FIGURE 39-5 Hollenhorst plaque lodged at the bifurcation of a retinal arteriole proves that a patient is shedding emboli from the carotid artery, great vessels, or heart.
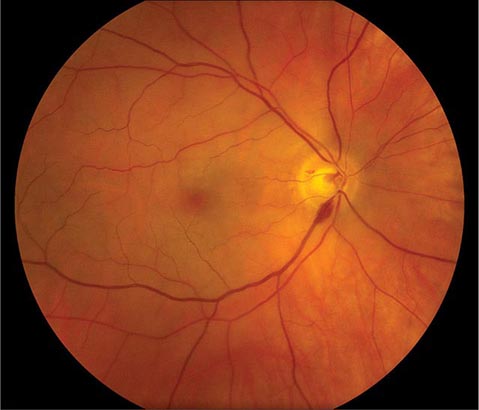
FIGURE 39-6 Central retinal artery occlusion in a 78-year-old man reducing acuity to counting fingers in the right eye. Note the splinter hemorrhage on the optic disc and the slightly milky appearance to the macula with a cherry-red fovea.
In rare instances, amaurosis fugax results from low central retinal artery perfusion pressure in a patient with a critical stenosis of the ipsilateral carotid artery and poor collateral flow via the circle of Willis. In this situation, amaurosis fugax develops when there is a dip in systemic blood pressure or a slight worsening of the carotid stenosis. Sometimes there is contralateral motor or sensory loss, indicating concomitant hemispheric cerebral ischemia.
Retinal arterial occlusion also occurs rarely in association with retinal migraine, lupus erythematosus, anticardiolipin antibodies, anticoagulant deficiency states (protein S, protein C, and antithrombin deficiency), pregnancy, IV drug abuse, blood dyscrasias, dysproteinemias, and temporal arteritis.
Marked systemic hypertension causes sclerosis of retinal arterioles, splinter hemorrhages, focal infarcts of the nerve fiber layer (cotton-wool spots), and leakage of lipid and fluid (hard exudate) into the macula (Fig. 39-7). In hypertensive crisis, sudden visual loss can result from vasospasm of retinal arterioles and retinal ischemia. In addition, acute hypertension may produce visual loss from ischemic swelling of the optic disc. Patients with acute hypertensive retinopathy should be treated by lowering the blood pressure. However, the blood pressure should not be reduced precipitously, because there is a danger of optic disc infarction from sudden hypoperfusion.
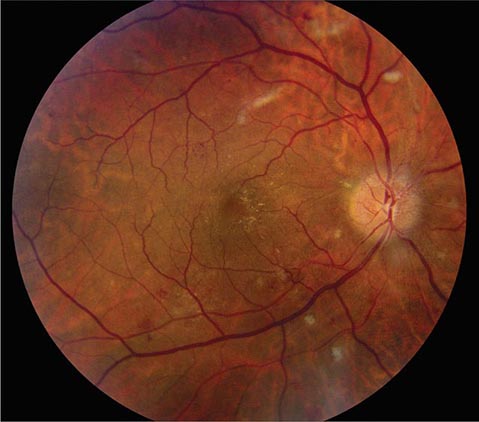
FIGURE 39-7 Hypertensive retinopathy with blurred optic disc, scattered hemorrhages, cotton-wool spots (nerve fiber layer infarcts), and foveal exudate in a 62-year-old man with chronic renal failure and a systolic blood pressure of 220.
Impending branch or central retinal vein occlusion can produce prolonged visual obscurations that resemble those described by patients with amaurosis fugax. The veins appear engorged and phlebitic, with numerous retinal hemorrhages (Fig. 39-8). In some patients, venous blood flow recovers spontaneously, whereas others evolve a frank obstruction with extensive retinal bleeding (“blood and thunder” appearance), infarction, and visual loss. Venous occlusion of the retina is often idiopathic, but hypertension, diabetes, and glaucoma are prominent risk factors. Polycythemia, thrombocythemia, or other factors leading to an underlying hypercoagulable state should be corrected; aspirin treatment may be beneficial.
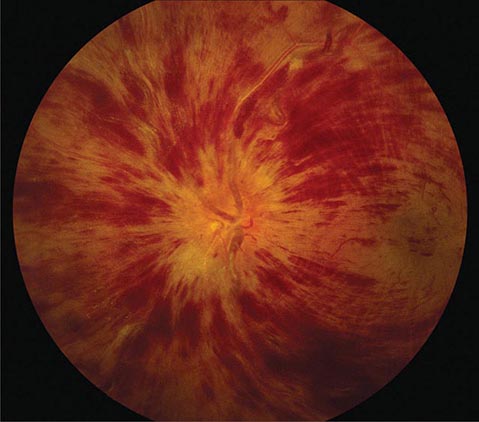
FIGURE 39-8 Central retinal vein occlusion can produce massive retinal hemorrhage (“blood and thunder”), ischemia, and vision loss.
Anterior Ischemic Optic Neuropathy (AION) This is caused by insufficient blood flow through the posterior ciliary arteries that supply the optic disc. It produces painless monocular visual loss that is sudden in onset, followed sometimes by stuttering progression. The optic disc appears swollen and surrounded by nerve fiber layer splinter hemorrhages (Fig. 39-9). AION is divided into two forms: arteritic and nonarteritic. The nonarteritic form is most common. No specific cause can be identified, although diabetes and hypertension are common risk factors. A crowded disc architecture and small optic cup predispose to the development of nonarteritic AION. No treatment is available. About 5% of patients, especially those age >60, develop the arteritic form of AION in conjunction with giant-cell (temporal) arteritis (Chap. 385). It is urgent to recognize arteritic AION so that high doses of glucocorticoids can be instituted immediately to prevent blindness in the second eye. Symptoms of polymyalgia rheumatica may be present; the sedimentation rate and C-reactive protein level are usually elevated. In a patient with visual loss from suspected arteritic AION, temporal artery biopsy is mandatory to confirm the diagnosis. Glucocorticoids should be started immediately, without waiting for the biopsy to be completed. The diagnosis of arteritic AION is difficult to sustain in the face of a negative temporal artery biopsy, but such cases do occur rarely. It is important to biopsy an arterial segment of at least 3 cm and to examine a sufficient number of tissue sections prepared from the specimen.
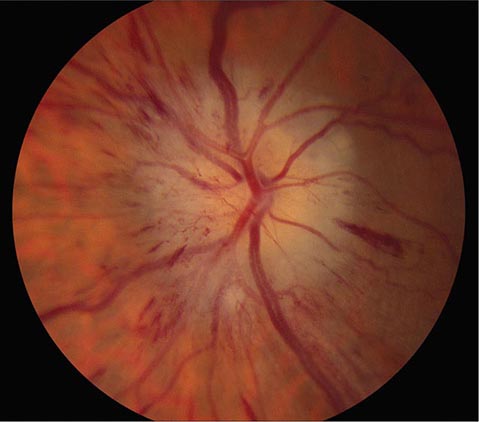
FIGURE 39-9 Anterior ischemic optic neuropathy from temporal arteritis in a 67-year-old woman with acute disc swelling, splinter hemorrhages, visual loss, and an erythrocyte sedimentation rate of 70 mm/h.
Posterior Ischemic Optic Neuropathy This is an uncommon cause of acute visual loss, induced by the combination of severe anemia and hypotension. Cases have been reported after major blood loss during surgery (especially in patients undergoing cardiac or lumbar spine operations), exsanguinating trauma, gastrointestinal bleeding, and renal dialysis. The fundus usually appears normal, although optic disc swelling develops if the process extends anteriorly far enough to reach the globe. Vision can be salvaged in some patients by prompt blood transfusion and reversal of hypotension.
Optic Neuritis This is a common inflammatory disease of the optic nerve. In the Optic Neuritis Treatment Trial (ONTT), the mean age of patients was 32 years, 77% were female, 92% had ocular pain (especially with eye movements), and 35% had optic disc swelling. In most patients, the demyelinating event was retrobulbar and the ocular fundus appeared normal on initial examination (Fig. 39-10), although optic disc pallor slowly developed over subsequent months.
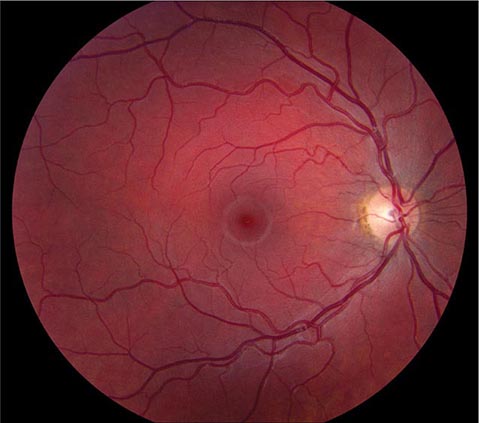
FIGURE 39-10 Retrobulbar optic neuritis is characterized by a normal fundus examination initially, hence the rubric “the doctor sees nothing, and the patient sees nothing.” Optic atrophy develops after severe or repeated attacks.
Virtually all patients experience a gradual recovery of vision after a single episode of optic neuritis, even without treatment. This rule is so reliable that failure of vision to improve after a first attack of optic neuritis casts doubt on the original diagnosis. Treatment with high-dose IV methylprednisolone (250 mg every 6 h for 3 days) followed by oral prednisone (1 mg/kg per day for 11 days) makes no difference in ultimate acuity 6 months after the attack, but the recovery of visual function occurs more rapidly. Therefore, when visual loss is severe (worse than 20/100), IV followed by PO glucocorticoids are often recommended.
For some patients, optic neuritis remains an isolated event. However, the ONTT showed that the 15-year cumulative probability of developing clinically definite multiple sclerosis after optic neuritis is 50%. A brain magnetic resonance (MR) scan is advisable in every patient with a first attack of optic neuritis. If two or more plaques are present on initial imaging, treatment should be considered to prevent the development of additional demyelinating lesions (Chap. 458).
LEBER’S HEREDITARY OPTIC NEUROPATHY
This disease usually affects young men, causing gradual, painless, severe central visual loss in one eye, followed weeks to years later by the same process in the other eye. Acutely, the optic disc appears mildly plethoric with surface capillary telangiectasias but no vascular leakage on fluorescein angiography. Eventually optic atrophy ensues. Leber’s optic neuropathy is caused by a point mutation at codon 11778 in the mitochondrial gene encoding nicotinamide adenine dinucleotide dehydrogenase (NADH) subunit 4. Additional mutations responsible for the disease have been identified, most in mitochondrial genes that encode proteins involved in electron transport. Mitochondrial mutations that cause Leber’s neuropathy are inherited from the mother by all her children, but usually only sons develop symptoms.
Toxic Optic Neuropathy This can result in acute visual loss with bilateral optic disc swelling and central or cecocentral scotomas. Such cases have been reported to result from exposure to ethambutol, methyl alcohol (moonshine), ethylene glycol (antifreeze), or carbon monoxide. In toxic optic neuropathy, visual loss also can develop gradually and produce optic atrophy (Fig. 39-11) without a phase of acute optic disc edema. Many agents have been implicated as a cause of toxic optic neuropathy, but the evidence supporting the association for many is weak. The following is a partial list of potential offending drugs or toxins: disulfiram, ethchlorvynol, chloramphenicol, amiodarone, monoclonal anti-CD3 antibody, ciprofloxacin, digitalis, streptomycin, lead, arsenic, thallium, D-penicillamine, isoniazid, emetine, sildenafil, tadalafil, vardenafil, and sulfonamides. Deficiency states induced by starvation, malabsorption, or alcoholism can lead to insidious visual loss. Thiamine, vitamin B12, and folate levels should be checked in any patient with unexplained bilateral central scotomas and optic pallor.
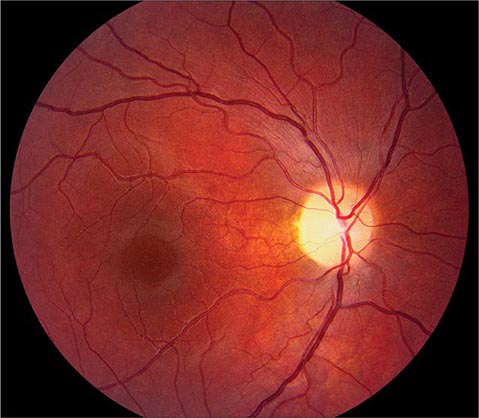
FIGURE 39-11 Optic atrophy is not a specific diagnosis but refers to the combination of optic disc pallor, arteriolar narrowing, and nerve fiber layer destruction produced by a host of eye diseases, especially optic neuropathies.
Papilledema This connotes bilateral optic disc swelling from raised intracranial pressure (Fig. 39-12). Headache is a common but not invariable accompaniment. All other forms of optic disc swelling (e.g., from optic neuritis or ischemic optic neuropathy) should be called “optic disc edema”. This convention is arbitrary but serves to avoid confusion. Often it is difficult to differentiate papilledema from other forms of optic disc edema by fundus examination alone. Transient visual obscurations are a classic symptom of papilledema. They can occur in only one eye or simultaneously in both eyes. They usually last seconds but can persist longer. Obscurations follow abrupt shifts in posture or happen spontaneously. When obscurations are prolonged or spontaneous, the papilledema is more threatening. Visual acuity is not affected by papilledema unless the papilledema is severe, long-standing, or accompanied by macular edema and hemorrhage. Visual field testing shows enlarged blind spots and peripheral constriction (Fig. 39-3F). With unremitting papilledema, peripheral visual field loss progresses in an insidious fashion while the optic nerve develops atrophy. In this setting, reduction of optic disc swelling is an ominous sign of a dying nerve rather than an encouraging indication of resolving papilledema.
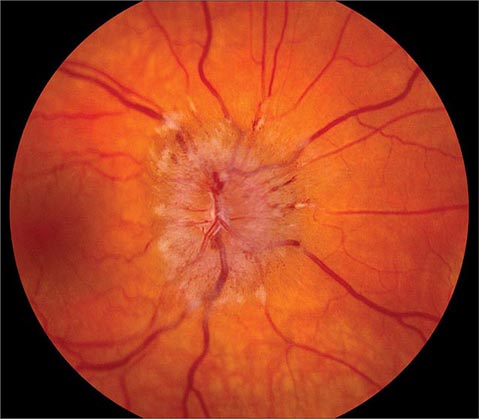
FIGURE 39-12 Papilledema means optic disc edema from raised intracranial pressure. This young woman developed acute papilledema, with hemorrhages and cotton-wool spots, as a rare side effect of treatment with tetracycline for acne.
Evaluation of papilledema requires neuroimaging to exclude an intracranial lesion. MR angiography is appropriate in selected cases to search for a dural venous sinus occlusion or an arteriovenous shunt. If neuroradiologic studies are negative, the subarachnoid opening pressure should be measured by lumbar puncture. An elevated pressure, with normal cerebrospinal fluid, points by exclusion to the diagnosis of pseudotumor cerebri (idiopathic intracranial hypertension). The majority of patients are young, female, and obese. Treatment with a carbonic anhydrase inhibitor such as acetazolamide lowers intracranial pressure by reducing the production of cerebrospinal fluid. Weight reduction is vital: bariatric surgery should be considered in patients who cannot lose weight by diet control. If vision loss is severe or progressive, a shunt should be performed without delay to prevent blindness. Occasionally, emergency surgery is required for sudden blindness caused by fulminant papilledema.
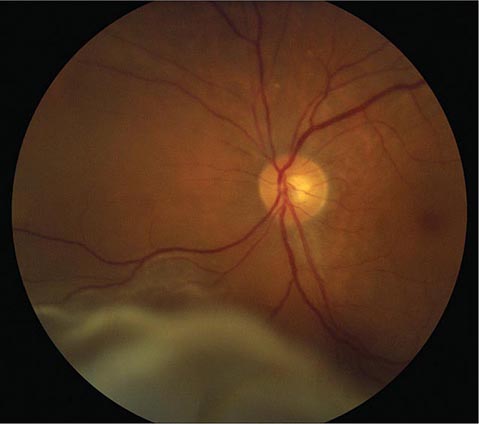
Optic Disc Drusen These are refractile deposits within the substance of the optic nerve head (Fig. 39-13). They are unrelated to drusen of the retina, which occur in age-related macular degeneration. Optic disc drusen are most common in people of northern European descent. Their diagnosis is obvious when they are visible as glittering particles on the surface of the optic disc. However, in many patients they are hidden beneath the surface, producing pseudopapilledema. It is important to recognize optic disc drusen to avoid an unnecessary evaluation for papilledema. Ultrasound or computed tomography (CT) scanning is sensitive for detection of buried optic disc drusen because they contain calcium. In most patients, optic disc drusen are an incidental, innocuous finding, but they can produce visual obscurations. On perimetry they give rise to enlarged blind spots and arcuate scotomas from damage to the optic disc. With increasing age, drusen tend to become more exposed on the disc surface as optic atrophy develops. Hemorrhage, choroidal neovascular membrane, and AION are more likely to occur in patients with optic disc drusen. No treatment is available.
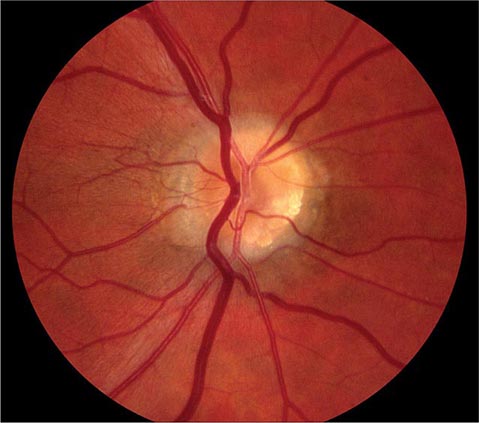
FIGURE 39-13 Optic disc drusen are calcified, mulberry-like deposits of unknown etiology within the optic disc, giving rise to “pseudopapilledema.”
Vitreous Degeneration This occurs in all individuals with advancing age, leading to visual symptoms. Opacities develop in the vitreous, casting annoying shadows on the retina. As the eye moves, these distracting “floaters” move synchronously, with a slight lag caused by inertia of the vitreous gel. Vitreous traction on the retina causes mechanical stimulation, resulting in perception of flashing lights. This photopsia is brief and is confined to one eye, in contrast to the bilateral, prolonged scintillations of cortical migraine. Contraction of the vitreous can result in sudden separation from the retina, heralded by an alarming shower of floaters and photopsia. This process, known as vitreous detachment, is a common involutional event in the elderly. It is not harmful unless it damages the retina. A careful examination of the dilated fundus is important in any patient complaining of floaters or photopsia to search for peripheral tears or holes. If such a lesion is found, laser application can forestall a retinal detachment. Occasionally a tear ruptures a retinal blood vessel, causing vitreous hemorrhage and sudden loss of vision. On attempted ophthalmoscopy the fundus is hidden by a dark haze of blood. Ultrasound is required to examine the interior of the eye for a retinal tear or detachment. If the hemorrhage does not resolve spontaneously, the vitreous can be removed surgically. Vitreous hemorrhage also results from the fragile neovascular vessels that proliferate on the surface of the retina in diabetes, sickle cell anemia, and other ischemic ocular diseases.
Retinal Detachment This produces symptoms of floaters, flashing lights, and a scotoma in the peripheral visual field corresponding to the detachment (Fig. 39-14). If the detachment includes the fovea, there is an afferent pupil defect and the visual acuity is reduced. In most eyes, retinal detachment starts with a hole, flap, or tear in the peripheral retina (rhegmatogenous retinal detachment). Patients with peripheral retinal thinning (lattice degeneration) are particularly vulnerable to this process. Once a break has developed in the retina, liquefied vitreous is free to enter the subretinal space, separating the retina from the pigment epithelium. The combination of vitreous traction on the retinal surface and passage of fluid behind the retina leads inexorably to detachment. Patients with a history of myopia, trauma, or prior cataract extraction are at greatest risk for retinal detachment. The diagnosis is confirmed by ophthalmoscopic examination of the dilated eye.
Classic Migraine (See also Chap. 447) This usually occurs with a visual aura lasting about 20 min. In a typical attack, a small central disturbance in the field of vision marches toward the periphery, leaving a transient scotoma in its wake. The expanding border of migraine scotoma has a scintillating, dancing, or zigzag edge, resembling the bastions of a fortified city, hence the term fortification spectra. Patients’ descriptions of fortification spectra vary widely and can be confused with amaurosis fugax. Migraine patterns usually last longer and are perceived in both eyes, whereas amaurosis fugax is briefer and occurs in only one eye. Migraine phenomena also remain visible in the dark or with the eyes closed. Generally they are confined to either the right or the left visual hemifield, but sometimes both fields are involved simultaneously. Patients often have a long history of stereotypic attacks. After the visual symptoms recede, headache develops in most patients.
FIGURE 39-14 Retinal detachment appears as an elevated sheet of retinal tissue with folds. In this patient, the fovea was spared, so acuity was normal, but an inferior detachment produced a superior scotoma.
Transient Ischemic Attacks Vertebrobasilar insufficiency may result in acute homonymous visual symptoms. Many patients mistakenly describe symptoms in the left or right eye when in fact the symptoms are occurring in the left or right hemifield of both eyes. Interruption of blood supply to the visual cortex causes a sudden fogging or graying of vision, occasionally with flashing lights or other positive phenomena that mimic migraine. Cortical ischemic attacks are briefer in duration than migraine, occur in older patients, and are not followed by headache. There may be associated signs of brainstem ischemia, such as diplopia, vertigo, numbness, weakness, and dysarthria.
Stroke Stroke occurs when interruption of blood supply from the posterior cerebral artery to the visual cortex is prolonged. The only finding on examination is a homonymous visual field defect that stops abruptly at the vertical meridian. Occipital lobe stroke usually is due to thrombotic occlusion of the vertebrobasilar system, embolus, or dissection. Lobar hemorrhage, tumor, abscess, and arteriovenous malformation are other common causes of hemianopic cortical visual loss.
Factitious (Functional, Nonorganic) Visual Loss This is claimed by hysterics or malingerers. The latter account for the vast majority, seeking sympathy, special treatment, or financial gain by feigning loss of sight. The diagnosis is suspected when the history is atypical, physical findings are lacking or contradictory, inconsistencies emerge on testing, and a secondary motive can be identified. In our litigious society, the fraudulent pursuit of recompense has spawned an epidemic of factitious visual loss.
CHRONIC VISUAL LOSS
Cataract Cataract is a clouding of the lens sufficient to reduce vision. Most cataracts develop slowly as a result of aging, leading to gradual impairment of vision. The formation of cataract occurs more rapidly in patients with a history of ocular trauma, uveitis, or diabetes mellitus. Cataracts are acquired in a variety of genetic diseases, such as myotonic dystrophy, neurofibromatosis type 2, and galactosemia. Radiation therapy and glucocorticoid treatment can induce cataract as a side effect. The cataracts associated with radiation or glucocorticoids have a typical posterior subcapsular location. Cataract can be detected by noting an impaired red reflex when viewing light reflected from the fundus with an ophthalmoscope or by examining the dilated eye with the slit lamp.
The only treatment for cataract is surgical extraction of the opacified lens. Millions of cataract operations are performed each year around the globe. The operation generally is done under local anesthesia on an outpatient basis. A plastic or silicone intraocular lens is placed within the empty lens capsule in the posterior chamber, substituting for the natural lens and leading to rapid recovery of sight. More than 95% of patients who undergo cataract extraction can expect an improvement in vision. In some patients, the lens capsule remaining in the eye after cataract extraction eventually turns cloudy, causing secondary loss of vision. A small opening, called a posterior capsulotomy, is made in the lens capsule with a laser to restore clarity.
