174 Drug Therapy in Renal Failure
The incidence of acute kidney injury or insufficiency (AKI) in the intensive care unit (ICU) ranges from 5.9% to 25%, depending on how AKI is defined.1 Renal replacement therapy is required for 4.3% of all critically ill patients and up to 72.5% of patients with AKI.2 In-hospital mortality in ICU patients ranges from 5% to 10% in those with no renal dysfunction, 9% to 27% at risk of renal dysfunction, 11% to 30% with AKI, and 26% to 40% with overt kidney failure.3 Critically ill patients with chronic kidney disease (CKD) have poorer outcomes than patients with normal renal function on admission, and the presence of CKD is a significant predictor of hospital mortality in Acute Physiology, Age, and Chronic Health Evaluation (APACHE) III score tools.4,5 Renal insufficiency, whether acute or chronic, alters the absorption, distribution, metabolism, and elimination of many pharmacotherapeutic agents used in the treatment of critically ill patients. In this chapter, the drugs affected are tabulated, and the mechanisms responsible for the changes in disposition are discussed. A general construct for the individualization of drug therapy in patients with CKD or AKI is presented, along with dosage guidelines for the most commonly used ICU medications. Finally, the influence of continuous and intermittent renal replacement therapy on drug clearance is discussed, and dosage guidelines are tabulated for selected drugs for patients with severe CKD or AKI.
 Quantitation of Renal Function
Quantitation of Renal Function
Accurate assessment of renal function in critically ill patients is imperative. Serial estimates or measurements of renal function routinely are recommended to guide individualization of drug dosage regimens to optimize clinical outcomes. The calculation of creatinine clearance (CLcr) from a timed urine collection with creatinine measurement in serum and urine has been the standard clinical measure of renal function for decades. Urine is difficult to collect accurately in the ICU. Furthermore, many commonly used medications interfere with measurement of creatinine, especially when colorimetric assay methods such as the Jaffé method are used. Thus, measurement of CLcr by this approach is not always the best way to assess glomerular filtration rate (GFR).6,7 The administration of radioactive (125I iothalamate, 51Cr-EDTA, or technetium-99m DTPA) or nonradioactive (aminoglycosides, iohexol, iothalamate, and inulin) markers of GFR, although scientifically sound, is clinically impractical because intravenous (IV) or subcutaneous (SQ) administration of the marker and the collection of multiple timed blood and urine collections make the procedures expensive and difficult to perform.
Estimation of CLcr or GFR requires only routinely collected laboratory and demographic data and is inexpensive and clinically feasible. The Cockcroft and Gault (C-G) method for estimation of CLcr8 and the Modification of Diet in Renal Disease (MDRD) method for estimation of GFR9 correlate well with CLcr and GFR measurements in individuals with stable renal function.9 These methods lose their predictive performance, however, in patients with liver disease,10–12 unstable renal function,6,13,14 estimated GFR above 60 mL/min,15 or obesity.16,17 In critically ill patients with AKI, both MDRD and C-G overestimate estimated GFR by 33% and 80%, respectively.18 Finally, although several methods for CLcr estimation in patients with unstable renal function have been proposed,7 as well as equations for “adjusted” weights (for use in the C-G equation) in the obese,16,17 the accuracy of these methods has not been rigorously assessed, and at present their use cannot be recommended.
