CHAPTER 109 VITAMIN DEFICIENCIES AND OTHER NUTRITIONAL DISORDERS OF THE NERVOUS SYSTEM
A vitamin is a substance that serves as a cofactor for a biochemical reaction and whose absence causes some derangement of function (Fig. 109-1). Thiamine is a classic example, required by three enzyme systems that are essential for glucose metabolism. However, the term vitamin deficiency is too restrictive to account for all disorders of nutrition with neurological consequences. A number of syndromes are associated with megadoses of vitamins (pyridoxine, zinc) and dietary supplements (Chinese herbal remedies, St. John’s Wort, ephedra) (Table 109-1). The recognition of mineral deficiencies such as copper myeloneuropathy, mineral excess disorders such as zinc-induced copper deficiency, and polynutritional disturbances such as postgastroplasty neuropathy requires that a discussion of “vitamin deficiency” be broadened to include other nutritional syndromes of the nervous system.
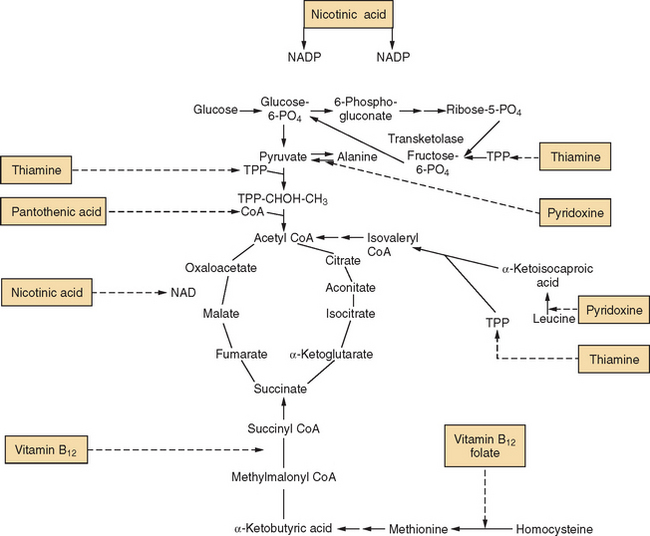
TABLE 109-1 Toxins Associated with Nutritional Syndromes
| Alcohol | Wernicke-Korsakoff syndrome, painful polyneuropathy |
| Zinc | Copper deficiency myelopathy |
| Pyridoxine | Large-fiber sensory neuropathy |
| Nitrous oxide | Cobalamin deficiency myelopathy |
| Methyltetrahydrofolate reductase (MTHFR) deficiency encephalopathy | |
| Vitamin A | Optic neuropathy, pseudotumor cerebri |
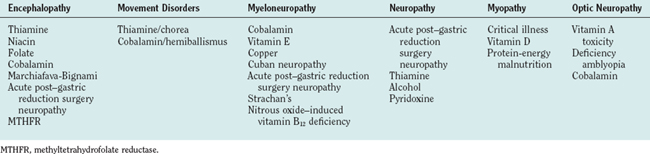
The diagnosis of a nutritional disorder of the nervous system may be challenging for a number of reasons. First, multiple deficiencies may coexist in the same patient, as in dietary malnutrition, postgastroplasty neuropathy, and malabsorption states. Some syndromes that manifest acutely may not be interpreted as nutritional (Tables 109-2 and 109-3). Instead of evolving in a slowly progressive manner, some deficiency states may have an explosive onset triggered by an environmental stressor or by a sudden increase in metabolic demands for the deficient nutrient.
TABLE 109-2 Acute and Subacute Manifestations of Nutritional Disorders
| Neurological Syndrome | Mechanism | Time Course |
|---|---|---|
| Acute post–gastric reduction surgery neuropathy | Excessive and prolonged vomiting, severe weight loss | Weeks to months after surgery |
| Wernicke-Korsakoff syndrome | IV glucose administration in a thiamine-deficient patient, inducing sudden demand for thiamine-dependent glycolytic enzymes | Hours |
| Wernicke-Korsakoff syndrome | Excessive and prolonged vomiting, severe weight loss (gastric surgery, hyperemesis gravidarum) | Weeks |
| Nitrous oxide–associated myeloneuropathy | N2O oxidizes cobalt core of cobalamin, affects vitamin B12–deficient patients acutely or vitamin B12–replete patients with multiple exposures | Hours |
| Pyridoxine neuropathy | Unknown, but probably multiple redox reactions | Hours to days |
IV, intravenous.
The underrecognition of nutritional disorders in industrialized countries has led to difficulties in diagnosis, and these deficiencies may be more common than has been clinically appreciated. Thiamine deficiency has been reported in up to 17% of hospitalized elderly individuals1 and is documented in 3% of autopsy series.2,3 One study of thiamine-deficient alcoholic subjects demonstrated that more than 50% were also riboflavin deficient, and 2% had a concomitant deficiency of pyridoxine.4 With the increasing popularity of obesity-related surgery, new neurological syndromes have emerged as a result of postoperative polynutritional deficiency states (such as acute post–gastric reduction surgery neuropathy, described later). Cobalamin deficiency occurs in 5% to 14% of ambulatory elderly persons,5,6 and up to 27% of hospitalized elderly people develop protein-energy malnutrition during their hospital stay.7
Finally, several inherited enzyme deficiency disorders, although not accompanied by a vitamin deficiency, may nonetheless be vitamin responsive (Table 109-4). Homocystinemia responds to pharmacological doses of folate, cobalamin, and pyridoxine, whereas methylmalonic acidemia responds to cobalamin. Patients with mitochondrial cytopathology may respond to large doses of thiamine (300 mg/day).
TABLE 109-4 Hereditary Disorders Responsive to Vitamin Therapy
| Thiamine | |
| Cobalamin, folate, pyridoxine | Methylmalonic acidemia |
| Homocystinemia | |
| Biotin | β-Methylcrotonyl glycinemia |
| Propionic acidemia | |
| Niacin | Hartnup’s disease (tryptophan metabolism) |
MELAS, mitochondrial encephalopathy, lactic acidosis, and strokelike episodes; MERRF, myoclonic epilepsy associated with ragged-red fibers.
