Disorders of growth
1. Summarize normal growth velocity for children until the pubertal growth spurt.

 Second 6 months: approximately 8 cm
Second 6 months: approximately 8 cm

 Third year: approximately 8 cm
Third year: approximately 8 cm

 Later childhood until puberty (5 to 10 years): growth averages 5 to 6 cm/year
Later childhood until puberty (5 to 10 years): growth averages 5 to 6 cm/year
2. Summarize growth velocity during the pubertal growth spurt.
Maximum growth rate is 11 to 13 cm/year.
In girls, growth spurt occurs early in puberty (breast Tanner stage II).
Growth spurt is later in boys (pubic hair Tanner stage III-IV, testicular volume 12-15 mL).
Some children may experience a transient period of slow growth just before the onset of puberty.
3. How is height measured accurately?
 The most essential tool for the detection of growth abnormalities is the ability to obtain accurate and reproducible measurements. This requires the availability of appropriate equipment as well as proper positioning of the patient.
The most essential tool for the detection of growth abnormalities is the ability to obtain accurate and reproducible measurements. This requires the availability of appropriate equipment as well as proper positioning of the patient.

 Children should be shoeless, and hair decorations or braids may need to be removed.
Children should be shoeless, and hair decorations or braids may need to be removed.

4. What technique is used for infants up to 2 years of age?
Supine length should be measured in infants. Accurate measurement requires a supine stadiometer, a boxlike structure with a headboard and movable footplate. Two people are needed, with one holding the infant’s head against the headboard while the other straightens the legs and places the ankles at 90 degrees against the movable footplate. The length is read from the attached measuring device, or marks are made for measurement by tape measure.
5. Describe the technique for children 2 years of age and older.
1. Standing height is measured. Accurate measurement requires a stadiometer with a rigid headboard, footplate, and backboard.
2. The child stands against the backboard, with heels, buttocks, thoracic spine, and head touching.
3. The measurer exerts upward pressure on the patient at the angle of the jaw to bring the spine into full stretch, and the headboard is lowered until it touches the top of the head. A counter reads the measurement.
4. If a stadiometer is not available, the child should stand against a wall in the same position as used for a stadiometer. A rigid right angle is moved downward to touch the top of the head, and a mark is made and measured.
5. Weight and head circumference (when appropriate) should be recorded.
The second critical tool for evaluation of growth is the standardized growth curve, and all measurements should be plotted rather than just recorded in the chart. A carefully constructed and up-to-date growth curve is critical to the recognition of growth abnormalities. Furthermore, the more points that are plotted on the curve, the greater the understanding of the child’s growth. Thus efforts should be made to obtain growth measurements at all patient contacts, including illness visits, because well-child visits are infrequent during the middle childhood years when growth abnormalities are most common.
7. List the common errors in plotting growth charts.
Errors in plotting of growth points are a frequent cause of apparent growth abnormalities. Common errors include:



8. What is meant by “appropriate growth chart”?
A number of growth charts are available, and careful consideration should be given to the appropriate chart for a particular patient at a particular time. Commonly available growth charts include:
 Charts for plotting supine length (the 0- to 36-month charts in common use)
Charts for plotting supine length (the 0- to 36-month charts in common use)
 Charts for plotting stature (i.e., standing height) (2- to 18-year charts)
Charts for plotting stature (i.e., standing height) (2- to 18-year charts)
Other specific growth charts are available and should be used when appropriate. These include:


9. How do age and position affect growth measurements?


10. What historic information is necessary for interpreting a growth chart?
 Birth history and birth weight
Birth history and birth weight
 Attainment of developmental milestones
Attainment of developmental milestones




 Height of biological parents and family history of significant short stature
Height of biological parents and family history of significant short stature
 Timing of parental puberty and family history of significant pubertal delay
Timing of parental puberty and family history of significant pubertal delay
11. What physical examination findings help interpret a growth chart?