There is considerable controversy at this time regarding whether the C-G or MDRD equation should be utilized to guide drug dosing adjustments in patients with CKD. The MDRD equation was developed to estimate GFR in patients with CKD.15 However, renal dosing recommendations for currently approved drugs are predominantly (>95%) based on relationships between drug clearance and CLcr estimated by the C-G equation.19 The literature now suggests that the two equations cannot be utilized interchangeably, as several studies have shown that the use of the MDRD equation results in discordant dosing recommendations (compared to dosing based on C-G) in up to 40% of patients.19–21 Complicating these comparisons even further is another problem: several versions of the MDRD equation (and most recently, an equation called Chronic Kidney Disease Epidemiology Collaboration [CKD-EPI]) have been reported since the publication of the original formula.22 As such, clinicians should continue to utilize CLcr estimated by C-G for drug dosing.23
 Altered Drug Disposition in Critically Ill Patients with Renal Insufficiency
Altered Drug Disposition in Critically Ill Patients with Renal Insufficiency
Effect On Drug Absorption
Absorption of drugs from the gastrointestinal (GI) tract is rarely altered in patients with CKD or AKI. Systemic availability of some drugs (i.e., some β-adrenergic blockers, dextropropoxyphene, and dihydrocodeine) is increased in CKD patients as a result of a decrease in metabolism during the drug’s first pass through the GI tract and liver.24,25 Some orally administered drugs that are extensively metabolized before reaching the systemic circulation may have increased bioavailability, but this phenomenon has been documented for relatively few drugs.26,27
Effect On Drug Distribution
The volume of distribution of several drugs is increased significantly in patients with AKI or severe CKD.25,28–30 Increases may result from fluid overload, decreased protein binding, or altered tissue binding (Table 174-1). The volume of distribution of only a few drugs is decreased in patients with CKD, and the mechanism proposed for this change is a reduction in tissue binding. Digoxin and pindolol are two prime examples, and for both of these drugs, a significant relationship exists between the decrease in distribution volume and CLcr.29,30
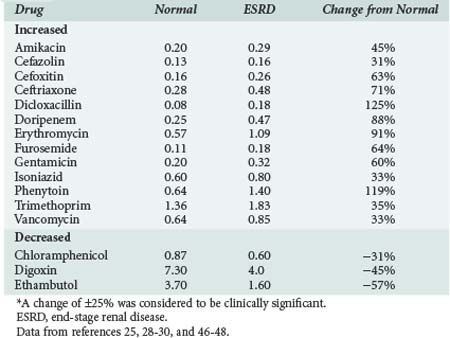
Effect On Drug Metabolism
Preliminary human data suggest a differential effect of CKD on cytochrome P450 (CYP) enzyme activity: the activities of CYP2C19 and CYP3A4 are reduced, whereas the activities of CYP2D6 and CYP2E1 are not affected.29,30 This differential effect on individual enzymes may help explain some of the conflicting data regarding changes in drug metabolism in the presence of severe CKD.
Reduction of nonrenal clearance of several drugs reported in patients with severe CKD supports the premise that alterations in hepatic cytochrome P450 enzyme expression and/or activity is responsible (Table 174-2).25,28–30 Prediction of the effect of CKD on metabolism of a particular drug is difficult even for drugs within the same pharmacologic class.29,30 Patients with CKD exhibit reductions in nonrenal clearance and alterations in bioavailability of predominantly hepatically metabolized drugs which are generally proportional to the reductions in GFR.
TABLE 174-2 Effect of End-Stage Renal Disease on Nonrenal Clearance of Selected Drugs Used in the ICU
| Decreased | ||
| Acyclovir | Erythromycin | Nitrendipine |
| Aztreonam | Imipenem | Procainamide |
| Bufuralol | Isoniazid | Propranolol |
| Cefotaxime | Ketorolac | Quinapril |
| Ceftriaxone | Metoclopramide | Vancomycin |
| Cilastatin | Morphine | Verapamil |
| Ciprofloxacin | Nicardipine | Warfarin |
| Doripenem | Nimodipine | |
| Increased | ||
| Bumetanide | Fosinopril | Phenytoin |
| Cefpiramide | Nifedipine | Sulfadimidine |
| Unchanged | ||
| Acetaminophen | Insulin | Nisoldipine |
| Chloramphenicol | Lidocaine | Pentobarbital |
| Clonidine | Metoprolol | Theophylline |
Data from references 25, 28–30.
Critically ill patients with AKI have been noted to have higher residual nonrenal clearance for three drugs—imipenem, meropenem, and vancomycin—than patients with CKD who have similar CLcr.31 This difference may be the result of less exposure to or accumulation of uremic waste products that alter hepatic function. Because patients with AKI may have a higher nonrenal clearance than patients with CKD, the resultant plasma concentrations will be lower than expected and possibly subtherapeutic if classic CKD-derived dosage guidelines are followed. Thus for these agents, initial dosing should be adjusted upward, and only after 7 to 10 days of persistent AKI do the dosing guidelines derived from CKD subjects likely become applicable.
