infant and family involved.
NEONATAL SEIZURES
Seizures may be the most frequent and often the only clinical sign of central nervous system dysfunction in the neonate.46,61 The occurrence of neonatal seizures typically prompts urgent medical attention. Seizures raise immediate concerns about the underlying cause of the brain disorder, associated clinical condition, the effect seizures may have on the developing brain, the need for anticonvulsant drugs, and the effect these drugs may have on the neonate with seizures. 46,61
Although the exact incidence of neonatal seizures is difficult to ascertain, Volpe noted marked differences in incidence associated with variations in birth weight, ranging from 57.5 per 1000 infants weighing less than 1500 g to 2.8 per 1000 infants weighing 2500 to 3999 g at birth. 5,61Seizures occur more frequently during the neonatal period than at other periods of life.31
Neonatal seizures increase the risk for impaired neurologic and developmental functioning in infancy and increase the risk for death. 22,45 Volpe notes that multiple or extended neonatal seizure activity is associated with significantly poorer prognosis than when seizures are controlled.61 There is also a suspected predisposition to cognitive, behavioral, or epileptic complications later in life. 31,46
Recognition of neonatal seizures with identification of etiology and prompt treatment is critical. Although not a disease entity, seizures are commonly related to significant disorders, which may require specific treatment. Untreated neonatal seizures may interfere with supportive therapies such as assisted ventilation and nutrition. Finally, experimental data suggest that seizures themselves may result in brain injury.61
Seizures result when an excessive synchronous electrical discharge of neurons within the CNS occurs (i.e., depolarization). 35,61Neonatal seizures are not a specific disease entity but, rather, a symptom.5,22 They may be associated with any disorder directly or indirectly affecting the CNS. Primary intracranial processes that may result in neonatal seizures include meningitis, intracranial hemorrhage (subdural, intraventricular, primary subarachnoid), encephalitis, and tumor.
However, seizures also occur secondary to systemic or metabolic disturbances including hypoglycemia, hypoxia-ischemia, hypocalcemia, hypomagnesemia, hyponatremia, and drug withdrawal. 4,35,61 A link between intrapartum fever and unexplained seizure activity in term infants also has been made in the literature. The presence of fever increased the likelihood of such seizure activity by four times the norm, even when the presence of infection was not found. 33
Seizures are signs of malfunctioning neuronal systems. Seizures occur when the neurons within the central nervous system excessively depolarize (extreme simultaneous electrical discharge). Volpe notes that a seizure is defined clinically as a paroxysmal alteration in neurologic function, that is, behavioral, motor, or autonomic function.61 These clinical signs may or may not be accompanied by abnormalities of the surface EEG. Clinical presentation of seizures is considerably different in the newborn period when compared with the well-organized seizure activity seen in older children and adults. The incomplete neurophysiologic development of a premature infant results in even less organized seizure activity than that seen with the term infant. 61
Etiology and Data Collection
Neonatal seizures may be caused by a variety of acute and chronic stresses on the brain. 4,22,35,46,61Table 26-4 lists the general groups of causes of neonatal seizures. The search for a cause proceeds in an orderly, methodical way. Most often, the known history of perinatal problems narrows the differential diagnosis to one or two likely causes. Acute metabolic changes that are likely to cause seizures should be rapidly investigated first. Blood glucose should be immediately checked both in the neonatal intensive care unit (NICU) (Accu-Chek with glucose meter reading) and in the laboratory, because hypoglycemia is a dangerous but very treatable cause of seizures (Table 26-5). 35,61
| Classification | Causes |
|---|---|
Acute metabolic conditions (Do blood gases, pH, 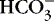 , Na, K, Ca, Mg, glucose, blood urea nitrogen [BUN]) , Na, K, Ca, Mg, glucose, blood urea nitrogen [BUN]) |
Hypocalcemia
Hypoglycemia; hyperglycemia
Hypomagnesemia
Pyridoxine dependency or deficiency
Hyponatremia; hypernatremia
|
| Inherited metabolic conditions (acidosis is common; assess urine amino acids, organic acids, NH 3, galactose) |
Maple syrup urine disease
Nonketotic hyperglycemia
Hyperprolinemia
Hyperglycinemia
Galactosemia
Urea cycle abnormalities
Organic acidemias
|
| Infections (12% of cases; assess cerebrospinal fluid [CSF]; culture blood, CSF; polymerase chain reaction assay in CSF; imaging) |
Viral encephalitis; herpes or enterovirus infection
Congenital infections
Bacterial meningitis
Sepsis
Brain abscess
Septic venous thrombosis
|
| Intracranial hemorrhage (15% of cases; imaging; CSF examination) |
Subdural hematoma
Cerebral contusion
Subarachnoid hemorrhage
Epidural hemorrhage
Intraventricular hemorrhage (premature)
|
| Hypoxic ischemia (0-3 days) most common (60%) | |
| Congenital malformations | |
| Neonatal drug withdrawal (see Chapter 11) (e.g., opiates) | |
| Local anesthetic intoxication | |
| Kernicterus | |
| Specific nongenetic syndromes | |
| Benign familial neonatal seizures | |
| Idiopathic (in only 10%, no cause is found) |
| *Appears to be preferred, although safety has not been clearly established. 61 | ||
| †Must be diluted in NS or D 5W to a concentration of 1.5 to 25 mg PE/mL for IV use. 69 |
||
| ‡Volpe cited 3-4 mg/kg/24 hr IV in divided doses every 12 hr, starting 12 hr after loading dose. 61 |
||
| Drug | Dose | Comments |
|---|---|---|
| Glucose |
10% solution
2 mL/kg bolus intravenously (IV) if hypoglycemic. 61
Maintenance: as high as 8 mg/kg/min IV61 (see Chapter 15).
|
Treat if hypoglycemic with glucose meter testing (e.g., Accu-Chek; One Touch II). 68 |
| Phenobarbital (drug of choice for neonatal seizures) | Loading: 20 mg/kg IV given slowly over 10-15 min; additional 5 mg/kg can be given 1 hour after dose to maximum of 40 mg/kg total for refractory seizures. 61,68,69 | Therapeutic level: 15-40 mcg/mL69 (obtain levels any time); respiratory depressant; incompatible with other drugs in solution. |
| Maintenance: 3-4 mg/kg/24 hr in 2 divided doses beginning no earlier than 12 hours after last loading dose. 61,69 | Maintain adequate oxygenation and ventilation. 61 | |
| Fosphenytoin (Cerebyx) preferred over phenytoin* (added if seizures not controlled by phenobarbital alone) |
Fosphenytoin advantages: high water solubility; pH value closer to neutral; faster, safe rate of administration; safe to give IM; absence of tissue injury with IV infusion; easy to prepare in IV solution. 61
Therapeutic level: measure trough serum phenytoin (not fosphenytoin) 48 hr after IV loading dose; 10-20 mcg/mL desirable level. 69
|
|
|
Maintenance: 4-8 mg PE/kg/24 hr IM or IV slow push (see above for dilution); infuse no faster than 1.5 mg/kg/min. Flush IV before/after with normal saline (NS). 69
Maintenance should be initiated 24 hr after loading dose. 69
Term infants greater than 1 wk of age may need up to 8 mg PE/kg/dose every 8-12 hr. 69
|
||
| Phenytoin used instead of Cerebyx to control seizures that are not controlled by phenobarbital alone |
Phenytoin disadvantages: incompatible with glucose and all other drugs; cannot be given IM (crystallizes in the muscle); rapid administration can result in bradycardia, dysrhythmias, hypotension. 69
Therapeutic level: measure trough level 48 hr after loading dose. Serum level 6-15 mcg/mL initially and 10-20 mcg/mL after the first few weeks. 69
|
|
| Pyridoxine (vitamin B 6) as indicated | 50-100 mg IV push or IM. 61,69 | |
| Lorazepam (Ativan) for seizures uncontrolled by phenobarbital and fosphenytoin (or phenytoin if used) 61 | 0.05-0.1 mg/kg IV slow push over several min. 61,69 | |
|
ADDITIONAL THERAPY AS INDICATED:
Calcium gluconate, 5% solution
Magnesium sulfate, 50% solution
IV antibiotics (bacterial infection) (see Chapter 22)
Acyclovir (herpes) (see Chapter 22)
|
||
Volpe61 also listed lumbar puncture as the other urgent laboratory test to be completed because bacterial meningitis is another dangerous but treatable cause of seizures. Sepsis should never be overlooked as a potential cause. The infectious agent (e.g., meningitis, encephalitis, empyema, abscess, septic thrombosis, ventriculitis) may directly affect the CNS. Systemic infection may cause seizures through the complication of shock, coagulopathy, impaired oxygenation, and multisystem organ failure. When the cerebrospinal fluid (CSF) is examined, not only should the changes associated with infection be identified but also evidence of bleeding (RBCs) or cell destruction (protein) may be found. 14,61
Structural studies are routinely performed as part of the evaluation. At present, the most useful studies are CT scans and cranial ultrasonographic examination, which can document intracranial hemorrhage. 22,46,68
The infant’s history should be carefully reviewed to narrow the possible causes to the most likely ones. Physical examination may further narrow the differential diagnosis. Once the history and physical examination are completed, blood should be drawn for assessment of arterial blood gases, electrolytes, glucose, calcium, and magnesium. 22,35,46,61,68
Appropriate cultures must be obtained. Usual culture sites or specimens include blood, urine, CSF, and pharyngeal or tracheal aspirate. The CSF should be examined for RBCs and white blood cells (WBCs), organisms (by Gram stain), protein, and sugar. 14,22,35,46,68
