Chapter 68 Wilms’ Tumor
Wilms’ tumor, or nephroblastoma, is the most common childhood renal tumor, accounting for about 6% of pediatric malignant diseases.1 It was first described in 1814, but Max Wilms first identified the true nature of the condition as a neoplasm with mixed cellular elements in 1899.2,3 His name has become synonymous with nephroblastoma. The early advances in managing these small patients with huge tumors were made by surgeons, who reduced the operative mortality from 25% to the current rate of less than 1%. The survival of children with Wilms’ tumor gradually increased from less than 10% to about 50% by the 1940s and 1950s. Postoperative RT became routine after results from Boston Children’s Hospital documented an increased cure rate. The advent of effective chemotherapy (dactinomycin and vincristine) also had an impact on the survival of these children.4 The systematic development of multidisciplinary management for Wilms’ tumor resulted in a striking improvement in survival. Thus the grim outlook at the beginning of the 20th century (90% mortality rate) has effectively inversed to a survival rate of 90% in the early 21st century. Randomized clinical trials in North America under the aegis of the National Wilms’ Tumor Study Group (NWTS) and those in Europe, largely by the International Society of Pediatric Oncology (SIOP), have resulted in Wilms’ tumor therapy becoming a paradigm for successful multidisciplinary integration in the treatment of cancer. Successive NWTS clinical trials have been based on postoperative therapies, whereas preoperative strategies have been the focus of SIOP investigators.5,6
The clinical risk factors such as tumor stage and histology were categorized so that treatment could be modulated in intensity according to risk groups, the objective being to achieve cure with minimum complications. As a result, children with low-risk tumors now receive minimal therapy whereas those at high risk benefit from more intensive therapy. The modern survivors of Wilms’ tumor enjoy a better quality of life owing to sequential reductions in the intensity of therapy.7 Current Wilms’ tumor studies in the United States and Canada are conducted by the Children’s Oncology Group (COG).
Etiology and Epidemiology
The cause of Wilms’ tumor is unknown. The peak incidence is between 3 and 4 years of age. Wilms’ tumor may arise as sporadic or hereditary tumors or in the setting of specific genetic disorders.8 The overall annual incidence of Wilms’ tumor is 7.1 per million children, and approximately 500 new cases can be expected each year in North America. Wilms’ tumor is found more often in black than in white children, with a ratio of 1.25 : 1. Girls are affected more than boys in the same ratio.1
Prevention and Early Detection
Prevention awaits an identified cause. Uncommon congenital anomalies (aniridia, genitourinary malformations, hemihypertrophy, or signs of overgrowth) may be seen in 13% to 28% of children with unilateral or bilateral disease, respectively.9 Syndromes associated with a higher risk for developing Wilms’ tumor include the “WAGR” syndrome (aniridia, genitourinary malformation, mental retardation), the Beckwith-Wiedemann syndrome, and the Denys-Drash syndrome (see next section on molecular biology). Children with such syndromes should be screened for the development of Wilms’ tumor using ultrasonographic examinations of both kidneys every 3 months until age 5 years (WAGR) or 7 years (Beckwith-Wiedemann syndrome).8,10
Biologic Characteristics and Molecular Biology
The biologic characterization of Wilms’ tumor has provided the foundation for our understanding of key concepts in cancer genetics, including the roles of tumor suppressor genes and relation of genomic imprinting in tumorigenesis. Although Wilms’ tumor was one of the original examples in Knudson’s two-hit model of cancer development, subsequent research has shown that multiple genes and several genetic events contribute to the formation of this malignant disorder.11 The molecular changes that have been described in Wilms’ tumor can be classified as primary events predisposing to the development of tumor or secondary events associated with tumor progression.
WT1
Initial insights into the molecular biology of Wilms’ tumor were derived from the observation that in patients with WAGR syndrome the risk for developing the tumor is more than 30%. Cytogenetic analysis of individuals with this syndrome showed deletions at chromosome 11p13, which was later found to be the locus of a contiguous set of genes including PAX6, the gene causing aniridia, and WT1, one of the Wilms’ tumor genes. The WT1 gene encodes a transcription factor that is crucial to normal kidney and gonadal development.8 The Denys-Drash syndrome, which is characterized by pseudohermaphroditism, glomerulopathy, renal failure, and a 95% chance of Wilms’ tumor development, is caused by point mutations in the zinc-finger DNA-binding region of the WT1 gene.10 Although WT1 has a clear role in tumorigenesis of Wilms’ tumor in patients with the WAGR and Denys-Drash syndromes, only a minority of patients with sporadic Wilms’ tumor carries WT1 mutations in the germline (<5%) or in tumor tissue (6% to 18%).8
WT2
The Beckwith-Wiedemann syndrome is an overgrowth disorder manifested by large birth weight, macroglossia, organomegaly, hemihypertrophy, neonatal hypoglycemia, abdominal wall defects, ear abnormalities, and predisposition to Wilms’ tumor and other malignant disorders. Approximately 5% of individuals with this syndrome develop Wilms’ tumor. Beckwith-Wiedemann syndrome maps to chromosome 11p15, a locus called WT2 because loss of heterozygosity (LOH) at this locus has been detected in Wilms’ tumor.12
The WT2 locus consists of several genetically imprinted genes that are in two clusters or imprinting centers. Several combinations of genetic and epigenetic alterations can give rise to Beckwith-Wiedemann syndrome, with increasing recognition of genotype-phenotype relationships.13,14 Imprinting Center 1, which contains the IGF2 and H19 genes, has been most implicated in Wilms’ tumor predisposition. Alterations in Imprinting Center 2, which contains the genes CDKN1C, KCNQ1 and KCNQ1OT1, are not strongly linked to Wilms’ tumor development.
