57 Wilms Tumor
Wilms tumor is the most common intra-abdominal tumor of childhood and the second most common extracranial solid tumor among children. It is a malignant neoplasm of the kidney believed to arise from persistent embryonal remnants known as nephrogenic rests (Fig. 57-1).1,2 Management of Wilms tumor in the United States is based on the National Wilms Tumor Study (NWTS) group trials; similarly, in Europe the International Society of Pediatric Oncology trials have formed the basis of care for children with the disease. Wilms represents a paradigm of multimodality treatment and multi-institutional study. The joint efforts of pediatric oncologists, radiation oncologists, surgeons, and pathologists have resulted in a dramatic improvement in survival of children with Wilms tumor. Whereas the disease was once uniformly fatal, high cure rates have been achieved across all stages for patients with favorable histologic (FH) disease, the most prevalent subtype. Advanced stages of Wilms tumor with unfavorable histologic (UH) findings continue to pose major therapeutic challenges.
Epidemiology
Wilms tumor accounts for 6% of all childhood cancers in the United States, where the estimated incidence is 470 cases per year,3 or 8 per million children younger than the age of 16 years.2,4 In the United States, the disease is slightly more prevalent among girls than boys (1 : .92 for unilateral tumors, 1 : .6 for bilateral tumors). This disparity is not maintained worldwide; the global incidence is 1 in 10,000 children with no gender predilection noted.4 The incidence of Wilms is three times higher among Africans and African Americans than among East Asians, with the incidence among the European and American white population falling between these two extremes.5
Age at presentation depends on gender and laterality. Males tend to present earlier than females, and bilateral tumors earlier than unilateral tumors. The median age at diagnosis of bilateral disease is 30 months, whereas for unilateral disease it is 40 months.6 Seventy-five percent of all patients present before 5 years of age.7 Bilateral disease is noted in 4% to 8% of patients at presentation8–10; 7% to 12% of patients with unilateral Wilms demonstrate multifocal disease in the affected kidney.11
The majority of cases are sporadic, but Wilms tumor is thought to have a hereditary basis in 15% to 20% of affected patients.12 Approximately 1.5% of patients have familial Wilms, wherein one or more family members has the disease (usually siblings or cousins, rarely parents); in these cases, the mode of inheritance is generally thought to be autosomal dominant with variable penetrance.13
Etiology, Genetics, and Cytogenetic Abnormalities
Investigation of the role of parental environmental exposures as a causal factor of Wilms tumor has yielded inconsistent findings. Case-control studies have suggested that paternal exposure to lead and hydrocarbons increases the incidence of Wilms tumor in offspring, and that fathers employed as auto mechanics, welders, and machinists demonstrate an increased odds ratio for Wilms tumor among their offspring.14–17 However, other studies have failed to find any such correlation with paternal exposure or occupation.18–20 Previously reported maternal associations (smoking during pregnancy, hypertension of pregnancy, tea consumption) were not confirmed in a large case-control study.16,21
Genetic explanations of the causal factors of Wilms tumor originated with Knudson’s “two-hit hypothesis,” originally developed for retinoblastoma.22,23 Briefly, the two-hit hypothesis proposes that tumors develop as a consequence of two mutational events in a single cell. If the first mutational event is prezygotic (or germ line), the tumor is potentially hereditary and affected individuals are at risk of developing multiple tumors. Conversely, sporadic cases occur as a result of two postzygotic (or somatic) mutations in a single cell; because two postzygotic events are unlikely to occur in more than one cell, these patients are unlikely to present with multiple tumors.21 This hypothesis explains the earlier age of onset and bilateral presentation of familial Wilms compared with sporadic cases.13,24
Whereas the two-hit hypothesis has been confirmed in retinoblastoma to occur via inactivation of both alleles of a tumor-suppressor gene on 13q, the genetic events that lead to the development of Wilms tumor are less well-defined and appear to be more complex. Multiple genetic loci appear to play a role in the development of Wilms. The best known of these are located on the short arm of chromosome 11.25 Designated Wilms Tumor-1 (WT-1, at 11p13) and WT-2 (at 11p15) mutations in these putative tumor suppressor genes are found in 30% to 40% of Wilms tumors21 and are associated with three well-described congenital anomaly syndromes:
Although evidence for the role of these genetic loci in the development of Wilms is compelling, studies of affected pedigrees have failed to confirm either 11p13 or 11p15 as the location of the familial Wilms tumor gene.28 Based on epidemiologic and clinicopathologic analyses, NWTS group investigators have suggested that mechanisms other than germ line mutations (e.g., somatic mosaicism) are responsible for some bilateral and multicentric tumors.6,29
Other genetic mutations appear to contribute to adverse outcomes in children with Wilms tumor, although they are not clearly causative of the disease. Alterations in chromosomes 1p and 16q are associated with poor prognosis in children with Wilms tumor, implying that these genes are involved with tumor progression rather than tumor initiation21; hence they are referred to as Wilms tumor progressor genes, to distinguish them from WT-1 and WT-2. In a study of 232 children registered on NWTS-3 and NWTS-4, loss of heterozygosity (LOH) at either locus was associated with a poorer 2-year relapse-free and overall survival (OS), even with adjustment for stage and histologic subgroup.30
Heterozygous mutations in p53 are strongly associated with the anaplastic phenotype as compared with FH findings,31 suggesting that p53 abnormalities result in more aggressive histologic features. Overall, however, the incidence of p53 mutations in Wilms tumor is relatively low.32,33
Anatomy
Wilms tumors can arise in any area of the renal parenchyma. There is no predilection for either side. Extrarenal Wilms tumors are rare but can occur in the retroperitoneum, usually in the vicinity of the kidney. The tumor has occasionally been known to occur in the pelvis, inguinal region, or thorax, arising from displaced metanephric elements or teratomas.34
Pathologic Conditions
Childhood kidney tumors are organized according to the NWTS pathologic classification system (Table 57-1), which includes favorable as well as focal and diffuse anaplastic Wilms tumors, as well as clear cell sarcoma of the kidney (CCSK) and rhabdoid tumor of the kidney. Although these are histologically distinct from Wilms tumor, children with these diseases are included in NWTS trials because of the rarity of their occurrence, and treatment recommendations are based on NWTS findings.
Table 57-1 National Wilms’ Tumor Study Pathologic Classification System of Pediatric Renal Tumors
| Congenital mesoblastic nephroma |
Congenital mesoblastic nephroma, which is included in the NWTS classification system, falls at the benign end of the spectrum of pediatric renal tumors. It is the most common renal tumor of infancy,24 presenting at a median age of 2 to 3 months and occurring predominantly in males.35 It is characterized by bundles of spindle cells resembling fibroblasts or smooth muscle; cells at the periphery frequently extend deeply into the parenchyma or perirenal tissues. Despite its tendency for local extension, local recurrence or distant metastasis is highly uncommon, and the tumor may be curable by surgery alone (radical nephrectomy with wide margins). The cellular (or atypical) subclass of mesoblastic nephroma demonstrates increased cellularity and high mitotic rates. In older infants, this has been occasionally associated with local recurrence or the development of distant metastases.36 However, in infants younger than 3 months, cellular congenital mesoblastic nephroma is not associated with a poorer prognosis.37 Because there is no established role for radiation therapy in the management of mesoblastic nephroma, the subject is not discussed further in this chapter.
