[level-membership-for-hematology-oncology-and-palliative-medicine-category]
2 Dna Damage and Repair
Introduction
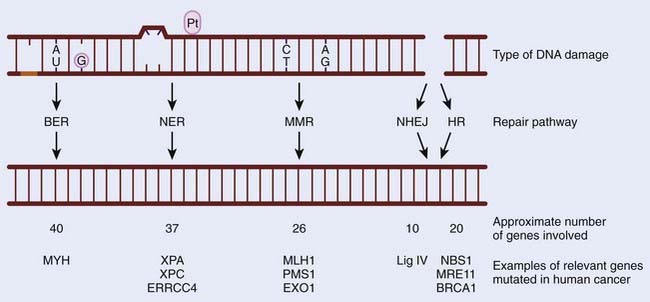
Preserving the integrity of the genome is of paramount importance to any organism. However, our DNA is under continual assault from both endogenous and exogenous damaging agents. These include the products of normal metabolism (reactive oxygen species [ROS]), ultraviolet (UV) and ionizing radiation, and genotoxic compounds such as those found in cigarettes. Inefficient or inadequate DNA repair can have dire consequences, which can lead to cell death, affect essential metabolic processes such as gene transcription, or result in the creation of a mutator phenotype.1 Decreased efficiency of DNA repair pathways have been linked with cancer predisposition in humans. This is clearly illustrated by studies that show that mutations in DNA repair pathways are associated with cancer-predisposing syndromes in humans. Furthermore, the loss of the ability to effectively repair damaged DNA leads to an increase in genomic instability and can contribute to tumorigenesis.2 DNA repair pathways are highly conserved throughout evolution, with many of the initial discoveries in the field having been made in Escherichia coli, but are still highly applicable to human cells. The purpose of this chapter is to give an overview of the five main repair pathways (BER, NER, MMR, NHEJ, and HR), and to highlight how mutations in these pathways affect tumor formation and response to therapy (Fig. 2-1).
DNA Damage Response Pathways
There are numerous mechanisms for the repair of damaged DNA, which are determined in part by the nature of the damage to which they respond and the presence of functioning repair pathways. For example, in the absence of functional BER, a single-strand break (SSB) can form a DNA double-strand break (DSB), which will then be processed by HR.3 In virtually all DNA repair pathways, DNA damage is detected by sensors that create a signal that is further transduced and amplified to initiate the repair pathway. The most critical DNA repair pathways are those that remove damaged bases that result as a consequence of cellular metabolism or genotoxic insult. BER, NER, and MMR pathways are involved in removing damaged bases and are discussed in the following text.
Base Excision Repair
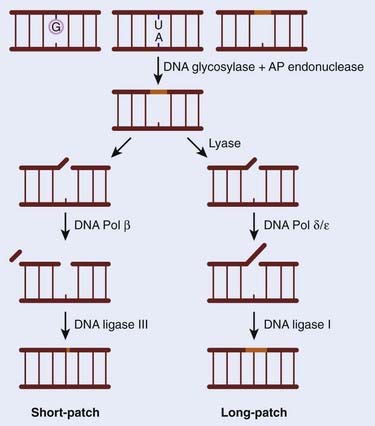
The BER pathway has a central role to play in preserving genomic integrity. Alterations in BER signaling are predicted to predispose humans to cancer and have been associated with colorectal adenomas and colon cancer. In contrast to other DNA repair pathways discussed in this chapter, only recently has an association between loss of BER and a human disease syndrome been identified. Patients affected by this syndrome, termed E. coli mutY human homolog (MYH)–associated polyposis (MAP), develop large numbers of colonic polyps (in the tens to hundreds range) by the age of 40, and nearly 50% present with colon cancer.4 The essential role of this pathway is supported by the finding that mice nullizygous for the genes involved were either lethal, indicating an absolute requirement for the gene product, or showed little to no phenotype because of a high degree of redundancy within the pathway. For example, loss of APE1, LIG1, LIG3, XRCC1, FEN1, and POLB all result in embryonic lethality, whereas deletion of NTH1, OGG1, UNG, AAAG, and MUTYH result in no discernible developmental defects.5 The absolute requirement for BER stems from its ability to repair DNA damage from both exogenous and endogenous sources. The majority of damage to the base portion of each nucleotide is the result of the products of normal metabolism that oxidize or alkylate DNA, such as ROS. Included in these lesions is the formation of 8-oxo-7,8-dihdroguanine (8-oxoG), which has mutagenic properties and also blocks DNA replication and transcription. BER is also used by the cell to handle the high rate of depurination (loss of purines from the DNA), and it has been estimated that a human being loses approximately a trillion guanines from his or her DNA every hour. In addition, BER is also used to remove a large number of cytosines that become spontaneously deaminated to form uracil. The BER mechanism has been extensively characterized in both E. coli and mammals, and includes five stages, shown schematically in Fig. 2-2. In brief, there is an initial recognition of the damaged base by DNA N-glycosylases. Different glycosylases recognize specific lesions, although there is considerable redundancy between them. The damaged base is then removed to give an apurinic/apyrimidinic (AP) site, which is processed by an AP-specific endonuclease (monofunctional glycosylase) or with an additional AP-lyase (bifunctional glycosylase), which leaves a single-strand interruption. These gaps are then filled by a DNA polymerase either with a single nucleotide (short patch) or polynucleotides (longer repair patch) before a ligation occurs. During BER, an SSB is formed as an intermediate. These SSBs are also generated by exogenous agents such as ionizing radiation and other oxidizing compounds. In response to ionizing radiation, BER effectively repairs the damage (SSB) although some additional enzymatic activities are thought to be required, such as PARP1. The control of BER has been recently described and indicates that BER proteins that are not actively involved in repair are ubiquitinated and degraded by the proteosome.6
Nucleotide Excision Repair
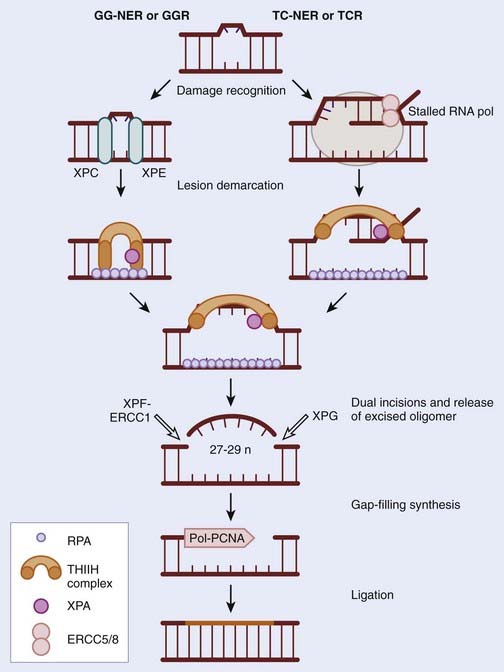
NER was discovered in the 1960s through elegant studies on the effects of UV irradiation on DNA synthesis and repair replication in bacteria. Since then it has been characterized extensively in mammals and has been described as the principle repair pathway for the removal of bulky adducts induced by UV radiation or other environmental carcinogens.7–9 In contrast to the BER pathway, damage detected by the NER proteins is not done in a specific manner because there is no equivalent to DNA N-glycosylases involved in BER, described previously. It is hard to imagine that a specific glycosylase could evolve for each type of DNA damage, especially in an environment in which exogenous damaging agents are numerous and constantly attack epithelial cells in the skin. Consequently, NER has been described as a more flexible version of BER. The major lesions recognized by NER are bulky adducts that result from intrastrand crosslinks, UV-induced cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts. When these lesions are not removed, they distort the DNA helix and in doing so prevent DNA replication and transcription, which can result in the employment of the error-prone translesion synthesis repair pathway. The process of NER can be subdivided into two pathways: global genome repair (GG-NER) or global genomic repair (GGR), and transcription coupled repair-nucleotide excision (TC-NER) or transcription-coupled repair (TCR).10 The process of GG-NER is genome wide (i.e., lesions can be removed from anywhere). In contrast, TC-NER only removes lesions in DNA strands of actively transcribed genes. When a DNA strand that is being actively transcribed becomes damaged, the ribonucleic acid (RNA) polymerase can block access to the site of damage and hence prevent DNA repair. TC-NER has evolved to prevent this by effectively removing the RNA polymerase from the site of damage to allow the repair proteins access. The mechanism of GG-NER and TC-NER differ only in the detection of the lesion; the subsequent pathway is the same. In general NER involves the recognition of the lesion, by proteins specific to GG-NER or TC-NER, followed by incision of the DNA strand near the damage, removal of the affected stretch of DNA, repair replication using the complementary strand as a template, and, finally, ligation to seal the 3′ end of the repair patch with the parental DNA (Fig. 2-3).11,12