Glaucoma Glaucoma is a slowly progressive, insidious optic neuropathy that usually is associated with chronic elevation of intraocular pressure. After cataract, it is the most common cause of blindness in the world. It is especially prevalent in people of African descent. The mechanism by which raised intraocular pressure injures the optic nerve is not understood. Axons entering the inferotemporal and superotemporal aspects of the optic disc are damaged first, producing typical nerve fiber bundle or arcuate scotomas on perimetric testing. As fibers are destroyed, the neural rim of the optic disc shrinks and the physiologic cup within the optic disc enlarges (Fig. 39-15). This process is referred to as pathologic “cupping.” The cup-to-disc diameter is expressed as a fraction (e.g., 0.2). The cup-to-disc ratio ranges widely in normal individuals, making it difficult to diagnose glaucoma reliably simply by observing an unusually large or deep optic cup. Careful documentation of serial examinations is helpful. In a patient with physiologic cupping the large cup remains stable, whereas in a patient with glaucoma it expands relentlessly over the years. Observation of progressive cupping and detection of an arcuate scotoma or a nasal step on computerized visual field testing is sufficient to establish the diagnosis of glaucoma. Optical coherence tomography reveals corresponding loss of fibers along the arcuate pathways in the nerve fiber layer.
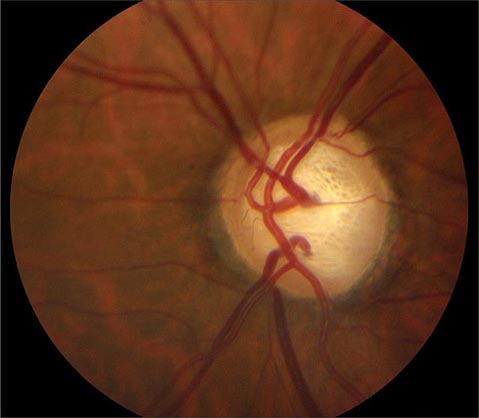
FIGURE 39-15 Glaucoma results in “cupping” as the neural rim is destroyed and the central cup becomes enlarged and excavated. The cup-to-disc ratio is about 0.8 in this patient.
About 95% of patients with glaucoma have open anterior chamber angles. In most affected individuals the intraocular pressure is elevated. The cause of elevated intraocular pressure is unknown, but it is associated with gene mutations in the heritable forms. Surprisingly, a third of patients with open-angle glaucoma have an intraocular pressure within the normal range of 10–20 mmHg. For this so-called normal or low-tension form of glaucoma, high myopia is a risk factor.
Chronic angle-closure glaucoma and chronic open-angle glaucoma are usually asymptomatic. Only acute angle-closure glaucoma causes a red or painful eye, from abrupt elevation of intraocular pressure. In all forms of glaucoma, foveal acuity is spared until end-stage disease is reached. For these reasons, severe and irreversible damage can occur before either the patient or the physician recognizes the diagnosis. Screening of patients for glaucoma by noting the cup-to-disc ratio on ophthalmoscopy and by measuring intraocular pressure is vital. Glaucoma is treated with topical adrenergic agonists, cholinergic agonists, beta blockers, and prostaglandin analogues. Occasionally, systemic absorption of beta blocker from eyedrops can be sufficient to cause side effects of bradycardia, hypotension, heart block, bronchospasm, or depression. Topical or oral carbonic anhydrase inhibitors are used to lower intraocular pressure by reducing aqueous production. Laser treatment of the trabecular meshwork in the anterior chamber angle improves aqueous outflow from the eye. If medical or laser treatments fail to halt optic nerve damage from glaucoma, a filter must be constructed surgically (trabeculectomy) or a drainage device placed to release aqueous from the eye in a controlled fashion.
Macular Degeneration This is a major cause of gradual, painless, bilateral central visual loss in the elderly. It occurs in a nonexudative (dry) form and an exudative (wet) form. Inflammation may be important in both forms of macular degeneration; susceptibility is associated with variants in the gene for complement factor H, an inhibitor of the alternative complement pathway. The nonexudative process begins with the accumulation of extracellular deposits called drusen underneath the retinal pigment epithelium. On ophthalmoscopy, they are pleomorphic but generally appear as small discrete yellow lesions clustered in the macula (Fig. 39-16). With time they become larger, more numerous, and confluent. The retinal pigment epithelium becomes focally detached and atrophic, causing visual loss by interfering with photoreceptor function. Treatment with vitamins C and E, beta-carotene, and zinc may retard dry macular degeneration.
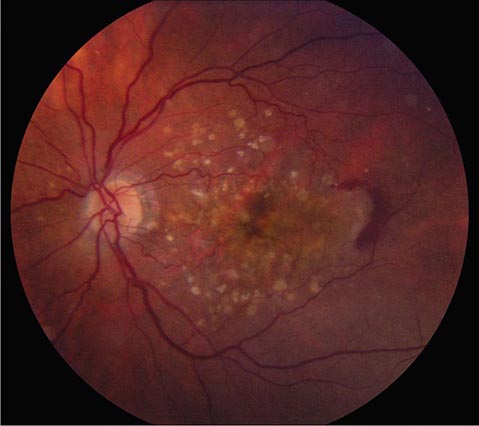
FIGURE 39-16 Age-related macular degeneration consisting of scattered yellow drusen in the macula (dry form) and a crescent of fresh hemorrhage temporal to the fovea from a subretinal neovascular membrane (wet form).
Exudative macular degeneration, which develops in only a minority of patients, occurs when neovascular vessels from the choroid grow through defects in Bruch’s membrane and proliferate underneath the retinal pigment epithelium or the retina. Leakage from these vessels produces elevation of the retina, with distortion (metamorphopsia) and blurring of vision. Although the onset of these symptoms is usually gradual, bleeding from a subretinal choroidal neovascular membrane sometimes causes acute visual loss. Neovascular membranes can be difficult to see on fundus examination because they are located beneath the retina. Fluorescein angiography and optical coherence tomography, a technique for acquiring images of the retina in cross-section, are extremely useful for their detection. Major or repeated hemorrhage under the retina from neovascular membranes results in fibrosis, development of a round (disciform) macular scar, and permanent loss of central vision.
A major therapeutic advance has occurred with the discovery that exudative macular degeneration can be treated with intraocular injection of antagonists to vascular endothelial growth factor. Bevacizumab, ranibizumab, or aflibercept is administered by direct injection into the vitreous cavity, beginning on a monthly basis. These antibodies cause the regression of neovascular membranes by blocking the action of vascular endothelial growth factor, thereby improving visual acuity.
Central Serous Chorioretinopathy This primarily affects males between the ages of 20 and 50 years. Leakage of serous fluid from the choroid causes small, localized detachment of the retinal pigment epithelium and the neurosensory retina. These detachments produce acute or chronic symptoms of metamorphopsia and blurred vision when the macula is involved. They are difficult to visualize with a direct ophthalmoscope because the detached retina is transparent and only slightly elevated. Optical coherence tomography shows fluid beneath the retina, and fluorescein angiography shows dye streaming into the subretinal space. The cause of central serous chorioretinopathy is unknown. Symptoms may resolve spontaneously if the retina reattaches, but recurrent detachment is common. Laser photocoagulation has benefited some patients with this condition.
Diabetic Retinopathy A rare disease until 1921, when the discovery of insulin resulted in a dramatic improvement in life expectancy for patients with diabetes mellitus, diabetic retinopathy is now a leading cause of blindness in the United States. The retinopathy takes years to develop but eventually appears in nearly all cases. Regular surveillance of the dilated fundus is crucial for any patient with diabetes. In advanced diabetic retinopathy, the proliferation of neovascular vessels leads to blindness from vitreous hemorrhage, retinal detachment, and glaucoma (Fig. 39-17). These complications can be avoided in most patients by administration of panretinal laser photocoagulation at the appropriate point in the evolution of the disease. For further discussion of the manifestations and management of diabetic retinopathy, see Chaps. 417–419.
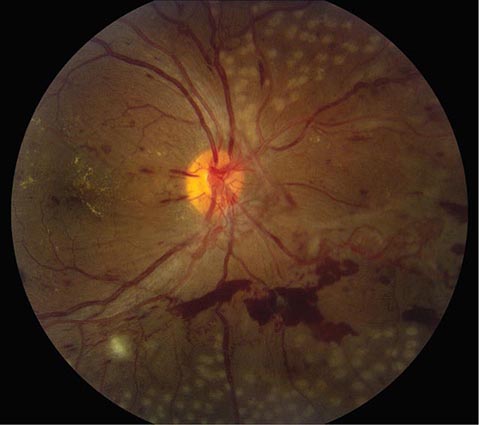
FIGURE 39-17 Proliferative diabetic retinopathy in a 25-year-old man with an 18-year history of diabetes, showing neovascular vessels emanating from the optic disc, retinal and vitreous hemorrhage, cotton-wool spots, and macular exudate. Round spots in the periphery represent recently applied panretinal photocoagulation.
Retinitis Pigmentosa This is a general term for a disparate group of rod-cone dystrophies characterized by progressive night blindness, visual field constriction with a ring scotoma, loss of acuity, and an abnormal electroretinogram (ERG). It occurs sporadically or in an autosomal recessive, dominant, or X-linked pattern. Irregular black deposits of clumped pigment in the peripheral retina, called bone spicules because of their vague resemblance to the spicules of cancellous bone, give the disease its name (Fig. 39-18). The name is actually a misnomer because retinitis pigmentosa is not an inflammatory process. Most cases are due to a mutation in the gene for rhodopsin, the rod photopigment, or in the gene for peripherin, a glycoprotein located in photoreceptor outer segments. Vitamin A (15,000 IU/d) slightly retards the deterioration of the ERG in patients with retinitis pigmentosa but has no beneficial effect on visual acuity or fields.
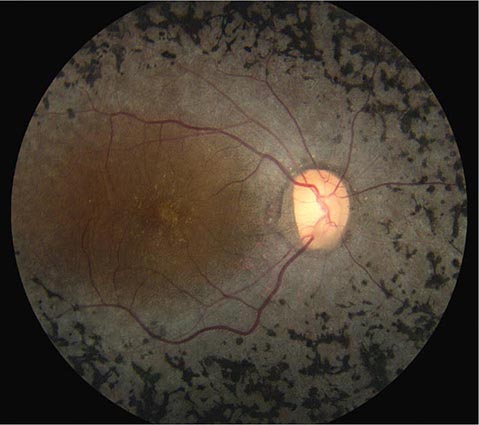
FIGURE 39-18 Retinitis pigmentosa with black clumps of pigment known as “bone spicules.” The patient had peripheral visual field loss with sparing of central (macular) vision.
Leber’s congenital amaurosis, a rare cone dystrophy, has been treated by replacement of the missing RPE65 protein through gene therapy, resulting in modest improvement in visual function. Some forms of retinitis pigmentosa occur in association with rare, hereditary systemic diseases (olivopontocerebellar degeneration, Bassen-Kornzweig disease, Kearns-Sayre syndrome, Refsum’s disease). Chronic treatment with chloroquine, hydroxychloroquine, and phenothiazines (especially thioridazine) can produce visual loss from a toxic retinopathy that resembles retinitis pigmentosa.
Epiretinal Membrane This is a fibrocellular tissue that grows across the inner surface of the retina, causing metamorphopsia and reduced visual acuity from distortion of the macula. A crinkled, cellophane-like membrane is visible on the retinal examination. Epiretinal membrane is most common in patients over 50 years of age and is usually unilateral. Most cases are idiopathic, but some occur as a result of hypertensive retinopathy, diabetes, retinal detachment, or trauma. When visual acuity is reduced to the level of about 6/24 (20/80), vitrectomy and surgical peeling of the membrane to relieve macular puckering are recommended. Contraction of an epiretinal membrane sometimes gives rise to a macular hole. Most macular holes, however, are caused by local vitreous traction within the fovea. Vitrectomy can improve acuity in selected cases.
Melanoma and Other Tumors Melanoma is the most common primary tumor of the eye (Fig. 39-19). It causes photopsia, an enlarging scotoma, and loss of vision. A small melanoma is often difficult to differentiate from a benign choroidal nevus. Serial examinations are required to document a malignant pattern of growth. Treatment of melanoma is controversial. Options include enucleation, local resection, and irradiation. Metastatic tumors to the eye outnumber primary tumors. Breast and lung carcinomas have a special propensity to spread to the choroid or iris. Leukemia and lymphoma also commonly invade ocular tissues. Sometimes their only sign on eye examination is cellular debris in the vitreous, which can masquerade as a chronic posterior uveitis. Retrobulbar tumor of the optic nerve (meningioma, glioma) or chiasmal tumor (pituitary adenoma, meningioma) produces gradual visual loss with few objective findings except for optic disc pallor. Rarely, sudden expansion of a pituitary adenoma from infarction and bleeding (pituitary apoplexy) causes acute retrobulbar visual loss, with headache, nausea, and ocular motor nerve palsies. In any patient with visual field loss or optic atrophy, CT or MR scanning should be considered if the cause remains unknown after careful review of the history and thorough examination of the eye.
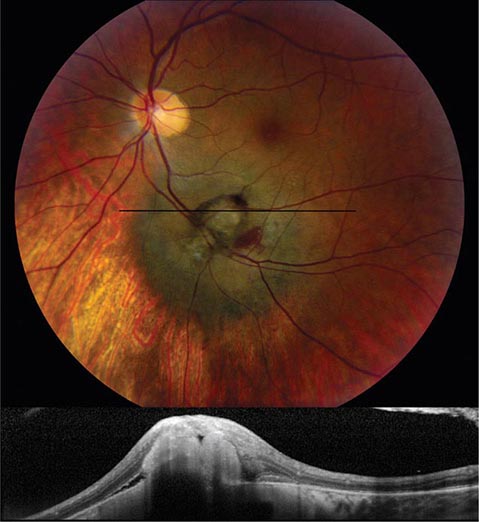
FIGURE 39-19 Melanoma of the choroid, appearing as an elevated dark mass in the inferior fundus, with overlying hemorrhage. The black line denotes the plane of the optical coherence tomography scan (below) showing the subretinal tumor.
PROPTOSIS
When the globes appear asymmetric, the clinician must first decide which eye is abnormal. Is one eye recessed within the orbit (enophthalmos), or is the other eye protuberant (exophthalmos, or proptosis)? A small globe or a Horner’s syndrome can give the appearance of enophthalmos. True enophthalmos occurs commonly after trauma, from atrophy of retrobulbar fat, or from fracture of the orbital floor. The position of the eyes within the orbits is measured by using a Hertel exophthalmometer, a handheld instrument that records the position of the anterior corneal surface relative to the lateral orbital rim. If this instrument is not available, relative eye position can be judged by bending the patient’s head forward and looking down upon the orbits. A proptosis of only 2 mm in one eye is detectable from this perspective. The development of proptosis implies a space-occupying lesion in the orbit and usually warrants CT or MR imaging.
Graves’ Ophthalmopathy This is the leading cause of proptosis in adults (Chap. 405). The proptosis is often asymmetric and can even appear to be unilateral. Orbital inflammation and engorgement of the extraocular muscles, particularly the medial rectus and the inferior rectus, account for the protrusion of the globe. Corneal exposure, lid retraction, conjunctival injection, restriction of gaze, diplopia, and visual loss from optic nerve compression are cardinal symptoms. Graves’ eye disease is a clinical diagnosis, but laboratory testing can be useful. The serum level of thyroid-stimulating immunoglobulins is often elevated. Orbital imaging usually reveals enlarged extraocular eye muscles, but not always. Graves’ ophthalmopathy can be treated with oral prednisone (60 mg/d) for 1 month, followed by a taper over several months. Worsening of symptoms upon glucocorticoid withdrawal is common. Topical lubricants, taping the eyelids closed at night, moisture chambers, and eyelid surgery are helpful to limit exposure of ocular tissues. Radiation therapy is not effective. Orbital decompression should be performed for severe, symptomatic exophthalmos or if visual function is reduced by optic nerve compression. In patients with diplopia, prisms or eye muscle surgery can be used to restore ocular alignment in primary gaze.
Orbital Pseudotumor This is an idiopathic, inflammatory orbital syndrome that is distinguished from Graves’ ophthalmopathy by the prominent complaint of pain. Other symptoms include diplopia, ptosis, proptosis, and orbital congestion. Evaluation for sarcoidosis, granulomatosis with polyangiitis, and other types of orbital vasculitis or collagen-vascular disease is negative. Imaging often shows swollen eye muscles (orbital myositis) with enlarged tendons. By contrast, in Graves’ ophthalmopathy, the tendons of the eye muscles usually are spared. The Tolosa-Hunt syndrome (Chap. 455) may be regarded as an extension of orbital pseudotumor through the superior orbital fissure into the cavernous sinus. The diagnosis of orbital pseudotumor is difficult. Biopsy of the orbit frequently yields nonspecific evidence of fat infiltration by lymphocytes, plasma cells, and eosinophils. A dramatic response to a therapeutic trial of systemic glucocorticoids indirectly provides the best confirmation of the diagnosis.
Orbital Cellulitis This causes pain, lid erythema, proptosis, conjunctival chemosis, restricted motility, decreased acuity, afferent pupillary defect, fever, and leukocytosis. It often arises from the paranasal sinuses, especially by contiguous spread of infection from the ethmoid sinus through the lamina papyracea of the medial orbit. A history of recent upper respiratory tract infection, chronic sinusitis, thick mucus secretions, or dental disease is significant in any patient with suspected orbital cellulitis. Blood cultures should be obtained, but they are usually negative. Most patients respond to empirical therapy with broad-spectrum IV antibiotics. Occasionally, orbital cellulitis follows an overwhelming course, with massive proptosis, blindness, septic cavernous sinus thrombosis, and meningitis. To avert this disaster, orbital cellulitis should be managed aggressively in the early stages, with immediate imaging of the orbits and antibiotic therapy that includes coverage of methicillin-resistant Staphylococcus aureus (MRSA). Prompt surgical drainage of an orbital abscess or paranasal sinusitis is indicated if optic nerve function deteriorates despite antibiotics.
Tumors Tumors of the orbit cause painless, progressive proptosis. The most common primary tumors are cavernous hemangioma, lymphangioma, neurofibroma, schwannoma, dermoid cyst, adenoid cystic carcinoma, optic nerve glioma, optic nerve meningioma, and benign mixed tumor of the lacrimal gland. Metastatic tumor to the orbit occurs frequently in breast carcinoma, lung carcinoma, and lymphoma. Diagnosis by fine-needle aspiration followed by urgent radiation therapy sometimes can preserve vision.
Carotid Cavernous Fistulas With anterior drainage through the orbit, these fistulas produce proptosis, diplopia, glaucoma, and corkscrew, arterialized conjunctival vessels. Direct fistulas usually result from trauma. They are easily diagnosed because of the prominent signs produced by high-flow, high-pressure shunting. Indirect fistulas, or dural arteriovenous malformations, are more likely to occur spontaneously, especially in older women. The signs are more subtle, and the diagnosis frequently is missed. The combination of slight proptosis, diplopia, enlarged muscles, and an injected eye often is mistaken for thyroid ophthalmopathy. A bruit heard upon auscultation of the head or reported by the patient is a valuable diagnostic clue. Imaging shows an enlarged superior ophthalmic vein in the orbits. Carotid cavernous shunts can be eliminated by intravascular embolization.
PTOSIS
Blepharoptosis This is an abnormal drooping of the eyelid. Unilateral or bilateral ptosis can be congenital, from dysgenesis of the levator palpebrae superioris, or from abnormal insertion of its aponeurosis into the eyelid. Acquired ptosis can develop so gradually that the patient is unaware of the problem. Inspection of old photographs is helpful in dating the onset. A history of prior trauma, eye surgery, contact lens use, diplopia, systemic symptoms (e.g., dysphagia or peripheral muscle weakness), or a family history of ptosis should be sought. Fluctuating ptosis that worsens late in the day is typical of myasthenia gravis. Examination should focus on evidence for proptosis, eyelid masses or deformities, inflammation, pupil inequality, or limitation of motility. The width of the palpebral fissures is measured in primary gaze to determine the degree of ptosis. The ptosis will be underestimated if the patient compensates by lifting the brow with the frontalis muscle.
Mechanical Ptosis This occurs in many elderly patients from stretching and redundancy of eyelid skin and subcutaneous fat (dermatochalasis). The extra weight of these sagging tissues causes the lid to droop. Enlargement or deformation of the eyelid from infection, tumor, trauma, or inflammation also results in ptosis on a purely mechanical basis.
Aponeurotic Ptosis This is an acquired dehiscence or stretching of the aponeurotic tendon, which connects the levator muscle to the tarsal plate of the eyelid. It occurs commonly in older patients, presumably from loss of connective tissue elasticity. Aponeurotic ptosis is also a common sequela of eyelid swelling from infection or blunt trauma to the orbit, cataract surgery, or contact lens use.
Myogenic Ptosis The causes of myogenic ptosis include myasthenia gravis (Chap. 461) and a number of rare myopathies that manifest with ptosis. The term chronic progressive external ophthalmoplegia refers to a spectrum of systemic diseases caused by mutations of mitochondrial DNA. As the name implies, the most prominent findings are symmetric, slowly progressive ptosis and limitation of eye movements. In general, diplopia is a late symptom because all eye movements are reduced equally. In the Kearns-Sayre variant, retinal pigmentary changes and abnormalities of cardiac conduction develop. Peripheral muscle biopsy shows characteristic “ragged-red fibers.” Oculopharyngeal dystrophy is a distinct autosomal dominant disease with onset in middle age, characterized by ptosis, limited eye movements, and trouble swallowing. Myotonic dystrophy, another autosomal dominant disorder, causes ptosis, ophthalmoparesis, cataract, and pigmentary retinopathy. Patients have muscle wasting, myotonia, frontal balding, and cardiac abnormalities.
Neurogenic Ptosis This results from a lesion affecting the innervation to either of the two muscles that open the eyelid: Müller’s muscle or the levator palpebrae superioris. Examination of the pupil helps distinguish between these two possibilities. In Horner’s syndrome, the eye with ptosis has a smaller pupil and the eye movements are full. In an oculomotor nerve palsy, the eye with the ptosis has a larger or a normal pupil. If the pupil is normal but there is limitation of adduction, elevation, and depression, a pupil-sparing oculomotor nerve palsy is likely (see next section). Rarely, a lesion affecting the small, central subnucleus of the oculomotor complex will cause bilateral ptosis with normal eye movements and pupils.
DOUBLE VISION (DIPLOPIA)
The first point to clarify is whether diplopia persists in either eye after the opposite eye is covered. If it does, the diagnosis is monocular diplopia. The cause is usually intrinsic to the eye and therefore has no dire implications for the patient. Corneal aberrations (e.g., keratoconus, pterygium), uncorrected refractive error, cataract, or foveal traction may give rise to monocular diplopia. Occasionally it is a symptom of malingering or psychiatric disease. Diplopia alleviated by covering one eye is binocular diplopia and is caused by disruption of ocular alignment. Inquiry should be made into the nature of the double vision (purely side-by-side versus partial vertical displacement of images), mode of onset, duration, intermittency, diurnal variation, and associated neurologic or systemic symptoms. If the patient has diplopia while being examined, motility testing should reveal a deficiency corresponding to the patient’s symptoms. However, subtle limitation of ocular excursions is often difficult to detect. For example, a patient with a slight left abducens nerve paresis may appear to have full eye movements despite a complaint of horizontal diplopia upon looking to the left. In this situation, the cover test provides a more sensitive method for demonstrating the ocular misalignment. It should be conducted in primary gaze and then with the head turned and tilted in each direction. In the above example, a cover test with the head turned to the right will maximize the fixation shift evoked by the cover test.
Occasionally, a cover test performed in an asymptomatic patient during a routine examination will reveal an ocular deviation. If the eye movements are full and the ocular misalignment is equal in all directions of gaze (concomitant deviation), the diagnosis is strabismus. In this condition, which affects about 1% of the population, fusion is disrupted in infancy or early childhood. To avoid diplopia, vision is suppressed from the nonfixating eye. In some children, this leads to impaired vision (amblyopia, or “lazy” eye) in the deviated eye.
Binocular diplopia results from a wide range of processes: infectious, neoplastic, metabolic, degenerative, inflammatory, and vascular. One must decide whether the diplopia is neurogenic in origin or is due to restriction of globe rotation by local disease in the orbit. Orbital pseudotumor, myositis, infection, tumor, thyroid disease, and muscle entrapment (e.g., from a blowout fracture) cause restrictive diplopia. The diagnosis of restriction is usually made by recognizing other associated signs and symptoms of local orbital disease. Omission of high-resolution orbital imaging is a common mistake in the evaluation of diplopia.
