CHAPTER 108 WILSON DISEASE
Wilson Disease is an autosomal recessive disorder of copper metabolism caused by mutations in the ATP7B gene. The gene product, a P-type adenosine triphosphatase (ATPase), is necessary for both the incorporation of copper into ceruloplasmin and its excretion into bile. The disease is associated with increased hepatic copper storage which leads to a clinically heterogeneous illness marked by cirrhosis of the liver and degenerative changes in the basal ganglia.
During the second half of the 19th century, a condition termed pseudosclerosis was distinguished from multiple sclerosis by the lack of ocular signs. In 1902, Kayser observed green corneal pigmentation in one such patient.1 Fleischer, who had also noted the green pigmentation of the cornea in 1903, commented on the association of the corneal rings with pseudosclerosis.2 In 1912, Wilson provided the classic description of the disease and its pathological anatomy but failed to mention the abnormal corneal pigmentation.3
CLINICAL FEATURES
Wilson disease is a progressive condition with a tendency toward temporary clinical improvement and arrest of symptoms.4 The presenting symptoms can be neurological, hepatic, psychiatric, or, less frequently, hematological. Symptoms at onset are listed in Table 108-1.
TABLE 108-1 Clinical Manifestations at Onset of Wilson’s Disease
| Symptoms | Percentage* |
|---|---|
| Hepatic or Hematologic Abnormalities | 35 |
| Behavioral Abnormalities | 25 |
| Neurological Abnormalities | 40 |
| “Pseudosclerotic” form: one or more of the following: | 40 |
| Resting or intentional tremor | |
| Dysarthria or scanning speech | |
| Diminished dexterity or mild clumsiness | |
| Unsteady gait | |
| Tremor alone | 33 |
| Dysarthria alone | 5 |
| “Dystonic” form: One or more of the following: | 60 |
| Hypophonic speech or mutism | |
| Drooling | |
| Rigid mouth, arms, or legs | |
| Seizures | 1 |
| Chorea or small-amplitude twitches | <1 |
Table prepared with the assistance of Drs. I. H. Scheinberg and I. Sternlieb, Department of Medicine, Albert Einstein College of Medicine, Bronx, New York.
* Percentages are approximate.
In the experience of Gow and colleagues,5 of symptomatic patients aged 7 to 58 years, 22% presented with fulminant hepatic failure, 54% presented with liver abnormalities, 10% with neurological features, and 4% with hemolysis. In 10% of patients, hepatic and neurological symptoms were concurrent.
In a fair number of cases, primarily in young children, initial symptoms are hepatic, such as jaundice or portal hypertension, and the disease can assume a rapidly fatal course without any detectable neurological abnormalities.6,7 In many of these patients, an attack of what appears to be acute viral hepatitis heralds the onset of Wilson disease.8 The manifestation with hepatic symptoms is common among affected children in the United States. In the series of Werlin and associates,9 who surveyed patients in the Boston area, the primary mode of manifestation was hepatic in 61% of patients younger than 21 years. In about 10% of affected children in the United States, Wilson disease manifests as an acute or intermittent, Coombs’ test–negative, nonspherocytic anemia that is accompanied by leukopenia and thrombocytopenia.9
Rigidity and spasms of the muscles are often present. In some instances, typical parkinsonian rigidity involves all muscles. Torticollis, tortipelvis, and other dystonic postures are not uncommon. Many patients have a fixed open-mouth smile with drooping of the lower jaw and excess salivation. Spasticity of the laryngeal and pharyngeal muscles can lead to dysarthria and dysphagia. A nearly pure Parkinson-like syndrome with progressive choreoathetosis or hemiplegia has also been described.10
When Wilson disease manifests during childhood, the first signs are usually bulbar; these can include indistinct speech and difficulty in swallowing. A rapidly progressive dystonic syndrome is not unusual. Such patients can present with acute dystonia, rigidity, and fever, with an occasional elevation of serum creatine phosphokinase level.11
Behavioral and personality disorders were noted in Wilson’s original description of the disease.3 These are almost invariable and predominate in about one third of patients.12,13 They include impaired school or work performance, depression, mood swings, and frank psychosis. Of patients who show primarily neurological symptoms, about two thirds have had psychiatric problems before the diagnosis of Wilson disease is made. Minor intellectual impairment can also be observed, but seizures and mental deterioration are not prominent features of the disease.