Three human syndromes that occur as a result of mutation of the NER pathways have been identified: xeroderma pigmentosum (XP), Cockayne syndrome (CS) and trichothiodystrophy (TTD).13 Many of the principle genes involved in NER are called XP, for example XPA, XPB, and so on. This is because they were identified in individuals suffering from XP.14 XP results from mutation in any one of seven complementation groups: XPA, XPB (ERCC3), XPC, XPD (ERCC2), XPE (DDB2), XPF and XPG. Interestingly XP patients with mutations in XPC or XPE are deficient in GG-NER, whereas the remaining are deficient in both GG-NER and TC-NER. XP presents early in life as extreme sensitivity to the sun; freckles appear at a young age (1 year) even after short exposure to the sun, and significant pigmentation abnormalities are common. The eyes of XP patients are photosensitive and often exhibit eye damage as a result of constant UV-induced conjunctivitis and keratitis. This extreme sensitivity to UV translates to a 1 in 4000-fold increased risk of skin cancer in XP individuals, often appearing as early as age 10.15 These malignancies are usually squamous and basal cell carcinomas, but melanomas have also been reported. Neurologic abnormalities also frequently occur, including peripheral neuropathy, sensorineural deafness, and progressive mental retardation. Similar to XP patients, those with CS are also very sensitive to UV, although they do not exhibit the increased risk of skin cancer. Instead CS patients have distinct developmental abnormalities, which include growth retardation (dwarfism), progressive cognitive impairment, skeletal abnormalities, severe mental retardation, cataracts, and retinitis pigmentosa. CS patients tend to die younger than the age of 20 (the average life span is 12 years) as a result of infectious or renal complications rather than cancer. The genetic determinants of CS are mutations in either the CSA or CSB genes, both of which are essential for recognition of DNA damage and initiation of the TC pathway of NER. CS individuals remain proficient in GG-NER. Finally, mutation of the XPB or XPD genes results in TTD. Again extreme sensitivity to the sun is a characteristic in some of these patients (approximately 50%), although they possess a low incidence of developing skin cancer. TTD individuals are similar to CS in presentation but have the additional symptom of brittle nails and hair; this is a result of reduced sulfur content in the component proteins. The differences and similarities between these three syndromes have proved intriguing, in that they all show UV sensitivity but only XP individuals have an increased risk of skin cancer. This has been attributed to the determination that loss of GG-NER is what predisposes individuals to skin cancer. In contrast, the developmental abnormalities associated with these syndromes (retardation, dwarfism, etc.) are the result of a loss of TC-NER. Given the essential role of NER in the removal of UV-induced damage it is not surprising that XP individuals succumb to skin cancer, but an increased risk of other malignancies would also be expected and this has not been noted. There have been reports documenting modest alteration in NER genes as a result of polymorphisms that contribute to the risk of solid tumors.
Mismatch Repair
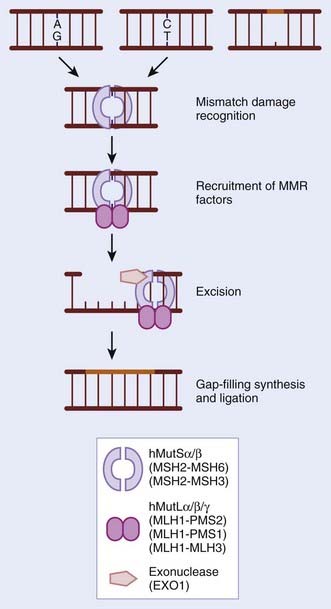
During normal replication and recombination, mistakes can be made that result in the insertion of an incorrect nucleotide leading to a mismatch. These mismatches also occur as a result of base modifications. If mismatches escape surveillance of the proofreading capabilities of DNA polymerases, there is a permanent change in sequence or mutation. The MMR pathway functions to identify such mistakes and correct them before they are propagated further. MMR also functions to remove small loops (insertion–deletion loops [IDL]) in the DNA that arise as a consequence of spontaneous slippage-dependent misalignment between primer and template DNA strands. These occur particularly in highly repetitive microsatellite regions that, in the absence of effective MMR, show a large increase in the frequency of spontaneously occurring mutations (microsatellite instability [MSI]).16 The process of MMR can be subdivided into four components: First, the mismatch must be identified by sensors that transduce a signal; second, MMR factors are recruited; third, the newly synthesized strand harboring the mismatch is identified and the incorrect or altered nucleotides are excised; and in the fourth stage, resynthesis and ligation of the excised tract occurs. MMR was first characterized in E. coli, which encodes MUT genes; homologues of these gene products have now been identified and extensively characterized in both yeast and humans.17 See Fig. 2-4 for a schematic representation and an indication of the critical gene products. The significance of MMR was highlighted when it became apparent in the 1990s that hereditary nonpolyposis colorectal cancer (HNPCC) and sporadic MSI-positive colon cancers were caused by defects in MMR. HNPCC, also known as Lynch syndrome, affects 1 in 1000 people and is characterized by early-onset colorectal cancers that have a high level of MSI. Individuals with HNPCC have an 80% risk of colorectal cancer in their lifetime, as well as an increased risk of other malignancies such as endometrial (50% risk), ovarian, and gastric malignancies. In 70% to 80% of the germline mutations found in HNPCC, the mutations occur in the MLH1 or MLH2 genes. In 10% of cases there is a mutation in MSH6, and, in rare cases, PMS1, PMS2, MLH3, and EXO1.18 Epigenetic silencing of the MMR genes has also been noted and shown to increase the mutation rate. In very rare cases, both copies of an MMR gene, such as MLH1, MSH2, MSH6, or PMS2 have been found mutated (homozygous germline mutations).19 These individuals have a reduced life span and succumb to malignancy of hematologic or brain origin.