Myasthenia Gravis (See also Chap. 461) This is a major cause of diplopia. The diplopia is often intermittent, variable, and not confined to any single ocular motor nerve distribution. The pupils are always normal. Fluctuating ptosis may be present. Many patients have a purely ocular form of the disease, with no evidence of systemic muscular weakness. The diagnosis can be confirmed by an IV edrophonium injection, which produces a transient reversal of eyelid or eye muscle weakness. Blood tests for antibodies against the acetylcholine receptor or the MuSK protein can establish the diagnosis but are frequently negative in the purely ocular form of myasthenia gravis. Botulism from food or wound poisoning can mimic ocular myasthenia.
After restrictive orbital disease and myasthenia gravis are excluded, a lesion of a cranial nerve supplying innervation to the extraocular muscles is the most likely cause of binocular diplopia.
Oculomotor Nerve The third cranial nerve innervates the medial, inferior, and superior recti; inferior oblique; levator palpebrae superioris; and the iris sphincter. Total palsy of the oculomotor nerve causes ptosis, a dilated pupil, and leaves the eye “down and out” because of the unopposed action of the lateral rectus and superior oblique. This combination of findings is obvious. More challenging is the diagnosis of early or partial oculomotor nerve palsy. In this setting any combination of ptosis, pupil dilation, and weakness of the eye muscles supplied by the oculomotor nerve may be encountered. Frequent serial examinations during the evolving phase of the palsy help ensure that the diagnosis is not missed. The advent of an oculomotor nerve palsy with a pupil involvement, especially when accompanied by pain, suggests a compressive lesion, such as a tumor or circle of Willis aneurysm. Neuroimaging should be obtained, along with a CT or MR angiogram. Occasionally, a catheter arteriogram must be done to exclude an aneurysm.
A lesion of the oculomotor nucleus in the rostral midbrain produces signs that differ from those caused by a lesion of the nerve itself. There is bilateral ptosis because the levator muscle is innervated by a single central subnucleus. There is also weakness of the contralateral superior rectus, because it is supplied by the oculomotor nucleus on the other side. Occasionally both superior recti are weak. Isolated nuclear oculomotor palsy is rare. Usually neurologic examination reveals additional signs that suggest brainstem damage from infarction, hemorrhage, tumor, or infection.
Injury to structures surrounding fascicles of the oculomotor nerve descending through the midbrain has given rise to a number of classic eponymic designations. In Nothnagel’s syndrome, injury to the superior cerebellar peduncle causes ipsilateral oculomotor palsy and contralateral cerebellar ataxia. In Benedikt’s syndrome, injury to the red nucleus results in ipsilateral oculomotor palsy and contralateral tremor, chorea, and athetosis. Claude’s syndrome incorporates features of both of these syndromes, by injury to both the red nucleus and the superior cerebellar peduncle. Finally, in Weber’s syndrome, injury to the cerebral peduncle causes ipsilateral oculomotor palsy with contralateral hemiparesis.
In the subarachnoid space the oculomotor nerve is vulnerable to aneurysm, meningitis, tumor, infarction, and compression. In cerebral herniation, the nerve becomes trapped between the edge of the tentorium and the uncus of the temporal lobe. Oculomotor palsy also can result from midbrain torsion and hemorrhages during herniation. In the cavernous sinus, oculomotor palsy arises from carotid aneurysm, carotid cavernous fistula, cavernous sinus thrombosis, tumor (pituitary adenoma, meningioma, metastasis), herpes zoster infection, and the Tolosa-Hunt syndrome.
The etiology of an isolated, pupil-sparing oculomotor palsy often remains an enigma even after neuroimaging and extensive laboratory testing. Most cases are thought to result from microvascular infarction of the nerve somewhere along its course from the brainstem to the orbit. Usually the patient complains of pain. Diabetes, hypertension, and vascular disease are major risk factors. Spontaneous recovery over a period of months is the rule. If this fails to occur or if new findings develop, the diagnosis of microvascular oculomotor nerve palsy should be reconsidered. Aberrant regeneration is common when the oculomotor nerve is injured by trauma or compression (tumor, aneurysm). Miswiring of sprouting fibers to the levator muscle and the rectus muscles results in elevation of the eyelid upon downgaze or adduction. The pupil also constricts upon attempted adduction, elevation, or depression of the globe. Aberrant regeneration is not seen after oculomotor palsy from microvascular infarct and hence vitiates that diagnosis.
Trochlear Nerve The fourth cranial nerve originates in the midbrain, just caudal to the oculomotor nerve complex. Fibers exit the brainstem dorsally and cross to innervate the contralateral superior oblique. The principal actions of this muscle are to depress and intort the globe. A palsy therefore results in hypertropia and excyclotorsion. The cyclotorsion seldom is noticed by patients. Instead, they complain of vertical diplopia, especially upon reading or looking down. The vertical diplopia also is exacerbated by tilting the head toward the side with the muscle palsy and alleviated by tilting it away. This “head tilt test” is a cardinal diagnostic feature.
Isolated trochlear nerve palsy results from all the causes listed above for the oculomotor nerve except aneurysm. The trochlear nerve is particularly apt to suffer injury after closed head trauma. The free edge of the tentorium is thought to impinge on the nerve during a concussive blow. Most isolated trochlear nerve palsies are idiopathic and hence are diagnosed by exclusion as “microvascular.” Spontaneous improvement occurs over a period of months in most patients. A base-down prism (conveniently applied to the patient’s glasses as a stick-on Fresnel lens) may serve as a temporary measure to alleviate diplopia. If the palsy does not resolve, the eyes can be realigned by weakening the inferior oblique muscle.
Abducens Nerve The sixth cranial nerve innervates the lateral rectus muscle. A palsy produces horizontal diplopia, worse on gaze to the side of the lesion. A nuclear lesion has different consequences, because the abducens nucleus contains interneurons that project via the medial longitudinal fasciculus to the medial rectus subnucleus of the contralateral oculomotor complex. Therefore, an abducens nuclear lesion produces a complete lateral gaze palsy from weakness of both the ipsilateral lateral rectus and the contralateral medial rectus. Foville’s syndrome after dorsal pontine injury includes lateral gaze palsy, ipsilateral facial palsy, and contralateral hemiparesis incurred by damage to descending corticospinal fibers. Millard-Gubler syndrome from ventral pontine injury is similar except for the eye findings. There is lateral rectus weakness only, instead of gaze palsy, because the abducens fascicle is injured rather than the nucleus. Infarct, tumor, hemorrhage, vascular malformation, and multiple sclerosis are the most common etiologies of brainstem abducens palsy.
After leaving the ventral pons, the abducens nerve runs forward along the clivus to pierce the dura at the petrous apex, where it enters the cavernous sinus. Along its subarachnoid course it is susceptible to meningitis, tumor (meningioma, chordoma, carcinomatous meningitis), subarachnoid hemorrhage, trauma, and compression by aneurysm or dolichoectatic vessels. At the petrous apex, mastoiditis can produce deafness, pain, and ipsilateral abducens palsy (Gradenigo’s syndrome). In the cavernous sinus, the nerve can be affected by carotid aneurysm, carotid cavernous fistula, tumor (pituitary adenoma, meningioma, nasopharyngeal carcinoma), herpes infection, and Tolosa-Hunt syndrome.
Unilateral or bilateral abducens palsy is a classic sign of raised intracranial pressure. The diagnosis can be confirmed if papilledema is observed on fundus examination. The mechanism is still debated but probably is related to rostral-caudal displacement of the brainstem. The same phenomenon accounts for abducens palsy from Chiari malformation or low intracranial pressure (e.g., after lumbar puncture, spinal anesthesia, or spontaneous dural cerebrospinal fluid leak).
Treatment of abducens palsy is aimed at prompt correction of the underlying cause. However, the cause remains obscure in many instances despite diligent evaluation. As was mentioned above for isolated trochlear or oculomotor palsy, most cases are assumed to represent microvascular infarcts because they often occur in the setting of diabetes or other vascular risk factors. Some cases may develop as a postinfectious mononeuritis (e.g., after a viral flu). Patching one eye, occluding one eyeglass lens with tape, or applying a temporary prism will provide relief of diplopia until the palsy resolves. If recovery is incomplete, eye muscle surgery nearly always can realign the eyes, at least in primary position. A patient with an abducens palsy that fails to improve should be reevaluated for an occult etiology (e.g., chordoma, carcinomatous meningitis, carotid cavernous fistula, myasthenia gravis). Skull base tumors are easily missed even on contrast-enhanced neuroimaging studies.
Multiple Ocular Motor Nerve Palsies These should not be attributed to spontaneous microvascular events affecting more than one cranial nerve at a time. This remarkable coincidence does occur, especially in diabetic patients, but the diagnosis is made only in retrospect after all other diagnostic alternatives have been exhausted. Neuroimaging should focus on the cavernous sinus, superior orbital fissure, and orbital apex, where all three ocular motor nerves are in close proximity. In a diabetic or immunocompromised host, fungal infection (Aspergillus, Mucorales, Cryptococcus) is a common cause of multiple nerve palsies. In a patient with systemic malignancy, carcinomatous meningitis is a likely diagnosis. Cytologic examination may be negative despite repeated sampling of the cerebrospinal fluid. The cancer-associated Lambert-Eaton myasthenic syndrome also can produce ophthalmoplegia. Giant cell (temporal) arteritis occasionally manifests as diplopia from ischemic palsies of extraocular muscles. Fisher’s syndrome, an ocular variant of Guillain-Barré, produces ophthalmoplegia with areflexia and ataxia. Often the ataxia is mild, and the reflexes are normal. Antiganglioside antibodies (GQ1b) can be detected in about 50% of cases.
Supranuclear Disorders of Gaze These are often mistaken for multiple ocular motor nerve palsies. For example, Wernicke’s encephalopathy can produce nystagmus and a partial deficit of horizontal and vertical gaze that mimics a combined abducens and oculomotor nerve palsy. The disorder occurs in malnourished or alcoholic patients and can be reversed by thiamine. Infarct, hemorrhage, tumor, multiple sclerosis, encephalitis, vasculitis, and Whipple’s disease are other important causes of supranuclear gaze palsy. Disorders of vertical gaze, especially downward saccades, are an early feature of progressive supranuclear palsy. Smooth pursuit is affected later in the course of the disease. Parkinson’s disease, Huntington’s disease, and olivopontocerebellar degeneration also can affect vertical gaze.
The frontal eye field of the cerebral cortex is involved in generation of saccades to the contralateral side. After hemispheric stroke, the eyes usually deviate toward the lesioned side because of the unopposed action of the frontal eye field in the normal hemisphere. With time, this deficit resolves. Seizures generally have the opposite effect: the eyes deviate conjugately away from the irritative focus. Parietal lesions disrupt smooth pursuit of targets moving toward the side of the lesion. Bilateral parietal lesions produce Bálint’s syndrome, which is characterized by impaired eye-hand coordination (optic ataxia), difficulty initiating voluntary eye movements (ocular apraxia), and visuospatial disorientation (simultanagnosia).
Horizontal Gaze Descending cortical inputs mediating horizontal gaze ultimately converge at the level of the pons. Neurons in the paramedian pontine reticular formation are responsible for controlling conjugate gaze toward the same side. They project directly to the ipsilateral abducens nucleus. A lesion of either the paramedian pontine reticular formation or the abducens nucleus causes an ipsilateral conjugate gaze palsy. Lesions at either locus produce nearly identical clinical syndromes, with the following exception: vestibular stimulation (oculocephalic maneuver or caloric irrigation) will succeed in driving the eyes conjugately to the side in a patient with a lesion of the paramedian pontine reticular formation but not in a patient with a lesion of the abducens nucleus.
INTERNUCLEAR OPHTHALMOPLEGIA This results from damage to the medial longitudinal fasciculus ascending from the abducens nucleus in the pons to the oculomotor nucleus in the midbrain (hence, “internuclear”). Damage to fibers carrying the conjugate signal from abducens interneurons to the contralateral medial rectus motoneurons results in a failure of adduction on attempted lateral gaze. For example, a patient with a left internuclear ophthalmoplegia (INO) will have slowed or absent adducting movements of the left eye (Fig. 39-20). A patient with bilateral injury to the medial longitudinal fasciculus will have bilateral INO. Multiple sclerosis is the most common cause, although tumor, stroke, trauma, or any brainstem process may be responsible. One-and-a-half syndrome is due to a combined lesion of the medial longitudinal fasciculus and the abducens nucleus on the same side. The patient’s only horizontal eye movement is abduction of the eye on the other side.
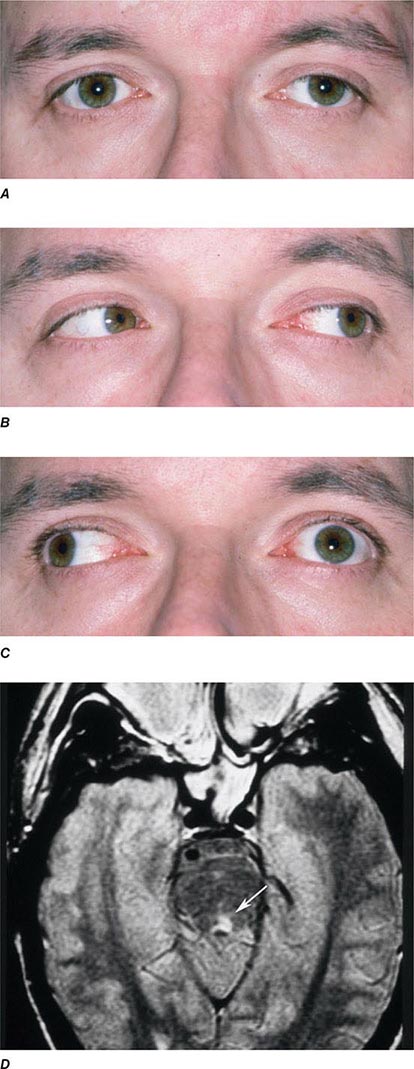
FIGURE 39-20 Left internuclear ophthalmoplegia (INO). A. In primary position of gaze, the eyes appear normal. B. Horizontal gaze to the left is intact. C. On attempted horizontal gaze to the right, the left eye fails to adduct. In mildly affected patients, the eye may adduct partially or more slowly than normal. Nystagmus is usually present in the abducted eye. D. T2-weighted axial magnetic resonance image through the pons showing a demyelinating plaque in the left medial longitudinal fasciculus (arrow).
Vertical Gaze This is controlled at the level of the midbrain. The neuronal circuits affected in disorders of vertical gaze are not fully elucidated, but lesions of the rostral interstitial nucleus of the medial longitudinal fasciculus and the interstitial nucleus of Cajal cause supranuclear paresis of upgaze, downgaze, or all vertical eye movements. Distal basilar artery ischemia is the most common etiology. Skew deviation refers to a vertical misalignment of the eyes, usually constant in all positions of gaze. The finding has poor localizing value because skew deviation has been reported after lesions in widespread regions of the brainstem and cerebellum.
PARINAUD’S SYNDROME Also known as dorsal midbrain syndrome, this is a distinct supranuclear vertical gaze disorder caused by damage to the posterior commissure. It is a classic sign of hydrocephalus from aqueductal stenosis. Pineal region or midbrain tumors, cysticercosis, and stroke also cause Parinaud’s syndrome. Features include loss of upgaze (and sometimes downgaze), convergence-retraction nystagmus on attempted upgaze, downward ocular deviation (“setting sun” sign), lid retraction (Collier’s sign), skew deviation, pseudoabducens palsy, and light-near dissociation of the pupils.
Nystagmus This is a rhythmic oscillation of the eyes, occurring physiologically from vestibular and optokinetic stimulation or pathologically in a wide variety of diseases (Chap. 28). Abnormalities of the eyes or optic nerves, present at birth or acquired in childhood, can produce a complex, searching nystagmus with irregular pendular (sinusoidal) and jerk features. Examples are albinism, Leber’s congenital amaurosis, and bilateral cataract. This nystagmus is commonly referred to as congenital sensory nystagmus. This is a poor term because even in children with congenital lesions, the nystagmus does not appear until weeks after birth. Congenital motor nystagmus, which looks similar to congenital sensory nystagmus, develops in the absence of any abnormality of the sensory visual system. Visual acuity also is reduced in congenital motor nystagmus, probably by the nystagmus itself, but seldom below a level of 20/200.
JERK NYSTAGMUS This is characterized by a slow drift off the target, followed by a fast corrective saccade. By convention, the nystagmus is named after the quick phase. Jerk nystagmus can be downbeat, upbeat, horizontal (left or right), and torsional. The pattern of nystagmus may vary with gaze position. Some patients will be oblivious to their nystagmus. Others will complain of blurred vision or a subjective to-and-fro movement of the environment (oscillopsia) corresponding to the nystagmus. Fine nystagmus may be difficult to see on gross examination of the eyes. Observation of nystagmoid movements of the optic disc on ophthalmoscopy is a sensitive way to detect subtle nystagmus.
GAZE-EVOKED NYSTAGMUS This is the most common form of jerk nystagmus. When the eyes are held eccentrically in the orbits, they have a natural tendency to drift back to primary position. The subject compensates by making a corrective saccade to maintain the deviated eye position. Many normal patients have mild gaze-evoked nystagmus. Exaggerated gaze-evoked nystagmus can be induced by drugs (sedatives, anticonvulsants, alcohol); muscle paresis; myasthenia gravis; demyelinating disease; and cerebellopontine angle, brainstem, and cerebellar lesions.
VESTIBULAR NYSTAGMUS Vestibular nystagmus results from dysfunction of the labyrinth (Ménière’s disease), vestibular nerve, or vestibular nucleus in the brainstem. Peripheral vestibular nystagmus often occurs in discrete attacks, with symptoms of nausea and vertigo. There may be associated tinnitus and hearing loss. Sudden shifts in head position may provoke or exacerbate symptoms.
DOWNBEAT NYSTAGMUS Downbeat nystagmus results from lesions near the craniocervical junction (Chiari malformation, basilar invagination). It also has been reported in brainstem or cerebellar stroke, lithium or anticonvulsant intoxication, alcoholism, and multiple sclerosis. Upbeat nystagmus is associated with damage to the pontine tegmentum from stroke, demyelination, or tumor.
Opsoclonus This rare, dramatic disorder of eye movements consists of bursts of consecutive saccades (saccadomania). When the saccades are confined to the horizontal plane, the term ocular flutter is preferred. It can result from viral encephalitis, trauma, or a paraneoplastic effect of neuroblastoma, breast carcinoma, and other malignancies. It has also been reported as a benign, transient phenomenon in otherwise healthy patients.
40e |
Use of the Hand-Held Ophthalmoscope |
Examination of the living human retina provides a unique opportunity for the direct study of nervous, vascular, and connective tissues. Many systemic disorders have retinal manifestations that are valuable for screening, diagnosis, and management of these conditions. Furthermore, retinal involvement in systemic disorders, such as diabetes mellitus, is a major cause of morbidity. Early recognition by ophthalmoscopic screening is a key factor in effective treatment. Ophthalmoscopy has the potential to be one of the most “high-yield” elements of the physical examination. Effective ophthalmoscopy requires a basic understanding of ocular structures and ophthalmoscopic techniques and recognition of abnormal findings.
OVERVIEW OF OCULAR STRUCTURES
The eye consists of a shell (cornea and sclera), lens, iris diaphragm, ciliary body, choroid, and retina. The anterior chamber is the space between the cornea and the lens, and it is filled with aqueous humor. The space between the posterior aspect of the lens and the retina is filled by vitreous gel. The choroid and the retina cover the posterior two-thirds of the sclera internally. The cornea and the lens form the focusing system of the eye, while the retina functions as the photoreceptor system, translating light to neuronal signals that are in turn transmitted to the brain via the optic nerve and visual pathways. The choroid is a layer of highly vascularized tissue that nourishes the retina and is located between the sclera and the retina. The retinal pigment epithelium (RPE) layer is a monolayer of pigmented cells that are adherent to the overlying retinal photoreceptor cells. RPE plays a major role in retinal photoreceptor metabolism.
NORMAL FUNDUS
The important areas that are visible by ophthalmoscopy include the macula, optic disc, retinal blood vessels, and retinal periphery (Fig. 40e-1).
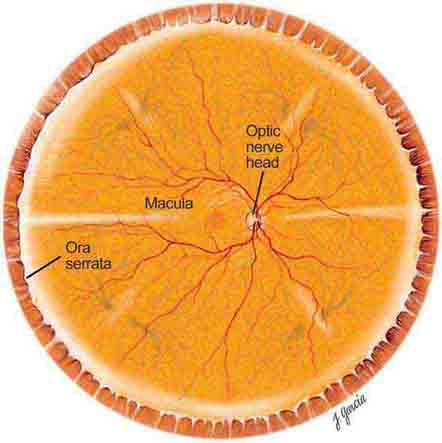
FIGURE 40e-1 Diagram showing the landmarks of the normal fundus. The macula is bounded by the superior and inferior vascular arcades and extends for 5 disc diameters (DD) temporal to the optic disc (optic nerve head). The central part of the macula (fovea) is located 2.5 DD temporal to the optic disc. The peripheral fundus is arbitrarily defined as the area extending anteriorly from the opening of the vortex veins to the ora serrata (the juncture between the retina and ciliary body). (Drawing courtesy of Juan R. Garcia. Used with permission from Johns Hopkins University.)
THE MACULA
The macula is the central part of the retina and is responsible for detailed vision (acuity) and perception of color. The macula is defined clinically as the area of the retina centered on the posterior pole of the fundus, measuring about 5 disc diameters (DD) (7–8 mm) and bordered by the optic disc nasally and the temporal vascular arcades superiorly and inferiorly. Temporally, the macula extends for about 2.5 DD from its center. The fovea, in the central part of the macula, corresponds to the site of sharpest visual acuity. It is approximately 1 DD in size and appears darker in color than the surrounding area. The center of the fovea, the foveola, has a depressed pit-like configuration measuring about 350 μm.
THE OPTIC DISC
The optic disc measures about 1.5 mm and is located about 4 mm (2.5 DD) nasal to the fovea. It contains the central retinal artery and vein as they branch, a central excavation (cup), and a peripheral neural rim. Normally, the cup-to-disc ratio is less than 0.6. The cup is located temporal to the entry of the disc vessels. The normal optic disc is yellow/pink in color. It has clear and well-defined margins and is in the same plane as the retina (Fig. 40e-2). Pathologic findings include pallor (atrophy), swelling, and enlarged cupping.
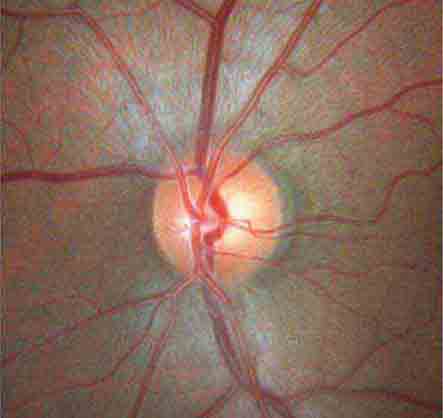
FIGURE 40e-2 Photograph of a normal left optic disc illustrating branching of the central retinal vein and artery, a physiologic cup, surface capillaries, and distinct margin. The cup is located temporal to the entry of the disc vessels. (From H Tabandeh, MF Goldberg: Retina in Systemic Disease: A Color Manual of Ophthalmoscopy. New York, Thieme, 2009.)
THE EQUATOR AND PERIPHERAL RETINA
The equator of the fundus is clinically defined as the area that includes the internal opening of the vortex veins. The peripheral retina extends from the equator anteriorly to the ora serrata.
OPHTHALMOSCOPY
There are a number of ways to visualize the retina, including direct ophthalmoscopy, binocular indirect ophthalmoscopy, and slit-lamp biomicroscopy. Most nonophthalmologists prefer direct ophthalmoscopy, performed with a hand-held ophthalmoscope, because the technique is simple to master and the device is very portable. Ophthalmologists often use slit-lamp biomicroscopy and indirect ophthalmoscopy to obtain a more extensive view of the fundus.
DIRECT OPHTHALMOSCOPE
Direct ophthalmoscopes are simple hand-held devices that include a small light source for illumination, a viewing aperture through which the examiner looks at the retina, and a lens dial used for correction of the examiner’s and the patient’s refractive errors. A more recent design, the PanOptic ophthalmoscope, provides a wider field of view.