Familial Wilms’ Tumor Genes
Familial predisposition to Wilms’ tumor is rare, identified in only 1.5% of patients. A few families were shown to carry constitutional mutations of the WT1 gene, but genetic linkage analysis has excluded WT1 in most familial Wilms’ tumor pedigrees. Linkage analysis has uncovered two familial Wilms’ tumor loci, FWT1 (17q) and FWT2 (19q), but the causative genes remain to be identified.8
Wnt Signaling Pathway Genes (WTX and β-Catenin)
Alterations of the Wnt signaling pathway have been implicated in several human malignancies. Central to the pathway is the activator, β-catenin, which is degraded in the absence of Wnt signaling. Activating mutations in the gene encoding β-catenin (CTNNB1) occur in approximately 15% of Wilms’ tumors.15 Interestingly, mutations in CTNNB1 occur mostly in tumors in which WT1 is also mutated, suggesting that these two events cooperate in the formation of Wilms tumor. WTX, a recently discovered novel Wilms tumor suppressor gene on the X chromosome, is inactivated in one third of sporadic Wilms’ tumor cases. In contrast to the “two-hit” model of inactivation of WT1, WTX is inactivated by a monoallelic “single hit” event that targets the X chromosome in tumors in males and the active X chromosome in tumors in females.16 WTX has been shown to be a negative regulator of Wnt/β-catenin signaling.17
Loss of Heterozygosity at 1p and 16q
One of the major goals of the NWTS-5 trial was to prospectively analyze the prognostic significance of LOH at chromosomes 1p and 16q. Analysis of these data revealed that the relative risks for relapse for patients with stage I to IV favorable histology tumors with LOH stratified by stage are 1.77 for LOH 1p (p <.01) and 1.39 for LOH 16q (p = .05). When the effects of LOH for both 1p and 16q were considered jointly, the relative risk for relapse in stage I and II favorable histology disease was 2.88 (p = .001) and that for stage III and IV favorable histology disease was 2.41 (p = .01). The relative risk for death for patients with stage I and II favorable histology disease with LOH for both regions is 4.25 (p = .01), and for stage III and IV it is 2.66 (p = .04).18
Pathology and Pathways of Spread
Most Wilms’ tumors are solitary lesions, although 6% involve both kidneys and 12% show multifocal involvement within a single kidney. The classic triphasic “favorable histology” Wilms’ tumor consists of varying proportions of three cell types—blastemal, stromal, and epithelial—recapitulating various stages of normal renal development (Fig. 68-1A, B). Less commonly, heterologous epithelial or stromal components are identified, including mucinous or squamous epithelium, skeletal muscle, cartilage, osteoid, or fat. Not all specimens are triphasic; biphasic and monophasic patterns are frequently encountered. Favorable histologic features characterize 87% of Wilms’ tumors.19,20
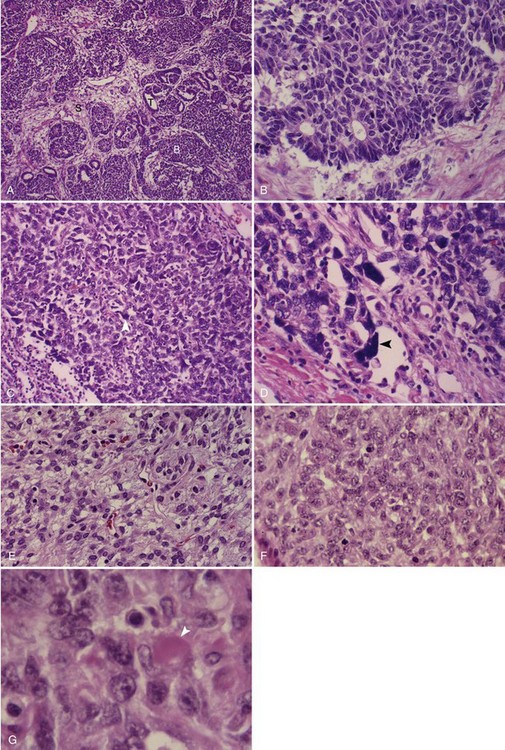
The histologic feature of greatest clinical significance is anaplasia, defined by the presence of greatly enlarged polyploid nuclei (see Fig. 68-1C, D). Anaplasia is present in approximately 8%; it is rare in the first 2 years of life and increases in frequency to approximately 13% in children older than age 5 years. Anaplasia is far more frequent in African-American than in white patients and has been strongly linked to the presence of TP53 mutations.21,22 The distinction between focal and diffuse anaplasia is prognostically significant.23
Correlations between histologic characteristics and clinical behavior have identified blastemal-rich tumors as typically invasive, presenting at an advanced stage, if often responsive to chemotherapy. By contrast, predominantly epithelial and rhabdomyomatous Wilms’ tumors are more likely to present at a low stage, yet many are relatively chemotherapy resistant. Analysis of the cohort of young children with small stage I intrarenal tumors assigned no adjuvant chemotherapy provides further support for the excellent prognosis of epithelial-differentiated Wilms’ tumors.24 Although much of the past success of the NWTS has relied on accurate histologic subclassification of Wilms’ tumors to define high-risk and low-risk subtypes, further risk classification is likely to depend on molecular genetic features.
The existence of precursor lesions (nephrogenic rests) is rather common. These lesions consist of abnormally persistent intrarenal embryonal nephroblastic tissue with small clusters of blastemal cells, tubules, or stromal cells. Nephrogenic rests can be subclassified by their positions within the kidney and histologic appearance: perilobar nephrogenic rests are limited to the periphery of the renal cortex, and intralobar nephrogenic rests occur randomly throughout the renal lobe. Nephroblastomatosis refers to the presence of multiple nephrogenic rests typically throughout the kidney. There are several possible fates for nephrogenic rests: most become dormant or involute spontaneously; only a small number develop clonal transformation into Wilms’ tumor. The presence of nephrogenic rests within a kidney resected for Wilms’ tumor indicates the need for monitoring the contralateral kidney for tumor development, particularly in young infants.25
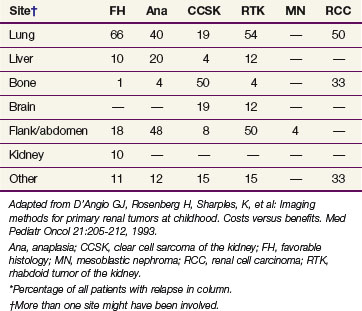
The other major cellular types of kidney tumors are outlined in Table 68-1, including:
Clinical Manifestations, Patient Evaluations, and Staging
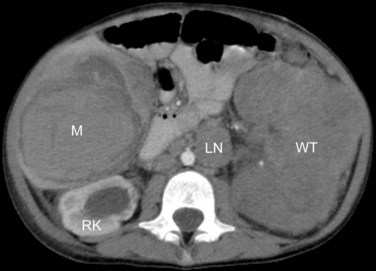
Laboratory evaluation after physical examination should include a complete blood cell count, routine hepatic and renal chemistries, and urinalysis, noting the presence or absence of urinary protein and white or red blood cells. Imaging studies before surgery are designed to evaluate the extent of the renal mass and differentiate primary intrarenal tumors from neuroblastoma (most often arising from the adjacent suprarenal gland or retroperitoneal structures), the presence of a normally functioning, morphologically normal contralateral kidney, the presence of a patent renal vein and inferior vena cava (i.e., free from thrombosis, most often tumor thrombosis when seen), and the presence or absence of metastases in the lungs. Diagnostic imaging studies include abdominal ultrasonography (particularly useful in the detection of tumor thrombosis) and CT or MRI of the abdomen (Fig. 68-2). A chest radiograph or CT scan of the chest can be used to detect lung metastasis. The clinical significance of metastatic tumors detected by chest CT but not on chest radiographs remains unclear, although evidence is increasing that some of these patients do not fare well with chemotherapy with vincristine and dactinomycin alone.28,29 MRI of the abdomen can help to distinguish between nephrogenic rests and Wilms’ tumor.30 Postoperatively, when the histology is established, a bone scintiscan and skeletal survey should be obtained for patients with CCSK, RTK, and RCC and MRI of the brain should be performed for those with RTK and RCC.
