[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 24
Water metabolism
1. What is the water composition of the human body?
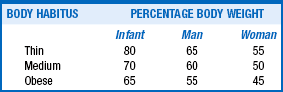
Water composition of the body depends on age, sex, muscle mass, body habitus, and fat content. Various body tissues have the following water percentages: lungs, heart, and kidneys (80%); skeletal muscle and brain (75%); skin and liver (70%); bone (20%); and adipose tissue (10%). Clearly, people with more muscle than fat have more water. Generally, thin people have less fat and more water. Men are 60% water and women 50% water by weight. Older people have more fat and less muscle. The average man and woman older than 60 years are made up of 50% and 45% water, respectively (see Table 24-1). Most discussions of total body water (TBW) consider a man who is 60% water, weighs 70 kg, and is 69 inches (175 cm) tall.
2. Where is water located within the body?
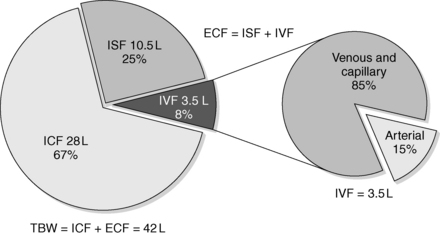
TBW comprises water located inside the cells (intracellular fluid [ICF]) and outside the cells (extracellular fluid [ECF]). TBW is 60% of body weight—40% ICF (⅔) and 20% ECF (⅓). Of the ECF, approximately ¾ is interstitial fluid (ISF) and ¼ is intravascular fluid (IVF). IVF is a major component of the total blood volume necessary to maintain effective vascular pressure. ISF is 15% of body weight, and IVF is 5% of body weight. In a 70-kg man, TBW = 42 L, ICF water = 28 L, and ECF water = 14 L. ISF is 10.5 L and IVF (plasma) is 3.5 L. Tight regulation of the relatively small volume of IVF maintains blood pressure and avoids symptomatic hypovolemia and congestive heart failure. Normal plasma is 93% water and 7% proteins and lipids. The arterial volume is only 15% of IVF. Although arterial volume is small, its integrity is most important for maintaining the effective circulation and preventing abnormalities of water balance (Fig. 24-1).
3. What is transcellular water (TCW)?
TCW is water formed by cellular transport activities and is located in various ducts and spaces throughout the body. This water includes cerebrospinal fluid (CSF) and aqueous humor; secretions in the sweat, salivary, and lacrimal glands; secretions in pancreas, liver, biliary, gastrointestinal, and respiratory tracts; and peritoneal, pleural, and synovial fluids.
4. Explain the significance of TCW.
TCW carries secretions to specific sites for enzymatic and lubricant activity and is normally quite small—1.5% of body weight. In disease states, excess or deficiency of TCW can cause dysfunction. Marked excess TCW formation—third spacing—may decrease effective circulating volume (ECV), stimulate antidiuretic hormone (ADH) and aldosterone release, increase retention of salt and water, and cause edema and hyponatremia.
5. What controls distribution of body water?
With few exceptions (e.g., ascending loop of Henle [LOH] and distal nephron), water moves freely across cell membranes, depending on tonicity. Because tonicity depends on impermeable solutes, such as sodium (Na), disorders of water metabolism are reflected by changes in solute concentrations. In addition to changes in water distribution, changes in TBW, blood volume, and ECV affect overall water balance. A thorough understanding of disorders of water metabolism requires a clear understanding of changes in plasma Na concentration (PNa), plasma osmolality (Posm), and ECV.
 KEY POINTS 1: WATER METABOLISM
KEY POINTS 1: WATER METABOLISM
1. Changes in body water or distribution are usually reflected by changes in plasma sodium concentration (PNa) and may occur in states of low, normal, or high total body sodium. Low PNa reflects high TBW, and high PNa reflects low total body water (TBW).
2. Water always moves across cell membranes from lower to higher osmolality. This movement is determined by the concentration of effective osmotic solute in the intracellular or extracellular fluid and is responsible for the neurologic symptoms and signs associated with changes in PNa.
3. Hyponatremia may occur with low, normal, or high osmolality, whereas hypernatremia is always associated with hyperosmolality and hypertonicity.
4. Water content of the body is a balance of input and output.
5. Water balance is controlled by thirst, access to water, solute intake, antidiuretic hormone (ADH), cortisol, aldosterone, natriuretic peptides, baroreceptor sensors, ADH receptors, renal water channels called aquaporins, level of kidney function, and drugs.
6. What is effective circulating volume (ECV)?
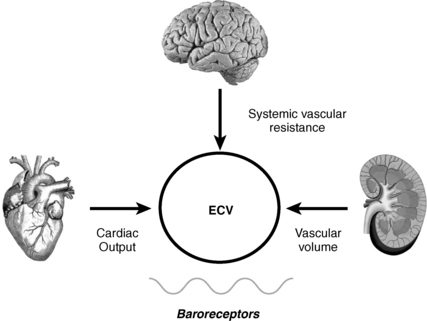
ECV is the arterial volume required to maintain normal baroreceptor pressure that is appropriate for a given level of vascular resistance. ECV is also called effective arterial blood volume (EABV). By inducing changes in baroreceptor tone, alterations in ECV have a major impact on water balance. Low ECV causes renal salt and water retention, whereas high ECV causes renal salt and water loss. Depending on the patient’s water intake, these changes may produce significant hyponatremia. Maintaining normal ECV preserves circulatory homeostasis.
7. How do baroreceptors affect ECV?
Baroreceptors are the major sensors of changes in ECV (Fig. 24-2). However, their main role is to maintain normal pressure (not volume) at the level of the baroreceptor sensors located primarily in the carotid sinus, aortic arch, atria, pulmonary veins, and afferent renal arterioles. These anatomic locations are important because perfusion to these areas affects the three main effectors of circulatory homeostasis and ECV: brain, heart, and kidneys.
8. How does vascular pressure, as sensed by the baroreceptors, relate to ECV and hyponatremia?
Baroreceptors normally maintain tonic inhibition of vasoconstrictor nerves and natriuretic hormone release but tonic stimulation of vagal cardiac nerves. A drop in ECV decreases effective vascular pressure (EVP), baroreceptor tone, tonic inhibition, and tonic stimulation. This causes vasoconstriction; increases heart rate; and increases renin, aldosterone, angiotensin II, and ADH secretion. It decreases atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) from brain and ventricles, and urodilatin (kidney). These alterations enhance renal Na and water retention. If the patient receives unlimited water, these changes may lead to hyponatremia. Hyponatremia cannot develop unless the patient retains more water than is excreted. Decreased ECV/EVP predisposes to water retention, but the patient must receive free water for hyponatremia to develop. The venous system, through atrial stretch receptors, has similar effects but responds to changes in ECV earlier than the arterial system.
9. Define osmolality and tonicity, and outline their effects on water movement.
Osmolality is the concentration of a substance in 1 L of water divided by its molecular weight. Tonicity is effective osmolality—the osmotic pressure caused by dissolved particles restricted to one side of the cell membrane. Because Na and glucose are partially restricted to the ECF, they are effective osmols and account for normal tonicity. Mannitol, sorbitol, glycerol, and glycine are also effective osmols. Urea freely crosses cell membranes and distributes evenly in TBW, and therefore it changes osmolality but not tonicity. Thus, except during early and rapid solute and water changes, urea is an ineffective osmol. Ethanol and methanol are other ineffective osmols. Water always moves across cell membranes from lower osmolality to higher osmolality until osmolality on the two sides is equal. At equilibrium, the following is always true:
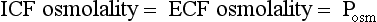
10. What formulas are useful in evaluating osmolality and tonicity?
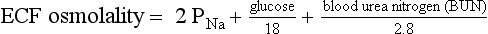
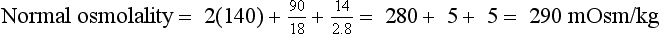
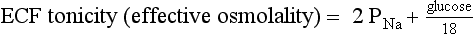
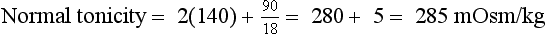
The normal range for Posm, 275 to 295 mOsm/kg, varies with the normal ranges for plasma Na, urea, and glucose. Correction factors for other effective solutes (osmols) are mannitol/18, sorbitol/18, and glycerol/9. Correction factors for other ineffective solutes (osmols) are ethanol/4.6 and methanol/3.2.
11. How does PNa relate to TBW, osmolality, and tonicity?
The following formulas are useful in understanding the relationship of PNa, plasma potassium (PK), total body sodium and potassium [Na+ + K+ ], and TBW. [Na+ + K+] estimates total body solute:
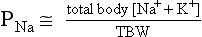
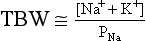
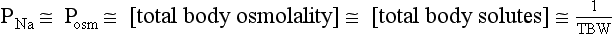
Thus PNa is proportional to [Na+ + K+] and inversely proportional to TBW. An increase or decrease in total plasma Na particles can proportionately change the PNa. However, in clinical medicine, changes in PNa usually reflect changes in plasma water. When PNa is high, plasma water is low. When PNa is low, plasma water is high. Low PNa may occur with low, normal, or high osmolality, whereas high PNa is always associated with hyperosmolality and hypertonicity.
12. How does PK relate to PNa and TBW?
Although 98% of K+ is intracellular, a K+ infusion increases PNa. This occurs as follows. In hypokalemia, infused K+ enters cells. To preserve electroneutrality, Na+ leaves or chloride (Cl−) enters cells. ECF water follows K+ and Cl− into cells because of increased ICF osmolality. Both mechanisms increase PNa. Hypokalemic patients infused with equal amounts of KCl or NaCl have equal increases in PNa. Thus addition of KCl to isotonic saline makes hypertonic saline, and infusion of saline with KCl may correct hyponatremia too rapidly (see questions 36 and 44).
13. Describe the input and output of water.
TBW is a balance of input (including endogenous production) and output. In an average adult, input approximates 1600 mL (liquids), 700 mL (foods), and 200 mL (metabolic oxidation of carbohydrate and fat) for a total of 2500 mL/day. Average water losses are 1500 mL (kidneys), 500 mL (skin [400 mL evaporation and 100 mL perspiration]), 300 mL (lung—respiration), and 200 mL from the gastrointestinal tract (stool) for a total of 2500 mL/day. Large losses of water (increased output) occur with excessive sweating, respiration (exercise), burns, diarrhea, vomiting, and diuresis. Decreased water input occurs when defects in thirst and altered mental or physical function (especially in the elderly) prevent access to water.
14. What are the normal limits of urine output?
Water intake and osmotic products of metabolism determine the usual daily output of urine. On a normal diet, a normal adult must excrete 800 to 1000 mOsm of solute per day. The range of normal renal concentrating function is 50 to 1200 mOsm/kg. On this basis, the obligate water excretion varies from 0.8 to 20 L/day. The calculations are as follows:
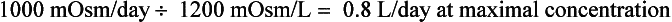
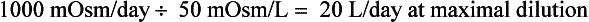
Note that higher solute loads (e.g., dietary) require more water excretion. For example, body builders consuming high-protein and high-carbohydrate diets with 1400 mOsm solute/day require a urine output of (1400/1200) to (1400/50) or 1.2 to 28 L/day. Alternatively, a low solute intake (starvation) with high water intake predisposes to water retention and water intoxication. This combination exists in binge beer drinkers, in whom the solute load may be only 300 mOsm/day. Low solute intake may also occur in starvation and in an elderly person on a “tea and toast diet.” The range of urine output would drop to (300/1200) – (300/50) or 0.25 to 6 L/day in such patients.