Wilms tumor is derived from primitive metanephric blastema and is classically triphasic in composition, consisting of varying proportions of blastemic, stromal, and epithelial elements.2 These elements appear to recapitulate normal renal embryogenesis. At the same time, Wilms tumors often contain tissues not found in the kidney, such as skeletal muscle, cartilage, and squamous epithelium, reflecting the pluripotent nature of primitive metanephric cells.21 Although triphasic composition is typical, Wilms tumor is characterized by tremendous structural and histologic diversity. Biphasic patterns are common, and tumors expressing predominantly or exclusively one element can still be categorized as Wilms tumor.38 Mixed composition is most common (41% of tumors), followed by blastemal predominant (39%) and epithelial predominant (18%); stromal predominant tumors are rare (1%). Although blastemal predominant tumors appear to be clinically more aggressive, these tumors are still classified as FH unless diffuse anaplasia is noted.39
Wilms tumors are classified as FH unless they meet clearly defined criteria for anaplasia. These criteria were established by Beckwith and Palmer40 following analysis of NWTS-1, and were refined by Faria and coworkers after analysis of NWTS-3 and 4 (Table 57-2). The original criteria did not account for location or multicentricity of anaplastic nests, and the distinction between focal and diffuse anaplasia lacked prognostic significance. In contrast, when Faria and colleagues’ strict criteria were applied in a review of NWTS-3 and four cases, patients with focal anaplasia were found to have a significantly superior 4-year survival as compared with patients with diffuse anaplasia; this difference was maintained stage for stage except for stage I tumors, in which survival was 100% for both focal and diffuse anaplasia.41 This prognostic significance highlights the importance of thorough sampling of the gross specimen during pathologic evaluation, as well as meticulous documentation of the exact specimen site from which each slide is obtained.
Table 57-2 Anaplastic Wilms’ Tumor:
| Definition of anaplasia | |
| Focal anaplasia: criteria | Anaplasia confined to a single region of the kidney |
| Diffuse anaplasia: criteria | Anaplasia present in more than one region of the kidney OR anaplasia in any extrarenal site (including vessels of the renal sinus, extracapsular infiltrates, or nodal or distant metastases) OR anaplasia in a random biopsy specimen |
Modified from the National Wilms’ Tumor Study-5: Therapeutic Trial and Biology Study. Unpublished. Activated June 16, 1995; last revised June 1998.
Eighty-nine percent of Wilms tumor cases in NWTS-3 were categorized as FH; 5% were anaplastic. The likelihood of anaplasia increased with age in NWTS-3; it was noted in only 2% of children younger than age 2 but increased to 13% in children ages 5 and older. Anaplastic tumors are significantly more frequent in black children than white children.42
Clear cell sarcomas and rhabdoid tumors of the kidney were originally believed to be monophasic variants of Wilms tumor; however, NWTS pathologists established both as unique tumors in 1978.40,43 They have since been analyzed separately but remain within the NWTS trials. Both entities are associated with strikingly poorer outcomes than Wilms tumor. CCSK is a primitive mesenchymal neoplasm distinguished by cells with poorly stained cytoplasm, prominent cytoplasmic vacuolation, and indistinct cell borders; groups of cells are often separated from one another by an arborizing network of thin-walled capillary vessels.24,39 CCSK accounts for 4% of all childhood renal tumors. Like Wilms, its most common site of distant spread is to the lungs; however, CCSK is distinguished by its propensity for bone and brain metastases as well. Twenty-three percent of children with CCSK in NWTS-3 developed bone metastases, as compared with 0.3% of all other children entered in the study.44,45
Rhabdoid tumor of the kidney, in contrast to CCSK, is marked by strongly acidophilic cytoplasmic staining. The tumor cells thus resemble myoblasts, but skeletal muscle markers are negative and the cell of origin is unknown.46,47 Like CCSK, rhabdoid tumors have a propensity for brain metastases; in addition, they can arise as primary CNS neoplasms,48 particularly primitive neuroectodermal tumors; this finding suggests a possible neural crest origin for rhabdoid tumors, a hypothesis that has been supported by histochemical analysis.47,49,50 Rhabdoid tumor occurs most commonly in male infants; median age at diagnosis is 13 months, and most cases are diagnosed in the first 2 years of life. In children older than 5 years diagnosed with rhabdoid tumor, pseudorhabdoid lesions (carcinomas, melanomas, histiocytic tumors, or myosarcomas) should be suspected.51 Rhabdoid tumors carry the poorest prognosis of all childhood renal tumors because of propensity for both local relapse and distant metastases.
Clinical Presentation
Eighty-three percent of children with Wilms tumor present with a painless abdominal mass.52 Other common presenting signs and symptoms include abdominal pain (37%), hypertension secondary to increased renin activity (25%), fever (23%), and hematuria (21%).53 Abdominal pain resulting from any cause (local distention, intralesional hemorrhage, peritoneal rupture) has been correlated with poorer 5-year survival.54 Acute subcapsular hemorrhage into the tumor results in a distinctive constellation of symptoms, including rapid abdominal enlargement, anemia, hypertension, and sometimes fever; plain radiographs of the abdomen may demonstrate a thin rim of eggshell-like calcification.55
Less commonly, children may present with varicocele, enlarged testicle, hernia, pleural effusion, congestive heart failure, or hydrocephalus.56 Several paraneoplastic syndromes have been reported in association with Wilms tumor, including Cushing syndrome, erythrocytosis, hypercalcemia, and acquired von Willebrand disease.57
Screening for children with known phenotypic or genotypic risk factors for the development of Wilms tumor is controversial. No randomized trials have been performed to address this question. Retrospective analyses of the NWTS registry and the National Childhood Cancer Registry of the United Kingdom failed to demonstrate a benefit to screening in children with aniridia, hemihypertrophy, or BWS.58,59 However, these studies were limited by the variation in incidence and time intervals of screening, as well as the methods used. In contrast, a case series analysis of children with BWS or hemihypertrophy, in which the case group had undergone systematic ultrasound screening at intervals of 4 months or less, suggested that screening sonograms every 4 months decreased the incidence of late-stage disease in these children (42% stage III to IV disease in unscreened children versus 0% in screened children, P < 0.003).60 A simulation model of sonogram screening three times yearly for children with BWS suggested that such a program would be comparably cost-effective to mammographic screening if applied from birth to age 4, largely because detection of earlier-stage disease would obviate the expense associated with radiation therapy.61 This hypothesis is supported by the finding that children with BWS in NWTS-3 and 4 were more likely to present with lower-stage tumors, although this may be a reflection of low biologic potential of Wilms tumor in these children in addition to the effect of increased surveillance screening.27
Routes of Spread
The most common location of local spread in Wilms is to the vessels of the renal sinus, which may be distended by tumor or demonstrate tumor thrombus. Penetration through the renal capsule is the second most common site of extrarenal spread. Not uncommonly, inflammation of the perirenal tissues can result in the formation of an inflammatory pseudocapsule, which may cause adhesions to adjacent organs such as the liver; yet on pathologic analysis, penetration of the true capsule by tumor is not demonstrated. Such cases are classified as stage I (limited to kidney, and completely resected, with negative margins).21 Even when tumor is adherent to or directly invades the liver, analysis of FH NWTS-3 cases demonstrated no detrimental effect on prognosis stage for stage.62 Conversely, patients with renal vein involvement were much more likely to have metastatic disease at diagnosis.63
Diagnostic and Staging Studies
Abdominal ultrasound can fulfill several functions. It can establish the renal origin of the mass; demonstrate whether the mass is solid or cystic; and identify the presence and extent of tumor thrombus in the renal vein and IVC.34 Evaluation of the contralateral kidney is also possible, although this function is largely supplanted by CT. Contrast-enhanced CT imaging of the abdomen is recommended for further evaluation of the mass and adjacent viscera. CT can provide additional information on renal function, retroperitoneal lymphadenopathy, and extension to or invasion of adjacent organs, particularly hepatic metastases. Of note, however, in most cases in which CT identifies hepatic invasion, subsequent surgical exploration reveals only compression by tumor.64
The use of preoperative CT or MRI to evaluate for tumor in the contralateral kidney has been advocated in some surgical literature. However, others believe that imaging cannot replace intraoperative exploration.24 The NWTS protocols require surgical exploration to rule out contralateral disease. Debate also exists concerning the use of abdominal CT versus abdominal MRI in diagnosis of children with suspected Wilms tumor because of concern regarding radiation exposure.65
In the past, there has been controversy over whether lung metastases detected by CT scan but not chest radiograph warrant whole lung radiotherapy. This question was addressed by Meisel et al.66 in an analysis of NWTS-3 and 4 data. The authors found no significant differences in event-free survival (EFS) or OS among chest x-ray–positive, CT-negative children who received intensive doxorubicin-containing chemotherapy and whole-lung irradiation (WLI) versus multidrug chemotherapy (with or without doxorubicin) and no WLI. Although WLI resulted in fewer pulmonary relapses, a greater number of deaths were attributed to pulmonary toxicity. The authors concluded that WLI was not clearly necessary in treatment of children with discordant chest x-ray, CT findings,66 noting, however, that the small study numbers prevented a definitive recommendation.
