[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 3
Vitamin D
From Photosynthesis, Metabolism, and Action to Clinical Applications
Origin of Vitamin D: Nutrition and Photosynthesis
General Characteristics of the Vitamin D Receptor
Noncalcemic or Nonclassic Actions of Vitamin D Endocrine System
Diagnostic and Therapeutic Aspects of Vitamin D
Historic Overview
Rickets as a bone disease of young children was clearly described by Whistler in 16451 and Glisson in 1650.2 The relationship of this disease with lack of exposure to sunlight was already suspected in the 19th century, since the incidence of rickets was higher in children living in large industrialized towns than in children living in rural districts (see Chapter 15).3,4 Early in the 20th century, Huldshinsky,5 Chick et al.,6 and Hess and Weinstock7 demonstrated that rachitic children were cured after exposure to sunlight. In the United Kingdom, following an independent line of research in search of essential nutritional factors, Mellanby and Cantag8 raised dogs on a diet of oatmeal (the basic food in parts of the United Kingdom where rickets was endemic) and observed that they developed rickets, curable by cod liver oil.9 However, McCollum et al.10 could demonstrate that cod liver oil made vitamin A-deficient by aeration and heating was still able to cure rickets and thus contained a new essential nutrient called vitamin D.10 The two discoveries of vitamin D were unified by the demonstration of Goldblatt and Soames11 that irradiation of 7-dehydrocholesterol in the skin could produce the antirachitic vitamin D. Similar observations were made by Hess and Weinstock.7 Windaus,12 a German chemist, then identified the structure of vitamins D2 and D3 after irradiation of plant sterols (ergosterol) or 7-dehydrocholesterol,12 which earned him the Nobel Prize in chemistry in 1928.
Origin of Vitamin D: Nutrition and Photosynthesis
Vitamin D can be obtained from dietary sources of vegetal (vitamin D2 or ergocalciferol) or animal origin (vitamin D3 or cholecalciferol). About 50% of dietary vitamin D is absorbed by the enterocytes and transported to the blood circulation via chylomicrons. Part of this vitamin D is taken up by a variety of tissues (fat and muscle) before the chylomicron remnants and its vitamin D finally reaches the hepatocytes. The best food sources are fatty fish or its liver oils, but it is also found in small amounts in butter, cream, and egg yolk. Both human and cow’s milk are poor sources of vitamin D, providing only 15 to 40 IU/L, and equally minimal concentrations of 25(OH)D or 1,25(OH)2D.13 Only an intake of pharmacologic amounts of vitamin D (6000 IU/d) can increase the vitamin D concentration of milk to a level equivalent to the daily requirements of an infant.14 Vitamin D intake is a poor predictor of serum 25(OH)D concentrations in subjects with an intake between 2 and 20 µg/d.15,16 It is very difficult to obtain adequate vitamin D from a natural diet. However, in North America, 98% of fluid and dried milk (≥400 IU/L), as well as some margarine, butter, and certain cereals, are fortified with vitamin D2 (irradiated ergosterol) or D3, but the real vitamin D content is frequently quite different from the labeling standard. Skim milk and even proprietary infant formula frequently do not have the stated vitamin D content.17,18 Vitamin D is remarkably stable and does not deteriorate when food is heated or stored for long periods. The Second National Health and Nutrition Survey (NHANES II) reported a median intake of about 3 µg/d in adults (range 0 to 49 µg),19 whereas a slightly lower median intake (2.3 µg) was recorded in older women.20 In view of the low vitamin D content of a vegetarian diet (natural vitamin D intake is indeed related to intake of animal fat), vitamin D deficiency and rickets is a risk factor for strictly vegetarian children with insufficient sun exposure or vitamin D supplementation.21
Nature probably intended that most vitamin D would be generated by photosynthesis in the skin, with minor contribution from food sources. However, exposure to sunlight also increases the risk of dermal photodamage and several skin cancers, including melanoma. This was no real problem during human evolution, but with increasing life expectancy, the benefits of UV light for the photosynthesis of vitamin D should be compared with the lifetime risk of skin damage, especially since vitamin D supplementation can safely replace the skin synthesis. The recommended dietary allowances by the U.S. Food and Nutrition Board of the National Research Council and the 1998 updated recommendations are given in Table 3-1, and similar recommendations are still valid in Europe.22,23 However, these recommendations were based on rudimentary knowledge of optimal vitamin D status and need to be revised upwards.
Table 3-1
Adequate Intake, Previous Recommended Dietary Allowance, Reasonable Daily Allowance, and Tolerable Upper Limit for Vitamin D

AI, Adequate intake; RDA, recommended dietary allowance; UL, upper limit.
*Data from the Food and Nutrition Board, National Research Council, NAS: Recommended Dietary Allowances, 10th ed. Washington, DC: National Academy Press, 1989.
†Data from the Food and Nutrition Board: Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D and Fluoride. Washington, DC: National Academy Press, 1997. Fairly similar advice is given by the European Food Safety Authority.23
‡Similar upper levels for vitamin D intake were defined by the European Food Safety Authority,23 defining 25 µg or 1000 IU/d as the upper limit for children 0 to10 years of age and 50 µg or 2000 IU/d for children older than 11 years and adults.
Hypervitaminosis can occur when pharmaceutical vitamin D is taken in excess, with a wide variety of symptoms and signs related to hypercalciuria, hypercalcemia, and metastatic calcifications (Table 3-2). The toxic dosage has not been established for all ages, but infants and children are more susceptible. Toxicity should always be monitored when daily doses markedly exceeding the present upper limit of more than 50 µg are given for a longer period. Overproduction of renal 1,25(OH)2D by abnormal hormonal stimuli (as seen in fibroblast growth factor-23 [FGF-23] or Klotho-null mice) or absence of CYP24A1 (see later), the main catabolizing enzyme, causes the same calcemic side effects, with severe multiple-organ calcification (especially kidney, vascular wall, and heart valves) leading to premature death.24
Table 3-2
Symptoms of Vitamin D Toxicity
Hypercalciuria
Kidney stones
Hypercalcemia
Hyperphosphatemia
Polyuria
Polydipsia
Decalcification of bone
Ectopic calcification of soft tissues (kidney and lung)
Nausea and vomiting
Anorexia
Constipation
Headache
Hypertension
The production of previtamin D3 is a nonenzymatic photochemical reaction which is not subject to regulation other than substrate (7DHC) availability and intensity of UVB irradiation. 7DHC is the last precursor in the de novo biosynthesis of cholesterol. The enzyme 7HDC-Δ7-reductase (or sterol Δ7-reductase) catalyses the production of cholesterol from 7DHC. Inactivating mutations of the 7DHC-Δ7-reductase gene25 are the hallmark of the autosomal recessive Smith-Lemli-Opitz syndrome, characterized by high tissue and serum 7DHC levels and multiple anomalies, including craniofacial dysmorphism and mental retardation due to the lack of cholesterol synthesis.26 These patients may exhibit sometimes increased serum vitamin D and 25(OH)D concentrations.27 Likewise, animals pretreated with a specific sterol-Δ7-reductase inhibitor also exhibit an augmented vitamin D synthesis following UVB irradiation.28 With increasing human age, cutaneous stores of provitamin D decrease, together with decreased photoproduction of vitamin D.16 In cats and the feline species in general, the high cutaneous sterol-Δ7-reductase activity hampers photoproduction of vitamin D, making it a true vitamin.29 Apart from substrate (7DHC) availability, the photochemical synthesis of vitamin D3 in the skin largely depends on the amount of UVB photons that strike the basal epidermal layers. Glass, sunscreen, clothes, and skin pigment absorb UVB and blunt vitamin D3 synthesis. Latitude, time of day, and season are factors that influence the intensity of solar radiation and the cutaneous production of vitamin D3. Therefore, there is a risk for a shortage of vitamin D supply during winter and spring. In both the Northern and Southern hemispheres above 40 degrees latitude, vitamin D3 synthesis of the skin decreases or disappears during winter months, owing to the low inclination of the sun and the atmospheric filtration of the shortest (but effective for vitamin D3 synthesis) UV waves of sunlight. The importance of skin synthesis of vitamin D3 to maintain normal vitamin D status is best reflected by the vitamin D deficiency observed in submarine personnel or inhabitants of Antarctica30 during prolonged absence of sun exposure, and also by the extremely high prevalence of vitamin D deficiency in countries where exposure to sunlight is extremely low for cultural and religious reasons, as in several Arabian countries with strict adherence to Islamic rules for body covering.31–34 Solar exposure of 2 hours per week of the face and hands is probably sufficient for maintaining normal 25(OH)D concentrations in children35 and adults but should be further fine-tuned according to the climate and latitude.36
Nature has built in several feedback mechanisms to minimize the risk that prolonged sun exposure would cause vitamin D intoxication. Cutaneous vitamin D and especially previtamin D are photosensitive and will be degraded to inactive sterols (lumisterol, tachysterol) before they are translocated to the circulation (Fig. 3-1). Only a maximum of 10% to 15% of the provitamin D will be converted to vitamin D. Sunlight-induced melanin synthesis, acting as a natural sunscreen, provides an additional negative feedback.
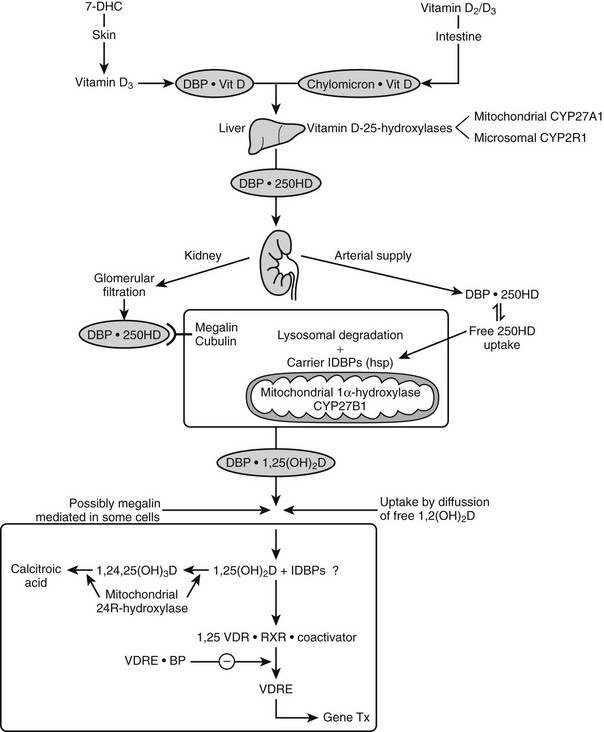
Metabolism of Vitamin D
Vitamin D is biologically inert and requires two successive hydroxylations in the liver (on C25) and kidney (on the α position of C1), using cytochrome P450 enzymes37,38 to form its hormonally active metabolite, 1α,25-dihydroxyvitamin D (see Fig. 3-1).
25-Hydroxylation
25(OH)D was the first metabolite identified after the availability of radiolabeled vitamin D3.39,40 Although the liver is probably the main tissue responsible for 25-hydroxylation of vitamin D, extrahepatic 25-hydroxylation has been observed in vitro in a large number of tissues. In vivo observations after hepatectomy in rats also revealed that the conversion rate of [3H]-vitamin D was still about 10% when compared with intact rats.40 The hepatic 25-hydroxylation step is probably performed by more than one enzyme, localized either in the inner mitochondrial membrane (CYP27A1 or sterol 27-hydroxylase) or in the microsomes (including CYP2D11, CYP2D25, CYP3A4, and especially CYP2R1).37,41,42 CYP27A1 is a multifunctional enzyme with broad substrate specificity and is mainly involved in the 26- or 27-hydroxylation of cholesterol and bile-acid precursors.37 The rather mild (if any) disturbance of vitamin D metabolism in animals or humans lacking this enzyme43 indicates that the 25-hydroxylation of vitamin D does not rely exclusively on the activity of CYP27A1. It is therefore likely that a microsomal enzyme is the more physiologic enzyme, as initially suspected on the basis of hepatic enzyme activity being much higher and with lower Km in the microsomal fraction when compared with mitochondrial 25-hydroxylase activity.44 The microsomal enzyme activity is up-regulated by vitamin D deficiency or by prior exposure to phenobarbital. The most important 25-hydroxylase is probably CYP2R1, since a homozygous mutation was identified in a patient with classical rickets with low 25(OH)D levels.42
1α-Hydroxylation
25(OH)D is biologically inactive and requires further hydroxylation in the kidney45,46 to the active hormone, 1,25(OH)2D, by 25-hydroxyvitamin D-1α-hydroxylase (CYP27B1). The production of 1,25(OH)2D is regulated primarily at this final step by several factors (vide infra). The rat, mouse, and human 1α-hydroxylase have been cloned by several groups47–51 and mapped on human chromosome 12q13.3 in close vicinity to the VDR gene. The proximal renal tubule is the principle site of 1α-hydroxylation, but high levels of 1α-hydroxylase mRNA have also been found in human keratinocytes,48 and its gene expression is also observed in mouse macrophages52 and about ten other tissues.24 1α-Hydroxylase activity is under tight control by 1,25(OH)2D (negative but probably indirect feedback); parathyroid hormone (PTH), calcitonin, and insulin-like growth factor 1 (all positive feedback); and phosphate, calcium, and especially FGF-23 (negative regulation).38,53,54 The promoters of the mouse and human 1α-hydroxylase genes have been characterized with a profound responsiveness to PTH and a negative regulation by 1,25(OH)2D55,56 by complex chromatin and DNA modifications.57
Pseudovitamin D–deficiency rickets (PDDR), also known as vitamin D-dependency rickets type I, is an autosomal recessive disease characterized by failure to thrive, muscle weakness, skeletal deformities, hypocalcemia, secondary hyperparathyroidism, normal to high serum levels of 25(OH)D, and low serum 1,25(OH)2D concentrations, all caused by impaired activity of the renal 1α-hydroxylase.58 These patients recover with supplementation of physiologic doses of 1,25(OH)2D. The human CYP27B1 maps to the previously identified PDDR locus and mutations found in this gene in patients with PDDR provide the molecular genetic basis for the disease.48,59
24-Hydroxylation: Catabolism or Specific Function?
An alternative hydroxylation of 25(OH)D occurs on carbon 24 by the multifunctional enzyme, 24-hydroxylase (CYP24A), mapped on human chromosome 20q13.60 This enzyme not only initiates the catabolic cascade of 25(OH)D and 1,25(OH)2D61,62 by 24-hydroxylation but catalyzes also the dehydrogenation of the 24-OH group and performs 23-hydroxylation, resulting in 24-oxo-1,23,25(OH)3D.62 This C24 oxidation pathway finally leads to calcitroic acid, which is the major end product of 1,25(OH)2D (Fig. 3-2). In vivo evidence for this catabolic role of 24-hydroxylase was provided by the generation of mice deficient in the 24-hydroxylase gene, resulting in pathology consistent with systemic excess of 1,25(OH)2D.63 The expression of the 24-hydroxylase gene has been detected in virtually all nucleated cells. The induction of CYP24A belongs to the most sensitive biomarkers for responsiveness and is explained by the presence of several vitamin D responsive elements in its promoter.64,65 As a consequence, CYP24A mRNA levels appear to fall under the detection limits in VDR knockout mice.50,66 No human mutation in the 24-hydroxylase gene have yet been identified, but local overexpression of the enzyme may be involved in cancer.67
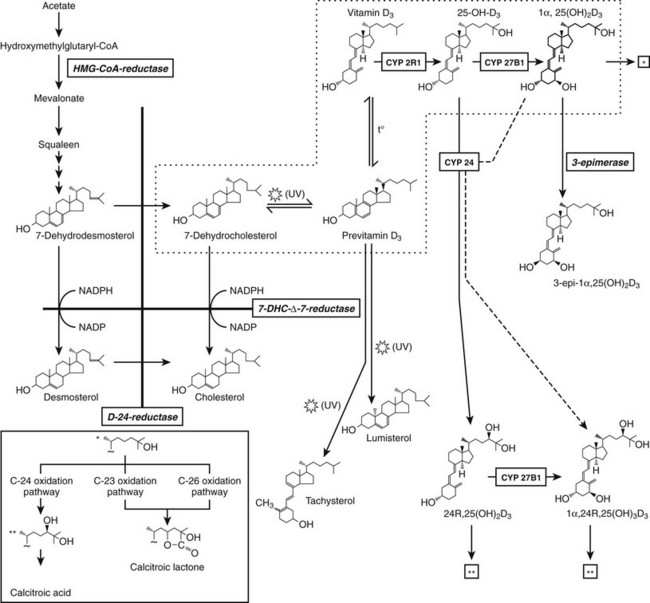
Other Metabolic Pathways For Vitamin D and Its Metabolites
Apart from the multifunctional 24-hydroxylation pathway, C23 and C26 hydroxylation of 1,25(OH)2D is also possible in the absence of prior 24-hydroxylation. The 23-hydroxylation probably only becomes important in the case of vitamin D excess; its major locus is the kidney. In contrast, 26-hydroxylation is mainly performed outside the kidney. Both activities are necessary for the formation of 25(OH)D- or 1,25(OH)2D-23,26-lactones (see Fig. 3-1). The A-ring metabolism involves the oxidation of C19 and the recently discovered 3-epimerisation. The latter, irreversible, reaction occurs only in a limited number of cells (e.g., keratinocytes, bone, and parathyroid cells) and is performed by hydroxysteroid dehydrogenases.68
Vitamin D Transport
Nutritional vitamin D is absorbed by the gut and then transported via the lymphatic system by chylomicrons69 and stored in several tissues (e.g., fat and muscle). Skin-produced vitamin D probably binds directly to an α-globulin known as vitamin D binding protein (DBP) and is then transported to the liver, where it is hydroxylated and thereafter released as 25(OH)D.
Human DBP,70 detected immunologically in 1959 as a group-specific component, or Gc-globulin,71 is a 43-amino-acid glycoprotein synthesized by the liver. Long before DBP’s functions had been characterized, its polymorphicity was already used in population genetics, parentage testing, and forensic medicine.72 Worldwide, over 120 Gc alleles have been detected,73 making the DBP locus one of the most polymorphic known. The Gc1F, Gc1S and Gc2 are the three most common alleles. Since in the many thousands of sera tested, none had been found of DBP deficiency, such a mutation was for a long time considered to be lethal; but this was contradicted by the generation of viable and fertile homozygous, DBP-deficient mice (DBP-null animals).74 The existence of similarity among the genes and protein structure of DBP, albumin, and α-fetoprotein is long recognized.75 The crystal structure, with or without actin, is now available and identified a surface cleft to bind 25(OH)D.76
Role of Vitamin D Binding Protein For Vitamin D Homeostasis
DBP, the major plasma carrier of vitamin D3, all its metabolites, and the vitamin D3 analogs, has one vitamin D sterol-specific binding site.75 The relative binding affinity is 25(OH)D-23,26-lactone > 25(OH)D = 24,25(OH)2D = 25,26(OH)2D (Ka = 5.108 mol/L at 4° C for human DBP) >> 1,25(OH)2D (4.107 mol/L) >> vitamin D >> previtamin D.77 The affinity for D2 metabolites is slightly lower than for D3 metabolites in mammals, but especially in birds. Since probably only non-DBP-bound vitamin D metabolites can readily cross the plasma membrane, and since the VDR has a much higher affinity for 1,25(OH)2D than for 25(OH)D (100-fold difference), while the opposite is true for DBP, it is clear that 1,25(OH)2D has substantially higher cellular uptake than 25(OH)D. This is also confirmed by the distribution space of (radiolabeled) metabolites: 25(OH)D has a distribution space similar to that of DBP and the plasma volume, whereas the distribution space of 1,25(OH)2D is closer to that of intracellular water. The half-life of 25(OH)D and 1,25(OH)2D in the human circulation is about 2 to 3 weeks and 4 to 6 hours, respectively.78,79 DBP’s function in the vitamin D endocrine system is assumed to reflect the “free hormone” hypothesis, which states that the unbound (free) rather than the protein-bound fraction of the active vitamin D hormone is responsible for the biological activity. The plasma concentration of DBP is increased by estrogens in most mammalian species and birds. In women, the DBP concentration therefore doubles at the end of pregnancy.80 Recent studies with megalin knockout mice indicate that megalin, a lipoprotein-like receptor present at the surface of the proximal tubular cells in the kidney, is responsible for the reabsorption of DBP and of DBP complexed with vitamin D sterols. This megalin reabsorption mechanism may control the availability of the 25(OH)D/DBP complex for the 25(OH)D-1α-hydroxylation enzyme and explain the severe bone disease of megalin-deficient mice.81
Other Functions of Vitamin D Binding Protein
DBP binds globular actin with a high affinity (Ka = 2 × 109 mmol/L).82,83 Actin is the most abundant intracellular protein. The cell motility, shape, and size depend on the ability of globular actin to polymerize into filaments (F-actin). Upon cell injury or cell necrosis, actin is released into extracellular space. However, when actin is released from cells, it may rapidly form filaments with detrimental effects for the microcirculation. Two plasma proteins, DBP and gelsolin, bind actin avidly, thereby acting as “actin-scavenger” system.84,85
DBP-null (KO) mice, however, develop normally. They are nevertheless more sensitive to vitamin D deficiency and less sensitive to vitamin D excess, probably by an enhanced urinary loss of vitamin D metabolites.74 The DBP and megalin KO mice, however, suggest that the main function of DBP is indeed to transport all vitamin D metabolites and preserve them from rapid clearance or urinary loss.