Effect On Renal Excretion
Renal clearance is the composite of GFR, renal tubular secretion, and reabsorption: renal clearance = (GFR × fu) + (renal tubular secretion − renal reabsorption), where fu is the fraction of the drug unbound to plasma proteins. An acute or chronic progressive reduction in GFR decreases renal clearance; historically, drug dosage guidelines for patients with AKI or CKD have been based on this phenomenon. The contribution of a reduction in renal clearance to the degree of change in the total body clearance of a drug is highly dependent, however, on the fraction of the dose eliminated unchanged by the normal kidney, the intrarenal pathways for drug elimination and transport, and the degree of functional impairment of each of these pathways.29,30
Drug elimination by GFR occurs by diffusion, but renal tubular secretion and renal reabsorption are bidirectional processes that involve carrier-mediated renal transport systems and passive diffusion. The important renal transport systems involved in the renal tubular excretion of multiple compounds include the organic anionic (i.e., ampicillin, cefazolin, and furosemide), organic cationic (i.e., famotidine, trimethoprim, and dopamine), nucleoside (i.e., zidovudine), and P-glycoprotein transporters (i.e., digoxin and steroids).32,33 Accordingly, the clearance of drugs that are extensively renally secreted (renal clearance > 300 mL/min) may be reduced significantly by drug interactions (i.e., probenecid with β-lactam antibiotics) and/or impaired function of one or more of the renal transporter systems. For example, AKI due to ischemia or toxicants results in a significant impairment in the function of renal solute carrier (SLC) 22A organic ion transporters, compounding the effects of decreased GFR on drug clearance.34
 Strategies for Drug Therapy Individualization
Strategies for Drug Therapy Individualization
Secondary references such as the American Hospital Formulary Service Drug Information,35 Drug Prescribing in Renal Failure by Aronoff and colleagues,36 and Goodman and Gilman’s The Pharmacological Basis of Therapeutics37 are excellent sources from which one can acquire information on the pharmacokinetic characteristics of drugs in subjects with normal renal as well as impaired renal function. However, these references often do not provide the explicit relationships of kinetic parameters with CLcr or GFR. In addition, since the references employ different definitions of renal impairment and utilize varying methodologies for deriving recommendations, drug dosing recommendations occasionally differ significantly.38 This section provides a practical approach for drug dosage individualization in critically ill patients with AKI or CKD and patients receiving continuous renal replacement therapy (CRRT) or intermittent hemodialysis (IHD). Basic pharmacokinetic principles (see Chapter 169) combined with the disposition properties of a particular drug and a quantitative measure of the patient’s degree of renal function enable the clinician to design an individualized therapeutic regimen.
If the relationship of a drug’s total body clearance with CLcr or GFR is not known, one can estimate the patient’s total body clearance, provided that the fraction of the drug that is eliminated renally unchanged (fe) in subjects with normal renal function is known. The following approach makes six assumptions: (1) the volume of distribution is unchanged, (2) the change in total body clearance is proportional to CLcr, (3) renal disease does not alter the drug’s metabolism, (4) metabolites, if formed, are inactive and nontoxic, (5) the drug obeys first-order (linear) kinetic principles, and (6) the drug’s pharmacokinetics can be described adequately by a one-compartment model. If these assumptions are valid, the kinetic parameter/dosage adjustment factor (Q) can be calculated as Q = 1 − (fe [1 − KF]), where KF is the ratio of the patient’s CLcr to an assumed normal value of 120 mL/min. The estimated total body clearance (CLPT) can be calculated as follows: CLPT = CLnormT × Q, where CLnormT is the value in patients with normal renal function (i.e., patients with a CLcr of ≥ 120 mL/min). The elimination rate constant of the drug can be calculated as the quotient of the estimated total body clearance and volume of distribution. When these three key kinetic parameters are estimated, the individualized dosage regimen can be calculated as described in Chapter 169.
The optimal dosage regimen for an ICU patient with AKI or CKD depends on the desired goal. If there is a significant relationship between maximal plasma concentration and clinical response39,40 (i.e., aminoglycosides) or toxicity40,41 (i.e., quinidine, phenobarbital, and phenytoin), the dose and dosing interval may have to be modified. If the dosing interval is increased, the maximal plasma concentration and minimal plasma concentration are similar to values in individuals with normal renal function, but the desired target concentrations may not be precisely attained. In this case, consultation with a clinical pharmacist/pharmacologist may be warranted to facilitate the design of a revised dosage regimen. If no specific target values for maximal plasma concentration or minimal plasma concentration have been reported, attaining the same average steady-state concentration may be appropriate (i.e., cephalosporins). This goal can be achieved by decreasing the dose (DPT = DNORM × Q) or prolonging the dosing interval (τ) (τPT = τNORM ÷ Q).* If the dose is reduced while the dosing interval remains unchanged, the maximal plasma concentration becomes lower and the minimal plasma concentration higher (Figure 174-1). This dosage adjustment method, if taken to the extreme, results in maintenance of the desired average steady-state concentration by continuous infusion of a parenteral product. These principles have been used to derive dosage recommendations for commonly used drugs in the ICU for patients with mild, moderate, and severe kidney injury (Table 174-3).