Ultrasonographic examinations are particularly useful for identifying and following the clinical course of intraventricular bleeding and hydrocephalus. The infant is not exposed to radiation, either immediately or long-term. Complications, either immediate or long-term, have not been identified. The test may be repeated as often as needed and usually is performed at the bedside. CT scan or MRI also may be indicated. 61
Clinical Seizure Types
Simultaneous EEG and video recording allow the accurate diagnosis of difficult-to-assess subtle behaviors, apneic and bradycardic spells, and the jerks and twitches commonly seen in preterm newborns. 41 Many have been surprised to find no correlation between events thought to be seizures and changes on the EEG. 61
Seizures result from excessive simultaneous electrical discharge or depolarization of neurons. 22,48,61 They are a manifestation of an underlying disorder rather than being an isolated disorder. As a paroxysmal alteration of neurologic function, behavioral, motor, or autonomic clinical phenomena are associated with EEG seizure activity, and there are also clinical phenomena not consistently correlated with EEG seizure activity. An increasing body of literature indicates that epileptic phenomena can be formed at subcortical levels and are therefore not detectable by surface-recorded EEG. 61 The Critical Findings box on p. 769 lists classification types and usual EEG findings. It has been noted also that many neonatal seizures identified by EEG are not correlated with motor and behavioral seizure activity—a phenomenon called electroclinical dissociation. The most immature infants are more prone to such seizures. 48,61
Traditional Categorization of Neonatal Seizures
| Classification/Types | Clinical Manifestations | Definition/Description |
|---|---|---|
|
Clonic
• Focal clonic
• Multifocal clonic
|
• Rhythmic jerks (1-3/sec)
• Rate slows during seizure
• + Electroencephalogram (EEG) seizure activity
|
• Focal: well-localized to a body part
• Multifocal: several body parts jerking simultaneously or in migrating order
|
|
Tonic
• Focal tonic
• Generalized tonic
|
• Characterized by posturing
• Focal: + EEG seizure activity
• Generalized: usually no EEG seizure activity
|
• Focal: continued posturing of limb or a posturing (asymmetric) of trunk or neck
• Generalized: extension of lower limbs with either upper limb extension (looks like decerebrate posturing) or upper limb flexion (looks like decorticate posturing)
|
|
Myoclonic
• Focal myoclonic
• Generalized myoclonic
|
• Faster jerking than in clonic seizures
• Flexor muscles (limbs) involved
• Focal: usually no EEG seizure activity
• Generalized: + EEG seizure activity
|
• Focal: flexor jerking of upper limbs
• Generalized: bilateral jerking of upper extremities; sometimes lower limbs are involved; often single or irregular jerks
|
| Subtle (more common in the premature infant) |
• Abnormal behavioral, autonomic, or motor activities that do not result from the other 3 seizure classifications
• + EEG seizure activity with only some of the seizure activities
|
• Ocular: nystagmus, horizontal or vertical deviation of eyes, staring episodes, eyelid flutter or blinking
• Facial: repetitive sucking, mouth movements, tongue protrusion, chewing, drooling
• Limb: bicycling, swimming movements, “boxing” or “hooking” motions, stepping
• Apnea: only 2% result from seizures
• Autonomic or vasomotor changes
|
Focal clonic and multifocal clonic seizures are the most likely to have true cortical origins. Eye blinking, a clonic manifestation, or nystagmus may be seen. Focal clonic seizures have been seen as an important manifestation of cerebral infarction in the neonate. 48,61Apnea with electrical seizure activity has been seen as an ictal manifestation but is more commonly seen in a full-term infant. The majority of apneic episodes in the premature population are not epileptic in origin. 48,61
The lack of ongoing monitoring of brain activity in most neonatal units makes accurate identification of seizures extremely difficult. The best correlation can be made by obtaining an EEG during periods of suspected seizure activity. The EEG may confirm clinical manifestations as true epileptic seizure activity. As discussed, however, there is evidence that epileptic discharges may be present without EEG detection. 48,61
The traditional categorization of neonatal seizures is presented in the Critical Findings box above. The classification does not have the same significance as that of the International Classification of Seizures in older individuals. Some general observations may pertain, even with the confusion surrounding the accurate diagnosis of neonatal seizures. However, seizures continue to be more difficult to recognize in neonates. Newborn jitteriness compounds this difficulty, so care must be taken to avoid mistaking this jitteriness for seizure activity (see the Critical Findings box on p. 770). 46,61
Episodes characterized as tonic and subtle are most likely to be seen in premature infants. Tonic episodes are quite commonly associated with IVH. Clonic and multifocal clonic seizures are more common in term infants. Myoclonic seizures often include a metabolic cause, such as nonketotic hyperglycemia or urea cycle disorder (see the Critical Findings box on p. 769 for a review).
Seizures Versus Jitteriness
| Clinical Observations | Seizure | Jitteriness |
|---|---|---|
| Ocular abnormalities (eye deviations or staring) | Yes | No |
| Gentle restraint of the involved body part halts the activity | No | Yes |
| Activity is easily elicited with stimulation (e.g., voice, motions) | No | Yes |
| Dominant movement is a slower clonic jerking having both fast and slow elements | Yes | No |
| Tremor in which the amplitude and rate of the alternating movements are equal | No | Yes |
| Autonomic changes are present (e.g., apnea, tachycardia, elevated blood pressure, pupil changes, increased salivation) | Yes | No |
Prevention
Many neonatal seizures can be successfully prevented through careful attention to possible metabolic changes expected on the basis of the infant’s condition. Hypoglycemia, hypocalcemia, hypomagnesemia, and often hypoxia can be anticipated and controlled.
Seizures resulting from intracranial malformations, infections, or prenatal injury most often cannot be prevented. Inherited metabolic disorders may not be identified until after initial symptoms, which may include seizures, appear. 48
Whether neonatal seizures can be prevented by pretreatment of the mother in high-risk situations contributing to neonatal seizures has not been adequately investigated. As progress in antenatal treatment of the fetus continues, this may become an area for further investigation. 61
Treatment
The rational treatment of neonatal seizures involves a vigorous attempt to achieve four specific goals: acute treatment, correction, prevention, and minimization.
ACUTE TREATMENT
The first goal is acute treatment of prolonged or multiple seizures and status epilepticus. Prolonged seizures and frequent, multiple seizures may result in metabolic changes and cardiorespiratory difficulties. Seizures are associated with increased energy consumption by the neurons and may interfere with adequate oxygenation. The neonatal brain appears to be less sensitive to seizure-induced injury than the adult brain; however, repeated seizures may be detrimental to the developing nervous system. 23Although it may not always be possible, vigorous efforts should be made to control the seizure activity. When the administration of a single drug does not result in lasting control, a second or third should be tried.48,61,63
The most common drugs used for the control of acute seizures and status epilepticus in the newborn are phenobarbital and phenytoin (see Table 26-5). In a randomized controlled study, these two drugs equally performed in controlling EEG-confirmed seizures; combined therapy was needed in over half of the infants, regardless of which drug was received first. 61 Both drugs are given in loading doses of 15 to 20 mg/kg. 48,63,69 In most infants, this load achieves a blood level within the therapeutic range.
Because both drugs are always given intravenously (IV) for this indication, the blood level is promptly achieved. 48,61,63,69When these antiepileptic drugs are unsuccessful in bringing the seizure(s) under control, alternative drugs, lorazepam or midazolam, may be used.63,69 Fosphenytoin (Cerebyx) has significantly decreased the adverse effects associated with phenytoin, but information is still limited regarding its safety and efficacy in neonates. 63,69 However, some important features of fosphenytoin differentiate it from phenytoin and should be appreciated if fosphenytoin is eventually approved for use in newborns. With fosphenytoin, potential is less for local toxicity such as phlebitis. An additional advantage of the use of fosphenytoin over phenytoin is the ability to administer fosphenytoin intramuscularly (IM) if no IV sites are available. A considerable advantage of fosphenytoin is that it can be infused safely at a much faster rate than that for phenytoin, but the safety of its use in newborns has not yet been proven. 61
CORRECTION
The second goal is correction of underlying remediable causes. This goal is often more important than the first goal, because some seizures induced by metabolic abnormalities cannot be controlled with antiepileptic drugs until the metabolic derangement is corrected. It is especially inappropriate to treat a newborn with antiepileptic drugs before correctable causes have been excluded.