How to Use a Direct Ophthalmoscope Good alignment is the key. The goal is to align the examiner’s eye with the viewing aperture of the ophthalmoscope, the patient’s pupil, and the area of interest on the retina. Both the patient and the examiner should be in a comfortable position (sitting or lying for the patient, sitting or standing for the examiner). Dilating the pupil and dimming the room lights make the examination easier. Steps for performing direct ophthalmoscopy are summarized in Table 40e-1.
|
GUIDELINES FOR PERFORMING DIRECT OPHTHALMOSCOPY |
PANOPTIC OPHTHALMOSCOPE
The PanOptic ophthalmoscope is a type of direct ophthalmoscope that is designed to provide a wider view of the fundus and has slightly more magnification than the standard direct ophthalmoscope. Steps for using the PanOptic Ophthalmoscope are summarized in Table 40e-2.
|
HOW TO USE A PANOPTIC OPHTHALMOSCOPE |
RETINAL SIGNS ASSOCIATED WITH SYSTEMIC DISEASES
AGE-RELATED CHANGES
Common age-related changes include diminished foveal light reflex, drusen (small yellow subretinal deposits), mild RPE atrophy, and pigment clumping.
RETINAL HEMORRHAGES
Retinal hemorrhages may take various shapes and sizes depending on their location within the retina (Figs. 40e-3 and 40e-4). Flame-shaped hemorrhages are located at the level of the superficial nerve fiber layer and represent bleeding from the inner capillary network of the retina. A white-centered hemorrhage is a superficial flame-shaped hemorrhage with an area of central whitening, often representing edema, focal necrosis, or cellular infiltration. Causes of white-centered hemorrhage include bacterial endocarditis and septicemia (Roth spots), lymphoproliferative disorders, diabetes mellitus, hypertension, anemia, and collagen vascular disorders. Dot hemorrhages are small, round, superficial hemorrhages that also originate from the superficial capillary network of the retina. They resemble microaneurysms. Blot hemorrhages are slightly larger in size, dark, and intraretinal. They represent bleeding from the deep capillary network of the retina. Subhyaloid hemorrhages are variable in shape and size and tend to be larger than other types of hemorrhages. They often have a fluid level (“boat-shaped” hemorrhage) and are located within the space between the vitreous and the retina. Subretinal hemorrhages are located deep (external) to the retina. The retinal vessels can be seen crossing over (internal to) such hemorrhages. Subretinal hemorrhages are variable in size and most commonly are caused by choroidal neovascularization (e.g., wet macular degeneration).
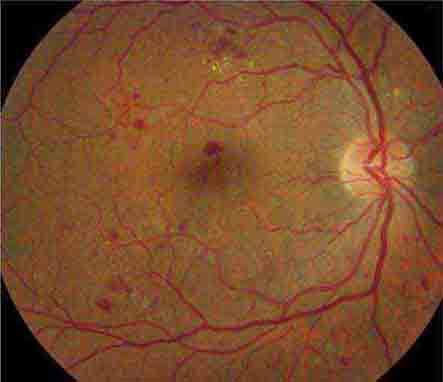
FIGURE 40e-3 Superficial flame-shaped hemorrhages, dot hemorrhages, and microaneurysms in a patient with nonproliferative diabetic retinopathy.
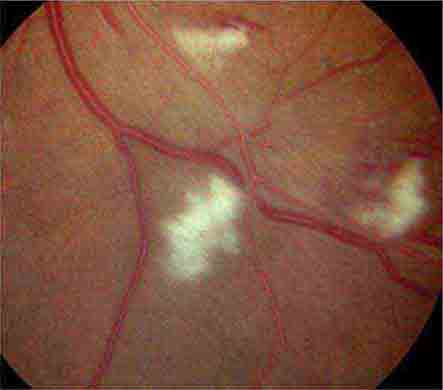
FIGURE 40e-4 Deep and superficial retinal hemorrhages in a patient with chronic leukemia.
Conditions associated with retinal hemorrhages include diseases causing retinal microvasculopathy (Table 40e-3), retinitis, retinal macroaneurysm, papilledema, subarachnoid hemorrhage (Terson’s syndrome), Valsalva retinopathy, trauma (ocular injury, head injury, compression injuries of chest and abdomen, shaken baby syndrome, strangulation), macular degeneration, and posterior vitreous detachment. Hyperviscosity states may produce dot and blot hemorrhages, dilated veins (“string of sausages” appearance), optic disc edema, and exudates; similar changes can occur with adaptation to high altitude in mountain climbers.
|
DISEASES ASSOCIATED WITH RETINAL MICROVASCULOPATHY |
MICROANEURYSMS
Microaneurysms are outpouchings of the retinal capillaries, appearing as red dots (similar to dot hemorrhages) and measuring 15–50 μm. Microaneurysms have increased permeability and may bleed or leak, resulting in localized retinal hemorrhage or edema. A microaneurysm ultimately thromboses and disappears within 3–6 months. Microaneurysms may occur in any condition that causes retinal microvasculopathy (Table 40e-3).
HARD EXUDATES
Hard exudates are well-circumscribed, shiny, yellow deposits located within the retina. They arise at the margins of areas of retinal edema and indicate increased capillary permeability. Hard exudates contain lipoproteins and lipid-laden macrophages. They may clear spontaneously or following laser photocoagulation, often within 6 months. Hard exudates may occur in isolation or may be scattered throughout the fundus. They may occur in a circular (circinate) pattern centered around an area of leaking microaneurysms. A macular star consists of a radiating, star-shaped pattern of hard exudates that is characteristically seen in severe systemic hypertension and in neuroretinitis associated with cat-scratch disease. Conditions associated with hard exudates include those causing retinal microvasculopathy (Table 40e–3), papilledema, neuroretinitis such as cat-scratch disease and Lyme disease, retinal vascular lesions (macroaneurysm, retinal capillary hemangioma, Coats’ disease), intraocular tumors, and wet age-related macular degeneration. Drusen may be mistaken for hard exudates on ophthalmoscopy. Unlike hard exudates, drusen are nonrefractile subretinal deposits with blurred margins. They are usually seen in association with age-related macular degeneration.
COTTON-WOOL SPOTS
Cotton-wool spots are yellow/white superficial retinal lesions with indistinct feathery borders measuring 0.25–1 DD in size (Fig. 40e-5). They represent areas of edema within the retinal nerve fiber layer due to focal ischemia. Cotton-wool spots usually resolve spontaneously within 3 months. If the underlying ischemic condition persists, new lesions can develop in different locations. Cotton-wool spots often occur in conjunction with retinal hemorrhages and microaneurysms and represent retinal microvasculopathy caused by a number of systemic conditions (Table 40e-3). They may occur in isolation in HIV retinopathy, systemic lupus erythematosus, anemia, bodily trauma, other systemic conditions (Purtscher’s/Purtscher’s-like retinopathy), and interferon therapy.
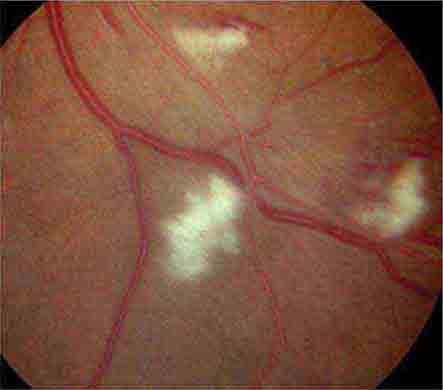
FIGURE 40e-5 Cotton-wool spots, yellow-white superficial lesions with characteristic feathery borders, in a patient with hypertensive retinopathy. (From H Tabandeh, MF Goldberg: Retina in Systemic Disease: A Color Manual of Ophthalmoscopy. New York, Thieme, 2009.)
RETINAL NEOVASCULARIZATION
Retinal neovascular complexes are irregular meshworks of fine blood vessels that grow in response to severe retinal ischemia or chronic inflammation (Fig. 40e-6). They may occur on or adjacent to the optic disc or elsewhere in the retina. Neovascular complexes are very fragile and have a high risk for hemorrhaging, often causing visual loss. Diseases associated with retinal neovascularization include conditions that cause severe retinal microvasculopathy, especially diabetic and sickle cell retinopathies (Table 40e-3), intraocular tumors, intraocular inflammation (sarcoidosis, chronic uveitis), and chronic retinal detachment.
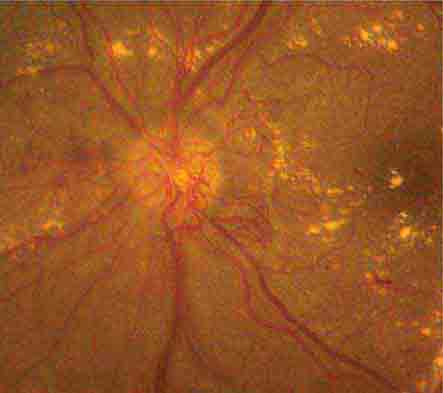
FIGURE 40e-6 Optic disc neovascularization in a patient with severe proliferative diabetic retinopathy. Multiple hard exudates are also present.
RETINAL EMBOLI
Common sources of retinal emboli include carotid artery atheromatous plaque, cardiac valve and septal abnormalities, cardiac arrhythmias, atrial myxoma, bacterial endocarditis, septicemia, fungemia, and intravenous drug abuse.
Platelet emboli are yellowish in appearance and conform to the shape of the blood vessel. They usually originate from an atheromatous plaque within the carotid artery and can cause transient loss of vision (amaurosis fugax). Cholesterol emboli, otherwise termed Hollenhorst plaques, are yellow crystalline deposits that are commonly found at the bifurcations of the retinal arteries and may be associated with amaurosis fugax. Calcific emboli have a pearly white appearance, are larger than the platelet and cholesterol emboli, and tend to lodge in the larger retinal arteries in or around the optic disc. Calcific emboli often result in retinal arteriolar occlusion. Septic emboli can cause white-centered retinal hemorrhages (Roth spots), retinal microabscesses, and endogenous endophthalmitis. Fat embolism and amniotic fluid embolism are characterized by multiple small vessel occlusions, typically causing cotton-wool spots and few hemorrhages (Purtscher’s-like retinopathy). Talc embolism occurs with intravenous drug abuse and is characterized by multiple refractile deposits within the small retinal vessels. Any severe form of retinal artery embolism may result in retinal ischemia and its sequelae, including retinal neovascularization.
CHERRY RED SPOT AT THE MACULA
Cherry red spot at the macula is the term used to describe the dark red appearance of the central foveal area in comparison to the surrounding macular region (Fig. 40e-7). This appearance is most commonly due to a relative loss of transparency of the parafoveal retina resulting from ischemic cloudy swelling or storage of macromolecules within the ganglion cell layer. Diseases associated with a cherry red spot at the macula include central retinal artery occlusion, sphingolipidoses, and mucolipidoses.
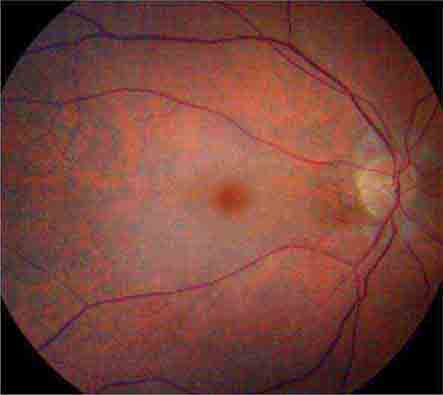
FIGURE 40e-7 Cherry red spot at the macula and cloudy swelling of the macula in a patient with central retinal artery occlusion due to embolus originating from a carotid artery atheromatous plaque.
RETINAL CRYSTAL DEPOSITION
Retinal crystals appear as fine, refractile, yellow-white deposits. Associated conditions include infantile cystinosis, primary hyperoxaluria, secondary oxalosis, Sjögren-Larson syndrome, intravenous drug abuse (talc retinopathy), and drugs such as tamoxifen, canthaxanthin, nitrofurantoin, methoxyflurane, and ethylene glycol. Crystals may also be seen in primary retinal diseases such as juxtafoveal telangiectasia, gyrate atrophy, and Bietti’s crystalline degeneration. Old microemboli may mimic retinal crystals.
RETINAL VASCULAR SHEATHING
Vascular sheathing appears as a yellow-white cuff surrounding a retinal artery or vein (Fig. 40e-8). Diseases associated with retinal vascular sheathing include sarcoidosis, tuberculosis, toxoplasmosis, syphilis, HIV, retinitis (cytomegalovirus, herpes zoster, and herpes simplex), Lyme disease, cat-scratch disease, multiple sclerosis, chronic leukemia, amyloidosis, Behçet’s disease, retinal vasculitis, retinal vascular occlusion, and chronic uveitis.
FIGURE 40e-8 Vascular sheathing over the optic disc in a patient with neurosarcoidosis.
RETINAL DETACHMENT
Retinal detachment is the separation of the retina from the underlying RPE. There are three main types: (1) serous/exudative, (2) tractional, and (3) rhegmatogenous retinal detachment.
In serous retinal detachment, the location of the subretinal fluid is position-dependent, characteristically gravitating to the lowermost part of the fundus (shifting fluid sign), and retinal breaks are absent. Diseases associated with serous/exudative retinal detachment include severe systemic hypertension, dural arteriovenous shunt, retinal vascular anomalies, hyperviscosity syndromes, papilledema, posterior uveitis, scleritis, orbital inflammation, and intraocular neoplasms such as choroidal melanoma, choroidal metastasis, lymphoma, and multiple myeloma.
Tractional retinal detachment is caused by internal traction on the retina in the absence of a retinal break. The retina in the area of detachment is immobile and concaved internally. Fibrovascular proliferation is a frequent associated finding. Conditions associated with tractional retinal detachment include vascular proliferative retinopathies such as severe proliferative diabetic retinopathy, branch retinal vein occlusion, sickle cell retinopathy, and retinopathy of prematurity. Ocular trauma, proliferative vitreoretinopathy, and intraocular inflammation are other causes of a tractional retinal detachment.
Rhegmatogenous retinal detachment is caused by the presence of a retinal break, allowing fluid from the vitreous cavity to gain access to the subretinal space. The surface of the retina is usually convex forward. Rhegmatogenous retinal detachment has a corrugated appearance, and undulates with eye movement. Causes of retinal breaks include posterior vitreous detachment, severe vitreoretinal traction, trauma, intraocular surgery, retinitis, and atrophic holes.
OPTIC DISC SWELLING
Optic disc swelling is abnormal elevation of the optic disc with blurring of its margins (Fig. 40e-9). The term “papilledema” is used to describe swelling of the optic disc secondary to elevation of intracranial pressure. In papilledema, the normal venous pulsation at the disc is characteristically absent. The differential diagnosis of optic disc swelling includes papilledema, anterior optic neuritis (papillitis), central retinal vein occlusion, anterior ischemic optic neuropathy, toxic optic neuropathy, hereditary optic neuropathy, neuroretinitis, diabetic papillopathy, hypertension (Fig. 40e-10), respiratory failure, carotid-cavernous fistula, optic disc nerve infiltration (glioma, lymphoma, leukemia, sarcoidosis, and granulomatous infections), ocular hypotony, chronic intraocular inflammation, optic disc drusen (pseudopapilledema), and high hypermetropia (pseudopapilledema).
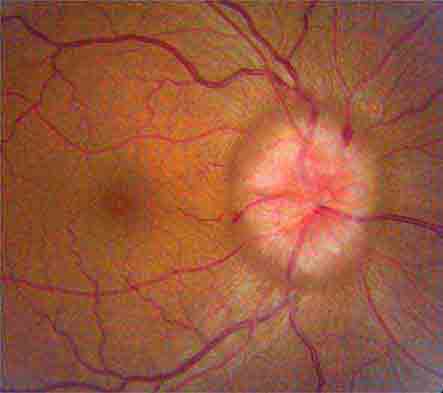
FIGURE 40e-9 Optic disc swelling in a patient with papilledema due to idiopathic intracranial hypertension. The optic disc is hyperemic, with indistinct margins. Superficial hemorrhages are present.
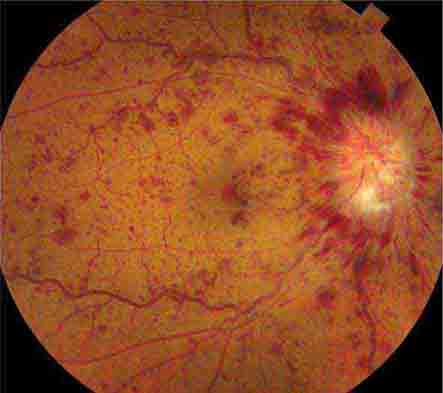
FIGURE 40e-10 Optic disc edema and retinal hemorrhages in a patient with malignant hypertension.
CHORIORETINAL MASS LESIONS
Choroidal mass lesions appear thickened and may or may not be associated with increased pigmentation. Pigmented mass lesions include choroidal nevus (usually flat), choroidal malignant melanoma (Fig. 40e-11), and melanocytoma. Nonpigmented lesions include amelanotic choroidal melanoma, choroidal metastasis, retinoblastoma, capillary hemangioma, granuloma (e.g., Toxocara canis), choroidal detachment, choroidal hemorrhage, and wet age-related macular degeneration. Other rare tumors that may be visible on ophthalmoscopy include osteoma, astrocytoma (e.g., tuberous sclerosis), neurilemmoma, and leiomyoma.
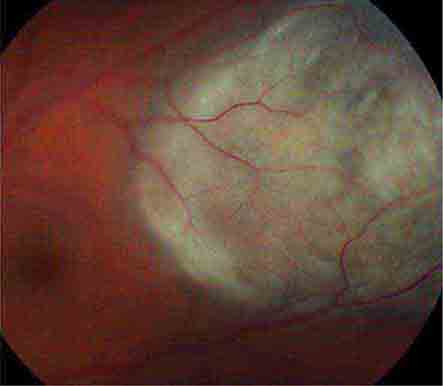
FIGURE 40e-11 Choroidal malignant melanoma. The lesion is highly elevated and pigmented, and has subretinal orange pigment deposits characteristic for malignant melanoma.
PIGMENTED LESIONS
The differential diagnosis of flat pigmented lesions of the fundus is summarized in Table 40e-4. The appearance of chorioretinal scarring from old Toxoplasma chorioretinitis is shown in Fig. 40e-12.
|
DIFFERENTIAL DIAGNOSIS OF FLAT PIGMENTED LESIONS OF THE FUNDUS |
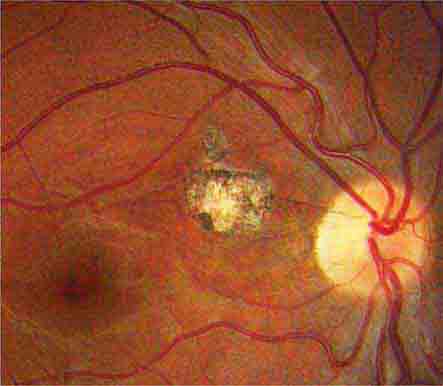
FIGURE 40e-12 Chorioretinal scarring due to old Toxoplasma chorioretinitis. The lesion is flat and pigmented. Areas of hypopigmentation are also present.
41e |
Video Library of Neuro-Ophthalmology |
The proper control of eye movements requires the coordinated activity of many different anatomic structures in the peripheral and central nervous system, and in turn, manifestations of a diverse array of neurologic and medical disorders are revealed as disorders of eye movement. In this remarkable video collection, an introduction to distinctive eye movement disorders encountered in the context of neuromuscular, paraneoplastic, demyelinating, neurovascular, and neurodegenerative disorders is presented.
Cases with Multiple Sclerosis
VIDEO 41e-1 Fisher’s One-and-a-Half Syndrome (ID164-2)
VIDEO 41e-2 A Case of Ocular Flutter (ID166-2)
VIDEO 41e-3 Downbeat Nystagmus and Periodic Alternating Nystagmus (ID168-6)
VIDEO 41e-4 Bilateral Internuclear Ophthalmoplegia (ID933-1)
Cases with Myasthenia Gravis or Mitochondrial Myopathy
VIDEO 41e-5 Unilateral Ptosis: Myasthenia Gravis (Thymic Tumor) (ID163-1)
VIDEO 41e-6 Progressive External Ophthalmoplegia (Progressive External Ophthalmoplegia: Mitochondrial Cytopathy) (ID906-2)
Cases with Paraneoplastic Disease
VIDEO 41e-7 Paraneoplastic Upbeat Nystagmus, Cancer of the Pancreas, Positive Anti-Hu Antibody (ID212-3)
VIDEO 41e-8 Paraneoplastic Ocular Flutter, Small-Cell Adenocarcinoma of the Lung, Negative Marker (ID936-7)
VIDEO 41e-9 Opsoclonus/Flutter, Bilateral Sixth Nerve Palsy, Adenocarcinoma of the Breast, Negative Marker (ID939-8)
Cases with Fisher’s Syndrome
VIDEO 41e-10 Bilateral Ptosis: Facial Diplegia, Total External Ophthalmoplegia, Positive Anti-GQ1b Antibody (ID944-1)
Cases with Vascular Disease
VIDEO 41e-11 Retinal Emboli (Film or Fundus) (ID16-1)
VIDEO 41e-12 Third Nerve Palsy (Microinfarct) (ID939-2)
Case with Neurodegenerative Disease
VIDEO 41e-13 Apraxia of Eyelid Opening (Progressive Supranuclear Palsy) (ID932-3)
Case of Thyroid-Associated Ophthalmopathy
VIDEO 41e-14 Restrictive Orbitopathy of Graves’ Disease, Bilateral Exophthalmos (ID925-4)
Case with Wernicke’s Encephalopathy
VIDEO 41e-15 Bilateral Sixth Nerve Palsies (ID 163-3)
Case with the Locked-in-Syndrome
VIDEO 41e-16 Ocular Dipping (ID 4-1)
Case in a Comatose Patient
VIDEO 41e-17 Downbeat Nystagmus (ID 166-11)
Case Two Years After a Pontine Hemorrhage
VIDEO 41e-18 Palatal Tremor (ID 936-4)
The Video Library of Neuro-Ophthalmology shows a number of cases with eye movement disorders. All the clips are taken from Dr. Shirley Wray’s collection on the NOVEL website.
To access go to:
http://Respitory.Countway.Harvard.edu/Wray
and/or to her book
Eye Movement Disorders in Clinical Practice
Shirley H. Wray, MD, PhD, Oxford University Press, 2014.
42 |
Disorders of Smell and Taste |
All environmental chemicals necessary for life enter the body by the nose and mouth. The senses of smell (olfaction) and taste (gustation) monitor such chemicals, determine the flavor and palatability of foods and beverages, and warn of dangerous environmental conditions, including fire, air pollution, leaking natural gas, and bacteria-laden foodstuffs. These senses contribute significantly to quality of life and, when dysfunctional, can have untoward physical and psychological consequences. A basic understanding of these senses in health and disease is critical for the physician, because thousands of patients present to doctors’ offices each year with complaints of chemosensory dysfunction. Among the more important recent developments in neurology is the discovery that decreased smell function is among the first signs, if not the first sign, of such neurodegenerative diseases as Parkinson’s disease (PD) and Alzheimer’s disease (AD), signifying their “presymptomatic” phase.
ANATOMY AND PHYSIOLOGY
Olfactory System Odorous chemicals enter the front of nose during inhalation and active sniffing, as well as the back of the nose (nasopharynx) during deglutition. After reaching the highest recesses of the nasal cavity, they dissolve in the olfactory mucus and diffuse or are actively transported by specialized proteins to receptors located on the cilia of olfactory receptor cells. The cilia, dendrites, cell bodies, and proximal axonal segments of these bipolar cells are located within a unique neuroepithelium covering the cribriform plate, the superior nasal septum, superior turbinate, and sectors of the middle turbinate (Fig. 42-1). Each of the ~6 million bipolar receptor cells expresses only one of ~450 receptor protein types, most of which respond to more than a single chemical. When damaged, the receptor cells can be replaced by stem cells near the basement membrane. Unfortunately, such replacement is often incomplete.