 KEY POINTS 1: GENERAL GROWTH
KEY POINTS 1: GENERAL GROWTH
1. Proper evaluation of growth depends on accurate measurement of height and correct plotting of measurements on the appropriate growth curve.
2. Common errors in plotting include plotting the wrong height, not plotting the patient’s height at the exact chronologic age, and use of an inappropriate growth chart.
3. An abnormal growth velocity for age generally distinguishes growth abnormalities from normal growth variants.
4. Apparent abnormalities in growth are most frequently due to normal growth variants. Poor growth secondary to chronic medical illness is the next most frequent cause. Hormonal causes are less frequent.
12. How does radiologic imaging help interpret a growth chart?
 A bone-age film can provide important information about skeletal maturity. The degree of skeletal maturity is an important determinant of remaining growth potential and can help estimate expected height in children developing more slowly or more rapidly than their peers.
A bone-age film can provide important information about skeletal maturity. The degree of skeletal maturity is an important determinant of remaining growth potential and can help estimate expected height in children developing more slowly or more rapidly than their peers.

13. Explain the significance of parental target height or “midparental height.”
Parental height helps determine expected adult height on the basis of genetic potential. Add the parents’ heights in centimeters; add 13 cm if the child is male, and subtract 13 cm if the child is female; then divide by two. The resulting midparental height ±5 cm gives the 10th to 90th percentile for offspring of those parents.
14. What is the most important factor in identifying an abnormal growth curve?
An abnormal growth velocity for age generally distinguishes growth abnormalities from normal growth variants. Although there are many causes of short stature, including genetic, short normal children grow normally, whereas children with a problem almost always have an abnormal growth velocity. For example, a child with stature in the fifth percentile who is growing with a normal growth velocity is less worrisome than the child whose stature has fallen from the 90th to the 75th percentile, even though the latter is taller than the former. Growth velocity abnormalities may, however, be subtle.
15. What causes abnormal growth in children?
Abnormalities in growth are most frequently due to either normal growth variants (familial short stature or constitutional delay of growth and puberty) or underlying chronic medical illness, either recognized or unrecognized. Hormonal causes are less frequent.
16. Which syndromes are associated with abnormal growth?





17. List nonendocrine diseases and treatments that may be associated with poor growth.

 Pulmonary disease (cystic fibrosis, asthma)
Pulmonary disease (cystic fibrosis, asthma)


 Gastrointestinal disease (Crohn’s disease, inflammatory bowel disease)
Gastrointestinal disease (Crohn’s disease, inflammatory bowel disease)