Staging
Wilms’ tumors are staged on the basis of anatomic tumor extent; therapy is currently based on stage and histology.31 Classifications based on tumor extent have evolved over the years. After analysis of the prognostic significance of several clinicopathologic factors in NWTS-1 and NWTS-2, an NWTS staging system has been in use from NWTS-3 onward. Patients with lymph node involvement, previously included with stage II disease, are now classified as having stage III disease, and those with local tumor spill were moved from stage III to stage II disease.32 Refinements to the inclusion criteria for stages I and II disease were introduced in the NWTS-5 study. Criteria for stage I were refined to accommodate an important subset of Wilms’ tumor currently managed by nephrectomy alone. Before NWTS-5, the distinction between stages I and II in the renal sinus was established by the hilar plane, which was an imaginary plane connecting the most medial aspects of the upper and lower poles of the kidney. This criterion was difficult to apply because of tumor distortion, and thus the hilar plane criterion has been replaced with renal sinus vascular or lymphatic invasion. The latter definition includes not only the involvement of vessels within the hilar soft tissue but also vessels located in the radial extensions of the renal sinus into the renal parenchyma.33,34
The current COG staging guidelines for Wilms tumor are shown in Table 68-2. These guidelines are essentially similar to those for NWTS-5 except for the fact that children with tumor spillage are upstaged from stage II to stage III because of the higher risk for relapse in these patients with two-drug chemotherapy absent use of RT.35
TABLE 68-2 Children’s Oncology Group Staging of Wilms’ Tumor (2010)
| Stage | Description |
|---|---|
| I | Tumor limited to kidney, completely resected. The renal capsule is intact. The tumor was not ruptured or sampled before removal. The vessels of the renal sinus are not involved. There is no evidence of tumor at or beyond the margins of resection.* |
| II | The tumor is completely resected and there is no evidence of tumor at or beyond the margins of resection. The tumor extends beyond kidney, as is evidenced by any one of the following criteria† : |
* For a tumor to qualify for certain therapeutic protocols as stage I, regional lymph nodes must be examined microscopically.
† Rupture or spillage confined to the flank, including biopsy of the tumor, is no longer included in stage II and is now included in stage III.
‡ Lymph node involvement in the thorax or other extra-abdominal sites is a criterion for stage IV.
Primary Therapy
Primary surgical resection of Wilms’ tumor remains the standard initial therapy undertaken in North America. A transabdominal transperitoneal approach is recommended to provide adequate exposure for complete locoregional staging.36 This procedure includes biopsy of hilar and regional nodes (even if normal appearing), which remains a crucial factors in staging. Although suspicious lymph nodes are excised irrespective of location, a formal lymph node dissection is neither beneficial nor recommended.
Tumor spillage remains an important concept in the surgery of Wilms’ tumor. Surgeons must be aware of any tumor-capsule violation with contamination of the peritoneal cavity during resection. The accurate assessment of a local spill from diffuse contamination is difficult; peritoneal contamination definitely increases the risk for local and abdominal recurrence, and both localized and diffuse peritoneal spill (or preoperative tumor rupture) are now considered stage III disease.35,37
Radiation Therapy
Flank/Abdominal Irradiation
RT continues to play an important role in the management of Wilms’ tumor. Successive NWTS trials have refined the indications for RT. In NWTS-1 and NWTS-2, an age-adjusted dose schedule was used for flank irradiation, ranging from 18 to 24 Gy for children younger than 18 months to 35 to 40 Gy for children older than 40 months. The abdominal relapse rate was 3% to 5% for group II and III tumors, and there was no dose-response relationship observed across these dose ranges.38,39 NWTS-3 proved that RT could be avoided in children with stage II tumors if vincristine and dactinomycin were both given. This study also showed that children with stage III favorable histology tumors who received 10.8 Gy and vincristine, dactinomycin, and doxorubicin had tumor control similar to that of those who received 20 Gy with vincristine and dactinomycin. This was an important finding because it eliminated the need for an age-adjusted dose schedule and significantly reduced the recommended radiation dose.40
NWTS-1, NWTS-2, and NWTS-3 showed that delay in initiating RT beyond 10 days was correlated with poor outcome primarily in cases with unfavorable histology.38–41 A study of the impact of RT delay on patients with favorable histology in NWTS-3 and NWTS-4 showed that a delay of more than 10 days did not significantly influence flank or abdominal tumor recurrence rates. It is currently recommended for children requiring RT that it be initiated without undue delay (within 14 days of nephrectomy) to eliminate abdominal recurrence as a variable in the analyses of outcomes in future Wilms’ tumor trials.41
The NWTS evaluated the frequency with which tumor spill of favorable histology produced intra-abdominal recurrence in NWTS-3 and NWTS-4. Flank irradiation but not doxorubicin reduced abdominal relapse rates. The odds ratio for the risk of recurrence was 0.35 for 10 Gy and 0.08 for 20 Gy when compared with those with no radiation therapy. Tumor spillage resulted in higher relapse and significantly lower survival among patients with stage II disease in NWTS-4 (i.e., those without RT). The 8-year relapse-free survival with and without spillage were 79% and 87% (p = .07), and overall survival rates were 90% and 95% (p = .04), respectively.35
Although diffuse anaplastic tumors are relatively resistant to chemotherapy, and presumably relatively resistant to RT, these tumors have not shown a radiation dose response between 10.8 and 40 Gy.42 The optimum radiation dose for anaplastic Wilms’ tumor and other unfavorable histology tumors remains unknown. In NWTS-5, children with stage I focal and diffuse anaplasia were treated with two-drug chemotherapy (vincristine and dactinomycin) alone without flank irradiation; the approach resulted in a poor survival rate.43 The current COG study recommends flank irradiation (10.8 Gy) with three-drug chemotherapy for these tumors. The survival rates for children with stage III diffuse anaplastic tumors and rhabdoid tumors continue to be poor (Table 68-3). The COG study will test the value of a higher dose of irradiation (19.8 Gy) together with intensified chemotherapy (Tables 68-4 and 68-5). The COG protocol will also prospectively study whether flank irradiation could be avoided in children with stage I CCSK who have undergone lymph node sampling and central pathology review (see Tables 68-4 and 68-5).