After blood has been drawn for glucose, calcium, magnesium, electrolytes, and blood gas determination, therapy may begin. It is always proper to administer glucose. Inspired oxygen concentration may be raised temporarily if hypoxia is suspected. In refractory seizure situations, the IV administration of 50 to 100 mg pyridoxine ideally should be performed under simultaneous EEG monitoring so that the true causes of pyridoxine dependency or deficiency can be detected. 48,61
PREVENTION
Prevention of future seizures is the third goal (see Table 26-5). Seizure prophylaxis is a worthwhile goal for patients of all ages, but it is often not easily achieved in newborns. Often, despite the appropriate and vigorous administration of several antiepileptic drugs, seizures persist for several days, only to remit spontaneously and never return. Despite this observation, attempts to provide adequate seizure prophylaxis seem justified.
Phenobarbital remains the most studied and used drug for seizure control in the newborn. 46,48,61,63 Because the half-life of phenobarbital is long and may vary from 40 to 200 hours, serum concentration is monitored and it may not be necessary to give routine maintenance doses on a fixed schedule. 69
Phenytoin also is used extensively in neonatal seizures. 48,61,63,69 Although it may be very useful in acute treatment, phenytoin often is quite difficult to use as a maintenance drug. The most frequently encountered problem is the extremely variable half-life in newborns. It is not predictable, and frequent blood level determinations are necessary to estimate a useful half-life for the individual infant. 61,63 This pharmacokinetic problem is dramatically compounded by oral administration in which the medication interacts with milk proteins and is absorbed erratically. 69 It is reasonable to avoid altogether the oral use of phenytoin in the newborn.
A second problem is the variability in binding of phenytoin to albumin in blood. Bilirubin competes for protein binding sites and results in an increase in free phenytoin levels. 69 The binding is affected by the amount of albumin, concurrent drugs, and other poorly understood factors. Because changes in binding alter the amount of drug available to enter the brain, there is often little control over the true unbound “level.”48,61,63
Maintenance doses of phenobarbital (3 to 5 mg/kg) should be given after blood level determinations indicate that the level is dropping.69 Phenytoin and fosphenytoin maintenance doses are difficult to predict. Frequent blood level assessments may be necessary. 48,61,63
If unbound levels of phenytoin are available, these are often easier to use to ensure that a therapeutic range is maintained. Because the characteristic signs of phenytoin toxicity are cerebellar, they are ordinarily not recognized in the newborn. One must rely on the accurate determination of blood levels to safeguard against excessive administration. Maintenance doses for phenytoin range from 4 to 8 mg/kg IV every 24 hours for the neonate. 63,69 Phenytoin should be given slowly when pushed IV (never exceeding 0.5 mg/kg/min). 69
MINIMIZATION
The fourth goal is minimization of the side effects of antiepileptic drug therapy. In the attempt to control seizures, the potential of antiepileptic drugs to produce side effects must not be ignored. Drug-induced encephalopathy may mimic the clinical changes seen in hypoxic-ischemic encephalopathy (HIE) or numerous metabolic derangements. The possibility that the pharmaceutical agent may be causing some of the findings being attributed to the underlying disorder always exists. Likewise, improvement in the underlying disorder may be masked by changes induced by the drug.
More significant side effects such as respiratory or cardiovascular depression produced by large doses of any of the drugs, hepatotoxic changes induced by valproic acid, or hyperbilirubinemia intensified by diazepam are rare but worthy of recognition. 61,63,69
The issue of long-term side effects of the antiepileptic drugs is far from being settled. Virtually all of the drugs used have been shown in animal or tissue culture studies to have detrimental effects on the growth or development of the brain. The extent to which any of this information can be transferred to the human situation remains the subject of extensive investigation. 48,61,63
Complications and Outcome
Studies to date have been unable to separate the effect of the seizure from the effect of the cause. Early hypocalcemia, not uncommonly found in stressed newborns, is an example of a relatively benign cause of neonatal seizures. These infants have an excellent chance of recovery without complications. For acute treatment, 1 to 2 mL/kg of a 10% solution of calcium gluconate (10 to 20 mg/kg of elemental calcium) should be diluted in an appropriate IV solution and infused IV over 10 to 30 minutes. The infant should be closely monitored by electrocardiogram (ECG) during infusion of IV calcium. 48,69 The infusion should be halted if the heart rate falls below 100 beats/min. 69
Separate from any discussion of the direct effect of seizures on the developing brain is the question of whether seizures in the newborn period predispose to later seizures. Again, cause seems to be the most important factor. Those seizures caused by transient metabolic changes that do not cause other permanent neurologic dysfunction are themselves likely to be transient and not occur outside the neonatal period. Seizures caused by congenital anomalies or those accompanied by obvious permanent brain damage are likely to persist.
Important prognostic findings and signs can be grouped to provide a general guide to assess newborns with seizures. Factors favoring a good prognosis include transient metabolic causes (hypocalcemia, hypomagnesemia), normal neurologic examination, normal EEG findings, and benign familial neonatal seizures.
Factors favoring a poorer prognosis include the presence of a congenital malformation, seizures persisting for more than several days, presence of a major IVH with seizure activity, severely abnormal EEG findings (burst suppression, extremely low voltage, isoelectric), or major signs on neurologic examination (hemi-syndrome, multiple brainstem signs, and severe hypotonia with unresponsiveness). 48,61
Parent Teaching
Lay terms such as “fit” or “spell” provide a hint of the fear that seizure activity can instill in parents. Parent teaching should focus not only on providing pertinent information but also on correcting existing misinformation. Parents may initially have difficulty believing that an infant is experiencing a seizure, because a neonatal seizure is difficult to recognize. It is also not unusual for parents to expect staff to insert items in the mouth, perform cardiopulmonary resuscitation (CPR), restrain or shake the infant, or institute other measures once they understand that the infant is having a seizure.
Parents may express an urgent and understandable need to know the cause and the long-term outcomes of the seizure activity. Supply careful explanations of tests being performed and their purpose in identification of the cause of the seizure. The long-term impact on the infant may be harder to predict for the parents, although the presence of certain factors can result in a poorer or better prognosis (see “Complications and Outcome” section). Close follow-up after discharge by both medical and developmental services is vital.
Once the parents are home with the infant, their ability to recognize seizures and appropriately intervene is crucial. Careful documentation of teaching and parent understanding will allow the nursing staff to build on previous knowledge and skills. Parent handouts should focus on the skills and goals listed on the teaching checklist (see the Parent Teaching box on p. 773). Care should be taken to use short, easy-to-understand sentences and to explain all terminology that parents might find confusing (e.g., what an EEG is). Without parent handouts, attempting to teach the volume of necessary information is more stressful on both the caregiver and the family. Handouts improve the parents’ retention of new and complex material and are available for reference after discharge.

Providing parents with a form for documenting seizures is also helpful. Documentation can guide the parents in making appropriate observations during seizure activity (e.g., date, time, duration, seizure activities observed, color changes, behavior after seizure). Parents should practice using this form while the infant is in the hospital so that staff members can assist them with their assessments and documentation. Parents should also administer the medications whenever possible during the infant’s hospitalization to establish skills and reinforce their confidence.
A multidisciplinary team approach to discharge planning is crucial for the provision of effective discharge teaching. This planning and parental education must be initiated early in the hospitalization for successful outcomes. After discharge, a multidisciplinary approach to follow-up is also necessary.
In summary, discharge from the hospital after the diagnosis and treatment of neonatal seizures is a period of both relief and increased anxiety for parents, other family members, and caregivers. For infants with a good prognosis, efforts should be made to help family members look past the neonatal seizures and view their newborn as “healthy.” For infants whose neurologic outcome will clearly not be normal, clinicians should stress the needs of the child with multiple handicaps. The clinician should take the lead in providing the basis for this discussion and guidance in the normalization of the lives of these infants and children.