VITAMIN DEFICIENCIES
Thiamine
Pathogenesis and Pathophysiology
The metabolically active form of thiamine, thiamine pyrophosphate (TPP), is crucial in the intermediary metabolism of carbohydrate. TPP is involved in three enzyme systems: (1) pyruvate dehydrogenase, which converts pyruvate to acetyl coenzyme A; (2) α-ketoglutarate dehydrogenase, which catalyzes the conversion of α-ketoglutarate to succinate in the Krebs cycle; and (3) transketolase, which catalyzes the pentose monophosphate shunt (see Fig. 109-1). A deficiency of TPP leads to elevated levels of serum pyruvate and lactate, reduced red blood cell transketolase activity, and a corresponding increase in transketolase activity in response to added TPP (“TPP effect”).8
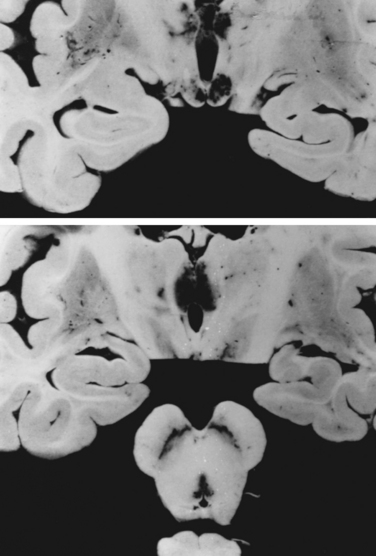
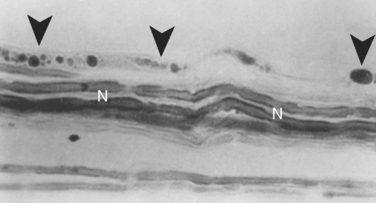
Pathologically, patients with Wernicke-Korsakoff syndrome show capillary proliferation and petechiae. Spongy degeneration of astrocytes with neuronal preservation occurs in midline structures of the brain, such as the medial thalamic nuclei, the mammillary bodies, the periaqueductal gray area of the mesencephalon, and the pontine tegmentum (Fig. 109-2). Degeneration of the superior cerebellar vermis is frequent. The lesions in the thalami and mammillary bodies probably account for the confusion, memory loss, and confabulation. The pontine tegmental lesions may cause the oculomotor palsies, and the truncal ataxia may result from the midline cerebellar degeneration.9 Cellular injury in these regions may be caused by inhibition of adenosine triphosphate synthesis and induction of abnormal carbohydrate metabolism. In thiamine deficiency polyneuropathy, nerves show axonal degeneration with secondary demyelination. The neuropathy of dry beriberi may be related to TPP deficiency–induced impairment of nerve excitability and conduction.10,11
Epidemiology and Risk Factors
Thiamine is most abundant in yeast, pork, legumes, cereal grains, and unpolished rice, and the recommended daily allowance of this vitamin is 0.5 mg/1000 kcal.12 The total body store is 30 to 100 mg, and thiamine is present in heart, skeletal muscle, liver, kidney, and brain tissue. Because the quantity stored is limited, the supply must be constantly replenished. The half-life of thiamine is approximately 2 weeks, and patients may suffer severe neurological complications and even death after 6 weeks of total thiamine depletion. Patients at high risk for deficiency include alcoholic persons, adults who derive most of their carbohydrate from white rice, and infants breastfed by malnourished mothers. Other potential causes of thiamine deficiency include prolonged total parenteral nutrition,13 defective baby formula,14 hyperemesis gravidarum,15 anorexia nervosa, gastric or jejunoileal bypass,16,17 intractable vomiting after gastric stapling for morbid obesity,18 and severe malabsorption. Thiamine deficiency is also found among prisoners of war19 and persons engaged in hunger strikes. In addition, thiamine deficiency has been reported after long-standing peritoneal or hemodialysis.20,21
Clinical Features and Associated Disorders
Wernicke-Korsakoff syndrome and polyneuropathy (dry beriberi) are the two neurological disorders resulting from thiamine deficiency. The classic triad of confusion, ataxia, and oculomotor palsies is uncommon in clinical practice. In Harper and colleagues’ autopsy series of 131 patients, only 16% had all three features premorbidly.3
The frequency of Wernicke-Korsakoff syndrome in autopsy series ranges from 0.8% to 2.8%. The disorder is probably underdiagnosed during life. Wernicke-Korsakoff syndrome is most common in alcoholic persons as a result of a combination of poor diet, inadequate intake, impaired absorption of thiamine, and overdependence on alcohol as a source of calories. Certain individuals may also have a genetic predisposition toward the development of this syndrome because of an abnormality of thiamine-dependent enzymes.22,23
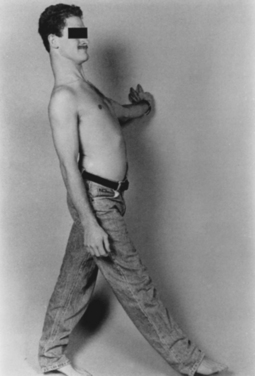
Polyneuropathy is present in more than 80% of patients with Wernicke-Korsakoff syndrome, but most cases are probably caused by alcoholism. Koike and associates demonstrated that thiamine deficiency and alcohol-induced neuropathy are distinct entities.11 Thiamine deficiency may manifest acutely with prominent motor weakness and large-fiber sensory loss. In contrast, alcoholic neuropathy manifests with slowly progressive muscular weakness and with sensory and reflex loss, accompanied by burning sensation in the feet and lancinating pains. Calf tenderness is a prominent feature. Bilateral footdrop and even wristdrop may be present (Fig. 109-3). Half of Koike and associates’ patients had evidence of autonomic neuropathy with orthostatic hypotension.