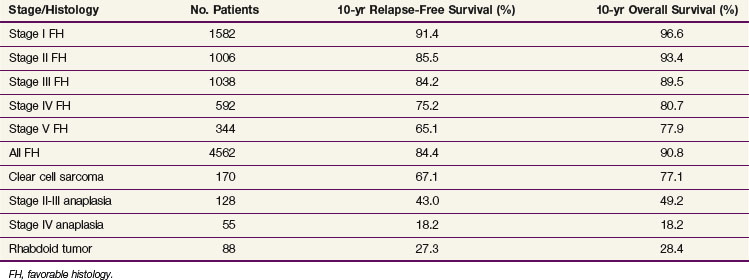
TABLE 68-4 Radiation Therapy Recommendations for the Children’s Oncology Group Renal Tumor Protocols
| Tumor Stage/Histology | RT Dose and Fields |
|---|---|
| Stage I and II FH | No RT |
| Stage III FH, stage I-III focal anaplasia, stage I-II diffuse anaplasia, stage I-III CCSK‡ | 10.8 Gy flank* RT† |
| Stage III diffuse anaplasia, stage I-III RTK | 19.8 Gy (infants 10.8 Gy) flank* RT† |
| Stage IV (lung metastases, FH) | 12 Gy (whole-lung irradiation in children who are not in complete remission at week 6 after induction chemotherapy) |
| Stage IV (lung metastases, UH) | 12 Gy whole-lung irradiation regardless of chemotherapy response |
| Stage IV (brain metastases) | 25.2 Gy (whole brain) + 10.6 Gy (local boost) |
| Stage IV (bone metastases) | 25.2 Gy (tumor + 3-cm margin) |
| Lymph node metastases not surgically resected | 19.8 Gy |
| Relapsed Wilms’ tumor (flank/abdomen) | 12.6-18 Gy (<age 12 mo) and 21.6 Gy in older children if previous radiation dose is ≤10.8 Gy 9 Gy boost to gross residual tumor after surgery |
CCSK, clear-cell sarcomas of the kidney; FH, favorable histology; RTK, rhabdoid tumors of the kidney; UH, unfavorable histology.
* Whole-abdomen irradiation is indicated when there is diffuse tumor spillage, intraperitoneal tumor rupture, peritoneal tumor seeding, and cytology-positive ascites. When dose is >10.8 Gy, renal shielding is required to limit the dose to the remaining kidney to <15 Gy.
† A boost of 10.8 Gy is to be administered to areas of gross residual tumor after surgery.
‡ Patients with stage I CCSK will not receive flank irradiation if lymph node sampling and central pathologic review were performed.
TABLE 68-5 Children’s Oncology Group Renal Tumor Protocol Schema
| Tumor Risk Classification | Multimodality Treatment |
|---|---|
| Very low-risk favorable histology Wilms’ tumor: <2 years, stage I FH, tumor weight <550 g | Nephrectomy without adjuvant therapy, only if central pathology review and lymph node sampling has been performed |
| Low-risk favorable histology Wilms’ tumor: | Nephrectomy, no RT, regimen EE4A |
| ≥2 years, stage I FH, tumor weight ≥550 g, stage II FH without LOH of 1p and 16q | |
| Standard risk favorable histology Wilms’ tumor: | |
| Stage I and II FH with LOH of 1p and 16q | Nephrectomy, regimen DD4A |
| Stage III FH without LOH of 1p and 16q | Nephrectomy, RT, regimen DD4A |
| High-risk favorable histology Wilms’ tumor: | |
| Stage III and IV FH with LOH of 1p and 16q, stage IV FH slow/incomplete responders | Nephrectomy, RT, regimen M, whole-lung irradiation |
| Stage IV FH: complete resolution of lung metastases at week 6 with regimen DD4A (rapid early responders) | Nephrectomy, RT, regimen DD4A. No whole-lung irradiation |
| Stage I-III focal anaplasia | Nephrectomy, RT, regimen DD4A |
| Stage I diffuse anaplasia | |
| Stage IV focal anaplasia | Nephrectomy, RT, regimen UH1 |
| Stage II-IV diffuse anaplasia | |
| Stage IV CCSK | |
| Stage I-IV RTK | |
| Stage I-III CCSK | Nephrectomy, RT, regimen I |
CCSK, clear cell sarcoma of kidney; FH, favorable histology; LOH, loss of heterozygosity; RTK, rhabdoid tumor of kidney.
Regimens: DD4A: vincristine/dactinomycin/doxorubicin; EE4A: vincristine/dactinomycin; I: vincristine/doxorubicin/cyclophosphamide, cyclophosphamide/etoposide; M: vincristine/dactinomycin/doxorubicin, cyclophosphamide/etoposide; UH1: cyclophosphamide/carboplatin/etoposide, vincristine/doxorubicin/cyclophosphamide.
Whole-Lung Irradiation
In children with lung metastases detected on chest radiographs, whole-lung irradiation (WLI) results in high cure rates. In NWTS-3, the 4-year relapse-free survival and overall survival were 72% and 78%, respectively, in children with favorable histology Wilms’ tumor and lung metastases.44 These results are superior to the survival rates reported by investigators from the United Kingdom Children’s Cancer Study Group (UKCCSG) study in which all patients did not receive WLI.45 In children with pulmonary metastases visible on CT but not chest radiographs, the role of WLI is unclear. In such patients treated on NWTS-3 and NWTS-4, the 4-year, event-free survival with WLI was 89% compared with 80% with chemotherapy alone, a difference that was not statistically significant.46 The current COG protocol recommends WLI based on the response of pulmonary metastatic lesions to one course of chemotherapy with vincristine, dactinomycin, and doxorubicin. Central radiology review of chest CT will be performed before and at week 6 after chemotherapy. WLI will be omitted only in patients with favorable histology tumors with no LOH at 1p and 16q and those who have achieved a complete response to chemotherapy on chest CT central radiology review at week 6. All other children will receive WLI (12 Gy).
The current RT recommendations of the COG Renal Tumors Committee are shown in Table 68-4. There are several differences in the current COG recommendations compared with those from NWTS-5: (1) patients with local tumor spillage are upstaged to stage III and will receive flank RT; (2) patients with stage I focal and diffuse anaplastic tumors will receive flank RT; (3) the radiation dose for children with stage III diffuse anaplasia and stage I to III RTK will be increased to 19.8 Gy; and (4), in an effort to study if irradiation could be omitted in stage I CCSK, patients who have undergone nodal sampling and central pathology review will not receive flank RT.