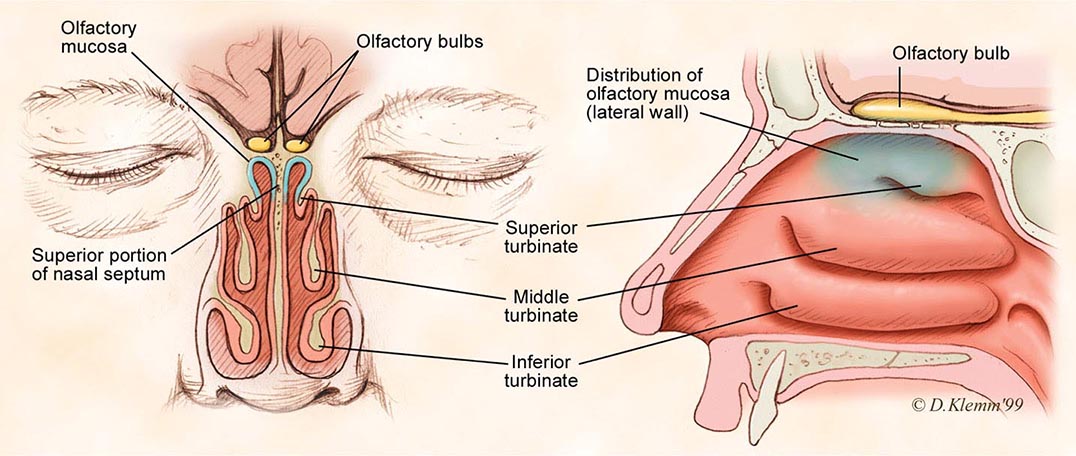
FIGURE 42-1 Anatomy of the olfactory neural pathways, showing the distribution of olfactory receptors in the roof of the nasal cavity. (Copyright David Klemm, Faculty and Curriculum Support [FACS], Georgetown University Medical Center; used with permission.)
After coalescing into bundles surrounded by glia-like ensheathing cells (termed fila), the receptor cell axons pass through the cribriform plate to the olfactory bulbs, where they synapse with dendrites of other cell types within the glomeruli (Fig. 42-2). These spherical structures, which make up a distinct layer of the olfactory bulb, are a site of convergence of information, because many more fibers enter than leave them. Receptor cells that express the same type of receptor project to the same glomeruli, effectively making each glomerulus a functional unit. The major projection neurons of the olfactory system—the mitral and tufted cells—send primary dendrites into the glomeruli, connecting not only with the incoming receptor cell axons, but with dendrites of periglomerular cells. The activity of the mitral/tufted cells is modulated by the periglomerular cells, secondary dendrites from other mitral/tufted cells, and granule cells, the most numerous cells of the bulb. The latter cells, which are largely GABAergic, receive inputs from central brain structures and modulate the output of the mitral/tufted cells. Interestingly, like the olfactory receptor cells, some cells within the bulb undergo replacement. Thus, neuroblasts formed within the anterior subventricular zone of the brain migrate along the rostral migratory stream, ultimately becoming granule and periglomerular cells.
FIGURE 42-2 Schematic of the layers and wiring of the olfactory bulb. Each receptor type (red, green, blue) projects to a common glomerulus. The neural activity within each glomerulus is modulated by periglomerular cells. The activity of the primary projection cells, the mitral and tufted cells, is modulated by granule cells, periglomerular cells, and secondary dendrites from adjacent mitral and tufted cells. (From www.med.yale.edu/neurosurg/treloar/index.html.)
The axons of the mitral and tufted cells synapse within the primary olfactory cortex (POC) (Fig. 42-3). The POC is defined as those cortical structures that receive direct projections from the olfactory bulb, most notably the piriform and entorhinal cortices. Although olfaction is unique in that its initial afferent projections bypass the thalamus, persons with damage to the thalamus can exhibit olfactory deficits, particularly ones of odor identification. Such deficits likely reflect the involvement of thalamic connections between the primary olfactory cortex and the orbitofrontal cortex (OFC), where odor identification occurs. The close anatomic ties between the olfactory system and the amygdala, hippocampus, and hypothalamus help to explain the intimate associations between odor perception and cognitive functions such as memory, motivation, arousal, autonomic activity, digestion, and sex.
FIGURE 42-3 Anatomy of the base of the brain showing the primary olfactory cortex.
Taste System Tastants are sensed by specialized receptor cells present within taste buds—small grapefruit-like segmented structures located on the lateral margins and dorsum of the tongue, roof of the mouth, pharynx, larynx, and superior esophagus (Fig. 42-4). Lingual taste buds are imbedded in well-defined protuberances, termed fungiform, foliate, and circumvallate papillae. After dissolving in a liquid, tastants enter the opening of the taste bud—the taste pore—and bind to receptors on microvilli, small extensions of receptor cells within each taste bud. Such binding changes the electrical potential across the taste cell, resulting in neurotransmitter release onto the first-order taste neurons. Although humans have ~7500 taste buds, not all harbor taste-sensitive cells; some contain only one class of receptor (e.g., cells responsive only to sugars), whereas others contain cells sensitive to more than one class. The number of taste receptor cells per taste bud ranges from zero to well over 100. A small family of three G-protein-coupled receptors (GPCRs), namely T1R1, T1R2, and T1R3, mediate sweet and umami taste sensations. Bitter sensations, on the other hand, depend on T2R receptors, a family of ~30 GPCRs expressed on cells different from those that express the sweet and umami receptors. T2Rs sense a wide range of bitter substances but do not distinguish among them. Sour tastants are sensed by the PKD2L1 receptor, a member of the transient receptor potential protein (TRP) family. Perception of salty sensations, such as induced by sodium chloride, arises from the entry of Na+ ions into the cells via specialized membrane channels, such as the amiloride-sensitive Na+ channel.
FIGURE 42-4 Schematic of the taste bud and its opening (pore), as well as the location of buds on the three major types of papillae: fungiform (anterior), foliate (lateral), and circumvallate (posterior).
Recent studies have found that both bitter and sweet taste-related receptors are also present elsewhere in the body, most notably in the alimentary and respiratory tracts. This important discovery generalizes the concept of taste-related chemoreception to areas of the body beyond the mouth and throat, with α-gustducin, the taste-specific G-protein α-subunit, expressed in so-called brush cells found specifically within the human trachea, lung, pancreas, and gallbladder. These brush cells are rich in nitric oxide (NO) synthase, known to defend against xenobiotic organisms, protect the mucosa from acid-induced lesions, and, in the case of the gastrointestinal tract, stimulate vagal and splanchnic afferent neurons. NO further acts on nearby cells, including enteroendocrine cells, absorptive or secretory epithelial cells, mucosal blood vessels, and cells of the immune system. Members of the T2R family of bitter receptors and the sweet receptors of the T1R family have been identified within the gastrointestinal tract and in enteroendocrine cell lines. In some cases, these receptors are important for metabolism, with the T1R3 receptors and gustducin playing decisive roles in the sensing and transport of dietary sugars from the intestinal lumen into absorptive enterocytes via a sodium-dependent glucose transporter and in regulation of hormone release from gut enteroendocrine cells. In other cases, these receptors may be important for airway protection, with a number of T2R bitter receptors in the motile cilia of the human airway that responded to bitter compounds by increasing their beat frequency. One specific T2R38 taste receptor is expressed in human upper respiratory epithelia and responds to acyl-monoserine lactone quorum-sensing molecules secreted by Pseudomonas aeruginosa and other gram-negative bacteria. Differences in T2R38 functionality, as related to TAS2R38 genotype, correlate with susceptibility to upper respiratory infections in humans.
Taste information is sent to the brain via three cranial nerves (CNs): CN VII (the facial nerve, which involves the intermediate nerve with its branches, the greater petrosal and chorda tympani nerves), CN IX (the glossopharyngeal nerve), and CN × (the vagus nerve) (Fig. 42-5). CN VII innervates the anterior tongue and all of the soft palate, CN IX innervates the posterior tongue, and CN × innervates the laryngeal surface of the epiglottis, larynx, and proximal portion of the esophagus. The mandibular branch of CN V (V3) conveys somatosensory information (e.g., touch, burning, cooling, irritation) to the brain. Although not technically a gustatory nerve, CN V shares primary nerve routes with many of the gustatory nerve fibers and adds temperature, texture, pungency, and spiciness to the taste experience. The chorda tympani nerve is famous for taking a recurrent course through the facial canal in the petrosal portion of the temporal bone, passing through the middle ear, and then exiting the skull via the petrotympanic fissure, where it joins the lingual nerve (a division of CN V) near the tongue. This nerve also carries parasympathetic fibers to the submandibular and sublingual glands, whereas the greater petrosal nerve supplies the palatine glands, thereby influencing saliva production.
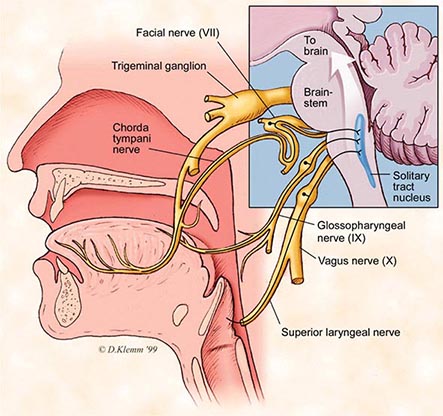
FIGURE 42-5 Schematic of the cranial nerves (CNs) that mediate taste function, including the chorda tympani nerve (CN VII), the glossopharyngeal nerve (CN IX), and the vagus nerve (CN X).
The axons of the projection cells, which synapse with taste buds, enter the rostral portion of the nucleus of the solitary tract (NTS) within the medulla of the brainstem (Fig. 42-5). From the NTS, neurons then project to a division of the ventroposteromedial thalamic nucleus (VPM) via the medial lemniscus. From here, projections are made to the rostral part of the frontal operculum and adjoining insula, a brain region considered the primary taste cortex (PTC). Projections from the PTC then go to the secondary taste cortex, namely the caudolateral OFC. This brain region is involved in the conscious recognition of taste qualities. Moreover, because it contains cells that are activated by several sensory modalities, it is likely a center for establishing “flavor.”
DISORDERS OF OLFACTION
The ability to smell is influenced, in everyday life, by such factors as age, gender, general health, nutrition, smoking, and reproductive state. Women typically outperform men on tests of olfactory function and retain normal smell function to a later age than do men. Significant decrements in the ability to smell are present in over 50% of the population between 65 and 80 years of age and in 75% of those 80 years of age and older (Fig. 42-6). Such presbyosmia helps to explain why many elderly report that food has little flavor, a problem that can result in nutritional disturbances. This also helps to explain why a disproportionate number of elderly die in accidental gas poisonings. A relatively complete listing of conditions and disorders that have been associated with olfactory dysfunction is presented in Table 42-1.
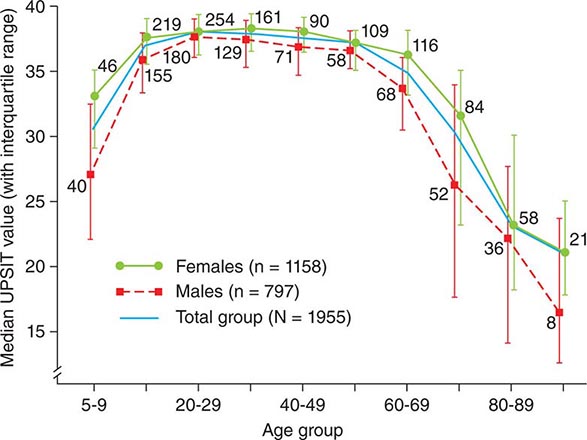
FIGURE 42-6 Scores on the University of Pennsylvania Smell Identification Test (UPSIT) as a function of subject age and sex. Numbers by each data point indicate sample sizes. Note that women identify odorants better than men at all ages. (From RL Doty et al: Science 226:1421, 1984. Copyright © 1984 American Association for the Advancement of Science.)
|
DISORDERS AND CONDITIONS ASSOCIATED WITH COMPROMISED OLFACTORY FUNCTION, AS MEASURED BY OLFACTORY TESTING |
Aside from aging, the three most common identifiable causes of long-lasting or permanent smell loss seen in the clinic are, in order of frequency, severe upper respiratory infections, head trauma, and chronic rhinosinusitis. The physiologic basis for most head trauma–related losses is the shearing and subsequent scarring of the olfactory fila as they pass from the nasal cavity into the brain cavity. The cribriform plate does not have to be fractured or show pathology for smell loss to be present. Severity of trauma, as indexed by a poor Glasgow Coma Scale score on presentation and the length of posttraumatic amnesia, is associated with higher risk of olfactory impairment. Less than 10% of posttraumatic anosmic patients will recover age-related normal function over time. This increases to nearly 25% of those with less-than-total loss. Upper respiratory infections, such as those associated with the common cold, influenza, pneumonia, or HIV, can directly and permanently harm the olfactory epithelium by decreasing receptor cell number, damaging cilia on remaining receptor cells, and inducing the replacement of sensory epithelium with respiratory epithelium. The smell loss associated with chronic rhinosinusitis is related to disease severity, with most loss occurring in cases where rhinosinusitis and polyposis are both present. Although systemic glucocorticoid therapy can usually induce short-term functional improvement, it does not, on average, return smell test scores to normal, implying that chronic permanent neural loss is present and/or that short-term administration of systemic glucocorticoids does not completely mitigate the inflammation. It is well established that microinflammation in an otherwise seemingly normal epithelium can influence smell function.
A number of neurodegenerative diseases are accompanied by olfactory impairment, including PD, AD, Huntington’s disease, Down’s syndrome, parkinsonism-dementia complex of Guam, dementia with Lewy bodies (DLB), multiple system atrophy, corticobasal degeneration, and frontotemporal dementia; smell loss can also occur in multiple sclerosis (MS) and idiopathic rapid eye movement (REM) behavioral sleep disorder (iRBD). Olfactory impairment in PD often predates the clinical diagnosis by at least 4 years. In staged cases, studies of the sequence of formation of abnormal α-synuclein aggregates and Lewy bodies suggest that the olfactory bulbs may be, along with the dorsomotor nucleus of the vagus, the first site of neural damage in PD. In postmortem studies of patients with very mild “presymptomatic” signs of AD, poorer smell function has been associated with higher levels of AD-related pathology. Smell loss is more marked in patients with early clinical manifestations of DLB than in those with mild AD. Interestingly, smell loss is minimal or nonexistent in progressive supranuclear palsy and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. In MS, the olfactory disturbance varies as a function of the plaque activity within the frontal and temporal lobes.
The smell loss seen in iRBD is of the same magnitude as that found in PD. This is of particular interest because patients with iRBD frequently develop PD and hyposmia. There is some evidence that iRBD may actually represent an early associated condition of PD. REM behavior disorder is not only seen in its idiopathic form, but can also be associated with narcolepsy. This led to a recent study of narcoleptic patients with and without REM behavior disorder, which demonstrated that narcolepsy, independent of REM behavior disorder, was associated with impairments in olfactory function. Orexin A, also known as hypocretin-1, is dramatically diminished or undetectable in the cerebrospinal fluid of patients with narcolepsy and cataplexy (Chap. 38). The orexin-containing neurons in the hypothalamus project throughout the entire olfactory system (from the olfactory epithelium to the olfactory cortex), and damage to these orexin-containing projections may be one underlying mechanism for impaired olfactory performance in narcoleptic patients. The administration of intranasal orexin A (hypocretin-1) appears to result in improved olfactory function, supporting the notion that mild olfactory impairment is not only a primary feature of narcolepsy with cataplexy, but that central nervous system orexin deficiency may be a fundamental part of the mechanism for this loss.
DISORDERS OF TASTE
The majority of patients who present with taste dysfunction exhibit olfactory, not taste, loss. This is because most flavors attributed to taste actually depend on retronasal stimulation of the olfactory receptors during deglutition. As noted earlier, taste buds only mediate basic tastes such as sweet, sour, bitter, salty, and umami. Significant impairment of whole-mouth gustatory function is rare outside of generalized metabolic disturbances or systemic use of some medications, because taste bud regeneration occurs and peripheral damage alone would require the involvement of multiple cranial nerve pathways. Nonetheless, taste can be influenced by (1) the release of foul-tasting materials from the oral cavity from oral medical conditions or appliances (e.g., gingivitis, purulent sialadenitis), (2) transport problems of tastants to the taste buds (e.g., drying of the orolingual mucosa, infections, inflammatory conditions), (3) damage to the taste buds themselves (e.g., local trauma, invasive carcinomas), (4) damage to the neural pathways innervating the taste buds (e.g., middle ear infections), (5) damage to central structures (e.g., multiple sclerosis, tumor, epilepsy, stroke), and (6) systemic disturbances of metabolism (e.g., diabetes, thyroid disease, medications). Unlike CN VII, CN IX is relatively protected along its path, although iatrogenic interventions such as tonsillectomy, bronchoscopy, laryngoscopy, endotracheal intubation, and radiation therapy can result in selective injury. CN VII damage commonly results from mastoidectomy, tympanoplasty, and stapedectomy, in some cases inducing persistent metallic sensations. Bell’s palsy (Chap. 455) is one of the most common causes of CN VII injury that results in taste disturbance. On rare occasions, migraines (Chap. 447) are associated with a gustatory prodrome or aura, and in some cases, tastants can trigger a migraine attack. Interestingly, dysgeusia occurs in some cases of burning mouth syndrome (BMS; also termed glossodynia or glossalgia), as do dry mouth and thirst. BMS is likely associated with dysfunction of the trigeminal nerve (CN V). Some of the etiologies suggested for this poorly understood syndrome are amenable to treatment, including (1) nutritional deficiencies (e.g., iron, folic acid, B vitamins, zinc), (2) diabetes mellitus (possibly predisposing to oral candidiasis), (3) denture allergy, (4) mechanical irritation from dentures or oral devices, (5) repetitive movements of the mouth (e.g., tongue thrusting, teeth grinding, jaw clenching), (6) tongue ischemia as a result of temporal arteritis, (7) periodontal disease, (8) reflux esophagitis, and (9) geographic tongue.
Although both taste and smell can be adversely influenced by pharmacologic agents, drug-related taste alterations are more common. Indeed, over 250 medications have been reported to alter the ability to taste. Major offenders include antineoplastic agents, antirheumatic drugs, antibiotics, and blood pressure medications. Terbinafine, a commonly used antifungal, has been linked to taste disturbance lasting up to 3 years. In a recent controlled trial, nearly two-thirds of individuals taking eszopiclone (Lunesta) experienced a bitter dysgeusia that was stronger in women, systematically related to the time since drug administration, and positively correlated with both blood and saliva levels of the drug. Intranasal use of nasal gels and sprays containing zinc, which are common over-the-counter prophylactics for upper respiratory viral infections, has been implicated in loss of smell function. Whether their efficacy in preventing such infections, which are the most common cause of anosmia and hyposmia, outweighs their potential detriment to smell function requires study. Dysgeusia occurs commonly in the context of drugs used to treat or minimize symptoms of cancer, with a weighted prevalence from 56–76% depending on the type of cancer treatment. Attempts to prevent taste problems from such drugs using prophylactic zinc sulfate or amifostine have proven to be minimally beneficial. Although antiepileptic medications are occasionally used to treat smell or taste disturbances, the use of topiramate has been reported to result in a reversible loss of an ability to detect and recognize tastes and odors during treatment.
As with olfaction, a number of systemic disorders can affect taste. These include chronic renal failure, end-stage liver disease, vitamin and mineral deficiencies, diabetes mellitus, and hypothyroidism (to name a few). In diabetes, there appears to be a progressive loss of taste beginning with glucose and then extending to other sweeteners, salty stimuli, and then all stimuli. Psychiatric conditions can be associated with chemosensory alterations (e.g., depression, schizophrenia, bulimia). A recent review of tactile, gustatory, and olfactory hallucinations demonstrated that no one type of hallucinatory experience is pathognomonic to any given diagnosis.
Pregnancy proves to be a unique condition with regard to taste function. There appears to be an increase in dislike and intensity of bitter tastes during the first trimester that may help to ensure that pregnant women avoid poisons during a critical phase of fetal development. Similarly, a relative increase in the preference for salt and bitter in the second and third trimesters may support the ingestion of much needed electrolytes to expand fluid volume and support a varied diet.
CLINICAL EVALUATION
In most cases, a careful clinical history will establish the probable etiology of a chemosensory problem, including questions about its nature, onset, duration, and pattern of fluctuations. Sudden loss suggests the possibility of head trauma, ischemia, infection, or a psychiatric condition. Gradual loss can reflect the development of a progressive obstructive lesion. Intermittent loss suggests the likelihood of an inflammatory process. The patient should be asked about potential precipitating events, such as cold or flu infections prior to symptom onset, because these often go underappreciated. Information regarding head trauma, smoking habits, drug and alcohol abuse (e.g., intranasal cocaine, chronic alcoholism in the context of Wernicke’s and Korsakoff’s syndromes), exposures to pesticides and other toxic agents, and medical interventions is also informative. A determination of all the medications that the patient was taking before and at the time of symptom onset is important, because many can cause chemosensory disturbances. Comorbid medical conditions associated with smell impairment, such as renal failure, liver disease, hypothyroidism, diabetes, or dementia, should be assessed. Delayed puberty in association with anosmia (with or without midline craniofacial abnormalities, deafness, and renal anomalies) suggests the possibility of Kallmann’s syndrome. Recollection of epistaxis, discharge (clear, purulent, or bloody), nasal obstruction, allergies, and somatic symptoms, including headache or irritation, may have localizing value. Questions related to memory, parkinsonian signs, and seizure activity (e.g., automatisms, blackouts, auras, déjà vu) should be posed. Pending litigation and the possibility of malingering should be considered. Modern forced-choice olfactory tests can detect malingering from improbable responses.
Neurologic and otorhinolaryngologic (ORL) examinations, along with appropriate brain and nasosinus imaging, aid in the evaluation of patients with olfactory or gustatory complaints. The neural evaluation should focus on cranial nerve function, with particular attention to possible skull base and intracranial lesions. Visual acuity, field, and optic disc examinations aid in the detection of intracranial mass lesions that induce intracranial pressure (papilledema) and optic atrophy, especially when considering Foster Kennedy syndrome. The ORL examination should thoroughly assess the intranasal architecture and mucosal surfaces. Polyps, masses, and adhesions of the turbinates to the septum may compromise the flow of air to the olfactory receptors, because less than a fifth of the inspired air traverses the olfactory cleft in the unobstructed state. Blood tests may be helpful to identify such conditions as diabetes, infection, heavy metal exposure, nutritional deficiency (e.g., vitamin B6 or B12), allergy, and thyroid, liver, and kidney disease.
As with other sensory disorders, quantitative sensory testing is advised. Self-reports of patients can be misleading, and a number of patients who complain of chemosensory dysfunction have normal function for their age and gender. Quantitative smell and taste testing provides valid information for worker’s compensation and other legal claims, as well as a way to accurately assess treatment interventions. A number of standardized olfactory and taste tests are commercially available. Most evaluate the ability of patients to detect and identify odors or tastes. For example, the most widely used of these tests, the 40-item University of Pennsylvania Smell Identification Test (UPSIT), uses norms based on nearly 4000 normal subjects. A determination is made of both absolute dysfunction (i.e., mild loss, moderate loss, severe loss, total loss, probable malingering) and relative dysfunction (percentile rank for age and gender). Although electrophysiologic testing is available at some smell and taste centers (e.g., odor event-related potentials), they require complex stimulus presentation and recording equipment and rarely provide additional diagnostic information. With the exception of electrogustometers, commercially available taste tests have only recently become available. Most use filter paper strips impregnated with tastants, so no stimulus preparation is required.