The intracorneal ring-shaped pigmentation, first noted by Kayser and Fleischer,1,2 might be evident to the naked eye or might appear only on slit-lamp examination. The color of the Kayser-Fleischer ring varies from yellow to green to brown. It is the consequence of copper deposition close to the endothelial surface of Descemet’s membrane. The ring can be complete or incomplete. It occurs in 79% of patients who present with hepatic symptoms and in all patients who present with cerebral or a combination of cerebral and hepatic symptoms.5,7 “Sunflower” cataracts are less commonly encountered, as are azure lunulae of the fingernails.14 Hypercalciuria and nephrocalcinosis are not uncommon and can be presenting signs of Wilson disease.15
NEUROIMAGING
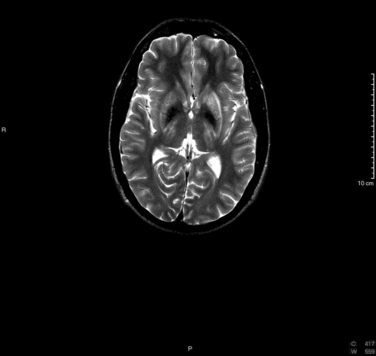
T2-weighted magnetic resonance imaging demonstrates symmetrical areas of increased signal intensity in the putamen, particularly in its outer rim; in the thalami; in the head of the caudate nucleus; and in the globus pallidus (Fig. 108-1). In the series of King and coworkers, the midbrain was abnormal in 77% of patients with Wilson disease.16 Involvement included primarily the tegmentum but also included the substantia nigra and the mesencephalic tectum. Abnormalities are also seen in the pons and the cerebellum. These areas are hypointense on T1-weighted images.17 Correlation with the clinical picture is not good in that magnetic resonance imaging can produce normal findings in patients with neurological symptoms and abnormal findings in patients with no neurological symptoms.16,18
Positron emission tomography demonstrates a widespread depression of glucose metabolism; the greatest focal hypometabolism is observed in the lenticular nucleus. This abnormality precedes any alteration visible on computed tomography19 and improves with chelation therapy.20 In addition, a reduction in dopamine D2 receptor binding and loss of striatal dopamine transporters have been documented.21,22
DIAGNOSIS
When Wilson disease manifests with neurological manifestations, the diagnostic features include progressive extrapyramidal symptoms, which commence after the first decade of life; abnormal liver function; aminoaciduria; cupriuria; and absence or decreased serum levels of ceruloplasmin. In the experience of Gow and colleagues5 and of Steindl and coworkers,23 the diagnosis is more difficult to establish in patients who present with hepatic symptoms, particularly those who present in fulminant hepatic failure.
The presence of a Kayser-Fleischer ring is the single most important diagnostic feature; its absence in a patient with neurological symptoms virtually rules out the diagnosis of Wilson disease. The ring is not seen in the majority of presymptomatic patients, nor is it seen in 33% of patients who present with hepatic symptoms.5
Absence or low serum level of ceruloplasmin is of lesser diagnostic importance; approximately 5% to 20% of patients with Wilson disease have normal levels of the copper protein. Ceruloplasmin levels are abnormally low in about 10% of persons heterozygous for the Wilson disease gene. Because gene carriers constitute about 1% of the population, there are 40 times more gene carriers with low ceruloplasmin values than patients with Wilson disease and low ceruloplasmin values. In affected families, the differential diagnosis between heterozygous and presymptomatic homozygous patients is of utmost importance, inasmuch as it is generally accepted that presymptomatic homozygous patients should be treated preventively.24
Several workers have stressed the diagnostic value of measuring 24-hour urine copper concentrations.25 In the experience of Gow and colleagues,5 however, 41% of patients who had a nonfulminant manifestation of Wilson disease, including all those with a neurological manifestation, had normal urinary copper excretion (<100 μg/24 hours).
The “gold standard” study for the diagnosis of Wilson disease is liver biopsy.25 When this procedure is undertaken, it is advisable to perform histological studies with stains for copper and copper-associated proteins and chemical quantitation for copper. In all confirmed cases of Wilson disease, hepatic copper levels are greater than 3.9 μmol/g dry weight (237.6 μg/g), in comparison with a normal range of 0.2 to 0.6 μmol/g (20 to 50 μg/g dry tissue).
Therefore, when low ceruloplasmin levels are found on routine screening and are unaccompanied by any abnormality of hepatic function or copper excretion, the patient is much more likely to be heterozygous for the Wilson disease gene than is a presymptomatic patient with Wilson disease.4 However, this supposition should be confirmed by liver biopsy.