Chemotherapy
Chemotherapy and Combined-Modality Treatment
Single-agent chemotherapy has been used for Wilms’ tumor since the 1950s, when both dactinomycin and vincristine were found to be active. The interplay between relatively more aggressive chemotherapy and locoregional irradiation was tested in the initial NWTS studies. NWTS-1 showed that RT conferred no advantage in children younger than age 24 months with group I tumors who also received 15 months of dactinomycin.47 NWTS-2 showed that RT could be avoided in all children with group I Wilms’ tumor if they received both vincristine and dactinomycin.48 The results of NWTS-3 showed that stage II tumors required only vincristine and dactinomycin without any RT. NWTS-3 also demonstrated an interaction between chemotherapy and RT: 10.8 Gy with three drugs (vincristine, dactinomycin, doxorubicin) was equivalent to 20 Gy with two drugs (vincristine, dactinomycin).44 In NWTS-4, two questions were posed concerning duration of chemotherapy (6 vs. 15 months) and the delivery of dactinomycin and doxorubicin (pulse-intensive, single-dose vs. 5-day course of dactinomycin or 3-day course of doxorubicin). Results showed no advantage to prolonged therapy or divided doses, which actually proved to be more toxic; the consolidated 2-year, relapse-free rate and survival results were 90% and 97%, respectively, for the low- and high-risk patients (see Table 68-3). Thus the shorter courses of the pulse-intensive regimens are now considered standard, reducing both clinic time and cost for both parents and staff.49
Anaplastic Histology
The first NWTS trial to stratify patients with anaplastic Wilms’ tumor into a distinct treatment group was NWTS-3. Both NWTS-3 and NWTS-4 called for 15 months of vincristine, dactinomycin, and doxorubicin, with randomization to receive cyclophosphamide or not. Patients with stages II to IV diffuse anaplastic tumors who were treated with cyclophosphamide had significant improvement in 4-year relapse-free survival (27% vs. 55%)42 (see Table 68-3). Patients with all stages of focal anaplastic histology or stage I diffuse anaplastic histology had excellent outcomes, regardless of treatment regimen. Based on these results, patients with anaplastic histology were treated in prospective single-arm studies in NWTS-5. Patients with stage I anaplastic tumors were treated with vincristine and dactinomycin for 18 weeks without RT. Patients with stage II to IV diffuse anaplastic histology were treated with vincristine, doxorubicin, cyclophosphamide, and etoposide for 24 weeks plus flank/abdominal RT. Among 2596 patients with Wilms tumor enrolled onto NWTS-5, 281 (10.8%) had anaplastic histology. The 4-year event-free and overall survival rates for stage I anaplastic histology tumors were 70% and 83%, respectively. The 4-year event-free and overall survival in stages II, III, and IV diffuse anaplastic tumors after immediate operative chemotherapy and RT were 83% and 82%, 65% and 67%, and 33% and 33%, respectively.43 These results form the basis for the current COG study that recommends augmentation of therapy for stage I, III, and IV anaplastic tumors. The new COG protocol will evaluate whether a treatment regimen containing cyclophosphamide/carboplatin/etoposide alternating with vincristine/doxorubicin/cyclophosphamide will improve the event-free and overall survival of patients with stage II to IV diffuse anaplasia (see Table 68-5).
Clear Cell Sarcoma of the Kidney
Data from NWTS-1 to NWTS-4 suggested that the addition of doxorubicin to vincristine and dactinomycin improved relapse-free survival rates of patients with CCSK.50,51 NWTS-3 demonstrated no improvement in outcomes of patients with CCSK with the addition of cyclophosphamide.44 In children in NWTS-4 with CCSK there was no significant difference between those patients initially randomized to standard and pulse-intensive chemotherapy with vincristine, dactinomycin, and doxorubicin. The 8-year relapse free and overall survival were 72% and 87% for pulse-intensive chemotherapy and 70% and 84% for standard chemotherapy, respectively. The second randomization was to then standard versus long-duration (additional 9 months) chemotherapy; there was no significant difference in survival between the two trial arms. The 8-year relapse-free and overall survival were 61% and 86% for the short-duration and 88% and 88% for the long-duration chemotherapy, respectively. Even though the relapse-free survival were not significantly different (p = .08), the long-duration arm resulted in a higher relapse-free survival. The overall survival of CCSK patients treated on NWTS-4 was significantly superior to that of NWTS-3 (83% vs. 67%). The pulse-intensive chemotherapy administration of dactinomycin and doxorubicin in NWTS-4 is presumed to be one of the reasons for the improvement in outcomes.52
Preliminary analysis of NWTS-5 has shown promising results with the new chemotherapy regimen (vincristine, doxorubicin, cyclophosphamide, and etoposide) and RT except for patients with stage IV disease (NWTS, unpublished data). Based on these results, the COG study will continue to treat patients with stages I to III CCSK as described for stage I. Patients with stage IV CCSK will be treated with cyclophosphamide/carboplatin/etoposide alternating with vincristine/doxorubicin/cyclophosphamide, also as just described (see Table 68-5).
Rhabdoid Tumor of the Kidney
Patients with RTK have been treated on NWTS trials with agents such as vincristine, dactinomycin, and doxorubicin, with or without cyclophosphamide. The outcomes attained with these agents have been consistently very poor44,53,54 (see Table 68-3). To try to improve on these results, NWTS-5 adopted a different treatment strategy consisting of carboplatin/etoposide alternating with cyclophosphamide. Preliminary analysis of patients treated with this regimen revealed an overall survival of 26%, not an improvement compared with previous studies, and thus the treatment arm was closed.54 The current COG protocol evaluates whether a treatment regimen containing cyclophosphamide/carboplatin/etoposide alternating with vincristine/doxorubicin/cyclophosphamide improves the event-free and overall survival in these patients.
Irradiation Techniques
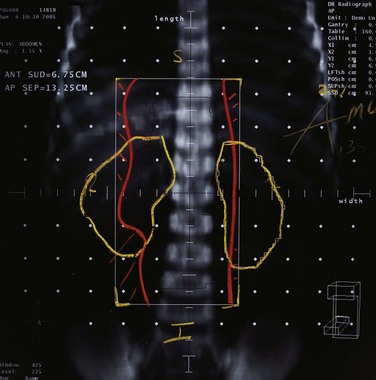
Parallel-opposed (anterior-posterior/posterior-anterior) treatment portals using 4-MV or 6-MV photons are recommended for flank, whole-abdomen, and whole-lung irradiation. The details regarding the planning techniques and setup for conventional RT are shown and described in Figures 68-3 to 68-5. If the lungs and either flank or abdomen have to be treated simultaneously, it is preferred that they be included as one radiation volume. However, if these two sites have to be treated sequentially, appropriate field gap calculations should be used for field matching, to avoid overlap, particularly over critical structures such as the liver and kidney.
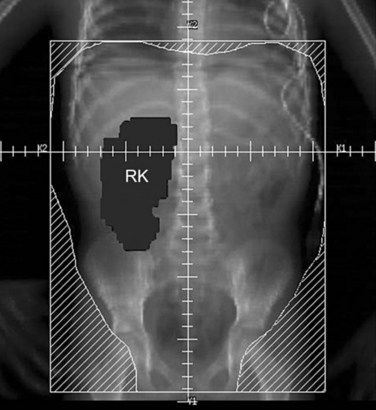
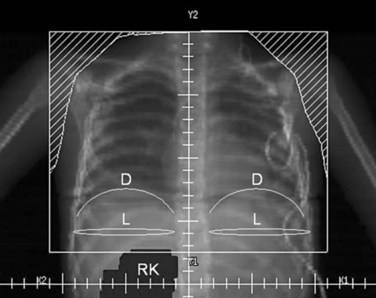
Current Clinical Recommendations
Based on the results of NWTS-5, the new COG renal tumor protocol will use LOH at both 1p and 16q in addition to tumor stage and pathologic classification for defining tumor-risk groups and assigning risk-adapted therapy (see Table 68-5). The goals of therapy in the COG study are to reduce the incidence of treatment-related morbidity in low-risk groups who have good survival rates and increase treatment intensity in high-risk tumors to improve cure rates.
