Chapter 471 von Willebrand Disease
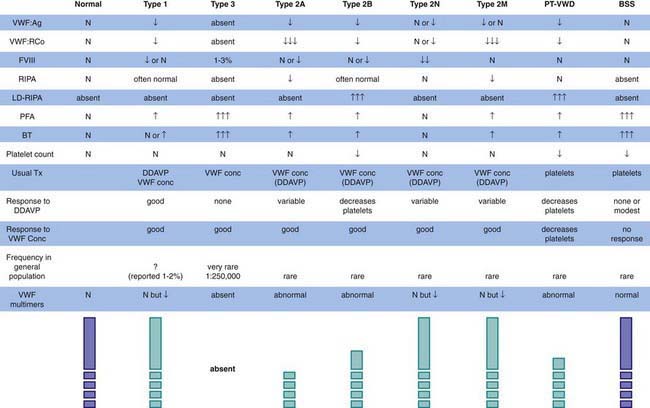
The most common hereditary bleeding disorder is von Willebrand disease (VWD), and some reports suggest that it is present in 1-2% of the general population. VWD is inherited autosomally, but most centers report more affected women than men. Because menorrhagia is a major symptom, women may be more likely to seek treatment and thus to be diagnosed. VWD is classified on the basis of whether the protein is quantitatively reduced, but not absent (type 1); qualitatively abnormal (type 2); or absent (type 3) (Fig. 471-1). Mutations in different loci that code for different functional domains of the von Willebrand factor (VWF) protein cause the different variants of VWD.
Laboratory Findings
Although patients with VWD were described historically as having a long bleeding time and a long partial thromboplastin time, these findings are frequently normal in patients with type 1 VWD. Normal results on screening tests do not preclude the diagnosis of VWD. Because there is no single assay that has demonstrated the ability to rule out VWD, if the history is suggestive of a mucocutaneous bleeding disorder, VWD testing should be undertaken, including a quantitative assay for VWF antigen, testing for VWF activity (ristocetin cofactor activity), testing for plasma factor VIII activity, determination of VWF structure (VWF multimers), and a platelet count. Although the platelet count is usually normal in most patients, those with type 2B disease or platelet-type disease (pseudo-VWD) may have lifelong thrombocytopenia. Figure 471-1 lists the variants of VWD and summarizes their laboratory findings. Levels of VWF vary with blood type (type O < A < B < AB), which can confound the clinical diagnosis of hereditary VWD, but most clinicians feel bleeding is related to the plasma level of VWF. In addition, there is controversy regarding the clinical definition of “true” VWD. Molecular genetics may clarify the diagnosis of type 1 VWD, but other genetic modifiers may exist outside the gene for VWF and significantly influence the diagnosis. The milder the patient’s phenotype, the greater the difficulty in diagnosis.
Genetics
Chromosome 12 contains the gene for VWF. In each of the type 2 variants listed in Figure 471-1, specific areas of the molecule are affected. The phenotype can guide the genetic diagnosis of the specific mutation. Investigations are underway to clarify whether all cases of type 1 VWD are related to mutations in the VWF gene on chromosome 12 or if there are genetic modifiers, such as blood type, that cause phenotypic VWD. Clinical genetic testing for VWD variants is only available in a few referral laboratories.
Von Willebrand Disease Variants
Diagnosis and Differential Diagnosis
The diagnosis of VWD is dependent on the finding of a low level of at least 1 of the laboratory measures of VWF noted in Figure 471-1. The differential diagnosis of mucocutaneous bleeding includes abnormalities of platelet number, platelet function, or the vessel wall (Chapter 478). In caring for children, it is important to remember that the most common cause of such findings is trauma, especially nonaccidental trauma—child abuse.
Favaloro EJ. Toward a new paradigm for the identification and functional characterization of von Willebrand disease. Semin Thromb Hemost. 2009;35:60-75.
Federici AB, Mannucci PM, Castaman G, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood. 2009;113:526-534.
Haberichter SL, Balistreri M, Christopherson P, et al. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood. 2006;108:3344-3351.
Mannucci PM. Treatment of von Willebrand’s disease. N Engl J Med. 2004;351:683-694.
Montgomery RR, Cox Gill J, Di Paola J. Hemophilia and von Willebrand disease. In: Orkin SH, Nathan DG, Ginsburg D, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders Elsevier; 2009:1487-1524.
Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008;14:171-232.
Sadler JE. von Willebrand factor: two sides of a coin. J Thromb Haemost. 2005;3:1702-1709.
Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103-2114.
Shankar M, Lee CA, Sabin CA, et al. von Willebrand disease in women with menorrhagia: a systematic review. BJOG. 2004;111:734-740.
Werner EJ. von Willebrand disease in children and adolescents. Pediatr Clin North Am. 1996;43:683-707.