15. What are the main factors controlling water metabolism?
Thirst, hormonal, and renal mechanisms are tightly integrated for control of water metabolism. This integration is strongly influenced by nervous system and baroreceptor control (see Fig. 24-2).
16. What are the stimuli of thirst?
Osmoreceptors in the organum vasculosum of the anterior hypothalamus control thirst. Increasing plasma tonicity stimulates thirst at a threshold about 5 mOsm/kg higher than the value that stimulates ADH release. However, oropharyngeal receptors are also important in thirst regulation. A dry mouth increases thirst. Drinking and swallowing water decrease thirst even without changing Posm. Volume depletion changes afferent baroreceptor input and increases angiotensin II—both changes increase thirst. An unusual idiosyncratic effect of angiotensin-converting enzyme (ACE) inhibitors causes central polydipsia, increased ADH release, and propensity to hyponatremia.
17. What hormonal mechanisms are involved in control of body water?
Although natriuretic peptides, aldosterone, angiotensin II, prostaglandins, and neurohumoral changes affect renal water retention and excretion, ADH is most important. ADH is also called arginine vasopressin (AVP). Supraoptic and paraventricular nuclei in the hypothalamus secrete ADH in response to increased osmolality and decreased volume. ADH attaches to vasopressin 2 receptors (V2-Rs) on the basolateral membrane of renal collecting tubular cells. This activates cyclic adenosine monophosphate (cAMP) and protein kinase A, causing intracellular water channels called aquaporins (AQPs) to insert into the luminal membrane. Water moves down osmotic gradients from tubular lumen through AQP channels into the cell and interstitium. At least seven AQP isoforms (AQP1-4, AQP6-8) are present in the kidney. AQP1 is constitutively expressed in the proximal tubule and descending loop of Henle and is important for isotonic fluid reabsorption and water conservation. The collecting duct has high concentrations of AQP2 that serve as the major target for ADH-mediated water reabsorption. Abnormalities of the V2-R cause most cases of nephrogenic diabetes insipidus (DI), but some are caused by abnormalities of AQP2. Increased AQP2 may cause water retention in conditions such as pregnancy and congestive heart failure. Twenty percent of ADH receptors in the collecting tubular cells are vasopressin 1 receptors (V1-Rs). ADH activates V1-Rs only at very high levels. This increases prostaglandin E2 and prostacyclin, which opposes the antidiuretic effects of excessive ADH.
18. What are the major conditions that influence ADH secretion?
ADH functions to maintain osmotic and volume homeostasis. Secretion starts at an osmotic threshold of 280 mOsm/kg and increases proportionately to further rises in tonicity. Maximum diuresis (urine dilution) occurs at ADH levels of 0.5 pmol/L, and maximum reabsorption (urine concentration) occurs at ADH levels of 3 to 4 pmol/L. A 1% to 2% increase in osmolality stimulates ADH secretion, whereas an 8% to 10% drop in vascular volume is required for the same effect. Through action on baroreceptors, increased ECV raises the osmotic threshold for ADH secretion, and decreased ECV lowers this threshold. Severe volume depletion and hypotension may completely override the hypo-osmotic inhibition of ADH secretion. This finding has been called the “law of circulating volume.” In severe volume depletion and hypotension, ADH secretion continues despite low osmolality, thereby worsening the hyponatremia. Nausea, pain, and stress (as seen postoperatively) are potent stimuli of ADH release and may cause lifethreatening hyponatremia if hypotonic fluid is given. This is particularly true if patients with these symptoms also receive hypotonic fluid and drugs that potentiate the release or action of ADH.
19. What are the major causes of ADH secretion?
Major causes of ADH secretion include hyperosmolality, hypovolemia, nausea, pain, stress, human chorionic gonadotropin as in pregnancy (reset osmostat), hypoglycemia, corticotropin-releasing hormone (CRH), central nervous system (CNS) infections, CNS tumors, vascular catastrophes (thrombosis, hemorrhage), and ectopic ADH of malignancy (carcinomas of lung [primarily small cell], duodenum, pancreas, ureter, bladder, and prostate, and lymphoma). ADH secretion may be increased by any major pulmonary disorder, including pneumonia, tuberculosis, asthma, atelectasis, cystic fibrosis, positive pressure ventilation, and adult respiratory distress syndrome. Human immunodeficiency virus (HIV) infection may have the multifactorial role of causing CNS dysfunction, pulmonary disease, and malignancy. Excessive exogenous ADH or desmopressin acetate (DDAVP) in patients with DI directly increases ADH effect. Oxytocin also has significant ADH activity in the large dosages used to induce labor. Other drugs that affect ADH secretion and action are listed in Table 24-2.
TABLE 24-2.
DRUGS THAT AFFECT ANTIDIURETIC HORMONE (ADH) SECRETION AND ACTION*
| Increase ADH secretion | Antidepressants Amitriptyline Protriptyline Desipramine Fluoxetine Selective serotonin reuptake inhibitors Duloxetine Antipsychotics* Fluphenazine Haloperidol Phenothiazines Butyrophenones Monoamine oxidase inhibitors Ecstasy Nicotine Bromocriptine Carbamazepine Chlorpropamide Clofibrate Cyclophosphamide Ifosfamide Morphine Nicotine Thioridazine Vincristine Angiotensin-converting enzyme inhibitors Amiodarone Methyldopa |
| Increase ADH effect | Acetaminophen Carbamazepine Chlorpropamide Cyclophosphamide Nonsteroidal anti-inflammatory drugs Tolbutamide |
| Decrease ADH secretion | EthanolPhenytoin |
| Decrease ADH effect | Demeclocycline Lithium Acetohexamide Tolazamide Glyburide Methoxyflurane Propoxyphene Colchicine Amphotericin Vinblastine Prostaglandin E2 Prostacyclin |
*Because psychosis itself may cause the syndrome of inappropriate secretion of ADH (SIADH), one must question the true ADH-stimulatory effect of the antipsychotic drugs. Changes in ADH secretion may be direct or indirect.
 KEY POINTS 2: SYNDROMES AND TREATMENT OF WATER DYSFUNCTION
KEY POINTS 2: SYNDROMES AND TREATMENT OF WATER DYSFUNCTION
1. Clinical syndromes of water dysfunction include syndrome of inappropriate secretion of antidiuretic hormone, diabetes insipidus, and changes in effective circulating volume that can cause marked retention of salt and water, pulmonary and peripheral edema, and severe neurologic dysfunction.
2. Effective correction of water problems requires correcting abnormalities of PNa and a clear understanding of changes in plasma and urine osmolality, urine sodium and potassium, and effective circulating volume (ECV). Additionally, a thorough assessment of patient volume and neurologic symptoms is essential.
3. If neurologic symptoms occur rapidly or are severe, correction of PNa toward normal should be rapid; if symptoms are absent, there is no urgency, and PNa correction should occur more slowly.
4. Depending on the water disturbance, treatment includes water restriction or administration; hypertonic, isotonic, or hypotonic saline; sodium; diuretics; antidiuretic hormone; and aquaretics or other medications.
20. How does the kidney handle salt and water?
To control excess or deficient water intake, there must be an adequate glomerular filtration rate (GFR) and delivery of filtrate to the LOH and distal nephron. Solute is separated from water in the ascending limb of the LOH, distal convoluted tubule (DCT), and cortical connecting segment; normal action of ADH allows controlled reabsorption of water in the cortical and medullary collecting tubules. The proximal convoluted tubule reabsorbs 65% and the descending limb of the LOH 25% of filtered solute and water isotonically. The ascending limb is impermeable to water but removes solute, resulting in dilution of the luminal filtrate, concentration of the interstitium (important for ADH action), and delivery of 10% of the filtrate to the cortical collecting tubules with an osmolality of 100 mOsm/kg. In the absence of ADH, this fluid ( 18 L/day) would be lost in the urine and cause marked dehydration. In the presence of ADH, the collecting duct becomes permeable to water and reabsorbs all but 1% of the filtrate. Thus the final urine volume is only 1.5 to 2.0 L/day. Because normal GFR is 125 mL/min, the normal kidneys filter 180 L of plasma each day and reabsorb 99%. In normal adults, 99% of all Na and H2O filtered is reabsorbed.
18 L/day) would be lost in the urine and cause marked dehydration. In the presence of ADH, the collecting duct becomes permeable to water and reabsorbs all but 1% of the filtrate. Thus the final urine volume is only 1.5 to 2.0 L/day. Because normal GFR is 125 mL/min, the normal kidneys filter 180 L of plasma each day and reabsorb 99%. In normal adults, 99% of all Na and H2O filtered is reabsorbed.
21. What are the causes and consequences of decreased renal water excretion?
Any reduction in water excretion predisposes to hyponatremia and hypo-osmolality. Conditions that impair GFR, delivery of tubular fluid to the distal nephron, or the ability of the distal nephron to separate solute from water, or that increase the permeability of the collecting tubule to water impair water excretion. Such conditions include renal failure, decreased ECV, diuretics (thiazides and loop), and excessive ADH or ADH action.
22. How do hypothyroidism and adrenal insufficiency cause hyponatremia?
Hypothyroidism and adrenal insufficiency reduce cardiac output and thereby decrease ECV and increase ADH release. A hypothyroidism-associated decrease in ECV reduces renal blood flow, glomerular filtration, and maximal solute-free water excretion. Failure to dilute the urine maximally results from nonosmotic ADH release and increased ADH-mediated AQP2 receptors and action. The main effect of glucocorticoid deficiency is altered systemic hemodynamics, and not salt and water loss. Low cortisol impairs cardiac output and the systemic vascular responses to catecholamines, reducing both blood pressure and ECV. The resulting drop in absolute and effective vascular filling pressure reduces stretch on the arterial baroreceptors and thereby decreases tonic vagal and glossopharyngeal inhibition of ADH release. These baroreceptor changes override the hypo-osmotic inhibition of ADH release, and consequently, ADH secretion increases. The decreased ECV also lowers GFR, thereby reducing delivery of filtrate to the distal nephron and enhancing proximal tubular water reabsorption. Normally, CRH and ADH are co-secreted from the same neurons in the paraventricular nuclei of the hypothalamus, and both hormones work synergistically to release adrenocorticotropic hormone (ACTH) from the anterior pituitary—ADH via the vasopressin V1b receptor. Cortisol feeds back negatively at the hypothalamus and pituitary to inhibit the release of both CRH and ADH. Cortisol deficiency decreases this negative feedback and increases ADH release to further enhance water reabsorption.