HYPOXIC-ISCHEMIC ENCEPHALOPATHY
Pathophysiology
A common cause of brain damage in newborns is HIE. 61Hypoxemia refers to a diminished amount of oxygen in the blood, and ischemia refers to a diminished amount of blood perfusing the brain. Either of these may result in the lack of a sufficient oxygen supply to the brain. Asphyxia refers to the impairment of the exchange of respiratory gases, implying low oxygen and high carbon dioxide in the blood. 29,35,60,61 When comparing hypoxemia and ischemia, ischemia is more important. With ischemia, the brain lacks a supply of both oxygen and glucose, which increases likelihood of brain injury. During the perinatal period, hypoxia or ischemia (or a combination of them both) is usually the outcome of asphyxia. 29,60,61
Injury to the brain is thought to occur at two time periods. The initial insult is caused by the hypoxemia and/or ischemia. Once cerebral circulation and oxygenation are restored, there is a resolution of the acute event and a return of cellular energy metabolism. 36,60 During the reperfusion, glutamate is cleared, lactate levels are reduced, and high energy phosphate levels are returned to normal. 61Eight to sixteen hours after this initial acute event, a second decrease in high energy phosphate levels can be seen and reaches a nadir at approximately 24 to 48 hours. 60,61
The ratio of phosphocreatine/inorganic phosphate decreases, nitric oxide vasodilates the cerebral circulation, and calcium shifts into the cell, disrupting the sodium-potassium-ATPase pump. Levels of glutamate, arachidonic acids, prostaglandins, and uric acid are increased. Adhesion molecules in platelets and leukocytes are activated by free radicals, promoting occlusion of the microvasculature. The reduced delivery of oxygen and glucose perpetuates the insult. 28,60,61As mitochondria become more dysfunctional, clinical deterioration including the presence of seizure activity becomes evident. Animal research has shown that neuronal protein loss can be noted by 6 hours and is very evident by 18 hours. 58The period of time before the occurrence of the secondary energy failure is the targeted therapeutic window in which an intervention may ameliorate injury. It has been demonstrated that the secondary energy collapse is the consequence of the hypoxic-ischemic event rather than the cause of it. 58,61
Etiology
HIE occurs in 1 to 2 of every 1000 live term births, and 0.3 per 1000 demonstrate significant neurologic sequelae. 30,35,61 Approximately 20% of these outcomes can be associated with primarily antepartum events (e.g., hypotension of the mother, intrauterine growth restriction [IUGR], maternal diabetes). 60,61 When hypoxic-ischemic insults are linked primarily to intrapartum events (e.g., cord prolapse, abruptio placentae, traumatic delivery), they occur in 35% of the cases. 60,61 Primarily postnatal occurrences (e.g., cardiac failure with congestive heart disease, pulmonary failure) have a 10% incidence. 60,61 Finally, 35% of HIE cases occur from a combination of both antepartum and intrapartum difficulties. 30,60,61
In a publication by the AAP and the American College of Obstetricians and Gynecologists (ACOG), the authors note that new information indicates that intrapartum hypoxia is usually not the only cause of neonatal encephalopathy or cerebral palsy (CP).1 The authors state that “less than a quarter of infants with neonatal encephalopathy have evidence of hypoxia or ischemia at birth, and, therefore it is inappropriate to label most newborns with encephalopathy as having hypoxic-ischemic neonatal encephalopathy.”1 Rather than occurring during labor and delivery, they state that the majority of cases of neonatal encephalopathy and CP can be linked to events that occur before the onset of labor. They also emphasize that HIE is just a single subset of neonatal encephalopathy and define HIE as neonatal encephalopathy with hypoxia occurring in the intrapartum period with no evidence of any other abnormality.1 Because of limitation in our current ability to determine the actual timing of the insult, it is often hard to identify/quantitate the antepartum contribution separately from the intrapartum.
The AAP and ACOG task force established criteria to be used in defining an acute intrapartum event that is adequate to produce CP.1 The task force listed four criteria that must all be present:
1. Metabolic acidosis in fetal cord arterial blood obtained at delivery (pH <7 and base deficit [BD] ≥12 mmol/L)
2. Severe or moderate neonatal encephalopathy with an early onset occurring in infants born at 34 weeks’ gestation or less
3. CP, either spastic quadriplegia or dyskinetic type
4. Other identifiable etiologies (trauma, infections, genetic or coagulation disorders) must be excluded
The task force also cited criteria “that collectively suggest an intrapartum timing (within close proximity to labor and delivery (e.g., 0 to 48 hours) but are nonspecific to asphyxial insults”3,60:
1. “A sentinel (signal) hypoxic event occurring immediately before or during labor”1
2. “A sudden and sustained fetal bradycardia or the absence of fetal heart rate variability in the presence of persistent, late, or variable decelerations, usually after a hypoxic sentinel event when the pattern was previously normal”1
3. Apgar scores of 0 to 3 occurring later than 5 minutes after birth
4. Multisystem involvement with an onset within 72 hours of birth
5. The presence of evidence of acute nonfocal cerebral abnormality shown on early imaging study
Prevention
Prevention of HIE requires the avoidance of hypoxic-ischemic insults. Anticipation of risk factors in the antepartum period, monitoring of the fetus during the intrapartum period, and prompt intervention when needed are discussed in the literature. 61 Chorioamnionitis has been linked to the development of encephalopathy and CP in the term infant. 67After delivery, management includes maintaining adequate oxygenation and carbon dioxide levels and preserving adequate perfusion while avoiding fluctuations in blood pressure or systemic hypotension.35,60,61Serum glucose levels should be kept stable (Volpe recommends 75 to 100 mg/dL61). Although neuronal injury may result from hypoglycemia, allowing marked hyperglycemia may result in hemorrhage or increase cerebral lactic acid levels. 61Promptly diagnose seizure activity and provide treatment.35,61Seizures have been linked to swift falls in glucose levels in the brain (secondary to heightened cerebral metabolic rate), lactic acid increases in the brain, apnea and hypoventilation (which worsens hypoxia and elevated CO2levels), and marked increases in arterial blood pressure (with resultant increased likelihood of IVH). When seizure activity of infants is poorly controlled, the incidence of severe neurologic sequelae rises. 46,48,61,63
Data Collection
HISTORY
Infants with intrapartum causes of HIE are symptomatic in the newborn period. These infants have fetal distress in utero, are depressed at birth, and have prolonged low Apgar scores. Most have evidence of systemic organ damage: renal, cardiac, and sometimes pulmonary dysfunction is readily apparent, accompanying the neurologic features of HIE. Diagnosis of HIE depends on careful prenatal and perinatal history-taking and a thorough postnatal neurologic examination. Severe and persistent neurologic findings suggest an unfavorable prognosis. Risk for more severe neurologic sequelae increases when the duration of the neurologic abnormalities in the neonate lengthens. Conversely, when neurologic abnormalities depart by week 1 or 2, the prognosis is excellent. 60,61
SIGNS AND SYMPTOMS
The neurologic findings vary by the stage of HIE (see theCritical Findings boxbelow). Seizures occur within the initial 12 hours after birth and are almost always subtle seizure activity.22,35,36,49,61Generalized tonic seizures occur in the preterm, whereas multifocal clonic seizure activity occurs in the term infant (see theCritical Findings box on p. 769) .22,35,36,61
LABORATORY DATA
Essential laboratory data to reflect liver, renal, or cardiopulmonary dysfunction include a complete blood count (CBC), sodium, potassium, calcium, phosphorus, magnesium, glucose, blood urea nitrogen (BUN) and creatinine, urinalysis, liver function studies, enzymes (aspartate transaminase [AST], serum creatine kinase), echocardiogram, blood gases, lumbar puncture with CSF analysis, and cultures of CSF and blood. 22,36,60,61 Acidosis, hypoxemia, and hypercarbia are frequent findings after asphyxia. Other parameters needing correction include a low blood sugar (hypoglycemia), hypocalcemia, hyponatremia, and, more rarely, hyperammonemia. 22,35,36,61
Stages of Hypoxic-Ischemic Encephalopathy and Neurologic Assessment
| Stage | Neurologic Assessment |
|---|---|
| Stage I: Mild encephalopathy | Hyperalert, with normal tone and activity, exaggerated response to stimulation, reactive pupils, no seizure activity |
| Stage II: Moderate encephalopathy |
Hypotonic, weak suck, constricted but reactive pupils; periodic breathing or apnea
Development of seizure activity or lethargy indicates deteriorated status
|
| Stage III: Severe encephalopathy | Stupor or coma, absent reflexes, pupils nonreactive, no spontaneous activity, requires mechanical ventilation |
One should sample the spinal fluid to assess bleeding and to rule out infectious possibilities. Chest radiograph is recommended. The infant should be monitored with an amplified EEG for seizures.22,36EEGs not only confirm or rule out the diagnosis of seizures but also may be used to identify the severity of hypoxic-ischemic brain injury and assess future prognosis.35,61 In a term infant, a severely abnormal pattern such as burst suppression, very low amplitude, or isoelectric EEG suggests an ominous prognosis. 22,36,48,61
Accurately performed ultrasonography may show periventricular increased echo densities, but most scans of the term neonate do not show such changes. 61MRI scans are most valuable in follow-up at 2 to 3 months, but early CT or MRI scans may demonstrate areas of cerebral edema or of ischemic injury. Hyperdense areas are characteristic of hemorrhagic infarction on CT scans. 22,36,61 MRI is the diagnostic tool of choice for the neonate with hypoxic-ischemic injury. 61 Even conventional MRI can display abnormal findings as early as the first day. Diffusion-weighted MRI has a greater sensitivity and can display abnormal findings within hours of birth. 61
Treatment
Treatment includes the broad principles of adequate ventilation, gas exchange, and perfusion; maintenance of normal glucose, calcium, and electrolyte levels; control of seizures; and prevention and control of brain swelling.22,36,50,60
Current clinical trials support possible benefits of “modest” hypothermia as a neuroprotective intervention for moderate hypoxic-ischemic encephalopathy. Although the exact mechanisms underlying the neuroprotective strategy of hypothermia is not totally elucidated, a Cochrane review has found it to be beneficial in term neonates suffering from HIE.24 Because the long-term efficacy and safety issues have not been established, hypothermia is considered to be an evolving therapy that should be used in NICUs that strictly follow the recommendations of the protocols used in the CoolCap and the National Institute of Child Health and Human Development (NICHD) trials.21,54
Eligibility criteria for neonatal cooling are listed inBox 26-2. The NICU must be well organized and prepared to screen the neonate and have equipment ready to initiate cooling within 6 hours after the initial insult because this is the window of opportunity. First, the baby is examined to determine if the eligibility criteria are met. Before or as the neonate is being screened, equipment can be assembled and the blanket is pre-cooled and placed at the bedside. The neonate is always admitted and/or transported in a warmed bed on servocontrol to ensure that a thorough physical and neurologic examination may be carried out. Care should be taken with provision of heat support since overheating is potentially very dangerous and has been shown to markedly add to the neurologic damage. After cooling, rewarming should be done gradually because rapid rewarming may cause vasodilatory shock and rebound seizures. A total body-cooling hypothermia protocol is outlined inBox 26-3.