Staging
Under the NWTS (now Children’s Oncology Group [COG]) system, the staging of Wilms tumor has continuously evolved to reflect prognostic information gained through successive clinical trials. The latest series of COG (AREN) protocols provide staging updated in 2008 (Table 57-3). The NWTS-3 and NWTS-4 staging system was significantly modified from NWTS-1 and NWTS-2, in which the categories were referred to as groups rather than stages. The NWTS-5 staging system was slightly modified from that used in NWTS-3 and 4 and the only change since NWTS-5 has been to upstage patients with spill or rupture confined to the flank, including tumor biopsy, to stage III.
| Stage I |
Standard Therapeutic Approaches
The treatment approach advocated by the NWTS involves upfront surgery consisting of a modified radical nephrectomy and selective lymph node sampling. Exceptions to this rule include cases with intravascular tumor thrombus above the level of the hepatic vein, tumor contiguous with other abdominal structures, or bilateral renal involvement. In these situations, initial surgical exploration is still recommended for accurate staging and to obtain biopsies (in these situations biopsy will not result in further upstaging), but definitive surgical resection is postponed until after the administration of preoperative chemotherapy.67 Preoperative biopsy is not encouraged and results in automatic upstaging unless performed via fine-needle aspiration.
Preoperative sonography is strongly recommended to identify extensive intravascular thrombus, which can extend into the IVC to the right atrium, or in the case of left-sided tumors, down the gonadal vein. IVC extension occurs in only 6% of cases, and is usually asymptomatic. Attempts at primary excision of tumor and thrombus were associated with excessive surgical morbidity in NWTS-468; however, if appropriate presurgical chemotherapy is given, even extensive intravascular thrombus does not adversely affect prognosis.21,69,70
Diagnostic imaging studies may reveal contiguity of tumor to adjacent structures such as the liver, spleen, or pancreas. If the surgeon is sure that all disease can be completely removed, radical en bloc resection of the affected kidney and adjacent nonessential structures is acceptable. However, if complete en bloc resection cannot be performed, the tumor will be considered spilled.21,67,71
Preoperative chemotherapy is recommended by the COG in three distinct circumstances: tumors deemed unresectable at initial surgical exploration,72 bilateral tumors at presentation,67 and tumor extension into the IVC above the level of the hepatic veins.70 Preoperative chemotherapy almost always reduces tumor bulk, which can facilitate complete removal of the tumor and decrease surgical complications. However, it has not been shown to improve OS and is not generally advocated by the COG because it results in loss of important staging information. Even in the circumstances outlined earlier in which upfront chemotherapy is recommended, the COG strongly encourages surgical exploration for diagnosis and staging before chemotherapy.73,74
NATIONAL WILMS TUMOR STUDY–1
NWTS-1 (1969 to 1974) asked three major treatment questions: whether postoperative radiation therapy could be eliminated in children with stage I disease, whether actinomycin D or vincristine or the combination of both was superior in treatment of children with stage II and III disease, and whether preoperative vincristine was beneficial in the management of children with stage IV disease.75
The results of NWTS-1 led to the following treatment recommendations. First, postoperative radiation could be eliminated in children younger than age 2 with stage I disease; among older children, radiation appeared to decrease the rate of abdominal recurrences. Second, combination chemotherapy with actinomycin-D and vincristine was preferred for children with stage II or III disease, rather than monotherapy with either agent alone. Lastly, preoperative vincristine was of no benefit to children with metastatic disease.75
Retrospective analyses of the NWTS-1 data demonstrated that initiation of radiation later than postoperative day 10 was associated with adverse outcomes,76 a finding that was confirmed in NWTS-2 and NWTS-3.71 Retrospective review also suggested that local spillage at surgery required flank irradiation only, rather than whole-abdomen irradiation.77
NATIONAL WILMS TUMOR STUDY–2
Radiation therapy was eliminated for all stage I children. Flank irradiation was given to all stage II to IV children according to the same age-dependent scale used in NWTS-1; whole-abdomen irradiation was reserved for children with diffuse peritoneal seeding. Children with lung metastases were initially treated with WLI to 14 Gy, but the dose was scaled back to 12 Gy after a 10% incidence of pneumonitis was noted. No randomization was performed with regard to radiation. Comparison of stage I outcomes with those recorded in NWTS-1 suggested that elimination of radiation in children age 2 and older did not adversely affect outcomes.78 Data analysis showed no statistically significant difference in 3-year survival between children receiving 6 months of chemotherapy (97%) and children receiving 15 months of chemotherapy (92%). Addition of doxorubicin was shown to benefit children with stages II to IV disease. When analyzed by histologic examination, this benefit was significant for FH patients. Patients with UH showed a trend toward improved survival with the addition of doxorubicin, but this did not reach statistical significance.78
NATIONAL WILMS TUMOR STUDY–3
NWTS-3 (1979 to 1985) was the first of the trials in which histologic anatomy was used to stratify patients into treatment arms. Results of randomization of stage I FH patients confirmed that these children could be successfully treated with surgery followed by an abbreviated course of chemotherapy (10 weeks versus 6 months). The 4-year recurrence-free survival (RFS) and OS for children treated with short-course chemotherapy was 89% and 96%, respectively; this did not differ significantly from the outcomes of children treated with long-course chemotherapy.9
Regarding stage II FH patients, NWTS-3 asked two questions: (1) whether flank irradiation could be safely eliminated, and (2) whether addition of doxorubicin to the standard regimen (actinomycin and vincristine) was beneficial. Children treated on the least intensive arm (actinomycin plus vincristine, without radiotherapy) demonstrated outcomes (87% 4-year RFS and 91% 4-year OS) that were statistically equivalent to those seen in the other three arms. Both doxorubicin and flank irradiation were thus shown to be unnecessary for children with stage II FH disease.9
Children with stage III FH disease were similarly randomized in a two-step fashion, to irradiation to 10 or 20 Gy and to chemotherapy with or without doxorubicin. In these children, addition of doxorubicin resulted in superior outcomes. There was no significant difference in rates of abdominal relapse between patients receiving 10 versus 20 Gy. However, a trend toward inferior outcomes was noted among patients receiving 10 Gy in the absence of doxorubicin. This suggested the necessity of including either doxorubicin or full dose (20 Gy) irradiation in the treatment of stage III FH patients9; NWTS-4 subsequently prescribed doxorubicin plus 10 Gy for all stage III FH patients.