Other Studies
In addition to the NWTSG, there have been randomized studies in SIOP, typically built on preoperative therapy (chemotherapy and, in defined instances, RT),55–57 and the UKCCSG.45,58
Retrieval Therapy
Children with relapsed favorable histology Wilms’ tumor have a variable prognosis, depending on the site of relapse, the time from initial diagnosis to relapse, and previous therapy. Favorable prognostic factors include no previous treatment with doxorubicin, relapse more than 12 months after diagnosis, and intra-abdominal relapse in a patient not previously treated with abdominal irradiation.59,60 Patients with relapsed or progressive disease after initial chemotherapy with vincristine and dactinomycin and no RT were treated on a specific stratum in NWTS-5, consisting of alternating courses of vincristine, doxorubicin, cyclophosphamide and etoposide/cyclophosphamide, plus surgery and RT. The 4-year event-free and overall survival was 71% and 81% for all patients, 68% and 81% for those who experienced relapse in the lung only, and 78% and 83% for those who had relapse in the operative bed with or without lung metastasis.61 Patients who had experienced relapse or whose disease progressed after initial chemotherapy that included vincristine, dactinomycin, and doxorubicin plus RT were treated with alternating courses of drug pairs (cyclophosphamide/etoposide and carboplatin/etoposide), surgery, and RT. The 4-year event-free and overall survival was 42% and 48% for all patients and 49% and 53% for those who had relapse in the lung only.62 In children who previously had not received RT, the recommended flank radiation dose is 12.6 to 18 Gy (birth to 12 months) and 21.6 Gy (≥13 months). Supplemental boost fields could be added for gross residual disease (see Table 68-4). The COG protocol recommends postoperative irradiation for all children with abdominal relapse because these tumors are generally large and infiltrative and surgical resection with negative margins is unlikely.
Late Effects
As the follow-up of successfully treated children increases, data are increasingly emerging on the late consequences of treatment. The types of late sequelae and their severity depend on the age and sex of the child, extent of surgery, chemotherapy drugs, and irradiation-related factors. The most common cause for renal failure in Wilms’ tumor patients is bilateral nephrectomy, whereas the second leading cause is irradiation-induced damage or surgical complications affecting the remaining kidney. The frequency of renal failure in bilateral Wilms’ tumor was 16.4% for NWTS-1 and NWTS-2, 9.9% for NWTS-3, and 3.8% for NWTS-4.63 The incidence of scoliosis ranges from 40% to 60% with radiation doses of 25 to 40 Gy. However, the rate of scoliosis should be low with the current doses of 10.8 Gy.64,65 The cumulative frequency of congestive heart failure among patients on NWTS-1 to NWTS-4 was 4.4% at 20 years among patients treated initially with doxorubicin and 17.4% among patients treated with doxorubicin for first or subsequent relapse. Factors significantly associated with heart failure were female sex, cumulative doxorubicin dose, lung irradiation, and left-sided abdominal irradiation.66 Women who are Wilms’ tumor survivors have significantly higher rates of adverse pregnancy outcomes, such as malposition of the fetus and premature labor, with the incidence greatest after flank irradiation to doses higher than 25 Gy. Their offspring more often are premature and of low birth weight compared with control cohorts; a trend toward an increased number of congenital malformations has been noted after flank irradiation.67,68 The cumulative 15-year risk of second malignant neoplasms was 1.6% among patients enrolled on the NWTS, after a mean follow-up of 7.5 years per patient. Higher doses of abdominal irradiation and doxorubicin increased the risk of another neoplasm.69
The standardized mortality ratio in an NWTS review was 24.3 within 5 years of diagnosis, 12.6 for the next 5 years, and more than 3.0 thereafter. The main cause of mortality within the first 5 years related to the original disease (91%). Beyond 5 years the mortality was equally related to the original disease (40%) and late effects of treatment (39%). The three most common treatment-related effects contributing to mortality were second malignant neoplasms, congestive heart failure, and end-stage renal disease. The risk of death particularly from treatment-related late effects remained elevated even 20 years after diagnosis.70
2 Rance TF. Case of fungus haematodes of the kidneys. Med Phys J. 1814;32:19.
3 Wilms M. Die Mischgeschwülste der Niere. Leipzig: Verlag von Arthur Georgi; 1899. pp 1-90
5 National Wilms’ Tumor Study Committee. Wilms’ tumor. Status report, 1990. J Clin Oncol. 1991;9:877-887.
8 Coppes MJ, Pritchard-Jones K. Principles of Wilms’ tumor biology. Urol Clin North Am. 2000;27:423-433.
10 Little MH, Williamson KA, Mannens M, et al. Evidence that WT1 mutations in Denys-Drash syndrome patients may act in a dominant negative fashion. Hum Mol Genet. 1993;2:259-264.
11 Dome JS, Coppes MJ. Recent advances in Wilms’ tumor genetics. Curr Opin Pediatr. 2002;14:5-11.
13 DeBaun MR, Niemitz EL, McNeil DE, et al. Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. Am J Hum Genet. 2002;70:604-611.
14 Weksberg R, Nishikawa J, Caluseriu O, et al. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum Mol Genet. 2001;10:2989-3000.
15 Maiti S, Alam R, Amos CI, et al. Frequent association of beta-catenin and WT1 mutations in Wilms’ tumors. Cancer Res. 2000;60:6288-6292.
19 Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms’ tumor. Results from the National Wilms’ Tumor Study. Cancer. 1978;41:1937-1948.
20 Beckwith JB. Wilms’ tumor and other renal tumors of childhood. A selective review from the National Wilms’ Tumor Study Pathology Center. Hum Pathol. 1983;14:481-492.
21 Bonadio JR, Storer B, Norkool P, et al. Anaplastic Wilms’ tumor. Clinical and pathologic studies. J Clin Oncol. 1985;3:513-520.
22 Bardeesy N, Falkoff D, Petruzzi MJ, et al. Anaplastic Wilms’ tumor, a subtype displaying poor prognosis, harbors p53 gene mutations. Nat Genet. 1994;7:91-97.
23 Faria P, Beckwith JB, Mishra K, et al. Focal versus diffuse anaplasia in Wilms’ tumor—new definitions with prognostic significance. A report from the National Wilms’ Tumor Study Group. Am J Surg Pathol. 1996;20:909-920.
26 Beckwith JB, Palmer NF. Wilms’ tumor and other renal tumors in childhood. In: Feingold M, editor. Pathology of Neoplasia in Children and Adolescents. Philadelphia: WB Saunders; 1986:313-332.
27 Marsden HB, Lawler W, Kumar PM. Bone metastasizing renal tumor of childhood. Morphological and clinical features and differences from Wilms’ tumor. Cancer. 1978;42:1922-1928.
31 National Wilms’ Tumor Study Group. NWTS-5 protocol. Seattle: NWTSG; 1995.
32 Farewell VT, D’Angio GJ, Breslow NE, et al. Retrospective validation of a new staging system for Wilms’ tumor. Cancer Clin Trials. 1981;4:168-171.