TREATMENT AND MANAGEMENT
Given the various mechanisms by which olfactory and gustatory disturbance can occur, management of patients tends to be condition specific. For example, patients with hypothyroidism, diabetes, or infections often benefit from specific treatments to correct the underlying disease process that is adversely influencing chemoreception. For most patients who present primarily with obstructive/transport loss affecting the nasal and paranasal regions (e.g., allergic rhinitis, polyposis, intranasal neoplasms, nasal deviations), medical and/or surgical intervention is often beneficial. Antifungal and antibiotic treatments may reverse taste problems secondary to candidiasis or other oral infections. Chlorhexidine mouthwash mitigates some salty or bitter dysgeusias, conceivably as a result of its strong positive charge. Excessive dryness of the oral mucosa is a problem with many medications and conditions, and artificial saliva (e.g., Xerolube) or oral pilocarpine treatments may prove beneficial. Other methods to improve salivary flow include the use of mints, lozenges, or sugarless gum. Flavor enhancers may make food more palatable (e.g., monosodium glutamate), but caution is advised to avoid overusing ingredients containing sodium or sugar, particularly in circumstances when a patient also has underlying hypertension or diabetes. Medications that induce distortions of taste can often be discontinued and replaced with other types of medications or modes of therapy. As mentioned earlier, pharmacologic agents result in taste disturbances much more frequently than smell disturbances, and over 250 medications have been reported to alter the sense of taste. It is important to note, however, that many drug-related effects are long lasting and not reversed by short-term drug discontinuance.
A recent study of endoscopic sinus surgery in patients with chronic rhinosinusitis and hyposmia revealed that patients with severe olfactory dysfunction prior to the surgery had a more dramatic and sustained improvement over time compared to patients with more mild olfactory dysfunction prior to intervention. In the case of intranasal and sinus-related inflammatory conditions, such as seen with allergy, viruses, and traumas, the use of intranasal or systemic glucocorticoids may also be helpful. One common approach is to use a tapering course of oral prednisone. The utility of restoring olfaction with either topical or systemic glucocorticoids has been studied. Topical intranasal administration was found to be less effective in general than systemic administration; however, the effects of different nasal administration techniques were not analyzed; for example, intranasal glucocorticoids are more effective if administered in the Moffett’s position (head in the inverted position such as over the edge of the bed with the bridge of the nose perpendicular to the floor). After head trauma, an initial trial of glucocorticoids may help to reduce local edema and the potential deleterious deposition of scar tissue around olfactory fila at the level of the cribriform plate.
Treatments are limited for patients with chemosensory loss or primary injury to neural pathways. Nonetheless, spontaneous recovery can occur. In a follow-up study of 542 patients presenting to our center with smell loss from a variety of causes, modest improvement occurred over an average time period of 4 years in about half of the participants. However, only 11% of the anosmic and 23% of the hyposmic patients regained normal age-related function. Interestingly, the amount of dysfunction present at the time of presentation, not etiology, was the best predictor of prognosis. Other predictors were age and the duration of dysfunction prior to initial testing.
A nonblinded study has reported that patients with hyposmia may benefit from smelling strong odors (e.g., eucalyptol, citronella, eugenol, and phyenyl ethyl alcohol) before going to bed and immediately upon awakening each day over the course of several months. The rationale for such an approach comes from animal studies demonstrating that prolonged exposure to odorants can induce increased neural activity within the olfactory bulb. In an uncontrolled study, α-lipoic acid (400 mg/d), an essential cofactor for many enzyme complexes with possible antioxidant effects, was reported to be beneficial in mitigating smell loss following viral infection of the upper respiratory tract; controlled studies are needed to confirm this observation. This agent has also been suggested to be useful in some cases of hypogeusia and BMS.
The use of zinc and vitamin A in treating olfactory disturbances is controversial, and there does not appear to be much benefit beyond replenishing established deficiencies. However, zinc has been shown to improve taste function secondary to hepatic deficiencies, and retinoids (bioactive vitamin A derivatives) are known to play an essential role in the survival of olfactory neurons. One protocol in which zinc was infused with chemotherapy treatments suggested a possible protective effect against developing taste impairment. Diseases of the alimentary tract can not only influence chemoreceptive function, but also occasionally influence vitamin B12 absorption. This can result in a relative deficiency of vitamin B12, theoretically contributing to olfactory nerve disturbance. Vitamin B2 (riboflavin) and magnesium supplements are reported in the alternative literature to aid in the management of migraine that, in turn, may be associated with smell dysfunction. Because vitamin D deficiency is a cofactor of chemotherapy-induced mucocutaneous toxicity and dysgeusia, adding vitamin D3, 1000–2000 units per day, may benefit some patients with smell and taste complaints during or following chemotherapy.
A number of medications have reportedly been used with success in ameliorating olfactory symptoms, although strong scientific evidence for efficacy is generally lacking. A report that theophylline improved smell function was uncontrolled and failed to account for the fact that some meaningful improvement occurs without treatment; indeed, the percentage of responders was about the same (~50%) as that noted by others to show spontaneous improvement over a similar time period. Antiepileptics and some antidepressants (e.g., amitriptyline) have been used to treat dysosmias and smell distortions, particularly following head trauma. Ironically, amitriptyline is also frequently on the list of medications that can ultimately distort smell and taste function, possibly from its anticholinergic effects. A recent study suggests that the use of the centrally acting acetylcholinesterase inhibitor donepezil in AD resulted in improvements on smell identification measures that correlated with overall clinician-based impressions of change in dementia severity scores.
Alternative therapies, such as acupuncture, meditation, cognitive-behavioral therapy, and yoga, can help patients manage uncomfortable experiences associated with chemosensory disturbance and oral pain syndromes and to cope with the psychosocial stressors surrounding the impairment. Additionally, modification of diet and eating habits is also important. By accentuating the other sensory experiences of a meal, such as food texture, aroma, temperature, and color, one can optimize the overall eating experience for a patient. In some cases, a flavor enhancer like monosodium glutamate (MSG) can be added to foods to increase palatability and encourage intake.
Proper oral and nasal hygiene and routine dental care are extremely important ways for patients to protect themselves from disorders of the mouth and nose that can ultimately result in chemosensory disturbance. Patients should be warned not to overcompensate for their taste loss by adding excessive amounts of sugar or salt. Smoking cessation and the discontinuance of oral tobacco use are essential in the management of any patient with smell and/or taste disturbance and should be repeatedly emphasized.
A major and often overlooked element of therapy comes from chemosensory testing itself. Confirmation or lack of conformation of loss is beneficial to patients who come to believe, in light of unsupportive family members and medical providers, that they may be “crazy.” In cases where the loss is minor, patients can be informed of the likelihood of a more positive prognosis. Importantly, quantitative testing places the patient’s problem into overall perspective. Thus, it is often therapeutic for an older person to know that, while his or her smell function is not what it used to be, it still falls above the average of his or her peer group. Without testing, many such patients are simply told they are getting old and nothing can be done for them, leading in some cases to depression and decreased self-esteem.
43 |
Disorders of Hearing |
Hearing loss is one of the most common sensory disorders in humans and can present at any age. Nearly 10% of the adult population has some hearing loss, and one-third of individuals age >65 years have a hearing loss of sufficient magnitude to require a hearing aid.
PHYSIOLOGY OF HEARING
The function of the external and middle ear is to amplify sound to facilitate conversion of the mechanical energy of the sound wave into an electrical signal by the inner ear hair cells, a process called mechanotransduction (Fig. 43-1). Sound waves enter the external auditory canal and set the tympanic membrane (eardrum) in motion, which in turn moves the malleus, incus, and stapes of the middle ear. Movement of the footplate of the stapes causes pressure changes in the fluid-filled inner ear, eliciting a traveling wave in the basilar membrane of the cochlea. The tympanic membrane and the ossicular chain in the middle ear serve as an impedance-matching mechanism, improving the efficiency of energy transfer from air to the fluid-filled inner ear.
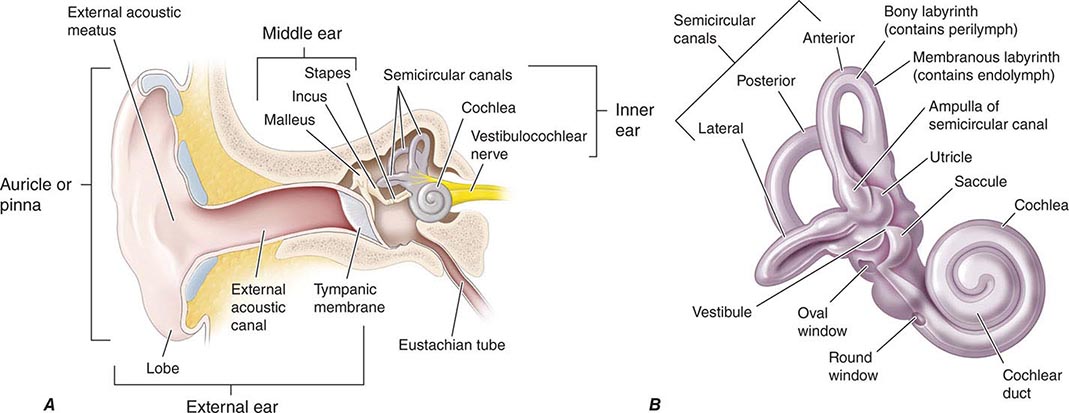
FIGURE 43-1 Ear anatomy. A. Drawing of modified coronal section through external ear and temporal bone, with structures of the middle and inner ear demonstrated. B. High-resolution view of inner ear.
Stereocilia of the hair cells of the organ of Corti, which rests on the basilar membrane, are in contact with the tectorial membrane and are deformed by the traveling wave. A point of maximal displacement of the basilar membrane is determined by the frequency of the stimulating tone. High-frequency tones cause maximal displacement of the basilar membrane near the base of the cochlea, whereas for low-frequency sounds, the point of maximal displacement is toward the apex of the cochlea.
The inner and outer hair cells of the organ of Corti have different innervation patterns, but both are mechanoreceptors. The afferent innervation relates principally to the inner hair cells, and the efferent innervation relates principally to outer hair cells. The motility of the outer hair cells alters the micromechanics of the inner hair cells, creating a cochlear amplifier, which explains the exquisite sensitivity and frequency selectivity of the cochlea.
Beginning in the cochlea, the frequency specificity is maintained at each point of the central auditory pathway: dorsal and ventral cochlear nuclei, trapezoid body, superior olivary complex, lateral lemniscus, inferior colliculus, medial geniculate body, and auditory cortex. At low frequencies, individual auditory nerve fibers can respond more or less synchronously with the stimulating tone. At higher frequencies, phase-locking occurs so that neurons alternate in response to particular phases of the cycle of the sound wave. Intensity is encoded by the amount of neural activity in individual neurons, the number of neurons that are active, and the specific neurons that are activated.
There is evidence that the right and left ears as well as the central nervous system may process speech asymmetrically. Generally, a sound is processed symmetrically from the peripheral to the central auditory system. However, a “right ear advantage” exists for dichotic listening tasks, in which subjects are asked to report on competing sounds presented to each ear. In most individuals, a perceptual right ear advantage for consonant-vowel syllables, stop consonants, and words also exists. Similarly, whereas central auditory processing for sounds is symmetric with minimal lateral specialization for the most part, speech processing is lateralized. There is specialization of the left auditory cortex for speech recognition and production, and of the right hemisphere for emotional and tonal aspects of speech. Left hemisphere dominance for speech is found in 95–98% of right-handed persons and 70–80% of left-handed persons.
DISORDERS OF THE SENSE OF HEARING
Hearing loss can result from disorders of the auricle, external auditory canal, middle ear, inner ear, or central auditory pathways (Fig. 43-2). In general, lesions in the auricle, external auditory canal, or middle ear that impede the transmission of sound from the external environment to the inner ear cause conductive hearing loss, whereas lesions that impair mechanotransduction in the inner ear or transmission of the electrical signal along the eighth nerve to the brain cause sensorineural hearing loss.
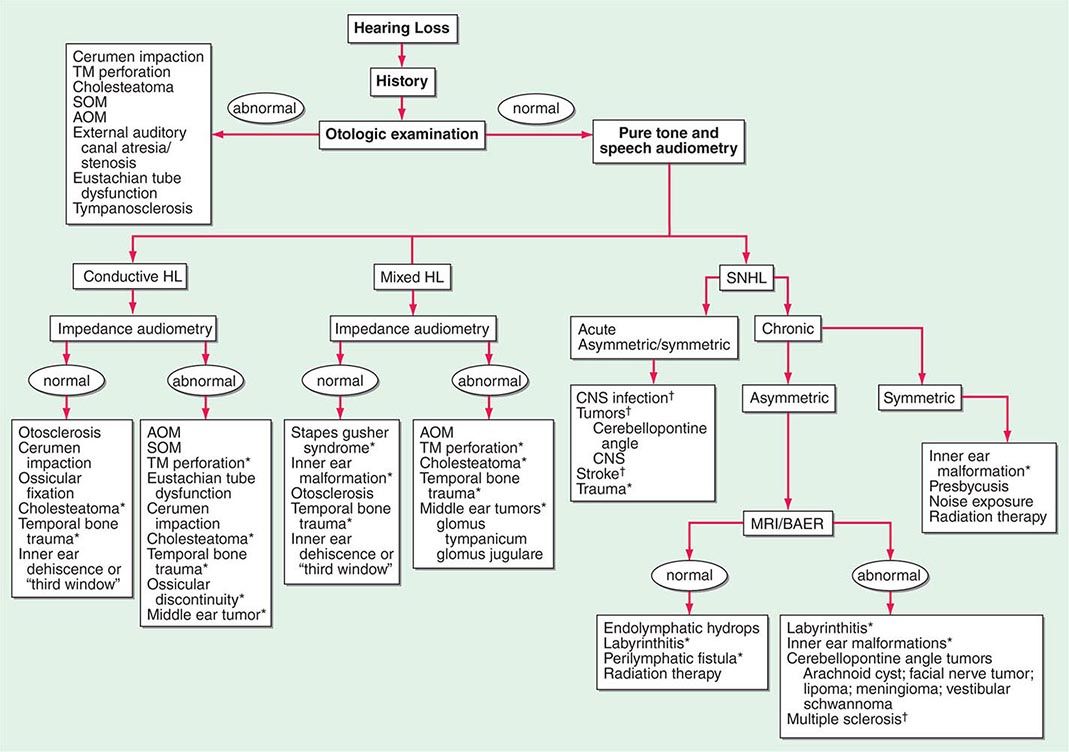
FIGURE 43-2 An algorithm for the approach to hearing loss. AOM, acute otitis media; BAER, brainstem auditory evoked response; CNS, central nervous system; HL, hearing loss; SNHL, sensorineural hearing loss; SOM, serous otitis media; TM, tympanic membrane. *Computed tomography scan of temporal bone. †Magnetic resonance imaging (MRI) scan.
Conductive Hearing Loss The external ear, the external auditory canal, and the middle ear apparatus is designed to collect and amplify sound and efficiently transfer the mechanical energy of the sound wave to the fluid-filled cochlea. Factors that obstruct the transmission of sound or serve to dampen the acoustical energy result in conductive hearing loss. Conductive hearing loss can occur from obstruction of the external auditory canal by cerumen, debris, and foreign bodies; swelling of the lining of the canal; atresia or neoplasms of the canal; perforations of the tympanic membrane; disruption of the ossicular chain, as occurs with necrosis of the long process of the incus in trauma or infection; otosclerosis; or fluid, scarring, or neoplasms in the middle ear. Rarely, inner ear malformations or pathologies, such as superior semicircular canal dehiscence, lateral semicircular canal dysplasia, incomplete partition of the inner ear, and large vestibular aqueduct, may also be associated with conductive hearing loss.
Eustachian tube dysfunction is extremely common in adults and may predispose to acute otitis media (AOM) or serous otitis media (SOM). Trauma, AOM, and chronic otitis media are the usual factors responsible for tympanic membrane perforation. While small perforations often heal spontaneously, larger defects usually require surgical intervention. Tympanoplasty is highly effective (>90%) in the repair of tympanic membrane perforations. Otoscopy is usually sufficient to diagnose AOM, SOM, chronic otitis media, cerumen impaction, tympanic membrane perforation, and eustachian tube dysfunction; tympanometry can be useful to confirm the clinical suspicion of these conditions.
Cholesteatoma, a benign tumor composed of stratified squamous epithelium in the middle ear or mastoid, occurs frequently in adults. This is a slowly growing lesion that destroys bone and normal ear tissue. Theories of pathogenesis include traumatic immigration and invasion of squamous epithelium through a retraction pocket, implantation of squamous epithelia in the middle ear through a perforation or surgery, and metaplasia following chronic infection and irritation. On examination, there is often a perforation of the tympanic membrane filled with cheesy white squamous debris. The presence of an aural polyp obscuring the tympanic membrane is highly suggestive of an underlying cholesteatoma. A chronically draining ear that fails to respond to appropriate antibiotic therapy should raise suspicion of a cholesteatoma. Conductive hearing loss secondary to ossicular erosion is common. Surgery is required to remove this destructive process.
Conductive hearing loss with a normal ear canal and intact tympanic membrane suggests either ossicular pathology or the presence of “third window” in the inner ear (see below). Fixation of the stapes from otosclerosis is a common cause of low-frequency conductive hearing loss. It occurs equally in men and women and is inherited as an autosomal dominant trait with incomplete penetrance; in some cases, it may be a manifestation of osteogenesis imperfecta. Hearing impairment usually presents between the late teens and the forties. In women, the otosclerotic process is accelerated during pregnancy, and the hearing loss is often first noticeable at this time. A hearing aid or a simple outpatient surgical procedure (stapedectomy) can provide adequate auditory rehabilitation. Extension of otosclerosis beyond the stapes footplate to involve the cochlea (cochlear otosclerosis) can lead to mixed or sensorineural hearing loss. Fluoride therapy to prevent hearing loss from cochlear otosclerosis is of uncertain value.
Disorders that lead to the formation of a pathologic “third window” in the inner ear can be associated with conductive hearing loss. There are normally two major openings, or windows, that connect the inner ear with the middle ear and serve as conduits for transmission of sound; these are, respectively, the oval and round windows. A third window is formed where the normally hard otic bone surrounding the inner ear is eroded; dissipation of the acoustic energy at the third window is responsible for the “inner ear conductive hearing loss.” The superior semicircular canal dehiscence syndrome resulting from erosion of the otic bone over the superior circular canal can present with conductive hearing loss that mimics otosclerosis. A common symptom is vertigo evoked by loud sounds (Tullio phenomenon), by Valsalva maneuvers that change middle ear pressure, or by applying positive pressure on the tragus (the cartilage anterior to the external opening of the ear canal). Patients with this syndrome also complain of being able to hear the movement of their eyes and neck. A large jugular bulb or jugular bulb diverticulum can create a “third window” by eroding into the vestibular aqueduct or posterior semicircular canal; the symptoms are similar to those of the superior semicircular canal dehiscence syndrome.
Sensorineural Hearing Loss Sensorineural hearing loss results from either damage to the mechanotransduction apparatus of the cochlea or disruption of the electrical conduction pathway from the inner ear to the brain. Thus, injury to hair cells, supporting cells, auditory neurons, or the central auditory pathway can cause sensorineural hearing loss. Damage to the hair cells of the organ of Corti may be caused by intense noise, viral infections, ototoxic drugs (e.g., salicylates, quinine and its synthetic analogues, aminoglycoside antibiotics, loop diuretics such as furosemide and ethacrynic acid, and cancer chemotherapeutic agents such as cisplatin), fractures of the temporal bone, meningitis, cochlear otosclerosis (see above), Ménière’s disease, and aging. Congenital malformations of the inner ear may be the cause of hearing loss in some adults. Genetic predisposition alone or in concert with environmental exposures may also be responsible (see below).
Presbycusis (age-associated hearing loss) is the most common cause of sensorineural hearing loss in adults. In the early stages, it is characterized by symmetric, gentle to sharply sloping high-frequency hearing loss (Fig. 43-3). With progression, the hearing loss involves all frequencies. More importantly, the hearing impairment is associated with significant loss in clarity. There is a loss of discrimination for phonemes, recruitment (abnormal growth of loudness), and particular difficulty in understanding speech in noisy environments such as at restaurants and social events. Hearing aids are helpful in enhancing the signal-to-noise ratio by amplifying sounds that are close to the listener. Although hearing aids are able to amplify sounds, they cannot restore the clarity of hearing. Thus, amplification with hearing aids may provide only limited rehabilitation once the word recognition score deteriorates below 50%. Cochlear implants are the treatment of choice when hearing aids prove inadequate, even when hearing loss is incomplete (see below).
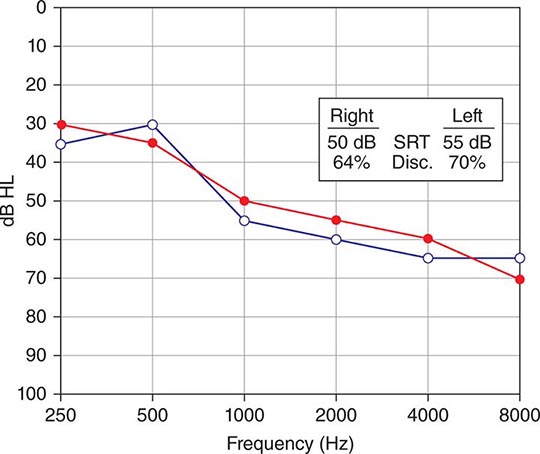
FIGURE 43-3 Presbyacusis or age-related hearing loss. The audiogram shows a moderate to severe downsloping sensorineural hearing loss typical of presbyacusis. The loss of high-frequency hearing is associated with a decreased speech discrimination score; consequently, patients complain of lack of clarity of hearing, especially in a noisy background. HL, hearing threshold level; SRT, speech reception threshold.
Ménière’s disease is characterized by episodic vertigo, fluctuating sensorineural hearing loss, tinnitus, and aural fullness. Tinnitus and/or deafness may be absent during the initial attacks of vertigo, but it invariably appears as the disease progresses and increases in severity during acute attacks. The annual incidence of Ménière’s disease is 0.5–7.5 per 1000; onset is most frequently in the fifth decade of life but may also occur in young adults or the elderly. Histologically, there is distention of the endolymphatic system (endolymphatic hydrops) leading to degeneration of vestibular and cochlear hair cells. This may result from endolymphatic sac dysfunction secondary to infection, trauma, autoimmune disease, inflammatory causes, or tumor; an idiopathic etiology constitutes the largest category and is most accurately referred to as Ménière’s disease. Although any pattern of hearing loss can be observed, typically, low-frequency, unilateral sensorineural hearing impairment is present. Magnetic resonance imaging (MRI) should be obtained to exclude retrocochlear pathology such as a cerebellopontine angle tumor or demyelinating disorder. Therapy is directed toward the control of vertigo. A 2-g/d low-salt diet is the mainstay of treatment for control of rotatory vertigo. Diuretics, a short course of glucocorticoids, and intratympanic gentamicin may also be useful adjuncts in recalcitrant cases. Surgical therapy of vertigo is reserved for unresponsive cases and includes endolymphatic sac decompression, labyrinthectomy, and vestibular nerve section. Both labyrinthectomy and vestibular nerve section abolish rotatory vertigo in >90% of cases. Unfortunately, there is no effective therapy for hearing loss, tinnitus, or aural fullness from Ménière’s disease.
Sensorineural hearing loss may also result from any neoplastic, vascular, demyelinating, infectious, or degenerative disease or trauma affecting the central auditory pathways. HIV leads to both peripheral and central auditory system pathology and is associated with sensorineural hearing impairment.
Primary diseases of the central nervous system can also present with hearing impairment. Characteristically, a reduction in clarity of hearing and speech comprehension is much greater than the loss of the ability to hear pure tone. Auditory testing is consistent with an auditory neuropathy; normal otoacoustic emissions (OAE) and an abnormal auditory brainstem response (ABR) is typical (see below). Hearing loss can accompany hereditary sensorimotor neuropathies and inherited disorders of myelin. Tumors of the cerebellopontine angle such as vestibular schwannoma and meningioma usually present with asymmetric sensorineural hearing loss with greater deterioration of speech understanding than pure tone hearing. Multiple sclerosis may present with acute unilateral or bilateral hearing loss; typically, pure tone testing remains relatively stable while speech understanding fluctuates. Isolated labyrinthine infarction can present with acute hearing loss and vertigo due to a cerebrovascular accident involving the posterior circulation, usually the anterior inferior cerebellar artery; it may also be the heralding sign of impending catastrophic basilar artery infarction (Chap. 446).
A finding of conductive and sensory hearing loss in combination is termed mixed hearing loss. Mixed hearing losses are due to pathology of both the middle and inner ear, as can occur in otosclerosis involving the ossicles and the cochlea, head trauma, chronic otitis media, cholesteatoma, middle ear tumors, and some inner ear malformations.