Patients with UH (any stage) or stage IV FH all underwent nephrectomy followed by tumor bed irradiation and radiotherapy to sites of metastatic disease. They were then randomized to receive actinomycin, vincristine, and doxorubicin with or without cyclophosphamide. Addition of cyclophosphamide did not result in a statistically significant improvement in RFS or OS.9 However, subset analysis later suggested that when UH patients were further categorized as having either focal or diffuse anaplasia, those with stages II to IV diffuse anaplasia or CCSK (but not rhabdoid tumor) did benefit from the addition of cyclophosphamide.79 For NWTS-4, the term favorable histology was thus amended to include focal anaplasia, and unfavorable histology was more strictly defined as diffuse anaplasia. Furthermore, stage I patients were grouped together in NWTS-4 regardless of histologic findings, as neither focal nor diffuse anaplasia had a negative prognostic effect in the earliest stage of the disease.
NATIONAL WILMS TUMOR STUDY–4
The primary intent of NWTS-4 (1986 to 1994) was to ascertain whether cost- and time-saving measures could be safely incorporated into the treatment regimens. To this end, 1687 patients across all stages were randomized to pulse-intensive (PI) versus standard-dose chemotherapy; for children with stages II to IV FH disease, a second randomization to 6 versus 15 months of chemotherapy was performed. As predicted, PI chemotherapy was both cost- and time-effective, resulting in fewer clinic visits and lower treatment costs. The 2-year RFS was equivalent between the PI and standard-dose arms (91.3% and 91.4%, respectively). Interestingly, the PI regimen also resulted in less hematologic toxicity.80 Analysis of short- versus long-course chemotherapy in stages II to IV FH patients similarly demonstrated no significant difference in 4-year RFS. The cost of treatment for patients treated with PI short-course chemotherapy was approximately one-half that of patients treated on the standard-dose, long-course regimen.81
The focus on socioeconomic effects in NWTS-4 was made possible by the excellent treatment outcomes achieved in previous trials for children with FH disease. However, outcomes for patients with stages II to IV diffuse anaplasia and children with CCSK remained relatively poor. Therefore, these children were also randomized to receive cyclophosphamide (in addition to actinomycin D, vincristine, and doxorubicin), in an attempt to improve outcomes and clarify the suggestion of benefit to cyclophosphamide, which was retrospectively noted in NWTS-3. Cyclophosphamide was shown to improve both RFS and OS for patients with stages II to IV diffuse anaplastic disease. The combined NWTS-3 and NWTS-4 results for this group of children revealed a 55% 4-year RFS and 52% 4-year OS for patients who received cyclophosphamide, approximately twice that of patients who did not receive the drug (27% 4-year RFS and OS) (P = 0.02).2 However, even the improved outcomes noted in the cyclophosphamide group compared poorly with those of patients with FH disease.
Patients with CCSK (all stages) were randomized to PI versus standard-dose chemotherapy, followed by a second randomization to short (24 to 26 weeks) versus long (54 to 65 weeks) courses of chemotherapy. There was no statistically significant difference in outcomes between randomization arms, leading to the conclusion that PI short-course chemotherapy was also acceptable for children with CCSK.80
NATIONAL WILMS TUMOR STUDY–5
The primary intent of NWTS-5 was to identify genetic prognostic factors (specifically, deoxyribonucleic acid [DNA] ploidy and LOH at 16q or 1p) that predict for adverse outcomes, with the goal of targeting intensified therapies to patients at risk for relapse and minimizing treatment for the remainder of patients. Urine, blood, and tumor tissue samples of enrolled patients were obtained for analysis, and blood samples were also obtained from both biologic parents of each child.67
The results of NWTS-5 have not yet been published, but outside of a current protocol, the NWTS-5 management guidelines by stage and histologic findings represent the current standard of care in the United States (Table 57-4). Treatment recommendations were modified from NWTS-4 for children with UH in an attempt to improve their historically poor outcomes. For children with stages II to IV diffuse anaplasia or CCSK, actinomycin was replaced by etoposide, and cyclophosphamide has been dose-intensified. Children with rhabdoid tumors were treated with a combination of cyclophosphamide, etoposide, and carboplatin, given that a neural crest origin has been postulated for rhabdoid tumor and that these agents have been previously employed for treatment of primitive neurogenic tumors.67,81
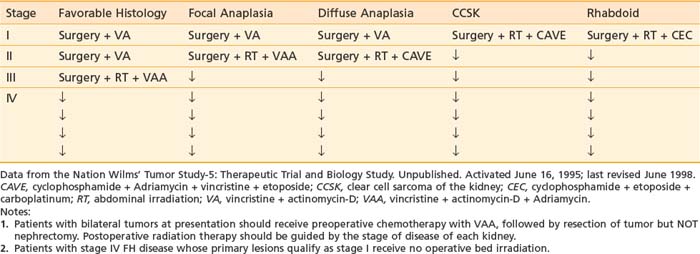
CURRENT CHILDREN’S ONCOLOGY GROUP PROTOCOLS
The most recent generation of Wilms tumor protocols were activated in 2008 and 2009. They comprise five protocols of, which one is a tumor biology and banking study (AREN03B2).82 The other four are experimental therapeutic trials designated as follows: high-risk renal tumors (AREN0321)83; very low- and standard-risk FH Wilms tumor (AREN 0532)84; treatment of newly diagnosed higher-risk FH Wilms tumors (AREN0533)85; treatment for patients with bilateral, multicentric, or bilaterally predisposed unilateral Wilms tumor (AREN0534).86
AREN0533 seeks to investigate two separate strategies for patients with higher-risk stage III and IV FH Wilms tumor. The first is to eliminate pulmonary radiotherapy for patients with lung metastases who have a favorable early response to chemotherapy. This is based on reports from European investigators who have had success with this approach, sparing 75% of patients with lung metastases from radiotherapy. In the current protocol, patients will receive 6 weeks of chemotherapy consisting of vincristine, dactinomycin, and doxorubicin followed by restaging. Only those patients who do not have complete resolution of pulmonary nodules by chest CT scan will undergo pulmonary irradiation. They will also receive intensified chemotherapy. It is imperative that this approach only be used for patients who have properly consented and enrolled in this investigational trial. A recent report from the English UKW trials have shown that omission of protocol-recommended pulmonary radiotherapy significantly decreased EFS for patients with metastatic Wilms tumor.87 Outside of a clinical trial, whole-lung radiotherapy to 12 Gy remains the standard of care for patients with pulmonary metastases.