Evaluation
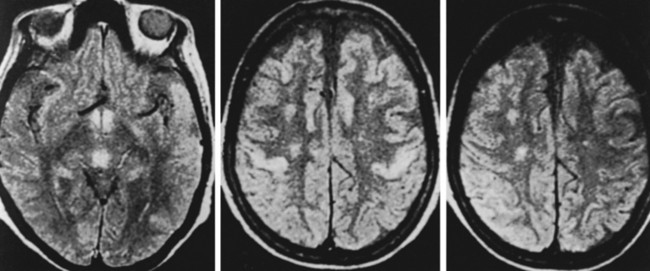
Serum thiamine levels lack sufficient sensitivity and specificity to be used alone. Red blood cell transketolase activity, with or without TPP challenge, is the most accurate assessment tool,8 but the test has become commercially unavailable. Magnetic resonance imaging may reveal abnormal signal in the midline nuclei corresponding to the pathological lesions described (Fig. 109-4).24
Niacin and Nicotinic Acid
Niacin includes both nicotinic acid and nicotinamide, which form the metabolically active nicotinamide adenine dinucleotide (NAD) and NAD phosphate (NADP), an end product of tryptophan metabolism. More than 200 enzymes are dependent on NAD and NADP to carry out oxidation and reduction reactions, and these enzymes are involved in the synthesis and breakdown of carbohydrates, lipids, and amino acids. Although niacin is endogenously produced in humans, exogenous intake is necessary to prevent deficiency.
Pellagra, or rough skin, continues to occur in parts of Africa and Asia, especially in populations dependent on corn as the principle source of carbohydrate. When corn is first soaked in lime water, as is done in Mexico when tortillas are prepared, niacin is liberated, and deficiency occurs less commonly. In the United States, niacin deficiency is seen in alcoholic persons and in patients taking isoniazid. Pregnant women are protected from niacin deficiency because of their enhanced ability to convert tryptophan to niacin endogenously, particularly in the third trimester.
Pellagra affects the skin, the gastrointestinal system, and the central nervous system; hence, the classic triad of the “three Ds”: dermatitis, diarrhea, and dementia. In industrialized countries, particularly among alcoholic persons, niacin deficiency may manifest only with encephalopathy.25–27 Patients may have altered sensorium, diffuse rigidity of the limbs, and grasping and sucking reflexes. Dementia and confusion are the most constant findings, followed by diarrhea (50%), and dermatitis (30%).27 Spinal cord and peripheral nerve defects have also been reported, particularly in prisoners of war.19 Coexisting deficiencies of thiamine and pyridoxine are common, especially in alcoholic persons.
Cobalamin (Vitamin B12)
Pathogenesis and Pathophysiology
Vitamin B12 deficiency produces neurological and hematological symptoms by impairing two enzyme systems (Fig. 109-5).5,28 Methylcobalamin is a cofactor of methionine synthase, a cytosolic enzyme that catalyzes the conversion of homocysteine and methyltetrahydrofolate to produce methionine and tetrahydrofolate. Methionine is further metabolized to S-adenosylmethionine, which is necessary for the methylation of myelin sheath phospholipids and proteins. Tetrahydrofolate is the required precursor for purine and pyrimidine synthesis. In the mitochondria, adenosylcobalamin catalyzes the conversion of L-methylmalonyl–coenzyme A to succinyl–coenzyme A.
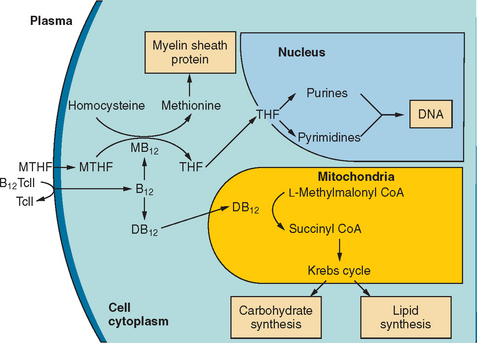
In deficiency states, serum levels of homocysteine and methylmalonic acid rise. Although the mechanism of megaloblastic changes in both folate and cobalamin deficiency is reasonably well understood, the biochemical basis of the neurological damage that occurs in cobalamin deficiency remains uncertain. Of the two reactions that require cobalamin, the methionine synthase reaction is considered more likely to play a critical role in nervous system function. In rare cases, neurological complications have also been reported in folate deficiency, because methionine synthase also requires this cosubstrate.29 It has been proposed that the accumulation of methylmalonate and propionate provides abnormal substrates for fatty acid synthesis, resulting in abnormal odd-carbon and branched-chain fatty acids, so-called funny fatty acids, which may be incorporated into the myelin sheath and interfere with impulse conduction.
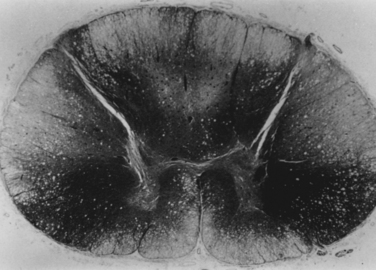
Vitamin B12 deficiency results in demyelination of the posterior columns, corticospinal tracts, and white matter of the cerebral hemispheres (Fig. 109-6).30 Less commonly, a sensorimotor and autonomic neuropathy that is axonal and demyelinating in nature may also be present.31,32 These lesions lead to a constellation of symptoms, including cognitive and affective disorders, ataxia, spasticity, and paresthesias.
Epidemiology and Risk Factors
In the Framingham Heart Study, 5% of elderly subjects had serum vitamin B12 levels less than 148 pmol/L, 40.5% had values less than 258 pmol/L, and 15% had serum methylmalonic acid levels greater than 376 nmol/L.33 Therefore, 15% of the elderly population demonstrate biochemical evidence of vitamin B12 deficiency. Folate deficiency is far more common in alcoholism than is cobalamin deficiency. Of all cases of folate deficiency, 87% occur in alcoholic persons, whereas only 11% to 13% of cobalamin-deficient patients are alcoholic.34,35 For unclear reasons, vitamin B12 deficiency is uncommon in alcoholics. Anemia and macrocytosis (mean cell volume >100 fL) occur in 72% and 83%, respectively, of patients with vitamin B12 deficiency and in 100% and 75%, respectively, of those with folate deficiency.35
Approximately 50% to 78% of patients with vitamin B12 deficiency have autoimmune parietal cell dysfunction (pernicious anemia).36 Another 10% to 40% have food-bound cobalamin malabsorption, caused by achlorhydria.37 The rest have a variety of causes, mainly malabsorption from medical, surgical, or pharmacological impairments of gastric acid or intrinsic factor secretion. These include patients after gastric surgery38 and after bypass procedures for weight reduction,39 as well as those taking long-term H2 blocker therapy, as a result of inhibition of acid secretion.40 Nitrous oxide administration may precipitate acute vitamin B12 deficiency in patients with asymptomatic low cobalamin levels.28,41 Other causes of vitamin B12 deficiency include ileal resection, parasitic infestations from Diphyllobothrium latum, Crohn’s disease and other malabsorption states, short gut syndrome, a vegan diet, chronic alcoholism with poor diet, and rare congenital enzyme deficiencies.