The MMR proteins have been described as having a role to play in cell cycle checkpoints and apoptosis. Specifically, both hMutSα- and hMutLα-deficient cells exhibit defective S-phase checkpoint responses.20 The proposed model behind this observation is that MMR proteins act as sensors of DNA damage and then recruit the checkpoint proteins and hence activate these signaling pathways. This is supported by the findings that hMutSα and hMutLα physically interact with ataxia-telangiectasia mutated (ATM), ataxia-telangiectasia Rad3-related (ATR), ATR-interacting protein (ATRIP), c-Abl, and p73 in cells exposed to DNA-damage-inducing agents. Also supportive of this model is the wealth of data indicating that cells that become defective in MMR gain resistance to cytotoxic agents. Interestingly, patients with MSI-positive tumors have a greater chance of survival than those that do not, although whether this is as a result in differences in tumorigenesis or therapeutic response is unclear.
Double-Strand Break Repair
Nonhomologous End-Joining
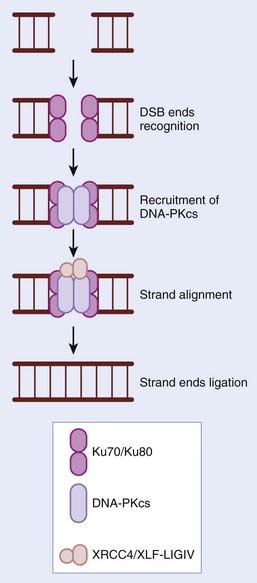
In contrast to HR, the NHEJ repair pathway does not make use of a template strand of DNA to maintain sequence fidelity during repair, but instead relies on short regions of microhomology (1-6 bp) (Fig. 2-5). NHEJ is predominantly active in the G1 phase of the cell cycle, but can function in all phases and is estimated to repair 90% of all DSBs in humans. NHEJ is also required for the development of both T- and B-cell repertoires. The essential role for NHEJ in the development of the immune system is highlighted by the finding that severe combined immunodeficient mice result from a loss of DNA-dependent protein kinase catalytic subunit (DNA-PKcs), one of the principle players in NHEJ repair. DNA-PK is composed of a cs and the Ku70/80 heterodimer. In the presence of a DSB, DNA ends are recognized by the Ku70/80 heterodimer, which in turn recruits DNA-PKcs as well as other proteins involved in strand alignment. The XRCC4-ligase IV complex then seals the breaks (see Fig. 2-5). Unless the DSB is blunt-ended, several nucleotides can be either gained or lost during the process, resulting in genetic errors. Numerous elegant mouse models have shown that by disrupting NHEJ in vivo, there are significant effects on radiosensitivity, genomic instability, immunodeficiency, development, and cancer predisposition.
Homologous Recombination
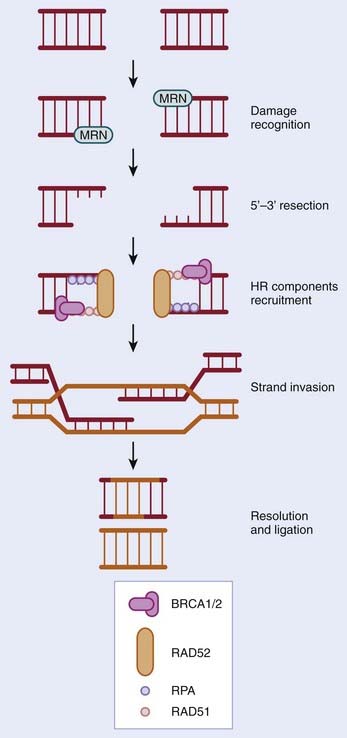
HR occurs predominantly in the S and G2 phases of the cell cycle and is estimated to repair 10% of DSBs acquired by human cells. The process of HR, which takes place in cells with a 2N DNA content, uses the presence of a second copy of each sequence to ensure that DNA repair occurs with no loss or alteration in genomic sequence. The process of HR is shown schematically in Fig. 2-6. In brief, DSBs are detected by the MRE11-NBS1-RAD50 (MRN) complex; the nuclease MRE11 processes the break to give a 3′ single-stranded DNA (ssDNA) tail. This region of ssDNA is then coated with replication protein A (RPA) to form a nucleoprotein filament. This filament then undergoes strand invasion (i.e., it invades the complementary sequence on a sister chromatid to form heteroduplex DNA). The genes involved in HR are numerous and include MRE11, NBS1, RAD50, BRCA1, BRCA2, and RAD51.
Syndromes Associated With a Failure in DNA Repair
Ataxia-Telangiectasia
The ATM gene has a critical role to play in DNA repair and is often referred to as sentinel kinase, meaning that it is one of the first kinases activated in response to DNA damage, and is responsible for the initial activation of repair signaling. ATM is a member of the PI3 kinase family and acts by phosphorylating serine or threonine residues that are followed by a glutamine residue (i.e., serine-glutamine or threonine-glutamine). Although the majority of ATM signaling has been described in response to agents that cause DSBs, more recent data is emerging to suggest that ATM may also respond to non-DNA damaging stresses.21,22 Currently a model has been proposed that suggests ATM exists within the cell as an inactive dimer which, upon activation in response to a DSB and with the aid of the mediator proteins and complexes, undergoes autophosphorylation to become active monomers. These monomeric, phosphorylated ATM molecules then in turn phosphorylate many downstream targets.23 However, mice bearing ATM mutated at the 1987 amino acid autophosphorylation site retain their ATM-mediated signaling pathways,24 suggesting a difference between human and mouse ATM activation in response to DNA damage. The downstream targets of ATM include many proteins involved in DNA repair, for example, NBS1, MRE11, CHK2, H2AX, FANC D2, and p53. Cells lacking ATM activity are extremely sensitive to DNA damage agents, fail to delay DNA replication, and continue to replicate in the presence of nonrepaired DNA (radio-resistant DNA synthesis). Normal tissue as well as cells derived from AT patients exhibit hypersensitivity to conventional doses of radiotherapy. AT patients have a 10% risk of malignancy, usually lymphoma or leukemia, which usually occurs at an early age. However, AT presents as a diverse group of symptoms prior to the formation of malignancies. These include progressive neurodegeneration, immune deficiency, ocular apraxia, and telangiectasia. Both Nijmegen breakage syndrome (NBS) and ataxia telangiectasia-like disorder show some overlap with AT patients, including defects in chromosome repair.25,26 This has been attributed to the finding that the genes mutated in these syndromes (NBS and MRE11, respectively) are both downstream targets of ATM and are also involved in the initial activation of ATM in response to DNA damage.