Unlike secondary adrenal insufficiency, mineralocorticoid deficiency associated with primary adrenal insufficiency causes a hyperkalemic non–anion gap metabolic acidosis. This is due to retention of K+ and H+ that are normally excreted under aldosterone influence. The aldosterone deficiency also causes renal NaCl loss and associated volume (ECF) depletion. The resulting low ECV stimulates ADH release. There is also upregulation of collecting duct AQP2 and AQP3, which enhances ADH action. The combination of increased ADH secretion and augmented ADH responsiveness promotes the development of hyponatremia. A high-sodium diet compensates for the mineralocorticoid deficiency and improves the hyponatremia. Although hyponatremia may occur with both primary and secondary adrenal insufficiency, it occurs more commonly in primary adrenal insufficiency. This fact emphasizes the importance of aldosterone deficiency in renal salt wasting, volume depletion, and ADH secretion. All of these events combined with continued water intake synergistically contribute to hyponatremia.
23. What PNa concentrations are causes for concern?
The seriousness of hyponatremia or hypernatremia depends on the rapidity of development. Acute changes in PNa (within 48 hours) are always of more concern. Normal PNa ranges from 136 to 145 mEq/L. Patients with a PNa value of 115 or 165 mEq/L may not show any clinical features if the problem develops over several days to weeks. However, both conditions may produce major neurologic dysfunction if they develop over hours to days. As a rule, however, Na concentrations of 120 to 155 mEq/L are not usually associated with symptoms. PNa values outside these limits and occasionally rapidly developing disturbances within these limits may be of major concern. With appropriate care, patients have been reported to survive with PNa as low as 85 mEq/L and as high as 274 mEq/L without permanent sequelae. Although elderly individuals with chronic PNa levels of 120 to 125 mEq/L may appear asymptomatic, they may have associated gait disturbances and may be at increased risk for falls and fractures. Thus treatment of this mild hyponatremia may benefit this group.
24. What causes the symptoms and signs of increased or decreased TBW?
The main symptoms and signs of TBS excess (decreased PNa) or TBW deficiency (increased PNa) result, respectively, from brain swelling or contraction. If TBW changes occur more rapidly than the brain can adapt, symptoms and signs occur. The severity of the symptoms and signs depends on the degree and rapidity of the TBW change. After adaptation occurs, correcting the disturbance in body water too rapidly may be more deleterious than the initial disturbance.
25. What are the symptoms and signs of hyponatremia and hypernatremia?
 Hyponatremia: headache, confusion, muscle cramps, weakness, lethargy, apathy, agitation, nausea, vomiting, anorexia, altered levels of consciousness, seizures, depressed deep tendon reflexes, hypothermia, Cheyne-Stokes respiration, respiratory depression, coma, and death.
Hyponatremia: headache, confusion, muscle cramps, weakness, lethargy, apathy, agitation, nausea, vomiting, anorexia, altered levels of consciousness, seizures, depressed deep tendon reflexes, hypothermia, Cheyne-Stokes respiration, respiratory depression, coma, and death.

26. How does the brain adapt to hyponatremia?
Because ICF and ECF osmolalities must always be equal, developing hyponatremia and decreased Posm immediately shift water into the brain, raising intracranial pressure (ICP). The increased ICP causes loss of NaCl into the CSF. Over the next several hours, there is also loss of intracellular K and, over the next few days, loss of organic solute. These changes lower ICF osmolality and return the brain volume to normal. However, if severe hyponatremia occurs too rapidly, there is not enough time for cerebral adaptation. Brain edema occurs, further increasing ICP; the brain herniates, and the patient dies.
27. How does the brain adapt to hypernatremia?
With acute hypernatremia and increased Posm, water immediately shifts out of the brain and decreases ICP. The decreased ICP promotes movement of CSF with NaCl into the brain ICF, partially correcting volume. Within hours, further brain adaptation occurs, increasing brain ICF K+, Na+, and Cl−. The resulting increase in osmolality pulls water from the ECF and restores about 60% of the brain volume. Over the next several days, the brain accumulates organic solutes (osmolytes), previously called idiogenic osmoles, that return the brain volume to a near-normal level. These solutes include glutamine, taurine, glutamate, myoinositol, and phosphocreatine. If the brain has no time to adapt to rapidly developing hypernatremia, it shrinks, retracts from the dura, and tears vessels, causing intracranial hemorrhage, increased ICP, compressive injury, herniation, and death.
28. How should you approach the patient with hyponatremia?
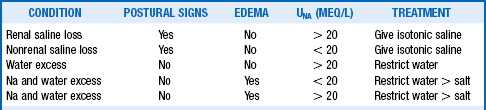
Hyponatremia occurs in 1% of outpatients, more than 4% to 15% of hospitalized patients, 18% of elderly nursing home residents, and nearly 30% of intensive care unit (ICU) patients. Hyponatremia always means too much ECF water relative to Na. Serum osmolality (reflective of Posm) should be measured, and volume status carefully assessed. With hyponatremia, the osmolality should be low. If Posm is elevated (hypertonic hyponatremia), the ECF is high in other osmotically active substances, such as glucose (uncontrolled diabetes) and mannitol (treatment of increased ICP). Large-volume bladder irrigation with mannitol and glycine is sometimes responsible as well. When Posm is normal (isotonic hyponatremia), there may be displacement of H2O in the assay volume by excess lipid (hypertriglyceridemia) or protein (multiple myeloma), causing pseudohyponatremia. With pseudohyponatremia, the osmolar gap is increased to more than 10 mOsm/kg, and measurement of PNa without dilution with a Na-selective electrode gives the true PNa concentration. Lastly, when Posm is appropriately low (hypotonic hyponatremia), the common causes of hyponatremia to consider are listed in Table 24-3. Measuring Uosm is helpful in this differential diagnosis; if Uosm is less than 100 mOsm/kg, primary polydipsia, beer potomania, or malnutrition may be present (see questions 45 and 48). If Uosm is greater than 100 mOsm/kg, there is usually a diluting defect and an ADH effect (appropriate or not). Because total body volume is proportional to total body Na, a thorough assessment of the patient’s volume status helps determine ECV and therapy. Patients whose neck veins are flat while they are supine and who have postural changes in blood pressure and pulse (standing blood pressure decreases more than 20 systolic/10 diastolic mm Hg and pulse increases greater than 20 beats/min) are hypovolemic and invariably saline (NaCl and H2O) depleted. Patients with distended neck veins and edema are hypervolemic and have salt and water (saline) excess. Hyponatremic patients with no postural changes and no edema are clinically euvolemic, but volume may be subclinically increased. If possible, always direct treatment to correct the underlying disorder (Tables 24-3 and 24-4). If patients have lost saline, give them saline. If they have retained too much water, restrict their water. If they have retained too much salt and water but more water than salt, restrict their salt and water but water more than salt. It sounds simple and it is in concept. However, sometimes it is difficult to determine the subtle changes in volume status that are key to this assessment (see question 29). Carefully use loop diuretics in hypervolemic patients and 3% saline in acutely symptomatic patients (see question 47).
TABLE 24-3.
| PATHOPHYSIOLOGY | ASSOCIATED CONDITIONS |
| Renal saline loss and decreased ECV UNa > 20 mEq/L |
Diuretics Osmotic diuresis (glucose, urea, mannitol) Primary adrenal insufficiency Renal tubular acidosis (NaHCO3 loss) Salt-losing nephritis Ketonuria Cerebral salt wasting |
| Nonrenal saline loss and decreased ECV UNa < 20 mEq/L |
Vomiting Diarrhea Pancreatitis, rhabdomyolysis, burns Peritonitis, bowel obstruction |
| Water excess UNa > 20 mEq/L |
SIADH Drugs (see Table 24-2) Secondary adrenal insufficiency Hypothyroidism |
| Na and H2O excess with decreased ECV UNa < 20 mEq/L |
Congestive heart failure Cirrhosis Nephrotic syndrome |
| Na and H2O excess with increased ECV UNa > 20 mEq/L |
Acute renal failure Chronic renal failure Pregnancy |
*Hyponatremia always means too much plasma water relative to Na. Thorough volume assessment is crucial. Volume loss (renal or nonrenal) usually means saline (salt > H2O) loss, and is associated with decreased ECV. Volume excess (hypervolemic) usually means saline (H2O > salt) excess with associated edema and may be associated with decreased or increased ECV. Water excess usually causes mild excess of volume that affects baroreceptor activity. UNa reflects renal perfusion, tubular integrity, and hormonal status. When UNa > 20 mEq/L, the kidney contributes to Na loss and; when UNa < 20 mEq/L, the kidney is conserving Na. ECV, effective circulating volume; SIADH, syndrome of inappropriate antidiuretic hormone.
TABLE 24-4.
*Marked hyperlipidemia or hyperproteinemia causes pseudohyponatremia and artifactually lowers the standard measurement for PNa. Measuring PNa undiluted with Na-selective electrodes corrects for the excess lipid and protein and provides a true PNa reading. However, routine measurement or prior dilution gives falsely low results for the PNa. Measuring Posm helps differentiate pseudohyponatremia. Osmometer-measured Posm measures only the osmotic activity of plasma water that excludes lipids and proteins. Measured Posm is normal in pseudohyponatremia, and the osmolar gap is increased to >10 mOsm/kg. Use loop diuretics carefully to treat edema and 3% saline for symptomatic acute hyponatremia.
29. What is the importance of an initial thorough volume assessment in patients with hyponatremia?
A thorough volume assessment is essential to help determine the underlying cause of hyponatremia (see Table 24-3) and to guide treatment (see Table 24-4). The volume status is best assessed by looking at neck veins, postural vital signs, and edema. At times the best clinician cannot get a good evaluation of ECV, but central venous catheterization is rarely necessary. Urinary Na and edema are other valuable ECV clues. Body weight should be measured daily, and postural vital signs should be assessed serially as necessary. Initial lab tests should include Posm, general blood chemistry panel (Na, K, Cl, CO2, Cr, BUN, glucose, albumin, Ca, Mg); urinary Na, Cl, Cr; Uosm; and fractional excretion of Na. The presence or absence of edema and the UNa value are most helpful.
30. How should you characterize and diagnose the patient with syndrome of inappropriate secretion of antidiuretic hormone?
Syndrome of inappropriate secretion of antidiuretic hormone (SIADH) has recently been called SIAD or syndrome of inappropriate antidiuresis because many of patients with the disorder do not have measurable ADH levels. Clinical euvolemia, hypotonic plasma, and less than maximally dilute urine are the clues to SIADH. The patient should be approached as discussed in question 28. It is important to establish normovolemia by physical examination. Then Posm, Uosm, PNa, UNa, and urinary potassium (UK) should be ordered. Finally, pituitary, adrenal, and thyroid dysfunction must be excluded before a diagnosis of SIADH can be made. Confirmatory criteria of SIADH include low PNa (<135 mEq/L), low Posm (<280 mOsm/kg), Uosm higher than 100 mOsm/kg, UNa higher than 40 mEq/L, and [UNa + UK] higher than PNa. Patients with SIADH are usually said to have normal volume status. However, they actually have excess TBW. Unlike excess saline, which is limited to ECF, excess water distributes ⅔ to the ICF and ⅓ to the ECF (see Fig. 24-1). Thus, the ECF excess is minor and not usually perceptible by clinical examination. Nonetheless, patients with SIADH have mildly increased ECV, which is sensed by the kidneys. The GFR is increased, causing low serum levels of uric acid, BUN, and creatinine. The increased ECV also increases atrial natriuretic peptide (ANP) and, along with increased GFR, promotes natriuresis. These are the classic findings in SIADH. Obviously, SIADH does not protect against dehydration and other conditions that can obscure the classic presentation. For example, a patient with ectopic ADH from lung cancer may present with dehydration from diarrhea, lack of food intake (solute), and lack of water intake from debilitation. In this instance, the UNa and UCl may be less than 20 mEq/L, and measuring ADH may help.