Clinical Features and Associated Disorders
Subacute combined degeneration of the spinal cord, peripheral nerve dysfunction, and cerebral dysfunction are classic features of the disorder. Many patients present without accompanying anemia or macrocytosis. Healton and associates reviewed 143 patients who had 153 episodes of cobalamin deficiency.42 On examination, 25% of patients demonstrated neuropathy; 12%, isolated myelopathy; and 41%, combined neuropathy and myelopathy. In 65% of patients, peripheral neuropathic symptoms and signs were combined with other manifestations such as myelopathy, cortical dysfunction, and autonomic dysfunction. In only 3% was neuropathy the only abnormality. In approximately 2% of all patients with peripheral neuropathies seen at referral centers, vitamin B12 deficiency was a primary cause.32 Memory dysfunction and affective and behavioral changes were seen in 8%. Cognitive deficits included psychosis, affective disturbances, and memory disturbances, as well as changes in personality. Orthostatic hypotension has been reported and may result from a disordered release of norepinephrine.43,44 Fourteen percent of patients with cobalamin deficiency had normal examination findings.
Patients may develop a myeloneuropathy after exposure to nitrous oxide (Table 109-5).28,41,45 This occurs in two populations: those who have normal cobalamin levels but chronically abuse the anesthetic gas for recreational purposes, and those with a subclinical vitamin B12 deficiency who, after a short exposure to nitrous oxide for a dental or surgical procedure, develop paresthesias, burning sensation in the feet, and ataxia. Nitrous oxide is a potent oxidizing agent, which irreversibly oxidizes the cobalt core of cobalamin from a 1+ to 3+ valence state, rendering methylcobalamin inactive. This effectively inhibits the conversion of homocysteine to methionine, thus blocking the supply of S-adenosylmethionine.
TABLE 109-5 Differential Diagnosis for a Myeloneuropathy (“Absent Ankle Jerks and Upgoing Toes”)
Low cobalamin levels have also been found in occasional patients with multiple sclerosis46 and human immunodeficiency virus infection47; however, no pathogenic relationship or treatment response has been established in either disorder.
Differential Diagnosis
Twenty-five percent of patients with vitamin B12 deficiency have no anemia or macrocystosis.42 The most characteristic manifestations are gait dysfunction and paresthesias in an elderly individual. The hallmark findings of myeloneuropathy, absence of ankle reflexes, and extensor plantar responses, are present in only 41% of patients.42
Vitamin B12 deficiency must be differentiated from other causes of myelopathy and neuropathy.
Evaluation
An algorithm for the diagnosis and treatment of cobalamin deficiency (Fig. 109-7) is based on two assumptions: (1) Normal results of a cobalamin assay do not fully rule out cobalamin deficiency, and (2) the normal range may vary, depending on the assay type. Many laboratories have switched from radioassay to chemiluminescence assay, which may have a higher normal reference range (250 to 1100 pg/mL).

When cobalamin deficiency is suspected, it is practical to measure methylmalonic acid at the same time as cobalamin. Methylmalonic acid has greater specificity for vitamin B12 deficiency than does homocysteine. If the vitamin B12 level is low and the methylmalonic acid level is elevated, the patient is likely to have true vitamin B12 deficiency. False-positive elevations of the methylmalonic acid level are most commonly caused by renal insufficiency.48 A diagnosis of pernicious anemia (autoimmune parietal cell destruction) can be made by finding intrinsic factor antibodies. Unfortunately, this test has low sensitivity (40% to 60%). An elevated serum gastrin level may indicate achlorhydria, pernicious anemia, or food-bound cobalamin malabsorption. A Schilling test helps distinguish the condition as pernicious anemia, food-bound cobalamin malabsorption, or an ileal malabsorption problem.59
Management
Treatment may begin with intramuscular injections of cobalamin, 1000 μg/day for 5 days and then 500 to 1000 μg intramuscularly every month. Oral replacement is an alternative for patients who cannot tolerate intramuscular injections or for whom they are impractical. Kuzminski and colleagues (1998) demonstrated that 2000 μg of oral vitamin B12 is as effective as or more so than intramuscular shots given monthly for maintaining normal serum vitamin B12 level and for correcting elevation in serum methylmalonic acid level, with a comparable onset of action.50 It may be practical to replenish cobalamin stores first by using injections of cyanocobalamin for 1 week and then shifting to maintenance with 1000-μg daily oral supplements.
Folate (Vitamin B9)
Although folic acid deficiency has long been recognized as an important contributor to neural tube defects, less well appreciated is its role in cognition and depression. Although both folate and vitamin B12 deficiencies may cause megaloblastic anemia, the neuropsychiatric disturbances may differ. Whereas vitamin B12 deficiency is more likely to result in subacute combined degeneration and neuropathy, folate deficiency is more likely to cause depression and dementia.
Folate deficiency may result more frequently in elderly persons from a combination of factors, including poor diet, malabsorption, medications, and increased demand. Of the patients admitted to a geriatric unit, 16% showed evidence of folate deficiency, and this was correlated with both depression and cognitive decline.52
In 1998, folate supplementation of the food supply became mandatory in order to prevent neural tube defects. An unexpected benefit has been a 50% decline in the prevalence of homocysteine elevations in the United States.53 What effect this may have on the incidence of atherosclerosis, cerebrovascular events, and dementia remains to be seen. The link between folate deficiency and elevated homocysteine levels and vascular disease is well established. Less clear is whether aggressive suppression of homocysteine levels by folate supplementation reduces cardiovascular and cerebrovascular events. The Vitamin Intervention in Stroke Prevention (VISP) trial demonstrated that moderate reduction of total homocysteine through the use of folate, cobalamin, and pyridoxine supplements in patients with nondisabling cerebral infarction had no effect on vascular outcomes after 2 years.54
Pyridoxine (Vitamin B6)
Pyridoxal phosphate is the active biochemical form of pyridoxine. It is a coenzyme of amino acid metabolism, particularly tryptophan and methionine. Inhibition of methionine metabolism as a result of pyridoxine deficiency results in excessive S-adenosylmethionine accumulation, which in turn inhibits nerve lipid and myelin synthesis.12
Pyridoxine is unique in that both the deficiency and toxic states result in a peripheral neuropathy (described later). The deficiency affects the blood, skin, and nervous system. The skin changes are indistinguishable from those of pellagra, probably because of the close interaction of niacin and pyridoxine. Pyridoxine improves the microcytic anemia of alcoholic persons, as well as the anemia associated with pyridoxine-responsive seizures in infants. Pyridoxine-deficient peripheral neuropathy is seen primarily in patients taking isoniazid or hydralazine, and it is characterized by small-fiber sensory loss in distal limbs, weakness, and reflex changes. Patients describe burning sensation in the feet and painful paresthesias. Central nervous system manifestations include depression, irritability, and confusion.55 Up to 10% of patients taking isoniazid may develop a peripheral sensory neuropathy. Isoniazid promotes increased pyridoxine excretion in the urine, which results in a deficiency state. Daily intake of vitamin B6 (10-50 mg) prevents the neuropathy induced by isoniazid treatment, and thus vitamin B6 should be taken by patients receiving isoniazid. Once established, the neuropathy does not entirely resolve but may improve with replacement vitamin B6.