Trauma resulting in temporal bone fractures may be associated with conductive, sensorineural, or mixed hearing loss. If the fracture spares the inner ear, there may simply be conductive hearing loss due to rupture of the tympanic membrane or disruption of the ossicular chain. These abnormalities can be surgically corrected. Profound hearing loss and severe vertigo are associated with temporal bone fractures involving the inner ear. A perilymphatic fistula associated with leakage of inner ear fluid into the middle ear can occur and may require surgical repair. An associated facial nerve injury is not uncommon. Computed tomography (CT) is best suited to assess fracture of the traumatized temporal bone, evaluate the ear canal, and determine the integrity of the ossicular chain and the involvement of the inner ear. Cerebrospinal fluid leaks that accompany temporal bone fractures are usually self-limited; the value of prophylactic antibiotics is uncertain.
Tinnitus is defined as the perception of a sound when there is no sound in the environment. It may have a buzzing, roaring, or ringing quality and may be pulsatile (synchronous with the heartbeat). Tinnitus is often associated with either a conductive or sensorineural hearing loss. The pathophysiology of tinnitus is not well understood. The cause of the tinnitus can usually be determined by finding the cause of the associated hearing loss. Tinnitus may be the first symptom of a serious condition such as a vestibular schwannoma. Pulsatile tinnitus requires evaluation of the vascular system of the head to exclude vascular tumors such as glomus jugulare tumors, aneurysms, dural arteriovenous fistulas, and stenotic arterial lesions; it may also occur with SOM. It is most commonly associated with some abnormality of the jugular bulb such as a large jugular bulb or jugular bulb diverticulum.
GENETIC CAUSES OF HEARING LOSS
![]() More than half of childhood hearing impairment is thought to be hereditary; hereditary hearing impairment (HHI) can also manifest later in life. HHI may be classified as either nonsyndromic, when hearing loss is the only clinical abnormality, or syndromic, when hearing loss is associated with anomalies in other organ systems. Nearly two-thirds of HHIs are nonsyndromic, and the remaining one-third are syndromic. Between 70 and 80% of nonsyndromic HHI is inherited in an autosomal recessive manner and designated DFNB; another 15–20% is autosomal dominant (DFNA). Less than 5% is X-linked (DFNX) or maternally inherited via the mitochondria.
More than half of childhood hearing impairment is thought to be hereditary; hereditary hearing impairment (HHI) can also manifest later in life. HHI may be classified as either nonsyndromic, when hearing loss is the only clinical abnormality, or syndromic, when hearing loss is associated with anomalies in other organ systems. Nearly two-thirds of HHIs are nonsyndromic, and the remaining one-third are syndromic. Between 70 and 80% of nonsyndromic HHI is inherited in an autosomal recessive manner and designated DFNB; another 15–20% is autosomal dominant (DFNA). Less than 5% is X-linked (DFNX) or maternally inherited via the mitochondria.
More than 150 loci harboring genes for nonsyndromic HHI have been mapped, with recessive loci outnumbering dominant; numerous genes have now been identified (Table 43-1). The hearing genes fall into the categories of structural proteins (MYH9, MYO7A, MYO15, TECTA, DIAPH1), transcription factors (POU3F4, POU4F3), ion channels (KCNQ4, SLC26A4), and gap junction proteins (GJB2, GJB3, GJB6). Several of these genes, including GJB2, TECTA, and TMC1, cause both autosomal dominant and recessive forms of nonsyndromic HHI. In general, the hearing loss associated with dominant genes has its onset in adolescence or adulthood, varies in severity, and progresses with age, whereas the hearing loss associated with recessive inheritance is congenital and profound. Connexin 26, product of the GJB2 gene, is particularly important because it is responsible for nearly 20% of all cases of childhood deafness; half of genetic deafness in children is GJB2-related. Two frameshift mutations, 35delG and 167delT, account for >50% of the cases; however, screening for these two mutations alone is insufficient, and sequencing of the entire gene is required to diagnose GJB2-related recessive deafness. The 167delT mutation is highly prevalent in Ashkenazi Jews; ~1 in 1765 individuals in this population are homozygous and affected. The hearing loss can also vary among the members of the same family, suggesting that other genes or factors influence the auditory phenotype.
|
HEREDITARY HEARING IMPAIRMENT GENES |
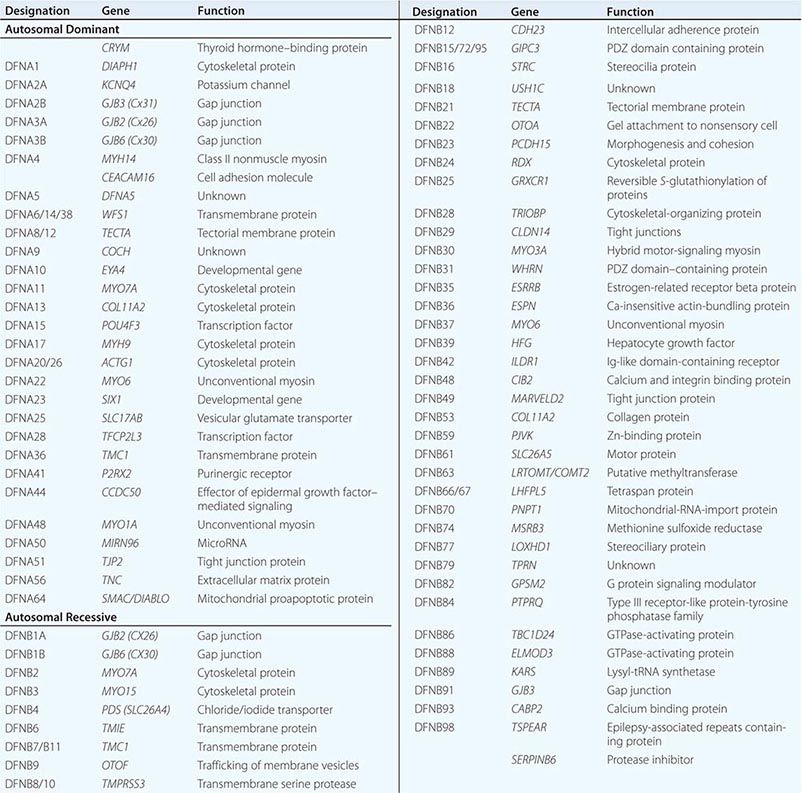
In addition to GJB2, several other nonsyndromic genes are associated with hearing loss that progresses with age. The contribution of genetics to presbycusis is also becoming better understood. Sensitivity to aminoglycoside ototoxicity can be maternally transmitted through a mitochondrial mutation. Susceptibility to noise-induced hearing loss may also be genetically determined.
There are >400 syndromic forms of hearing loss. These include Usher’s syndrome (retinitis pigmentosa and hearing loss), Waardenburg’s syndrome (pigmentary abnormality and hearing loss), Pendred’s syndrome (thyroid organification defect and hearing loss), Alport’s syndrome (renal disease and hearing loss), Jervell and Lange-Nielsen syndrome (prolonged QT interval and hearing loss), neurofibromatosis type 2 (bilateral acoustic schwannoma), and mitochondrial disorders (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes [MELAS]; myoclonic epilepsy and ragged red fibers [MERRF]; progressive external ophthalmoplegia [PEO]) (Table 43-2).
|
SYNDROMIC HEREDITARY HEARING IMPAIRMENT GENES |
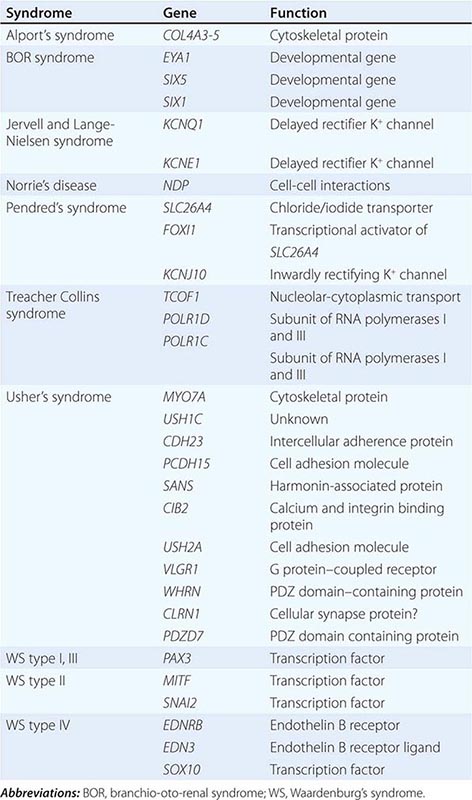
LABORATORY ASSESSMENT OF HEARING
Audiologic Assessment The minimum audiologic assessment for hearing loss should include the measurement of pure tone air-conduction and bone-conduction thresholds, speech reception threshold, word recognition score, tympanometry, acoustic reflexes, and acoustic-reflex decay. This test battery provides a screening evaluation of the entire auditory system and allows one to determine whether further differentiation of a sensory (cochlear) from a neural (retrocochlear) hearing loss is indicated.
Pure tone audiometry assesses hearing acuity for pure tones. The test is administered by an audiologist and is performed in a sound-attenuated chamber. The pure tone stimulus is delivered with an audiometer, an electronic device that allows the presentation of specific frequencies (generally between 250 and 8000 Hz) at specific intensities. Air- and bone-conduction thresholds are established for each ear. Air-conduction thresholds are determined by presenting the stimulus in air with the use of headphones. Bone-conduction thresholds are determined by placing the stem of a vibrating tuning fork or an oscillator of an audiometer in contact with the head. In the presence of a hearing loss, broad-spectrum noise is presented to the nontest ear for masking purposes so that responses are based on perception from the ear under test.
The responses are measured in decibels. An audiogram is a plot of intensity in decibels of hearing threshold versus frequency. A decibel (dB) is equal to 20 times the logarithm of the ratio of the sound pressure required to achieve threshold in the patient to the sound pressure required to achieve threshold in a normal-hearing person. Therefore, a change of 6 dB represents doubling of sound pressure, and a change of 20 dB represents a tenfold change in sound pressure. Loudness, which depends on the frequency, intensity, and duration of a sound, doubles with approximately each 10-dB increase in sound pressure level. Pitch, on the other hand, does not directly correlate with frequency. The perception of pitch changes slowly in the low and high frequencies. In the middle tones, which are important for human speech, pitch varies more rapidly with changes in frequency.
Pure tone audiometry establishes the presence and severity of hearing impairment, unilateral versus bilateral involvement, and the type of hearing loss. Conductive hearing losses with a large mass component, as is often seen in middle ear effusions, produce elevation of thresholds that predominate in the higher frequencies. Conductive hearing losses with a large stiffness component, as in fixation of the footplate of the stapes in early otosclerosis, produce threshold elevations in the lower frequencies. Often, the conductive hearing loss involves all frequencies, suggesting involvement of both stiffness and mass. In general, sensorineural hearing losses such as presbycusis affect higher frequencies more than lower frequencies (Fig. 43-3). An exception is Ménière’s disease, which is characteristically associated with low-frequency sensorineural hearing loss. Noise-induced hearing loss has an unusual pattern of hearing impairment in which the loss at 4000 Hz is greater than at higher frequencies. Vestibular schwannomas characteristically affect the higher frequencies, but any pattern of hearing loss can be observed.
Speech recognition requires greater synchronous neural firing than is necessary for appreciation of pure tones. Speech audiometry tests the clarity with which one hears. The speech reception threshold (SRT) is defined as the intensity at which speech is recognized as a meaningful symbol and is obtained by presenting two-syllable words with an equal accent on each syllable. The intensity at which the patient can repeat 50% of the words correctly is the SRT. Once the SRT is determined, discrimination or word recognition ability is tested by presenting one-syllable words at 25–40 dB above the SRT. The words are phonetically balanced in that the phonemes (speech sounds) occur in the list of words at the same frequency that they occur in ordinary conversational English. An individual with normal hearing or conductive hearing loss can repeat 88–100% of the phonetically balanced words correctly. Patients with a sensorineural hearing loss have variable loss of discrimination. As a general rule, neural lesions produce greater deficits in discrimination than do cochlear lesions. For example, in a patient with mild asymmetric sensorineural hearing loss, a clue to the diagnosis of vestibular schwannoma is the presence of greater than expected deterioration in discrimination ability. Deterioration in discrimination ability at higher intensities above the SRT also suggests a lesion in the eighth nerve or central auditory pathways.
Tympanometry measures the impedance of the middle ear to sound and is useful in diagnosis of middle ear effusions. A tympanogram is the graphic representation of change in impedance or compliance as the pressure in the ear canal is changed. Normally, the middle ear is most compliant at atmospheric pressure, and the compliance decreases as the pressure is increased or decreased (type A); this pattern is seen with normal hearing or in the presence of sensorineural hearing loss. Compliance that does not change with change in pressure suggests middle ear effusion (type B). With a negative pressure in the middle ear, as with eustachian tube obstruction, the point of maximal compliance occurs with negative pressure in the ear canal (type C). A tympanogram in which no point of maximal compliance can be obtained is most commonly seen with discontinuity of the ossicular chain (type Ad). A reduction in the maximal compliance peak can be seen in otosclerosis (type As).
During tympanometry, an intense tone elicits contraction of the stapedius muscle. The change in compliance of the middle ear with contraction of the stapedius muscle can be detected. The presence or absence of this acoustic reflex is important in determining the etiology of hearing loss as well as in the anatomic localization of facial nerve paralysis. The acoustic reflex can help differentiate between conductive hearing loss due to otosclerosis and that caused by an inner ear “third window”: it is absent in otosclerosis and present in inner ear conductive hearing loss. Normal or elevated acoustic reflex thresholds in an individual with sensorineural hearing impairment suggest a cochlear hearing loss. An absent acoustic reflex in the setting of sensorineural hearing loss is not helpful in localizing the site of lesion. Assessment of acoustic reflex decay helps differentiate sensory from neural hearing losses. In neural hearing loss, such as with vestibular schwannoma, the reflex adapts or decays with time.
OAEs generated by outer hair cells only can be measured with microphones inserted into the external auditory canal. The emissions may be spontaneous or evoked with sound stimulation. The presence of OAEs indicates that the outer hair cells of the organ of Corti are intact and can be used to assess auditory thresholds and to distinguish sensory from neural hearing losses.
Evoked Responses Electrocochleography measures the earliest evoked potentials generated in the cochlea and the auditory nerve. Receptor potentials recorded include the cochlear microphonic, generated by the outer hair cells of the organ of Corti, and the summating potential, generated by the inner hair cells in response to sound. The whole nerve action potential representing the composite firing of the first-order neurons can also be recorded during electrocochleography. Clinically, the test is useful in the diagnosis of Ménière’s disease, where an elevation of the ratio of summating potential to action potential is seen.
Brainstem auditory evoked responses (BAERs), also known as auditory brainstem responses (ABRs), are useful in differentiating the site of sensorineural hearing loss. In response to sound, five distinct electrical potentials arising from different stations along the peripheral and central auditory pathway can be identified using computer averaging from scalp surface electrodes. BAERs are valuable in situations in which patients cannot or will not give reliable voluntary thresholds. They are also used to assess the integrity of the auditory nerve and brainstem in various clinical situations, including intraoperative monitoring, and in determination of brain death.
The vestibular-evoked myogenic potential (VEMP) test elicits a vestibulocolic reflex whose afferent limb arises from acoustically sensitive cells in the saccule, with signals conducted via the inferior vestibular nerve. VEMP is a biphasic, short-latency response recorded from the tonically contracted sternocleidomastoid muscle in response to loud auditory clicks or tones. VEMPs may be diminished or absent in patients with early and late Ménière’s disease, vestibular neuritis, benign paroxysmal positional vertigo, and vestibular schwannoma. On the other hand, the threshold for VEMPs may be lower in cases of superior canal dehiscence, other inner ear dehiscence, and perilymphatic fistula.
Imaging Studies The choice of radiologic tests is largely determined by whether the goal is to evaluate the bony anatomy of the external, middle, and inner ear or to image the auditory nerve and brain. Axial and coronal CT of the temporal bone with fine 0.3- to 0.6-mm cuts is ideal for determining the caliber of the external auditory canal, integrity of the ossicular chain, and presence of middle ear or mastoid disease; it can also detect inner ear malformations. CT is also ideal for the detection of bone erosion with chronic otitis media and cholesteatoma. Pöschl reformatting in the plane of the superior semicircular canal is required for the identification of dehiscence or absence of bone over the superior semicircular canal. MRI is superior to CT for imaging of retrocochlear pathology such as vestibular schwannoma, meningioma, other lesions of the cerebellopontine angle, demyelinating lesions of the brainstem, and brain tumors. Both CT and MRI are equally capable of identifying inner ear malformations and assessing cochlear patency for preoperative evaluation of patients for cochlear implantation.
|
TREATMENT |
DISORDERS OF THE SENSE OF HEARING |
In general, conductive hearing losses are amenable to surgical correction, whereas sensorineural hearing losses are usually managed medically. Atresia of the ear canal can be surgically repaired, often with significant improvement in hearing. Tympanic membrane perforations due to chronic otitis media or trauma can be repaired with an outpatient tympanoplasty. Likewise, conductive hearing loss associated with otosclerosis can be treated by stapedectomy, which is successful in >95% of cases. Tympanostomy tubes allow the prompt return of normal hearing in individuals with middle ear effusions. Hearing aids are effective and well tolerated in patients with conductive hearing losses.
Patients with mild, moderate, and severe sensorineural hearing losses are regularly rehabilitated with hearing aids of varying configuration and strength. Hearing aids have been improved to provide greater fidelity and have been miniaturized. The current generation of hearing aids can be placed entirely within the ear canal, thus reducing any stigma associated with their use. In general, the more severe the hearing impairment, the larger the hearing aid required for auditory rehabilitation. Digital hearing aids lend themselves to individual programming, and multiple and directional microphones at the ear level may be helpful in noisy surroundings. Because all hearing aids amplify noise as well as speech, the only absolute solution to the problem of noise is to place the microphone closer to the speaker than the noise source. This arrangement is not possible with a self-contained, cosmetically acceptable device. A significant limitation of rehabilitation with a hearing aid is that although it is able to enhance detection of sound with amplification, it cannot restore clarity of hearing that is lost with presbycusis.
Patients with unilateral deafness have difficulty with sound localization and reduced clarity of hearing in background noise. They may benefit from a CROS (contralateral routing of signal) hearing aid in which a microphone is placed on the hearing-impaired side and the sound is transmitted to the receiver placed on the contralateral ear. The same result may be obtained with a bone-anchored hearing aid (BAHA), in which a hearing aid clamps to a screw integrated into the skull on the hearing-impaired side. Like the CROS hearing aid, the BAHA transfers the acoustic signal to the contralateral hearing ear, but it does so by vibrating the skull. Patients with profound deafness on one side and some hearing loss in the better ear are candidates for a BICROS hearing aid; it differs from the CROS hearing aid in that the patient wears a hearing aid, and not simply a receiver, in the better ear. Unfortunately, while CROS and BAHA devices provide benefit, they do not restore hearing in the deaf ear. Only cochlear implants can restore hearing (see below). Increasingly, cochlear implants are being investigated for the treatment of patients with single-sided deafness; early reports show great promise in not only restoring hearing but also improving sound localization and performance in background noise.
In many situations, including lectures and the theater, hearing-impaired persons benefit from assistive devices that are based on the principle of having the speaker closer to the microphone than any source of noise. Assistive devices include infrared and frequency-modulated (FM) transmission as well as an electromagnetic loop around the room for transmission to the individual’s hearing aid. Hearing aids with telecoils can also be used with properly equipped telephones in the same way.
In the event that the hearing aid provides inadequate rehabilitation, cochlear implants may be appropriate (Fig. 43-4). Criteria for implantation include severe to profound hearing loss with open-set sentence cognition of ≤40% under best aided conditions. Worldwide, more than 300,000 hearing-impaired individuals have received cochlear implants. Cochlear implants are neural prostheses that convert sound energy to electrical energy and can be used to stimulate the auditory division of the eighth nerve directly. In most cases of profound hearing impairment, the auditory hair cells are lost but the ganglionic cells of the auditory division of the eighth nerve are preserved. Cochlear implants consist of electrodes that are inserted into the cochlea through the round window, speech processors that extract acoustical elements of speech for conversion to electrical currents, and a means of transmitting the electrical energy through the skin. Patients with implants experience sound that helps with speech reading, allows open-set word recognition, and helps in modulating the person’s own voice. Usually, within the first 3–6 months after implantation, adult patients can understand speech without visual cues. With the current generation of multichannel cochlear implants, nearly 75% of patients are able to converse on the telephone.
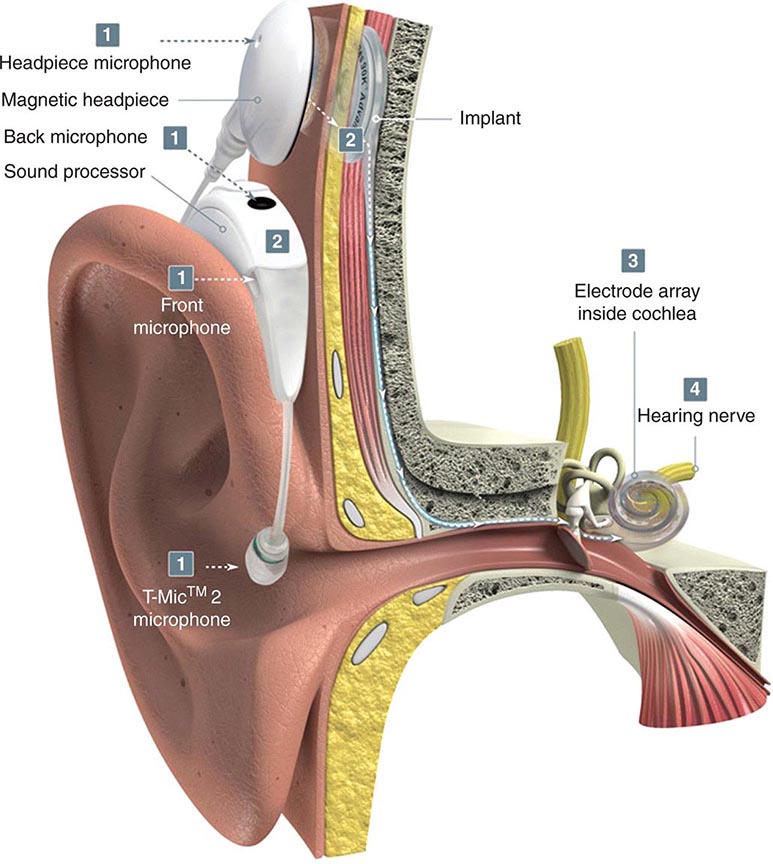
FIGURE 43-4 A cochlear implant is composed of an external microphone and speech processor worn on the ear and a receiver implanted underneath the temporalis muscle. The internal receiver is attached to an electrode that is placed surgically in the cochlea.
The U.S. Food and Drug Administration recently approved the first hybrid cochlear implant for the treatment of high-frequency hearing loss. Patients with presbyacusis typically have normal low-frequency hearing, while suffering from high-frequency hearing loss associated with loss of clarity that cannot always be adequately rehabilitated with a hearing aid. However, these patients are not candidates for conventional cochlear implants because they have too much residual hearing. The hybrid implant has been specifically designed for this patient population; it has a shorter electrode than a conventional cochlear implant and can be introduced into the cochlea atraumatically, thus preserving low-frequency hearing. Individuals with a hybrid implant use their own natural low-frequency “acoustic” hearing and rely on the implant for providing “electrical” high-frequency hearing. Patients who have received the hybrid implant perform better on speech testing in both quiet and noisy backgrounds.
For individuals who have had both eighth nerves destroyed by trauma or bilateral vestibular schwannomas (e.g., neurofibromatosis type 2), brainstem auditory implants placed near the cochlear nucleus may provide auditory rehabilitation.