TABLE 174-3 Dosing Guidelines for Drugs Commonly Used in the ICU by Patients with Renal Insufficiency



Continuous Renal Replacement Therapy
The three most commonly used forms of CRRT are continuous venovenous hemofiltration (CVVH), continuous venovenous hemodialysis (CVVHD), and continuous venovenous hemodiafiltration (CVVHDF). During CVVH, drugs are removed primarily by convection/ultrafiltration.42 The clearance of a drug is a function of the permeability of the hemofilter, which is called the sieving coefficient, and the ultrafiltrate flow rate. The sieving coefficient can be approximated by dividing the concentration of the drug in the ultrafiltrate (Cuf) by the concentration in the plasma entering the hemofilter (Ca): sieving coefficient = Cuf/Ca. The sieving coefficient is often approximated by the fraction unbound to plasma proteins (fu) because plasma concentrations of the drug of interest may not be readily available. The clearance by CVVH can be estimated as ultrafiltrate flow rate × fu. Drug clearance by CVVHDF can be estimated, provided that the blood flow rate is over 100 mL/min and dialysate flow rate is less than 33 mL/min, as (ultrafiltrate flow rate + dialysate flow rate) × (fu or sieving coefficient). If ultrafiltrate flow rate is negligible (<3 mL/min), as is often the case with CVVHD, CVVHD clearance can be estimated as the product of dialysate flow rate and fu or sieving coefficient.
Individualization of therapy for a patient receiving CRRT depends on the patient’s residual renal function and clearance of the drug by the mode of CRRT the patient is receiving. The patient’s residual drug clearance can be predicted based on the CLcr and the relationship between total body clearance of the drug and CLcr as described previously. The CRRT type (CVVH, CVVHD, or CVVHDF) and recommended initial dosage regimens of selected drugs that are used frequently in ICU patients receiving CRRT are listed in Table 174-4.43,44 In cases in which specific data on drug clearance are not available and the patient receiving CRRT is functionally anuric (i.e., urine output is <300 mL/d), drug dosing can be initiated at a level consistent with the individual having a CLcr of 30 to 50 mL/min, if their ultrafiltration/dialysate or ultrafiltrate flow rate is 2 L/h or greater.


Hemodialysis
Drug-related factors that influence hemodialyzability include the molecular weight, protein binding, and volume of distribution of a drug.25 The dialysis prescription factors include the composition of the dialyzer, surface area, and blood and dialysate flow rates. The semisynthetic and synthetic dialyzers used in high-flux hemodialysis have the largest ultrafiltration rates and more closely mimic the filtration characteristics of the human kidney. These dialyzers allow the passage of most drugs that have a molecular weight of ≤15,000 D.45 High-molecular-weight drugs such as vancomycin are significantly cleared by this mode of dialysis, and the clearance of many smaller drugs is significantly increased, as reviewed by Matzke.46 In order to obtain therapeutic plasma concentrations, patients receiving high-flux dialysis often require larger drug doses than the doses recommended in most references.
Quantification of the impact of hemodialysis on drug disposition can be calculated in several ways, and this contributes to the variability of values in the literature.25 The difference between the half-life during dialysis and the half-life of the drug when the patient is off dialysis provides a crude guide to the impact of dialysis. The half-life during dialysis may not be interpretable in AKI patients because declining plasma drug concentrations during dialysis represent elimination by the patient, which may be considerable. The most accurate means of assessing the effect of hemodialysis is to calculate the dialyzer clearance of the drug.25 Because drug concentrations generally are determined in plasma, the calculation of plasma clearance by the dialyzer (CLpD) can be calculated as: CLpD = Qp ([Ap − Vp]/Ap), where Ap is the concentration of drug in plasma going into the dialyzer, Vp is the concentration of drug in the plasma leaving the dialyzer, and Qp is plasma flow, which equals blood flow through the dialyzer (1 hematocrit). This method accounts for clearance due to diffusion, convection, and adsorption to the dialyzer. The recovery clearance (CLrD) approach also has been used for the determination of dialyzer clearance: CLrD = R/AUC0−t, where R is the total amount of drug recovered unchanged in the dialysate and AUC0−t is the area under the predialyzer plasma concentration-time curve during hemodialysis.25 This method yields lower clearance values than the previous method if there is a significant degree of binding of the drug to the dialyzer. If adsorption contributes minimally to clearance, the two methods are likely to correlate well.