Vitamin E
α-Tocopherol is the most active form of vitamin E present in humans. Tocopherol is absorbed and incorporated into chylomicrons in the small intestine. It is carried in portal blood to the liver, and α-tocopherol transfer protein binds and recycles vitamin E in the liver for incorporation into low-density lipoproteins and very-low-density lipoproteins.56 Once it is delivered to the cells, α-tocopherol serves as an antioxidant, preventing free radical peroxidation and injury to cell membranes. It is stored in adipose tissue, the liver, and muscles. Deficiency can occur at any stage of tocopherol metabolism: reduced intake, fat malabsorption, inhibition of enterohepatic circulation, mutation of α-tocopherol transport protein, and abetalipoproteinemia. Vitamin E deficiency leads to axonal membrane injury, with resultant axonal degeneration of peripheral nerves, dorsal root ganglia, and posterior columns (Figs. 109-8 and 109-9).57 Superficially, the spinal cord abnormalities bear a striking resemblance to those found with subacute combined degeneration of vitamin B12 deficiency. However, the lesions of vitamin B12 deficiency are caused by spongy demyelination of the posterior columns and lateral tracts, whereas those of vitamin E deficiency are the result of swollen and dystrophic axons, or spheroids, and astrocytosis in the posterior columns, dorsal root ganglia, and Clarke’s column.
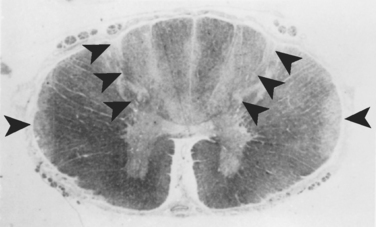
Vitamin E is fat-soluble and found in abundance in vegetable oils and wheat germ. The recommended daily allowance is 15 mg (22.5 IU) for adults. Patients at risk for the development of vitamin E deficiency include those who have the clinical conditions listed in Table 109-6.
From Jackson CE, Amato AA, Barohn RJ: Isolated vitamin E deficiency. Muscle Nerve 1996; 19:1162.
Patients develop areflexia, cerebellar ataxia, cutaneous sensory impairment, position and vibratory sense abnormalities, and, less commonly, ophthalmoplegia, muscle weakness, nystagmus, extensor plantar responses, ptosis, and dysarthria (Table 109-7).58 Acanthocytosis and pigmentary retinopathy is seen primarily in patients with abetalipoproteinemia. Patients with abetalipoproteinemia and congenital malabsorption develop symptoms in childhood and adolescence. On occasion, adults with acquired malabsorption present with progressive ataxia. The clinical phenotype that accompanies familial vitamin E deficiency phenotype is indistinguishable from that of Friedreich’s ataxia (Fig. 109-10). Autosomal recessive familial vitamin E deficiency has been studied extensively and found to be caused by a frameshift mutation within the α-tocopherol transfer protein gene on chromosome 8. Defective α-tocopherol transfer protein prevents incorporation of vitamin E into very-low-density lipoproteins.59,60
TABLE 109-7 Neurologic Findings in Vitamin E deficiency
From Jackson CE, Amato AA, Barohn RJ: Isolated vitamin E deficiency. Muscle Nerve 1996; 19:1162.
Friedreich’s ataxia, Machado-Joseph disease, and other familial spinocerebellar ataxias61 should be considered in the differential diagnosis of patients presenting with this constellation of signs and symptoms.