Techniques of Radiotherapy
Indications and doses for flank–versus–whole abdomen irradiation are outlined in Table 57-5. Both regimens are delivered in an anterior-posterior fashion and are prescribed to midplane. For flank irradiation, the medial field border is extended across the midline to completely include the vertebral bodies at the levels concerned, but not far enough to overlap any portion of the contralateral kidney.67 The other borders are defined as the preoperative outline of the kidney and tumor, plus a 1-cm margin. Using this technique, the superior border should rarely approach the diaphragmatic dome.
| Extent of Disease | Radiotherapy Field | Dose |
|---|---|---|
| Renal hilar nodes | Flank, crossing midline to include entire vertebral bodies | 1.8 Gy × 6 = 10.8 Gy |
| Microscopic residuum confined to flank | ||
| Positive para-aortic nodes | Include entire length of bilateral para-aortic chains | 1.8 Gy × 6 = 10.8 Gy |
| Peritoneal seeding | Whole abdomen | 1.8 Gy × 6 = 10.8 Gy or 1.5 Gy × 7 = 10.5 Gy |
| Preoperative intraperitoneal rupture | ||
| Diffuse operative spill | ||
| Gross residual disease >3 cm | Disease + 1 cm margin | 10.8 Gy boost |
Adapted from Grundy PE, Green DM, Coppes MJ, et al: In Pizzo PA, Poplack DG, eds: Principles and practice of pediatric oncology, ed 4, Philadelphia, 2002, Lippincott Williams & Wilkins, p 865; and National Wilms’ tumor study-5: therapeutic trial and biology study. Unpublished. Activated June 16, 1995; last revised June 1998.
Abdominal irradiation should be initiated no later than 10 days postoperatively, provided the patient is stable, free of ileus or diarrhea, and has an absolute neutrophil count of at least 1000 and hemoglobin of at least 10.5. Rhabdoid tumor is the exception to this rule with abdominal irradiation delayed until week 6 of chemotherapy. The recommended schedule for flank irradiation is 1.8 Gy daily to 10.8 Gy; higher doses may be employed for CCSK and rhabdoid tumor. (Note that this represents a significant modification from all prior NWTS protocols, in which patients with UH received higher-dose radiotherapy [up to 40 Gy] on an age-dependent scale.) This recommendation is based in part on retrospective analysis of prior NWTS studies, which failed to show a dose-response relationship for anaplastic Wilms tumor.79 However, doses as low as 10.8 Gy have never been studied in this subgroup; the prior minimum dose was 18 Gy, for children 18 months or younger. An additional 10.8 Gy should be delivered to regions of gross (>3 cm) residual disease, with a 1-cm margin.
The treatment schedule may be modified to 1.5 Gy daily to 10.5 Gy in whole-abdomen irradiation if the treating clinician is concerned about large-volume fields. This modification can also be used in either flank or whole-abdomen therapy when simultaneous WLI is required, to avoid disparate fractionation schedules to the two regions. Fig. 57-2 displays an example of such a modification.
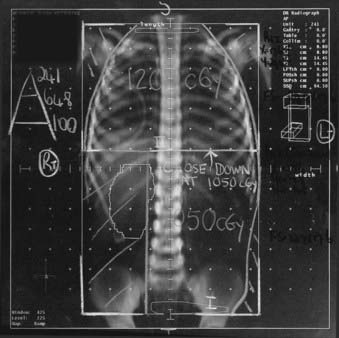
Whole-lung radiotherapy is delivered via anterior-posterior portals and is prescribed to midplane. The superior border extends above the clavicles to encompass the apices; the inferior border includes the posterior inferior portions of the lung. The humeral heads and extrathoracic scapulae and clavicles are shielded. No other shielding is employed. The recommended schedule is 1.5 Gy daily to 12 Gy. Localized foci of lung disease persisting 2 weeks after radiation may be either excised or treated with an additional 7.5 Gy (1.5 Gy/day × 5),67 although caution is advised given the potentially fatal risk of pulmonary toxicity in patients receiving combined modality therapy. Initial reports show promise of significant cardiac sparing when intensity-modulated radiation therapy is used to deliver whole-lung radiotherapy.88
Other metastatic sites requiring radiation include lymph nodes outside of the abdomen or pelvis, bone, and brain. Metastatic nodes receive 19.8 Gy; bone metastases are treated with at least a 3-cm margin to a dose of 30.6 Gy. Cerebral metastases are treated with whole-brain irradiation to 30.6 Gy.67
Radiotherapeutic management of metastatic Wilms tumor is outlined in Table 57-6. In all cases, care must be taken to limit the dose received by the normal kidney. NWTS-5 specifies that the dose to more than one-third of the contralateral kidney (or to the residual normal kidney in children with bilateral Wilms) should not exceed 14.4 Gy. Use of brachytherapy in the management of Wilms tumor has been anecdotally reported in fewer than 20 patients,89–92 all of whom had either recurrent disease or residual tumor after standard therapy.
Table 57-6 Radiotherapy for Metastatic Disease
| Disease Site | Radiotherapy Field | Dose |
|---|---|---|
| Lung | ||
| Age 18 mo or older | WLI ± boost to residual foci post-RT | |
CCSK, Clear cell sarcoma of the kidney; FH, favorable histology; WLI, whole lung irradiation.
From Grundy PE, Green DM, Coppes MJ, et al: In Pizzo PA, Poplack DG, eds: Principles and practice of pediatric oncology, ed 4, Philadelphia, 2002, Lippincott Williams & Wilkins, p 865; and National Wilms’ tumor study-5: therapeutic trial and biology study. Unpublished. Activated June 16, 1995; last revised June 1998.
Management of Inoperable Disease
As previously described, patients with unresectable disease should undergo surgical exploration and biopsies for diagnosis and staging, followed by chemotherapy with vincristine, actinomycin, and doxorubicin. Radiographic re-evaluation should be performed at week 5, with surgery performed shortly thereafter. If a persistent inadequate response is noted, preoperative irradiation with concurrent vincristine may be considered. The suggested dose is 12 to 12.6 Gy in 1.5- to 1.8-Gy daily fractions. Postoperative radiotherapy should be administered to all children who did not receive it preoperatively. After completion of radiotherapy, the chemotherapy regimen for stage III disease should be completed.34
Management of Synchronous Bilateral Disease
After surgical exploration, bilateral biopsies, and staging of each kidney, children with bilateral Wilms tumor should receive preoperative chemotherapy according to the highest stage of disease identified. Radiographic evaluation of response to therapy should begin at week 5, followed by a second-look procedure when the disease appears resectable; if serial imaging studies fail to show reduction in tumor, second-look surgery should still be performed to distinguish between persistent viable tumor and residual benign tissue. At that time, bilateral partial nephrectomies or wedge excisions should be performed only if negative margins can be obtained.93 If one kidney has extensive tumor precluding partial resection, complete excision of tumor from the least involved kidney is performed; if this procedure leaves the partially resected kidney viable and functioning, the extensively involved kidney can be removed by radical nephrectomy.67
Postoperatively, patients in whom complete resection was achieved should complete the chemotherapy regimen that corresponds to the highest stage of disease identified either initially or at second-look surgery. Radiotherapy is not indicated in the management of these patients.67
Patients with persistent gross disease after a second-look procedure should be considered for reoperation after further chemotherapy. If repeat abdominal explorations (the number and extent of which are at the discretion of the treating surgeon) fail to eradicate gross disease, patients should receive radiation to the involved kidney and tumor bed in the manner described earlier.67
CRITICAL NORMAL TISSUES—RADIATION INJURY
In the past when higher radiation doses were used, musculoskeletal late effects in patients with Wilms tumor could be directly attributed to radiation therapy.94,95 These included scoliosis, kyphosis, muscular hypoplasia, and iliac wing hypoplasia in patients who received flank or whole-abdomen irradiation. Severe musculoskeletal complications are unlikely when doses less than 24 Gy are used. Even in the setting of lower doses, however, radiation fields should be designed to minimize asymmetric development (e.g., treating the entire vertebral bodies in flank irradiation to avoid scoliosis).