The p53 Tumor Suppressor Gene and Li-Fraumeni Syndrome
The p53 tumor suppressor gene, once dubbed the “guardian of the genome,” is the most mutated gene in human cancers. It has been estimated that it, or its downstream pathways, are mutated in more than 75% of cancers. The p53 gene encodes a protein that is at the heart of the cellular response to stress, including DNA damage, hypoxia, UV damage, and starvation.27 In response to stress, the levels of p53 protein, which are maintained at low levels in unstressed cells, increases rapidly because of increased protein translation and stabilization. The function of p53 is primarily as a transcription factor, and as such it binds to a consensus sequence in a sequence specific manner to target genes to regulate their expression (recently reviewed in Riley, 200828). The number of p53-activated and repressed genes is steadily increasing and, relevant to this chapter, include genes involved in cell cycle checkpoints, DNA repair, and apoptosis. Examples of genes that fall into these three categories are the cell-cycle inhibitory gene p21; the proapoptotic genes BAX and PERP; and the DNA repair genes DDB2, DDIT4, TRIM22, GADD45α, and XPC. More recently, there have been reports documenting a nontranscriptional role of p53, in particular in the induction of apoptosis at the mitochondria.29,30 Included in this nontraditional role for p53 is also the ability to modulate BER though direct interactions with DNA polymerase beta and OGG1, HR through modulation of the recQ helicases, and NER through a direct interaction with XPB.31 Consequently, in response to stress, p53 functions to arrest the cell cycle and signal to the repair pathways to restore genomic integrity unless the damage is too severe, in which case the cell is directed toward apoptosis. Without functional p53, cells are allowed to progress through the cell cycle before repair has taken place or escape apoptosis, and therefore accumulate genomic instability, which can contribute to tumorigenesis.
Li-Fraumeni syndrome is an autosomal-dominant inherited disorder, which is caused by mutation of the p53 gene in 70% to 85% of cases. Affected individuals inherit one mutated copy of the p53 gene and subsequently lose the remaining allele by mutation or loss of heterozygosity. Individuals with Li-Fraumeni syndrome usually present at an early age with a bone or soft tissue sarcoma, but may also develop breast cancer, a brain tumor, lymphoma, leukemia, and adrenocortical carcinoma.32–34
Familial Breast Cancer, BRCA1 and BRCA2
The breast cancer susceptibility genes (BRCA1 and BRCA2) are mutated in between 5% and 10% of all breast cancer cases and are also associated with familial ovarian cancers. Familial breast cancer is distinct from sporadic cases in that it has an earlier age of onset, an increased frequency of bilateral tumors, and can be associated with other tumor types within the affected family. However, familial and sporadic cases of breast cancer are indistinguishable at the histologic level and in their metastatic patterns. Interestingly, BRCA1 and BRCA2 mutations in sporadic cases of breast cancer are a rare occurrence. Both BRCA1 and BRCA2 have been associated with a plethora of cellular responses, including DNA damage, repair, cell cycle progression, transcription, ubiquitination, apoptosis; most recently BRCA1 and BRCA2 have been found to play a role in the determination of stem-cell fate.35–38 The links between both BRCA1 and BRCA2 and the DNA repair pathways are numerous and extend beyond the HR repair pathway, although their role in HR is most characterized. Cells lacking either BRCA1 or BRCA2 have a high degree of genomic instability, which is highly indicative of the important role of HR in maintaining genomic integrity. This was highlighted more than 10 years ago by the finding that mice nullizygous for BRCA1 have the same phenotype as those null for the HR essential protein RAD51.39 Functionally, BRCA1 acts as a sensor of DNA damage and replication stress and mediates HR through BRCA2. Biochemically, in response to DNA damaging agents, BRCA1 is phosphorylated at numerous sites by ATM, ATR, and CHK2, and subsequently associates with many other repair components.40,41 These include MSH2, MSH6, ATM, RAD51, and the MRN complex. BRCA2 plays a more direct role in HR by interacting with RAD51 and facilitating the formation of aggregates of RAD51 at the sites of DNA breaks.42 Therefore, in the absence of either BRCA1 or BRCA2, repair relies on the error-prone NHEJ pathways. The resulting accumulation of mutations undoubtedly contributes to tumorigenesis. BRCA1 has also been described as having a role to play in the regulation of genes involved in NER, including XPC and DDB2.43
Fanconi Anemia
Fanconi anemia (FA) was first described by Fanconi in 1927 when he studied a family with three boys, all with reduced blood cell number (pancytopenia) and birth defects. Since this initial description, it has become clear that the phenotype of this disease is highly variable and as such is difficult to diagnosis clinically. FA patients are described as having an increased risk of acute myeloid leukemia; squamous cell carcinomas of the head, neck, and esophagus; gynecologic carcinomas; and liver tumors at a young age as well as a range of congenital anatomical abnormalities. The range of congenital abnormalities seen in 60% to 75% of FA patients include characteristic thumb malformations and abnormal skin pigmentation.44,45 At the molecular level FA is diagnosed as an extreme sensitivity to agents that induce DNA crosslinks, for example mitomycin C, diepoxybutane, cisplatin, and to a lesser degree ionizing radiation. In contrast to the majority of familial cancer syndromes, FA is not the result of the mutation of a single gene; instead 13 genetic complementation groups have been identified to date.46,47 Mutations in any one of these can cause FA. The FANC genes all encode proteins that are involved in the repair of DNA crosslinks. This repair mechanism is somewhat uncharacterized and seems to by a hybrid of both excision repair and DSB repair systems, in particular HR. Of the 13 FA genes that have been identified, 8 have been shown to form a nuclear complex in response to DNA damage, and there is evidence to suggest that this complex has ubiquitin-E3 ligase activity.48 During the DNA damage response to crosslinking agents, this core nuclear complex monoubiquitinates FANCD2,49 which then translocates to co-localize with proteins involved in HR such as BRCA1,50 RAD51,51 and BRCA2/FANCD152 in nuclear foci. Both the ATM53 and ATR54 kinases have been demonstrated to regulate the FANC/BRCA1 pathway in response to DNA damage and replication stress respectively. Recently, a critical role for the Fanconi pathway in the survival of neural stem and progenitor cells in both development and adult neurogenesis has been reported.55,56
Homologs of RECQ: Bloom Syndrome, Werner Syndrome, and Rothmund-Thompson Syndrome