ETIOLOGY AND PATHOPHYSIOLOGY
Pathological Anatomy
The abnormalities in copper metabolism result in a deposition of the metal in several tissues. Anatomically, the liver exhibits focal necrosis that leads to a coarsely nodular, postnecrotic cirrhosis. The nodules vary in size and are separated by bands of fibrous tissues of different widths. Some hepatic cells are enlarged and contain fat droplets, intranuclear glycogen, and clumped pigment granules; other cells are necrotic, with regenerative changes in the surrounding parenchyma.26
Electron microscopic studies indicate that copper is initially spread diffusely within the cytoplasm, probably as the monomeric metallothioneine complex. Later in the course of the disease, the metal is sequestered within lysosomes, which become increasingly sensitive to rupture.4 Copper probably initiates and catalyzes oxidation of the lysosomal membrane lipids, which results in lipofuscin accumulation. Within the kidneys, the tubular epithelial cells can degenerate, and their cytoplasm can contain copper deposits.
In the brain, particularly in patients whose symptoms commenced before the onset of puberty, the basal ganglia have a brick-red pigmentation; spongy degeneration of the putamen frequently leads to the formation of small cavities.3 Lesser degenerative changes are seen in the brainstem, the dentate nucleus, the substantia nigra, and the convolutional white matter.
Molecular Genetics and Biochemical Pathology
Cellular copper transport consists of three processes: copper uptake, intracellular distribution and use, and copper excretion.27
After its intestinal uptake, copper enters the plasma, where it is bound to albumin in the form of cupric ion. Within 2 hours, the absorbed copper is incorporated into a liver protein. Cellular copper uptake is facilitated by Ctr1, a membrane protein that transports the metal in a high-affinity, metal-specific manner. The concentration of copper in normal liver ranges from 20 to 50 μg/g dry weight. Once within the hepatocyte, copper is bound to metallochaperones, a family of proteins that deliver it to various specific sites. The chaperone Atox1, through direct interaction with the P-type ATPase (ATP7B) of Wilson disease, delivers copper to the hepatic secretory pathway for excretion into bile. Within the hepatocyte cytoplasm, copper is complexed to what is probably a polymeric form of metallothionein. Last, copper can combine with apoceruloplasmin to form ceruloplasmin, which then reenters the circulation.28 More than 95% of serum copper is in this form.27
The gene for Wilson disease has been mapped to chromosome 13q14.3-q21.1. It has been cloned and encodes a copper-transporting P-type ATPase that is expressed in many tissues, including the brain.29 The ATPase is present in two forms: one form is probably localized to the late endosomes, in which it is involved in the delivery of copper to apoceruloplasmin.30 The other form, believed to represent a cleavage product, is found in mitochondria.31,32 More than 300 gene mutations have been described. Some mutations are population-specific; others are common in many nationalities. The majority are missense mutations, small insertions, or deletions.33 Most patients with Wilson disease have compound heterozygosity.34 A complete list of mutations is maintained at http://www.medicalgenetics.med.ualberta.ca/wilson/index.php. Although several workers have attempted to correlate genotype and phenotype, the same mutation can present vastly different clinical pictures.35
The genetic mutation induces extensive changes in copper homeostasis. Normally, the amount of copper in the body is kept constant through excretion from the liver into the bile. The two fundamental defects in Wilson disease are (1) reduced biliary transport and excretion of copper and (2) impaired formation of plasma ceruloplasmin.27 Biliary excretion of copper is between 20% and 40% of normal, and fecal output of copper is also reduced.36 Apoceruloplasmin is present in the livers of patients with Wilson disease, but because of a lack of copper available for incorporation, apoceruloplasmin is rapidly degraded.
Of most importance, copper accumulates within liver. At first, it is firmly bound to copper proteins, such as ceruloplasmin and superoxide dismutase, or is complexed with metallothionein in the cupric form. When the copper load overwhelms the binding capacity of metallothionein, cytotoxic cupric copper is released, which causes damage to hepatocyte mitochondria and peroxisomes.37,38 Ultimately, copper leaks from the liver into blood, where it is taken up by other tissues, including the brain, which in turn are damaged by copper.