Because there is marked variability among dialyzers in the clearance of some drugs, it is recommended that dialyzer clearance data for a cellulose dialyzer-drug pair not be extrapolated directly to a synthetic dialyzer. If there are no data regarding high-flux dialysis for a given drug, one should anticipate that the dialyzer clearance by the synthetic dialyzer will be 60% to 100% greater than that of the cellulose dialyzer. If there are no published data on dialyzer clearance, or reference sources do not identify the dialyzer that was used, prospective plasma concentration monitoring is recommended to guide therapy. Table 174-4 lists initial dosage recommendations for several drugs commonly administered to patients in the ICU.
 Summary
SummaryKey Points
Heintz BH, Matzke GR, Dager WE. Antimicrobial dosing concepts and recommendations for critically ill adult patients receiving continuous renal replacement therapy or intermittent hemodialysis. Pharmacotherapy. 2009;29:562-577.
Aronoff GR, Bennett WM, Berns JS, et al. Drug prescribing in renal failure: dosing guidelines for adults and children, 5th ed. Philadelphia: American College of Physicians; 2007.
Levey AS, Greene T, Kusek JW, Beck GJ. A simplified equation to predict glomerular filtration rate from serum creatinine. J Am Soc Nephrol. 2000;11:A0828.
Matzke GR. Status of hemodialysis of drugs in 2002. J Pharm Practice. 2002;15:405-418.
Churchwell MD, Mueller BA. Drug dosing during continuous renal replacement therapy. Semin Dial. 2009;22:185-188.
Nolin TD. Altered nonrenal drug clearance in ESRD. Curr Opin Nephrol Hypertens. 2008;17:555-559.
1 Tillyard A, Keays R, Soni N. The diagnosis of acute renal failure in intensive care: mongrel or pedigree? Anaesthesia. 2005;60:903-914.
2 Uchino S, Kellum JA, Bellomo R, et alfor the Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators. Acute renal failure in critically ill patients: a multinational, mutlicenter study. JAMA. 2005;294:813-818.
3 Brochard L, Abroug F, Brenner M, et al. An official ATS/ERS/ESICM/SCCM/SRLF statement: prevention and management of acute renal failure in the ICU patient. Am J Respir Crit Care Med. 2010;181:1128-1155.
4 Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: A severity of disease classification system. Crit Care Med. 1985;13:818-829.
5 Knaus WA, Wagner DP, Draper EA, et al. The APACHE III prognostic system: Risk prediction of hospital mortality for critically ill hospitalized adults. Chest. 1991;100:1619-1636.
6 Robert S, Zarowitz BJ. Is there a reliable index of glomerular filtration rate in critically ill patients? DICP. 1991;25:169-178.
7 Dowling TC. Quantification of renal function. In: Dipiro JT, Talbert RL, Yee GC, et al, editors. Pharmacotherapy: A Pathophysiologic Approach. New York: McGraw-Hill; 2008:705-722.
8 Cockroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31-41.
9 Levey AS, Greene T, Kusek JW, Beck GJ. A simplified equation to predict glomerular filtration rate from serum creatinine. J Am Soc Nephrol. 2000;11:A0828.
10 Orlando R, Floreani M, Padrini R, Palatini P. Evaluation of measured and calculated creatinine clearances as glomerular filtration markers in different stages of liver cirrhosis. Clin Nephrol. 1999;51:341-347.
11 Caregaro L, Menon F, Angeli P. Limitations of serum creatinine level and creatinine clearance as filtration markers in cirrhosis. Arch Intern Med. 1994;154:201-205.