RECQ was first described in E. coli and has been extensively characterized in that system.57 Since then, five human homologs of RECQ have been described, three of which have clearly been shown to be mutated in cancer-prone syndromes.58 The RECQ homologs are DNA helicases that have two enzymatic activities: they unwind DNA 3′ to 5′ in an ATP-dependent manner59,60 and also act 3′ to 5′ as an exonuclease.61 The RECQ helicases have been described as the “guardians of the DNA replication fork” and are thought to act primarily at the interface between replication and recombination.62 There is significant evidence to support this claim, which includes the following observations: First, cells with perturbed RECQ activity are extremely sensitive to agents such as hydroxyurea (Hu) and aphidicolin (APH), both of which inhibit replication. Second, even in the absence of agents such as APH and Hu, cells with RECQ mutations did not progress through S phase normally and instead accumulated abnormal DNA structures. Third, the activity and expression of the RECQ helicases is seen to peak in the S phase of the cell cycle, indicating this is the period in which they function. Finally, the helicase are localized to sites of DNA replication and also seem to protect replication forks during periods of fork stalling, and regulate fragile site stability.63 With regard to human disease, the most well-studied human recQ helicases are Bloom (BLM) syndrome, Werner (WRN) syndrome and Rothmund-Thompson (RT) syndrome. Loss of the BLM helicase results in BLM syndrome, which is associated with a high incidence of cancer and in particular lymphomas, leukemias, and solid tumors of the gastrointestinal tract and breast. Skin disorders, sunlight sensitivity, dwarfism, immunodeficiency, male sterility, and elevated levels of sister chromatid exchange are also all associated with BLM syndrome.64,65 Individuals with WRN syndrome also show an increased risk of cancers, but also characteristically demonstrate premature aging. This puts patients at risk for age-related diseases such as cancer, diabetes, osteoporosis, and atherosclerotic cardiovascular disease. Increased genomic instability is also manifested in WS, as high levels of chromosomal deletions and translocations.66–68 Finally, RT syndrome, caused by a loss of the RECQ4 helicase, displays growth deficiency, photosensitivity with poikilodermatous skin changes, early graying and hair loss, juvenile cataracts, skeletal dysplasias, and a predisposition to malignancy, especially osteogenic sarcomas and chromosomal instability.69,70
DNA Repair Deficiency: A Therapeutic Target
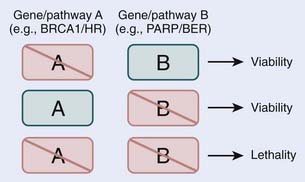
Loss, by either mutation or epigenetic silencing, of DNA repair pathways sensitizes cells to genotoxic agents such as chemotherapy and radiotherapy. The finding that in comparison with normal, nontumorigenic cells, rapidly proliferating cancer cells exhibit increased sensitivity to exogenously induced DNA damage and can be selectively killed forms the base of modern cancer therapies. Unfortunately, the treatment of human cancers is rarely this straightforward, as tumor cells acquire mutations in genes that control the cell-cycle and activate apoptotic programs. For these reasons, there is an effort to selectively kill tumor cells by exploiting deficient DNA repair. Examples include inhibitors of DNA-PK, ATM, and ERCC1 (reviewed in Helleday and colleagues, 200871; and Martin and colleagues, 200872). More recently, this concept of selectivity has been expanded to the targeting of specific repair pathways in tumor cells with specific genetic backgrounds. This approach is likely to receive more attention for cancer therapy, and at present is best illustrated by the use of poly (ADP-ribose) polymerase (PARP) inhibitors. PARP is involved in BER and specifically in the sensing of SSBs.73 When an SSB goes unrepaired by BER, a DSB is formed after replication, which is repaired by HR. However in tumor cells that lack BRCA1 or BRCA2, HR is severely compromised, making them sensitive to PARP inhibition. It has been effectively demonstrated that BRCA1– and BRCA2-deficient cells are sensitive to PARP inhibition, whereas normal or heterozygote BRCA1 and BRCA2 cells are not.74,75 The deficiency of HR in BRCA1– and BRCA2-deficient cells presents an ideal therapeutic window to treat cancer cells with PARP inhibitors, and to minimize the effect on the normal surrounding tissues. The success of PARP inhibitors is the result of the synthetic lethality of inhibiting two repair pathways, in this case BER and HR. Thus, although the loss of the HR repair pathway may be tolerated in BRCA1– and BRCA2-deficient cells, the loss of a second repair pathway BER, which alone may also be compatible with viability, cannot be tolerated in the HR-deficient background, and leads to cell death (Fig. 2-7).76–79
Conclusions
Recently it was shown that during the very early stages of tumorigenesis, DNA damage signaling is initiated by the activation of oncogenes. It was further proposed that at this stage tumor cells are kept in check by the activation of cell cycle checkpoints and in some cases apoptosis. These studies led to DNA damage signaling being described as acting as a barrier to tumorigenesis.80,81 However, once the neoplastic cells take on a mutator phenotype, they start losing essential components of damage signaling and repair. These findings highlight just how reliant human cells are on effective means of both sensing and repairing DNA damage. Further study of DNA repair pathways will undoubtedly affect the way we treat, diagnose, and potentially prevent disease.
1 Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268-274.
2 Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci U S A. 2003;100(3):776-781.
3 Tutt AN, Lord CJ, McCabe N, et al. Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer. Cold Spring Harb Symp Quant Biol. 2005;70:139-148.
4 Cheadle JP, Sampson JR. MUTYH-associated polyposis—from defect in base excision repair to clinical genetic testing. DNA Repair (Amst). 2007;6(3):274-279.
5 Hakem R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008;27(4):589-605.
6 Parsons JL, Tait PS, Finch D, et al. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol Cell. 2008;29(4):477-487.
7 Boyce RP, Howard-Flanders P. Release of ultraviolet light-induced thymine dimers from DNA in E. Coli K-12. Proc Natl Acad Sci U S A. 1964;51:293-300.
8 Pettijohn D, Hanawalt P. Evidence for repair-replication of ultraviolet damaged DNA in bacteria. J Mol Biol. 1964;9:395-410.
9 Setlow RB, Carrier WL. The disappearance of thymine dimers from DNA: an error-correcting mechanism. Proc Natl Acad Sci U S A. 1964;51:226-231.
10 Hanawalt PC. Subpathways of nucleotide excision repair and their regulation. Oncogene. 2002;21(58):8949-8956.
11 de Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13(7):768-785.
12 Wood RD. Nucleotide excision repair in mammalian cells. J Biol Chem. 1997;272(38):23465-23468.
13 Thoms KM, Baesecke J, Emmert B, et al. Functional DNA repair system analysis in haematopoietic progenitor cells using host cell reactivation. Scand J Clin Lab Invest. 2007;67(6):580-588.