BOX 26-2
Screening Inclusion Criteria
• Postmenstrual age ≥36 weeks
• Admitted ≤6 hours of age with a diagnosis of encephalopathy
• pH ≤7 or base deficit >16 mmol/L on cord blood or blood gas within first hour of life (for head cooling)
• pH 7.01-7.15 and base deficit 10-15.9 mmol/L in the first hour of life (for whole body cooling)
• If no blood gas available in the first hour, there also must be:
• Evidence of an acute perinatal event, or
• 10-minute Apgar score <5, or
• Assisted ventilation (PPV or CPAP) initiated at birth and continued for a minimum of 10 minutes
Inclusion Criteria
• Seizure activity present
• Diagnosis of moderate or severe encephalopathy, which includes any one of the following:
• Lethargy
• Decreased tone (may have normal peripheral tone but have central hypotonia), abnormal tendon reflexes, myoclonus, weak suck, abnormal Moro reflex
• Any evidence of seizures
• Abnormal breathing
• Moderate to severe EEG amplitude reduction (lower margin <5 microvolts and/or upper margin <10 microvolts) on a 20-minute aEEG or evidence of seizures
Exclusion Criteria
• >6 hours of age
• Severe intrauterine growth restriction (<1.8 kg)
• Major congenital anomaly
• Head trauma resulting in severe intracranial hemorrhage (head cooling)
• Prophylactic high-dose anticonvulsants (head cooling)
• Parents do not grant consent
• Inability to initiate cooling by 6 hours of age
aEEG, Amplitude-integrated electroencephalogram; CPAP, continuous positive airway pressure; EEG, electroencephalogram; HIE, hypoxic-ischemic encephalopathy; PPV, positive pressure ventilation.
Data from Gluckman PD, Wyatt JS, Azzopardi D, et al: Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial, Lancet 365:663, 2005; Higgins RD, Tonse NKR, Perlman J, et al: Hypothermia and perinatal asphyxia: executive summary of the National Institute of Child Health and Human Development workshop, J Pediatr 148:2, 170, 2006; and Shankaran S, Laptook AR, Ehrenkranz RA, et al: Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy, N Engl J Med 353:1574, 2005.
BOX 26-3
• Place the precooled blanket, covered with one regular hospital baby blanket, under the baby.
• Insert (nasally) an esophageal temperature probe to T6-9, or approximately 2 centimeters above the diaphragm.
• After an x-ray film confirms the probe’s position, the radiant warmer is turned to manual control with the heater in the off position and the radiant warmer temperature probe is left in place.
• The cooling machine (Blanketrol II [or III]) is placed on auto control to maintain the esophageal temperature at 33.5° C (92.3° F) for 72 hours. Although some fluctuation is expected, it should be no more than 1° C higher or lower than the target (32.5° to 34.5° C or 90.5° to 94.1° F). If using the Blanketrol II, the water temperature in the blanket is better stabilized when two blankets are connected together, such that one blanket is under the neonate and the other is hung next to the radiant warmer.
• The set point, patient esophageal temperature, skin temperature, and water (blanket) temperature are recorded on a specific hypothermia flow sheet, while a standard nursing flow sheet documents axillary temperature, blood pressure, heart rate, and skin condition.
• Effects of cooling may include the following:
• Decreased perfusion to the extremities (caused by cold stress) may affect reliability of pulse oximetry values.
• Decrease in the resting heart rate, which may result in blood pressure fluctuations.
• It may be necessary to adjust the temperature of the blood gas analyzer (to the baby’s temperature) to increase accuracy of blood gas results.
• At exactly 72 hours of hypothermia, the cooling machine is turned to the warming mode. Auto control is increased by 0.5° C every hour for 6 hours until the set temperature is 36.5° C.
• The blanket is turned off, the esophageal probe is removed, and the heater on the radiant warmer is turned on with the servocontrol set to 0.5° C warmer than the neonate’s skin temperature. The servocontrol set point is increased by 0.5° C each hour until the axillary or skin temperature reaches 36.5° C. Wide fluctuations in heart rate and blood pressure should be anticipated.
• As the neonate approaches discharge, a Whole Body Hypothermia Discharge Checklist is completed to ensure that a brain MRI, developmental evaluations, and follow-up appointments with the high-risk clinic and neurologist have been done.
Complications
A variety of sequelae may result from selective neuronal necrosis. Outcome depends on the degree of insult to the brain. When asphyxiated infants exhibit HIE, there is a 20% to 50% mortality rate within the newborn period. Sequelae include hyperactivity, attention deficits, spastic diplegia, motor difficulties, intellectual deficits, and impairments of vision. 35 Other complications include CP, mental retardation, deafness, epilepsy, and learning disabilities. 61
Cooling reduces mortality without increasing major disability in survivors. 24 Selective head cooling has shown a significant reduction in major neurodevelopmental disability but not a significant decrease in mortality. 24 Total body cooling has demonstrated significant reductions in both mortality and major neurodevelopmental disability. 24,53 The benefits of cooling on survival and neurodevelopment outweigh short-term adverse effects. Adverse effects of neonatal cooling are transient and minimal: (1) bradycardia, (2) hypotension, (3) thrombocytopenia, (4) coagulopathy, and (5) renal impairment. Overcooling (especially <28 ° C) is associated with added risks of dysrhythmias and bradycardia.
INTRAVENTRICULAR HEMORRHAGE
Physiology
The problem of bleeding into and around the ventricular system has received more attention in the past two decades than any other neurologic problem in neonates. In large measure, this relates to the frequency with which the problem develops, generally estimated to be 15% in premature infants weighing 1500 g or less.22,36,61 The growth of routine cranial ultrasonographic examination in premature infants has resulted directly from the need to evaluate this common problem. With the advent of CT and ultrasonographic scanning, a large number of infants who were not otherwise suspected of having intraventricular bleeding are diagnosed on scans.
Although the bleeding is regularly spoken of as intraventricular and intracranial hemorrhage, these terms do not accurately reflect its causes. Highly vascularized areas, which have relatively fragile and poorly supported blood vessels, are the source of bleeding. In a premature infant, the most common source of hemorrhage is the subependymal germinal matrix.26,43,61 The width of the matrix is 2.5 mm at 23 to 24 weeks but decreases to 1.4 mm at 32 weeks. By 36 weeks, almost complete involution has occurred. Volpe lists principal clinical features in a premature baby requiring a ventilator secondary to respiratory distress syndrome. Approximately 90% of bleeding events occur in the first 72 hours of life, and at least one in two affected infants experiences hemorrhage in the first 24 hours.36,61
The incidence of IVH in term infants is approximately 3.5%, making IVH both an uncommon and unanticipated diagnosis. Approximately 50% of cases of IVH in the term newborn are caused by asphyxia or trauma with symptoms occurring within the first 2 days after delivery. About 25% of these infants with IVH have no significant risk factors (e.g., delivery without occurrence of trauma or asphyxia, a neonatal history without complications before the hemorrhage). Symptoms in this group may appear as late as 3 to 4 weeks after delivery. 36,61
In term infants, the choroid plexus of the lateral ventricles is the most common site in which bleeding originates. 3,22,36,61In premature infants, the germinal matrix, in the subependymal area adjacent to the caudate nucleus, is the primary site of bleeding.3,11,43,61 Both of these are areas of high arterial and capillary blood flow; in addition, they use an anatomically awkward venous drainage system, eventually draining into the internal cerebral vein (Figure 26-2).
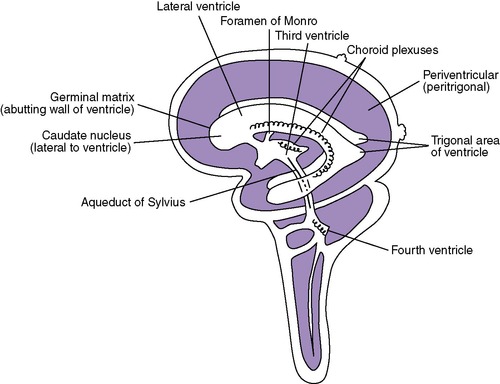 |
| FIGURE 26-2 |
The extent of bleeding generally predicts the likelihood of complications and sequelae. Bleeding may be confined to the germinal matrix or the choroid plexus, or it may enter the ventricular system. When filled under pressure, the ventricular system may dilate. Blood may also extravasate out into the brain parenchyma (more likely with germinal matrix bleeding than with choroid plexus bleeding).