Females who receive whole abdominal irradiation are at increased risk of late reproductive sequelae including ovarian failure96 and adverse pregnancy outcomes such as fetal loss, premature delivery, and possibly birth defects.96–98 In the largest series to date, Li and coworkers reported an excess risk of low birth weight (relative risk 4.0) and perinatal mortality (relative risk 7.9) in females who had received abdominal irradiation, as compared with white women in the United States.98
Radiation pneumonitis can be a life-threatening consequence of WLI, and is potentiated by concurrent chemotherapies. Twelve percent of patients on NWTS-3 who received pulmonary irradiation developed pneumonitis not caused by Pneumocystis carinii, with an associated 75% mortality.99 Reduction of the dose of concurrently administered doxorubicin and actinomycin-D has reduced the current incidence of radiation pneumonitis, although the risk of fatal pulmonary complications remains a major concern for children who receive supplemental radiation for residual lung disease after the standard 12 Gy.67
Chronic renal failure can occur as a consequence of radiation nephritis, but the reported incidence is less than 0.25% among children with unilateral disease and an apparently normal contralateral kidney at the time of diagnosis.100 The risk of renal failure is higher in patients with bilateral disease, although the incidence has decreased over the span of the NWTS trials from 9.9% in NWTS-1 to 3.8% in NWTS-4. Most cases of renal failure in children with bilateral Wilms are caused by either surgery or the sequelae of the Denys-Drash syndrome.
Second malignancies are a potential complication of both chemotherapy and radiation therapy. The cumulative incidence of second malignancies among children followed by the NWTS is 1.6% at 15 years, and the risk appears to continuously increase.101,102 Reviewers of the International Society of Pediatric Oncology experience reported a 0.65% cumulative incidence of second cancer at 15 years, with all second cancers occurring in the first 10 years after treatment. No excess risk was observed after 10 years, but this may have been a function of the length of follow-up.103 The single most important risk factor appears to be radiation with doxorubicin potentiating the radiation-associated risk. Notably, however, even patients who received only vincristine and actinomycin-D, without doxorubicin or radiation therapy, have an increased risk of second cancers as compared with the U.S. population when adjusted for age, sex, and race.102 Furthermore, inherited predisposition to multiple cancers may contribute to the excess risk of second malignancies among children treated for Wilms tumor.
Although late congestive heart failure is primarily due to doxorubicin toxicity, the risk of congestive heart failure is increased from 1% to 5.4% in patients receiving WLI103 and has been anecdotally reported in patients whose left ventricles received radiation during the course of left-sided abdominal therapy.104
Small-bowel obstruction occurred in 6.9% of children enrolled in NWTS-3, primarily as a result of bowel adhesions after surgery. This incidence is comparable to that of other children undergoing major abdominal operations. A review of the NWTS-3 experience found that the incidence of small bowel obstruction was not increased by postoperative abdominal radiation therapy.105
1 Paulino AC. Current issues in the diagnosis and management of Wilms’ tumor. Oncology (Huntington). 1996;10:1553.
2 Spierer M, Tereffe W, Wolden S. Neuroblastoma and Wilms’ tumor. In: Leibel SA, Phillips TL, editors. Textbook of radiation oncology. ed 2. Philadelphia: WB Saunders; 2002:1281.
3 Green DM. Wilms’ tumor. Eur J Cancer. 1997;33:409.
4 Breslow N, Olshan A, Beckwith JB, et al. Epidemiology of Wilms’ tumor. Med Pediatr Oncol. 1993;21:172.
5 Stiller C, Parker DM. International variations in the incidence of childhood renal tumors. Br J Cancer. 1990;62:1026.
6 Breslow N, Beckwith JB, Ciol M, et al. Age distribution of Wilms tumor: a report from the National Wilms Tumor Study. Cancer Res. 1988;48:1653.
7 D’Angio GJ. Informational bulletin #19. National Wilms’ Tumor Study. Seattle: NWTS Data and Statistical Center; 1991.
8 Blute ML, Kelalis PP, Offord KP, et al. Bilateral Wilms’ tumor. J Urol. 1991;146:514.
9 D’Angio GJ, Breslow N, Beckwith JB, et al. Treatment of Wilms’ tumor: results of the third national Wilms’ tumor study. Cancer. 1989;64:349.
10 Montgomery BT, Kelalis PP, Blute MD, et al. Extended follow-up of bilateral Wilms’ tumor: results of the National Wilms’ Tumor Study. J Urol. 1991;146:514.
11 Bond JV. Bilateral Wilms’ tumour: age at diagnosis, associated congenital abnormalities, and possible pattern of inheritance. Lancet. 1975;2:482.
12 Knudson AG. Genetics and the child cured of cancer. In: van Eys J, Sullivan MD, editors. Status of the curability of childhood cancers. New York: Raven Press; 1980:295.
13 Matsunaga E. Genetics of Wilms’ tumor. Hum Genet. 1981;57:231.
14 Kantor AF, Curnen MG, Meigs JW, et al. Occupations of fathers of patients with Wilms’ tumour. J Epidemiol Community Health. 1979;33:253.
15 Sanders BM, White GC, Draper GJ. Occupations of fathers of children dying from neoplasms. J Epidemiol Community Health. 1981;35:245.
16 Olshan AF, Breslow NE, Daling JR, et al. Wilms’ tumor and paternal occupation. Cancer Res. 1990;50:3212.
17 Bunin GR, Nass C, Kramer S, et al. Parental occupation and Wilms’ tumor: results of a case-control study. Cancer Res. 1989;49:725.
18 Fabia J, Thuy TD. Occupation of father at time of birth of children dying of malignant disease. Br J Prev Soc Med. 1974;28:98.
19 Hicks N, Zack M, Caldwell GG, et al. Childhood cancer and occupational radiation exposure in parents. Cancer. 1984;53:1637.
20 Zack M, Cannon S, Lloyd D, et al. Cancer in children of parents exposed to hydrocarbon-related industries and occupations. Am J Epidemiol. 1980;111:329.
21 Grundy PE, Green DM, Coppes MJ, et al. Renal tumors. In: Pizzo PA, Poplack DG, editors. Principles and practice of pediatric oncology. ed 4. Philadelphia, Lippincott: Williams & Wilkins; 2002:865.
22 Knudson AG. Mutation and cancer: Statistical study of retinoblastoma. Proc Nat Acad Sci USA. 1971;68:820.
23 Knudson AG, Strong LC. Mutation and cancer: A model for Wilms’ tumor of the kidney. J Natl Cancer Inst. 1972;48:313.
24 Halperin E, Constine LS, Tarbell NJ, et al. Pediatric radiation oncology, ed 3. Philadelphia, Lippincott: Williams & Wilkins; 1999.