14 Cleaver JE. Defective repair replication of DNA in xeroderma pigmentosum. Nature. 1968;218(5142):652-656.
15 Kraemer KH, Lee MM, Scotto J. DNA repair protects against cutaneous and internal neoplasia: evidence from xeroderma pigmentosum. Carcinogenesis. 1984;5(4):511-514.
16 Peltomaki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003;21(6):1174-1179.
17 Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18(1):85-98.
18 Vasen HF. Review article: the Lynch syndrome (hereditary nonpolyposis colorectal cancer). Aliment Pharmacol Ther. 2007;26(Suppl 2):113-126.
19 Felton KE, Gilchrist DM, Andrew SE. Constitutive deficiency in DNA mismatch repair. Clin Genet. 2007;71(6):483-498.
20 Brown KD, Rathi A, Kamath R, et al. Nat Genet. 2003;33(1):80-84.
21 Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499-506.
22 Hammond EM, Kaufmann MR, Giaccia AJ. Oxygen sensing and the DNA-damage response. Curr Opin Cell Biol. 2007;19(6):680-684.
23 Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499-506.
24 Pellegrini M, Celeste A, Difilippantonio S, et al. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature. 2006;443(7108):222-225.
25 Carney JP, Maser RS, Olivares H, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93(3):477-486.
26 Stewart GS, Maser RS, Stankovic T, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99(6):577-587.
27 Giaccia AJ, Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev. 1998;12(19):2973-2983.
28 Riley T, Sontag E, Chen P, et al. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9(5):402-412.
29 Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303(5660):1010-1014.
30 Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11(3):577-590.
31 Chang YC, Jan KY, Cheng CA, et al. Direct involvement of the tumor suppressor p53 in nucleotide excision repair. DNA Repair (Amst). 2008;7(5):751-761.
32 Frebourg T, Barbier N, Yan YX, et al. Germ-line p53 mutations in 15 families with Li-Fraumeni syndrome. Am J Hum Genet. 1995;56(3):608-615.
33 Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250(4985):1233-1238.
34 Srivastava S, Zou ZQ, Pirollo K, et al. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature. 1990;348(6303):747-749.
35 Hartman AR, Ford JM. BRCA1 induces DNA damage recognition factors and enhances nucleotide excision repair. Nat Genet. 2002;32(1):180-184.
36 Liu S, Ginestier C, Charafe-Jauffret E, et al. BRCA1 regulates human mammary stem/progenitor cell fate. Proc Natl Acad Sci U S A. 2008;105(5):1680-1685.
37 Smalley MJ, Reis-Filho JS, Ashworth A. BRCA1 and stem cells: tumour typecasting. Nat Cell Biol. 2008;10(4):377-379.
38 Zhang J, Powell SN. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol Cancer Res. 2005;3(10):531-539.
39 Lim DS, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol. 1996;16(12):7133-7143.
40 Wang Y, Cortez D, Yazdi P, et al. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14(8):927-939.
41 Xu B, Kim S, Kastan MB. Involvement of BRCA1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol. 2001;21(10):3445-3450.
42 Moynahan ME, Chiu JW, Koller BH, et al. BRCA1 controls homology-directed DNA repair. Mol Cell. 1999;4(4):511-518.
43 Hartman AR, Ford JM. BRCA1 and p53: compensatory roles in DNA repair. J Mol Med. 2003;81(11):700-707.
44 Alter BP. Cancer in Fanconi anemia, 1927–2001. Cancer. 2003;97(2):425-440.
45 D’Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. 2003;3(1):23-34.
46 Gurtan AM, D’Andrea AD. Dedicated to the core: understanding the Fanconi anemia complex. DNA Repair (Amst). 2006;5(9–10):1119-1125.
47 Kennedy RD, D’Andrea AD. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes. J Clin Oncol. 2006;24(23):3799-3808.
48 Grompe M, van de Vrugt H. The Fanconi family adds a fraternal twin. Dev Cell. 2007;12(5):661-662.
49 Wang X, D’Andrea AD. The interplay of Fanconi anemia proteins in the DNA damage response. DNA Repair (Amst). 2004;3(8–9):1063-1069.
50 Garcia-Higuera I, Taniguchi T, Ganesan S, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7(2):249-262.
51 Taniguchi T, Garcia-Higuera I, Andreassen PR, et al. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood. 2002;100(7):2414-2420.
52 Wang X, Andreassen PR, D’Andrea AD. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol Cell Biol. 2004;24(13):5850-5862.
53 Taniguchi T, Garcia-Higuera I, Andreassen PR, et al. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood. 2002;100(7):2414-2420.
54 Wang X, D’Andrea AD. The interplay of Fanconi anemia proteins in the DNA damage response. DNA Repair (Amst). 2004;3(8–9):1063-1069.
55 Sii-Felice K, Barroca V, Etienne O, et al. Role of Fanconi DNA repair pathway in neural stem cell homeostats. Cell Cycle. 2008;7(13):1911-1915.
56 Sii-Felice K, Etienne O, Hoffschir F, et al. Fanconi DNA repair pathway is required for survival and long-term maintenance of neural progenitors. EMBO J. 2008;27(5):770-781.
57 Nakayama H, Nakayama K, Nakayama R, et al. Isolation and genetic characterization of a thymineless death-resistant mutant of Escherichia coli K12: identification of a new mutation (recQ1) that blocks the RecF recombination pathway. Mol Gen Genet. 1984;195(3):474-480.
58 Sharma S, Brosh RMJr. Human RECQ1 is a DNA damage responsive protein required for genotoxic stress resistance and suppression of sister chromatid exchanges. PLoS ONE. 2007;2(12):e1297.
59 Gray MD, Shen JC, Kamath-Loeb AS, et al. The Werner syndrome protein is a DNA helicase. Nat Genet. 1997;17(1):100-103.
60 Suzuki N, Shimamoto A, Imamura O, et al. DNA helicase activity in Werner’s syndrome gene product synthesized in a baculovirus system. Nucleic Acids Res. 1997;25(15):2973-2978.
61 Huang S, Li B, Gray MD, et al. The premature ageing syndrome protein, WRN, is a 3′→5′ exonuclease. Nat Genet. 1998;20(2):114-116.
62 Bachrati CZ, Hickson ID. RecQ helicases: guardian angels of the DNA replication fork. Chromosoma. 2008;117(3):219-233.
63 Pirzio LM, Pichierri P, Bignami M, et al. Werner syndrome helicase activity is essential in maintaining fragile site stability. J Cell Biol. 2008;180(2):305-314.
64 Ellis NA, German J. Molecular genetics of Bloom’s syndrome. Hum Mol Genet. 1996;5 Spec No:1457-1463.
65 Gruber SB, Ellis NA, Scott KK, et al. BLM heterozygosity and the risk of colorectal cancer. Science. 2002;297(5589):2013.