Action and Mode of Action
General Characteristics of The Vitamin D Receptor
Protein
1,25(OH)2D, the hormonally active form of vitamin D, exerts its effects mainly by activating the nuclear VDR, a member of the nuclear-receptor superfamily of ligand-activated transcription factors. Based on structure and function similarities between members of this family, different functional domains can be distinguished in these nuclear-receptor proteins. The short A/B domain at the N-terminus of VDR lacks the usual ligand-independent activation function (AF1). Two highly conserved zinc finger DNA binding motifs constitute the DNA-binding C domain, which also harbors the nuclear localization signal. The D domain or hinge region may regulate the receptor’s flexibility between DNA-binding and ligand-binding domains and may be crucial to allowing the heterodimer complex of the ligand-binding domains to interact with two differently oriented response elements (direct repeat or palindrome orientation with variable number of spacer nucleotides). The large multifunctional E region contains the ligand-binding domain, as well as a dimerization surface and a ligand-dependent activation function (AF2) at the extreme C-terminus, represented by helix 12.86
Gene
The human VDR gene, consisting of 14 exons, spans more than 60 kb on chromosome 12.87,88 The major VDR transcript is a 4.8 kb mRNA species, but multiple promoters and alternative splicing give rise to a multitude of less abundant transcripts that mostly vary in their 5′ untranslated region but encode the same 427-amino-acid protein.88 However, two of these mRNAs are translated into VDR proteins that contain an additional 23 or 50 amino acids at the N-terminus.88
Genomic Actions
Binding of 1,25(OH)2D to VDR generates conformational changes of VDR followed by heterodimerization with unliganded RXR and binding to vitamin D response elements (VDREs) in the promoter region of vitamin D target genes, with subsequent release of corepressors and recruitment of coactivators and general transcription factors for the assembly of an active transcriptional complex.89 A putative crucial event in this respect is the mousetrap-like intramolecular folding of helix 12, closing off the ligand-binding pocket and exposing the AF2 domain for interaction with coactivators.90 Corepressors bind and silence unliganded steroid receptors by recruitment of histone deacetylases, maintaining chromatin in a transcriptional repressive state.91 Coactivators are a group of proteins that allow gene transcription in several waves of activities. First, coactivators of the CBP/p300 family and of the p160 protein family, including the steroid receptor coactivators (SRCs), are recruited.92–94 These proteins possess intrinsic histone acetyltransferase (HAT) activity and by acetylating histone tails open up the chromatin structure, creating a chromatin environment permissive for gene transcription.95 In a second wave, the vitamin D receptor interacting protein (DRIP) multimeric complex is recruited, followed by recruitment of basal transcription factors, as well as RNA polymerase II. Finally, target gene transcription is induced.96 Gene expression can also be mediated by ATP-dependent chromatin remodeling complexes such as SWI/SNF-type and ISWI-type complexes and the multiprotein complex WINAC.97–99 Distinct regulation of transcriptional coregulators may provide species-specific, tissue-specific, or developmental stage–specific regulation of nuclear receptor function.100 Furthermore, the expression or the recruitment of these regulatory proteins is regulated by several intracellular signaling pathways101 and by steroids themselves,100 with receptor agonists or antagonists inducing preferential recruitment of coactivators or corepressors, respectively.101
A hexanucleotide direct repeat spaced by three nucleotides (DR3) is the cognate vitamin D response element (VDRE) to which RXR and VDR bind the 5′ and 3′ half-site, respectively, although alternative options appear to be possible, both with respect to dimer formation (VDR/VDR, VDR/RAR) and target-gene VDRE structure (DR4, DR6; IP9).102
Nongenomic Actions
Aside from the VDR transcriptional or genomic effects, several research groups have described rapid effects by 1,25(OH)2D that are independent of transcription and would be mediated by a membrane receptor for 1,25(OH)2D or by the localization of the nuclear VDR near the membrane.103 These so-called nongenomic effects include the opening of calcium or chloride channels and the activation of second messenger signaling pathways (phosphoinositide turnover, activation of protein kinase C, and the Ras/Raf/ERK/MAPK pathway). A wide variety of rapid and transient modifications in the second messenger signaling system have also been observed for other steroid hormones.104 At the tissue or cellular level, however, nongenomic activity of vitamin D and its analogs or metabolites have only been described for intestinal calcium absorption (transcaltachia) or cellular differentiation of leukemia cells.105–107 This pathway seems to prefer 6-s-cis to the 6-s-trans configuration of vitamin D.105 Moreover, the agonist/antagonist specificity differs for that of the genomic pathway.108
Classic Target Tissues
The action of 1,25(OH)2D on bone, intestine, kidney, and parathyroid glands and its role in mineral metabolism is the result of a complex interplay between calcium and phosphate 1,25(OH)2D, PTH, and phosphatonins. PTH induces calcium mobilization from bone and stimulates 1,25(OH)2D production, but its secretion is inhibited by the action of 1,25(OH)2D on the parathyroid glands (negative feedback). In a second negative feedback loop, 1,25(OH)2D limits its own availability by inhibition of 1α-hydroxylase and stimulation of 24-hydroxylase, inducing 1,25(OH)2D catabolism. In the last few years, considerable progress has been made in the understanding of phosphate homeostasis.109 The phosphaturic hormone phosphatonin, or FGF-23, is produced by osteocytes and osteoblasts and inhibits the activity of the NPT2 protein. The NPT2 gene encodes a renal sodium/phosphate cotransporter responsible for reabsorption of phosphate and represents a newly identified target gene for 1,25(OH)2D.110 Phosphatonin can be indirectly inactivated by a protease encoded by the PEX gene, which was identified as the gene that is defective in X-linked hypophosphatemic rickets. FGF-23 secretion is stimulated by 1,25(OH)2D and impairs renal 1α-hydroxylase, creating an additional feedback system so that the production of 1,25(OH)2D is tightly feedback regulated (Fig. 3-3).
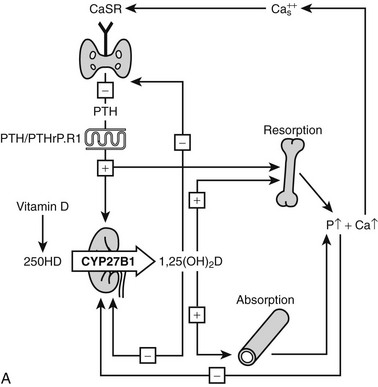
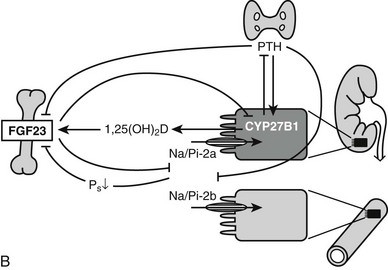
FIGURE 3-3 Feedback regulation of renal synthesis of 1,25(OH)2D. A, Regulation of renal 1,25(OH)2D synthesis by parathyroid hormone and calcium, with multiple feedback control mechanisms. CaSR, Calcium-sensing receptor. B, Regulation of renal 1,25(OH)2D synthesis by FGF23 and phosphate, with several feedback control mechanisms.
Effects on Intestine
The absorption capacity of calcium along the gastrointestinal tract of the rat is dependent on the segment and follows the order ileum > jejunum > duodenum. The efficiency of the small intestine to absorb dietary calcium is increased by 1,25(OH)2D,111 and the abundance of the vitamin D receptor is highest in the duodenum, followed by jejunum and ileum. Although the exact mechanism by which 1,25(OH)2D alters the flux of calcium across the intestinal absorptive cell is not known, 1,25(OH)2D increases the production and activity of several proteins in the small intestine, including TRPV6 and V5, calbindin-D9K, alkaline phosphatase, and low-affinity Ca-ATPase (PMCA). The entry of Ca2+ from the intestinal lumen across the brush border membrane into the enterocyte is mainly regulated by the epithelial channels TRPV6 and V5.112 The intracellular calcium transfer is considered to be dependent mainly on calbindin-D9K.113 The transfer of Ca2+ from the cytoplasm to the extracellular space requires energy input because of an uphill concentration gradient and an unfavorable electrochemical gradient. Both the plasma membrane calcium pump and a sodium-calcium exchanger play important roles in this process. The stimulatory effect of 1,25(OH)2D on the ATP-dependent uptake of Ca2+ at the basolateral membrane involves an increase in PMCA gene expression.114 The essential role of the intestine for calcium and phosphate homeostasis was clearly demonstrated by the phenotype of VDR KO mice. Such VDR-null mice are phenotypically normal at birth, but after weaning, they develop hypocalcemia, secondary hyperparathyroidism, and hypophosphatemia despite very high levels of 1,25(OH)2D. They become growth retarded and develop severe rickets.66,112,115,116 Mice deficient in 1α-hydroxylase display a similar phenotype.117,118 This bone and calcium phenotype can be largely corrected by a high dietary calcium intake (especially in combination with high lactose intake) in both knockout models or 1,25(OH)2D treatment of 1α hydroxylase–null mice.112,119–125 These findings confirm previous observations in humans.59,126,127 The data strongly suggest that the intestine is the primary target for 1,25(OH)2D’s action on calcium/bone homeostasis. This is largely confirmed by genetic mouse models of selective rescue or deletion of VDR in the intestine of transgenic mice.128 The primary molecular targets, however, merit further exploration; ablation of CaBP-9k or TRPV6 or even their combined deficiency have shown no major effects on basal intestinal calcium absorption or serum calcium levels when calcium intake is normal.129 Paracellular intestinal calcium transport may also be part of the picture of vitamin D’s action in that the expression of claudin 2 and claudin 12, both known to form paracellular calcium channels, are induced by 1,25(OH)2D and decreased in the intestine of VDR-null mice.130
Effects on Kidney
The kidney is important both for the metabolism of 1,25(OH)2D and the reabsorption of calcium and phosphate, processes regulated by 1,25(OH)2D. The kidney and more specifically the proximal tubule is the central tissue for 1α-hydroxylation of 25(OH)D. Chronic renal failure reduces 1α-hydroxylase activity, which ultimately results in renal osteodystrophy or uremic bone disease. 1,25(OH)2D also increased the distal tubular reabsorption of calcium; as in the intestine, TRP channels (now TRPV5), calbindin-D9K and 28K, and the plasma membrane calcium ATPase are involved. Whereas in the intestine, active calcium absorption in the duodenum takes place before the less-regulated diffusion process in the ileum, reabsorption of filtered calcium follows a more logical sequence of massive calcium-sodium reabsorption in the proximal convoluted tubuli, followed by specific, actively regulated calcium reabsorption in the distal parts of the nephron. The crucial role of 1,25(OH)2D-regulated renal calcium reabsorption was demonstrated by persistent hypercalciuria and reduction in bone mass in TRPV5-deficient mice.131 The kidney is also the major component in phosphate homeostasis, as both PTH and FGF-23, in complex interplay with 1,25(OH)2D, are able to reduce renal phosphate reabsorption (see Fig. 3-3).
Effects on Bone
1,25(OH)2D has dual effects on bone: it can stimulate osteoclastogenesis and bone resorption as well as modify osteoblast function and bone mineralization. The overall effects of vitamin D metabolites on bone are thus extremely complex. From observations in man and animals, it is clear that vitamin D deficiency or resistance impairs bone-matrix mineralization, whereas osteoblast activity and matrix synthesis are even stimulated. Excess 1,25(OH)2D can clearly enhance osteoclastogenesis and bone resorption (Fig. 3-4). Because bone mineralization and bone structure can be largely normalized in vitamin D- or 1,25(OH)2D-deficient or resistant mice by sufficient supply of minerals via active or passive intestinal calcium absorption, it seems that direct effects of vitamin D metabolites on chondrocytes and bone cells are redundant if calcium and phosphate supply are guaranteed. However, most of the genes and proteins typically expressed in osteoblasts and osteoclasts are vitamin D regulated, so it is likely that 1,25(OH)2D can fine-tune bone mineral homeostasis. Moreover, pharmacologic use of vitamin D metabolites or analogs might positively influence bone balance, as shown by human and animal experiments132,133 and transgenic mice overexpressing osteoblast VDR.134
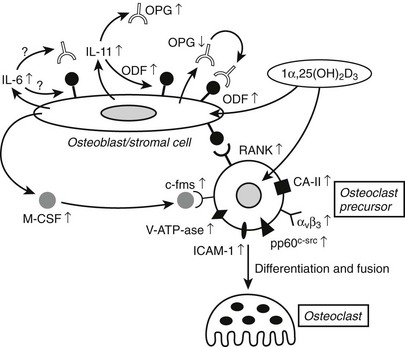
FIGURE 3-4 Effect of 1,25(OH)2D on bone cells and osteoclastogenesis. In osteoblast/stromal cells, 1,25(OH)2D induces ODF expression, down-regulates OPG, and stimulates M-CSF production. It also stimulates production of IL-6 and IL-11, which represent distinct signals in osteoclastogenesis. On osteoclast precursors, 1,25(OH)2D induces the expression of RANK (or ODF receptor) and several osteoclast differentiation markers, such as the vitronectin receptor αvβ3 and carbonic anhydrase-II. CA-II, Carbonic anhydrase-II; c-fms, M-CSF receptor; M-CSF, macrophage colony-stimulating factor; ODF, osteoclast differentiation factor; OPG, osteoprotegerin; V-ATP-ase, vacuolar adenosine triphosphatase.
Effects on Growth Plate
The absence of VDR or 1α-hydroxylase creates no detectable phenotype in overall growth or growth plate of prenatal animals, but the longitudinal growth of long bones is impaired after weaning. X-ray analysis reveals advanced rickets, including widening of the epiphyseal growth plate, with an increased width and marked disorganization of the growth plate on histology, including impaired mineralization of hypertrophic chondrocytes.66,115–118 This increased growth-plate width in VDR or 1α hydroxylase–null mice cannot be explained by their (normal) chondrocyte proliferation and differentiation, including collagen X and osteopontin expression. The expansion of the growth plate can be largely explained by decreased apoptosis of hypertrophic chondrocytes.135 Based on analysis of several genetic models with abnormal phosphate homeostasis, serum phosphate levels are probably crucial for hypertrophic chondrocyte apoptosis in vivo. This is confirmed in vitro: apoptosis of hypertrophic chondrocytes is regulated by phosphate levels via the activation of the caspase-9-mediated mitochondrial pathway.136
In accordance with these findings, chondrocyte-specific inactivation of the VDR did not cause a growth-plate phenotype and certainly not rickets.137 Critical analysis of these mice, however, revealed that VDR action in chondrocytes regulates bone development and phosphate homeostasis by inducing expression of paracrine factors such as vascular endothelial growth factor and receptor activator of nuclear factor κB (NFκB) ligand expression, leading to impaired vascular invasion and decreased osteoclast number in the metaphysic, as well as increased bone mass of long bones of juvenile chondrocyte-specific VDR-null mice. In addition, FGF-23 expression in osteoblasts was decreased, probably linked to the increased gene expression profile of NPT2 and 1α-hydroxylase in the kidney and resulting in increased serum levels of phosphate and 1,25(OH)2D.137
Noncalcemic or Nonclassic Actions of Vitamin D Endocrine System
The virtual ubiquitous expression of the VDR in all nucleated cells, the presence of a functional 1α-hydroxylase in at least 10 different tissues apart from the kidney, and the very large number of genes that are under direct or indirect control of 1,25(OH)2D all point toward a more universal role for the vitamin D endocrine system than just regulation of calcium/phosphate/bone metabolism. This is not totally unexpected; most other ligands for nuclear receptors also have a very wide spectrum of activities such as androgens, estrogens, glucocorticoids, and retinoids.24 Based on controlled observations in cells, tissues, and transgenic mice and on observational studies in humans, it seems that the functioning of nearly all major tissues or systems of the organism is modulated by vitamin D.
Skin
The combined presence of vitamin D production, 25-hydroxylase, 1α-hydroxylase, and VDR expression in the epidermis suggests the existence of a unique vitamin D intracrine system in which UVB-irradiated keratinocytes may supply their own needs for 1,25(OH)2D. A role for vitamin D in epidermal homeostasis can also be expected from the prominent effects of vitamin D compounds on keratinocyte growth and differentiation.138 The epidermal keratinocyte represents the major cell type in the epidermis and most likely the major cutaneous target cell for vitamin D, but many other cell types present in the epidermis are also vitamin D targets.
Based on studies of 1α hydroxylase–deficient mice, the repair of the essential barrier function of the skin is impaired in the absence of vitamin D action.139 The major skin phenotype of both VDR-null mice and children with VDR mutations is, however, the development of total alopecia. Hair development at birth is normal, but hair loss starts after the first catagen and ultimately leads to alopecia totalis associated with large dermal cysts. The absence of alopecia in vitamin D–deficient WT mice or in mice with CYP27B1 mutations clearly suggests that the absence of receptor and not its ligand is the cause of the skin phenotype. Mutations in the NR corepressor, hairless, or keratinocyte-specific loss of interaction of Lefl with β-catenin (part of the Wnt signaling pathway) produce a strikingly similar alopecia. There is therefore little doubt that ligand-independent effects of the VDR are required for normal keratinocyte stem cell function.
As expected from animal studies, no clear skin disorders are linked to vitamin D deficiency or insufficiency in humans. The very same photons that can generate the photoconversion of 7-dehydrocholesterol into previtamin D are also able to cause DNA damage and, ultimately, photoaging and increase the risk for skin cancer, so exposure to the UVB or sunlight needed to produce vitamin D always involves a small but cumulative risk of skin damage. This risk is especially relevant for humans with a fair skin type (phototypes 1 and 2).140 Although 1,25(OH)2D is able to generate a strong photoprotective effect against UVB-mediated events in cultured keratinocytes,141,142 the overall effect is negative.
Cell Proliferation and Cancer
Exposure to 1,25(OH)2D of virtually all normal cells and even most malignant cells results in an accumulation in the G0/G1 phase of the cell cycle.143–145 This inhibition of cell proliferation involves a large number of mechanisms and genes, and the exact sequence of events between VDR-mediated transactivation of genes and the actual G0/G1 arrest is probably cell-type specific. A general downstream effect is the regulation of the E2F family of transcription factors, which act as master switch for a very large number of genes involved in cell-cycle progression. These EF factors are under the control of the retinoblastoma protein members (especially pocket proteins p107 and p130), and their phosphorylation state is regulated by cyclins and cyclin-dependent kinases (p18, p19, p21, or p27), many of which are regulated by 1,25(OH)2D.143,146 However, 1,25(OH)2D may also inhibit cell growth by interfering with signaling pathways initiated by TGF-β, epidermal growth factor (IGF), prostaglandins,147 and Wnt ligands,148 as well as by intervening in other mitogenic signaling pathways (e.g., ERK/MAPK pathway and c-myc) (Fig. 3-5).149–156 Moreover, 1,25(OH)2D can regulate apoptosis and angiogenesis, mechanisms well known to be important for cancer cell expansion. In view of these well-established in vitro effects, one might expect a greater sensitivity for carcinogenesis in VDR-null mice. Epidermal, mammary, and intestinal cells of such animals do indeed show signs of hyperproliferation. Moreover, when exposed to chemocarcinogens or oncogens, VDR-null mice develop more mammary cancer–type lesions, skin tumors, and lymphomas.157,158
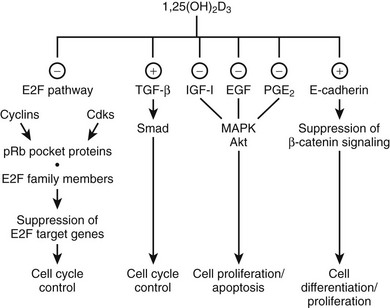
FIGURE 3-5 Effect of 1,25(OH)2D on cell-cycle progression. 1,25(OH)2D treatment leads to a cell cycle phase–specific effect characterized by an accumulation of cells in G1 through modulation of different signaling pathways. (−), Inhibitory effect; (+), stimulatory effect; EGF, epidermal growth factor; IGF-1, insulin-like growth factor 1; PGE2, prostaglandin E2; pRb, retinoblastoma tumor-suppressor gene; TGF-β, transforming growth factor β.
In humans, absolute VDR or CYP27B1 deficiencies are rare, but vitamin D deficiency is highly frequent. This obviously raises the question whether such vitamin D deficiency is associated with increased risk of cancer in humans. Such a hypothesis was originally reinforced by observations of higher cancer prevalence in areas of the United States and Japan with lower UVB exposure. Serum concentrations of 25(OH)D are, of course, a much better indication of the real vitamin D status, and a vast literature links lower levels of 25(OH)D with higher prevalence of the major cancers, especially colon and breast cancer, with more mixed results for prostate cancer. The inverse association between colorectal cancer or breast cancer and serum 25(OH)D levels was confirmed by the results of the Third National Health and Nutrition Examination Survey (NHANES III),159 although 25(OH)D levels were not related to overall cancer mortality. In most of the vast number of cross-sectional or observational studies, the higher cancer risk was found in subjects with serum 25(OH)D levels below 20 ng/mL, but most studies also revealed a significant trend across the different 25(OH)D subgroups and risk of cancer. Meta-analysis of studies addressing the association between 25(OH)D levels suggest that women with serum 25(OH)D of approximately 48 ng/mL (median of the top quintile) had a 50% lower risk of breast cancer than those with serum less than 13 ng/mL in the lowest quintile,160 and that individuals with serum 25(OH)D levels greater than 32 ng/mL had a 50% lower incidence of colorectal cancer than those with relatively low levels (≤12 ng/mL).161 However, a number of studies also link higher vitamin D nutritional status with a higher prevalence or more aggressive type of cancer.24,162
The final question is, of course, whether serum 25(OH)D is a predictor or has a causative relation with the overall cancer risk. Intervention studies should be able to provide the answer. In the Women’s Health Initiative (WHI) study, a significant inverse relationship was found between baseline levels of serum 25(OH)D and subsequent colorectal cancer incidence, but postmenopausal women receiving calcium (1 g) plus vitamin D (400 IU) did not develop less colon cancer than control patients.163 In a much smaller 4-yr study in postmenopausal women, higher doses of calcium (1.4 to 1.5 g) and vitamin D (1100 IU), which raised serum 25(OH)D to mean levels above 80 nmol/L, did significantly reduce overall cancer risk.164 The small number of cancer deaths and a major confounding factor of calcium intake, however, limit the value of this study to hypothesis, and much larger prospective studies with substantial vitamin D supplementation are essential.
Immune Function and Vitamin D
All immune cells (antigen-presenting cells, T and B cells, natural killer [NK] cells, and even mast cells) express at certain stages of their differentiation a functional VDR. Antigen-presenting cells (dendritic cells and equivalent resident cells, as well as monocytes/macrophages) can synthesize 1,25(OH)2D using the same enzyme as in the kidney but controlled by immune stimuli instead of calciotropic hormones.52,165–167 Finally, 1,25(OH)2D regulates a wide range of genes that play crucial roles in the immune system. The overall effects are different for the innate immune system (largely mediated by monocytes/macrophages) than for the acquired immune system. The innate immune system, upon exposure to bacterial agents, is first stimulated to produce 1,25(OH)2D (Fig. 3-6A) and thereafter activated by this paracrine 1,25(OH)2D to become a more active macrophage, including the local production of a number of defensins (including cathelicidin). Defects in immune functions indispensable for antimicrobial activity have been observed in vitamin D–deficient mice.168,169 The overall effects suggest that 1,25(OH)2D enhances the natural defense against bacterial infection. In the human situation, low serum levels of 25(OH)D were repeatedly associated with increased susceptibility to and more rapid disease progression of tuberculosis.170–174 This hypothesis is confirmed by the lower induction of cyclic adenosine monophosphate (cAMP) by monocytes incubated with 25(OH)D-deficient serum from sunlight-deprived African Americans.175 Moreover, oral administration of vitamin D markedly improved tuberculosis outcome in small-scale studies.176,177 Prospective clinical trials are ongoing to evaluate the effect of vitamin D supplementation on the evolution of various infections, including tuberculosis, to confirm the cause/effect relationship between vitamin D status and the native immune defense system.