33 Beckwith JB. National Wilms’ Tumor Study. An update for pathologists. Pediatr Dev Pathol. 1998;1:79-84.
53 Weeks DA, Beckwith B, Mierau GW, et al. Rhabdoid tumor of kidney. Am J Surg Pathol. 1989;13:439-458.
1 Gurney JG, Severson RK, Davis S, et al. Incidence of cancer in children in the United States. Sex-, race-, and 1-year age-specific rates by histologic type. Cancer. 1995;75:2186-2195.
2 Rance TF. Case of fungus haematodes of the kidneys. Med Phys J. 1814;32:19.
3 Wilms M. Die Mischgeschwülste der Niere. Leipzig: Verlag von Arthur Georgi; 1899. pp 1-90
4 Gross RE, Neuhauser BD. Treatment of mixed tumors of the kidney in childhood. Pediatrics. 1950;6:843-852.
5 National Wilms’ Tumor Study Committee. Wilms’ tumor. Status report, 1990. J Clin Oncol. 1991;9:877-887.
6 Tournade MF, Com-Nougue C, Voüte PA, et al. Results of the Sixth International Society of Pediatric Oncology Wilms’ tumor trial and study. A risk-adapted therapeutic approach in Wilms’ tumor. J Clin Oncol. 1993;11:1014-1023.
7 Kalapurakal JA, Dome JS, Perlman EJ, et al. Management of Wilms’ tumor. Current practice and future goals. Lancet Oncol. 2003;5:37-46.
8 Coppes MJ, Pritchard-Jones K. Principles of Wilms’ tumor biology. Urol Clin North Am. 2000;27:423-433.
9 Coppes MJ, de Kraker J, van Dijken PJ, et al. Bilateral Wilms’ tumor. Long-term survival and some epidemiological features. J Clin Oncol. 1989;7:310-315.
10 Little MH, Williamson KA, Mannens M, et al. Evidence that WT1 mutations in Denys-Drash syndrome patients may act in a dominant negative fashion. Hum Mol Genet. 1993;2:259-264.
11 Dome JS, Coppes MJ. Recent advances in Wilms’ tumor genetics. Curr Opin Pediatr. 2002;14:5-11.
12 Koufos A, Grundy P, Morgan K, et al. Familial Wiedemann-Beckwith syndrome and a second Wilms’ tumor locus both map to 11p15.5. Am J Hum Genet. 1989;44:711-719.
13 DeBaun MR, Niemitz EL, McNeil DE, et al. Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. Am J Hum Genet. 2002;70:604-611.
14 Weksberg R, Nishikawa J, Caluseriu O, et al. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum Mol Genet. 2001;10:2989-3000.
15 Maiti S, Alam R, Amos CI, et al. Frequent association of beta-catenin and WT1 mutations in Wilms’ tumors. Cancer Res. 2000;60:6288-6292.
16 Rivera MN, Kim WJ, Wells J, et al. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science. 2007;315:642-645.
17 Major MB, Camp ND, Berndt JD, et al. Wilms’ tumor suppressor WTX negatively regulates WNT/β-catenin signaling. Science. 2007;316:1043-1046.
18 Grundy PE, Breslow NE, Li S, et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable histology Wilms’ tumor. A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 2005;23:7312-7321.
19 Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms’ tumor. Results from the National Wilms’ Tumor Study. Cancer. 1978;41:1937-1948.
20 Beckwith JB. Wilms’ tumor and other renal tumors of childhood. A selective review from the National Wilms’ Tumor Study Pathology Center. Hum Pathol. 1983;14:481-492.
21 Bonadio JR, Storer B, Norkool P, et al. Anaplastic Wilms’ tumor. Clinical and pathologic studies. J Clin Oncol. 1985;3:513-520.
22 Bardeesy N, Falkoff D, Petruzzi MJ, et al. Anaplastic Wilms’ tumor, a subtype displaying poor prognosis, harbors p53 gene mutations. Nat Genet. 1994;7:91-97.
23 Faria P, Beckwith JB, Mishra K, et al. Focal versus diffuse anaplasia in Wilms’ tumor—new definitions with prognostic significance. A report from the National Wilms’ Tumor Study Group. Am J Surg Pathol. 1996;20:909-920.
24 Green DM, Breslow NE, Beckwith JB, et al. Treatment with nephrectomy only for small, stage I/favorable histology Wilms’ tumor. A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 2001;19:3719-3721.
25 Beckwith JB. Precursor lesions of Wilms’ tumor. Clinical and biological implications. Med Pediatr Oncol. 1993;21:158-168.
26 Beckwith JB, Palmer NF. Wilms’ tumor and other renal tumors in childhood. In: Feingold M, editor. Pathology of Neoplasia in Children and Adolescents. Philadelphia: WB Saunders; 1986:313-332.
27 Marsden HB, Lawler W, Kumar PM. Bone metastasizing renal tumor of childhood. Morphological and clinical features and differences from Wilms’ tumor. Cancer. 1978;42:1922-1928.
28 Owens CM, Veys PA, Pritchard J, et al. Role of chest computed tomography at diagnosis in the management of Wilms’ tumor. A study by the United Kingdom Children’s Cancer Study Group. J Clin Oncol. 2002;20:2768-2773.
29 Green DM. Use of chest computed tomography for staging and treatment of Wilms’ tumor in children. J Clin Oncol. 2002;20:2763-2764.
30 Gylys-Morin V, Hoffer FA, Kozakewich H, et al. Wilms’ tumor and nephroblastomatosis. Imaging characteristics at gadolinium-enhanced MR imaging. Radiology. 1993;188:517-521.
31 National Wilms’ Tumor Study Group. NWTS-5 protocol. Seattle: NWTSG; 1995.
32 Farewell VT, D’Angio GJ, Breslow NE, et al. Retrospective validation of a new staging system for Wilms’ tumor. Cancer Clin Trials. 1981;4:168-171.
33 Beckwith JB. National Wilms’ Tumor Study. An update for pathologists. Pediatr Dev Pathol. 1998;1:79-84.
34 Weeks DA, Beckwith JB, Luckey DW. Relapse-associated variables in stage I favorable histology Wilms’ tumor. A report of the National Wilms’ Tumor Study. Cancer. 1987;60:1204-1212.
35 Kalapurakal JA, Li SM, Breslow NE, et al. Intraoperative spillage of favorable histology Wilms tumor cells. Influence of irradiation and chemotherapy on abdominal recurrence. A report from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys. 2010;76:201-206.
36 Blakely ML, Ritchey ML. Controversies in the management of Wilms’ tumor. Semin Pediatr Surg. 2001;10:127-131.
37 Shamberger RC, Guthrie KA, Ritchey ML, et al. Surgery-related factors and local recurrence of Wilms’ tumor in National Wilms’ Tumor Study-4. Ann Surg. 1999;229:292-297.