25 Coppes MJ, Williams BR. The molecular genetics of Wilms’ tumor. Cancer Invest. 1994;12:57.
26 Coppes MJ, Haber DA, Grundy PE. Genetic events in the development of Wilms’ tumor. N Eng J Med. 1994;331:586.
27 Porteus MH, Narkool P, Neuberg D, et al. Characteristics and outcome of children with Beckwith-Wiedemann syndrome and Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol. 2000;18:2026.
28 Grundy P, Koufos A, Morgan K, et al. Familial predisposition to Wilms’ tumour does not map to the short arm of chromosome 11. Nature. 1988;336:374.
29 Breslow NE, Beckwith JB. Epidemiological features of Wilms’ tumor: results of the National Wilms’ Tumor Study. J Natl Cancer Inst. 1982;68:429.
30 Coppes MJ, Ritchey ML, D’Angio GJ. Preface. Hematol Oncol Clin North Am. 1995;9:xiii.
31 Bardeesy N, Falkoff D, Petruzzi MJ, et al. Anaplastic Wilms’ tumour, a subtype displaying poor prognosis, harbours p53 mutations. Nature Genet. 1994;7:91.
32 Malkin D, Sexsmith E, Yeger H, et al. Mutations of the p53 tumor suppressor gene occur infrequently in Wilms’ tumor. Cancer Res. 1994;54:2077.
33 Waber PG, Chen J, Nisen PD. Infrequency of ras, p53, WT1, or RB gene alterations in Wilms tumors. Cancer. 1993;72:3732.
34 Grundy PE, Green DM, Coppes MJ, et al. Principles and practice of pediatric oncology. In: Pizzo PA, Poplack DG, editors. Principles and practice of pediatric oncology. ed 4. Philadelphia: Lippincott Williams & Wilkins; 2002:865.
35 Pettinato G, Manivel JC, Wick MR, et al. Classical and cellular (atypical) congenital mesoblastic nephroma: a clinicopathologic, ultrastructural, immunohistochemical, and flow cytometric study. Hum Pathol. 1989;20:682.
36 Joshi VV, Kasnicka J, Walter TR. Atypical congenital mesoblastic nephroma: pathologic characterization of a potentially aggressive variant of conventional congenital mesoblastic nephroma. Arch Pathol Lab Med. 1986;110:100.
37 Beckwith JB, Weeks DA. Congenital mesoblastic nephroma: when should we worry? Arch Pathol Lab Med. 1986;110:98.
38 Murphy WM, Beckwith JB, Farrow GM. Tumours of the kidney, bladder, and related urinary structures. In Atlas of tumor pathology, 3rd series, fascicle 11. Washington DC: Armed Forces Institute of Pathology; 1994.
39 Schmidt D, Beckwith JB. Histopathology of childhood renal tumors. Hematol Oncol Clin North Am. 1995;9:1179.
40 Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms’ Tumor Study. Cancer. 1978;41:1937.
41 Faria P, Beckwith JB, Mishra K, et al. Focal versus diffuse anaplasia in Wilms tumor—new definitions with prognostic significance: a report from the National Wilms’ Tumor Study Group. Am J Surg Pathol. 1996;20:909.
42 Bonadio JF, Storer B, Norkool P, et al. Anaplastic Wilms’ tumor: clinical and pathologic studies. J Clin Oncol. 1985;3:513.
43 Marsden HB, Lawler W, Kumar PM. Bone metastasizing renal tumor of childhood: morphological and clinical features and differences from Wilms’ tumor. Cancer. 1978;42:1922.
44 Beckwith JB, Larson E. Clear cell sarcoma of the kidney. Pediatr Pathol. 1989;9:211.
45 Green DM, Breslow NE, Beckwith JB, et al. The treatment of children with clear cell sarcoma of the kidney: a report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1994;12:2132.
46 Haas JE, Palmer NF, Weinberg AG, et al. Ultrastructure of malignant rhabdoid tumor of the kidney: a distinctive renal tumor of children. Hum Pathol. 1981;12:646.
47 Weeks DA, Beckwith JB, Mierau GW, et al. Rhabdoid tumor of the kidney: a report of 111 cases from the National Wilms’ Tumor Study pathology center. Am J Surg Pathol. 1989;13:439.
48 Bonnin JM, Rubinstein LJ, Palmer NF, et al. The association of embryonal tumors originating in the kidney and in the brain: a report of seven cases. Cancer. 1984;54:2137.
49 Fischer HP, Thomsen H, Altmannsberger M, et al. Malignant rhabdoid tumor of the kidney expressing neurofilament proteins: immunohistochemical findings and histogenic aspects. Pathol Res Pract. 1989;184:541.
50 Gururangan S, Bowman LC, Parham DM, et al. Primary extracranial rhabdoid tumors: clinicopathologic features and response to ifosfamide. Cancer. 1993;71:2653.
51 Vogel AM, Gown AM, Caughlan J, et al. Rhabdoid tumors of the kidney contain mesenchymal specific and epithelial specific intermediate filament proteins. Lab Invest. 1984;50:232.
52 Green DM. Diagnosis and management of malignant solid tumors in infants and children. Boston: Martinus Nijhoff; 1985.
53 Ganguly A, Gribble J, Tune B, et al. Renin-secreting Wilms’ tumor with severe hypertension: Report of a case and brief review of renin-secreting tumors. Ann Intern Med. 1973;79:835.
54 LLeape, Breslow N, Bishop H. Surgical resection of Wilms’ tumor: results of the National Wilms’ Tumor Study. Ann Surg. 1978;181:351.
55 Ramsay NKC, Dehner LP, Coccia PF, et al. Acute hemorrhage into Wilms’ tumor. J Pediatr. 1977;91:763.
56 Green DM, Jaffe N. Wilms’ tumor—model of a curable pediatric malignant solid tumor. Cancer Treat Rev. 1978;5:143.
57 Coppes MJ. Serum biological markers and paraneoplastic syndromes in Wilms’ tumor. Med Pediatr Oncol. 1993;21:213.
58 Green DM, Breslow NE, Beckwith JB, et al. Screening of children with hemihypertrophy, aniridia, and Beckwith-Weidemann syndrome in patients with Wilms tumor: a report from the National Wilms Tumor Study. Med Pediatr Oncol. 1993;21:188.
59 Craft AW, Parker L, Stiller C, et al. Screening for Wilms’ tumour in patients with aniridia, Beckwith syndrome, or hemi hypertrophy. Med Pediatr Oncol. 1995;24:231.
60 Choyke PL, Siegel MJ, Craft AW, et al. Screening for Wilms tumor in children with Beckwith-Wiedemann syndrome or idiopathic hemihypertrophy. Med Pediatr Oncol. 1999;32:196.
61 McNeil DE, Brown M, Ching A, et al. Screening for Wilms tumor and hepatoblastoma in children with Beckwith-Wiedemann syndromes: a cost effective model. Med Pediatr Oncol. 2001;37:349.
62 Thomas PR, Shochat SJ, Norkool P, et al. Prognostic implications of hepatic adhesions, invasion, and metastases at diagnosis of Wilms’ tumor. The National Wilms’ Tumor Study Group. Cancer. 1991;68:2486.
63 Breslow NE, Churchill G, Nesmith B, et al. Clinicopathologic features and prognosis for Wilms’ tumor patients with metastases at diagnosis. Cancer. 1986;58:2501.