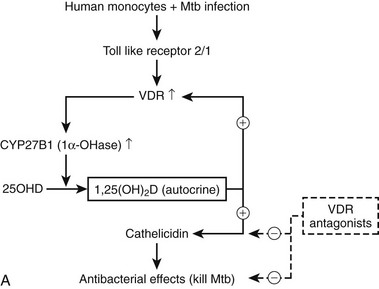
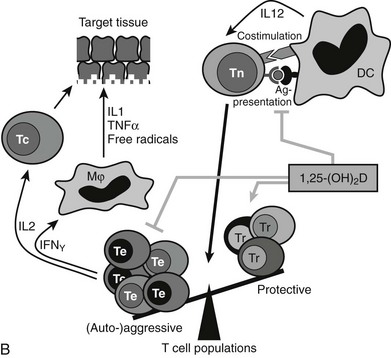
FIGURE 3-6 Immune effects of the vitamin D endocrine system. In the innate immune system, 1,25-(OH)2D strengthens the antimicrobial function of monocytes/macrophages, for example, through enhanced expression of the cathelicidin antimicrobial peptide, eventually leading to better clearance of pathogenic microorganisms. In the acquired immune system, the immunomodulatory effects of 1,25(OH)2D on players of the adaptive immune system can lead to the protection of target tissues in autoimmune diseases and transplantation. 1,25(OH)2D inhibits the surface expression of MHC II–complexed antigen and of costimulatory molecules, as well as the production of the cytokine IL-12 in antigen-presenting cells (such as dendritic cells), thereby shifting the polarization of T cells from an (auto-)aggressive effector (Te) towards a protective or regulatory (Tr) phenotype. 1,25(OH)2D also directly exerts its immunomodulatory effects at the level of T cells.
The acquired immune system reacts in opposite ways to the native immune system (see Fig. 3-6B). Indeed, 1,25(OH)2D inhibits dendritic cell maturation and generates a coordinated action on T cell gene expression of key cytokines (IL-1, IL-2, IL-12, IL-17, INF-γ) and genes needed for antigen presentation to T cells (MHC class II and cosignaling proteins). The global effect of these immune-modulating actions is thus a down-regulation of the acquired immune system. This should have beneficial effects on the occurrence or evolution of autoimmune diseases. Vitamin D–deficient mice more easily develop a more severe type of such autoimmune diseases. For example, genetically predisposed NOD mice exposed to transient vitamin D deficiency early in life have a much higher incidence of type 1 diabetes than vitamin D–replete mice,169,178 and 1α hydroxylase–deficient mice are more prone to several types of inflammatory bowel disease.179 Moreover, 1,25(OH)2D and more potent and selective analogs can significantly reduce spontaneous or experimental autoimmune diseases in rodents, such as type 1 diabetes in NOD mice, experimental allergic encephalitis, nephritis, or inflammatory bowel disease in rodents.16,24,180
Studies in human autoimmune diseases are, however, more complicated. VDR polymorphism is not clearly related to the risk for type 1 diabetes, according to a large meta-analysis.181 However, polymorphism of the 1α-hydroxylase gene is associated with the risk of type 1 diabetes in cross-sectional as well as in family studies.182 Consistent with data from NOD mice, several epidemiologic studies in humans report that vitamin D intake in early life may reduce later risk of type 1 diabetes. Risk reduction varied between 26% with cod liver oil to 78% with 2000 IU/d, with an overall effect of 30% reduction in five published reports.180 Since 25(OH)D serum levels have not been measured in the cohorts of children, a threshold cannot be defined for optimal reduction of type 1 diabetes.
A low vitamin D status has also been repeatedly associated with a higher risk for multiple sclerosis. A large prospective, nested, case-control study among more than 7 million U.S. military personnel revealed that low 25(OH)D level was a strong risk factor for later occurrence of multiple sclerosis (odds ratio of about 2 for serum 25[OH]D <20 ng/mL, with possibly even greater “protection” by higher levels).183 However, as for type 1 diabetes, no controlled, randomized intervention studies have yet proved a causal relation between vitamin D (deficiency or insufficiency) and later occurrence of autoimmune diseases, but randomized trials (especially in genetically at-risk groups) should receive high priority. All these observations, however, suggest that preventing vitamin D deficiency in the perinatal period, early childhood, or adolescence may have long-lasting effects on autoimmune diseases. Moreover, there are several observations in mouse models that could explain such effects, such as the increased apoptosis of (autoreactive?) thymocytes or lymphocytes, the generation of regulatory T cells, and NKT cells after exposure to 1,25(OH)2D.180,184
Cardiovascular System
VDR-null mice, as well as 1α hydroxylase–null mice, develop high-renin hypertension and cardiac hypertrophy that can be prevented by treatment with an angiotensin blocker.185 In vitro or in vivo exposure to 1,25(OH)2D decreases renin production, probably by direct regulation of the gene expression via a VDRE in the promoter of renin. Observational studies in humans found an inverse association between 1,25(OH)2D levels and blood pressure or plasma renin levels in normotensive or hypertensive individuals. Prospective cohort studies reported that incident hypertension over a 4-year follow-up is lowest when the serum 25(OH)D is 30 ng/mL or higher.186 Also, in the NHANES III population, a significant negative relation between serum 25(OH)D concentrations and systolic, diastolic, and pulse pressure among the total adult population was observed.187 The results of these observational studies are supported by two randomized controlled trials in which vitamin D treatment reduced blood pressure in hypertensive subjects and elderly community-dwelling women.
VDR-null mice display increased thrombogenicity and decreased fibrinolysis when exposed to inflammatory stimuli,188 whereas 1,25(OH)2D has beneficial effects on most cells of the vascular wall. In humans, a low vitamin D status is associated with a number of cardiovascular risk factors,189 including the metabolic syndrome. The largest prospective intervention trial (WHI) with calcium (1g/d) and vitamin D (400 IU/d), however, revealed no increased nor decreased coronary or cerebrovascular risk after 7 years of follow-up.190 Several large-scale observational studies demonstrated that survival and especially cardiovascular events were lower in patients on chronic renal replacement therapy treated with 1,25(OH)2D analogs, compared with either untreated or 1,25(OH)2D-treated groups.191 However, a large meta-analysis casts doubt on this conclusion.192 In addition, vitamin D excess can have deleterious effects on all structures of the vascular wall, with ectopic calcification and organ failure of kidney, cardiac valves, myocardium, and most other soft tissues. These data suggest a beneficial effect of the vitamin endocrine system (within specific optimal limits) on cardiovascular targets but certainly need confirmation by a proper prospective, large-scale randomized trial.
Muscle and Muscle Function
VDR is expressed in myoblasts and is also present in low concentrations in mature striated muscle cells. VDR-null mice, even on a high calcium diet, show maturation problems of their muscle fibers, with smaller muscle fibers and expression of embryonic markers even after weaning.193 Genes that are typically expressed early in life (e.g., myf-5) are under negative control by 1,25(OH)2D. Evaluation of muscle performance in VDR-resistant or vitamin D–deficient mice is difficult to interpret, because hypocalcemia may have major effects on calcium fluxes in muscle cells. Patients with chronic renal failure and vitamin D deficiency (thus combined deficiency of 25[OH]D and 1,25[OH]2D) can develop severe myopathy and inability to walk that can be promptly restored by appropriate vitamin D and/or analog treatment. Sarcopenia (progressive loss of muscle mass and strength) is highly prevalent in the elderly and frequently associated with vitamin D deficiency. Vitamin D supplementation can modestly and inconsistently improve muscle function and improve body sway. Meta-analysis of several prospective intervention studies revealed that vitamin D supplementation in vitamin D–deficient elderly subjects can modestly reduce the risk of falls;194 this may explain, together with beneficial effects on bone, a reduced fracture risk.195
Glucose and Energy Metabolism
Several tissues which are important for energy and glucose metabolism, such as endocrine β cells, muscle, and fat cells, are also targets for vitamin D, so the obvious question is whether vitamin regulates or modulates overall metabolism apart from the effects of vitamin D on the immune system and autoimmune diabetes. Vitamin D deficiency in experimental animals (rodents and rabbits) impairs glucose tolerance.180,196,197 VDR-null mice, however, did not have a consistently abnormal glucose tolerance; one strain differs from another strain.24,180 This discrepancy between effects of ligand and receptor deficiency is not unique (see alopecia and immune effect) nor specific for vitamin D. Similar effects are known for thyroid hormone/thyroid receptor function. 1,25(OH)2D has modest stimulatory effects on insulin production and secretion, probably mediated by the well-known effects of calcium on β-cell functions.
Most observational studies in humans link vitamin D insufficiency with nearly all aspects of the metabolic syndrome—including obesity, insulin resistance, fasting blood glucose or type 2 diabetes, hypertension, and hyperlipidemia.24,180 This was confirmed in the NHANES study198 and a Scandinavian cohort study,199 as well as in a prospective British cohort study.200 Baseline 25(OH)D levels were inversely associated with a 10-year risk of fasting hyperglycemia and insulin resistance, with the greatest risk in subjects with 25(OH)D levels below 20 ng/mL and the lowest risk in subjects with above 30 ng/mL.200 Short-term interventional studies to correct severe vitamin D deficiency in a few subjects indicate improved glycemic control, but larger studies using 400 IU/d, such as in the WHI trial, could not demonstrate an effect on glucose levels.201 A much smaller study was also negative, but post hoc analysis revealed a modest effect of a higher dose of vitamin D (700 IU/d) on subjects with fasting hyperglycemia at randomization.202 In view of the high prevalence of vitamin D insufficiency and of metabolic syndrome and their association, it is highly desirable to demonstrate a causal link and then take appropriate actions.
Mortality
Because vitamin D status is associated with so many major diseases, it is worthwhile to explore whether vitamin D deficiency is associated with increased mortality. Indeed, in a prospective 8-year cohort study, all-cause and cardiovascular mortality was twofold higher when 25(OH)D levels were in the lower 2 quartiles (<17 ng/mL), compared with the upper quartile.203 Similar results were obtained from the NHANES III study: mortality was about 1.5-fold higher when 25(OH)D levels were well below 20 ng/mL, and the lowest mortality was observed in subjects with 25(OH)D levels between 20 and 50 ng/mL.204 Also, in patients on chronic hemodialysis, all-cause mortality was greater (significant 1.6-fold increase) when 25(OH)D levels fell below 10 ng/mL.205 In addition, a meta-analysis of all studies using vitamin D supplementation for the prevention of fractures revealed a modest but significant (−8%) reduction in overall mortality in the vitamin D supplemented groups.206
Diagnostic and Therapeutic Aspects of Vitamin D
Assays For Vitamin D and Metabolites: Methodology and Applications
Vitamin D and about 30 of its metabolites are found in plasma. Measurements of their concentrations may be essential for clinical or research purposes.207,208 Most techniques require a lipid extraction to free these compounds from their binding proteins (especially DBP). In view of the high molar extinction of vitamin D, UV absorptiometry can be used for measurement of vitamin D2, vitamin D3, or 25(OH)D after high-performance liquid chromatography (HPLC). However, competitive-binding assays or radioimmunoassay (RIA) are preferred for measurements of 25(OH)D, 1,25(OH)2D, or 24,25(OH)2D.209 These assays remain difficult, as demonstrated by remarkably poor intralaboratory and especially interlaboratory quality-control studies.210,211 Non-chromatographic assays using DBP overestimate the true 25(OH)D concentration by 10% to 20%, but nonspecific interferences (DBP, lipids?) in some assays can result in up to 100% higher values. The quality control of routine 25(OH)D assays revealed extreme problems with accuracy,210 and since the definition of vitamin D deficiency or insufficiency is defined in absolute concentration (see later), there is urgent need for improved quality assurance.210 The use of tandem mass spectrometry after liquid chromatography and serum extraction is now considered the gold standard, but this assay is not yet routinely available for purposes other than research.212
The measurement of serum concentrations of vitamin D2 or D3 is of little clinical value. Indeed, because of their short half-life in plasma, it reflects only recent exposure to UV light or nutritional intake. Serum 25(OH)D concentration is, however, an excellent reflection of the vitamin D status because of the rapid conversion of vitamin D into 25(OH)D and its long plasma half-life.208,209 Its plasma concentration varies widely in normal subjects because of large variations in endogenous and exogenous supply of vitamin D (Table 3-3).
Table 3-3
Plasma Concentration of 25(OH)D
Normal Fluctuation According to:
Dietary intake (+)*
Sun (UV light) exposure (+) influenced by seasonal life style and cultural habits
Age (−)
Skin pigmentation (−)
Latitude (−)
Sunscreen use (−)
Increased 25(OH)D Concentration
Exposure to pharmaceutical vitamin D†
Excess exposure to nutritional vitamin D
Excess exposure to UV light
Decreased 25(OH)D Concentration‡
Combined deficiency of access/exposure to nutritional vitamin D and UV light
Major risk groups include:
Infants, especially when born in late winter
Women and children of immigrants with pigmented skin living in temperate climates
Elderly population with limited mobility
Subset of population with low exposure to sunshine because of socioeconomic, religious, or cultural reasons
Decreased intestinal absorption of vitamin D associated with fat malabsorption (e.g., associated with biliary cirrhosis)
Short bowel syndrome
Exocrine pancreas insufficiency
Gluten enteropathy
Increased loss or catabolism of vitamin D
Nephrotic syndrome
Chronic liver P450 activation by drugs (e.g., barbiturates or antiepileptic drugs)
Low calcium intake or absorption
*Positive or negative effects are indicated by + or −, respectively.
†Vitamin D toxicity with hypercalciuria, hypercalcemia, nephrocalcinosis, kidney stones, metastatic calcification, etc., are only observed if 25(OH)D concentrations exceed 100 ng/mL. Without access to pharmaceutical vitamin D, it is therefore unlikely to acquire clinical vitamin D toxicity.
‡For definition of vitamin D insufficiency or deficiency, see chapter-recommended daily intake and Table 3-5.
Plasma 25(OH)D concentration thus behaves as a true vitamin whose concentration depends on nutritional supply or synthesis in the skin after exposure to UVB light. Low exposure to UV light and low vitamin D intake is quite common in infants and elderly subjects if food sources are not supplemented with vitamin D. Plasma 25(OH)D concentrations are indeed low at birth (about half the maternal concentration because of the 2:1 ratio of maternal to fetal DBP concentration), and the natural vitamin D content of milk is low. Sun exposure was therefore evolution’s solution to prevent rickets. However, in view of the relation between exposure to UVB light (especially in young children) and subsequent risk for skin malignancies, it is probably wise to advocate systematic vitamin D supplementation of all infants and young children. Whereas widespread vitamin D deficiency in infants was recognized and prevented in the beginning of the 20th century, a similar endemic deficiency in the elderly was only recognized and addressed at the end of the same century (see Treatment, later). Intestinal malabsorption of fat-soluble vitamins interrupts the absorption of exogenous (probably also endogenous) hepatobiliary excretion of vitamin D and therefore requires either substitution with large amounts of vitamin D or more physiologic doses (10 to 20 µg/d) of the more soluble 25(OH)D. A low calcium intake can markedly (twofold) increase the catabolism of 25(OH)D and will thus facilitate substrate deficiency if the “nutritional” supply is marginal.213
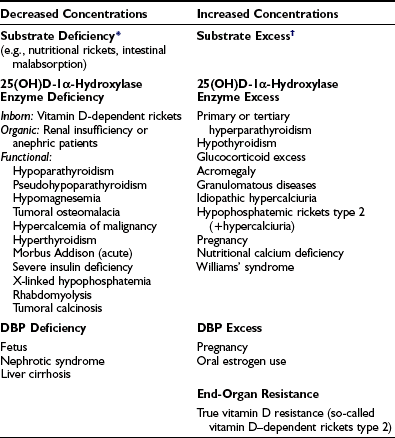
The metabolism of 25(OH)D into 1,25(OH)2D and 24,25(OH)2D is tightly controlled by hormones, ions, and humoral factors. The plasma concentration of 1,25(OH)2D is therefore regulated as a true hormone (Table 3-4), and measurements of its concentration can be useful for clinical exploration of unusual cases of rickets, osteopenia, and hypo- or hypercalcemia.
Table 3-4
Plasma Concentration of 1,25(OH)2D
DBP, Vitamin D-binding protein.
*In many cases of rickets or osteomalacia, 1,25(OH)2D concentrations are still measurable or even nearly normal. This may be due to recent (and insufficient) access to vitamin D after long-term vitamin D deficiency. Nevertheless, such concentration is too low in comparison with the degree of secondary hyperparathyroidism. In any way, 25(OH)D is a better marker for vitamin D deficiency than 1,25(OH)2D. A similar situation is observed in hypothyroidism when precursor hormone T4 is a better marker for clinical hypothyroidism than the real hormone, T3.
†Vitamin D excess only increases serum 1,25(OH)2D when renal function remains normal and/or PTH secretion is elevated. Frequently, 1,25(OH)2D levels are low or normal in vitamin D toxicity.
All vitamin D metabolites are tightly bound to DBP. Since the hepatic 25-hydroxylase activity is not feedback regulated, the free (or total) 25(OH)D concentration is largely fluctuating according to substrate supply. In contrast, renal 25(OH)D-1α-hydroxylase is tightly controlled, and since the access to VDR in target tissues is dependent on the circulating free concentrations, free and not total 1,25(OH)2D concentration is important.80,214 The circulating DBP concentration is fairly stable, except when stimulated by estrogens (or pregnancy) or decreased by reduced synthesis (liver cirrhosis) or increased urinary loss (nephrotic syndrome). The major arguments for the importance of free rather than total 1,25(OH)2D are (1) in vitro experiments (biological activity of 1,25[OH]2D on cultured cells)215,216 and (2) in vivo observations such as increased steady-state concentration of 1,25(OH)2D without signs of increased action during chronic estrogen use or in animals immunized against 1,25(OH)2D-hapten-protein complex.217
Clinical Aspects of Vitamin D
Recommended Daily Intake and Clinical Use of Vitamin D
In contrast to some rare inborn diseases related to vitamin D production, metabolism, or action (see Chapter 11), vitamin D deficiency and insufficiency are extremely frequent worldwide, and the full scope of their prevalence has only recently been appreciated.16,24,32 Vitamin D excess is also a serious disease but seems to occur rarely without the context of excess intake of vitamin D supplementation and is thus usually iatrogenic.
Before the discovery of the dual origin of vitamin D and the introduction of vitamin D supplementation of infants and children, rickets was highly prevalent among the poor in many European cities but also in children of wealthy families (see Chapter 15). The optimal dosage was determined over time, largely on a trial-and-error basis, since this happened well before even the concept of randomized clinical trials was conceived. The vitamin D content of 1 teaspoon of cod liver oil (later found to contain the equivalent of 400 IU of vitamin D3) was found to be efficient and sufficient to prevent endemic rickets. Subsequently, a dose of 200 to 400 IU/d was considered protective. The recommended treatment dose for a child with established rickets was usually larger, partly because of the need for a loading dose. Most recommendations by various official nutritional boards around the world also suggested, up to recently, a daily intake of 5 or 10 µg of vitamin D for infants and children. Indeed, the standing committee on the scientific evaluation of dietary reference intakes (by the U.S. National Academy of Sciences Panel for vitamin D in partnership with the Institute of Medicine) carefully evaluated vitamin D requirements and recommendations in 1997 and suggested 5 µg as the daily supplement (see Table 3-1),218 with similar recommendation by the European Food and Safety Authority.23 Only one real randomized trial with regard to preventive action of vitamin D was ever published and found no cases of new rickets in young Turkish children treated with 400 IU/d for 18 months.219,220 The recommended daily supplement for children, long set at 5 µg or 200 IU/d, was increased in 2008 to 10 µg or 400 IU of vitamin D3/d by the American Academy of Pediatrics.221 No well-designed prospective or randomized trials have ever used serum levels of 25(OH)D to define the minimal threshold for preventing or curing rickets. In clinical case studies, serum 25(OH)D levels found in simple vitamin D–deficiency rickets are well below 10 ng/mL and usually even below 5 ng/mL. Serum 25(OH)D levels are frequently low in newborn sera (cord sera); the mean level in newborns is usually only slightly above 50% of the 25(OH)D levels in the mother’s serum.80 Despite the cheap and effective strategies to prevent rickets, many countries or regions around the world are still facing endemic rickets affecting even a few percent of the infants or children,222 especially in many Islamic countries, rural China, in children of immigrants in Western Europe, or in children born to mothers living in areas of high frequency of vitamin D insufficiency in general.223 The disease prevalence or severity may be aggravated by simultaneous poor dietary calcium intake, such as in many African countries.224 Poor vitamin D status in perinatal life or during childhood may also predispose to lower bone mass much later in life.225
Vitamin D is also known to be a major factor in maintaining bone integrity later in life, and most studies have dealt with the elderly or postmenopausal women. The idea for this role came from repeated and well-documented cross-sectional studies linking increasing PTH serum levels with increasing age and decreasing 25(OH)D levels. To define an optimal 25(OH)D level for optimal bone health, numerous studies have looked at surrogate endpoints (Table 3-5), but fortunately several intervention studies are now also available, including several meta-analyses of these data. Indeed, several surrogate endpoints have been evaluated to define a minimal or optimal threshold for 25(OH)D. Serum concentrations of 1,25(OH)2D are not related to the substrate concentration if serum 25(OH)D exceeds 20 ng/mL in adults, but a positive relation with serum 25(OH)D has been observed when subjects with low 25(OH)D were included. Intervention studies reveal that 1,25(OH)2D rapidly increases, even transiently above the normal level, when vitamin D supplementation is given to vitamin D–insufficient patients (25[OH]D levels < 12 to 20 ng/mL).226 Thus, it seems that the plasma level of 1,25(OH)2D is no longer substrate dependent once 25(OH)D levels exceed 20 ng/mL. PTH has been very extensively used as a surrogate marker for defining optimal vitamin D status. Undoubtedly PTH increases in groups of patients with low 25(OH)D levels; however, PTH concentrations increase only to levels above the normal range when 25(OH)D is very low, whereas in about a third of patients with low 25(OH)D levels, PTH remains normal. The threshold of 25(OH)D below which serum PTH starts to increase varies between 12 and 40 ng/mL in a large number of cross-sectional studies.32 Such differences may partly be due to differences in accuracy of the 25(OH)D assay and differences in nutritional calcium intake and kidney function. Intervention studies are therefore more reliable. Serum PTH decreases after vitamin D supplementation when baseline levels are below 20 ng/mL.32,227 Active intestinal calcium absorption is the primary target for vitamin D action and would thus represent an ideal surrogate endpoint for defining 25(OH)D levels. However, measuring intestinal calcium absorption is difficult because only dual-isotope techniques allow accurate estimations. Other methods, such as plasma concentrations of a calcium isotope after oral intake, provide a more crude estimation, whereas change in total serum calcium concentrations after a large oral calcium loading is a very poor estimation of active calcium absorption. Cross-sectional data suggested a minimal 25(OH)D level of 32 ng/mL for optimal calcium absorption but used a poor procedure to measure calcium absorption.228,229 Other large cross-sectional studies could not identify a true 25(OH)D threshold but only a relation with serum 1,25(OH)2D, either in adults230 or adolescents.231 Again, intervention studies with vitamin D supplementation revealed no232 or only a minimal233 increase in active intestinal calcium absorption in subjects with baseline 25(OH)D levels above 20 ng/mL. Cross-sectional data on 25(OH)D and bone mineral density (BMD) values could not demonstrate a strong correlation; this may be due to a long lag time between vitamin D intake and bone turnover or bone mass. Lower BMD levels, however, were observed in subjects with the lowest 25(OH)D levels (<12 ng/mL).32,234 Fracture prevalence in cross-sectional or prospective studies according to 25(OH)D levels revealed that a higher fracture risk is associated with single point measurements of 25(OH)D levels below 20 ng/mL.235 Finally, intervention studies with vitamin D and/or calcium supplements are most relevant to define the optimal vitamin D status. A very large number of studies have addressed this question, but only a limited number can be classified as well-designed randomized trials. Several meta-analyses came to slightly different conclusions. Vitamin D alone given to postmenopausal or elderly subjects cannot reliably reduce hip-fracture incidence,219,236 and oral calcium supplementation alone also has no clear benefit on hip-fracture risk, with one study even revealing an increased risk.194,237 Combined vitamin D and calcium supplementation, however, can reduce hip-fracture risk by about 20%, with a similar reduction on other nonvertebral fractures.236,238 In these studies, a vitamin D dosage of 800 IU/d was more efficient than 400 IU/d.195
Table 3-5
Strategies and Clinical Studies to Define Optimal Vitamin D Status for Bone Health
Hard Endpoints
Placebo-controlled intervention studies
Vitamin D/25(OH)D and fractures
Prospective/cross-sectional studies
25(OH)D and fractures
Surrogate Endpoints
Prospective/cross-sectional studies
25(OH)D and BMC/BMD
25(OH)D and bone turnover markers
Prospective/cross-sectional studies
25(OH)D and calcium absorption
Cross-sectional/intervention studies
25(OH)D and PTH
Cross-sectional/intervention studies
25(OH)D and 1,25(OH)2D
BMC, Bone mineral content; BMD, bone mineral density; PTH, parathyroid hormone.