Several classification schemes have been used, each trying to assess the degree of bleeding or amount of blood present. Ideally, a classification should relate to pathophysiology, treatment, or outcome; however, with present knowledge, this is not possible.
Volpe listed three grades of germinal matrix IVH using ultrasonographic scanning to identify the presence and extent of blood in the germinal matrix and lateral ventricles (Table 26-6). A “separate notation” is made for the existence of “periventricular hemorrhage infarction or of other parenchymal lesions.” He clarified the use of this separate notation by noting that these abnormalities are not usually the result of simple “extension” of matrix or IVH hemorrhage into “normal brain parenchyma.”61 Others also use this classification system. 25
| Severity | Description |
|---|---|
| Grade I | Germinal matrix hemorrhage with no or minimal intraventricular hemorrhage (10% of ventricular area on parasagittal view) |
| Grade II | Intraventricular hemorrhage (10%-50% of ventricular area on parasagittal view) |
| Grade III | Intraventricular hemorrhage (greater than 50% of ventricular area on parasagittal view; usually distends lateral ventricle) |
| Separate notation | Periventricular echodensity (location and extent) |
An older classification system based on the extent of hemorrhage seen on the CT scan grades germinal matrix hemorrhages as follows (Figure 26-3). Because both grading systems are still cited in the literature, we have included each.
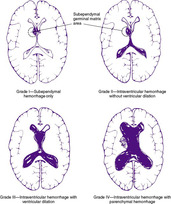 |
| FIGURE 26-3
(From Rozmus C: Periventricular-intraventricular hemorrhage in the newborn, Matern Child Nurs 17:79, 1992.)
|
0—No bleeding
I—Germinal matrix only
II—Germinal matrix with blood in the ventricles
III—Germinal matrix with blood in the ventricles and hydrocephalus (ventricular dilation)
IV—Intraventricular and parenchymal bleeding (other than germinal matrix)
Etiology
The etiologic factors identified in infants who have experienced IVH are multiple. 11,45,59 These include asphyxia, severe respiratory distress, pneumothorax, hypoglycemia, shock, acidosis, blood transfusions, seizures, and rapid volume expansion (see the Critical Findings box on p. 781). What appears to be the common factor underlying the pathologic condition is a fluctuation/alteration in cerebral blood flow61 that causes the numerous and thin-walled blood vessels in the germinal matrix to bleed. 22,36,61
Factors that Predispose the Premature Infant to Intraventricular Hemorrhage
Prematurity
• Birth weight less than 1500 g
• Less than 34 weeks’ gestation
Asphyxia (see Chapters 4 and 8)
• Before, during, after birth
Respiratory (see Chapters 8 and 23)
• Idiopathic respiratory distress syndrome
• Hypoxia
• Positive pressure ventilation
• Pneumothorax
• Apnea
Cardiovascular (see Chapter 4)
• Rapid volume expansion
• Elevated venous pressure
• Elevated or lowered arterial pressure (shock; transfusions)
Hematologic (see Chapter 20)
• Hyperosmolarity
• Coagulation disorders
• Hyperviscosity
Metabolic (see Chapters 814 and 15)
• Hypoglycemia/hyperglycemia
• Hypernatremia/hyponatremia
• Metabolic acidosis
• Rapid pH shifts
Miscellaneous
• Hypothermia (see Chapter 6)
• Acetylsalicylic acid ingestion by mother
• Neonatal pain (see Chapter 12)
• Neonatal environmental stressors (see Chapter 13)
Intraventricular bleeding tends to occur in the first few hours or days of life. The profound physiologic changes normally seen after birth are coupled with the multiple problems (primarily cardiorespiratory) typically experienced by the premature infant, making intraventricular bleeding common. The degree to which the aggressive management of premature newborns has a role in the development of bleeding cannot be accurately assessed. Generally, although not exclusively, sicker infants both require more intervention and have a greater likelihood of bleeding. In a recent study of 3721 premature infants, the authors concluded that a significant reduction in the incidence of IVH would occur if “extremely premature infants, the vast majority of patients suffering from IVH, didn’t have to be transferred postnatally to another hospital.”18
Data Collection
Some infants, generally those who are term, have IVH associated with severe asphyxia. Premature infants often show one or more of the following: birth weight less than 1500 g, gestational age less than 34 weeks, shock, respiratory distress syndrome (RDS), need for blood transfusions, coagulopathy, hyperviscosity, hypoxia, and birth asphyxia. 22,36,61
Because premature infants, particularly those weighing less than 1500 g, tend to have multiple problems, it is not surprising that the clinical presentation of germinal matrix hemorrhage may range from subtle (or even undetectable) to catastrophic.
Deterioration in clinical condition followed by apnea, flaccid quadriparesis, unresponsiveness, and death from circulatory collapse is a recognizable syndrome. Common signs of germinal matrix hemorrhage include apnea, hypotension, drop in hematocrit, flaccidity, areflexia, full fontanel, tonic posturing, and oculomotor disturbances.
When intracranial bleeding is suspected, appropriate studies of intracranial structures should be performed as soon as possible. For periventricular-intraventricular hemorrhage, both ultrasonography and CT scanning are useful tools for defining the presence of bleeding and for following its evolution. Ultrasonographic scanning is listed by Volpe as “the procedure of choice in the diagnosis of germinal matrix-IVH.”61 Because ultrasonography is the safer procedure and uses no radiation, it should be used for follow-up.
Treatment and Intervention
The primary treatment of IVH is supportive care. Ventilatory support, maintenance of oxygenation, regulation of acid-base balance, suppression of seizures, and treatment of any attendant coagulopathy are all extremely important in reducing mortality and morbidity. The role that successful management has in the amelioration or prevention of complications is unclear.
Many therapies have been proposed or used but remain unproven. Therapies are listed not to recommend their general clinical use but, rather, to suggest ways the problem has been approached.
When the hemorrhage is confined to the germinal matrix, little can be done, even from a theoretic standpoint.
Intraventricular fibrinolytics (streptokinase/urokinase) have been used to dissolve the clot and prevent hydrocephalus. After intraventricular blood clots were successfully dissolved by urokinase in the adult population with subsequent improved outcomes, several neonatal studies were completed. Initial favorable results were followed by two studies that showed no benefit from the use of fibrinolytic therapy. 61
Care must be given to reduce the risk for continued bleeding, to maintain perfusion of the brain, and to reduce wide fluctuations in blood pressure, oxygenation, and pH.22,35,36
Medications to reduce intracranial pressure and treat secondary effects of the bleeding include furosemide, acetazolamide, and steroidal agents. Mannitol and glycerol also have been used.
Helpful pharmacologic preventive agents (glucocorticoids and possibly phenobarbital administered prenatally and indomethacin, vitamin E, and phenobarbital postnatally) show varying success in the literature. Antepartal prophylactic use of phenobarbital to prevent IVH or neurologic disabilities in the preterm infant is not supported by research. 10,63 Vitamin K administration to women before preterm delivery is not preventive of periventricular hemorrhage in the preterm neonate. 10 Postnatal prophylactic use of phenobarbital in preterm infants to prevent IVH results in a greater requirement for mechanical ventilation, and it is recommended that phenobarbital not be used for this purpose. 66
Complications
The complications from IVH relate to the underlying causes and the extent of bleeding. Massive bleeding with dilation of the ventricular system is much more likely to cause an acute change in brain function, with increased intracranial pressure, brainstem abnormalities, and apnea. Milder degrees of hemorrhage may be asymptomatic or associated with seizurelike events, changes in muscle tone, or apnea.
When bleeding extends into the parenchyma, porencephaly may result from liquefactive necrosis or ischemia-induced encephalomalacia. Follow-up structural brain studies may show hypodense areas in which blood was present; later they may show areas of porencephaly.
The most common complication is posthemorrhagic hydrocephalus. The risk is directly related to the severity of the hemorrhage, with up to 10% of survivors of mild IVH and 65% to 100% of survivors of severe IVH showing progressive ventricular dilation. 5 Evidence of posthemorrhagic hydrocephalus should be investigated in all survivors of germinal matrix hemorrhage. CT scanning or ultrasonography to assess ventricular size should be used because clinical signs alone are not reliable. 5,22,61 CP, visual and hearing difficulties, problems with fine or gross motor control, seizures, and mental disabilities are other possible complications. 5,25,61
With the hope of avoiding the necessity of placing a shunt, some attempts at control of the hydrocephalus have been made. Osmotic and diuretic agents, including furosemide, isosorbide, and acetazolamide, have been used to reduce the formation of CSF, although the practice has not gained widespread acceptance.