18. Using the tools of growth curve, bone age, and height, how does one distinguish between familial (genetic) short stature and other causes?
Children with familial short stature grow at a normal velocity for age but with stature below the normal curve. They also grow within the expected target height percentile (i.e., they are as tall as expected for their genetic potential). If a child’s projected height (by extrapolation of the growth curve) falls within the target range, the likelihood is high that current height is explained by genetic factors. Children with familial short stature also have a bone age approximately equal to chronologic age.
19. Give an example of distinguishing familial short stature from other causes of short stature.
A 5-year-old whose height is below the third percentile, whose growth has traced a line parallel to the third percentile, whose height projects within the parental target range, and whose bone age is also 5 years is likely to have familial short stature. However, if the growth velocity is abnormal or projected height falls below the predicted range, other factors may be involved in the short stature (Figs. 25-1 and 25-2).
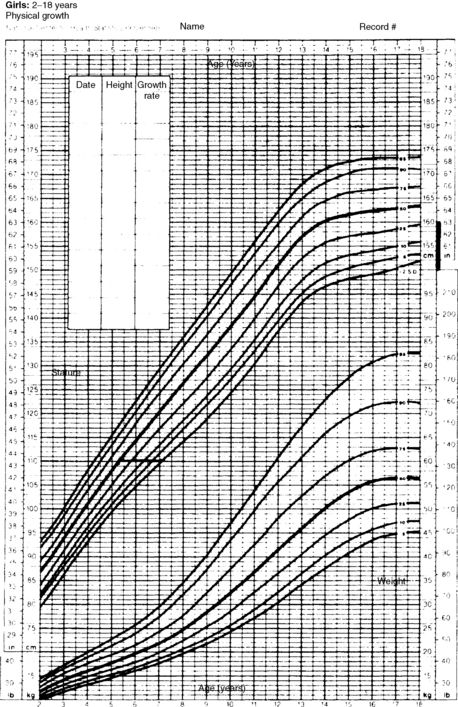
Figure 25-1. Growth chart for a 7-year-old girl with a height of 110 cm. Height age = 5 years, 3 months; bone age = 7 years; father’s height = 65 inches (165 cm); mother’s height = 62 inches (157 cm); corrected midparental height (± 1 SD) = 155 ± 5 cm; predicted adult height = 60 inches. The child has a predicted adult height within genetic potential and a bone age equal to chronologic age. She has genetic or familial short stature.
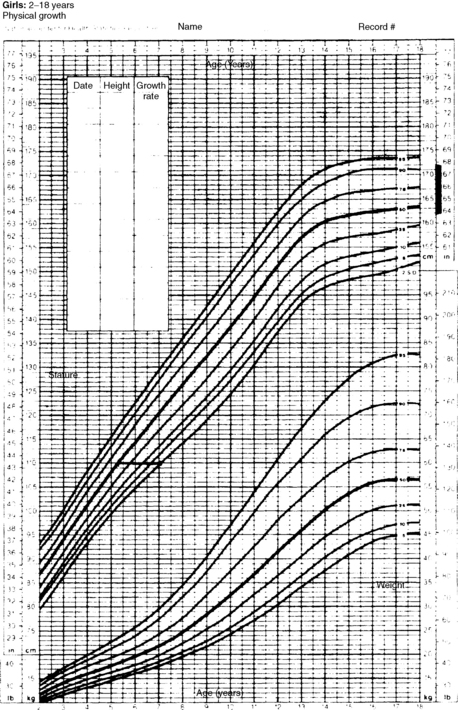
Figure 25-2. Growth chart for a 7-year-old girl with a height of 110 cm. Height age = 5 years, 3 months; bone age = 5 years; father’s height = 70 inches (178 cm); mother’s height = 66 inches (168 cm); corrected midparental height (± 1 SD) = 167. 5 ± 5 cm. The child is growing below the fifth percentile, but extrapolation of her growth curve to adult height gives a final height below genetic potential. Clearly her height cannot be attributed to genetic short stature alone.
20. Other than familial short stature, what is the most common cause of short stature?
Constitutional delay of growth (constitutional short stature), which affects up to 2% of children, is characterized by short stature and delayed bone age and represents a normal growth pattern simply shifted to a later age. Affected children typically have a period of subnormal growth between 18 and 30 months of age, followed by normal growth velocity throughout the remainder of childhood. In accord with the delayed developmental pattern, bone age is delayed. The continuing growth delay also results in a delay in pubertal development and physical maturity. Such children (often boys) generally have a family history of a similar growth pattern and may have a more dramatic deceleration of growth velocity before they enter puberty than normal children. They complete their growth at a later age, reaching an adult height within the expected genetic potential (Fig. 25-3).
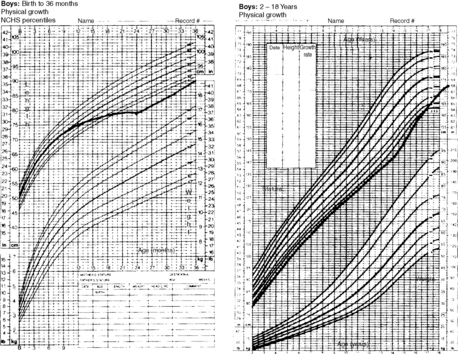
21. How is the diagnosis of constitutional delay of growth made?
The diagnosis of constitutional delay of growth based on the following criteria does not require further laboratory support:





22. What is the effect of testosterone therapy on boys with constitutional delay of growth?
Short-term testosterone therapy for boys with constitutional delay (75-100 mg of long-acting testosterone esters given once a month for 6 months) accelerates growth and stimulates pubertal development without compromising final adult height or advancing bone age. Clinically the boys experience pubertal changes, including genital enlargement (but not testicular growth), growth of pubic and axillary hair, deepening of voice, body odor, and acne. There may be personality changes characteristic of early puberty as well.
23. List the endocrine causes for short stature in children, in order of prevalence.
 Hypothyroidism: congenital or acquired
Hypothyroidism: congenital or acquired

 Glucocorticoid excess: iatrogenic or endogenous (less common)
Glucocorticoid excess: iatrogenic or endogenous (less common)

24. What laboratory measurements should be considered in evaluating a patient for short stature?
Laboratory tests should be designed to achieve two goals: (1) exclusion of undiagnosed chronic illness and (2) exclusion of specific disorders associated with poor growth.
25. Which laboratory tests help exclude undiagnosed chronic illness?