It thus seems that only combined high calcium intake (>1 g/d) and vitamin D supplementation (≥800 IU/d) has shown to be efficient for fracture reduction in target populations of elderly subjects, with the greatest effect in institutionalized patients. The corresponding 25(OH)D level is more disputed, mainly owing to lack of accuracy of 25(OH)D assays in older studies and the confusion about optimal (minimal) 25(OH)D in individual subjects and mean population levels. Because most intervention studies conclude that serum 25(OH)D levels increase by 1 ng/mL for each additional 100 IU of vitamin D supplement per day,239 serum 25(OH)D levels (calculated from baseline population level and expected increase of 8 ng/mL for a 800 IU/d supplement) reached during the vitamin D intervention studies with positive fracture effects can be estimated as over 20 ng/mL in most adults, with mean levels closer to 30 ng/mL.240 The amount of vitamin D needed to obtain a minimal 25(OH)D level above 20 ng/mL and a population mean closer to 30 ng/mL, of course, depends on the baseline 25(OH)D level and accesses to UVB and dietary vitamin D. Intervention studies, however, revealed that 400 IU/d is not sufficient to raise 25(OH)D above 20 ng/mL in more than 95% of the target population of postmenopausal or elderly Caucasians, whereas this can be better achieved by 800 IU/d.
What are the options for vitamin D supplementation based on the available information? For infants and children, a daily intake of 400 IU/d should be assured from early life till adolescence, since this can prevent vitamin D–deficiency rickets. Real-life implementation is far from ideal in at-risk groups in Western countries and in many regions of the world where exposure to sunlight is minimal for geographic or sociocultural reasons. For elderly subjects and probably also for all adults, the minimal 25(OH)D level associated with the lowest risk for fractures, falls (based on intervention studies), and a number of major diseases (based on epidemiologic surveys) is above 20 ng/mL. This can be achieved by increasing vitamin D intake by 800 IU/d or equivalent per week or month.241 In populations with better 25(OH)D baseline levels, 400 IU/d can be sufficient, and in some countries or regions of the world with mean 25(OH)D levels of 30 ng/mL, no vitamin D supplements may be needed. Alternative sources of vitamin D such as fatty fish are not a practical solution for many millions of mildly vitamin D–deficient subjects. Higher exposure to UVB light could certainly improve the vitamin D status but cannot be recommended to subjects with a fair-skin phototype (phototypes 1, 2) because of lifetime risk of photodamage or skin cancer. Of course, for a number of patients with specific diseases (see Table 3-3) a higher dose of vitamin D or 25(OH)D or 1,25(OH)2D is needed because of poor intestinal absorption, increased catabolism, or impaired metabolism.
There are a number of arguments that 25(OH)D levels over 30 to 40 ng/mL may provide additional benefits for bone, muscle, and noncalcemic endpoints. To reach such levels in over 97% of the population, daily supplements of at least 2000 IU/d (and usually more) would be needed, with or without substantially greater exposure to UVB.229 Since severe vitamin D toxicity is usually observed only when 25(OH)D levels exceed 100 ng/mL, such levels of 30 to 40 ng/mL are probably safe. However, randomized, large-scale, long-term studies with supplements of 2000 IU/d or more do not exist; such doses were only evaluated in a few hundred subjects for a maximum of 6 months.242 As a reminder of possible toxicity, it is worth remembering that a mild but significant increase in kidney stones was observed in the WHI trial when calcium supplements were combined with at least 400 IU/d of vitamin D for 7 years.163 A causal relationship between vitamin D status and more major diseases has still to be proven, so it seems wise to defer recommendations for a generalized vitamin D intake above the present upper limits. However, in view of the solid hypotheses generated by observational studies, appropriate randomized controlled trials with multiple end points deserve a great priority.
Worldwide Vitamin D Status
Serum 25(OH)D levels, as the best marker for vitamin D status, vary widely in different populations around the world, and the frequency of vitamin D deficiency or insufficiency (Table 3-6) varies accordingly. In an extensive meta-analysis of cross-sectional studies on serum 25(OH)D levels in healthy subjects around the world (394 studies), average serum 25(OH)D levels were 21 ng/mL. Caucasians had slightly higher levels than non-Caucasians,243 but this may be biased, since 25(OH)D levels are lower in subjects with darker skin when living in moderate climate zones. Older subjects (>75 yrs), as well as young children (<15 yr), had lower 25(OH)D levels. Latitude had only a minimal effect, demonstrating that apart from potential exposure to UVB light, many other factors—skin pigmentation, lifestyle, and nutritional factors—define vitamin D status. In more homogenous populations, the expected North-South gradient has been confirmed (e.g., in France),244 whereas for all European populations, the North-South gradient was reversed, probably because of high fish intake in Scandinavian countries and differences in sun-seeking behavior.34,245 In North America, NHANES data confirm that 25(OH)D levels remain relatively stable over time, with mean levels of 30 ng/mL187 and thus substantially higher than in Europe, probably related to the widespread use of vitamin D–enriched food. Non-Hispanic U.S. blacks had mean levels of 20 ng/mL. In some countries, however, mean levels can be quite low.246,247 Lower levels are usually observed in obese subjects or those with special risk factors (see Table 3-3). Low levels are also frequent in pregnant women and their infants, and this poses an additional risk in view of the potential late consequences of perinatal vitamin D deficiency.248
Table 3-6
Vitamin D Nutritional Status as Described by Circulating Levels of 25(OH)D
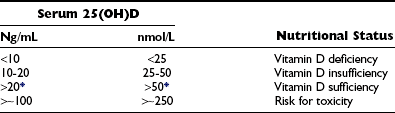
Optimal vitamin D status defined by serum 25(OH)D levels.
*Different opinions exist for defining the minimal threshold for optimal 25(OH)D levels; 30 ng/mL or 50 nmol/L is suggested by others.272
It is therefore obvious that mild vitamin D deficiency (see Table 3-6), even when defined conservatively as 25(OH)D levels under 20 ng/mL, is very widespread worldwide and can affect about one third to half of the world population. When insufficiency is defined by 25(OH)D levels below 30 ng/mL, then half of the healthy U.S. population (based on NHANES data) and about two thirds of the European population (and even more in most Muslim countries) would be vitamin D deficient.16,249 It is therefore imperative that the health consequences of vitamin D status should be better evaluated and that the most severe forms of vitamin D deficiency or insufficiency (see Table 3-2) should be corrected by appropriate strategies.
Therapeutic Potential of 1,25(OH)2D Analogs
The combined presence of 25(OH)D-1α-hydroxylase48 and VDR in several tissues introduced the concept of a paracrine role for 1,25(OH)2D.250 These newly discovered functions of 1,25(OH)2D create possible new therapeutic applications for immune modulation (e.g., for the treatment of autoimmune diseases or prevention of graft rejection), inhibition of cell proliferation (e.g., psoriasis), and induction of cell differentiation (cancer). To achieve growth inhibition or cell differentiation, supraphysiologic doses of 1,25(OH)2D are needed, causing calcemic side effects. Therefore, new analogs of 1,25(OH)2D have been developed to dissociate the antiproliferative and prodifferentiating effects from the calcemic and bone-metabolism effects.251
The secosteroid 1,25(OH)2D, with its open B ring and side chain of 8 carbon atoms, is a very flexible molecule. Different modifications have already been introduced in the A, B, C, and D rings and in the side chain by addition or transposition of hydroxyl groups, introducing unsaturation, replacing a carbon atom with a hetero atom, inverting the stereochemistry, and/or shortening or lengthening the side chain.251 During the last decade, over 500 analogs were synthesized and their biological potency reported in the non-patent literature.251,252 Moreover, several thousands of analogs have been synthesized by pharmaceutical and academic research groups and reported briefly in patent literature. Some of these analogs demonstrate a clear dissociation between antiproliferative and calcemic effects. In the meantime, nonsteroidal analogs were synthesized with a totally new structure lacking the full CD region of 1,25(OH)2D.253,254
No single or simple mechanism can explain the exact mechanism of superagonistic and selective activity profile (calcemic versus noncalcemic effects) of the new vitamin D analogs. Most of the biological effects of 1,25(OH)2D are believed to be mediated via binding to the VDR, but surprisingly the binding affinity of the analogs to VDR does not always correlate with their potency. Some analogs extend the VDR half-life and induce different conformational changes to the VDR-ligand complex as assessed by limited proteolytic digestion and site-directed mutagenesis.255–257 However, most analogs bind to the ligand-binding pocket of the VDR without major modifications of the surface of the ligand-binding domain of the receptor.252,254,258 Nevertheless, the superagonists are able to enhance gene transcription at the level of coactivator recruitment or activity. The selective action of analogs, however, requires different mechanisms, such as different metabolism in different target cells or cell- or gene-specific regulation, depending on the VDR (e.g., homo-heterodimer configuration induced by specific analogs, presence or selective interaction with coactivators or repressor protein, etc).251,259,260 Some analogs are selective agonists or antagonists for nongenomic rapid actions while being devoid of significant genomic activity.105 Since for other steroid hormones (e.g., estrogens, androgens, glucocorticoids), it is now well established that analog-specific gene regulation can be generated by chemical modification of the parent ligand molecule, it is likely that among the many powerful selective vitamin D analogs, at least some will be found to be clinically useful for noncalcemic indications.
Bone Disorders
The role of vitamin D or its metabolites in the treatment of bone disorders characterized by defective mineralization, such as rickets and renal osteodystrophy, is well established. Presently only a few analogs are being evaluated in preclinical or human phase II trials for the prevention or cure of osteoporosis. The vitamin D analog, 2β-(3-hydroxypropoxy)-1,25(OH)2D (ED-71), was found to be more potent than 1,25(OH)2D in rodents and increased bone mineral mass and density in a phase III trial in Japanese postmenopausal women.261 Another vitamin D analog, 2MD, was also very efficient for treatment of ovariectomy-induced bone loss in rats,262 but clinical development was interrupted.
Renal Osteodystrophy
Bone disease in patients with chronic renal failure is due to a complex set of mechanisms such as impaired 1,25(OH)2D synthesis, vitamin D resistance, secondary hyperparathyroidism, increased FGF-23, and abnormal mineral handling (hyperphosphatemia, aluminum or fluoride excess, acidosis). While 1α(OH)D3 and 1,25(OH)2D are widely used for the prevention and cure of renal osteodystrophy, several analogs have been evaluated for this indication, with the aim of better PTH suppression with less risk for inducing hypercalcemia or hyperphosphatemia. Paracalcitol is widely used in the United States for control of secondary hyperparathyroidism. Its use was also associated with a lower rate of cardiovascular and overall mortality in comparison with patients receiving 1,25(OH)2D or no vitamin D treatment.191 These results have been confirmed in a large number of similar nonrandomized studies, possibly biased by unequal patient selection, but prospective randomized clinical trials have not yet addressed this question.
Cancer
A large number of 1,25(OH)2D analogs have been developed with potent antiproliferative and prodifferentiating effects on cancer cells in vitro145 and reduced effects on calcium and bone metabolism.251 Several potent analogs have already been tested in animal models for the treatment of different cancers.158
Although oral seocalcitol (EB 1089) was initially very promising in animal models and in early human studies, it did not provide clear benefits in later studies involving patients with advanced pancreatic and hepatocellular carcinoma. Large doses of oral 1,25(OH)2D given in combination with taxol to patients with advanced prostate cancer was found to increase survival and decrease the risk of thrombosis,263 but a phase III trial in similar patients was stopped early for as yet unknown reasons. Some other analogs are still in early clinical development for a variety of cancers, usually in combination with conventional chemotherapeutics.
Skin
The epidermis is a unique tissue in that it can fully produce and activate the full vitamin D synthesis and metabolism pathways and is very sensitive to 1,25(OH)2D. Calcitriol at pharmacologic concentrations potently induces growth arrest and differentiation of the epidermal keratinocyte.138 The profound effects of calcitriol on keratinocyte proliferation and differentiation have led to the application of vitamin D analogs for skin diseases with disturbed keratinocyte proliferation and differentiation, primarily psoriasis.264 Topical vitamin D analogs that display decreased calcemic activity (calcipotriol and tacalcitol) are now widely used for mild to moderate forms of psoriasis. Monotherapy with these vitamin D compounds achieves an equal effectiveness as topical medium-potency glucocorticoids, without a risk for skin atrophy. Mild irritation is the only frequently observed side effect.264 During treatment with vitamin D analogs, the abnormal epidermal homeostasis is fully restored: keratinocyte proliferation is inhibited; the perturbed psoriatic differentiation profile is normalized, with a decrease of the premature expression of involucrin and type I transglutaminase and enhancement of filaggrin expression;265 and the aberrant expression of cell adhesion molecules (integrins, ICAM1) also returns to normal.266 The concomitant decrease of the inflammatory infiltrate is, however, incomplete,265,266 which may account for the residual redness of the lesions after completion of therapy. This can, however, be further improved by combination with topical usage of corticosteroids.
Immunology
The detection of VDR in almost all cells of the immune system, especially antigen-presenting cells (macrophages and dendritic cells) and activated T lymphocytes, led to the investigation of a potential for 1,25(OH)2D as an immunomodulator.267–270 Vitamin D supplementation is now actively explored in several clinical trials for the stimulation of the natural immune defense and as adjuvant treatment of tuberculosis or other infectious diseases. Similar studies have started for the prevention of major autoimmune diseases. However, vitamin D analogs, though effective in animal models of autoimmunity, have not reached the stage of clinical trials in humans.
Prostate Hyperplasia
Since the prostate is both a VDR- and CYP27B1-expressing tissue, a vitamin D analog, elocalcitol, was evaluated in animal models of inflammatory prostate diseases and subsequently in patients with benign prostate hyperplasia, with modest beneficial effects requiring confirmation in large-scale clinical trials.271
Summary
Vitamin D was discovered in the beginning of the 20th century, and vitamin D supplementation of 200 to 400 IU/d led to eradication of the widespread endemic disease, rickets. At the end of the same century, it became clear that vitamin D deficiency or insufficiency was widely present, especially so in the elderly and subjects with poor exposure to sunshine and nutritional vitamin D (immigrants with dark skin, people with poor skin exposure to sunlight because of sociocultural regions; see Chapter 15).
References
1. Whistler, D. Morbo puerili Anglorum, quem patrio idiomate indigenae vocant. The Rickets. Lugduni Batavorum; 1645.
2. Glisson, F. De Rachitide sive morbo puerili, qui vulgo. London: The Rickets diciteur; 1650.
3. Mozolowski, W. Jedrzej Sniadecki (1768–1883) on the cure of rickets. Nature. 1939;143:121.
4. Palm, TA. The geographic distribution and etiology of rickets. Practitioner. 1890;45:321–342.
5. Huldshinsky, K. Heilung von Rachitis durch künstliche Höhensonne. Dtsch Med Wochenschr. 1919;45:712–713.
6. Chick, HP, Dalyell, EJ, Hume, EM, et al. Studies of rickets in Vienna 1919–1922. Medical Research Council Special Report Series, No. 77. London: Medical Research Council; 1923.
7. Hess, AF, Weinstock, M. Antirachitic properties imparted to lettuce and to growing wheat by ultraviolet irradiation. Proc Soc Exp Biol Med. 1924;22:5–6.
8. Mellanby, E, Cantag, MD. Experimental investigation on rickets. Lancet. 1919;196:407–412.
9. McCollum, EV, Simmonds, N, Pitz, W. The relation of unidentified dietary factors, the fat-soluble A and water-soluble B of the diet to the growth promoting properties of milk. J Biol Chem. 1916;27:33–38.
10. McCollum, EV, Simmonds, N, Becker, JE, et al. Studies on experimental rickets. XXI. An experimental demonstration of the existence of a vitamin which promotes calcium deposition. J Biol Chem. 1922;53:293–312.
11. Goldblatt, H, Soames, KN. A study of rats on a normal diet irradiated daily by the mercury vapor quartz lamp or kept in darkness. Biochem J. 1923;17:294–297.
12. Windaus, A, Linsert, O. Vitamin D1. Ann Chem. 1928;465:148.
13. Specker, BL, Tsang, RC, Hollis, BW. Effect of race and diet on human-milk vitamin-D and 25-hydroxyvitamin D. Am J Dis Child. 1985;139:1134–1137.
14. Hollis, BW. Vitamin D requirement during pregnancy and lactation. J Bone Miner Res. 2007;22:V39–V44.
15. Holick, MF. Vitamin D and the kidney. Kidney Intl. 1987;32:912–929.
16. Holick, MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281.
17. Chen, TC, Shao, Q, Heath, H, et al. An update on the vitamin-D content of fortified milk from the United States and Canada. N Engl J Med. 1993;329:1507.
18. Holick, MF, Shao, Q, Liu, WW, et al. The vitamin D content of fortified milk and infant formula. N Engl J Med. 1992;326:1178–1181.
19. Murphy, SP, Calloway, DH. Nutrient intakes of women in NHANES II, emphasizing trace minerals, fiber, and phytate. J Am Diet Assoc. 1986;86:1366–1372.
20. Krall, EA, Sahyoun, N, Tannenbaum, S, et al. Effect of vitamin D intake on seasonal variations in parathyroid hormone secretion in postmenopausal women. N Engl J Med. 1989;321:1777–1783.
21. Lamberg-Allardt, C, Karkkainen, M, Seppanen, R, et al. Low serum 25-hydroxyvitamin D concentrations and secondary hyperparathyroidism in middle-aged white strict vegetarians. Am J Clin Nutr. 1993;58:684–689.
22. Norman, AW, Bouillon, R, Whiting, SJ, et al. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol. 2007;103:204–205.
23. European Food Safety Authority Scientific Committee on FoodScientific Panel on Dietetic Products, Nutrition and Allergies. Tolerable upper intake levels for vitamin D and minerals. http://www.efsa.europa.eu/, 2006.
24. Bouillon, R, Carmeliet, G, Verlinden, L, et al. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev. 2008;29:726–776.
25. Kelley, RI. RXH/Smith-Lemli-Opitz syndrome: mutations and metabolic morphogenesis. Am J Hum Genet. 1998;63:322–326.
26. Cunniff, C, Kratz, LE, Moser, A, et al. Clinical and biochemical spectrum of patients with RSH/Smith-Lemli-Opitz syndrome and abnormal cholesterol metabolism. Am J Med Genet. 1997;68:263–269.
27. Rossi, M, Federico, G, Corso, G, et al. Vitamin D status in patients affected by Smith-Lemli-Opitz syndrome. J Inherit Metab Dis. 2005;28:69–80.
28. Bonjour, JP, Trechsel, U, Granzer, E, et al. The increase in skin 7-dehydrocholesterol induced by an hypocholesterolemic agent is associated with elevated 25-hydroxyvitamin D3 plasma level. Pflügers Arch. 1987;410:165–168.
29. Morris, JG. Ineffective synthesis of vitamin D in kittens exposed to sun or UV light is reversed by an inhibitor of 7-dehydrocholesterol-delta7-reductase. In: Norman AW, Bouillon R, Thomasset M, eds. Vitamin D: Chemistry, Biology and Clinical Applications of the Steroid Hormone. Riverside, CA: University of California; 1997:721–722.