31. How do you treat the patient with SIADH?
SIADH should be treated initially with water restriction (500-1500 mL/day). The fluid restriction needed to correct the hyponatremia is often not tolerated, especially when [UNa + UK] is much greater PNa. The combination of a high-sodium diet (4-8 g), a loop diuretic, and water restriction may be more practical. The treatment of markedly symptomatic hyponatremia is discussed in question 40. It is also important to correct the underlying abnormality (see questions 18 and 19). If the patient has unresectable cancer and water restriction is not tolerated, demeclocycline, 600 to 1200 mg/day, or lithium carbonate, 600 to 1200 mg/day, in 2 to 4 divided doses, may be given. Because lithium carbonate can cause neurologic, cardiovascular, and other toxicities, it should be avoided unless there are no other therapeutic options. Demeclocycline may cause severe renal failure in patients with cirrhosis. Thus it is contraindicated in patients with cirrhosis and severe liver disease. Oral and intravenous (IV) V2-R antagonists have also proved useful for SIADH treatment (see question 42). Although careful monitoring of PNa is important and the expense of these aquaretics may be prohibitive, the V2-R antagonists are probably the agents of choice.
32. What are the four patterns of SIADH?
The four patterns of SIADH are distinguished according to responses of ADH to Posm:

 30% of cases).
30% of cases).
 30% of cases).
30% of cases).
 30% of cases).
30% of cases).
 10% of cases).
10% of cases).33. Define polyuria and list its main causes.
Polyuria is a urine output greater than 3.0 L/day. Four main disorders cause polyuria: central neurogenic DI (defect in ADH secretion), nephrogenic DI (defect in ADH action on the kidney), psychogenic polydipsia (psychosis), and dipsogenic DI (defect in thirst center). All forms of DI may be partial or complete. In general, patients with DI have Uosm less than Posm and often the Uosm is lower than 100 mOsm/kg. Polyuria also may occur from osmotic diuresis in such conditions as diabetes mellitus (glucose), recovery from renal failure (urea), and IV infusions (saline, mannitol). In the last cases, the diagnosis is usually clear from the history and Uosm is greater than Posm. See Table 24-2 for drugs and conditions that decrease ADH secretion and action. Causes of acquired nephrogenic DI include chronic renal disease, electrolyte abnormalities (hypokalemia, hypomagnesemia, hypercalcemia), drugs (lithium, demeclocycline, cisplatinum), sickle-cell disease (damaged medullary interstitium), diet (increased water and decreased solute—beer, starvation), and inflammatory or infiltrative renal disease (multiple myeloma, amyloidosis, sarcoidosis). DI may be associated with specific genetic abnormalities. Hereditary central DI is usually autosomal dominant and expresses itself in childhood rather than at birth. Wolfram’s syndrome results from a familial defect on the short arm of chromosome 4 and has associated central DI, diabetes mellitus, optic atrophy, and deafness (DIDMOAD). Congenital nephrogenic DI results from abnormalities of V2-R or AQP2 channels, and symptoms of polyuria and dehydration appear in the first week of life. Most cases of nephrogenic DI (90%) are related to abnormalities of the V2-R and are X-linked and therefore almost always limited to expression in males. More than 150 mutations are noted to cause DI related to V2-R abnormalities. Nephrogenic DI related to abnormalities of AQP2 (10%) may be autosomal dominant or recessive. When recessive and in a female, the DI is likely due to a mutation on chromosome 12.
34. How do you distinguish polyuric patients with the various forms of DI from patients with excessive water drinking?
In excessive water drinking, the PNa, BUN, and uric acid are relatively low. In DI, the PNa and uric acid are relatively high, and the BUN is relatively low. Central DI often has an abrupt onset resulting from loss of a critical amount of AVP due to destruction of more than 80% to 90% of the ADH-secreting hypothalamic neurons at a critical point in time. Affected patients also have a preference for ice-cold water. Often the cause of polyuria is clear from the history and basic lab values. If the diagnosis is in doubt, a water restriction test (WRT) can be performed. Other names for the WRT are dehydration test and water deprivation test. The test may take 6 to 18 hours, depending on the initial state of hydration.
1. Office testing is acceptable unless the patient cannot be watched closely, in which case hospitalization may be required.
2. Measure baseline weight, Posm, PNa, PBUN, Pglucose, Uvolume, Uosm, UNa, and UK. Measure hourly weight and Uosm.
4. Watch the patient closely for signs of dehydration and surreptitious water drinking.
5. End the WRT when Uosm has not increased more than 30 mOsm/kg for 3 consecutive hours, Posm has reached 295 to 300 mOsm/kg, or the patient has lost 3% to 5% of body weight. If weight loss exceeds 3% to 5% of body weight, further dehydration is unsafe.
6. At Posm of 295 to 300 mOsm/kg, endogenous ADH levels should be 5 pg/mL or greater, and the kidney should respond with maximal urinary concentration.
7. Repeat all baseline measurements toward and at the end of the WRT.
8. Give 5 units of aqueous AVP or 2 μg of desmopressin (DDAVP) subcutaneously.
9. Repeat the baseline tests at 30, 60, and 120 minutes.
10. Calculate Uosm/Posm and [UNa + UK]/PNa as a check on measured Uosm/Posm.
36. How do you interpret the results of the WRT?
Table 24-5 summarizes the expected results of the WRT. The WRT stimulates maximal endogenous ADH release by increasing Posm and evaluates the kidneys’ concentrating ability by measuring Uosm. Giving exogenous ADH allows evaluation of the renal concentrating response to ADH if dehydration-induced ADH production is impaired. Baseline and end-test plasma samples should be saved and frozen for later ADH measurement if the WRT results are equivocal. Expected normal values for PADH are less than 0.5 pg/mL for Posm less than 280 mOsm/kg, and more than 5 pg/mL for Posm more than 295 mOsm/kg.
TABLE 24-5.
VALUES BEFORE AND AFTER WATER RESTRICTION*
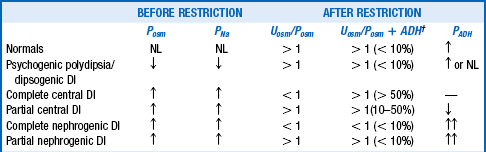
*Recall that when Uosm > Posm, there is antidiuresis, and the kidney is retaining free water. The same is true when [UNa + UK] > PNa, and these measurements are more easily obtainable. When Uosm < Posm or [UNa + UK] < PNa, there is net loss of free water with little net clinical ADH effect.
†The values in parentheses indicate the percentage changes in Uosm (not the Uosm/Posm ratios) after 5 units of subcutaneous aqueous vasopressin or 2 μg of desmopressin acetate.
37. What are the expected plasma ADH concentrations and urinary osmolality in polyuric patients after water restriction?
TABLE 24-6.
EXPECTED VALUES FOR ADH AND Uosm AFTER WATER RESTRICTION
| CAUSE OF POLYURIA | ADH (PG/ML) | UOSM (MOSM/KG) |
| Normal | > 2 | > 800 |
| Primary polydipsia | < 5 | > 500 |
| Complete central DI | Undetectable | < 300 |
| Partial central DI | < 1.5 | 300-800 |
| Nephrogenic DI | > 5 | 300-500 |
38. How should you approach the patient with hypernatremia?
Hypernatremia is uncommon in comparison with hyponatremia, occurring in less than 1% of general hospitalized patients. Indeed, unless patients have an abnormality of thirst or do not have access to water, they usually maintain near-normal PNa by drinking water in proportion to losses. However, as many as 5% to 10% of ICU patients may have some degree of hypernatremia. Loss of water is the usual cause of hypernatremia, and almost all patients require water for treatment (Table 24-7). As in questions 28 and 29, the patient’s volume status must be assessed carefully. After lab studies are obtained, the patient should be approached as outlined in Table 24-8. If the patient has polyuria, the approach described in questions 33 and 34 should be included.
TABLE 24-7.
| PATHOPHYSIOLOGY | ASSOCIATED CONDITION |
| Renal H2O loss > Na loss, UNa > 20 mEq/L | Osmotic diuretics Loop diuretics Renal disease Postobstructive diuresis |
| Non-renal H2O loss > Na loss, UNa < 20 mEq/L | Osmotic diarrhea Vomiting Sweating Diarrhea Burns |
| Excess Na > H2O, UNa > 20 mEq/L | Cushing’s syndrome Primary hyperaldosteronism Excessive intake of NaCl or NaHCO3 Hypertonic saline and bicarbonate Hypertonic dialysis |
| Renal H2O loss, UNa > 20 mEq/L | Central diabetes insipidus Nephrogenic diabetes insipidus |
| Non-renal H2O loss UNa < 20 mEq/L | Increased sensible loss No access to H2O |
*Hypernatremia always means too little plasma water relative to Na. With access to water, hypernatremia usually does not occur or is mild. However, unattended patients who are too old, too young, or too sick may not have adequate access to water, and hypernatremia may be severe. Thorough volume assessment is crucial. Volume loss (hypovolemic) usually means renal or nonrenal saline (H2O > salt) loss and is usually treated with 0.9% to 0.45% saline to correct the volume deficit, followed by water. Volume excess (hypervolemic) usually means saline (salt > H2O) excess with high total body Na and is treated with water and restriction of salt. A loop diuretic may also be necessary to treat volume overload. Euvolemic hypernatremia results from water loss and is treated with free water replacement, and vasopressin is used if water loss is caused by diabetes insipidus.
TABLE 24-8.
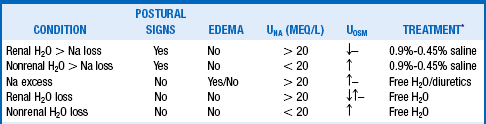
↑, hypertonic; ↓, hypotonic; –, isosmotic.
*Free H2O = 5% dextrose in water infusion or water orally. Infuse saline to restore the volume deficit when patients show signs of severe volume depletion, such as hypotension or postural changes in blood pressure and pulse. This is appropriate when isotonic (0.9%) saline with an osmolality of 308 mOsm/kg is lower than plasma osmolality. This corrects both the volume deficit and the hypernatremia. After the volume deficit has improved, switch to 0.45% saline and eventually 5% dextrose in water. Loop diuretics are used to treat Na excess.