26. Which laboratory tests help exclude gastrointestinal disorders associated with poor growth?


27. List the laboratory tests for genetic disorders associated with poor growth.
 Karyotype (Turner’s syndrome): consider in all short girls
Karyotype (Turner’s syndrome): consider in all short girls
 Fluorescence in situ hybridization (FISH), for Prader-Willi syndrome
Fluorescence in situ hybridization (FISH), for Prader-Willi syndrome

28. Which hormonal disorders should be excluded by laboratory results?


29. Describe the causes of GH deficiency.
Most cases of GH deficiency are isolated and idiopathic. Idiopathic GH deficiency affects as many as 1:10,000 to 1:15,000 children. It is sporadic in the great majority of cases, but a rising number of specific gene mutations involved in the synthesis of GH or the regulation of its secretion is being reported. The other important underlying causes are listed in the following sections.
30. How is GH deficiency diagnosed?
The diagnosis of GH deficiency is primarily a clinical one, aided by laboratory support, rather than a diagnosis based on definitive testing. Most important is identifying the patient in whom such a diagnosis would be appropriate. Children with subnormal growth should be evaluated for GH deficiency only after a thorough search fails to reveal any other cause for growth delay.
31. List the components of the laboratory evaluation for GH deficiency.



32. Why is the serum level of IGF-1 important?
IGF-1 is a GH-dependent protein that is produced in target tissues in response to GH. Serum level of IGF-1 reflects production of the protein by the liver and gives an indirect indication of GH secretion. The following characteristics of IGF-1 should be kept in mind when its serum level is assessed:



33. Does a normal level of IGF-1 exclude GH deficiency?
A normal IGF-1 level is reassuring but does not rule out partial GH deficiency in the appropriate clinical context.
34. Does a low serum level of IGF-1 confirm the diagnosis of GH deficiency?
No. Poor nutrition, chronic disease, and hypothyroidism suppress IGF-1 concentrations. In addition, before the age of 6 years, the IGF-level is low, and the overlap between normal and GH-deficient levels renders its measurement highly insensitive.
Because secretion of GH is episodic, random measurements are not helpful for the diagnosis of GH deficiency. GH must be formally measured in response to a series of stimuli. Various pharmacologic agents are used, but there is no consensus about which is optimal. The child must have fasted overnight, must be euthyroid, and must have no underlying chronic disease. In addition, at least two tests using different stimulating agents are generally performed.
36. How are the results of GH testing interpreted?
The normal GH response to stimulation testing depends on the stimulation test and the type of GH assay used. Failure of response to all tests with values equal to or greater than those expected for normal children is consistent with the diagnosis of classic GH deficiency.
Criteria for the diagnosis of partial GH deficiency and of neurosecretory dysfunction (normal pituitary response to stimuli, but low IGF-1, suggesting that endogenous GH secretion is impaired) are less well established.
37. How is idiopathic GH deficiency diagnosed?
GH deficiency can be isolated or associated with other pituitary hormone deficiencies. It can be congenital or can result from trauma or an intracranial neoplasm. All patients diagnosed with GH deficiency should undergo cranial imaging, unless the cause of the deficiency is previously known. Isolated GH deficiency without identifiable etiology is considered idiopathic.
38. How is GH deficiency treated?
GH for administration is available through recombinant DNA technology; the majority of children are treated with 6 or 7 daily shots per week at a total weekly dose of approximately 0.30 mg/kg administered subcutaneously. Because the effect of GH wanes after several years of therapy, it is common to see dramatic catch-up growth (<10-12 cm/year) in the first or second year of therapy, followed by velocities ranging from normal to 1.5 times normal in subsequent years.
39. What is the prognosis for adult height in treated children with GH deficiency?
Although nearly all treated children reach an adult height significantly better than predicted before therapy is initiated, many do not reach their predicted genetic potential. Children diagnosed and treated at earlier ages have better height prognoses than those whose therapy is initiated later. Similarly, the more mature the skeleton at diagnosis, the poorer the final outcome.
40. When is GH therapy discontinued?
In children with GH deficiency, the point of diminishing benefit of therapy correlates with skeletal maturity rather than chronologic age or duration of therapy. Therapy often is discontinued at a bone age of 15 years (96% of growth) to 16 years (98% of growth) in boys and 14 years (98% of growth) in girls. However, given what is now known about the effects of GH deficiency in adulthood, some patients with severe deficiencies may require lifelong hormonal replacement.
41. What other syndromes are considered indications for GH therapy?
GH is now approved by the U.S. Food and Drug Administration (FDA) for the treatment of short stature in the following conditions:
1. Chronic renal insufficiency before transplant
2. Turner’s syndrome (45,XO or mosaic variants)
3. Acquired immunodeficiency syndrome–related wasting syndrome
6. Short stature due to intrauterine growth retardation in the absence of catch-up growth
7. Idiopathic short stature in boys with predicted adult height less than 63 inches and girls with predicted height less than 59 inches (normal GH secretion)
Indications 2 through 6 do not require demonstration of GH deficiency. The use of GH for treatment of idiopathic short stature remains controversial among pediatric endocrinologists.
42. What is the prognosis for girls with Turner’s syndrome treated with GH?
Girls with Turner’s syndrome generally demonstrate a significant increase in predicted adult height, with an average increase of 8.8 cm. The overall effectiveness of therapy, like that in GH deficiency, depends on chronologic age and bone age at initiation of treatment and on duration of treatment. Because GH therapy in Turner’s syndrome normalizes height in younger girls, estrogen replacement therapy can be initiated at an age similar to the age of puberty of the patient’s peers.
43. What are the potential risks of GH therapy?
The side effects of GH therapy can be divided into three categories: (1) common but clinically unimportant, (2) uncommon with potential clinical importance, and (3) rare or theoretical.
44. List the common but clinically unimportant side effects of GH therapy.
 Acute correction of body water deficit after initiation of GH in deficient patients may lead to transient peripheral edema, headache, and joint aches and stiffness.
Acute correction of body water deficit after initiation of GH in deficient patients may lead to transient peripheral edema, headache, and joint aches and stiffness.


45. List the uncommon side effects with potential clinical importance.




46. What rare or theoretical side effects may be associated with GH therapy?
 Increased risk for development of a secondary neoplasm: Reports now suggest a small increase in the long-term risk of secondary development of meningioma in childhood cancer survivors treated with GH.
Increased risk for development of a secondary neoplasm: Reports now suggest a small increase in the long-term risk of secondary development of meningioma in childhood cancer survivors treated with GH.

47. Should children with idiopathic short stature (without GH deficiency) be treated with GH?
The FDA has approved the use of GH in children with idiopathic short stature with a predicted adult height less than 63 inches for boys and less than 59 inches for girls. However, the use of GH in children in whom no hormonal abnormality can be demonstrated continues to be intensely controversial among pediatric endocrinologists. Short-term studies involving small cohorts have demonstrated a consistent increase in growth velocity with GH therapy in such children. Several studies that monitored children to final height disagreed about the overall effectiveness of therapy. However, most studies agree that the increase in final adult height is limited and can be obtained only at significant financial cost. The decision to use GH in such children should be carefully considered and requires a thoughtful dialogue among child, family, and an experienced pediatric endocrinologist who knows the child well.
48. How does the pattern of growth in children with excessive glucocorticoids differ from the pattern in children with exogenous obesity?
Glucocorticoid excess, whether iatrogenic (common) or intrinsic (rare), results in impairment of linear growth. The mechanism reflects increased protein catabolism, increased lipolysis, and a decline in collagen synthesis. Glucocorticoids also suppress the pulsatile release of GH from the pituitary gland and the production of IGF-1 at the target organ. The net result is that children with steroid excess are frequently short. They also have an increased weight-to-height ratio and appear obese. Children with exogenous obesity, on the other hand, generally show accelerated linear growth; thus, they are not only obese but also tall for age.
49. What conditions are associated with excessive growth in childhood?
Relatively few conditions result in overgrowth during childhood. These include familial tall stature (stature appropriate for parental target), constitutional advanced growth, hormonal causes, and genetic syndromes.
50. Explain constitutional advanced growth.
Constitutional advanced growth is associated with advanced bone age, accelerated growth, and early puberty, with predicted adult height appropriate for parental target (see question 21). Obesity and familial factors may be involved.
51. List the hormonal causes of excessive growth.