30. Oliveri, B, Zeni, S, Lorenzetti, MP, et al. Effect of one year residence in Antarctica on bone mineral metabolism and body composition. Eur J Clin Nutr. 1999;53:88–91.
31. Du, X, Greenfield, H, Fraser, DR, et al. Vitamin D deficiency and associated factors in adolescent girls in Beijing. Am J Clin Nutr. 2001;74:494–500.
32. Lips, P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: consequences for bone loss and fractures and therapeutic implications. Endocr Rev. 2001;22:477–501.
33. Lips, P. Which circulating level of 25-hydroxyvitamin D is appropriate? J Steroid Biochem Mol Biol. 2004;89–90:611–614.
34. Lips, P. Vitamin D status and nutrition in Europe and Asia. J Steroid Biochem Mol Biol. 2007;103:620–625.
35. Munns, C, Zacharin, MR, Rodda, CP, et al. Prevention and treatment of infant and childhood vitamin D deficiency in Australia and New Zealand: a consensus statement. Med J Aust. 2006;185:268–272.
36. Working Group of the Australian and New Zealand Bone and Mineral Society ESoAaOA. Vitamin D and adult bone health in Australia and New Zealand: a position statement. Med J Aust. 2005;182:281–285.
37. Gascon-Barré, M. The vitamin D 25-hydroxylase. In: Feldman D, Pike JW, Glorieux FH, eds. Vitamin D. San Diego: Academic Press; 2005:47–67.
38. Henry, HL. The 25-hydroxyvitamin D 1α-hydroxylase. In: Feldman D, Pike JW, Glorieux FH, eds. Vitamin D. San Diego: Academic Press; 2005:69–83.
39. Blunt, JW, DeLuca, HF, Schnoes, HK. 25-Hydroxycholecalciferol. A biologically active metabolite of vitamin D3. Biochemistry. 1968;7:3317–3322.
40. Ponchon, G, Kennan, AL, DeLuca, HF. “Activation” of vitamin D by the liver. J Clin Invest. 1969;48:2032–2037.
41. Postlind, H, Axen, E, Bergman, T, et al. Cloning, structure, and expression of a cDNA encoding vitamin D3 25-hydroxylase. Biochem Biophys Res Commun. 1997;241:491–497.
42. Cheng, JB, Levine, MA, Bell, NH, et al. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci U S A. 2004;101:7711–7715.
43. Maeda, N, Reshef, A, Lippoldt, A, et al. Markedly reduced bile acid synthesis but maintained levels of cholesterol and vitamin D metabolites in mice with disrupted sterol 27-hydroxylase gene. J Biol Chem. 1998;273:14805–14812.
44. Bhattacharyya, MH, DeLuca, HF. The regulation of calciferol-25-hydroxylase in the chick. Biochem Biophys Res Commun. 1974;59:734–741.
45. Fraser, DR, Kodicek, E. Unique biosynthesis by kidney of a biologically active vitamin D metabolite. Nature. 1970;228:764–766.
46. Nicolaysen, R, Eeglarsen, N, Malm, J. Physiology of calcium metabolism. Physiol Rev. 1953;33:424–444.
47. Monkawa, T, Yoshida, T, Wakino, S, et al. Molecular cloning of cDNA and genomic DNA for human 25-hydroxyvitamin D3 1α-hydroxylase. Biochem Biophys Res Commun. 1997;239:527–533.
48. Fu, GK, Lin, D, Zhang, MYH, et al. Cloning of human 25-hydroxyvitamin D-1α-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol. 1997;11:1961–1970.
49. St-Arnaud, R, Messerlian, S, Moir, JM, et al. The 25-hydroxyvitamin D 1-alpha-hydroxylase gene maps to the pseudovitamin D-deficiency rickets (PDDR) disease locus. J Bone Miner Res. 1997;12:1552–1559.
50. Takeyama, K, Kitanaka, S, Sato, T, et al. 25-Hydroxyvitamin D3 1α-hydroxylase and vitamin D synthesis. Science. 1997;277:1827–1830.
51. Shinki, T, Shimada, H, Wakino, S, et al. Cloning and expression of rat 25-hydroxyvitamin D3–1α-hydroxylase cDNA. Proc Natl Acad Sci U S A. 1997;94:12920–12925.
52. Overbergh, L, Decallonne, B, Valckx, D, et al. Identification and immune regulation of 25-hydroxyvitamin D-1-alpha-hydroxylase in murine macrophages. Clin Exp Immunol. 2000;120:139–146.
53. Bell, NH. 25-Hydroxyvitamin D-1α-hydroxylases and their clinical significance. J Bone Miner Res. 1998;13:350–353.
54. Razzaque, MS, Sitara, D, Taguchi, T, et al. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J. 2006;20:720–722.
55. Brenza, HL, Kimmel-Jehan, C, Jehan, F, et al. Parathyroid hormone activation of the 25-hydroxylation D3–1α-hydroxylase gene promotor. Proc Natl Acad Sci U S A. 1998;95:1387–1391.
56. Murayama, A, Takeyama, K, Kitanaka, S, et al. The promoter of the human 25-hydroxyvitamin D3 1α-hydroxylase gene confers positive and negative responsiveness to PTH, Calcitonin, and 1α,25(OH)2D3. Biochem Biophys Res Commun. 1998;249:11–16.
57. Kato, S, Fujiki, R, Kim, MS, et al. Ligand-induced transrepressive function of VDR requires a chromatin remodeling complex, WINAC. J Steroid Biochem Mol Biol. 2007;103:372–380.
58. Fraser, D, Kooh, SW, Kind, HP, et al. Pathogenesis of hereditary vitamin-D-dependent rickets, An inborn error of vitamin D metabolism involving defective conversion of 25-hydroxyvitamin D to 1α,25-dihydroxyvitamin D. N Engl J Med. 1973;289:817–822.
59. Kitanaka, S, Takeyama, K, Murayama, A, et al. Inactivating mutations in the 25-hydroxyvitamin D3 1α-hydroxylase gene in patients with pseudovitamin D-deficient rickets. N Engl J Med. 1998;338:653–661.
60. Ohyama, Y, Noshiro, M, Eggertsen, G, et al. Structural characterization of the gene encoding rat 25-hydroxyvitamin-D(3) 24-hydroxylase. Biochemistry. 1993;32:76–82.
61. Lohnes, D, Jones, G. Side chain metabolism of vitamin D3 in osteosarcoma cell line UMR-106. J Biol Chem. 1987;262:14394–14401.
62. Akiyoshi-Shibata, M, Sakaki, T, Ohyama, Y, et al. Further oxidation of hydroxycalcidiol by calcidiol 24-hydroxylase. A study with the mature enzyme expressed in Escherichia coli. Eur J Biochem. 1994;224:335–343.
63. St Arnaud, R, Arabian, A, Travers, R, et al. Deficient mineralization of intramembranous bone in vitamin D-24-hydroxylase-ablated mice is due to elevated 1,25-dihydroxyvitamin D and not to the absence of 24,25-dihydroxyvitamin D. Endocrinology. 2000;141:2658–2666.
64. Meyer, MB, Zella, LA, Nerenz, RD, et al. Characterizing early events associated with the activation of target genes by 1,25-dihydroxyvitamin D3 in mouse kidney and intestine in vivo. J Biol Chem. 2007;282:22344–22352.
65. Omdahl, J, May, B. The 25-hydroxyvitamin D 24-hydroxylase. In: Feldman D, Pike JW, Glorieux FH, eds. Vitamin D. San Diego: Academic Press; 2005:85–104.
66. Yoshizawa, T, Handa, Y, Uematsu, Y, et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet. 1997;16:391–396.
67. Albertson, DG, Ylstra, B, Segraves, R, et al. Quantitative mapping of amplicon structure by array CGH identifies CYP24 as a candidate oncogene. Nat Genet. 2000;25:144–146.
68. Reddy, GS, Siucaldera, ML, Schuster, I, et al. Target tissue specific metabolism of 1,25(OH)2D3 through A-ring modification. In: Norman AW, Bouillon R, Thomasset M, eds. Vitamin D: Chemistry, Biology and Clinical Applications of the Steroid Hormone. Riverside, CA: University of California; 1997:139–146.
69. Dueland, S, Pedersen, JI, Helgerud, P, et al. Absorption, distribution, and transport of vitamin-D3 and 25-hydroxyvitamin-D3 in the rat. Am J Physiol. 1983;245:E463–E467.
70. Bouillon, R, Van Baelen, H, Rombauts, W, et al. The purification and characterisation of the human-serum binding protein for the 25-hydroxycholecalciferol (transcalciferin). Identity with group-specific component. Eur J Biochem. 1976;66:285–291.
71. Hirschfeld, J. Immune-electrophoretic demonstration of qualitative differences in human sera and their relation to the haptoglobins. Acta Pathol Microbiol. 1959;47:160–168.
72. Westwood, WAWDJ. Group-specific component: a review of the isoelectric focusing methods and auxiliary methods available for the separation of its phenotypes. Forensic Sci Int. 1986;32:135–150.
73. Cleve, H, Constants, J. The mutants of the vitamin-D-binding protein: more than 120 variants of the GC/DBP system. Vox Sang. 1988;54:215–225.
74. Cooke, NE, Safadi, FF, Magiera, HM, et al. Biological consequences of vitamin D binding protein deficiency in a mouse model. In: Norman AW, Bouillon R, Thomasset M, eds. Vitamin D: Chemistry, Biology and Clinical Applications of the Steroid Hormone. Riverside, CA: University of California; 1997:105–111.
75. Laing, CJ, Cooke, NE. Vitamin D binding protein. In: Feldman D, Pike JW, Glorieux FH, eds. Vitamin D. San Diego: Academic Press; 2005:117–134.
76. Verboven, C, Rabijns, A, De, MM, et al. A structural basis for the unique binding features of the human vitamin D-binding protein. Nat Struct Biol. 2002;9:131–136.
77. Bouillon, R, Van Baelen, H. The transport of vitamin D: significance of free and total concentrations of vitamin D metabolites. In: Norman AW, Schaefer K, von Herrath D, et al, eds. Vitamin D: Chemical, Biochemical and Clinical Endocrinology of Calcium Metabolism. Berlin: Walter de Gruyter; 1982:1181–1186.
78. Vicchio, D, Yergey, A, Obrien, K, et al. Quantification and kinetics of 25-hydroxyvitamin-D3 by isotope dilution liquid chromatography/thermospray mass spectrometry. Biol Mass Spectrom. 1993;22:53–58.
79. Kumar, R. The metabolism and mechanism of action of 1,25-dihydroxyvitamin-D3. Kidney Intl. 1986;30:793–803.
80. Bouillon, R, Van Assche, FA, Van Baelen, H, et al. Influence of the vitamin D-binding protein on the serum concentration of 1,25-dihydroxyvitamin D3. J Clin Invest. 1981;67:589–596.
81. Nykjaer, A, Dragun, D, Walther, D, et al. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell. 1999;96:507–515.
82. McLeod, JF, Kowalski, MA, Haddad, JG. Interactions among serum vitamin D binding protein, monomeric actin, profilin, and profilactin. J Biol Chem. 1989;264:1260–1267.
83. Van Baelen, H, Bouillon, R, De Moor, P. Vitamin D-binding protein (Gc-globulin) binds actin. J Biol Chem. 1980;255:2270–2272.
84. Goldschmidt-Clermont, PJ, Van Baelen, H, Bouillon, R, et al. Role of group-specific component (vitamin D binding protein) in clearance of actin from the circulation in the rabbit. J Clin Invest. 1988;81:1519–1527.
85. Lee, WM, Galbraith, RM. The extracellular actin-scavenger system and actin toxicity. N Engl J Med. 1992;326:1335–1341.
86. Whitfield, GK, Jurutka, PW, Haussler, CA, et al. Nuclear vitamin D receptor: structure-function, molecular control of gene transcription, and novel bioactions. In: Feldman D, Pike JW, Glorieux FH, eds. Vitamin D. San Diego: Academic Press; 2005:219–261.
87. Miyamoto, K, Kesterson, RA, Yamamoto, H, et al. Structural organization of the human vitamin D receptor chromosomal gene and its promoter. Mol Endocrinol. 1997;11:1165–1179.
88. Crofts, LA, Hancock, MS, Morrison, NA, et al. Multiple promoters direct the tissue-specific expression of novel N-terminal variant human vitamin D receptor gene transcripts. Proc Natl Acad Sci USA. 1998;95:10529–10534.
89. Haussler, MR, Whitfield, GK, Haussler, CA, et al. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13:325–349.
90. Masuyama, H, Jefcoat, SC, Jr., MacDonald, PN. The N-terminal domain of transcription factor IIB is required for direct interaction with the vitamin D receptor and participates in vitamin D-mediated transcription. Mol Endocrinol. 1997;11:218–228.
91. Nagy, L, Kao, J-YCD, Lin, RJ, et al. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89:373–380.
92. Chakravarti, D, LaMorte, VJ, Nelson, MC, et al. Role of CBP/P300 in nuclear receptor signalling. Nature. 1996;383:99–103.
93. Kamei, Y, Xu, L, Heinzel, T, et al. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414.
94. Rachez, C, Freedman, LP. Mechanisms of gene regulation by vitamin D(3) receptor: a network of coactivator interactions. Gene. 2000;246:9–21.
95. Spencer, TE, Jenster, G, Burcin, MM, et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389:194–198.
96. Rachez, C, Gamble, M, Chang, CP, et al. The DRIP complex and SRC-1/p160 coactivators share similar nuclear receptor binding determinants but constitute functionally distinct complexes. Mol Cell Biol. 2000;20:2718–2726.
97. Kitagawa, H, Fujiki, R, Yoshimura, K, et al. The chromatin-remodeling complex WINAC targets a nuclear receptor to promoters and is impaired in Williams syndrome. Cell. 2003;113:905–917.
98. Li, B, Carey, M, Workman, JL. The role of chromatin during transcription. Cell. 2007;128:707–719.
99. Villagra, A, Cruzat, F, Carvallo, L, et al. Chromatin remodeling and transcriptional activity of the bone-specific osteocalcin gene require CCAAT/enhancer-binding protein beta-dependent recruitment of SWI/SNF activity. J Biol Chem. 2006;281:22695–22706.
100. Li, H, Chen, JD. The receptor-associated coactivator 3 activates transcription through CREB-binding protein recruitment and autoregulation. J Biol Chem. 1998;273:5948–5954.
101. Lavinsky, RM, Jepsen, K, Heinzel, T, et al. Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc Natl Acad Sci U S A. 1998;95:2920–2925.
102. Carlberg, C. The concept of multiple vitamin D signaling pathways. J Invest Dermatol Symp Proc. 1996;1:10–14.
103. Norman, AW, Mizwicki, MT, Norman, DP. Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Discov. 2004;3:27–41.
104. Revelli, A, Massobrio, M, Tesarik, J. Nongenomic effects of 1α,25-dihydroxyvitamin D3. Trends Endocrinol Metab. 1998;9:419–422.
105. Norman, AW, Zanello, LP, De Song, X, et al. Effectiveness of 1α,25(OH)2-vitamin D3-mediated signal transduction for genomic and rapid biological responses is dependent upon the conformation of the signaling ligand. In: Norman AW, Bouillon R, Thomasset M, eds. Vitamin D: Chemistry, Biology and Clinical Applications of the Steroid Hormone. Riverside, CA: University of California; 1997:331–333.
106. Norman, AW, Bouillon, R, Farach-Carson, MC, et al. Demonstration that 1b,25-dihydroxyvitamin D3 is an antagonist of the nongenomic but not genomic biological responses and biological profile of the three A-ring diastereomers of 1α,25-dihydroxyvitamin D3. J Biol Chem. 1993;268:20022–20030.
107. Song, X, Bishop, JE, Okamura, WH, et al. Stimulation of phosphorylation of mitogen-activated protein kinase by 1α,25-dihydroxyvitamin D3 in promyelocytic NB4 leukemia cells: a structure-function study. Endocrinology. 1998;139:457–468.
108. Norman, AW, Okamura, WH, Farach-Carson, MC, et al. Structure-function studies of 1,25-dihydroxyvitamin D3 and the vitamin-D endocrine system. 1,25-Dihydroxy-pentadeuterio-previtamin D3 (as a 6-s-cis analog) stimulates nongenomic but not genomic biological responses. J Biol Chem. 1993;268:13811–13819.
109. Strom, TM, Juppner, H. PHEX, FGF23, DMP1 and beyond. Curr Opin Nephrol Hypertens. 2008;17:357–362.
110. Taketani, T, Miyamoto, K-I, Tanaka, K, et al. Gene structure and functional analysis of the human Na+/phosphate co-transporter. Biochem J. 1997;324:927–937.
111. Wasserman, RH. Vitamin D and the intestinal absorption of calcium: a view and overview. In: Feldman D, Pike JW, Glorieux FH, eds. Vitamin D. San Diego: Academic Press; 2005:411–428.
112. Van Cromphaut, SJ, Dewerchin, M, Hoenderop, JG, et al. Duodenal calcium absorption in vitamin D receptor-knockout mice: functional and molecular aspects. Proc Natl Acad Sci U S A. 2001;98:13324–13329.
113. Feher, JJ. Facilitated calcium diffusion by intestinal calcium-binding protein. Cell Physiol. 1983;13:C303–C307.
114. Pannabecker, TL, Chandler, JS, Wasserman, RH. Vitamin D-dependent transcriptional regulation of the intestinal plasma membrane calcium pump. Biochem Biophys Res Commun. 1995;213:499–505.
115. Li, YC, Pirro, AE, Amling, M, et al. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94:9831–9835.
116. Erben, RG, Soegiarto, DW, Weber, K, et al. Deletion of deoxyribonucleic acid binding domain of the vitamin D receptor abrogates genomic and nongenomic functions of vitamin D. Mol Endocrinol. 2002;16:1524–1537.
117. Dardenne, O, Prud’homme, J, Arabian, A, et al. Targeted inactivation of the 25-hydroxyvitamin D(3)-1(alpha)-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142:3135–3141.
118. Panda, DK, Miao, D, Tremblay, ML, et al. Targeted ablation of the 25-hydroxyvitamin-D 1-alpha-hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci U S A. 2001;98:7498–7503.
119. Dardenne, O, Prud’homme, J, Hacking, SA, et al. Correction of the abnormal mineral ion homeostasis with a high-calcium, high-phosphorus, high-lactose diet rescues the PDDR phenotype of mice deficient for the 25-hydroxyvitamin-D 1-alpha-hydroxylase (CYP27B1). Bone. 2003;32:332–340.
120. Amling, M, Priemel, M, Holzmann, T, et al. Rescue of the skeletal phenotype of vitamin D receptor–ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analyses. Endocrinology. 1999;140:4982–4987.
121. Dardenne, O, Prudhomme, J, Hacking, SA, et al. Rescue of the pseudo-vitamin D deficiency rickets phenotype of CYP27B1-deficient mice by treatment with 1,25-dihydroxyvitamin D3: biochemical, histomorphometric, and biomechanical analyses. J Bone Miner Res. 2003;18:637–643.
122. Hoenderop, JG, Dardenne, O, Van, AM, et al. Modulation of renal Ca2+ transport protein genes by dietary Ca2+ and 1,25-dihydroxyvitamin D3 in 25-hydroxyvitamin D3–1α-hydroxylase knockout mice. FASEB J. 2002;16:1398–1406.
123. Li, YC, Amling, M, Pirro, AE, et al. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. 1998;139:4391–4396.
124. Rowling, MJ, Gliniak, C, Welsh, J, et al. High dietary vitamin D prevents hypocalcemia and osteomalacia in CYP27B1 knockout mice. J Nutr. 2007;137:2608–2615.
125. Song, Y, Kato, S, Fleet, JC. Vitamin D receptor (VDR) knockout mice reveal VDR-independent regulation of intestinal calcium absorption and ECaC2 and calbindin D9k mRNA. J Nutr. 2003;133:374–380.
126. Balsan, S, Garabedian, M, Larchet, M, et al. Long-term nocturnal calcium infusions can cure rickets and promote normal mineralization in hereditary resistance to 1,25-dihydroxyvitamin D. J Clin Invest. 1986;77:1661–1667.
127. Hochberg, Z, Tiosano, D, Even, L. Calcium therapy for calcitriol-resistant rickets. J Pediatr. 1992;121:803–808.
128. Lieben, L, Masuyama, R, Moermans, K, et al. Intestinal-specific vitamin D receptor null mice maintain normal calcemia but display severe bone loss. J Bone Miner Res. 2008.
129. Benn, BS, Ajibade, D, Porta, A, et al. Active intestinal calcium transport in the absence of transient receptor potential vanilloid type 6 and calbindin-D9k. Endocrinology. 2008;149:3196–3205.
130. Fujita, H, Sugimoto, K, Inatomi, S, et al. Tight junction proteins claudin-2 and -12 are critical for vitamin D-dependent Ca2+ absorption between enterocytes. Mol Biol Cell. 2008;19:1912–1921.
131. Renkema, KY, Nijenhuis, T, van der Eerden, BC, et al. Hypervitaminosis D mediates compensatory Ca2+ hyperabsorption in TRPV5 knockout mice. J Am Soc Nephrol. 2005;16:3188–3195.
132. Okano, T, Tsugawa, N, Masuda, S, et al. Regulatory activities of 2b-(3-hydroxypropoxy)-1α,25-dihydroxy- vitamin D3, a novel synthetic vitamin D3 derivative, on calcium metabolism. Biochem Biophys Res Commun. 1989;163:1444–1449.
133. Tilyard, MW, Spears, GFS, Thomson, J, et al. Treatment of postmenopausal osteoporosis with calcitriol or calcium. N Engl J Med. 1992;326:357–362.
134. Gardiner, EM, Sims, NA, Thomas, GP, et al. Elevated osteoblastic vitamin D receptor in transgenic mice yields stronger bones. Bone. 1998;23:176.
135. Donohue, MM, Demay, MB. Rickets in VDR null mice is secondary to decreased apoptosis of hypertrophic chondrocytes. Endocrinology. 2002;143:3691–3694.
136. Sabbagh, Y, Carpenter, TO, Demay, MB. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci U S A. 2005;102:9637–9642.
137. Masuyama, R, Stockmans, I, Torrekens, S, et al. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest. 2006;116:3150–3159.