38 D’Angio GJ, Tefft M, Breslow NE, et al. Radiation therapy of Wilms’ tumor. Results according to dose, field, post-operative timing, and histology. Int J Radiat Oncol Biol Phys. 1978;4:769-780.
39 Thomas PRM, Tefft M, Farewell VT, et al. Abdominal relapses in irradiated Second National Wilms’ Tumor Study patients. J Clin Oncol. 1984;2:1098-1101.
40 Thomas PRM, Tefft M, Compaan PJ, et al. Results of two radiotherapy randomizations in the Third National Wilms’ Tumor Study (NWTS-3). Cancer. 1991;68:1703-1707.
41 Kalapurakal JA, Li SM, Breslow NE, et al. Influence of radiation therapy delay on abdominal tumor recurrence in patients with favorable histology Wilms’ tumor treated on NWTS-3 and NWTS-4. A report from the National Wilms’ Tumor Study Group. Int J Radiat Oncol Biol Phys. 2003;57:495-499.
42 Green DM, Breslow NE, Beckwith NE, et al. Treatment of children with stages II to IV anaplastic Wilms’ tumor. A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1994;12:2126-2131.
43 Dome JS, Cotton CA, Perlman EJ, et al. Treatment of anaplastic histology Wilms’ tumor. Results from the fifth National Wilms’ Tumor Study. J Clin Oncol. 2006;24:2352-2358.
44 D’Angio GJ, Breslow N, Beckwith JB, et al. Treatment of Wilms’ tumor. Results of the Third National Wilms’ Tumor Study. Cancer. 1989;64:349-360.
45 Pritchard J, Imeson J, Barnes J, et al. Results of the United Kingdom Children’s Cancer Study Group First Wilms’ Tumor Study. J Clin Oncol. 1995;13:124-133.
46 Meisel JA, Guthrie KA, Breslow NE, et al. Significance and management of computed tomography detected pulmonary nodules: A report from the National Wilms’ Tumor Study Group. Int J Radiat Oncol Biol Phys. 1999;44:579-585.
47 D’Angio GJ, Evans AE, Breslow N, et al. The treatment of Wilms’ tumor. Results of the National Wilms’ Tumor Study. Cancer. 1976;38:633-646.
48 D’Angio GJ, Evans A, Breslow N, et al. The treatment of Wilms tumor. Results of the Second National Wilms’ Tumor Study. Cancer. 1981;47:2302-2311.
49 Green DM, Breslow NE, Beckwith JB, et al. A comparison between single dose and divided dose administration of dactinomycin and doxorubicin. J Clin Oncol. 1998;16:237-245.
50 Green DM, Breslow NE, Beckwith JB, et al. Treatment of children with clear-cell sarcoma of the kidney. A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1994;12:2132-2137.
51 Argani P, Perlman EJ, Breslow NE, et al. Clear cell sarcoma of the kidney. A review of 351 cases from the National Wilms’ Tumor Study Group Pathology Center. Am J Surg Pathol. 2000;24:4-18.
52 Seibel N, Li S, Breslow NE, et al. Effect of duration of treatment on treatment outcome for patients with clear-cell sarcoma of the kidney. A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 2004;22:468-473.
53 Weeks DA, Beckwith B, Mierau GW, et al. Rhabdoid tumor of kidney. Am J Surg Pathol. 1989;13:439-458.
54 Tomlinson GE, Breslow NE, Dome J, et al. Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study. Age at diagnosis as a prognostic factor. J Clin Oncol. 2005;23:7641-7645.
55 Graf N, Tournade MF, de Kraker J. The role of preoperative chemotherapy in the management of Wilms’ tumor. The SIOP studies. Urol Clin North Am. 2000;27:443-454.
56 Tournade MF, Com-Nogue C, de Kraker J, et al. Optimal duration of preoperative therapy in unilateral and nonmetastatic Wilms’ tumor in children older than 6 months. Results of the Ninth International Society of Pediatric Oncology Wilms’ Tumor Trial and Study. J Clin Oncol. 2001;19:488-500.
57 De Kraker J, Graf N, van Tinteren H, et al. Reduction of postoperative chemotherapy in children with stage I intermediate risk and anaplastic Wilms’ tumor (SIOP 93-01). A randomized trial. Lancet. 2004;364:1229-1235.
58 Mitchell C, Jones PM, Kelsey A, et al. The treatment of Wilms’ tumor. Results of the United Kingdom Children’s Cancer Study Group (UKCCSG) second Wilms’ tumor study. Br J Cancer. 2000;83:602-608.
59 Grundy P, Breslow NE, Green DM, et al. Prognostic factors of children with recurrent Wilms’ tumor. Results from the second and third National Wilms’ Tumor Study. J Clin Oncol. 1989;7:638-647.
60 Tannous R, Giller G, Holmes E, et al. Intensive therapy for high risk (HR) relapsed Wilms’ tumor (WT). A CCG-4921/POG-9445 study report. Proc Am Soc Clin Oncol. 2000;19:588a.
61 Green DM, Cotton CA, Malogolowkin M, et al. Treatment of Wilms’ tumor relapsing after initial treatment with vincristine and Actinomycin D. A report from the National Wilms’ Tumor Study Group. Pediatr Blood Cancer. 2007;48:493-499.
62 Malogolowkin M, Cotton CA, Green DM, et al. Treatment of Wilms’ tumor relapsing after initial treatment with vincristine, actinomycin D and doxorubicin. A report from the National Wilms’ Tumor Study Group. Pediatr Blood Cancer. 2008;50:236-241.
63 Ritchey ML, Green DM, Thomas PRM, et al. Renal failure in Wilms’ tumor patients. A report from the National Wilms’ Tumor Study Group. Med Pediatr Oncol. 1996;26:75-80.
64 Paulino AC, Wen BC, Brown CK, et al. Late effects in children treated with radiation therapy for Wilms’ tumor. Int J Radiat Oncol Biol Phys. 2000;19:488-500.
65 Thomas PRM, Griffith KD, Fineberg BB, et al. Late effects of treatment for Wilms’ tumor. Int J Radiat Oncol Biol Phys. 1983;9:651-657.
66 Green DM, Grigoriev YA, Nan B, et al. Congestive heart failure after treatment for Wilms’ tumor. A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 2001;19:1926-1934.
67 Green DM, Peabody EM, Nan B, et al. Pregnancy outcome after treatment for Wilms’ tumor. A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 2002;20:2506-2513.
68 Kalapurakal JA, Peterson S, Peabody EM, et al. Pregnancy outcomes after abdominal irradiation that included or excluded the pelvis in childhood Wilms’ tumor survivors. A report from the National Wilms’ Tumor Study. Int J Radiat Oncol Biol Phys. 2004;58:1364-1368.
69 Breslow NE, Takashima JR, Whitton JA, et al. Second malignant neoplasms following treatment for Wilms’ tumor. A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1995;13:1851-1859.
70 Cotton CA, Peterson S, Norkool PA, et al. Early and late mortality after diagnosis of Wilms’ tumor. J Clin Oncol. 2009;27:1304-1309.