Two other biochemical abnormalities are consistently found in patients with Wilson disease:
TREATMENT
Even though it is clear that all patients with Wilson disease, whether symptomatic or asymptomatic, require treatment, there is no current consensus as to the optimal means of treating Wilson disease. The aims of treatment are initially to remove the toxic amounts of copper and secondarily to prevent tissue reaccumulation of the metal.25,42
Triethylene tetramine dihydrochloride (trientine) (500 mg twice a day, given at least 1 hour before or 2 hours after meals) is another chelator that increases urinary excretion of copper. It is less effective than penicillamine or ammonium tetrathiomolybdate, but the incidence of toxicity and hypersensitivity reactions is lower than in the former.42a
Zinc acetate (50 mg of elemental zinc acetate three times a day) acts by inducing intestinal metallothionein, which has a high affinity for copper and prevents its entrance into blood. Zinc is far less toxic than penicillamine but is much slower acting. Diet does not play an important role in the management of Wilson disease, although Brewer recommended restriction of liver and shellfish during the first year of treatment.42
Liver transplantation can be helpful in the patient who presents in end-stage liver disease. The procedure appears to correct the metabolic defect and can reverse neurological symptoms.43 Schumacher and colleagues44 also recommended its use for patients with normal liver function but whose neurological symptoms have not responded to the various chelating agents.
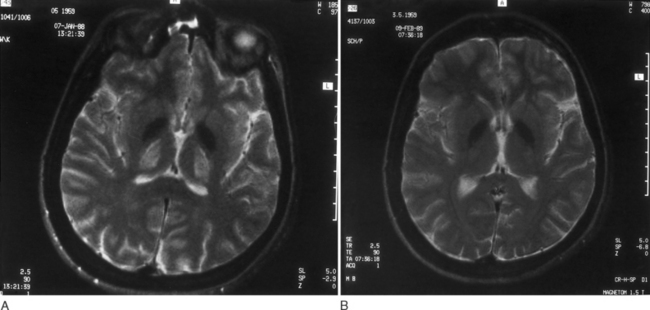
With these regimens, gradual improvement in neurological symptoms occurs. As a rule, brainstem auditory evoked potentials improve within 1 month of the onset of therapy; the somatosensory evoked responses return to normal somewhat more slowly.45 The Kayser-Fleischer ring begins to fade within 6 to 10 weeks after the onset of therapy and disappears completely in a couple of years.46 Neurological symptoms start improving 5 to 6 months after therapy begins and are generally gone in 24 months. As shown by serial neuroimaging studies, a significant regression of lesions occurs within thalamus and basal ganglia (Fig. 108-2A and B).18 Successive biopsies reveal a reduction in the amount of hepatic copper. Total serum copper and ceruloplasmin levels decrease, and the aminoaciduria and phosphaturia diminish.
As a rule, patients who begin therapy before the evolution of symptoms remain healthy. Patients who have had exclusively hepatic disease do well, and in 80%, hepatic functions return to normal. Approximately 40% of children who present with neurological symptoms become completely asymptomatic and remain so for 10 or more years. Patients with the mixed hepatocerebral picture do poorly. Fewer than 25% recover completely, and approximately 25% continue to deteriorate, often with the appearance of seizures. In all forms of the disease, the earlier therapy begins, the better the outlook is.7
When symptom-free patients with Wilson disease discontinue chelation therapy, their hepatic function deteriorates in 9 months to 3 years, a rate that is far more rapid than deterioration after birth.47
There is accumulating evidence that oxidative damage caused by free radical formation can play a significant role in producing cell damage in Wilson disease. Gu and coworkers37 have noted severe mitochondrial dysfunction in the livers of patients with Wilson disease, with significant reduction in the activities of all enzyme complexes involved in oxidative phosphorylation. Other findings pointing to oxidative stress are the reduction in the plasma levels of various antioxidants, such as ascorbate and urate, and the increase of allantoin, a possible marker of free radical generation.48 These findings provide experimental support for the addition of antioxidants such as ascorbate or vitamin E to the therapeutic regimen of all patients with Wilson disease.
Akil M, Brewer GJ. Psychiatric and behavioral abnormalities in Wilson disease. In: Weiner WJ, Lang AE, editors. Advances in Neurology, vol 65: Behavioral Neurology of Movement Disorders. New York: Raven Press; 1995:171-178.
Arima M, Takeshita K, Yoshino K, et al. Prognosis of Wilson disease in childhood. Eur J Pediatr. 1977;126:147-154.
Gitlin JD. Wilson disease. Gastroenterology. 2003;125:1868-1877.