138. Bikle, DD, Pillai, S. Vitamin D, calcium, and epidermal differentiation. Endocr Rev. 1993;14:3–19.
139. Bikle, DD, Chang, S, Crumrine, D, et al. 25 Hydroxyvitamin D 1 alpha-hydroxylase is required for optimal epidermal differentiation and permeability barrier homeostasis. J Invest Dermatol. 2004;122:984–992.
140. Gilchrest, BA. Sun exposure and vitamin D sufficiency. Am J Clin Nutr. 2008;88:570S–577S.
141. De HP, Garmyn, M, Verstuyf, A, et al. 1,25-Dihydroxyvitamin D3 and analogues protect primary human keratinocytes against UVB-induced DNA damage. J Photochem Photobiol B. 2005;78:141–148.
142. Gupta, R, Dixon, KM, Deo, SS, et al. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J Invest Dermatol. 2007;127:707–715.
143. Jensen, SS, Madsen, MW, Lukas, J, et al. Inhibitory effects of 1-alpha,25-dihydroxyvitamin D(3) on the G(1)-S phase-controlling machinery. Mol Endocrinol. 2001;15:1370–1380.
144. Colston, K, Colston, MJ, Feldman, D. 1,25-dihydroxyvitamin D3 and malignant melanoma: the presence of receptors and inhibition of cell growth in culture. Endocrinology. 1981;108:1083–1086.
145. Abe, E, Miyaura, C, Sakagami, H, et al. Differentiation of mouse myeloid leukemia cells induced by 1 alpha,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1981;78:4990–4994.
146. Verlinden, L, Eelen, G, Beullens, I, et al. Characterization of the condensin component Cnap1 and protein kinase Melk as novel E2F target genes down-regulated by 1,25-dihydroxyvitamin D3. J Biol Chem. 2005;280:37319–37330.
147. Moreno, J, Krishnan, AV, Peehl, DM, et al. Mechanisms of vitamin D-mediated growth inhibition in prostate cancer cells: inhibition of the prostaglandin pathway. Anticancer Res. 2006;26:2525–2530.
148. Aguilera, O, Pena, C, Garcia, JM, et al. The Wnt antagonist DICKKOPF-1 gene is induced by 1-alpha,25-dihydroxyvitamin D3 associated to the differentiation of human colon cancer cells. Carcinogenesis. 2007;28:1877–1884.
149. Tong, WM, Kallay, E, Hofer, H, et al. Growth regulation of human colon cancer cells by epidermal growth factor and 1,25-dihydroxyvitamin D3 is mediated by mutual modulation of receptor expression. Eur J Cancer. 1998;34:2119–2125.
150. Vink-van Wijngaarden, T, Pols, HA, Buurman, CJ, et al. Inhibition of insulin- and insulin-like growth factor-I-stimulated growth of human breast cancer cells by 1,25-dihydroxyvitamin D3 and the vitamin D3 analogue EB1089. Eur J Cancer. 1996;32A:842–848.
151. Verlinden, L, Verstuyf, A, Convents, R, et al. Action of 1,25(OH)2D3 on the cell cycle genes, cyclin D1, p21 and p27 in MCF-7 cells. Mol Cell Endocrinol. 1998;142:57–65.
152. Reitsma, PH, Rothberg, PG, Astrin, SM, et al. Regulation of myc gene expression in HL-60 leukaemia cells by a vitamin D metabolite. Nature. 1983;306:492–494.
153. Matsumoto, K, Hashimoto, K, Nishida, Y, et al. Growth-inhibitory effects of 1,25-dihydroxyvitamin D3 on normal human keratinocytes cultured in serum-free medium. Biochem Biophys Res Commun. 1990;166:916–923.
154. Mercier, T, Chaumontet, C, Gaillard-Sanchez, I, et al. Calcitriol and lexicalcitol (KH1060) inhibit the growth of human breast adenocarcinoma cells by enhancing transforming growth factor-beta production. Biochem Pharmacol. 1996;52:505–510.
155. Wu, Y, Haugen, JD, Zinsmeister, AR, et al. 1 alpha,25-dihydroxyvitamin D3 increases transforming growth factor and transforming growth factor receptor type I and II synthesis in human bone cells. Biochem Biophys Res Commun. 1997;239:734–739.
156. Rozen, F, Pollak, M. Inhibition of insulin-like growth factor I receptor signaling by the vitamin D analogue EB1089 in MCF-7 breast cancer cells: A role for insulin-like growth factor binding proteins. Int J Oncol. 1999;15:589–594.
157. Welsh, J. Targets of vitamin D receptor signaling in the mammary gland. J Bone Miner Res. 2007;22:V86–V90.
158. Bouillon, R, Eelen, G, Verlinden, L, et al. Vitamin D and cancer. J Steroid Biochem Mol Biol. 2006;102:156–162.
159. Freedman, DM, Looker, AC, Chang, SC, et al. Prospective study of serum vitamin D and cancer mortality in the United States. J Natl Cancer Inst. 2007;99:1594–1602.
160. Garland, CF, Gorham, ED, Mohr, SB, et al. Vitamin D and prevention of breast cancer: pooled analysis. J Steroid Biochem Mol Biol. 2007;103:708–711.
161. Giovannucci, E. Strengths and limitations of current epidemiologic studies: vitamin D as a modifier of colon and prostate cancer risk. Nutr Rev. 2007;65:S77–S79.
162. Bouillon, R, Bischoff-Ferrari, H, Willett, W. Vitamin D and health: perspectives from mice and man. J Bone Miner Res. 2008;23:974–979.
163. Wactawski-Wende, J, Kotchen, JM, Anderson, GL, et al. Calcium plus vitamin D supplementation and the risk of colorectal cancer. N Engl J Med. 2006;354:684–696.
164. Lappe, JM, Travers-Gustafson, D, Davies, KM, et al. Vitamin D and calcium supplementation reduces cancer risk: results of a randomized trial. Am J Clin Nutr. 2007;85:1586–1591.
165. Overbergh, L, Stoffels, K, Waer, M, et al. Immune regulation of 25-hydroxyvitamin D-1-alpha-hydroxylase in human monocytic THP1 cells: mechanisms of interferon-gamma-mediated induction. J Clin Endocrinol Metab. 2006;91:3566–3574.
166. Stoffels, K, Overbergh, L, Giulietti, A, et al. Immune regulation of 25-hydroxyvitamin-D3–1α-hydroxylase in human monocytes. J Bone Miner Res. 2006;21:37–47.
167. Stoffels, K, Overbergh, L, Bouillon, R, et al. Immune regulation of 1α-hydroxylase in murine peritoneal macrophages: unravelling the IFNgamma pathway. J Steroid Biochem Mol Biol. 2007;103:567–571.
168. Kankova, M, Luini, W, Pedrazzoni, M, et al. Impairment of cytokine production in mice fed a vitamin D3-deficient diet. Immunology. 1991;73:466–471.
169. Giulietti, A, Gysemans, C, Stoffels, K, et al. Vitamin D deficiency in early life accelerates type 1 diabetes in non-obese diabetic mice. Diabetologia. 2004;47:451–462.
170. Chan, TY. Vitamin D deficiency and susceptibility to tuberculosis. Calcif Tissue Int. 2000;66:476–478.
171. Davies, PDO, Brown, RC, Woodhead, JS. Serum concentrations of vitamin D metabolites in untreated tuberculosis. Thorax. 1985;40:187–190.
172. Grange, JM, Davies, PDO, Brown, RC, et al. A study of vitamin D levels in Indonesian patients with untreated pulmonary tuberculosis. Tubercle. 1985;66:187–191.
173. Waters, WR, Palmer, MV, Nonnecke, BJ, et al. Mycobacterium bovis infection of vitamin D-deficient NOS2-/- mice. Microb Pathog. 2004;36:11–17.
174. Wilkinson, RJ, Llewelyn, M, Toossi, Z, et al. Influence of vitamin D deficiency and vitamin D receptor polymorphisms on tuberculosis among Gujarati Asians in west London: a case-control study. Lancet. 2000;355:618–621.
175. Liu, PT, Stenger, S, Li, H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–1773.
176. Morcos, MM, Gabr, AA, Samuel, S, et al. Vitamin D administration to tuberculous children and its value. Boll Chim Farm. 1998;137:157–164.
177. Nursyam, EW, Amin, Z, Rumende, CM. The effect of vitamin D as supplementary treatment in patients with moderately advanced pulmonary tuberculous lesion. Acta Med Indones. 2006;38:3–5.
178. Zella, JB, DeLuca, HF. Vitamin D and autoimmune diabetes. J Cell Biochem. 2003;88:216–222.
179. Liu, N, Nguyen, L, Chun, RF, et al. Altered endocrine and autocrine metabolism of vitamin D in a mouse model of gastrointestinal inflammation. Endocrinology. 2008;149:4799–4808.
180. Mathieu, C, Gysemans, C, Giulietti, A, et al. Vitamin D and diabetes. Diabetologia. 2005;48:1247–1257.
181. Guo, SW, Magnuson, VL, Schiller, JJ, et al. Meta-analysis of vitamin D receptor polymorphisms and type 1 diabetes: a HuGE review of genetic association studies. Am J Epidemiol. 2006;164:711–724.
182. Bailey, R, Cooper, JD, Zeitels, L, et al. Association of the vitamin D metabolism gene CYP27B1 with type 1 diabetes. Diabetes. 2007;56:2616–2621.
183. Munger, KL, Levin, LI, Hollis, BW, et al. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006;296:2832–2838.
184. Yu, S, Cantorna, MT. The vitamin D receptor is required for iNKT cell development. Proc Natl Acad Sci U S A. 2008;105:5207–5212.
185. Li, YC, Kong, J, Wei, M, et al. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110:229–238.
186. Forman, JP, Giovannucci, E, Holmes, MD, et al. Plasma 25-hydroxyvitamin D levels and risk of incident hypertension. Hypertension. 2007;49:1063–1069.
187. Scragg, R, Sowers, M, Bell, C. Serum 25-hydroxyvitamin D, ethnicity, and blood pressure in the Third National Health and Nutrition Examination Survey. Am J Hypertens. 2007;20:713–719.
188. Aihara, K, Azuma, H, Akaike, M, et al. Disruption of nuclear vitamin D receptor gene causes enhanced thrombogenicity in mice. J Biol Chem. 2004;279:35798–35802.
189. Wang, TJ, Pencina, MJ, Booth, SL, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation. 2008;117:503–511.
190. Hsia, J, Heiss, G, Ren, H, et al. Calcium/vitamin D supplementation and cardiovascular events. Circulation. 2007;115:846–854.
191. Teng, M, Wolf, M, Lowrie, E, et al. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med. 2003;349:446–456.
192. Palmer, SC, McGregor, DO, Macaskill, P, et al. Meta-analysis: vitamin D compounds in chronic kidney disease. Ann Intern Med. 2007;147:840–853.
193. Endo, I, Inoue, D, Mitsui, T, et al. Deletion of vitamin D receptor gene in mice results in abnormal skeletal muscle development with deregulated expression of myoregulatory transcription factors. Endocrinology. 2003;144:5138–5144.
194. Bischoff-Ferrari, HA, wson-Hughes, B, Baron, JA, et al. Calcium intake and hip fracture risk in men and women: a meta-analysis of prospective cohort studies and randomized controlled trials. Am J Clin Nutr. 2007;86:1780–1790.
195. Bischoff-Ferrari, HA, wson-Hughes, B, Willett, WC, et al. Effect of vitamin D on falls: a meta-analysis. JAMA. 2004;291:1999–2006.
196. Norman, AW, Frankel, BJ, Heldt, AM, et al. Vitamin D deficiency inhibits pancreatic secretion of insulin. Science. 1980;209:823–825.
197. Nyomba, BL, Bouillon, R, De, MP. Influence of vitamin D status on insulin secretion and glucose tolerance in the rabbit. Endocrinology. 1984;115:191–197.
198. Ford, ES, Ajani, UA, McGuire, LC, et al. Concentrations of serum vitamin D and the metabolic syndrome among U.S. adults. Diabetes Care. 2005;28:1228–1230.
199. Hypponen, E, Boucher, BJ, Berry, DJ, et al. 25-hydroxyvitamin D, IGF-1, and metabolic syndrome at 45 years of age: a cross-sectional study in the 1958 British Birth Cohort. Diabetes. 2008;57:298–305.
200. Forouhi, NG, Luan, J, Cooper, A, et al. Baseline serum 25-hydroxy vitamin D is predictive of future glycemic status and insulin resistance: the Medical Research Council Ely Prospective Study 1990–2000. Diabetes. 2008;57:2619–2625.
201. de Boer, IH, Tinker, LF, Connelly, S, et al. Calcium plus vitamin D supplementation and the risk of incident diabetes in the Women’s Health Initiative. Diabetes Care. 2008;31:701–707.
202. Pittas, AG, Harris, SS, Stark, PC, et al. The effects of calcium and vitamin D supplementation on blood glucose and markers of inflammation in nondiabetic adults. Diabetes Care. 2007;30:980–986.
203. Dobnig, H, Pilz, S, Scharnagl, H, et al. Independent association of low serum 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D levels with all-cause and cardiovascular mortality. Arch Intern Med. 2008;168:1340–1349.
204. Melamed, ML, Michos, ED, Post, W, et al. 25-hydroxyvitamin D levels and the risk of mortality in the general population. Arch Intern Med. 2008;168:1629–1637.
205. Wolf, M, Shah, A, Gutierrez, O, et al. Vitamin D levels and early mortality among incident hemodialysis patients. Kidney Int. 2007;72:1004–1013.
206. Autier, P, Gandini, S. Vitamin D supplementation and total mortality: a meta-analysis of randomized controlled trials. Arch Intern Med. 2007;167:1730–1737.
207. Porteous, CE, Coldwell, RD, Trafford, DJH, et al. Recent developments in the measurement of vitamin D and its metabolites in human body fluids. J Steroid Biochem. 1987;28:785–801.
208. Bouillon, R. Radiochemical assays for vitamin D metabolites: technical possibilities and clinical applications. J Steroid Biochem. 1983;19:921–927.
209. Schmidt-Gayk, H, Bouillon, R, Roth, HJ. Measurement of vitamin D and its metabolites (calcidiol and calcitriol) and their clinical significance. Scand J Clin Lab Invest. 1997;57:35–45.
210. Binkley, N, Krueger, D, Gemar, D, et al. Correlation among 25-hydroxy-vitamin D assays. J Clin Endocrinol Metab. 2008;93:1804–1808.
211. Lips, P, Chapuy, MC, Dawson-Hughes, B, et al. An international comparison of serum 25-hydroxyvitamin D measurements. Osteoporos Int. 1999;9:394–397.
212. Maunsell, Z, Wright, DJ, Rainbow, SJ. Routine isotope-dilution liquid chromatography-tandem mass spectrometry assay for simultaneous measurement of the 25-hydroxy metabolites of vitamins D2 and D3. Clin Chem. 2005;51:1683–1690.
213. Clements, MR, Johnson, L, Fraser, DR. A new mechanism for induced vitamin-D deficiency in calcium deprivation. Nature. 1987;325:62–65.
214. Bouillon, R, Van Baelen, H. Transport of vitamin D: significance of free and total concentrations of the vitamin D metabolites. Calcif Tissue Int. 1981;33:451–453.
215. Bikle, DD, Gee, E. Free, and not total, 1,25-dihydroxyvitamin D regulates 25-hydroxyvitamin D metabolism by keratinocytes. Endocrinology. 1989;124:649–654.
216. Vanham, G, Van Baelen, H, Tan, BK, et al. The effect of vitamin D analogs and of vitamin D-binding protein on lymphocyte proliferation. J Steroid Biochem. 1988;29:381–386.
217. Bouillon, R, Van Baelen, H. The transport of vitamin D. In: Norman AW, Schaefer K, von Herrath D, et al, eds. Vitamin D: Basic Research and Its Clinical Application. Berlin: Walter de Gruyter; 1979:137–143.
218. Dietary reference intakes for calcium, phosphorus, magnesium, vitamin D and fluoride. In: Food and Nutrition Board. Washington: National Academy Press; 1997:1–30.
219. Lerch C, Meissner T: Interventions for the prevention of nutritional rickets in term born children, Cochrane Database Syst Rev CD00:61–64, 2007.
220. Beser, E, Cakmakci, T. Factors affecting the morbidity of vitamin D deficiency rickets and primary protection. East Afr Med J. 1994;71:358–362.
221. Wagner, CL, Greer, F. R, the section on breastfeeding and committee on nutrition: Prevention of rickets and vitamin D deficiency in infants, children, and adolescents. Pediatrics. 2008;122:1142–1152.
222. Baroncelli, GI, Bereket, A, El, KM, et al. Rickets in the Middle East: role of environment and genetic predisposition. J Clin Endocrinol Metab. 2008;93:1743–1750.
223. van der Meer, I, Karamali, NS, Boeke, AJ, et al. High prevalence of vitamin D deficiency in pregnant non-Western women in The Hague, Netherlands. Am J Clin Nutr. 2006;84:350–353.
224. Pettifor, JM. Rickets and vitamin D deficiency in children and adolescents. Endocrinol Metab Clin North Am. 2005;34:537–553.
225. Javaid, MK, Crozier, SR, Harvey, NC, et al. Maternal vitamin D status during pregnancy and childhood bone mass at age 9 years: a longitudinal study. Lancet. 2006;367:36–43.
226. Bouillon, RA, Auwerx, JH, Lissens, WD, et al. Vitamin D status in the elderly: seasonal substrate deficiency causes 1,25-dihydroxycholecalciferol deficiency. Am J Clin Nutr. 1987;45:755–763.
227. Malabanan, A, Veronikis, IE, Holick, MF. Redefining vitamin D insufficiency. Lancet. 1998;351:805–806.
228. Heaney, RP, Dowell, MS, Hale, CA, et al. Calcium absorption varies within the reference range for serum 25-hydroxyvitamin D. J Am Coll Nutr. 2003;22:142–146.
229. Heaney, RP. The case for improving vitamin D status. J Steroid Biochem Mol Biol. 2007;103:635–641.
230. Need, AG, O’Loughlin, PD, Morris, HA, et al. Vitamin D metabolites and calcium absorption in severe vitamin D deficiency. J Bone Miner Res. 2008;23:1859–1863.
231. Abrams, SA, Griffin, IJ, Hawthorne, KM, et al. Relationships among vitamin D levels, parathyroid hormone, and calcium absorption in young adolescents. J Clin Endocrinol Metab. 2005;90:5576–5581.
232. Zhu, K, Bruce, D, Austin, N, et al. Randomized controlled trial of the effects of calcium with or without vitamin D on bone structure and bone-related chemistry in elderly women with vitamin D insufficiency. J Bone Miner Res. 2008;23:1343–1348.
233. Hansen, KE, Jones, AN, Lindstrom, MJ, et al. Vitamin D insufficiency: disease or no disease? J Bone Miner Res. 2008;23:1052–1060.
234. Ooms, ME, Lips, P, Roos, JC, et al. Vitamin D status and sex hormone binding globulin: determinants of bone turnover and bone mineral density in elderly women. J Bone Miner Res. 1995;10:1177–1184.
235. Cauley, JA, LaCroix, AZ, Wu, L, et al. Serum 25-hydroxyvitamin D concentrations and risk for hip fractures. Ann Intern Med. 2008;149:242–250.
236. Boonen, S, Lips, P, Bouillon, R, et al. Need for additional calcium to reduce the risk of hip fracture with vitamin D supplementation: evidence from a comparative metaanalysis of randomized controlled trials. J Clin Endocrinol Metab. 2007;92:1415–1423.
237. Reid, IR, Bolland, MJ, Grey, A. Effect of calcium supplementation on hip fractures. Osteoporos Int. 2008;19:1119–1123.
238. Bischoff-Ferrari, HA, Rees, JR, Grau, MV, et al. Effect of calcium supplementation on fracture risk: a double-blind randomized controlled trial. Am J Clin Nutr. 2008;87:1945–1951.
239. Heaney, RP, Armas, LA, Shary, JR, et al. 25-Hydroxylation of vitamin D3: relation to circulating vitamin D3 under various input conditions. Am J Clin Nutr. 2008;87:1738–1742.
240. Roux, C, Bischoff-Ferrari, HA, Papapoulos, SE, et al. New insights into the role of vitamin D and calcium in osteoporosis management: an expert roundtable discussion. Curr Med Res Opin. 2008;24:1363–1370.
241. Chel, V, Wijnhoven, HA, Smit, JH, et al. Efficacy of different doses and time intervals of oral vitamin D supplementation with or without calcium in elderly nursing home residents. Osteoporos Int. 2008;19:663–671.
242. Vieth, R. Vitamin D toxicity, policy, and science. J Bone Miner Res. 2007;22(Suppl 2)):V64–V68.
243. Hagenau, T, Vest, R, Gissel, TN, et al. Global vitamin D levels in relation to age, gender, skin pigmentation and latitude: an ecologic meta-regression analysis. Osteoporos Int. 2008;20:133–140.
244. Chapuy, MC, Preziosi, P, Maamer, M, et al. Prevalence of vitamin D insufficiency in an adult normal population. Osteoporos Int. 1997;7:439–443.
245. van der Wielen, RP, Lowik, MR, van den, BH, et al. Serum vitamin D concentrations among elderly people in Europe. Lancet. 1995;346:207–210.
246. Rahman, SA, Chee, WS, Yassin, Z, et al. Vitamin D status among postmenopausal Malaysian women. Asia Pac J Clin Nutr. 2004;13:255–260.
247. Fraser, DR. Vitamin D-deficiency in Asia. J Steroid Biochem Mol Biol. 2004;89–90:491–495.
248. van der Mei, I, Ponsonby, AL, Engelsen, O, et al. The high prevalence of vitamin D insufficiency across Australian populations is only partly explained by season and latitude. Environ Health Perspect. 2007;115:1132–1139.
249. Bouillon, R, Norman, AW, Lips, P. Vitamin D deficiency. N Engl J Med. 2007;357:1980–1981.
250. Bouillon, R, Garmyn, M, Verstuyf, A, et al. Paracrine role for calcitriol in the immune system and skin creates new therapeutic possibilities for vitamin D analogs. Eur J Endocrinol. 1995;133:7–16.
251. Bouillon, R, Okamura, WH, Norman, AW. Structure-function relationships in the vitamin D endocrine system. Endocr Rev. 1995;16:200–257.