66 Oshima J. The Werner syndrome protein: an update. Bioessays. 2000;22(10):894-901.
67 Shen JC, Loeb LA. The Werner syndrome gene: the molecular basis of RecQ helicase-deficiency diseases. Trends Genet. 2000;16(5):213-220.
68 Yu CE, Oshima J, Fu YH, et al. Positional cloning of the Werner’s syndrome gene. Science. 1996;272(5259):258-262.
69 Kitao S, Shimamoto A, Goto M, et al. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet. 1999;22(1):82-84.
70 Wang LL, Gannavarapu A, Kozinetz CA, et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst. 2003;95(9):669-674.
71 Helleday T, Petermann E, Lundin C, et al. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8(3):193-204.
72 Martin SA, Lord CJ, Ashworth A. DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev. 2008;18(1):80-86.
73 Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. 2008;13:3046-3082.
74 Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913-917.
75 Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917-921.
76 Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913-917.
77 Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917-921.
78 Hartwell LH, Szankasi P, Roberts CJ, et al. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278(5340):1064-1068.
79 Iorns E, Lord CJ, Turner N, Ashworth A. Utilizing RNA interference to enhance cancer drug discovery. Nat Rev Drug Discov. 2007;6(7):556-568.
80 Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434(7035):864-870.
81 Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434(7035):907-913.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
2 Dna Damage and Repair
Introduction
Preserving the integrity of the genome is of paramount importance to any organism. However, our DNA is under continual assault from both endogenous and exogenous damaging agents. These include the products of normal metabolism (reactive oxygen species [ROS]), ultraviolet (UV) and ionizing radiation, and genotoxic compounds such as those found in cigarettes. Inefficient or inadequate DNA repair can have dire consequences, which can lead to cell death, affect essential metabolic processes such as gene transcription, or result in the creation of a mutator phenotype.1 Decreased efficiency of DNA repair pathways have been linked with cancer predisposition in humans. This is clearly illustrated by studies that show that mutations in DNA repair pathways are associated with cancer-predisposing syndromes in humans. Furthermore, the loss of the ability to effectively repair damaged DNA leads to an increase in genomic instability and can contribute to tumorigenesis.2 DNA repair pathways are highly conserved throughout evolution, with many of the initial discoveries in the field having been made in Escherichia coli, but are still highly applicable to human cells. The purpose of this chapter is to give an overview of the five main repair pathways (BER, NER, MMR, NHEJ, and HR), and to highlight how mutations in these pathways affect tumor formation and response to therapy (Fig. 2-1).
DNA Damage Response Pathways
There are numerous mechanisms for the repair of damaged DNA, which are determined in part by the nature of the damage to which they respond and the presence of functioning repair pathways. For example, in the absence of functional BER, a single-strand break (SSB) can form a DNA double-strand break (DSB), which will then be processed by HR.3 In virtually all DNA repair pathways, DNA damage is detected by sensors that create a signal that is further transduced and amplified to initiate the repair pathway. The most critical DNA repair pathways are those that remove damaged bases that result as a consequence of cellular metabolism or genotoxic insult. BER, NER, and MMR pathways are involved in removing damaged bases and are discussed in the following text.
Base Excision Repair
The BER pathway has a central role to play in preserving genomic integrity. Alterations in BER signaling are predicted to predispose humans to cancer and have been associated with colorectal adenomas and colon cancer. In contrast to other DNA repair pathways discussed in this chapter, only recently has an association between loss of BER and a human disease syndrome been identified. Patients affected by this syndrome, termed E. coli mutY human homolog (MYH)–associated polyposis (MAP), develop large numbers of colonic polyps (in the tens to hundreds range) by the age of 40, and nearly 50% present with colon cancer.4 The essential role of this pathway is supported by the finding that mice nullizygous for the genes involved were either lethal, indicating an absolute requirement for the gene product, or showed little to no phenotype because of a high degree of redundancy within the pathway. For example, loss of APE1, LIG1, LIG3, XRCC1, FEN1, and POLB all result in embryonic lethality, whereas deletion of NTH1, OGG1, UNG, AAAG, and MUTYH result in no discernible developmental defects.5 The absolute requirement for BER stems from its ability to repair DNA damage from both exogenous and endogenous sources. The majority of damage to the base portion of each nucleotide is the result of the products of normal metabolism that oxidize or alkylate DNA, such as ROS. Included in these lesions is the formation of 8-oxo-7,8-dihdroguanine (8-oxoG), which has mutagenic properties and also blocks DNA replication and transcription. BER is also used by the cell to handle the high rate of depurination (loss of purines from the DNA), and it has been estimated that a human being loses approximately a trillion guanines from his or her DNA every hour. In addition, BER is also used to remove a large number of cytosines that become spontaneously deaminated to form uracil. The BER mechanism has been extensively characterized in both E. coli and mammals, and includes five stages, shown schematically in Fig. 2-2. In brief, there is an initial recognition of the damaged base by DNA N-glycosylases. Different glycosylases recognize specific lesions, although there is considerable redundancy between them. The damaged base is then removed to give an apurinic/apyrimidinic (AP) site, which is processed by an AP-specific endonuclease (monofunctional glycosylase) or with an additional AP-lyase (bifunctional glycosylase), which leaves a single-strand interruption. These gaps are then filled by a DNA polymerase either with a single nucleotide (short patch) or polynucleotides (longer repair patch) before a ligation occurs. During BER, an SSB is formed as an intermediate. These SSBs are also generated by exogenous agents such as ionizing radiation and other oxidizing compounds. In response to ionizing radiation, BER effectively repairs the damage (SSB) although some additional enzymatic activities are thought to be required, such as PARP1. The control of BER has been recently described and indicates that BER proteins that are not actively involved in repair are ubiquitinated and degraded by the proteosome.6
Nucleotide Excision Repair