39. How should you diagnose and manage the patient with DI?
Patients with DI have excessive renal water loss owing to decreased ADH secretion (central DI) or renal unresponsiveness to ADH (nephrogenic DI). The hallmark of DI is polyuria with hypotonic urine. Mild hypernatremia, low BUN, and relatively high uric acid are suggestive of DI. ADH and oxytocin neurosecretory vesicles are responsible for the normal finding of a high-signal-intensity posterior pituitary lobe that appears as a bright spot on T1-weighted magnetic resonance images. In patients with idiopathic central DI, this normal posterior pituitary bright spot is absent. However, the posterior pituitary bright spot also decreases with age and may be absent in a majority of elderly patients without DI. Abrupt onset of polyuria is also suggestive of central DI. Little ADH is necessary for urinary concentration, and therefore 80% to 90% of the ADH-secreting neurons must be lost before polyuria develops. As described in questions 33 and 34, DI must first be distinguished from primary polydipsia and identified as being central or nephrogenic. Water should then be given to prevent dehydration until further evaluation suggests definitive therapy. Mild cases of DI require no treatment other than adequate fluid intake. A patient with DI will probably self-treat with water unless there is a thirst deficit or there is no access to water. Central DI is treated with DDAVP as a nasal spray or oral tablet. DDAVP is available for oral use (0.1 or 0.2 mg tablets) with a starting dose of 0.05 mg once or twice daily, and increasing to a maximum of 0.4 mg every 8 hours as necessary. The tablet is 5% absorbed, and absorption is further decreased as much as 50% with meals. At least one dose should be given at bedtime. Oral DDAVP is preferred for patients with sinusitis from the nasal preparation. DDAVP nasal spray (100 μg/mL solution) is given every 12 to 24 hours as needed for thirst and polyuria. It may be administered by metered-dose nasal inhaler (0.1 mL/spray) or by a plastic calibrated tube. The starting dosage is 0.05 to 0.1 mL once or twice daily, and the dosage is titrated to an acceptable urine output. Parenteral DDAVP (4 μg/mL) may be given intravenously, intramuscularly, or subcutaneously at 1 to 2 μg every 12 to 24 hours to hospitalized patients. Nephrogenic DI may be partial or incomplete and therefore may respond to DDAVP. If possible, the underlying cause should be corrected or ameliorated (see question 33).
A low-sodium and low-protein diet that does not compromise nutritional needs should be advised. Regular voiding to avoid overdistending the bladder and bladder dysfunction should be emphasized. Both central DI and nephrogenic DI respond partially to hydrochlorothiazide (25 mg once or twice daily). Amiloride 5 to 10 mg once or twice daily may be additive to thiazides and is especially useful for lithium nephrotoxicity. Nephrogenic DI may respond to combination therapy if one agent is ineffective. Combinations may include indomethacin with hydrochlorothiazide, indomethacin with DDAVP, or indomethacin with amiloride. Although oral indomethacin 25 to 50 mg every 8 hours has been effective, other NSAIDs (tolmetin and ibuprofen) may be less effective.
40. How quickly should you correct states of water excess or deficiency?
The main concern in therapy for abnormal TBW is to prevent devastating neurologic complications. Understanding brain adaptation to changes in TBW, as outlined in questions 25 and 26, emphasizes the need for urgent therapy only in the symptomatic patient. The three useful rules in treating disturbances of water (measured by changes in PNa) are as follows:
1. Return the PNa to normal at a speed relative to that by which it became abnormal. If the change in PNa was slow (days), correct it slowly (days). If the change was rapid (minutes to hours), correct it rapidly (minutes to hours).
2. If there are no symptoms of water or Na imbalance (see question 24), there is no immediate urgency. If there are symptoms, there is urgency. Questions 25 and 26 outline the brain adaptations to altered tonicity that may cause devastating changes in brain volume. These adaptations also cause the patient’s symptoms. Thus symptoms should drive the clinician to correct the altered tonicity rapidly.
3. The degree of rapid PNa correction should be toward normal (until symptoms abate), not to normal.
These concepts—speed, symptoms, and degree of PNa correction—apply for both hyponatremia and hypernatremia (see question 47).
41. What is the significance of the [UNa + UK]/PNa ratio?
The ratio [UNa + UK]/PNa allows the calculation of electrolyte-free water excreted per day and is useful in deciding how much water can be consumed without lowering the PNa. If [UNa + UK]/PNa is greater than 1, the patient is excreting no free water. Thus, all water given to the patient is being retained, and free water clearance is negative. Any water consumed would lower PNa. If the ratio is less than 1, the patient is excreting free water and may consume some free water without decreasing PNa. For example, if patient A makes 2 L of urine daily and has a UNa value of 20 mEq/L, a UK value of 20 mEq/L, and a PNa value of 135 mEq/L, the patient excretes 1.4 L of electrolyte-free urine. If insensible loss is 800 mL, then patient A could consume 1.4 L + 0.8 L = 2.2 L without a change in the PNa. If patient A had a 1000-mL fluid restriction, the net fluid balance would be 1000 mL – 2200 mL = 1200 mL lost.
However, if patient B makes 2 L of urine and has a UNa value of 130 mEq/L, a UK value of 60 mEq/L, and a PNa value of 125 mEq/L, patient B makes −1.04 L or retains free water—no free water loss. If patient B has the same insensible loss of 800 mL, patient B would retain 204 mL if no additional fluid were allowed. Given the same 1000-mL fluid restriction, patient B would have 1000 mL + 204 mL = 1204 mL net gain in fluid. This net free water retention would make the hyponatremia worse. The calculations for free water clearance in patient A and B are as follows.
The formula for free water clearance is 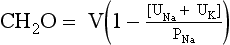 .
.
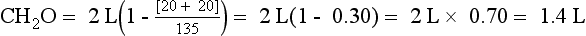
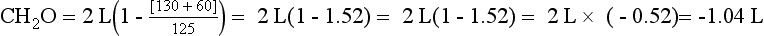
42. What are vasopressin receptor antagonists, and when would you use them for hyponatremia therapy?
The conventional treatment of hyponatremia, water restriction or saline administration, is still appropriate therapy for most patients with hyponatremia. Conivaptan is a first-in-class vasopressin receptor antagonist (VRA) available for treatment of hospitalized patients with hyponatremia and normal extracellular fluid volume (SIADH). Conivaptan prevents AVP binding to V1a and V2 receptors located within the vasculature and renal tubules, respectively. Blocking the V2-R decreases free water reabsorption and increases excretion. Blocking the V1a receptor may cause vasodilatation, reducing afterload in CHF, but also may cause negative hemodynamics in cirrhosis. Conivaptan is available in 20-mg/5-mL glass ampules. The recommended dosage is a 20-mg loading dose administered intravenously over 30 minutes followed by 20 mg infused continuously over 24 hours for an additional 1 to 3 days. If the serum sodium fails to rise at the desired rate, the dosage is increased to 40 mg/day by continuous infusion. The infusion should not exceed 4 days in duration. Tolvaptan is a pure V2-R antagonist available in 15-mg and 30-mg tablets for once-daily oral dosing. The dose can be increased by 15 to 30 mg daily to a maximum dose of 60 mg daily. Like conivaptan, tolvaptan produces selective water diuresis with no effect on Na and K excretion. The term “aquaretic drugs” (aquaretics) has been coined for these medications to highlight the fact that they have different mechanisms of action from those of the saluretic diuretic furosemide. They are proven to be beneficial in SIADH and in hyponatremic patients with CHF and cirrhosis. Blocking the ADH effect may allow rapid correction of hyponatremia to occur; therefore, judicious monitoring of PNa changes is important to prevent excessively rapid correction of PNa.
43. What is the appropriate PNa correction factor for hyperglycemia?
The standard correction factor is a 1.6-mEq/L decrease in PNa for each 100-mg/dL increase in plasma glucose concentration above 100 mg/dL. For glucose values greater than 400 mg/dL, data now suggest a correction factor as high as a 4.0-mEq/L decrease in PNa for each 100-mg/dL increase in plasma glucose and an average correction factor of 2.4 mEq/L.
Clinical problems in water metabolism
44. A 75-year-old woman presents with confusion but no focal neurologic signs. She has type 2 diabetes mellitus. Blood pressure is 110/54 mm Hg. Pulse is 96 beats/min. Neck veins are not visualized in the supine position. Pglucose = 900 mg/dL, PNa = 135 mEq/L, plasma creatinine = 3.0 mg/dL, BUN = 50 mg/dL, UNa = 40 mEq/L; urine glucose is 4+ and urine ketones 3+. Describe her fluid and volume status and treatment.
Glucose remains in the ECF because of insulin deficiency and increases ECF tonicity. Greater tonicity pulls water from the ICF to the ECF, concentrating the ICF and diluting the ECF until ICF and ECF osmolalities are equal. The osmotic pressure of 900 mg/dL glucose (900/18 = 50 mOsm/kg) is the driving force for water movement from ICF to ECF. Water movement from ICF to ECF dilutes the ECF and decreases PNa (translocation hyponatremia). Each 100-mg/dL rise in Pglucose above 100 mg/dL decreases the PNa by 1.6 mEq/L. In this patient, the predicted decrease in PNa = (900 − 100)/100 × 1.6 = 13 mEq/L. The predicted PNa would be 140 − 13 = 127 mEq/L. However, a more accurate correction factor for the elevated glucose would be 2.4 mEq/L (see question 43). Thus the predicted decrease in PNa would be (900 − 100)/100 × 2.4 = 19 mEq/L. The predicted PNa would be 140 − 19 = 121 mEq/L. But this patient’s PNa is 135 mEq/L, suggesting that there has been further water loss from osmotic diuresis and significant dehydration. The Posm—2(135) + 900/18 + 56/2.8 = 340 mOsm/kg—is compatible with hyperosmolar coma. Because this woman has decreased TBW, decrease blood pressure, and a BUN/Creatinine ratio that suggests prerenal azotemia, you might expect her to have low UNa and high Uosm. However, osmotic diuresis caused by urine glucose, ketones, and urea increases urinary Na and water, making UNa and Uosm less useful markers of dehydration. Flatness of neck veins in the supine position is usually due to intravascular volume depletion. Rapid lowering of her glucose to 100 mg/dL will quickly decrease Posm, shift water to the ICF, increase PNa by 13 to 19 mEq/L, and potentially cause cerebral edema and cardiovascular collapse. Thus therapy is normal saline to replace volume and judicious lowering of Pglucose with IV insulin.
45. You admit a 35-year-old schizophrenic patient because of a change in mental function and excessive urine output. Uosm = 70 mOsm/kg. Posm = 280 mOsm/kg, 24-hour urine output = 12 L/day. How much free water is the patient excreting each day?
Free water clearance (Ch2o) is the amount of solute-free water excreted per day. Osmolar clearance (Cosm) is the amount of urine excreted per day that contains all the solute that is isosmotic to plasma. When the urine is hypotonic to plasma, the total urine volume consists of two components: one part that is free of solute (Ch2o) and the other that is isosmotic to plasma (Cosm). To measure how much of the urine is pure (free) water, calculate the Ch2o. To do so, you need to know the Cosm and the urine volume (V). The formula for clearance of any substance (including osmoles) is always the same:
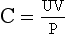
where C is volume of plasma cleared of the substance per unit time, U is urinary concentration of the substance, P is plasma concentration of the substance, and V is total urinary volume per unit time. The calculations for this patient follow:
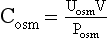
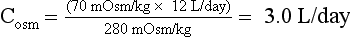
Manipulation of formula (2) offers another means of calculating free water clearance, as follows:
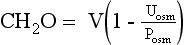
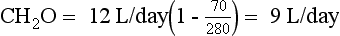
Thus the patient’s daily urine output contains 9 L/day of pure (free) water, and 3 L/day that is isotonic to plasma. This information does not distinguish primary polydipsia from DI. However, the low Posm, 280 mOsm/kg, suggests primary polydipsia.