252. Eelen, G, Gysemans, C, Verlinden, L, et al. Mechanism and potential of the growth-inhibitory actions of vitamin D and analogs. Curr Med Chem. 2007;14:1893–1910.
253. Verstuyf, A, Verlinden, L, Van Baelen, H, et al. The biological activity of nonsteroidal vitamin D hormone analogs lacking both the C- and D-rings. J Bone Miner Res. 1998;13:549–558.
254. Eelen, G, Valle, N, Sato, Y, et al. Superagonistic fluorinated vitamin D(3) analogs stabilize helix 12 of the vitamin D receptor. Chem Biol. 2008;15:1029–1034.
255. Peleg, S, Nguyen, C, Woodard, BT, et al. Differential use of transcription activation function 2 domain of the vitamin D receptor by 1,25-dihydroxyvitamin D3 and its A ring-modified analogs. Mol Endocrinol. 1998;12:525–535.
256. Liu, Y-Y, Collins, ED, Norman, AW, et al. Differential interaction of 1α,25-dihydroxyvitamin D3 analogues and their 20-epi homologues with the vitamin D receptor. J Biol Chem. 1997;272:3336–3345.
257. van den Bemd, GJCM, Pols, HAP, Birkenhager, JC, et al. Conformational change and enhanced stabilization of the vitamin D receptor by the 1,25-dihydroxyvitamin D3 analog KH1060. Proc Natl Acad Sci U S A. 1996;93:10685–10690.
258. Rochel, N, Wurtz, JM, Mitschler, A, et al. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5:173–179.
259. Bouillon, R, Allewaert, K, Xiang, DZ, et al. Vitamin D analogs with low affinity for the vitamin D binding protein: enhanced in vitro and decreased in vivo activity. J Bone Miner Res. 1991;6:1051–1057.
260. Dusso, AS, Negrea, L, Gunawardhana, S, et al. On the mechanisms for the selective action of vitamin D analogs. Endocrinology. 1991;128:1687–1692.
261. Matsumoto, T, Miki, T, Hagino, H, et al. A new active vitamin D, ED-71, increases bone mass in osteoporotic patients under vitamin D supplementation: a randomized, double-blind, placebo-controlled clinical trial. J Clin Endocrinol Metab. 2005;90:5031–5036.
262. Ke, HZ, Qi, H, Crawford, DT, et al. A new vitamin D analog, 2MD, restores trabecular and cortical bone mass and strength in ovariectomized rats with established osteopenia. J Bone Miner Res. 2005;20:1742–1755.
263. Beer, TM, Eilers, KM, Garzotto, M, et al. Weekly high-dose calcitriol and docetaxel in metastatic androgen-independent prostate cancer. J Clin Oncol. 2003;21:123–128.
264. Fogh, K, Kragballe, K. Vitamin D3 analogues. Clin Dermatol. 1997;15:705–713.
265. Van de Kerkhof, PCM. Reduction of epidermal abnormalities and inflammatory changes in psoriatic plaques during treatment with vitamin D3 analogs. J Invest Dermatol Symp Proc. 1996;1:78–81.
266. Lu, I, Gilleaudeau, P, McLane, JA, et al. Modulation of epidermal differentiation, tissue inflammation, and T-lymphocyte infiltration in psoriatic plaques by topical calcitriol. J Cutan Pathol. 1996;23:419–430.
267. Casteels, K, Bouillon, R, Waer, M, et al. Immunomodulatory effects of 1,25-dihydroxyvitamin D3. Curr Opin Nephrol Hypertens. 1995;4:313–318.
268. Baeke, F, Van Etten, E, Overbergh, L, et al. Vitamin D3 and the immune system: maintaining the balance in health and disease. Nutr Res Rev. 2007;20:106–118.
269. Van Etten, E, Mathieu, C. Immunoregulation by 1,25-dihydroxyvitamin D3: basic concepts. J Steroid Biochem Mol Biol. 2005;97:93–101.
270. Mathieu, C, Adorini, L. The coming of age of 1,25-dihydroxyvitamin D(3) analogs as immunomodulatory agents. Trends Mol Med. 2002;8:174–179.
271. Adorini, L, Penna, G, Amuchastegui, S, et al. Inhibition of prostate growth and inflammation by the vitamin D receptor agonist BXL-628 (elocalcitol). J Steroid Biochem Mol Biol. 2007;103:689–693.
272. Dawson-Hughes, B, Heaney, RP, Holick, MF, et al. Estimates of optimal vitamin D status. Osteoporos Int. 2005;16:713–716.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 3
Vitamin D
From Photosynthesis, Metabolism, and Action to Clinical Applications
Origin of Vitamin D: Nutrition and Photosynthesis
General Characteristics of the Vitamin D Receptor
Noncalcemic or Nonclassic Actions of Vitamin D Endocrine System
Diagnostic and Therapeutic Aspects of Vitamin D
Historic Overview
Rickets as a bone disease of young children was clearly described by Whistler in 16451 and Glisson in 1650.2 The relationship of this disease with lack of exposure to sunlight was already suspected in the 19th century, since the incidence of rickets was higher in children living in large industrialized towns than in children living in rural districts (see Chapter 15).3,4 Early in the 20th century, Huldshinsky,5 Chick et al.,6 and Hess and Weinstock7 demonstrated that rachitic children were cured after exposure to sunlight. In the United Kingdom, following an independent line of research in search of essential nutritional factors, Mellanby and Cantag8 raised dogs on a diet of oatmeal (the basic food in parts of the United Kingdom where rickets was endemic) and observed that they developed rickets, curable by cod liver oil.9 However, McCollum et al.10 could demonstrate that cod liver oil made vitamin A-deficient by aeration and heating was still able to cure rickets and thus contained a new essential nutrient called vitamin D.10 The two discoveries of vitamin D were unified by the demonstration of Goldblatt and Soames11 that irradiation of 7-dehydrocholesterol in the skin could produce the antirachitic vitamin D. Similar observations were made by Hess and Weinstock.7 Windaus,12 a German chemist, then identified the structure of vitamins D2 and D3 after irradiation of plant sterols (ergosterol) or 7-dehydrocholesterol,12 which earned him the Nobel Prize in chemistry in 1928.
Origin of Vitamin D: Nutrition and Photosynthesis
Vitamin D can be obtained from dietary sources of vegetal (vitamin D2 or ergocalciferol) or animal origin (vitamin D3 or cholecalciferol). About 50% of dietary vitamin D is absorbed by the enterocytes and transported to the blood circulation via chylomicrons. Part of this vitamin D is taken up by a variety of tissues (fat and muscle) before the chylomicron remnants and its vitamin D finally reaches the hepatocytes. The best food sources are fatty fish or its liver oils, but it is also found in small amounts in butter, cream, and egg yolk. Both human and cow’s milk are poor sources of vitamin D, providing only 15 to 40 IU/L, and equally minimal concentrations of 25(OH)D or 1,25(OH)2D.13 Only an intake of pharmacologic amounts of vitamin D (6000 IU/d) can increase the vitamin D concentration of milk to a level equivalent to the daily requirements of an infant.14 Vitamin D intake is a poor predictor of serum 25(OH)D concentrations in subjects with an intake between 2 and 20 µg/d.15,16 It is very difficult to obtain adequate vitamin D from a natural diet. However, in North America, 98% of fluid and dried milk (≥400 IU/L), as well as some margarine, butter, and certain cereals, are fortified with vitamin D2 (irradiated ergosterol) or D3, but the real vitamin D content is frequently quite different from the labeling standard. Skim milk and even proprietary infant formula frequently do not have the stated vitamin D content.17,18 Vitamin D is remarkably stable and does not deteriorate when food is heated or stored for long periods. The Second National Health and Nutrition Survey (NHANES II) reported a median intake of about 3 µg/d in adults (range 0 to 49 µg),19 whereas a slightly lower median intake (2.3 µg) was recorded in older women.20 In view of the low vitamin D content of a vegetarian diet (natural vitamin D intake is indeed related to intake of animal fat), vitamin D deficiency and rickets is a risk factor for strictly vegetarian children with insufficient sun exposure or vitamin D supplementation.21
Nature probably intended that most vitamin D would be generated by photosynthesis in the skin, with minor contribution from food sources. However, exposure to sunlight also increases the risk of dermal photodamage and several skin cancers, including melanoma. This was no real problem during human evolution, but with increasing life expectancy, the benefits of UV light for the photosynthesis of vitamin D should be compared with the lifetime risk of skin damage, especially since vitamin D supplementation can safely replace the skin synthesis. The recommended dietary allowances by the U.S. Food and Nutrition Board of the National Research Council and the 1998 updated recommendations are given in Table 3-1, and similar recommendations are still valid in Europe.22,23 However, these recommendations were based on rudimentary knowledge of optimal vitamin D status and need to be revised upwards.
Table 3-1
Adequate Intake, Previous Recommended Dietary Allowance, Reasonable Daily Allowance, and Tolerable Upper Limit for Vitamin D
AI, Adequate intake; RDA, recommended dietary allowance; UL, upper limit.
*Data from the Food and Nutrition Board, National Research Council, NAS: Recommended Dietary Allowances, 10th ed. Washington, DC: National Academy Press, 1989.
†Data from the Food and Nutrition Board: Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D and Fluoride. Washington, DC: National Academy Press, 1997. Fairly similar advice is given by the European Food Safety Authority.23
‡Similar upper levels for vitamin D intake were defined by the European Food Safety Authority,23 defining 25 µg or 1000 IU/d as the upper limit for children 0 to10 years of age and 50 µg or 2000 IU/d for children older than 11 years and adults.
Hypervitaminosis can occur when pharmaceutical vitamin D is taken in excess, with a wide variety of symptoms and signs related to hypercalciuria, hypercalcemia, and metastatic calcifications (Table 3-2). The toxic dosage has not been established for all ages, but infants and children are more susceptible. Toxicity should always be monitored when daily doses markedly exceeding the present upper limit of more than 50 µg are given for a longer period. Overproduction of renal 1,25(OH)2D by abnormal hormonal stimuli (as seen in fibroblast growth factor-23 [FGF-23] or Klotho-null mice) or absence of CYP24A1 (see later), the main catabolizing enzyme, causes the same calcemic side effects, with severe multiple-organ calcification (especially kidney, vascular wall, and heart valves) leading to premature death.24
Table 3-2
Symptoms of Vitamin D Toxicity
Hypercalciuria
Kidney stones
Hypercalcemia
Hyperphosphatemia
Polyuria
Polydipsia
Decalcification of bone
Ectopic calcification of soft tissues (kidney and lung)
Nausea and vomiting
Anorexia
Constipation
Headache
Hypertension
The production of previtamin D3 is a nonenzymatic photochemical reaction which is not subject to regulation other than substrate (7DHC) availability and intensity of UVB irradiation. 7DHC is the last precursor in the de novo biosynthesis of cholesterol. The enzyme 7HDC-Δ7-reductase (or sterol Δ7-reductase) catalyses the production of cholesterol from 7DHC. Inactivating mutations of the 7DHC-Δ7-reductase gene25 are the hallmark of the autosomal recessive Smith-Lemli-Opitz syndrome, characterized by high tissue and serum 7DHC levels and multiple anomalies, including craniofacial dysmorphism and mental retardation due to the lack of cholesterol synthesis.26 These patients may exhibit sometimes increased serum vitamin D and 25(OH)D concentrations.27 Likewise, animals pretreated with a specific sterol-Δ7-reductase inhibitor also exhibit an augmented vitamin D synthesis following UVB irradiation.28 With increasing human age, cutaneous stores of provitamin D decrease, together with decreased photoproduction of vitamin D.16 In cats and the feline species in general, the high cutaneous sterol-Δ7-reductase activity hampers photoproduction of vitamin D, making it a true vitamin.29 Apart from substrate (7DHC) availability, the photochemical synthesis of vitamin D3 in the skin largely depends on the amount of UVB photons that strike the basal epidermal layers. Glass, sunscreen, clothes, and skin pigment absorb UVB and blunt vitamin D3 synthesis. Latitude, time of day, and season are factors that influence the intensity of solar radiation and the cutaneous production of vitamin D3. Therefore, there is a risk for a shortage of vitamin D supply during winter and spring. In both the Northern and Southern hemispheres above 40 degrees latitude, vitamin D3 synthesis of the skin decreases or disappears during winter months, owing to the low inclination of the sun and the atmospheric filtration of the shortest (but effective for vitamin D3 synthesis) UV waves of sunlight. The importance of skin synthesis of vitamin D3 to maintain normal vitamin D status is best reflected by the vitamin D deficiency observed in submarine personnel or inhabitants of Antarctica30 during prolonged absence of sun exposure, and also by the extremely high prevalence of vitamin D deficiency in countries where exposure to sunlight is extremely low for cultural and religious reasons, as in several Arabian countries with strict adherence to Islamic rules for body covering.31–34 Solar exposure of 2 hours per week of the face and hands is probably sufficient for maintaining normal 25(OH)D concentrations in children35 and adults but should be further fine-tuned according to the climate and latitude.36
Nature has built in several feedback mechanisms to minimize the risk that prolonged sun exposure would cause vitamin D intoxication. Cutaneous vitamin D and especially previtamin D are photosensitive and will be degraded to inactive sterols (lumisterol, tachysterol) before they are translocated to the circulation (Fig. 3-1). Only a maximum of 10% to 15% of the provitamin D will be converted to vitamin D. Sunlight-induced melanin synthesis, acting as a natural sunscreen, provides an additional negative feedback.
Metabolism of Vitamin D
Vitamin D is biologically inert and requires two successive hydroxylations in the liver (on C25) and kidney (on the α position of C1), using cytochrome P450 enzymes37,38 to form its hormonally active metabolite, 1α,25-dihydroxyvitamin D (see Fig. 3-1).
25-Hydroxylation
25(OH)D was the first metabolite identified after the availability of radiolabeled vitamin D3.39,40 Although the liver is probably the main tissue responsible for 25-hydroxylation of vitamin D, extrahepatic 25-hydroxylation has been observed in vitro in a large number of tissues. In vivo observations after hepatectomy in rats also revealed that the conversion rate of [3H]-vitamin D was still about 10% when compared with intact rats.40 The hepatic 25-hydroxylation step is probably performed by more than one enzyme, localized either in the inner mitochondrial membrane (CYP27A1 or sterol 27-hydroxylase) or in the microsomes (including CYP2D11, CYP2D25, CYP3A4, and especially CYP2R1).37,41,42 CYP27A1 is a multifunctional enzyme with broad substrate specificity and is mainly involved in the 26- or 27-hydroxylation of cholesterol and bile-acid precursors.37 The rather mild (if any) disturbance of vitamin D metabolism in animals or humans lacking this enzyme43 indicates that the 25-hydroxylation of vitamin D does not rely exclusively on the activity of CYP27A1. It is therefore likely that a microsomal enzyme is the more physiologic enzyme, as initially suspected on the basis of hepatic enzyme activity being much higher and with lower Km in the microsomal fraction when compared with mitochondrial 25-hydroxylase activity.44 The microsomal enzyme activity is up-regulated by vitamin D deficiency or by prior exposure to phenobarbital. The most important 25-hydroxylase is probably CYP2R1, since a homozygous mutation was identified in a patient with classical rickets with low 25(OH)D levels.42
1α-Hydroxylation
25(OH)D is biologically inactive and requires further hydroxylation in the kidney45,46 to the active hormone, 1,25(OH)2D, by 25-hydroxyvitamin D-1α-hydroxylase (CYP27B1). The production of 1,25(OH)2D is regulated primarily at this final step by several factors (vide infra). The rat, mouse, and human 1α-hydroxylase have been cloned by several groups47–51 and mapped on human chromosome 12q13.3 in close vicinity to the VDR gene. The proximal renal tubule is the principle site of 1α-hydroxylation, but high levels of 1α-hydroxylase mRNA have also been found in human keratinocytes,48 and its gene expression is also observed in mouse macrophages52 and about ten other tissues.24 1α-Hydroxylase activity is under tight control by 1,25(OH)2D (negative but probably indirect feedback); parathyroid hormone (PTH), calcitonin, and insulin-like growth factor 1 (all positive feedback); and phosphate, calcium, and especially FGF-23 (negative regulation).38,53,54 The promoters of the mouse and human 1α-hydroxylase genes have been characterized with a profound responsiveness to PTH and a negative regulation by 1,25(OH)2D55,56 by complex chromatin and DNA modifications.57
Pseudovitamin D–deficiency rickets (PDDR), also known as vitamin D-dependency rickets type I, is an autosomal recessive disease characterized by failure to thrive, muscle weakness, skeletal deformities, hypocalcemia, secondary hyperparathyroidism, normal to high serum levels of 25(OH)D, and low serum 1,25(OH)2D concentrations, all caused by impaired activity of the renal 1α-hydroxylase.58 These patients recover with supplementation of physiologic doses of 1,25(OH)2D. The human CYP27B1 maps to the previously identified PDDR locus and mutations found in this gene in patients with PDDR provide the molecular genetic basis for the disease.48,59
24-Hydroxylation: Catabolism or Specific Function?
An alternative hydroxylation of 25(OH)D occurs on carbon 24 by the multifunctional enzyme, 24-hydroxylase (CYP24A), mapped on human chromosome 20q13.60 This enzyme not only initiates the catabolic cascade of 25(OH)D and 1,25(OH)2D61,62 by 24-hydroxylation but catalyzes also the dehydrogenation of the 24-OH group and performs 23-hydroxylation, resulting in 24-oxo-1,23,25(OH)3D.62 This C24 oxidation pathway finally leads to calcitroic acid, which is the major end product of 1,25(OH)2D (Fig. 3-2). In vivo evidence for this catabolic role of 24-hydroxylase was provided by the generation of mice deficient in the 24-hydroxylase gene, resulting in pathology consistent with systemic excess of 1,25(OH)2D.63 The expression of the 24-hydroxylase gene has been detected in virtually all nucleated cells. The induction of CYP24A belongs to the most sensitive biomarkers for responsiveness and is explained by the presence of several vitamin D responsive elements in its promoter.64,65 As a consequence, CYP24A mRNA levels appear to fall under the detection limits in VDR knockout mice.50,66 No human mutation in the 24-hydroxylase gene have yet been identified, but local overexpression of the enzyme may be involved in cancer.67
Other Metabolic Pathways For Vitamin D and Its Metabolites
Apart from the multifunctional 24-hydroxylation pathway, C23 and C26 hydroxylation of 1,25(OH)2D is also possible in the absence of prior 24-hydroxylation. The 23-hydroxylation probably only becomes important in the case of vitamin D excess; its major locus is the kidney. In contrast, 26-hydroxylation is mainly performed outside the kidney. Both activities are necessary for the formation of 25(OH)D- or 1,25(OH)2D-23,26-lactones (see Fig. 3-1). The A-ring metabolism involves the oxidation of C19 and the recently discovered 3-epimerisation. The latter, irreversible, reaction occurs only in a limited number of cells (e.g., keratinocytes, bone, and parathyroid cells) and is performed by hydroxysteroid dehydrogenases.68
Vitamin D Transport
Nutritional vitamin D is absorbed by the gut and then transported via the lymphatic system by chylomicrons69 and stored in several tissues (e.g., fat and muscle). Skin-produced vitamin D probably binds directly to an α-globulin known as vitamin D binding protein (DBP) and is then transported to the liver, where it is hydroxylated and thereafter released as 25(OH)D.
Human DBP,70 detected immunologically in 1959 as a group-specific component, or Gc-globulin,71 is a 43-amino-acid glycoprotein synthesized by the liver. Long before DBP’s functions had been characterized, its polymorphicity was already used in population genetics, parentage testing, and forensic medicine.72 Worldwide, over 120 Gc alleles have been detected,73 making the DBP locus one of the most polymorphic known. The Gc1F, Gc1S and Gc2 are the three most common alleles. Since in the many thousands of sera tested, none had been found of DBP deficiency, such a mutation was for a long time considered to be lethal; but this was contradicted by the generation of viable and fertile homozygous, DBP-deficient mice (DBP-null animals).74 The existence of similarity among the genes and protein structure of DBP, albumin, and α-fetoprotein is long recognized.75 The crystal structure, with or without actin, is now available and identified a surface cleft to bind 25(OH)D.76
Role of Vitamin D Binding Protein For Vitamin D Homeostasis
DBP, the major plasma carrier of vitamin D3, all its metabolites, and the vitamin D3 analogs, has one vitamin D sterol-specific binding site.75 The relative binding affinity is 25(OH)D-23,26-lactone > 25(OH)D = 24,25(OH)2D = 25,26(OH)2D (Ka = 5.108 mol/L at 4° C for human DBP) >> 1,25(OH)2D (4.107 mol/L) >> vitamin D >> previtamin D.77 The affinity for D2 metabolites is slightly lower than for D3 metabolites in mammals, but especially in birds. Since probably only non-DBP-bound vitamin D metabolites can readily cross the plasma membrane, and since the VDR has a much higher affinity for 1,25(OH)2D than for 25(OH)D (100-fold difference), while the opposite is true for DBP, it is clear that 1,25(OH)2D has substantially higher cellular uptake than 25(OH)D. This is also confirmed by the distribution space of (radiolabeled) metabolites: 25(OH)D has a distribution space similar to that of DBP and the plasma volume, whereas the distribution space of 1,25(OH)2D is closer to that of intracellular water. The half-life of 25(OH)D and 1,25(OH)2D in the human circulation is about 2 to 3 weeks and 4 to 6 hours, respectively.78,79 DBP’s function in the vitamin D endocrine system is assumed to reflect the “free hormone” hypothesis, which states that the unbound (free) rather than the protein-bound fraction of the active vitamin D hormone is responsible for the biological activity. The plasma concentration of DBP is increased by estrogens in most mammalian species and birds. In women, the DBP concentration therefore doubles at the end of pregnancy.80 Recent studies with megalin knockout mice indicate that megalin, a lipoprotein-like receptor present at the surface of the proximal tubular cells in the kidney, is responsible for the reabsorption of DBP and of DBP complexed with vitamin D sterols. This megalin reabsorption mechanism may control the availability of the 25(OH)D/DBP complex for the 25(OH)D-1α-hydroxylation enzyme and explain the severe bone disease of megalin-deficient mice.81
Other Functions of Vitamin D Binding Protein
DBP binds globular actin with a high affinity (Ka = 2 × 109 mmol/L).82,83 Actin is the most abundant intracellular protein. The cell motility, shape, and size depend on the ability of globular actin to polymerize into filaments (F-actin). Upon cell injury or cell necrosis, actin is released into extracellular space. However, when actin is released from cells, it may rapidly form filaments with detrimental effects for the microcirculation. Two plasma proteins, DBP and gelsolin, bind actin avidly, thereby acting as “actin-scavenger” system.84,85
DBP-null (KO) mice, however, develop normally. They are nevertheless more sensitive to vitamin D deficiency and less sensitive to vitamin D excess, probably by an enhanced urinary loss of vitamin D metabolites.74 The DBP and megalin KO mice, however, suggest that the main function of DBP is indeed to transport all vitamin D metabolites and preserve them from rapid clearance or urinary loss.