12 Gines P, Schrier RW. Renal failure in cirrhosis. N Engl J Med. 2009;361:1279-1290.
13 Pesola GR, Akhavan I, Madu A, et al. Prediction equation estimates of creatinine clearance in the intensive care unit. Intensive Care Med. 1993;19:39-43.
14 Martin C, Alaya M, Bras J, et al. Assessment of creatinine clearance in intensive care patients. Crit Care Med. 1990;18:1224-1226.
15 Levey AS, Coresh J, Greene T, Stevens LA, Zhang Y, Hendriksen S, et al. Using standardized serum creatinine values in the Modification of Diet in Renal Disease Study equation for estimating glomerular filtration rate. Ann Intern Med. 2006;145:247-254.
16 Demirovic JA, Pai AB, Pai MP. Estimation of creatinine clearance in morbidly obese patients. Am J Health-Syst Pharm. 2009;66:642-648.
17 Duffull SB, Dooley MJ, Green B, Poole SG, Kirkpatrick CM. A standard weight descriptor for dose adjustment in the obese patient. Clin Pharmacokinet. 2004;43:1167-1178.
18 Bouchard J, Macedo E, Soroko S, Chertow GM, et al. Comparison of methods for estimating glomerular filtration rate in critically ill patients with acute kidney injury. Nephrol Dial Transplant. 2010;25:102-107.
19 Golik MV, Lawrence KR. Comparison of dosing recommendations for antimicrobial drugs based on two methods for assessing kidney function: Cockcroft-Gault and modification of diet in renal disease. Pharmacotherapy. 2008;28:1125-1132.
20 Hermsen ED, Maiefski M, Florescu MC, et al. Comparison of the modification of diet in renal disease and Cockcroft-Gault equations for dosing antimicrobials. Pharmacotherapy. 2009;29:649-655.
21 Wargo KA, Eiland EH, Hamm W, et al. Comparison of the modification of diet in renal disease and Cockcroft-Gault equations for antimicrobial dosing adjustments. Ann Pharmacother. 2006;40:1248-1253.
22 Spruill WJ, Wade WE, Cobb HH3rd. Continuing the use of the Cockcroft-Gault equation for drug dosing in patients with impaired renal function. Clin Pharmacol Ther. 2009;86:468-470.
23 Dowling TC, Matzke GR, Murphy GR, Burckart GJ. Evaluation of renal drug dosing: prescribing information and clinical pharmacist approaches. Pharmacotherapy. 2010;30:776-786.
24 Ritschel WA, Denson DD. Influence of disease on bioavailability. In: Ritschel WA, editor. Pharmacokinetics: Regulatory, Industrial, Academic Perspectives. New York: Marcel Dekker, 1995.
25 Matzke GR, Frye RF. Drug therapy individualization for patients with renal insufficiency. In: Dipiro JT, Talbert RL, Yee GC, et al, editors. Pharmacotherapy: A Pathophysiologic Approach. 7th ed. New York: McGraw Hill; 2008:833-844.
26 Gibson TP, Giacomini KM, Briggs WA, et al. Propoxyphene and norpropoxyphene plasma concentrations in the anephric patient. Clin Pharmacol Ther. 1980;27:665-670.
27 Barnes JN, Williams AJ, Tomson MJ, et al. Dihydrocodeine in renal failure: Further evidence for an important role of the kidney in the handling of opioid drugs. BMJ. 1985;290:740-742.
28 Verbeeck RK, Musuamba FT. Pharmacokinetics and dosage adjustment in patients with renal dysfunction. Eur J Clin Pharmacol. 2009;65:757-773.
29 Dreisbach AW. The influence of chronic renal failure on drug metabolism and transport. Clin Pharmacol Ther. 2009;86:553-556.
30 Nolin TD. Altered nonrenal drug clearance in ESRD. Curr Opin Nephrol Hypertens. 2008;17:555-559.
31 Vilay AM, Churchwell MD, Mueller BA. Drug metabolism and clearance in acute kidney injury. Crit Care. 2008;12:235.
32 Masereeuw R, Russel FGM. Therapeutic implications of renal anionic drug transporters. Pharmacol Ther. 2010;126:200-216.
33 Sun H, Frassetto L, Benet LZ. Effects of renal failure on drug transport and metabolism. Pharmacol Ther. 2006;109:1-11.
34 Saito H. Pathophysiological regulation of renal SLC22A organic ion transporters in acute kidney injury: pharmacological and toxicological implications. Pharmacol Ther. 2010;125:79-91.