Outcome studies have been difficult to assess. Clearly, the sickest infants tend to do poorly. They also tend to have more complications, including CNS complications. There is a clear correlation between the grade of bleed and the likelihood of significant neurologic residua, but the correlation is far from perfect. 22,36,61 The influence of other factors on neurologic outcome may be more significant than that of the actual bleed itself. Hypoxia, hypoperfusion, and other conditions known to damage the developing nervous system cannot easily be separated as individual factors in outcome. Patients with grades I and II hemorrhages usually do as well as patients with no hemorrhage. Patients with grades III and IV hemorrhages most often have moderate to severe developmental disability.
Parent Teaching
Parents of an infant with IVH should be involved with their infant’s care plan. The rationale for a minimal handling protocol needs to be explained. Encouraging parents to participate in setting “time out” and “touch me” times will facilitate their ability to visit and assist with care. During visits, they should be encouraged to recognize signs of overstimulation and become knowledgeable about the appropriate interventions to take to calm the infant.
The infant with IVH has varying degrees of problems. 5,11,61 Often the acute situation resolves without ongoing problems. In these cases, parents should understand the possible complications such as hydrocephalus that may occur in the short term. Teaching the parents to measure head circumference and alerting them to the signs of increased intracranial pressure such as poor feeding, posturing, eye movement difficulties, full fontanel, and lethargy enable them to participate more fully in the medical follow-up (see the Parent Teaching box on pp. 754–755). Up to 80% to 90% of infants with hydrocephalus need a shunt because of ventricular dilation. 4,5
Parents must understand the risk for long-term neurologic sequelae. Despite the difficulty of predicting sequelae with any degree of certainty, parents should understand that mental and motor handicaps, delays in the acquisition of milestones, seizures, and problems associated with hydrocephalus and potential shunt placement may occur.5,11,45,61 Specific preparation for these potential problems should begin in the nursery but will be increased during follow-up visits if the possibility for such problems seems greater. Prompt and appropriate referral to medical specialists and supportive services is important in both inpatient and outpatient settings. Parents may find support and information from national and state organizations (see the “Parent Resources for Neurologic Disorders” section).
REFERENCES
1. American Academy of Pediatrics and American College of Obstetricians and Gynecologists, Neonatal encephalopathy and cerebral palsy: defining the pathogenesis and pathophysiology. ( 2003)The College, Washington, DC.
2. Back, S., Congenital malformations of the central nervous system, In: (Editors: Taeusch, H.; Ballard, R.; Gleason, C.) Avery’s diseases of the newborned 8 ( 2005)Saunders, Philadelphia.
3. Barks, J.D.; Donn, S.M., Intracranial hemorrhage, In: (Editor: Donn, S.M.) Michigan manual of neonatal intensive careed 3 ( 2003)Hanley & Belfus, Philadelphia.
4. Blackburn, S.T., Maternal, fetal and neonatal physiology: a clinical perspective. ed 3 ( 2007)Saunders, Philadelphia.
5. Blackburn, S.T.; Ditzenberger, G.R., Neurologic system, In: (Editors: Kenner, C.; Lott, J.W.) Comprehensive neonatal care: an interdisciplinary approached 4 ( 2007)Saunders, Philadelphia.
6. Bol, K.A.; Collins, J.S.; Kirby, R.S.; , National Birth Defects Prevention Network, Survival of infants with neural tube defects in the presence of folic acid fortification, Pediatrics 117 (2006) 803.
7. Brent, R.L.; Oakley, G.P.; Mattison, D.R., The unnecessary epidemic of folic acid-preventable spina bifida and anencephaly, Pediatrics 106 (2000) 825.
8. Bruner, J.P.; Tulipan, N.; Reed, G.; et al., Intrauterine repair of spina bifida: preoperative predictors of shunt-dependent hydrocephalus, Am J Obstet Gynecol 190 (2004) 1305.
9. Centers for Disease Control and Prevention (CDC), Folic acid now frequently asked questions, www.cdc.gov/ncbddd/folicacid/faqs.htm; (last review January 30, 2008). Accessed November 1, 2008, from.
10. Crowther, C.A.; Henderson-Smart, D.J., Vitamin K prior to preterm birth for preventing neonatal periventricular haemorrhage, Cochrane Database of Syst Rev 3 (2008); CD000229.
11. deVries, L.; Rennie, J.M., Preterm cerebral hemorrhage, In: (Editors: Rennie, J.M.; Roberton, N.R.C.) Roberton’s textbook of neonatologyed 4 ( 2005)Elsevier Health Sciences, Edinburgh.
12. Donnelly, V.; Foran, A.; Murphy, J.; et al., Neonatal brachial plexus palsy: an unpredictable injury, Am J Obstet Gynecol 187 (2002) 1209.
13. Dunham, E.A., Obstetrical brachial plexus palsy, Orthop Nurs 22 (2003) 106.
14. Edwards, M.S., Postnatal bacterial infections, In: ed 7 (Editors: Fanaroff, A.A.; Martin, R.J.) Neonatal-perinatal medicine: diseases of the fetus and infant, ( 2002)Mosby, St Louis; vol 2.
15. Fichter, M.A.; Dornseifer, U.; Henke, J.; et al., Fetal spina bifida repair: current trends and prospects of intrauterine neurosurgery, Fetal Diagn Ther 24 (2008) 318.
16. Geisel, J., Folic acid and neural tube defects in pregnancy, J Perinat Neonatal Nurs 17 (2003) 268.
17. Gleeson, J.G.; Dobyns, W.B.; Plawner, L.; et al., Congenital structural defects, In: (Editors: Swaiman, K.F.; Ashwal, S.; Ferriero, D.M.) Pediatric neurology: principles and practiceed 4 ( 2006)Mosby, St Louis.
18. Gleissner, M.; Jorch, G.; Avenarius, S., Risk factors for intraventricular hemorrhage in a birth cohort of 3721 premature infants, J Perinat Med 28 (2000) 104.
19. Gunes, T.; Ozturk, M.A.; Koklu, E.; et al., Effect of allopurinol supplementation on nitric oxide levels in asphyxiated newborns, Pediatr Neurol 36 (1) ( 2007) 17.
20. Hauser, K.W.; Lilly, C.M.; Frias, J.L., Florida health care providers’ knowledge of folic acid for the prevention of neural tube defects, South Med J 97 (2004) 437.
21. Higgins, R.D.; Tonse, N.K.R.; Perlman, J.; et al., Hypothermia and perinatal asphyxia: executive summary of the National Institute of Child Health and Human Development workshop, J Pediatr 148 (2) ( 2006) 170.
22. Hill, A., Neurological and neuromuscular disorders, In: (Editors: MacDonald, M.G.; Mullett, M.D.; Seshia, M.M.K.) Avery’s Neonatology: pathophysiology and management of the newborned 6 ( 2005)Lippincott Williams & Wilkins, Philadelphia.
23. Holmes, G.L.; Ben-Ari, Y., The neurobiology and consequences of epilepsy in the developing brain, Pediatr Res 49 (2001) 320.
24. Jacobs, S.; Hunt, R.; Tarnow-Mordi, W.; et al., Cooling for newborns with hypoxic ischaemic encephalopathy, Cochrane Database Syst Rev 4 (2007); CD003311.
25. Jones, M.W.; Bass, W.T., Perinatal brain injury in the premature infant, Neonatal Netw 22 (2003) 61.
26. Kirby, C.L., Posthemorrhagic hydrocephalus: a complication of intraventricular hemorrhage, Neonatal Netw 21 (2002) 59.
27. Klusman, A.; Heinrich, B.; Stopler, H.; et al., A decreasing rate of neural tube defects following the recommendations for periconceptional folic acid supplementation, Acta Paediatr 94 (2005) 1538.
28. Kumar, A.; Ramakrishna, V.K.; Basu, S.; et al., Oxidative stress in perinatal asphyxia, Pediatr Neurol 38 (3) ( 2008) 181.
29. Laptook, A.R., Hypoxic-ischemic-encephalopathy, In: (Editors: Lawson, E.E.; Lehmann, C.U.; Nogee, L.M.; et al.)eNeonatal Rev, vol 2 ( 2008), p. 1; (5).
30. Lavery, S.V.; Randall, K.S., Cerebral monitoring of the term infant, Neonatal Netw 27 (5) ( 2008) 329.
31. Levene, M., The clinical conundrum of neonatal seizures, Arch Dis Child Fetal Neonatal Ed 86 (2002) F75.
32. Levene, M.I., Intracranial haemorrhage at term, In: (Editors: Rennie, J.M.; Roberton, N.R.C.) Roberton’s textbook of neonatologyed 4 ( 2005)Elsevier Health Sciences, Edinburgh.
33. Lieberman, E.; Eichenwald, E.; Mathur, G.; et al., Intrapartum fever and unexplained seizures in term infants, Pediatrics 106 (2000) 983.
34. Lumley, J.; Watson, L.; Watson, M.; et al., Periconceptional supplementation with folate and/or multivitamins for preventing neural tube defects, Cochrane Database Syst Rev 3 (2008); CD001056.
35. Lynam, L.; Verklan, M.T., Neurologic disorders, In: (Editors: Verklan, M.T.; Walden, M.) Core curriculum for neonatal intensive care nursing,ed 4 ( 2009)Saunders, Philadelphia.