Action and Mode of Action
General Characteristics of The Vitamin D Receptor
Protein
1,25(OH)2D, the hormonally active form of vitamin D, exerts its effects mainly by activating the nuclear VDR, a member of the nuclear-receptor superfamily of ligand-activated transcription factors. Based on structure and function similarities between members of this family, different functional domains can be distinguished in these nuclear-receptor proteins. The short A/B domain at the N-terminus of VDR lacks the usual ligand-independent activation function (AF1). Two highly conserved zinc finger DNA binding motifs constitute the DNA-binding C domain, which also harbors the nuclear localization signal. The D domain or hinge region may regulate the receptor’s flexibility between DNA-binding and ligand-binding domains and may be crucial to allowing the heterodimer complex of the ligand-binding domains to interact with two differently oriented response elements (direct repeat or palindrome orientation with variable number of spacer nucleotides). The large multifunctional E region contains the ligand-binding domain, as well as a dimerization surface and a ligand-dependent activation function (AF2) at the extreme C-terminus, represented by helix 12.86
Gene
The human VDR gene, consisting of 14 exons, spans more than 60 kb on chromosome 12.87,88 The major VDR transcript is a 4.8 kb mRNA species, but multiple promoters and alternative splicing give rise to a multitude of less abundant transcripts that mostly vary in their 5′ untranslated region but encode the same 427-amino-acid protein.88 However, two of these mRNAs are translated into VDR proteins that contain an additional 23 or 50 amino acids at the N-terminus.88
Genomic Actions
Binding of 1,25(OH)2D to VDR generates conformational changes of VDR followed by heterodimerization with unliganded RXR and binding to vitamin D response elements (VDREs) in the promoter region of vitamin D target genes, with subsequent release of corepressors and recruitment of coactivators and general transcription factors for the assembly of an active transcriptional complex.89 A putative crucial event in this respect is the mousetrap-like intramolecular folding of helix 12, closing off the ligand-binding pocket and exposing the AF2 domain for interaction with coactivators.90 Corepressors bind and silence unliganded steroid receptors by recruitment of histone deacetylases, maintaining chromatin in a transcriptional repressive state.91 Coactivators are a group of proteins that allow gene transcription in several waves of activities. First, coactivators of the CBP/p300 family and of the p160 protein family, including the steroid receptor coactivators (SRCs), are recruited.92–94 These proteins possess intrinsic histone acetyltransferase (HAT) activity and by acetylating histone tails open up the chromatin structure, creating a chromatin environment permissive for gene transcription.95 In a second wave, the vitamin D receptor interacting protein (DRIP) multimeric complex is recruited, followed by recruitment of basal transcription factors, as well as RNA polymerase II. Finally, target gene transcription is induced.96 Gene expression can also be mediated by ATP-dependent chromatin remodeling complexes such as SWI/SNF-type and ISWI-type complexes and the multiprotein complex WINAC.97–99 Distinct regulation of transcriptional coregulators may provide species-specific, tissue-specific, or developmental stage–specific regulation of nuclear receptor function.100 Furthermore, the expression or the recruitment of these regulatory proteins is regulated by several intracellular signaling pathways101 and by steroids themselves,100 with receptor agonists or antagonists inducing preferential recruitment of coactivators or corepressors, respectively.101
A hexanucleotide direct repeat spaced by three nucleotides (DR3) is the cognate vitamin D response element (VDRE) to which RXR and VDR bind the 5′ and 3′ half-site, respectively, although alternative options appear to be possible, both with respect to dimer formation (VDR/VDR, VDR/RAR) and target-gene VDRE structure (DR4, DR6; IP9).102
Nongenomic Actions
Aside from the VDR transcriptional or genomic effects, several research groups have described rapid effects by 1,25(OH)2D that are independent of transcription and would be mediated by a membrane receptor for 1,25(OH)2D or by the localization of the nuclear VDR near the membrane.103 These so-called nongenomic effects include the opening of calcium or chloride channels and the activation of second messenger signaling pathways (phosphoinositide turnover, activation of protein kinase C, and the Ras/Raf/ERK/MAPK pathway). A wide variety of rapid and transient modifications in the second messenger signaling system have also been observed for other steroid hormones.104 At the tissue or cellular level, however, nongenomic activity of vitamin D and its analogs or metabolites have only been described for intestinal calcium absorption (transcaltachia) or cellular differentiation of leukemia cells.105–107 This pathway seems to prefer 6-s-cis to the 6-s-trans configuration of vitamin D.105 Moreover, the agonist/antagonist specificity differs for that of the genomic pathway.108
Classic Target Tissues
The action of 1,25(OH)2D on bone, intestine, kidney, and parathyroid glands and its role in mineral metabolism is the result of a complex interplay between calcium and phosphate 1,25(OH)2D, PTH, and phosphatonins. PTH induces calcium mobilization from bone and stimulates 1,25(OH)2D production, but its secretion is inhibited by the action of 1,25(OH)2D on the parathyroid glands (negative feedback). In a second negative feedback loop, 1,25(OH)2D limits its own availability by inhibition of 1α-hydroxylase and stimulation of 24-hydroxylase, inducing 1,25(OH)2D catabolism. In the last few years, considerable progress has been made in the understanding of phosphate homeostasis.109 The phosphaturic hormone phosphatonin, or FGF-23, is produced by osteocytes and osteoblasts and inhibits the activity of the NPT2 protein. The NPT2 gene encodes a renal sodium/phosphate cotransporter responsible for reabsorption of phosphate and represents a newly identified target gene for 1,25(OH)2D.110 Phosphatonin can be indirectly inactivated by a protease encoded by the PEX gene, which was identified as the gene that is defective in X-linked hypophosphatemic rickets. FGF-23 secretion is stimulated by 1,25(OH)2D and impairs renal 1α-hydroxylase, creating an additional feedback system so that the production of 1,25(OH)2D is tightly feedback regulated (Fig. 3-3).
FIGURE 3-3 Feedback regulation of renal synthesis of 1,25(OH)2D. A, Regulation of renal 1,25(OH)2D synthesis by parathyroid hormone and calcium, with multiple feedback control mechanisms. CaSR, Calcium-sensing receptor. B, Regulation of renal 1,25(OH)2D synthesis by FGF23 and phosphate, with several feedback control mechanisms.
Effects on Intestine
The absorption capacity of calcium along the gastrointestinal tract of the rat is dependent on the segment and follows the order ileum > jejunum > duodenum. The efficiency of the small intestine to absorb dietary calcium is increased by 1,25(OH)2D,111 and the abundance of the vitamin D receptor is highest in the duodenum, followed by jejunum and ileum. Although the exact mechanism by which 1,25(OH)2D alters the flux of calcium across the intestinal absorptive cell is not known, 1,25(OH)2D increases the production and activity of several proteins in the small intestine, including TRPV6 and V5, calbindin-D9K, alkaline phosphatase, and low-affinity Ca-ATPase (PMCA). The entry of Ca2+ from the intestinal lumen across the brush border membrane into the enterocyte is mainly regulated by the epithelial channels TRPV6 and V5.112 The intracellular calcium transfer is considered to be dependent mainly on calbindin-D9K.113 The transfer of Ca2+ from the cytoplasm to the extracellular space requires energy input because of an uphill concentration gradient and an unfavorable electrochemical gradient. Both the plasma membrane calcium pump and a sodium-calcium exchanger play important roles in this process. The stimulatory effect of 1,25(OH)2D on the ATP-dependent uptake of Ca2+ at the basolateral membrane involves an increase in PMCA gene expression.114 The essential role of the intestine for calcium and phosphate homeostasis was clearly demonstrated by the phenotype of VDR KO mice. Such VDR-null mice are phenotypically normal at birth, but after weaning, they develop hypocalcemia, secondary hyperparathyroidism, and hypophosphatemia despite very high levels of 1,25(OH)2D. They become growth retarded and develop severe rickets.66,112,115,116 Mice deficient in 1α-hydroxylase display a similar phenotype.117,118 This bone and calcium phenotype can be largely corrected by a high dietary calcium intake (especially in combination with high lactose intake) in both knockout models or 1,25(OH)2D treatment of 1α hydroxylase–null mice.112,119–125 These findings confirm previous observations in humans.59,126,127 The data strongly suggest that the intestine is the primary target for 1,25(OH)2D’s action on calcium/bone homeostasis. This is largely confirmed by genetic mouse models of selective rescue or deletion of VDR in the intestine of transgenic mice.128 The primary molecular targets, however, merit further exploration; ablation of CaBP-9k or TRPV6 or even their combined deficiency have shown no major effects on basal intestinal calcium absorption or serum calcium levels when calcium intake is normal.129 Paracellular intestinal calcium transport may also be part of the picture of vitamin D’s action in that the expression of claudin 2 and claudin 12, both known to form paracellular calcium channels, are induced by 1,25(OH)2D and decreased in the intestine of VDR-null mice.130
Effects on Kidney
The kidney is important both for the metabolism of 1,25(OH)2D and the reabsorption of calcium and phosphate, processes regulated by 1,25(OH)2D. The kidney and more specifically the proximal tubule is the central tissue for 1α-hydroxylation of 25(OH)D. Chronic renal failure reduces 1α-hydroxylase activity, which ultimately results in renal osteodystrophy or uremic bone disease. 1,25(OH)2D also increased the distal tubular reabsorption of calcium; as in the intestine, TRP channels (now TRPV5), calbindin-D9K and 28K, and the plasma membrane calcium ATPase are involved. Whereas in the intestine, active calcium absorption in the duodenum takes place before the less-regulated diffusion process in the ileum, reabsorption of filtered calcium follows a more logical sequence of massive calcium-sodium reabsorption in the proximal convoluted tubuli, followed by specific, actively regulated calcium reabsorption in the distal parts of the nephron. The crucial role of 1,25(OH)2D-regulated renal calcium reabsorption was demonstrated by persistent hypercalciuria and reduction in bone mass in TRPV5-deficient mice.131 The kidney is also the major component in phosphate homeostasis, as both PTH and FGF-23, in complex interplay with 1,25(OH)2D, are able to reduce renal phosphate reabsorption (see Fig. 3-3).
Effects on Bone
1,25(OH)2D has dual effects on bone: it can stimulate osteoclastogenesis and bone resorption as well as modify osteoblast function and bone mineralization. The overall effects of vitamin D metabolites on bone are thus extremely complex. From observations in man and animals, it is clear that vitamin D deficiency or resistance impairs bone-matrix mineralization, whereas osteoblast activity and matrix synthesis are even stimulated. Excess 1,25(OH)2D can clearly enhance osteoclastogenesis and bone resorption (Fig. 3-4). Because bone mineralization and bone structure can be largely normalized in vitamin D- or 1,25(OH)2D-deficient or resistant mice by sufficient supply of minerals via active or passive intestinal calcium absorption, it seems that direct effects of vitamin D metabolites on chondrocytes and bone cells are redundant if calcium and phosphate supply are guaranteed. However, most of the genes and proteins typically expressed in osteoblasts and osteoclasts are vitamin D regulated, so it is likely that 1,25(OH)2D can fine-tune bone mineral homeostasis. Moreover, pharmacologic use of vitamin D metabolites or analogs might positively influence bone balance, as shown by human and animal experiments132,133 and transgenic mice overexpressing osteoblast VDR.134
FIGURE 3-4 Effect of 1,25(OH)2D on bone cells and osteoclastogenesis. In osteoblast/stromal cells, 1,25(OH)2D induces ODF expression, down-regulates OPG, and stimulates M-CSF production. It also stimulates production of IL-6 and IL-11, which represent distinct signals in osteoclastogenesis. On osteoclast precursors, 1,25(OH)2D induces the expression of RANK (or ODF receptor) and several osteoclast differentiation markers, such as the vitronectin receptor αvβ3 and carbonic anhydrase-II. CA-II, Carbonic anhydrase-II; c-fms, M-CSF receptor; M-CSF, macrophage colony-stimulating factor; ODF, osteoclast differentiation factor; OPG, osteoprotegerin; V-ATP-ase, vacuolar adenosine triphosphatase.
Effects on Growth Plate
The absence of VDR or 1α-hydroxylase creates no detectable phenotype in overall growth or growth plate of prenatal animals, but the longitudinal growth of long bones is impaired after weaning. X-ray analysis reveals advanced rickets, including widening of the epiphyseal growth plate, with an increased width and marked disorganization of the growth plate on histology, including impaired mineralization of hypertrophic chondrocytes.66,115–118 This increased growth-plate width in VDR or 1α hydroxylase–null mice cannot be explained by their (normal) chondrocyte proliferation and differentiation, including collagen X and osteopontin expression. The expansion of the growth plate can be largely explained by decreased apoptosis of hypertrophic chondrocytes.135 Based on analysis of several genetic models with abnormal phosphate homeostasis, serum phosphate levels are probably crucial for hypertrophic chondrocyte apoptosis in vivo. This is confirmed in vitro: apoptosis of hypertrophic chondrocytes is regulated by phosphate levels via the activation of the caspase-9-mediated mitochondrial pathway.136
In accordance with these findings, chondrocyte-specific inactivation of the VDR did not cause a growth-plate phenotype and certainly not rickets.137 Critical analysis of these mice, however, revealed that VDR action in chondrocytes regulates bone development and phosphate homeostasis by inducing expression of paracrine factors such as vascular endothelial growth factor and receptor activator of nuclear factor κB (NFκB) ligand expression, leading to impaired vascular invasion and decreased osteoclast number in the metaphysic, as well as increased bone mass of long bones of juvenile chondrocyte-specific VDR-null mice. In addition, FGF-23 expression in osteoblasts was decreased, probably linked to the increased gene expression profile of NPT2 and 1α-hydroxylase in the kidney and resulting in increased serum levels of phosphate and 1,25(OH)2D.137
Noncalcemic or Nonclassic Actions of Vitamin D Endocrine System
The virtual ubiquitous expression of the VDR in all nucleated cells, the presence of a functional 1α-hydroxylase in at least 10 different tissues apart from the kidney, and the very large number of genes that are under direct or indirect control of 1,25(OH)2D all point toward a more universal role for the vitamin D endocrine system than just regulation of calcium/phosphate/bone metabolism. This is not totally unexpected; most other ligands for nuclear receptors also have a very wide spectrum of activities such as androgens, estrogens, glucocorticoids, and retinoids.24 Based on controlled observations in cells, tissues, and transgenic mice and on observational studies in humans, it seems that the functioning of nearly all major tissues or systems of the organism is modulated by vitamin D.
Skin
The combined presence of vitamin D production, 25-hydroxylase, 1α-hydroxylase, and VDR expression in the epidermis suggests the existence of a unique vitamin D intracrine system in which UVB-irradiated keratinocytes may supply their own needs for 1,25(OH)2D. A role for vitamin D in epidermal homeostasis can also be expected from the prominent effects of vitamin D compounds on keratinocyte growth and differentiation.138 The epidermal keratinocyte represents the major cell type in the epidermis and most likely the major cutaneous target cell for vitamin D, but many other cell types present in the epidermis are also vitamin D targets.
Based on studies of 1α hydroxylase–deficient mice, the repair of the essential barrier function of the skin is impaired in the absence of vitamin D action.139 The major skin phenotype of both VDR-null mice and children with VDR mutations is, however, the development of total alopecia. Hair development at birth is normal, but hair loss starts after the first catagen and ultimately leads to alopecia totalis associated with large dermal cysts. The absence of alopecia in vitamin D–deficient WT mice or in mice with CYP27B1 mutations clearly suggests that the absence of receptor and not its ligand is the cause of the skin phenotype. Mutations in the NR corepressor, hairless, or keratinocyte-specific loss of interaction of Lefl with β-catenin (part of the Wnt signaling pathway) produce a strikingly similar alopecia. There is therefore little doubt that ligand-independent effects of the VDR are required for normal keratinocyte stem cell function.
As expected from animal studies, no clear skin disorders are linked to vitamin D deficiency or insufficiency in humans. The very same photons that can generate the photoconversion of 7-dehydrocholesterol into previtamin D are also able to cause DNA damage and, ultimately, photoaging and increase the risk for skin cancer, so exposure to the UVB or sunlight needed to produce vitamin D always involves a small but cumulative risk of skin damage. This risk is especially relevant for humans with a fair skin type (phototypes 1 and 2).140 Although 1,25(OH)2D is able to generate a strong photoprotective effect against UVB-mediated events in cultured keratinocytes,141,142 the overall effect is negative.
Cell Proliferation and Cancer
Exposure to 1,25(OH)2D of virtually all normal cells and even most malignant cells results in an accumulation in the G0/G1 phase of the cell cycle.143–145 This inhibition of cell proliferation involves a large number of mechanisms and genes, and the exact sequence of events between VDR-mediated transactivation of genes and the actual G0/G1 arrest is probably cell-type specific. A general downstream effect is the regulation of the E2F family of transcription factors, which act as master switch for a very large number of genes involved in cell-cycle progression. These EF factors are under the control of the retinoblastoma protein members (especially pocket proteins p107 and p130), and their phosphorylation state is regulated by cyclins and cyclin-dependent kinases (p18, p19, p21, or p27), many of which are regulated by 1,25(OH)2D.143,146 However, 1,25(OH)2D may also inhibit cell growth by interfering with signaling pathways initiated by TGF-β, epidermal growth factor (IGF), prostaglandins,147 and Wnt ligands,148 as well as by intervening in other mitogenic signaling pathways (e.g., ERK/MAPK pathway and c-myc) (Fig. 3-5).149–156 Moreover, 1,25(OH)2D can regulate apoptosis and angiogenesis, mechanisms well known to be important for cancer cell expansion. In view of these well-established in vitro effects, one might expect a greater sensitivity for carcinogenesis in VDR-null mice. Epidermal, mammary, and intestinal cells of such animals do indeed show signs of hyperproliferation. Moreover, when exposed to chemocarcinogens or oncogens, VDR-null mice develop more mammary cancer–type lesions, skin tumors, and lymphomas.157,158
FIGURE 3-5 Effect of 1,25(OH)2D on cell-cycle progression. 1,25(OH)2D treatment leads to a cell cycle phase–specific effect characterized by an accumulation of cells in G1 through modulation of different signaling pathways. (−), Inhibitory effect; (+), stimulatory effect; EGF, epidermal growth factor; IGF-1, insulin-like growth factor 1; PGE2, prostaglandin E2; pRb, retinoblastoma tumor-suppressor gene; TGF-β, transforming growth factor β.
In humans, absolute VDR or CYP27B1 deficiencies are rare, but vitamin D deficiency is highly frequent. This obviously raises the question whether such vitamin D deficiency is associated with increased risk of cancer in humans. Such a hypothesis was originally reinforced by observations of higher cancer prevalence in areas of the United States and Japan with lower UVB exposure. Serum concentrations of 25(OH)D are, of course, a much better indication of the real vitamin D status, and a vast literature links lower levels of 25(OH)D with higher prevalence of the major cancers, especially colon and breast cancer, with more mixed results for prostate cancer. The inverse association between colorectal cancer or breast cancer and serum 25(OH)D levels was confirmed by the results of the Third National Health and Nutrition Examination Survey (NHANES III),159 although 25(OH)D levels were not related to overall cancer mortality. In most of the vast number of cross-sectional or observational studies, the higher cancer risk was found in subjects with serum 25(OH)D levels below 20 ng/mL, but most studies also revealed a significant trend across the different 25(OH)D subgroups and risk of cancer. Meta-analysis of studies addressing the association between 25(OH)D levels suggest that women with serum 25(OH)D of approximately 48 ng/mL (median of the top quintile) had a 50% lower risk of breast cancer than those with serum less than 13 ng/mL in the lowest quintile,160 and that individuals with serum 25(OH)D levels greater than 32 ng/mL had a 50% lower incidence of colorectal cancer than those with relatively low levels (≤12 ng/mL).161 However, a number of studies also link higher vitamin D nutritional status with a higher prevalence or more aggressive type of cancer.24,162
The final question is, of course, whether serum 25(OH)D is a predictor or has a causative relation with the overall cancer risk. Intervention studies should be able to provide the answer. In the Women’s Health Initiative (WHI) study, a significant inverse relationship was found between baseline levels of serum 25(OH)D and subsequent colorectal cancer incidence, but postmenopausal women receiving calcium (1 g) plus vitamin D (400 IU) did not develop less colon cancer than control patients.163 In a much smaller 4-yr study in postmenopausal women, higher doses of calcium (1.4 to 1.5 g) and vitamin D (1100 IU), which raised serum 25(OH)D to mean levels above 80 nmol/L, did significantly reduce overall cancer risk.164 The small number of cancer deaths and a major confounding factor of calcium intake, however, limit the value of this study to hypothesis, and much larger prospective studies with substantial vitamin D supplementation are essential.
Immune Function and Vitamin D
All immune cells (antigen-presenting cells, T and B cells, natural killer [NK] cells, and even mast cells) express at certain stages of their differentiation a functional VDR. Antigen-presenting cells (dendritic cells and equivalent resident cells, as well as monocytes/macrophages) can synthesize 1,25(OH)2D using the same enzyme as in the kidney but controlled by immune stimuli instead of calciotropic hormones.52,165–167 Finally, 1,25(OH)2D regulates a wide range of genes that play crucial roles in the immune system. The overall effects are different for the innate immune system (largely mediated by monocytes/macrophages) than for the acquired immune system. The innate immune system, upon exposure to bacterial agents, is first stimulated to produce 1,25(OH)2D (Fig. 3-6A) and thereafter activated by this paracrine 1,25(OH)2D to become a more active macrophage, including the local production of a number of defensins (including cathelicidin). Defects in immune functions indispensable for antimicrobial activity have been observed in vitamin D–deficient mice.168,169 The overall effects suggest that 1,25(OH)2D enhances the natural defense against bacterial infection. In the human situation, low serum levels of 25(OH)D were repeatedly associated with increased susceptibility to and more rapid disease progression of tuberculosis.170–174
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