35 McEvoy GK, Snow EK. American Hospital Formulary Service Drug Information. Bethesda, MD: American Society of Hospital Pharmacists; 2010.
36 Aronoff GR, Bennett WM, Berns JS, et al. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults and Children, 5th ed. Philadelphia: American College of Physicians; 2007.
37 Thummel KE, Shen DD, Isoherranen N, Smith HE. Appendix II. Design and optimization of dosage regimens: Pharmacokinetic data. Brunton LL, Lazo JS, Parker KL, editors. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 11th ed. New York: McGraw-Hill; 2005:1787-1888.
38 Vidal L, Shavit M, Fraser A, Paul M, Leibovici L. Systematic comparison of four sources of drug information regarding adjustment of dose for renal function. BMJ. 2005;331:263.
39 Adembri C, Novelli A. Pharmacokinetic and pharmacodynamic parameters of antimicrobials: potential for providing dosing regimens that are less vulnerable to resistance. Clin Pharmacokinet. 2009;48:517-528.
40 Czock D, Markert C, Hartman B, Keller F. Pharmacokinetics and pharmacodynamics of antimicrobial drugs. Expert Opin Drug Metab Toxicol. 2009;55:475-487.
41 Murphy JE. Clinical Pharmacokinetics Pocket Reference, 4th ed. Bethesda, MD: American Society of Hospital Pharmacists; 2008.
42 Joy MS, Matzke GR, Armstrong DK, et al. A primer on continuous renal replacement therapy for critically ill patients. Ann Pharmacother. 1998;32:362-375.
43 Churchwell MD, Mueller BA. Drug dosing during continuous renal replacement therapy. Semin Dial. 2009;22:185-188.
44 Heintz BH, Matzke GR, Dager WE. Antimicrobial dosing concepts and recommendations for critically ill adult patients receiving continuous renal replacement therapy or intermittent hemodialysis. Pharmacotherapy. 2009;29:562-577.
45 Konstantin P. Newer membranes: Cuprophane versus polysulfone versus polyacrylonitrile. In: Bosch JP, editor. Contemporary Issues in Nephrology. Hemodialysis: High Efficiency Treatments. New York: Churchill Livingstone; 1993:63-78.
46 Matzke GR. Status of hemodialysis of drugs in 2002. J Pharm Pract. 2002;15:405-418.
47 Simon N, Dussol B, Sampol E, et al. Population pharmacokinetics of ceftriaxone and pharmacodynamic considerations in haemodialysed patients. Clin Pharmacokinet. 2006;45:493-501.
48 Cirillo I, Vaccaro N, Tian H, et al. Pharmacokinetics of doripenem in subjects with varying degrees of renal impairment. Poster presentation: The 48th Interscience Conference on Antimicrobial Agents and Chemotherapy; Washington DC; 2008. A-1886.
49 Micromedex® Healthcare Series. Thomson Reuters (Healthcare) Inc. http://www.thomsonhc.com. (accessed June 28, 2010)
50 Mandell GL, Bennett JE, Dolin R. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Disease, 7th ed. Philadelphia: Churchill Livingstone Elsevier; 2010.
51 Drusano GL, Weir M, Forrest A, et al. Pharmacokinetics of intravenously administered ciprofloxacin in patients with various degrees of renal function. Antimicrob Agents Chemother. 1987;31:860-864.
52 Shah A, Lettieri J, Blum R, et al. Pharmacokinetics of intravenous ciprofloxacin in normal and renally impaired subjects. J Antimicrob Chemother. 1996;38:103-116.
53 Sprandel KA, Drusano GL, Hecht DW, et al. Population pharmacokinetic modeling and Monte Carlo simulation of varying doses of intravenous metronidazole. Diagn Microbiol Infect Dis. 2006;55:303-309.
54 Gilbert B, Robbins P, Livornese LLJr. Use of antibacterial agents in renal failure. Infect Dis Clin North Am. 2009;23:899-924.
55 Rybak M, Lomaestro B, Rotschafer JC, et al. Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American society of health-system pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am J Health Syst Pharm. 2009;66:82-98.
56 Warkentin TE, Greinacher A, Koster A, Lincoff AM. Treatment and prevention of heparin-induced thrombocytopenia. Chest. 2008;133:340S-380S.