52. Summarize the characteristics of GH excess in childhood.
GH excess is rare in children, in whom it causes tall stature (gigantism) rather than the bony overgrowth seen in adults (acromegaly). Diagnosis is based on the following laboratory results:



53. With what findings is androgen excess associated?



54. With what findings is estrogen excess associated?


55. List the genetic syndromes associated with excessive growth.
 Klinefelter’s syndrome (47,XXY): tall stature, small testes, delay of puberty
Klinefelter’s syndrome (47,XXY): tall stature, small testes, delay of puberty

 Marfan’s syndrome: tall stature, arachnodactyly, joint laxity, lens displacement
Marfan’s syndrome: tall stature, arachnodactyly, joint laxity, lens displacement


 Beckwith-Wiedemann syndrome: macroglossia, umbilical hernia, hypoglycemia, macrosomia in infancy
Beckwith-Wiedemann syndrome: macroglossia, umbilical hernia, hypoglycemia, macrosomia in infancy
 Homocystinuria: arachnodactyly, retardation, homocystine in urine
Homocystinuria: arachnodactyly, retardation, homocystine in urine
 KEY POINTS 2: GROWTH VARIANTS
KEY POINTS 2: GROWTH VARIANTS
1. Children with familial short stature grow at a normal velocity for age and within their expected target height percentile and have a bone age approximately equal to chronologic age.
2. Children with constitutional delay of growth have a period of slow growth in the second year of life but then grow with a normal growth velocity.
3. Children with constitutional delay of growth also have delayed bone age, height prediction appropriate for family, and delayed entry into puberty.
4. The diagnosis of a growth variant does not require laboratory confirmation, but growth should be followed over time to confirm the initial impression.
 KEY POINTS 3: GROWTH HORMONE DEFICIENCY
KEY POINTS 3: GROWTH HORMONE DEFICIENCYCarel, JC, Management of short stature with GnRH agonist and co-treatment with growth hormone. a controversial issue. Mol Cell Endocrinol 2006;254:226–233.
Clayton, PE, Cianfarani, S, Czernichow, P, et al, Management of the child born small for gestational age through to adulthood. a consensus statement of the International Societies of Pediatric Endocrinology and the Growth Hormone Research Society. J Clin Endocrinol Metab 2007;92:804–810.
Cytrynbaum, CS, Smith, AC, Rubin, T, et al, Advances in overgrowth syndromes. clinical classification to molecular delineation in Sotos syndrome and Beckwith-Wiedemann syndrome. Curr Opin Pediatr 2005;17:740–746.
Davenport, ML. Evidence for early initiation of growth hormone and transdermal estradiol therapies in girls with Turner syndrome. Growth Horm IGF Res. 2006;16:591–597.
Lee, MM. Clinical practice. Idiopathic short stature. N Engl J Med. 2006;354:2576–2582.
Myers, SE, Carrel, AL, Whitman, BY, et al. Sustained benefit after 2 years of growth hormone on body composition, fat utilization, physical strength and agility, and growth in Prader-Willi syndrome. J Pediatr. 2000;137:42–49.
Quigley, CA. Growth hormone treatment of non-growth hormone deficient disorders. Endocrinol Metab Clin North Am. 2007;36:131–186.
Rosenbloom, AL, Connor, EL. Hypopituitarism and other disorders of the growth hormone-insulin like growth factor-1 axis. In: Lifshitz, F, eds. Pediatric endocrinology, vol. 2. New York: Informa Healthcare; 2007:65–100.
Zeitler, PS, Meacham, LR, Allen, DB, et al, Principles and Practice of Pediatric Endocrinology. Springfield (IL): Charles C Thomas; 2005;857–910.