Gow PJ, Smallwood RA, Angus PW, et al. Diagnosis of Wilson disease: an experience over three decades. Gut. 2000;46:415-419.
His G, Cox DW. A comparison of the mutation spectra of Menkes disease and Wilson disease. Hum Genet. 2004;114:165-172.
1 Kayser B. Ueber einen Fall von angeborener grünlicher Verfärbung der Cornea. Klin Monatsbl Augenheilkd. 1902;40:22-25.
2 Fleischer B. Über einer der “Pseudosklerose” nahestehende bisher unbekannte Krankheit (gekennzeichnet durch Tremor, psychische Störungen, bräunliche Pigmentierung bestimmter Gewebe, insbesondere auch der Hornhautperipherie, Lebercirrhose). Deutsch Z Nervenheilk. 1912;44:179-201.
3 Wilson SAK. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain. 1912;34:295-509.
4 Scheinberg IH, Sternlieb I. Wilson’s Disease, 2nd ed. Philadelphia: WB Saunders, 1999.
5 Gow PJ, Smallwood RA, Angus PW, et al. Diagnosis of Wilson disease: an experience over three decades. Gut. 2000;46:415-419.
6 Ferlan-Marolt V, Stepec S. Fulminant wilsonian hepatitis unmasked by disease progression: report of a case and review of the literature. Dig Dis Sci. 1999;44:1054-1058.
7 Arima M, Takeshita K, Yoshino K, et al. Prognosis of Wilson disease in childhood. Eur J Pediatr. 1977;126:147-154.
8 Scott J, Golan JL, Samourian S, et al. Wilson disease presenting as chronic active hepatitis. Gastroenterology. 1978;74:645-651.
9 Werlin SL, Grand RJ, Perman JA, et al. Diagnostic dilemmas of Wilson disease: diagnosis and treatment. Pediatrics. 1978;62:47-51.
10 Lingan S, Wilson J, Nazer H, et al. Neurological abnormalities in Wilson disease are reversible. Neuropediatrics. 1987;18:11-12.
11 Kontaxakis V, Stefanis C, Markidis M, et al. Neuroleptic malignant syndrome in a patient with Wilson disease [Letter]. J Neurol Neurosurg Psychiatr. 1988;51:1001-1002.
12 Akil M, Brewer GJ. Psychiatric and behavioral abnormalities in Wilson disease. In: Weiner WJ, Lang AE, editors. Advances in Neurology, vol 65: Behavioral Neurology of Movement Disorders. New York: Raven Press; 1995:171-178.
13 Portala K, Westermark K, Ekselius L, et al. Personality traits in treated Wilson disease determined by means of the Karolinska Scales of Personality (KSP). Eur Psychiatry. 2001;16:362-371.
14 Cairns JE, Williams HP, Walshe JM. “Sunflower cataract” in Wilson disease. BMJ. 1969;3:95-96.
15 Azizi E, Eshel G, Aladjem M. Hypercalciuria and nephrolithiasis as a presenting sign in Wilson disease. Eur J Pediatr. 1989;148:548-549.
16 King AD, Walshe JM, Kendall BE, et al. Cranial MR imaging in Wilson disease. AJR Am J Roentgenol. 1996;167:1579-1584.
17 Van Wassenaer–van Hall HN, van den Heuvel AG, Algra A, et al. Wilson disease: findings at MR imaging and CT of the brain: clinical correlation. Radiology. 1996;198:531-536.
18 Prayer L, Wimberger D, Kramer J, et al. Cranial MRI in Wilson disease. Neuroradiology. 1990;32:211-214.
19 Hawkins RA, Mazziotta JC, Phelps ME. Wilson disease studied with FDG and positron emission tomography. Neurology. 1987;37:1707-1711.
20 Schlaug G, Hefter H, Engelbrecht V, et al. Neurological impairment and recovery in Wilson disease: evidence from PET and MRI. J Neurol Sci. 1996;136:129-139.
21 Oder W, Brucke T, Kollegger H, et al. Dopamine D2 receptor binding is reduced in Wilson disease: a correlation of neurological deficits with striatal 123I-iodobenzamide binding. J Neural Transm. 1996;103:1093-1103.
22 Jeon B, Kim JM, Jeong JM, et al. Dopamine transporter imaging with [123I]-β-CIT demonstrates presynaptic nigrostriatal dopaminergic damage in Wilson disease. J Neurol Neurosurg Psychiatry. 1998;65:60-64.