46. A 45-year-old-man with a 30-pack-year history of smoking presents with cough, dyspnea, fatigue, and a 15-lb weight loss. Chest x-ray shows mediastinal adenopathy and right atelectasis with pleural effusion. Posm = 270 mOsm/kg, PNa = 125 mEq/L, Uosm = 470 mOsm/kg, UNa = 130 mEq/L, UK = 60 mEq/L, and urine volume = 1 L/day. How much free water is the patient excreting each day? What is the likely pulmonary lesion?
Urine is hypertonic to plasma if the Uosm exceeds Posm or the U[Na + K] exceeds PNa. Urine hypertonic to plasma contains two parts: the volume that would be required to contain all solute and remain isosmotic to plasma is the osmolar clearance (Cosm); the volume of free water that was removed from the isotonic glomerular filtrate to make Uosm higher than Posm or U[Na + K] higher than PNa is the negative free water clearance (Tch2o; see next paragraph). There are two ways to calculate free water clearance: one method uses osmolality, as in question 45; the other uses electrolytes (Na and K). Electrolyte-free water clearance more accurately estimates free water clearance and negative free water clearance, especially when urine contains large numbers of nonelectrolyte osmolytes, such as urea, that increase osmolality unrelated to free water clearance. To calculate electrolyte free-water clearance, the urinary Na and K concentrations and the plasma Na are used. Because U[Na + K] is greater than PNa—(130 + 60) > 130—the net urinary excretion of free water is negative, and therefore free water clearance is negative. Calculations for osmolar and electrolyte-free water clearances in this patient follow:
Calculations for classic osmolar (negative) free water clearance:
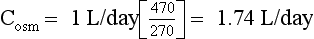
Manipulation of formula 2 offers another means of calculating negative free water clearance, as follows:
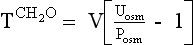
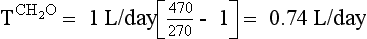
Calculations for electrolyte-free water clearance (negative free water clearance) are as follows:
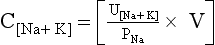
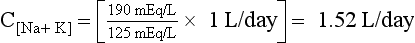
Thus the patient’s kidneys add (by water reabsorption) a net of 520 to 740 mL of free water to plasma each day. With a low Posm, it is usually inappropriate to retain water in excess of output. This finding suggests SIADH. One must exclude volume depletion, adrenal insufficiency, and hypothyroidism before making the diagnosis of SIADH. This patient had small cell carcinoma of the lung with ectopic ADH secretion. SIADH develops in 15% of patients with small cell carcinoma of the lung. This tumor is highly associated with smoking and accounts for 15% to 25% of lung cancer. Other lung cancers rarely secrete ADH.
47. A 34-year-old, 60-kg woman presents 12 hours after discharge following cholecystectomy. She has headache, confusion, muscle cramps, weakness, lethargy, agitation, nausea, and vomiting. She had no symptoms at discharge. PNa was 110 mEq/L. What has caused the hyponatremia? How quickly should you treat it?
According to this history, hyponatremia developed rapidly and was symptomatic. Treatment is ICU admission and administration of 3% saline and furosemide at rates sufficient to increase PNa 1.5 to 2.0 mEq/L/h for 2 to 4 hours on the basis of symptom resolution. PNa should be measured every 2 to 4 hours to follow progress and guide therapy. UNa and UK measurements should be ordered as needed if PNa is not changing at the expected rate, to guide replacement of urine water and electrolyte losses. After serious signs and symptoms improve, the rate of correction should be decreased to 0.5 to 1.0 mEq/L/h until symptoms further improve or the PNa is at least 120 mEq/L. A net increase in PNa greater than 10 mEq/L in the first 24 hours and 18 mEq/L over 48 hours must be avoided. For chronic hyponatremia without symptoms, the appropriate rate of correction is 0.5 mEq/L/h or less with similar net daily increases in PNa. Acute symptomatic hyponatremia requires expeditious correction of the PNa because the symptomatic patient has cerebral edema caused by “normal” brain-cell solute content that pulls water from the hypotonic ECF into the brain. Acute raising of the PNa increases ECF tonicity, pulls water out of the swollen brain, and reduces the brain volume toward normal. The brain has no room in the skull to swell more than 8% to 10% before herniation. Therefore, there is no benefit to acute correction of the Na more than 8%—in this case to a PNa greater than 119 mEq/L. Conversely, the patient with chronic asymptomatic hyponatremia has adapted by loss of brain solute and has near-normal brain volume. Increasing such a patient’s PNa too rapidly (> 0.5 mEq/L/h) would shrink the brain and predispose to the osmotic demyelination syndrome (previously called central pontine myelinolysis). The risks of not correcting acute symptomatic hyponatremia include increased cerebral edema, seizures, coma, tentorial herniation, and death. Calculations for water excess and for 3% saline necessary to correct the PNa to 120 mEq/L are as follows”
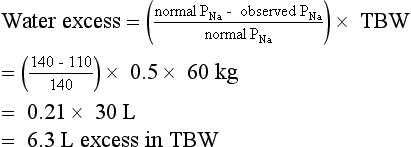

Knowing the Na deficit is useful clinically because it can be replaced at a controlled rate to improve the hyponatremia. The Na in 3% saline is 513 mEq/L:
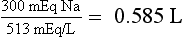
Thus, assuming no Na or water loss, giving 585 mL of 3% saline will correct the PNa to 120 mEq/L. Make a similar calculation for 3% saline to infuse over 3 to 4 hours to increase the PNa by 6 mEq/L. The answer is 350 mL. However, the patient will lose sodium and water during the infusion. If PNa does not correct at the desired rate, one may need to measure PNa, UNa, and UK to estimate loss and gain of Na and water during therapy and to replace those losses. An empiric rate of 3% saline infusion for rapid treatment of symptomatic hyponatremia is 2 mL/kg/h. Ideal body weight should be used unless the patient weighs less than ideal body weight, in which case actual weight is preferable. In this patient, 3% saline infusion would be 60 kg × 2 mL/kg/h or 120 mL/h × 4 h = 480 mL.
48. An 80-year-old woman who rarely leaves her home is brought to the hospital after being found confused. Three weeks ago, she saw her physician, who started a diuretic for systolic hypertension. On arrival, her PNa is 110 mEq/L. What is the cause of her hyponatremia?
As a consequence of aging, elderly patients lose GFR, concentrating ability, and diluting ability. Thus an 80-year-old woman may have a normal (for age) renal-concentrating range of 100 to 700 mOsm/kg. However, maximal Uosm in the elderly may be as low as 350 mOsm/kg. This woman’s average diet may generate only 600 mOsm/day. Her normal range of urine output would then be 0.9 to 6.0 L/day. If her dietary intake fell to 300 mOsm/day, her maximal urine output would fall to 3 L/day, calculated as follows:
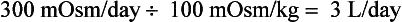
Given free access to water and a thiazide diuretic, which impairs urinary dilution, she could easily become water intoxicated and hyponatremic. The mechanism of hyponatremia in beer potomania and the “tea-and-toast diet” is low total osmolar intake and relatively increased water intake. The decreased osmotic load for excretion limits the amount of water excreted. This patient’s hyponatremia is probably chronic; however, she is symptomatic. Thus, it is not clear whether the hyponatremia is truly chronic or acute. Therefore, how to proceed with therapy in this severely hyponatremic patient is unclear. Computed tomography (CT) or magnetic resonance imaging may help by showing the presence or absence of cerebral edema. If cerebral edema is present, treatment for acute hyponatremia is necessary. If cerebral edema is absent, judicious treatment for chronic hyponatremia is appropriate. Symptoms, signs, and PNa should be assessed frequently. Remember: Elderly women taking thiazide diuretics, alcoholics, and malnourished, hypokalemic, or burned patients are at particular risk for the demyelination syndrome.
 WEBSITES
WEBSITES
American Academy of Family Physicians: Hyponatremia and hypernatremia in the elderly. http://www .aafp.org/afp/20000615/3623.html.
EMedicine: SIADH review. http://www.emedicine.com/ped/topic2190.htm.
EMedicine: Hyponatremia review. http://www.emedicine.com/med/topic1130.htm.
EMedicine: Lithium nephropathy review. http://www.emedicine.com/med/topic1313.htm.
EMedicine: Diabetes insipidus review. http://www.emedicine.com/med/topic543.htm.
Adrogue, HJ, Madias, NE. Hypernatremia. N Engl J Med. 2000;342:1493–1499.
Adrogue, HJ, Madias, NE. Hyponatremia. N Engl J Med. 2000;342:1581–1589.
Berl, T, Schrier, RW. Disorders of water metabolism. In: Schrier RW, ed. Renal and Electrolyte Disorders. ed 7. Philadelphia: Lippincott Williams & Wilkins; 2010:1.
Chawla, A, Sterns, RH, Nigwekar, SU, et al, Mortality and serum sodium. do patients die from or with hyponatremia. Clin J Am Soc Nephrol 2011;6:960–965.
Decaux, G, Soupart, A, Vassart, G, Non-peptide arginine-vasopressin antagonists. the vaptans. Lancet 2008;371:1624–1632.
Ellison, DH, Berl, T. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064–2072.
Gutierrez, OM, Lin, HY. Refractory hyponatremia. Kidney Int. 2007;71:79–82.
Hannon, MJ, Finucane, FM, Sherlock, M, et al. Disorders of water homeostasis in neurosurgical patients. J Clin Endocrinol Metab. 2012;97:1423–1433.
Hillier, TA, Abbott, RD, Barrett, EJ, Hyponatremia. evaluating the correction factor for hyperglycemia. Am J Med 1999;106:399–403.
Hoorn, EJ, van der Lubbe, N, Zietse, R, SIADH and hyponatraemia. why does it matter. NDT Plus. 2009;2(Suppl 3):iii5–iii11.
Liamis, G, Milionis, H, Elisaf, M. A review of drug-induced hyponatremia. Am J Kidney Dis. 2008;52:144–153.
Loh, JA, Verbalis, JG. Disorders of water and salt metabolism associated with pituitary disease. Endocrinol Metab Clin N Am. 2008;37:212–234.
Piper, GL, Lewis, JK. Fluid and electrolyte management for the surgical patient. Surg Clin N Am. 2012;92:189–205.
Robinson, AG, Verbalis, JG, et al. Posterior pituitary. In: Melmed S, Polonsky KS, Larsen PR, eds. Williams Textbook of Endocrinology. ed 12. Philadelphia: Elsevier Saunders; 2011:291.
Rosner, MH, Kirven, J. In-depth review. Exercise-associated hyponatremia. Clin J Am Soc Nephrol. 2007;2:151–161.
Sanghvi, SR, Kellerman, PS, Nanovic, L, Beer potomania. an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis 2007;50:673–680.
Schrier, RW, Review. Body water homeostasis. clinical disorders of urinary dilution and concentration. J Am Soc Nephrol 2006;17:1820–1832.
Shea, AM, Hammill, BG, Curtis, LH, et al. Medical costs of abnormal serum sodium levels. J Am Soc Nephrol. 2008;19:764–770.