Approximately 90% or more of vitamin E is α-tocopherol, which can be measured directly in serum. Occasional patients with hyperlipidemia may have a falsely low α-tocopherol level. Vitamin E deficiency may result in electrophysiological abnormalities, including low-amplitude sensory nerve action potentials, slowed conductions, and abnormal somatosensory evoked potentials.62
Vitamin E is fat soluble and found in abundance in vegetable oils and wheat germ. It is carried in portal blood to the liver, where α-tocopherol transfer protein binds it and recycles vitamin E for incorporation into low-density and very-low-density lipoproteins. The patients at risk for development of vitamin E deficiency include those with hypobetalipoproteinemia or abetalipoproteinemia; other disorders of the pancreas and liver, such as cystic fibrosis; protein-calorie malnutrition; familial vitamin E deficiency; and other malabsorption states.63 Symptoms include areflexia, cerebellar ataxia, cutaneous sensory impairment, position and vibratory sense abnormalities, and, less commonly, ophthalmoplegia, muscle weakness, nystagmus, extensor plantar responses, ptosis, and dysarthria. The peripheral neuropathy is usually limited to the legs and is mild, axonal, and sensorimotor in nature.62 Vitamin E supplementation has been found to be protective from chemotherapy-induced neuropathy when administered before a patient receives cisplatin or paclitaxel.64
OTHER NUTRITIONAL SYNDROMES
Postgastroplasty Polyneuropathy
Acute post–gastric restriction surgery neuropathy may affect up to 7% of patients after weight-reduction surgery.65 Some patients undergoing bariatric, or weight-reduction, surgery develop a syndrome of acute or subacute sensory loss, weakness, and areflexia, usually after a period of dramatic weight loss and repeated bouts of protracted vomiting.66 Some have also developed a type of encephalopathy that is clinically and pathologically identical to Wernicke-Korsakoff syndrome, with or without an associated polyneuropathy.18 Indeed, Wernicke and Korsakoff each described young women with intractable vomiting in their original reports: One woman had attempted suicide by drinking sulfuric acid, whereas the other had hyperemesis gravidarum.15
Thaisetthawatkul and colleagues (2004) compared 435 patients who underwent bariatric surgery with 126 control subjects who underwent gallbladder surgery. They found that 71 (16% of) those undergoing bariatric surgery developed some form of neuropathy, in comparison with 3% who underwent gallbladder surgery. Of the 71, more than half had entrapment neuropathy, mostly carpal tunnel syndrome. Twenty-seven had a polyneuropathy, and 5 had a radiculoplexus neuropathy. Sural nerve biopsies in five patients (four with polyneuropathy, one with radiculoplexus neuropathy) revealed prominent axonal degeneration with variable degrees of perivascular mononuclear cell infiltration. No definite vasculitis was seen. Risk factors identified for neuropathic complications of bariatric surgery were acute weight loss, excessive vomiting, postoperative complications, poor vitamin supplementation, and jejunoileal bypass procedure.65
Intractable vomiting is a constant feature. The syndrome may manifest suddenly several months after surgical procedures that include gastrojejunostomy, gastric stapling, vertical banding gastroplasty, and gastrectomy with Roux-en-Y anastomosis.67 After a period of recurrent vomiting and precipitous weight loss, patients develop numbness and tingling in the soles of the feet, calves, and thighs. Distal or proximal weakness may develop, and the patient may have difficulty arising from a chair or climbing stairs. Examination reveals symmetrical sensory loss in the legs more than in the arms, muscle weakness, and areflexia. Patients may develop quadriparesis and prolonged or permanent disability. Autonomic disturbances and orthostatic hypotension have also been recognized. When the condition is accompanied by encephalopathy, patients may show confusion, memory loss, and affective disturbances. Many patients have been mistakenly diagnosed early in their course as having a conversion disorder.18
The polyneuropathy is axonal and demyelinating in type, acute in onset, and slow to resolve. Pathological studies have demonstrated axonal degeneration. One study demonstrating lipid-laden neurons and Schwann cells surrounding demyelinating and degenerating axons has not been replicated.66
Copper Deficiency Myeloneuropathy
Subacute combined degeneration has been reported in patients with copper deficiency.68 In several instances, the cause was excessive zinc consumption in the form of remedies for prevention of colds and upper respiratory infections. Other patients have acquired the disorder as the result of malabsorption from gastric bypass or gastric reduction surgery. In a series of 25 patients, Kumar and associates found that all had gait dysfunction, probably as a result of sensory ataxia caused by posterior column dysfunction.68 Corticospinal signs and evidence of peripheral neuropathy were present in many. Seventeen had elevated serum zinc levels. Ten of 25 had a history of prior gastric surgery. Copper supplementation did not appear to reverse the clinical findings. Copper may now be added to other nutrient deficiencies, those of vitamin B12 and vitamin E, that may result in a myeloneuropathy.
Pyridoxine Toxicity Neuropathy
Excess pyridoxine also results in a peripheral neuropathy. Megadoses of pyridoxine (generally in excess of 2 g/day) produce large-fiber sensory neuropathy,69 which, however, has also been reported with long-standing use of as little as 200 mg/day.70,71 Paresthesias, ataxia, and burning sensation in the feet may occur abruptly after megadoses or from 1 month to 3 years after lower doses. Sural nerve biopsies reveal reduced myelin fiber density and myelin debris, suggestive of axonal degeneration. After they stop taking pyridoxine, all patients improve, but the condition resolves entirely in only a few. Contrary to common misconceptions, such cases demonstrate that a water-soluble B vitamin may be toxic when taken in megadoses.
Strachan’s Syndrome
The term Strachan’s syndrome was coined by M. Fisher, honoring Henry Strachan, a British medical officer stationed in Jamaica, who in 1888 described a syndrome of painful peripheral neuropathy, ataxia, optic neuropathy, and stomatitis among sugar cane workers.72 Symptoms included sensorineural deafness, dizziness, confusion, spastic leg weakness, footdrop, Wernicke’s encephalopathy, and rare cases of neck extensor weakness and myasthenic bulbar weakness. Poor nutrition, hard physical labor, and concurrent infection were thought to be exacerbating factors. In Fisher’s autopsy series of Canadian prisoners of war, the most prominent pathological finding was demyelination of the posterior columns of the thoracic and cervical spinal cord.73 This demyelination accounted for the loss of vibratory and position sense and sensory ataxia. Pathologically, the optic and auditory nerves showed moderate to severe demyelination.
An outbreak of optic and peripheral neuropathy closely resembling Strachan’s syndrome occurred in Cuba from 1992 to 1993.74 Fifty thousand people developed variable degrees of optic neuropathy, painful sensory neuropathy, dorsolateral myelopathy, sensorineural deafness, spastic paraparesis, dysphonia, and dysautonomia. Almost half (45%) developed only centrocecal scotoma and optic neuropathy, often after a period of weight loss. Optic neuropathy and myeloneuropathy were seen in 24%, optic neuropathy and sensorineural hearing loss in 14%, and peripheral and optic neuropathies with hearing loss in 7%.74 Proposed mechanisms included deficiencies of vitamin B complex and thiamine, cyanide intoxication, viral infection, and mitochondrial DNA mutations. Infections appeared to precipitate or exacerbate symptoms. Almost all patients responded to early supplementation with B complex vitamins. Evidence of peripheral nerve involvement has been inconsistent. Clinical evidence of neuropathy is often lacking despite severe symptoms.
Carmel R, Green R, Rosenblatt DS, et al. Update on cobalamin, folate, and homocysteine. Hematology (Am Soc Hematol Educ Program). 2003:62-81.
Kuzminski AM, Del Giacco EJ, Allen RH, et al. Effective treatment of cobalamin deficiency with oral cobalamin. Blood. 1998;92:1191-1198.
Thaisetthawatkul P, Collazo-Clavell ML, Sarr MG, et al. A controlled study of peripheral neuropathy after bariatric surgery. Neurology. 2004;63:1462-1470.