36. Madan, A.; Hamrick, S.E.G.; Ferriero, D.M., Central nervous system injury and neuroprotection, In: (Editors: Taeusch, H.; Ballard, R.; Gleason, C.) Avery’s diseases of the newborned 8 ( 2005)Saunders, Philadelphia.
37. March of Dimes, New folic acid seal helps women choose enriched grain foods to help prevent birth defect, www.marchofdimes.com/cgi-bin/; last review. January 29, 2008). Accessed November 1, 2008, from.
38. Miller, M.; Elixhauser, A.; Zhan, C., Patient safety events during pediatric hospitalizations, Pediatrics 111 (2003) 1358.
39. Mitchell, L.E., Epidemiology of neural tube defects, Am J Med Genet C Semin Med Genet 135 (2005) 88.
40. Molloy, A.M.; Kirke, P.N.; Troendle, J.F.; et al., Maternal vitamin B 12 status and risk of neural tube defects in a population with high neural tube defect prevalence and no folic acid fortification, Pediatrics 123 (2009) 917.
41. Moore, K.L.; Persaud, T.V.N., The nervous system, In: (Editors: Moore, K.L.; Persaud, T.V.N.) Before we are born: essentials of embryology and birth defects,ed 7 ( 2007)Saunders, Philadelphia.
42. Reference deleted in proofs.
43. Papile, L., Intracranial hemorrhage and vascular lesions, In: (Editors: Fanaroff, A.A.; Martin, R.J.; Walsh, M.C.) Fanaroff and Martin’s neonatal-perinatal medicine: diseases of the fetus and infanted 8, ( 2005)Mosby, St Louis.
44. Rathnau, C.H., The ABCs of genetics, Central Lines 17 (2001) 21.
45. Rennie, J.M., Assessment of the neonatal nervous system, In: (Editors: Rennie, J.M.; Roberton, N.R.C.) Roberton’s textbook of neonatologyed 4 ( 2005)Elsevier Health Sciences, Edinburgh.
46. Rennie, J.M., Seizures in the newborn, In: (Editors: Rennie, J.M.; Roberton, N.R.C.) Roberton’s textbook of neonatologyed 4 ( 2005)Elsevier Health Sciences, Edinburgh.
47. Sarnat, H.B., Embryology and malformations of the central nervous system, In: ed 7 (Editors: Fanaroff, A.A.; Martin, R.J.) Neonatal-perinatal medicine: diseases of the fetus and infant, ( 2002)Mosby, St Louis; vol 2.
48. Scher, M.S., Neonatal seizures, In: (Editors: Taeusch, H.; Ballard, R.; Gleason, C.) Avery’s diseases of the newborned 8 ( 2005)Saunders, Philadelphia.
49. Shalak, L.F.; Laptook, A.R.; Velaphi, S.C.; et al., Amplitude-integrated electroencephalography coupled with an early neurologic examination enhances prediction of term infants at risk for persistent encephalopathy, Pediatrics 111 (2003) 351.
50. Shankaran, S., The postnatal management of the asphyxiated term infant, Clin Perinatol 29 (2002) 675.
51. Reference deleted in proofs.
52. Reference deleted in proofs.
53. Shankaran, S.; Pappas, A.; Laptook, A.R.; et al., Outcomes of safety and effectiveness in a multicenter randomized, controlled trial of whole-body hypothermia for neonatal hypoxic-ischemic-encephalopathy, Pediatrics 122 (2008) e791.
54. Shaw, P.S.; Ohlsson, A.; Perlman, M., Hypothermia to treat neonatal hypoxic ischemic encephalopathy, Arch Pediatr Adolesc Med 161 (2007) 951.
55. Smith, S.A.; Ouvrier, R., Peripheral neuropathies, In: (Editors: Swaiman, K.F.; Ashwal, S.; Ferriero, D.M.) Pediatric neurology: principles and practiceed 4 ( 2006)Mosby, St Louis.
56. Thompson, D.N.P., Postnatal management and outcome for neural tube defects including spina bifida and encephalocoeles, Prenat Diagn 29 (2009) 412.
57. Vannucci, R.C.; Palmer, C., Hypoxia-ischemia: neuropathology, pathogenesis, and management, In: ed 7 (Editors: Fanaroff, A.A.; Martin, R.J.) Neonatal-perinatal medicine: diseases of the fetus and infant, vol 2 ( 2002)Mosby, St Louis.
58. Vannucci, R.C.; Towfighi, J.; Vannucci, S.J., Secondary energy failure after cerebral hypoxia-ischemia in the immature rat, J Cereb Blood Flow Metab 24 (2004) 1090.
59. Vergani, P.; Patane, L.; Doria, P.; et al., Risk factors for neonatal intraventricular haemorrhage in spontaneous prematurity at 32 weeks gestation or less, Placenta 21 (2000) 402.
60. Verklan, M.T., The chilling details: hypoxic ischemic encephalopathy, J Perinat Neonatal Nurs 23 (2009) 59.
61. Volpe, J.J., Neurology of the newborn. ed 5 ( 2008)Saunders, Philadelphia.
62. Wald, N.J., Folic acid and the prevention of neural-tube defects, N Engl J Med 350 (2004) 101.
63. Ward, R.M.; Lugo, R.A., Drug therapy in the newborn, In: (Editors: MacDonald, M.G.; Mullett, M.D.; Seshia, M.M.K.) Avery’s neonatology: pathophysiology & management of the newborned 6 ( 2005)Lippincott Williams & Wilkins, Philadelphia.
64. Watkins, M.L.; Rasmussen, S.A.; Honein, M.A.; et al., Maternal obesity and risk for birth defects, Pediatrics 111 (2003) 1152.
65. Whitby, E.H.; Griffiths, P.D.; Rutter, S.; et al., Frequency and natural history of subdural haemorrhages in babies and relation of obstetric factors, Lancet 362 (2004) 846.
66. Whitelaw, A.; Odd, D., Postnatal phenobarbital for the prevention of intraventricular hemorrhage in preterm infants, Cochrane Database Syst Rev 3 (2008); CD001691.
67. Willoughby, R.E.; Nelson, K.B., Chorioamnionitis and brain injury, Clin Perinatol 29 (2002) 603.
68. Yager, J.Y.; Vannucci, R.C., Seizures in neonates, In: ed 7 (Editors: Fanaroff, A.A.; Martin, R.J.) Neonatal-perinatal medicine: diseases of the fetus and infant, vol 2 ( 2002)Mosby, St Louis.
69. Young, T.E.; Mangum, B., Neofax. ed 21 ( 2008)Thomson Reuters Publishing, Montvale, NJ.
PARENT RESOURCES FOR NEUROLOGIC DISORDERS
The Hydrocephalus Association: 870 Market Street, Suite 705, San Francisco, CA 94102; Phone: (888) 598-3789 toll free; Phone: (415) 732-7040; Fax: (415) 732-7044; Website: www.hydroassoc.org; E-mail: info@hydroassoc.org
Support Center of New Jersey: 2516 Route 35, North Manasquan, NJ 08736; Phone: (732) 528-8080; Fax: (732) 528-4744; NJ only: (800) 372-6510; Website: www.familysupportnj.com
National Organization for Rare Disorders (NORD): PO Box 1968, Danbury, CT 06813-1968; Phone: (800) 999-6673 (help line); Phone: (203) 744-0100; Fax: (203) 798-2291; Website: www.rarediseases.org; E-mail: orphan@rarediseases.org
National Dissemination Center for Children with Disabilities (NICHCY): PO Box 1492, Washington, DC 20013; Phone: (800) 695-0285 (V/TTY); Fax: (202) 884-8441; Website: www.nichcy.org; E-mail: nichcy@aed.org
Epilepsy Foundation of America: 4351 Garden City Drive, Landover, MD 20785-7223; Phone: (800) 332-1000 toll free; Website: www.epilepsyfoundation.org
National Hydrocephalus Foundation: 12413 Centralia, Lakewood, CA 90715-1623; Phone: (888) 857-3434 toll free; Phone: (562) 402-3523; Fax: (562) 924-6666; Website: www.nhfonline.org; E-mail: hydrobrat@earthlink.net
Spina Bifida Association of America: 4590 MacArthur Blvd, NW, Suite 250, Washington, DC 20007-4226; Phone: (800) 621-3141 toll free; Phone: (202) 944-3285; Fax: (202) 944-3295; Website: www.sbaa.org; E-mail: sbaa@sbaa.org
American Self-Help Group Clearing House: 100 East Hanover Avenue, Suite 202, Cedar Knolls, NJ 07927-2020; Phone: (800) 367-6274 toll free (NJ only); Phone: (973) 326-6789; Website: www.selfhelpgroups.org
American Epilepsy Society: 342 North Main St., West Hartford, CT 06117; Phone: (860) 586-7505; Website: www.aesnet.org
Medic Alert: 2323 Colorado Ave., Turlock, CA 95382; Phone: (888) 633-4298 toll free; Fax: (209) 669-2450; Website: www.medicalert.org