Sherlock, M, O’Sullivan, E, Agha, A, et al. The incidence and pathophysiology of hyponatraemia after subarachnoid haemorrhage. Clin Endocrinol (Oxf). 2006;64:250–254.
Singer, I, Oster, JR, Fishman, LM. The management of diabetes insipidus in adults. Arch Intern Med. 1997;157:1293–1301.
Sterns, RH, Hix, JK, Silver, S, Treating profound hyponatremia. a strategy for controlled correction. Am J Kidney Dis 2010;56:774–779.
Verbalis, JG, et al. Disorders of water balance. In: Taal MW, Chertow GM, Marsden PA, eds. Brenner & Rector’s The Kidney. ed 9. Philadelphia: Elsevier Saunders; 2011:540.
Wald, R, Jaber, BL, Price, LL. Impact of hospital-associated hyponatremia on selected outcomes. Arch Intern Med. 2010;170:294–302.
Yee, AH, Burns, JD, Wijdicks, EFM, Cerebral salt wasting. pathophysiology, diagnosis, and treatment. Neurosurg Clin N Am 2010;21:339–352.
Zeidel, ML, Hyponatremia. mechanisms and newer treatments. Endocr Pract 2010;16:882–887.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 24
Water metabolism
1. What is the water composition of the human body?
Water composition of the body depends on age, sex, muscle mass, body habitus, and fat content. Various body tissues have the following water percentages: lungs, heart, and kidneys (80%); skeletal muscle and brain (75%); skin and liver (70%); bone (20%); and adipose tissue (10%). Clearly, people with more muscle than fat have more water. Generally, thin people have less fat and more water. Men are 60% water and women 50% water by weight. Older people have more fat and less muscle. The average man and woman older than 60 years are made up of 50% and 45% water, respectively (see Table 24-1). Most discussions of total body water (TBW) consider a man who is 60% water, weighs 70 kg, and is 69 inches (175 cm) tall.
2. Where is water located within the body?
TBW comprises water located inside the cells (intracellular fluid [ICF]) and outside the cells (extracellular fluid [ECF]). TBW is 60% of body weight—40% ICF (⅔) and 20% ECF (⅓). Of the ECF, approximately ¾ is interstitial fluid (ISF) and ¼ is intravascular fluid (IVF). IVF is a major component of the total blood volume necessary to maintain effective vascular pressure. ISF is 15% of body weight, and IVF is 5% of body weight. In a 70-kg man, TBW = 42 L, ICF water = 28 L, and ECF water = 14 L. ISF is 10.5 L and IVF (plasma) is 3.5 L. Tight regulation of the relatively small volume of IVF maintains blood pressure and avoids symptomatic hypovolemia and congestive heart failure. Normal plasma is 93% water and 7% proteins and lipids. The arterial volume is only 15% of IVF. Although arterial volume is small, its integrity is most important for maintaining the effective circulation and preventing abnormalities of water balance (Fig. 24-1).
3. What is transcellular water (TCW)?
TCW is water formed by cellular transport activities and is located in various ducts and spaces throughout the body. This water includes cerebrospinal fluid (CSF) and aqueous humor; secretions in the sweat, salivary, and lacrimal glands; secretions in pancreas, liver, biliary, gastrointestinal, and respiratory tracts; and peritoneal, pleural, and synovial fluids.
4. Explain the significance of TCW.
TCW carries secretions to specific sites for enzymatic and lubricant activity and is normally quite small—1.5% of body weight. In disease states, excess or deficiency of TCW can cause dysfunction. Marked excess TCW formation—third spacing—may decrease effective circulating volume (ECV), stimulate antidiuretic hormone (ADH) and aldosterone release, increase retention of salt and water, and cause edema and hyponatremia.
5. What controls distribution of body water?
With few exceptions (e.g., ascending loop of Henle [LOH] and distal nephron), water moves freely across cell membranes, depending on tonicity. Because tonicity depends on impermeable solutes, such as sodium (Na), disorders of water metabolism are reflected by changes in solute concentrations. In addition to changes in water distribution, changes in TBW, blood volume, and ECV affect overall water balance. A thorough understanding of disorders of water metabolism requires a clear understanding of changes in plasma Na concentration (PNa), plasma osmolality (Posm), and ECV.
KEY POINTS 1: WATER METABOLISM
1. Changes in body water or distribution are usually reflected by changes in plasma sodium concentration (PNa) and may occur in states of low, normal, or high total body sodium. Low PNa reflects high TBW, and high PNa reflects low total body water (TBW).
2. Water always moves across cell membranes from lower to higher osmolality. This movement is determined by the concentration of effective osmotic solute in the intracellular or extracellular fluid and is responsible for the neurologic symptoms and signs associated with changes in PNa.
3. Hyponatremia may occur with low, normal, or high osmolality, whereas hypernatremia is always associated with hyperosmolality and hypertonicity.
4. Water content of the body is a balance of input and output.
5. Water balance is controlled by thirst, access to water, solute intake, antidiuretic hormone (ADH), cortisol, aldosterone, natriuretic peptides, baroreceptor sensors, ADH receptors, renal water channels called aquaporins, level of kidney function, and drugs.
6. What is effective circulating volume (ECV)?
ECV is the arterial volume required to maintain normal baroreceptor pressure that is appropriate for a given level of vascular resistance. ECV is also called effective arterial blood volume (EABV). By inducing changes in baroreceptor tone, alterations in ECV have a major impact on water balance. Low ECV causes renal salt and water retention, whereas high ECV causes renal salt and water loss. Depending on the patient’s water intake, these changes may produce significant hyponatremia. Maintaining normal ECV preserves circulatory homeostasis.
7. How do baroreceptors affect ECV?
Baroreceptors are the major sensors of changes in ECV (Fig. 24-2). However, their main role is to maintain normal pressure (not volume) at the level of the baroreceptor sensors located primarily in the carotid sinus, aortic arch, atria, pulmonary veins, and afferent renal arterioles. These anatomic locations are important because perfusion to these areas affects the three main effectors of circulatory homeostasis and ECV: brain, heart, and kidneys.
8. How does vascular pressure, as sensed by the baroreceptors, relate to ECV and hyponatremia?
Baroreceptors normally maintain tonic inhibition of vasoconstrictor nerves and natriuretic hormone release but tonic stimulation of vagal cardiac nerves. A drop in ECV decreases effective vascular pressure (EVP), baroreceptor tone, tonic inhibition, and tonic stimulation. This causes vasoconstriction; increases heart rate; and increases renin, aldosterone, angiotensin II, and ADH secretion. It decreases atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) from brain and ventricles, and urodilatin (kidney). These alterations enhance renal Na and water retention. If the patient receives unlimited water, these changes may lead to hyponatremia. Hyponatremia cannot develop unless the patient retains more water than is excreted. Decreased ECV/EVP predisposes to water retention, but the patient must receive free water for hyponatremia to develop. The venous system, through atrial stretch receptors, has similar effects but responds to changes in ECV earlier than the arterial system.
9. Define osmolality and tonicity, and outline their effects on water movement.
Osmolality is the concentration of a substance in 1 L of water divided by its molecular weight. Tonicity is effective osmolality—the osmotic pressure caused by dissolved particles restricted to one side of the cell membrane. Because Na and glucose are partially restricted to the ECF, they are effective osmols and account for normal tonicity. Mannitol, sorbitol, glycerol, and glycine are also effective osmols. Urea freely crosses cell membranes and distributes evenly in TBW, and therefore it changes osmolality but not tonicity. Thus, except during early and rapid solute and water changes, urea is an ineffective osmol. Ethanol and methanol are other ineffective osmols. Water always moves across cell membranes from lower osmolality to higher osmolality until osmolality on the two sides is equal. At equilibrium, the following is always true:
10. What formulas are useful in evaluating osmolality and tonicity?
The normal range for Posm, 275 to 295 mOsm/kg, varies with the normal ranges for plasma Na, urea, and glucose. Correction factors for other effective solutes (osmols) are mannitol/18, sorbitol/18, and glycerol/9. Correction factors for other ineffective solutes (osmols) are ethanol/4.6 and methanol/3.2.
11. How does PNa relate to TBW, osmolality, and tonicity?
The following formulas are useful in understanding the relationship of PNa, plasma potassium (PK), total body sodium and potassium [Na+ + K+ ], and TBW. [Na+ + K+] estimates total body solute:
Thus PNa is proportional to [Na+ + K+] and inversely proportional to TBW. An increase or decrease in total plasma Na particles can proportionately change the PNa. However, in clinical medicine, changes in PNa usually reflect changes in plasma water. When PNa is high, plasma water is low. When PNa is low, plasma water is high. Low PNa may occur with low, normal, or high osmolality, whereas high PNa is always associated with hyperosmolality and hypertonicity.
12. How does PK relate to PNa and TBW?
Although 98% of K+ is intracellular, a K+ infusion increases PNa. This occurs as follows. In hypokalemia, infused K+ enters cells. To preserve electroneutrality, Na+ leaves or chloride (Cl−) enters cells. ECF water follows K+ and Cl− into cells because of increased ICF osmolality. Both mechanisms increase PNa. Hypokalemic patients infused with equal amounts of KCl or NaCl have equal increases in PNa. Thus addition of KCl to isotonic saline makes hypertonic saline, and infusion of saline with KCl may correct hyponatremia too rapidly (see questions 36 and 44).
13. Describe the input and output of water.
TBW is a balance of input (including endogenous production) and output. In an average adult, input approximates 1600 mL (liquids), 700 mL (foods), and 200 mL (metabolic oxidation of carbohydrate and fat) for a total of 2500 mL/day. Average water losses are 1500 mL (kidneys), 500 mL (skin [400 mL evaporation and 100 mL perspiration]), 300 mL (lung—respiration), and 200 mL from the gastrointestinal tract (stool) for a total of 2500 mL/day. Large losses of water (increased output) occur with excessive sweating, respiration (exercise), burns, diarrhea, vomiting, and diuresis. Decreased water input occurs when defects in thirst and altered mental or physical function (especially in the elderly) prevent access to water.
14. What are the normal limits of urine output?
Water intake and osmotic products of metabolism determine the usual daily output of urine. On a normal diet, a normal adult must excrete 800 to 1000 mOsm of solute per day. The range of normal renal concentrating function is 50 to 1200 mOsm/kg. On this basis, the obligate water excretion varies from 0.8 to 20 L/day. The calculations are as follows:
Note that higher solute loads (e.g., dietary) require more water excretion. For example, body builders consuming high-protein and high-carbohydrate diets with 1400 mOsm solute/day require a urine output of (1400/1200) to (1400/50) or 1.2 to 28 L/day. Alternatively, a low solute intake (starvation) with high water intake predisposes to water retention and water intoxication. This combination exists in binge beer drinkers, in whom the solute load may be only 300 mOsm/day. Low solute intake may also occur in starvation and in an elderly person on a “tea and toast diet.” The range of urine output would drop to (300/1200) – (300/50) or 0.25 to 6 L/day in such patients.
15. What are the main factors controlling water metabolism?
Thirst, hormonal, and renal mechanisms are tightly integrated for control of water metabolism. This integration is strongly influenced by nervous system and baroreceptor control (see Fig. 24-2).