1 O’Keefe ST, Tormey WP, Glasgow R, et al. Thiamin deficiency in hospitalized elderly patients. Gerontology. 1994;40:18-24.
2 Harper C. The incidence of Wernicke’s encephalopathy in Australia—a neuropathological study of 131 cases. J Neurol Neurosurg Psychiatry. 1983;46:593-598.
3 Harper C, Giles M, Finlay-Jones R. Clinical signs of the Wernicke-Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry. 1986;49:341-345.
4 Langohr HD, Petruch F, Schroth J. Vitamin B1, B2, and B6 deficiency and neurological disorders. J Neurol. 1981;225:95-108.
5 Joosten E, Van Den Berg A, Riezler R, et al. Metabolic evidence of deficiencies of vitamin B12, folate, and vitamin B6 occur commonly in elderly people. Am J Nutr. 1993;58:468-476.
6 Green R, Kinsella LJ. Current concepts in the diagnosis of cobalamin deficiency. Neurology. 1995;45:1430-1435.
7 Incalzi RA, Gemma A, Capparella O, et al. Energy intake and in-hospital starvation, a clinically relevant relationship. Arch Intern Med. 1996;156:425-429.
8 Leigh D, McBurney A, McIlwain H. Erythrocyte transketolase activity in the Wernicke-Korsakoff syndrome. Br J Psychol. 1981;138:153-156.
9 Rueler JB, Girard DE, Cooney TG. Wernicke’s encephalopathy. N Engl J Med. 1985;312:1035-1039.
10 Haas RH. Thiamine and the brain. Annu Rev Nutr. 1988;8:483-515.
11 Koike H, Iijima M, Sugiura M, et al. Alcoholic neuropathy is clinicopathologically distinct from thiamine-deficiency neuropathy. Ann Neurol. 2003;54:19-29.
12 Marcus R, Coulston AM. Water soluble vitamin, the vitamin B-complex and ascorbic acid. In: Shils ME, Olson JA, Shike M, editors. Modern Nutrition in Health and Disease. 8th ed. Philadelphia: Lea & Febiger; 1994:1547-1590.
13 Vortmeyer AO, Hagel C, Laas R. Hemorrhagic thiamine deficient encephalopathy following prolonged parenteral nutrition. J Neurol Neurosurg Psychiatry. 1992;55:826-829.
14 Fattal-Valevski A, Kessler A, Sela BA, et al. Outbreak of lifethreatening thiamine deficiency in infants in Israel caused by a defective soy-based formula. Pediatrics. 2005;115:e233-e238.
15 Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff Syndrome. Philadelphia: FA Davis, 1971.
16 Seehra H, Macdermott N, Lascelles RG, et al. Wernicke’s encephalopathy after vertical banded gastroplasty for morbid obesity. BMJ. 1996;312:434.
17 Haid RW, Gutman L, Crosby TN. Wernicke Korsakoff encephalopathy after gastric plication. JAMA. 1992;247:2566-2577.
18 Paulson GW, Martin EW, Mojzisik C, et al. Neurologic complications of gastric partitioning. Arch Neurol. 1985;42:675-677.
19 Denny-Brown D. Neurological conditions resulting from prolonged and severe dietary restriction. Case reports in prisoners of war, and general review. Medicine. 1947;26:41-113.
20 Jagadha V, Deck JHN, Halliday WC, et al. Wernicke’s encephalopathy in patients on peritoneal dialysis or hemodialysis. Ann Neurol. 1987;21:78-84.
21 Descombes E, Dessibourg CA, Felly G. Acute encephalopathy due to thiamine deficiency (Wernicke’s encephalopathy) in a chronic hemodialyzed patient: a case report. Clin Nephrol. 1991;35:171-175.
22 Martin PR, McCool BA, Singleton CK. Molecular genetics of transketolase in the pathogenesis of the Wernicke-Korsakoff syndrome. Metab Brain Dis. 1995;10:45-55.
23 Nixon PF. Is there a genetic component to the pathogenesis of the Wernicke-Korsakoff syndrome? Alcohol Alcohol. 1984;19:219-221.
24 Yamashita M, Yamamoto T. Wernicke’s encephalopathy with symmetric pericentral involvement: MR findings. J Comput Assist Tomogr. 1995;19:306-308.
25 Jollife N, Bowman KN, Rosenblum LA, et al. Nicotinic acid deficiency encephalopathy. JAMA. 1940;114:307-312.
26 Teare JP, Hyamas G, Pollock S. Acute encephalopathy due to co-existent nicotinic acid and thiamine deficiency. Br J Clin Pract. 1993;47:343-344.
27 Ishii N, Nishihara Y. Pellagra among chronic alcoholics: clinical and pathologic study of 20 necropsy cases. J Neurol Neurosurg Psychiatry. 1981;44:209-215.
28 Flippo TS, Holder WDJr. Neurologic degeneration associated with nitrous oxide anesthesia in patients with vitamin B12 deficiency. Arch Surg. 1993;128:1391-1395.
29 Lever EG, Elwes RDC, Williams A, et al. Subacute combined degeneration of the cord due to folate deficiency: response to methyl folate treatment. J Neurol Neurosurg Psychiatry. 1986;49:1203-1207.
30 Pant SS, Ashbury AK, Richardson EP. The myelopathy of pernicious anemia: a neuropathological reappraisal. Acta Neurol Scand. 1968;44(Suppl 35):1-36.
31 McCombe PA, McLeod JC. The peripheral neuropathy of vitamin B12 deficiency. J Neurol Sci. 1984;66:117-126.
32 Saperstein DS, Wofe GI, Gronseth GS, et al. Challenges in the identification of cobalamin deficiency polyneuropathy. Arch Neurol. 2003;60:1296-1301.
33 Lindenbaum J, Rosenberg IH, Wilson PW, et al. Prevalence of cobalamin deficiency in the Framingham elderly population. Am J Clin Nutr. 1994;60:2-11.
34 Savage DG, Lindenbaum J, Stabler SP, et al. Sensitivity of serum methylmalonic acid and total homocysteine determinations for diagnosing cobalamin and folate deficiencies. Am J Med. 1994;96:239-246.
35 Fernando OV, Grimsley EW. Prevalence of folate deficiency and macrocytosis in patients with and without alcohol-related illness. South Med J. 1999;92:841.