23 Steindl P, Ferenci P, Dienes HP, et al. Wilson disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology. 1997;113:212-218.
24 Walshe JM. Diagnosis and treatment of presymptomatic Wilson disease. Lancet. 1988;2:435-437.
25 Brewer GJ, Yusbasiyan-Gurkan V. Wilson disease. Medicine. 1992;71:139-164.
26 Strohmeyer FW, Ishak KG. Histology of the liver in Wilson disease: a study of 34 cases. Am J Clin Pathol. 1980;73:12-24.
27 Gitlin JD. Wilson disease. Gastroenterology. 2003;125:1868-1877.
28 Tapia L, Gonzalez-Aguerre M, Cisternas MF, et al. Metallothionein is crucial for safe intracellular copper storage and cell survival at normal and supra-physiological exposure levels. Biochem J. 2004;378:617-624.
29 Tanzi RE, Petrukhin K, Chernov I, et al. The Wilson disease gene is a copper-transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344-350.
30 Harada M, Kawaguchi T, Kumemura H, et al. The Wilson disease protein ATP7B resides in the late endosomes with Rab7 and the Niemann-Pick C1 protein. Am J Pathol. 2005;166:499-510.
31 Lutsenko S, Cooper MJ. Localization of the Wilson disease protein product to mitochondria. Proc Natl Acad Sci U S A. 1998;95:6004-6009.
32 Schaefer M, Hopkins RG, Failla ML, et al. Hepatocyte-specific localization and copper-dependent trafficking of the Wilson disease protein in the liver. Am J Physiol. 1999;276:G639-G646.
33 His G, Cox DW. A comparison of the mutation spectra of Menkes disease and Wilson disease. Hum Genet. 2004;114:165-172.
34 Thomas GR, Forbes JR, Roberts EA, et al. The Wilson disease gene: spectrum of mutations and their consequence. Nat Genet. 1995;9:210-217.
35 Palsson R, Jonasson JG, Kristjansson M, et al. Genotypephenotype interactions in Wilson disease: insights from an Icelandic mutation. Eur J Gastroenterol Hepatol. 2001;13:433-436.
36 Frommer DJ. Defective biliary excretion of copper in Wilson disease. Gut. 1974;15:125-129.
37 Gu M, Cooper JM, Butler P, et al. Oxidative-phosphorylation defects in liver of patients with Wilson disease. Lancet. 2000;356:469-474.
38 Sheline CT, Choi DW. Cu2+ toxicity inhibition of mitochondrial dehydrogenases in vitro and in vivo. Ann Neurol. 2004;55:645-653.
39 Vulpe CD, Packman S. Cellular copper transport. Annu Rev Nutr. 1995;15:293-322.
40 Shiono Y, Hayashi H, Wakusawa S, et al. Ultrastructural identification of iron and copper accumulation in the liver of a male patient with Wilson disease. Med Electron Microsc. 2001;34:54-60.
41 Bruehlmeier M, Leenders KL, Vontobel P, et al. Increased cerebral iron uptake in Wilson disease: a 52Fe-citrate PET study. J Nucl Med. 2000;41:781-787.
42 Brewer GJ. Practical recommendations and new therapies for Wilson disease. Drugs. 1995;50:240-249.
42a Brewer GJ, Askari F, Lorincz MT, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol. 2006;63:521-527.
43 Emre S, Atillasoy EO, Ozdemir S, et al. Orthotopic liver transplantation for Wilson disease: a single-center experience. Transplantation. 2001;72:1232-1236.
44 Schumacher G, Platz KP, Mueller AR, et al. Liver transplantation: treatment of choice for hepatic and neurological manifestations of Wilson disease. Clin Transplant. 1997;11:217-224.
45 Grimm G, Oder W, Prayer L, et al. Evoked potentials in assessment and follow-up of patients with Wilson disease. Lancet. 1990;336:963-964.
46 Mitchell AM, Heller GL. Changes in Kayser-Fleischer ring during treatment of hepatolenticular degeneration. Arch Ophthalmol. 1968;80:622-631.
47 Walshe JM, Dixon AK. Dangers of non-compliance in Wilson disease. Lancet. 1986;1:845-847.
48 Ogihara H, Ogihara T, Miki M, et al. Plasma copper and antioxidant status in Wilson disease. Pediatr Res. 1995;37:219-226.