16. What are the stimuli of thirst?
Osmoreceptors in the organum vasculosum of the anterior hypothalamus control thirst. Increasing plasma tonicity stimulates thirst at a threshold about 5 mOsm/kg higher than the value that stimulates ADH release. However, oropharyngeal receptors are also important in thirst regulation. A dry mouth increases thirst. Drinking and swallowing water decrease thirst even without changing Posm. Volume depletion changes afferent baroreceptor input and increases angiotensin II—both changes increase thirst. An unusual idiosyncratic effect of angiotensin-converting enzyme (ACE) inhibitors causes central polydipsia, increased ADH release, and propensity to hyponatremia.
17. What hormonal mechanisms are involved in control of body water?
Although natriuretic peptides, aldosterone, angiotensin II, prostaglandins, and neurohumoral changes affect renal water retention and excretion, ADH is most important. ADH is also called arginine vasopressin (AVP). Supraoptic and paraventricular nuclei in the hypothalamus secrete ADH in response to increased osmolality and decreased volume. ADH attaches to vasopressin 2 receptors (V2-Rs) on the basolateral membrane of renal collecting tubular cells. This activates cyclic adenosine monophosphate (cAMP) and protein kinase A, causing intracellular water channels called aquaporins (AQPs) to insert into the luminal membrane. Water moves down osmotic gradients from tubular lumen through AQP channels into the cell and interstitium. At least seven AQP isoforms (AQP1-4, AQP6-8) are present in the kidney. AQP1 is constitutively expressed in the proximal tubule and descending loop of Henle and is important for isotonic fluid reabsorption and water conservation. The collecting duct has high concentrations of AQP2 that serve as the major target for ADH-mediated water reabsorption. Abnormalities of the V2-R cause most cases of nephrogenic diabetes insipidus (DI), but some are caused by abnormalities of AQP2. Increased AQP2 may cause water retention in conditions such as pregnancy and congestive heart failure. Twenty percent of ADH receptors in the collecting tubular cells are vasopressin 1 receptors (V1-Rs). ADH activates V1-Rs only at very high levels. This increases prostaglandin E2 and prostacyclin, which opposes the antidiuretic effects of excessive ADH.
18. What are the major conditions that influence ADH secretion?
ADH functions to maintain osmotic and volume homeostasis. Secretion starts at an osmotic threshold of 280 mOsm/kg and increases proportionately to further rises in tonicity. Maximum diuresis (urine dilution) occurs at ADH levels of 0.5 pmol/L, and maximum reabsorption (urine concentration) occurs at ADH levels of 3 to 4 pmol/L. A 1% to 2% increase in osmolality stimulates ADH secretion, whereas an 8% to 10% drop in vascular volume is required for the same effect. Through action on baroreceptors, increased ECV raises the osmotic threshold for ADH secretion, and decreased ECV lowers this threshold. Severe volume depletion and hypotension may completely override the hypo-osmotic inhibition of ADH secretion. This finding has been called the “law of circulating volume.” In severe volume depletion and hypotension, ADH secretion continues despite low osmolality, thereby worsening the hyponatremia. Nausea, pain, and stress (as seen postoperatively) are potent stimuli of ADH release and may cause lifethreatening hyponatremia if hypotonic fluid is given. This is particularly true if patients with these symptoms also receive hypotonic fluid and drugs that potentiate the release or action of ADH.
19. What are the major causes of ADH secretion?
Major causes of ADH secretion include hyperosmolality, hypovolemia, nausea, pain, stress, human chorionic gonadotropin as in pregnancy (reset osmostat), hypoglycemia, corticotropin-releasing hormone (CRH), central nervous system (CNS) infections, CNS tumors, vascular catastrophes (thrombosis, hemorrhage), and ectopic ADH of malignancy (carcinomas of lung [primarily small cell], duodenum, pancreas, ureter, bladder, and prostate, and lymphoma). ADH secretion may be increased by any major pulmonary disorder, including pneumonia, tuberculosis, asthma, atelectasis, cystic fibrosis, positive pressure ventilation, and adult respiratory distress syndrome. Human immunodeficiency virus (HIV) infection may have the multifactorial role of causing CNS dysfunction, pulmonary disease, and malignancy. Excessive exogenous ADH or desmopressin acetate (DDAVP) in patients with DI directly increases ADH effect. Oxytocin also has significant ADH activity in the large dosages used to induce labor. Other drugs that affect ADH secretion and action are listed in Table 24-2.
TABLE 24-2.
DRUGS THAT AFFECT ANTIDIURETIC HORMONE (ADH) SECRETION AND ACTION*
| Increase ADH secretion | Antidepressants Amitriptyline Protriptyline Desipramine Fluoxetine Selective serotonin reuptake inhibitors Duloxetine Antipsychotics* Fluphenazine Haloperidol Phenothiazines Butyrophenones Monoamine oxidase inhibitors Ecstasy Nicotine Bromocriptine Carbamazepine Chlorpropamide Clofibrate Cyclophosphamide Ifosfamide Morphine Nicotine Thioridazine Vincristine Angiotensin-converting enzyme inhibitors Amiodarone Methyldopa |
| Increase ADH effect | Acetaminophen Carbamazepine Chlorpropamide Cyclophosphamide Nonsteroidal anti-inflammatory drugs Tolbutamide |
| Decrease ADH secretion | EthanolPhenytoin |
| Decrease ADH effect | Demeclocycline Lithium Acetohexamide Tolazamide Glyburide Methoxyflurane Propoxyphene Colchicine Amphotericin Vinblastine Prostaglandin E2 Prostacyclin |
*Because psychosis itself may cause the syndrome of inappropriate secretion of ADH (SIADH), one must question the true ADH-stimulatory effect of the antipsychotic drugs. Changes in ADH secretion may be direct or indirect.
KEY POINTS 2: SYNDROMES AND TREATMENT OF WATER DYSFUNCTION
1. Clinical syndromes of water dysfunction include syndrome of inappropriate secretion of antidiuretic hormone, diabetes insipidus, and changes in effective circulating volume that can cause marked retention of salt and water, pulmonary and peripheral edema, and severe neurologic dysfunction.
2. Effective correction of water problems requires correcting abnormalities of PNa and a clear understanding of changes in plasma and urine osmolality, urine sodium and potassium, and effective circulating volume (ECV). Additionally, a thorough assessment of patient volume and neurologic symptoms is essential.
3. If neurologic symptoms occur rapidly or are severe, correction of PNa toward normal should be rapid; if symptoms are absent, there is no urgency, and PNa correction should occur more slowly.
4. Depending on the water disturbance, treatment includes water restriction or administration; hypertonic, isotonic, or hypotonic saline; sodium; diuretics; antidiuretic hormone; and aquaretics or other medications.
20. How does the kidney handle salt and water?
To control excess or deficient water intake, there must be an adequate glomerular filtration rate (GFR) and delivery of filtrate to the LOH and distal nephron. Solute is separated from water in the ascending limb of the LOH, distal convoluted tubule (DCT), and cortical connecting segment; normal action of ADH allows controlled reabsorption of water in the cortical and medullary collecting tubules. The proximal convoluted tubule reabsorbs 65% and the descending limb of the LOH 25% of filtered solute and water isotonically. The ascending limb is impermeable to water but removes solute, resulting in dilution of the luminal filtrate, concentration of the interstitium (important for ADH action), and delivery of 10% of the filtrate to the cortical collecting tubules with an osmolality of 100 mOsm/kg. In the absence of ADH, this fluid (18 L/day) would be lost in the urine and cause marked dehydration. In the presence of ADH, the collecting duct becomes permeable to water and reabsorbs all but 1% of the filtrate. Thus the final urine volume is only 1.5 to 2.0 L/day. Because normal GFR is 125 mL/min, the normal kidneys filter 180 L of plasma each day and reabsorb 99%. In normal adults, 99% of all Na and H2O filtered is reabsorbed.
21. What are the causes and consequences of decreased renal water excretion?
Any reduction in water excretion predisposes to hyponatremia and hypo-osmolality. Conditions that impair GFR, delivery of tubular fluid to the distal nephron, or the ability of the distal nephron to separate solute from water, or that increase the permeability of the collecting tubule to water impair water excretion. Such conditions include renal failure, decreased ECV, diuretics (thiazides and loop), and excessive ADH or ADH action.
22. How do hypothyroidism and adrenal insufficiency cause hyponatremia?
Hypothyroidism and adrenal insufficiency reduce cardiac output and thereby decrease ECV and increase ADH release. A hypothyroidism-associated decrease in ECV reduces renal blood flow, glomerular filtration, and maximal solute-free water excretion. Failure to dilute the urine maximally results from nonosmotic ADH release and increased ADH-mediated AQP2 receptors and action. The main effect of glucocorticoid deficiency is altered systemic hemodynamics, and not salt and water loss. Low cortisol impairs cardiac output and the systemic vascular responses to catecholamines, reducing both blood pressure and ECV. The resulting drop in absolute and effective vascular filling pressure reduces stretch on the arterial baroreceptors and thereby decreases tonic vagal and glossopharyngeal inhibition of ADH release. These baroreceptor changes override the hypo-osmotic inhibition of ADH release, and consequently, ADH secretion increases. The decreased ECV also lowers GFR, thereby reducing delivery of filtrate to the distal nephron and enhancing proximal tubular water reabsorption. Normally, CRH and ADH are co-secreted from the same neurons in the paraventricular nuclei of the hypothalamus, and both hormones work synergistically to release adrenocorticotropic hormone (ACTH) from the anterior pituitary—ADH via the vasopressin V1b receptor. Cortisol feeds back negatively at the hypothalamus and pituitary to inhibit the release of both CRH and ADH. Cortisol deficiency decreases this negative feedback and increases ADH release to further enhance water reabsorption.
Unlike secondary adrenal insufficiency, mineralocorticoid deficiency associated with primary adrenal insufficiency causes a hyperkalemic non–anion gap metabolic acidosis. This is due to retention of K+ and H+ that are normally excreted under aldosterone influence. The aldosterone deficiency also causes renal NaCl loss and associated volume (ECF) depletion. The resulting low ECV stimulates ADH release. There is also upregulation of collecting duct AQP2 and AQP3, which enhances ADH action. The combination of increased ADH secretion and augmented ADH responsiveness promotes the development of hyponatremia. A high-sodium diet compensates for the mineralocorticoid deficiency and improves the hyponatremia. Although hyponatremia may occur with both primary and secondary adrenal insufficiency, it occurs more commonly in primary adrenal insufficiency. This fact emphasizes the importance of aldosterone deficiency in renal salt wasting, volume depletion, and ADH secretion. All of these events combined with continued water intake synergistically contribute to hyponatremia.
23. What PNa concentrations are causes for concern?
The seriousness of hyponatremia or hypernatremia depends on the rapidity of development. Acute changes in PNa (within 48 hours) are always of more concern. Normal PNa ranges from 136 to 145 mEq/L. Patients with a PNa value of 115 or 165 mEq/L may not show any clinical features if the problem develops over several days to weeks. However, both conditions may produce major neurologic dysfunction if they develop over hours to days. As a rule, however, Na concentrations of 120 to 155 mEq/L are not usually associated with symptoms. PNa
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