36 Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318:1720-1728.
37 Carmel R. Cobalamin, the stomach, and aging. Am J Clin Nutr. 1997;66:750-759.
38 Sumner AE, Chin MM, Abrahm JL, et al. Elevated methylmalonic acid and total homocysteine levels show high prevalence of vitamin B12 deficiency after gastric surgery. Ann Intern Med. 1996;124:469-476.
39 Halverson JD. Micronutrient deficiencies after gastric bypass for morbid obesity. Am Surg. 1986;52:594-598.
40 Marcuard SP, Albernaz L, Khazanie PG. Omeprazole therapy causes malabsorption of cyanocobalamin (vitamin B12). Ann Intern Med. 1994;120:211-215.
41 Kinsella LJ, Green R. Anesthesia paresthetica: nitrous oxide–induced cobalamin deficiency. Neurology. 1995;45:1608-1610.
42 Healton EV, Savage DG, Brust JCN, et al. Neurologic aspects of cobalamin deficiency. Medicine. 1991;70:229-244.
43 Eisenhofer G, Lambie DG, Johnson RH, et al. Deficient catecholamine release as the basis of orthostatic hypotension in pernicious anemia. J Neurol Neurosurg Psychiatry. 1982;45:1053-1055.
44 White WB, Reik LJr, Cutlip DE. Pernicious anemia seen initially as orthostatic hypotension. Arch Intern Med. 1991;141:1543-1544.
45 Layzer RB. Myeloneuropathy after prolonged exposure to nitrous oxide. Lancet. 1988;2:1227-1230.
46 Reynolds EH, Bottiglieri T, Laundy M, et al. Vitamin B12 metabolism in multiple sclerosis. Arch Neurol. 1992;49:649.
47 Herbert V. Vitamin B12 deficiency neuropsychiatric damage in acquired immunodeficiency syndrome. Arch Neurol. 1993;50:569.
48 Hvas AM, Juul S, Gerdes LU, et al. The marker of cobalamin deficiency, plasma methylmalonic acid, correlates to plasma creatinine. J Intern Med. 2000;247:507-512.
49 Carmel R, Green R, Rosenblatt DS, et al. Update on cobalamin, folate, and homocysteine. Hematology (Am Soc Hematol Educ Program). 2003:62-81.
50 Kuzminski AM, Del Giacco EJ, Allen RH, et al. Effective treatment of cobalamin deficiency with oral cobalamin. Blood. 1998;92:1191-1198.
51 Savage DG, Lindenbaum J. Neurological complications of acquired cobalamin deficiency: clinical aspects. Ballieres Clin Haematol. 1995;8:657-678.
52 Reynolds EH. Folic acid, ageing, depression and dementia. BMJ. 2002;324:1512-1515.
53 Rader JI. Folic acid fortification, folate status and plasma homocysteine. J Nutr. 2002;132(Suppl):2466S-2470S.
54 Toole JF, Malinow MR, Chambless LE, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) trial. JAMA. 2004;291:565-575.
55 Brent J, Vo N. Reversal of prolonged isoniazid-induced coma by pyridoxine. Arch Intern Med. 1990;150:1751-1753.
56 Bieri JG, Corash L, Hubbard VS. Medical uses of vitamin E. N Engl J Med. 1983;308:1063-1071.
57 Rosenblum JL, Keating JP, Prensky AL, et al. A progressive neurologic syndrome in children with chronic liver disease. N Engl J Med. 1981;304:503-508.
58 Muller DPR, Lloyd JK, Wolff OH. Vitamin E and neurological function. Lancet. 1983;1:225-227.
59 Gotoda T, Arita M, Arai H, et al. Adult onset spinocerebellar dysfunction caused by a mutation in the gene for the alphatocopherol transfer protein. N Engl J Med. 1995;333:1313-1318.
60 Harding AE, Matthews S, Jones S, et al. Spinocerebellar degeneration associated with a selective defect of vitamin E absorption. N Engl J Med. 1985;313:32-35.
61 Rosenberg RN. Spinocerebellar ataxias and ataxins. N Engl J Med. 1995;333:1351-1352.
62 Brin MF, Pedley TA, Lovelace RE, et al. Electrophysiologic features of abetalipoproteinemia: functional consequences of vitamin E deficiency. Neurology. 1986;36:669-673.
63 Jackson CE, Amato AA, Barohn RJ. Isolated vitamin E deficiency. Muscle Nerve. 1996;19:1161-1165.
64 Argyriou AA, Chroni E, Koutras A, et al. Vitamin E for prophylaxis against chemotherapy-induced neuropathy: a randomized controlled trial. Neurology. 2005;64:26-31.
65 Thaisetthawatkul P, Collazo-Clavell ML, Sarr MG, et al. A controlled study of peripheral neuropathy after bariatric surgery. Neurology. 2004;63:1462-1470.
66 Feit H, Glasberg MR, Ireton C, et al. Peripheral neuropathy and starvation after gastric partitioning for morbid obesity. Ann Intern Med. 1982;96:453-455.
67 Peltier G, Hermreck AS, Moffat RE, et al. Complications following gastric bypass procedure for morbid obesity. Surgery. 1979;86:648-654.
68 Kumar N, Gross JB, Ahlskog JE. Copper deficiency myelopathy produces a clinical picture like subacute combined degeneration. Neurology. 2004;63:33-39.
69 Schaumburg H, Kaplan J, Windebank A, et al. Sensory neuropathy from pyridoxine abuse. A new megavitamin syndrome. N Engl J Med. 1983;309:445-448.
70 Dalton K, Dalton MJT. Characteristics of pyridoxine overdose neuropathy syndrome. Acta Neurol Scand. 1987;76:8-11.
71 Parry GJ, Bredesen DE. Sensory neuropathy with low-dose pyridoxine. Neurology. 1985;35:1466-1468.
72 Strachan H. On a form of multiple neuritis prevalent in the West Indies. Practitioner. 1897;59:477-484.
73 Fisher M. Residual neuropathological changes in Canadians held prisoner of war by the Japanese (Strachan’s disease). Can Serv Med J. 1955;11:157-199.
74 Roman GC. An epidemic in Cuba of optic neuropathy, sensorineural deafness, peripheral sensory neuropathy and dorsal lateral myeloneuropathy. J Neurol Sci. 1994;127:11-28.