NER was discovered in the 1960s through elegant studies on the effects of UV irradiation on DNA synthesis and repair replication in bacteria. Since then it has been characterized extensively in mammals and has been described as the principle repair pathway for the removal of bulky adducts induced by UV radiation or other environmental carcinogens.7–9 In contrast to the BER pathway, damage detected by the NER proteins is not done in a specific manner because there is no equivalent to DNA N-glycosylases involved in BER, described previously. It is hard to imagine that a specific glycosylase could evolve for each type of DNA damage, especially in an environment in which exogenous damaging agents are numerous and constantly attack epithelial cells in the skin. Consequently, NER has been described as a more flexible version of BER. The major lesions recognized by NER are bulky adducts that result from intrastrand crosslinks, UV-induced cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts. When these lesions are not removed, they distort the DNA helix and in doing so prevent DNA replication and transcription, which can result in the employment of the error-prone translesion synthesis repair pathway. The process of NER can be subdivided into two pathways: global genome repair (GG-NER) or global genomic repair (GGR), and transcription coupled repair-nucleotide excision (TC-NER) or transcription-coupled repair (TCR).10 The process of GG-NER is genome wide (i.e., lesions can be removed from anywhere). In contrast, TC-NER only removes lesions in DNA strands of actively transcribed genes. When a DNA strand that is being actively transcribed becomes damaged, the ribonucleic acid (RNA) polymerase can block access to the site of damage and hence prevent DNA repair. TC-NER has evolved to prevent this by effectively removing the RNA polymerase from the site of damage to allow the repair proteins access. The mechanism of GG-NER and TC-NER differ only in the detection of the lesion; the subsequent pathway is the same. In general NER involves the recognition of the lesion, by proteins specific to GG-NER or TC-NER, followed by incision of the DNA strand near the damage, removal of the affected stretch of DNA, repair replication using the complementary strand as a template, and, finally, ligation to seal the 3′ end of the repair patch with the parental DNA (Fig. 2-3).11,12
Three human syndromes that occur as a result of mutation of the NER pathways have been identified: xeroderma pigmentosum (XP), Cockayne syndrome (CS) and trichothiodystrophy (TTD).13 Many of the principle genes involved in NER are called XP, for example XPA, XPB, and so on. This is because they were identified in individuals suffering from XP.14 XP results from mutation in any one of seven complementation groups: XPA, XPB (ERCC3), XPC, XPD (ERCC2), XPE (DDB2), XPF and XPG. Interestingly XP patients with mutations in XPC or XPE are deficient in GG-NER, whereas the remaining are deficient in both GG-NER and TC-NER. XP presents early in life as extreme sensitivity to the sun; freckles appear at a young age (1 year) even after short exposure to the sun, and significant pigmentation abnormalities are common. The eyes of XP patients are photosensitive and often exhibit eye damage as a result of constant UV-induced conjunctivitis and keratitis. This extreme sensitivity to UV translates to a 1 in 4000-fold increased risk of skin cancer in XP individuals, often appearing as early as age 10.15 These malignancies are usually squamous and basal cell carcinomas, but melanomas have also been reported. Neurologic abnormalities also frequently occur, including peripheral neuropathy, sensorineural deafness, and progressive mental retardation. Similar to XP patients, those with CS are also very sensitive to UV, although they do not exhibit the increased risk of skin cancer. Instead CS patients have distinct developmental abnormalities, which include growth retardation (dwarfism), progressive cognitive impairment, skeletal abnormalities, severe mental retardation, cataracts, and retinitis pigmentosa. CS patients tend to die younger than the age of 20 (the average life span is 12 years) as a result of infectious or renal complications rather than cancer. The genetic determinants of CS are mutations in either the CSA or CSB genes, both of which are essential for recognition of DNA damage and initiation of the TC pathway of NER. CS individuals remain proficient in GG-NER. Finally, mutation of the XPB or XPD genes results in TTD. Again extreme sensitivity to the sun is a characteristic in some of these patients (approximately 50%), although they possess a low incidence of developing skin cancer. TTD individuals are similar to CS in presentation but have the additional symptom of brittle nails and hair; this is a result of reduced sulfur content in the component proteins. The differences and similarities between these three syndromes have proved intriguing, in that they all show UV sensitivity but only XP individuals have an increased risk of skin cancer. This has been attributed to the determination that loss of GG-NER is what predisposes individuals to skin cancer. In contrast, the developmental abnormalities associated with these syndromes (retardation, dwarfism, etc.) are the result of a loss of TC-NER. Given the essential role of NER in the removal of UV-induced damage it is not surprising that XP individuals succumb to skin cancer, but an increased risk of other malignancies would also be expected and this has not been noted. There have been reports documenting modest alteration in NER genes as a result of polymorphisms that contribute to the risk of solid tumors.
Mismatch Repair
During normal replication and recombination, mistakes can be made that result in the insertion of an incorrect nucleotide leading to a mismatch. These mismatches also occur as a result of base modifications. If mismatches escape surveillance of the proofreading capabilities of DNA polymerases, there is a permanent change in sequence or mutation. The MMR pathway functions to identify such mistakes and correct them before they are propagated further. MMR also functions to remove small loops (insertion–deletion loops [IDL]) in the DNA that arise as a consequence of spontaneous slippage-dependent misalignment between primer and template DNA strands. These occur particularly in highly repetitive microsatellite regions that, in the absence of effective MMR, show a large increase in the frequency of spontaneously occurring mutations (microsatellite instability [MSI]).16 The process of MMR can be subdivided into four components: First, the mismatch must be identified by sensors that transduce a signal; second, MMR factors are recruited; third, the newly synthesized strand harboring the mismatch is identified and the incorrect or altered nucleotides are excised; and in the fourth stage, resynthesis and ligation of the excised tract occurs. MMR was first characterized in E. coli, which encodes MUT genes; homologues of these gene products have now been identified and extensively characterized in both yeast and humans.17 See Fig. 2-4 for a schematic representation and an indication of the critical gene products. The significance of MMR was highlighted when it became apparent in the 1990s that hereditary nonpolyposis colorectal cancer (HNPCC) and sporadic MSI-positive colon cancers were caused by defects in MMR. HNPCC, also known as Lynch syndrome, affects 1 in 1000 people and is characterized by early-onset colorectal cancers that have a high level of MSI. Individuals with HNPCC have an 80% risk of colorectal cancer in their lifetime, as well as an increased risk of other malignancies such as endometrial (50% risk), ovarian, and gastric malignancies. In 70% to 80% of the germline mutations found in HNPCC, the mutations occur in the MLH1 or MLH2 genes. In 10% of cases there is a mutation in MSH6, and, in rare cases, PMS1, PMS2, MLH3, and EXO1.18 Epigenetic silencing of the MMR genes has also been noted and shown to increase the mutation rate. In very rare cases, both copies of an MMR gene, such as MLH1, MSH2, MSH6, or PMS2 have been found mutated (homozygous germline mutations).19 These individuals have a reduced life span and succumb to malignancy of hematologic or brain origin.
The MMR proteins have been described as having a role to play in cell cycle checkpoints and apoptosis. Specifically, both hMutSα- and hMutLα-deficient cells exhibit defective S-phase checkpoint responses.20 The proposed model behind this observation is that MMR proteins act as sensors of DNA damage and then recruit the checkpoint proteins and hence activate these signaling pathways. This is supported by the findings that hMutSα and hMutLα physically interact with ataxia-telangiectasia mutated (ATM), ataxia-telangiectasia Rad3-related (ATR), ATR-interacting protein (ATRIP), c-Abl, and p73 in cells exposed to DNA-damage-inducing agents. Also supportive of this model is the wealth of data indicating that cells that become defective in MMR gain resistance to cytotoxic agents. Interestingly, patients with MSI-positive tumors have a greater chance of survival than those that do not, although whether this is as a result in differences in tumorigenesis or therapeutic response is unclear.
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