64 Ng YY, Hall-Graggs MA, Dicks-Mireaux C, et al. Pre- and post-chemotherapy CT appearances. Clin Radiol. 1991;43:255.
65 Radiation and pediatric computed tomography: a guide for health care providers. Published by the National Cancer Institute in concert with the Society for Pediatric Radiology. Summer 2002.
66 Meisel JA, Guthrie KA, Breslow NE, et al. Significance and management of computed tomography detected pulmonary nodules: a report from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys. 1999;44:579.
67 National Wilms’ tumor study-5: therapeutic trial and biology study. Unpublished. Activated June 16, 1995; last revised June 1998.
68 Ritchey ML, Kelalis PP, Breslow N, et al. Surgical complications after nephrectomy for Wilms’ tumor. Surg Gynecol Obstet. 1992;175:507.
69 Ritchey ML, Kelalis PP, Breslow N, et al. Intracaval and atrial involvement with nephroblastoma: review of National Wilms’ Tumor Study-3. J Urol. 1988;140:1113.
70 Ritchey ML, Kelalis PP, Haase GM, et al. Preoperative therapy for intracaval and atrial extension of Wilms’ tumor. Cancer. 1993;71:4104.
71 Thomas PR, Tefft M, Compaan PJ, et al. Results of two radiotherapy randomizations in the Third National Wilms’ Study (NWTS-3). Cancer. 1991;68:1703.
72 D’Angio GJ, Evans AE, Breslow N, et al. The treatment of Wilms’ tumor: Results of the National Wilms’ Tumor Study. Cancer. 1976;38:633.
73 Rutigliano DN, Kayton ML, Steinherz P, et al. The use of preoperative chemotherapy in Wilms’ tumor with contained retroperineal rupture. J Pediatr Surg. 2007;42:1595.
74 Su WT, Rutigliano DN, Gholizadeh M, et al. Hepatic metastasectomy in children. Cancer. 2007;109:2089.
75 D’Angio GJ, Tefft M, Breslow N, et al. Radiation therapy of Wilms’ tumor: results according to dose, field, post-operative timing and histology. Int J Radiat Oncol Biol Phys. 1978;4:769.
76 Tefft M, D’Angio GJ, Grant WIII. Postoperative radiation therapy for residual Wilms’ tumor: review of Group III patients in the National Wilms’ Tumor Study. Cancer. 1976;37:2768.
77 D’Angio GJ, Evans A, Breslow N, et al. The treatment of Wilms’ tumor: results of the second National Wilms’ Tumor Study. Cancer. 1981;47:2302.
78 Green DM, Beckwith JB, Breslow NE, et al. Treatment of children with stages II to IV anaplastic Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1994;12:2126.
79 Green DM, Breslow NE, Beckwith JB, et al. Comparison between single-dose and divided-dose administration of dactinomycin and doxorubicin for patients with Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1998;16:237.
80 Green DM, Breslow NE, Beckwith JB, et al. Effect of duration of treatment on treatment outcome and cost of treatment for Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1998;16:3744.
81 Villablanca JG, Matthay KK, Swift PS, et al. Phase I trial of carboplatin, etoposide, melphalan and local irradiation (CEM-LI) with purged autologous bone marrow transplantation for children with high-risk neuroblastoma. Med Pediatr Oncol. 1999;33:170.
82 Children’s Oncology Group (COG) current study #AREN03B2.
83 Children’s Oncology Group (COG) current study #AREN0321.
84 Children’s Oncology Group (COG) current study #AREN0532.
85 Children’s Oncology Group (COG) current study #AREN0533.
86 Children’s Oncology Group (COG) current study #AREN0534.
87 Nicolin G, Taylor R, Baughan C, et al. Outcome after pulmonary radiotherapy in Wilms’ tumor patients with pulmonary metastases at diagnosis: a UK Children’s Cancer Study Group, Wilms’ Tumor Working Group Study. Int J Radiat Oncol Biol Phys. 2008;70:175.
88 Kalapurakal JA, Gopalakrishnan M, Shore R, et al. Feasibility and potential utility of cardiac-sparing lung IMRT in children with Wilms tumor: a dosimetric study. Int J Radiat Oncol Biol Phys. 2009;75:s509. (abs)
89 Duckett CP, Zderic S, Goldwein J, et al. Brachytherapy for residual intra-renal Wilms’ tumor. Med Pediatr Oncol. 1997;28:316.
90 Goodman KA, Wolden SL, LaQuaglia MP, et al. Intraoperative high-dose rate brachytherapy for pediatric solid tumors: a 10-year experience. Brachytherapy. 2003;2:139.
91 McManus MC, Silliman C, Koyle MA. Combined endoscopic resection and brachytherapy for recurrent intrapelvic Wilms tumor. J Urol. 2002;167:2540.
92 Thoms WW, Goldwein JW, D’Angio G. A technique for the use of afterloading 137Cs brachytherapy in renal-sparing irradiation of bilateral Wilms tumor. Int J Radiat Oncol Biol Phys. 1997;39:1121.
93 Cooper CS, Jaffe WI, Huff D, et al. The role of renal salvage procedures for bilateral Wilms’ tumor: a 15-year review. J Urol. 2000;163:265.
94 Evans AE, Norkool P, Evans I, et al. Late effects of treatment for Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. Cancer. 1991;67:331.
95 Shalet SM, Beardwell CG, Jones PH, et al. Ovarian failure following abdominal irradiation in childhood. Br J Cancer. 1976;33:655.
96 Byrne J, Mulvihill JJ, Conelly RR, et al. Reproductive problems and birth defects in survivors of Wilms’ tumor and their relatives. Med Pediatr Oncol. 1988;16:233.
97 Green DM, Fine WE, Li FP. Offspring of patients treated for unilateral Wilms’ tumor in childhood. Cancer. 1982;49:2285.
98 Li FP, Gimbrere K, Gelber RD, et al. Outcome of pregnancy in survivors of Wilms’ tumor. JAMA. 1987;257:216.
99 Green DM, Finklestein JZ, Tefft ME, et al. Diffuse interstitial pneumonitis after pulmonary irradiation for metastatic Wilms’ tumor. Cancer. 1989;63:450.
100 Ritchey ML, Green DM, Thomas PR, et al. Renal failure in Wilms’ tumor patients: a report from the National Wilms’ Tumor Study Group [comment]. Med Pediatr Oncol. 1997;26:75.
101 Breslow NE, Takashima JR, Whitton JA, et al. Second malignant neoplasms following treatment for Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1995;13:1851.
102 Carli M, Frascella E, Tournade MF, et al. Second malignant neoplasms in patients treated on SIOP Wilms tumour studies and trials 1, 2, 5, and 6. Med Pediatr Oncol. 1997;29:329.
103 Green DM, Donckerwolcke R, Evans AE, et al. Late effects of treatment for Wilms’ tumor. Hematol Oncol Clin North Am. 1995;9:1317.
104 Pinkel D, Camitta B, Kun L, et al. Doxorubicin cardiomyopathy in children with left-sided Wilms’ tumor. Med Pediatr Oncol. 1982;10:483.
105 Ritchey ML, Kelalis PP, Etzioni R, et al. Small bowel obstruction after nephrectomy for Wilms’ tumor: a report of the National Wilms’ Tumor Study-3. Ann Surg. 1993;218:654.