Tinnitus often accompanies hearing loss. As for background noise, tinnitus can degrade speech comprehension in individuals with hearing impairment. Therapy for tinnitus is usually directed toward minimizing the appreciation of tinnitus. Relief of the tinnitus may be obtained by masking it with background music. Hearing aids are also helpful in tinnitus suppression, as are tinnitus maskers, devices that present a sound to the affected ear that is more pleasant to listen to than the tinnitus. The use of a tinnitus masker is often followed by several hours of inhibition of the tinnitus. Antidepressants have been shown to be beneficial in helping patients cope with tinnitus.
Hard-of-hearing individuals often benefit from a reduction in unnecessary noise in the environment (e.g., radio or television) to enhance the signal-to-noise ratio. Speech comprehension is aided by lip reading; therefore, the impaired listener should be seated so that the face of the speaker is well illuminated and easily seen. Although speech should be in a loud, clear voice, one should be aware that in sensorineural hearing losses in general and in hard-of-hearing elderly in particular, recruitment (abnormal perception of loud sounds) may be troublesome. Above all, optimal communication cannot take place without both parties giving it their full and undivided attention.
PREVENTION
Conductive hearing losses may be prevented by prompt antibiotic therapy of adequate duration for AOM and by ventilation of the middle ear with tympanostomy tubes in middle ear effusions lasting ≥12 weeks. Loss of vestibular function and deafness due to aminoglycoside antibiotics can largely be prevented by careful monitoring of serum peak and trough levels.
Some 10 million Americans have noise-induced hearing loss, and 20 million are exposed to hazardous noise in their employment. Noise-induced hearing loss can be prevented by avoidance of exposure to loud noise or by regular use of ear plugs or fluid-filled ear muffs to attenuate intense sound. Table 43-3 lists loudness levels for a variety of environmental sounds. High-risk activities for noise-induced hearing loss include use of electrical equipment for wood and metal working and target practice or hunting with small firearms. All internal-combustion and electric engines, including snow and leaf blowers, snowmobiles, outboard motors, and chainsaws, require protection of the user with hearing protectors. Virtually all noise-induced hearing loss is preventable through education, which should begin before the teenage years. Programs for conservation of hearing in the workplace are required by the Occupational Safety and Health Administration (OSHA) whenever the exposure over an 8-h period averages 85 dB. OSHA mandates that workers in such noisy environments have hearing monitoring and protection programs that include a preemployment screen, an annual audiologic assessment, and the mandatory use of hearing protectors. Exposure to loud sounds above 85 dB in the work environment is restricted by OSHA, with halving of allowed exposure time for each increment of 5 dB above this threshold; for example, exposure to 90 dB is permitted for 8 h; 95 dB for 4 h, and 100 dB for 2 h (Table 43-4).
|
DECIBEL (LOUDNESS) LEVEL OF COMMON ENVIRONMENTAL NOISE |
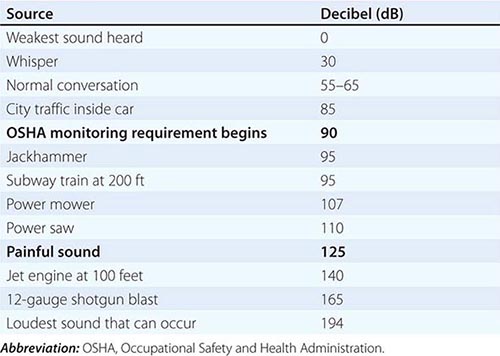
|
OSHA DAILY PERMISSIBLE NOISE LEVEL EXPOSURE |
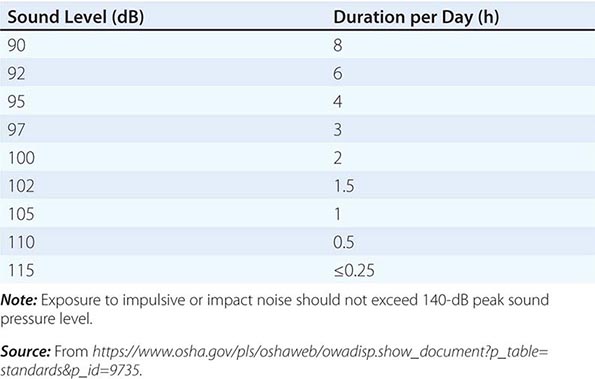
44 |
Sore Throat, Earache, and Upper Respiratory Symptoms |
Infections of the upper respiratory tract (URIs) have a tremendous impact on public health. They are among the most common reasons for visits to primary care providers, and although the illnesses are typically mild, their high incidence and transmission rates place them among the leading causes of time lost from work or school. Even though a minority (~25%) of cases are caused by bacteria, URIs are the leading diagnoses for which antibiotics are prescribed on an outpatient basis in the United States. The enormous consumption of antibiotics for these illnesses has contributed to the rise in antibiotic resistance among common community-acquired pathogens such as Streptococcus pneumoniae—a trend that in itself has an enormous influence on public health.
Although most URIs are caused by viruses, distinguishing patients with primary viral infection from those with primary bacterial infection is difficult. Signs and symptoms of bacterial and viral URIs are typically indistinguishable. Until consistent, inexpensive, and rapid testing becomes available and is used widely, acute infections will be diagnosed largely on clinical grounds. The judicious use and potential for misuse of antibiotics in this setting pose definite challenges.
NONSPECIFIC INFECTIONS OF THE UPPER RESPIRATORY TRACT
Nonspecific URIs are a broadly defined group of disorders that collectively constitute the leading cause of ambulatory care visits in the United States. By definition, nonspecific URIs have no prominent localizing features. They are identified by a variety of descriptive names, including acute infective rhinitis, acute rhinopharyngitis/nasopharyngitis, acute coryza, and acute nasal catarrh, as well as by the inclusive label common cold.
ETIOLOGY
The large assortment of URI classifications reflects the wide variety of causative infectious agents and the varied manifestations of common pathogens. Nearly all nonspecific URIs are caused by viruses spanning multiple virus families and many antigenic types. For instance, there are at least 100 immunotypes of rhinovirus (Chap. 223), the most common cause of URI (~30–40% of cases); other causes include influenza virus (three immunotypes; Chap. 224) as well as parainfluenza virus (four immunotypes), coronavirus (at least three immunotypes), and adenovirus (47 immunotypes) (Chap. 223). Respiratory syncytial virus (RSV), a well-established pathogen in pediatric populations, is also a recognized cause of significant disease in elderly and immunocompromised individuals. A host of additional viruses, including some viruses not typically associated with URIs (e.g., enteroviruses, rubella virus, and varicella-zoster virus), account for a small percentage of cases in adults each year. Although new diagnostic modalities (e.g., nasopharyngeal swab for polymerase chain reaction [PCR]) can assign a viral etiology, there are few specific treatment options, and no pathogen is identified in a substantial proportion of cases. A specific diagnostic workup beyond a clinical diagnosis is generally unnecessary in an otherwise healthy adult.
CLINICAL MANIFESTATIONS
The signs and symptoms of nonspecific URI are similar to those of other URIs but lack a pronounced localization to one particular anatomic location, such as the sinuses, pharynx, or lower airway. Nonspecific URI commonly presents as an acute, mild, and self-limited catarrhal syndrome with a median duration of ~1 week (range, 2–10 days). Signs and symptoms are diverse and frequently variable across patients, even when caused by the same virus. The principal signs and symptoms of nonspecific URI include rhinorrhea (with or without purulence), nasal congestion, cough, and sore throat. Other manifestations, such as fever, malaise, sneezing, lymphadenopathy, and hoarseness, are more variable, with fever more common among infants and young children. This varying presentation may reflect differences in host response as well as in infecting organisms; myalgias and fatigue, for example, sometimes are seen with influenza and parainfluenza infections, whereas conjunctivitis may suggest infection with adenovirus or enterovirus. Findings on physical examination are frequently nonspecific and unimpressive. Between 0.5% and 2% of colds are complicated by secondary bacterial infections (e.g., rhinosinusitis, otitis media, and pneumonia), particularly in higher-risk populations such as infants, elderly persons, and chronically ill or immunosuppressed individuals. Secondary bacterial infections usually are associated with a prolonged course of illness, increased severity of illness, and localization of signs and symptoms, often as a rebound after initial clinical improvement (the “double-dip” sign). Purulent secretions from the nares or throat often are misinterpreted as an indication of bacterial sinusitis or pharyngitis. These secretions, however, can be seen in nonspecific URI and, in the absence of other clinical features, are poor predictors of bacterial infection.
INFECTIONS OF THE SINUS
Rhinosinusitis refers to an inflammatory condition involving the nasal sinuses. Although most cases of sinusitis involve more than one sinus, the maxillary sinus is most commonly involved; next, in order of frequency, are the ethmoid, frontal, and sphenoid sinuses. Each sinus is lined with a respiratory epithelium that produces mucus, which is transported out by ciliary action through the sinus ostium and into the nasal cavity. Normally, mucus does not accumulate in the sinuses, which remain mostly sterile despite their adjacency to the bacterium-filled nasal passages. When the sinus ostia are obstructed or when ciliary clearance is impaired or absent, the secretions can be retained, producing the typical signs and symptoms of sinusitis. As these secretions accumulate with obstruction, they become more susceptible to infection with a variety of pathogens, including viruses, bacteria, and fungi. Sinusitis affects a tremendous proportion of the population, accounts for millions of visits to primary care physicians each year, and is the fifth leading diagnosis for which antibiotics are prescribed. It typically is classified by duration of illness (acute vs. chronic); by etiology (infectious vs. noninfectious); and, when infectious, by the offending pathogen type (viral, bacterial, or fungal).
ACUTE RHINOSINUSITIS
Acute rhinosinusitis—defined as sinusitis of <4 weeks’ duration—constitutes the vast majority of sinusitis cases. Most cases are diagnosed in the ambulatory care setting and occur primarily as a consequence of a preceding viral URI. Differentiating acute bacterial from viral sinusitis on clinical grounds is difficult. Therefore, it is perhaps not surprising that antibiotics are prescribed frequently (in 85–98% of all cases) for this condition.
Etiology The ostial obstruction in rhinosinusitis can arise from both infectious and noninfectious causes. Noninfectious etiologies include allergic rhinitis (with either mucosal edema or polyp obstruction), barotrauma (e.g., from deep-sea diving or air travel), and exposure to chemical irritants. Obstruction can also occur with nasal and sinus tumors (e.g., squamous cell carcinoma) or granulomatous diseases (e.g., granulomatosis with polyangiitis, rhinoscleroma), and conditions leading to altered mucus content (e.g., cystic fibrosis) can cause sinusitis through impaired mucus clearance. In ICUs, nasotracheal intubation and nasogastric tubes are major risk factors for nosocomial sinusitis.
Viral rhinosinusitis is far more common than bacterial sinusitis, although relatively few studies have sampled sinus aspirates for the presence of different viruses. In the studies that have done so, the viruses most commonly isolated—both alone and with bacteria—have been rhinovirus, parainfluenza virus, and influenza virus. Bacterial causes of sinusitis have been better described. Among community-acquired cases, S. pneumoniae and nontypable Haemophilus influenzae are the most common pathogens, accounting for 50–60% of cases. Moraxella catarrhalis causes disease in a significant percentage (20%) of children but a lesser percentage of adults. Other streptococcal species and Staphylococcus aureus cause only a small percentage of cases, although there is increasing concern about methicillin-resistant S. aureus (MRSA) as an emerging cause. It is difficult to assess whether a cultured bacterium represents a true infecting organism, an insufficiently deep sample (which would not be expected to be sterile), or—especially in the case of previous sinus surgeries—a colonizing organism. Anaerobes occasionally are found in association with infections of the roots of premolar teeth that spread to the adjacent maxillary sinuses. The role of atypical organisms like Chlamydia pneumoniae and Mycoplasma pneumoniae in the pathogenesis of acute sinusitis is unclear. Nosocomial cases commonly are associated with bacteria prevalent in the hospital environment, including S. aureus, Pseudomonas aeruginosa, Serratia marcescens, Klebsiella pneumoniae, and Enterobacter species. Often, these infections are polymicrobial and can involve organisms that are highly resistant to numerous antibiotics. Fungi also are established causes of sinusitis, although most acute cases are in immunocompromised patients and represent invasive, life-threatening infections. The best-known example is rhinocerebral mucormycosis caused by fungi of the order Mucorales, which includes Rhizopus, Rhizomucor, Mucor, Lichtheimia (formerly Mycocladus, formerly Absidia), and Cunninghamella (Chap. 242). These infections classically occur in diabetic patients with ketoacidosis but can also develop in transplant recipients, patients with hematologic malignancies, and patients receiving chronic glucocorticoid or deferoxamine therapy. Other hyaline molds, such as Aspergillus and Fusarium species, also are occasional causes of this disease.
Clinical Manifestations Most cases of acute sinusitis present after or in conjunction with a viral URI, and it can be difficult to discriminate the clinical features of one from the other, with timing becoming important in diagnosis (see below). A large proportion of patients with colds have sinus inflammation, although, as previously stated, true bacterial sinusitis complicates only 0.2–2% of these viral infections. Common presenting symptoms of sinusitis include nasal drainage and congestion, facial pain or pressure, and headache. Thick, purulent or discolored nasal discharge is often thought to indicate bacterial sinusitis but also occurs early in viral infections such as the common cold and is not specific to bacterial infection. Other nonspecific manifestations include cough, sneezing, and fever. Tooth pain, most often involving the upper molars, as well as halitosis are occasionally associated with bacterial sinusitis.
In acute sinusitis, sinus pain or pressure often localizes to the involved sinus (particularly the maxillary sinus) and can be worse when the patient bends over or is supine. Although rare, manifestations of advanced sphenoid or ethmoid sinus infection can be profound, including severe frontal or retroorbital pain radiating to the occiput, thrombosis of the cavernous sinus, and signs of orbital cellulitis. Acute focal sinusitis is uncommon but should be considered with severe symptoms involving the maxillary sinus and fever, regardless of illness duration. Similarly, patients with advanced frontal sinusitis can present with a condition known as Pott’s puffy tumor, with soft tissue swelling and pitting edema over the frontal bone from a communicating subperiosteal abscess. Life-threatening complications of sinusitis include meningitis, epidural abscess, and cerebral abscess.
Patients with acute fungal rhinosinusitis (such as mucormycosis; Chap. 242) often present with symptoms related to pressure effects, particularly when the infection has spread to the orbits and cavernous sinus. Signs such as orbital swelling and cellulitis, proptosis, ptosis, and decreased extraocular movement are common, as is retro- or periorbital pain. Nasopharyngeal ulcerations, epistaxis, and headaches are also common, and involvement of cranial nerves V and VII has been described in more advanced cases. Bony erosion may be evident on examination or endoscopy. Often the patient does not appear seriously ill despite the rapidly progressive nature of these infections.
Patients with acute nosocomial sinusitis are often critically ill and thus do not manifest the typical clinical features of sinus disease. This diagnosis should be suspected, however, when hospitalized patients with appropriate risk factors (e.g., nasotracheal intubation) develop fever without another apparent cause.
Diagnosis Distinguishing viral from bacterial rhinosinusitis in the ambulatory setting is usually difficult because of the relatively low sensitivity and specificity of the common clinical features. One clinical feature that has been used to help guide diagnostic and therapeutic decision-making is illness duration. Because acute bacterial sinusitis is uncommon in patients whose symptoms have lasted <10 days, expert panels now recommend reserving this diagnosis for patients with “persistent” symptoms (i.e., symptoms lasting >10 days in adults or >10–14 days in children) accompanied by the three cardinal signs of purulent nasal discharge, nasal obstruction, and facial pain (Table 44-1). Even among patients who meet these criteria, only 40–50% have true bacterial sinusitis. The use of CT or sinus radiography is not recommended for acute disease, particularly early in the course of illness (i.e., at <10 days) in light of the high prevalence of similar findings among patients with acute viral rhinosinusitis. In the evaluation of persistent, recurrent, or chronic sinusitis, CT of the sinuses becomes the radiographic study of choice.
|
GUIDELINES FOR THE DIAGNOSIS AND TREATMENT OF ACUTE SINUSITIS IN ADULTS |
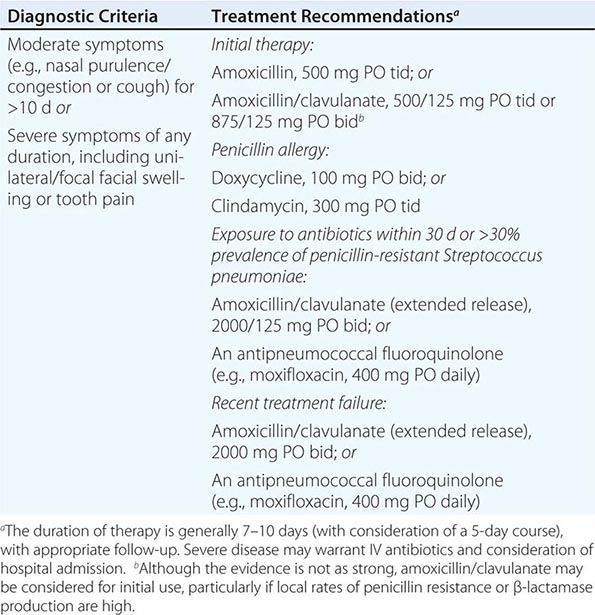
The clinical history and/or setting often can identify cases of acute anaerobic bacterial sinusitis, acute fungal sinusitis, or sinusitis from noninfectious causes (e.g., allergic rhinosinusitis). In the case of an immunocompromised patient with acute fungal sinus infection, immediate examination by an otolaryngologist is required. Biopsy specimens from involved areas should be examined by a pathologist for evidence of fungal hyphal elements and tissue invasion. Cases of suspected acute nosocomial sinusitis should be confirmed by sinus CT. Because therapy should target the offending organism, a sinus aspirate for culture and susceptibility testing should be obtained, whenever possible, before the initiation of antimicrobial therapy.
CHRONIC SINUSITIS
Chronic sinusitis is characterized by symptoms of sinus inflammation lasting >12 weeks. This illness is most commonly associated with either bacteria or fungi, and clinical cure in most cases is very difficult. Many patients have undergone treatment with repeated courses of antibacterial agents and multiple sinus surgeries, increasing their risk of colonization with antibiotic-resistant pathogens and of surgical complications. These patients often have high rates of morbidity, sometimes over many years.
In chronic bacterial sinusitis, infection is thought to be due to the impairment of mucociliary clearance from repeated infections rather than to persistent bacterial infection. The pathogenesis of this condition, however, is poorly understood. Although certain conditions (e.g., cystic fibrosis) can predispose patients to chronic bacterial sinusitis, most patients with chronic rhinosinusitis do not have obvious underlying conditions that result in the obstruction of sinus drainage, the impairment of ciliary action, or immune dysfunction. Patients experience constant nasal congestion and sinus pressure, with intermittent periods of greater severity, which may persist for years. CT can be helpful in determining the extent of disease, detecting an underlying anatomic defect or obstructing process (e.g., a polyp), and assessing the response to therapy. Management should involve an otolaryngologist to conduct endoscopic examinations and obtain tissue samples for histologic examination and culture. An endoscopy-derived culture not only has a higher yield but also allows direct visualization for abnormal anatomy.
Chronic fungal sinusitis is a disease of immunocompetent hosts and is usually noninvasive, although slowly progressive invasive disease is sometimes seen. Noninvasive disease, which typically is associated with hyaline molds such as Aspergillus species and dematiaceous molds such as Curvularia or Bipolaris species, can present as a number of different scenarios. In mild, indolent disease, which usually occurs in the setting of repeated failures of antibacterial therapy, only nonspecific mucosal changes may be seen on sinus CT. Although there is some controversy on this point, endoscopic surgery is usually curative in these cases, with no need for antifungal therapy. Another form of disease presents as long-standing, often unilateral symptoms and opacification of a single sinus on imaging studies as a result of a mycetoma (fungus ball) within the sinus. Treatment for this condition also is surgical, although systemic antifungal therapy may be warranted in the rare case in which bony erosion occurs. A third form of disease, known as allergic fungal sinusitis, is seen in patients with a history of nasal polyposis and asthma, who often have had multiple sinus surgeries. Patients with this condition produce a thick, eosinophil-laden mucus with the consistency of peanut butter that contains sparse fungal hyphae on histologic examination. These patients often present with pansinusitis.
INFECTIONS OF THE EAR AND MASTOID
Infections of the ear and associated structures can involve both the middle and the external ear, including the skin, cartilage, periosteum, ear canal, and tympanic and mastoid cavities. Both viruses and bacteria are known causes of these infections, some of which result in significant morbidity if not treated appropriately.
INFECTIONS OF EXTERNAL EAR STRUCTURES
Infections involving the structures of the external ear are often difficult to differentiate from noninfectious inflammatory conditions with similar clinical manifestations. Clinicians should consider inflammatory disorders as possible causes of external ear irritation, particularly in the absence of local or regional adenopathy. Aside from the more salient causes of inflammation, such as trauma, insect bite, and overexposure to sunlight or extreme cold, the differential diagnosis should include less common conditions such as autoimmune disorders (e.g., lupus or relapsing polychondritis) and vasculitides (e.g., granulomatosis with polyangiitis).
Auricular Cellulitis Auricular cellulitis is an infection of the skin overlying the external ear and typically follows minor local trauma. It presents as the typical signs and symptoms of cellulitis, with tenderness, erythema, swelling, and warmth of the external ear (particularly the lobule) but without apparent involvement of the ear canal or inner structures. Treatment consists of warm compresses and oral antibiotics such as cephalexin or dicloxacillin that are active against typical skin and soft tissue pathogens (specifically, S. aureus and streptococci). IV antibiotics such as a first-generation cephalosporin (e.g., cefazolin) or a penicillinase-resistant penicillin (e.g., nafcillin) occasionally are needed for more severe cases, with consideration of MRSA if either risk factors or failure of therapy point to this organism.
Perichondritis Perichondritis, an infection of the perichondrium of the auricular cartilage, typically follows local trauma (e.g., piercings, burns, or lacerations). Occasionally, when the infection spreads down to the cartilage of the pinna itself, patients may develop chondritis. The infection may closely resemble auricular cellulitis, with erythema, swelling, and extreme tenderness of the pinna, although the lobule is less often involved in perichondritis. The most common pathogens are P. aeruginosa and S. aureus, although other gram-negative and gram-positive organisms occasionally are involved. Treatment consists of systemic antibiotics active against both P. aeruginosa and S. aureus. An antipseudomonal penicillin (e.g., piperacillin) or a combination of a penicillinase-resistant penicillin and an antipseudomonal quinolone (e.g., nafcillin plus ciprofloxacin) is typically used. Incision and drainage may be helpful for culture and for resolution of infection, which often takes weeks. When perichondritis fails to respond to adequate antimicrobial therapy, clinicians should consider a noninfectious inflammatory etiology such as relapsing polychondritis.
Otitis Externa The term otitis externa refers to a collection of diseases involving primarily the auditory meatus. Otitis externa usually results from a combination of heat and retained moisture, with desquamation and maceration of the epithelium of the outer ear canal. The disease exists in several forms: localized, diffuse, chronic, and invasive. All forms are predominantly bacterial in origin, with P. aeruginosa and S. aureus the most common pathogens.
Acute localized otitis externa (furunculosis) can develop in the outer third of the ear canal, where skin overlies cartilage and hair follicles are numerous. As in furunculosis elsewhere on the body, S. aureus is the usual pathogen, and treatment typically consists of an oral antistaphylococcal penicillin (e.g., dicloxacillin or cephalexin), with incision and drainage in cases of abscess formation.
Acute diffuse otitis externa is also known as swimmer’s ear, although it can develop in patients who have not recently been swimming. Heat, humidity, and the loss of protective cerumen lead to excessive moisture and elevation of the pH in the ear canal, which in turn lead to skin maceration and irritation. Infection may then follow; the predominant pathogen is P. aeruginosa, although other gram-negative and gram-positive organisms—and rarely yeasts—have been recovered from patients with this condition. The illness often starts with itching and progresses to severe pain, which is usually elicited by manipulation of the pinna or tragus. The onset of pain is generally accompanied by the development of an erythematous, swollen ear canal, often with scant white, clumpy discharge. Treatment consists of cleansing the canal to remove debris and enhance the activity of topical therapeutic agents—usually hypertonic saline or mixtures of alcohol and acetic acid. Inflammation can also be decreased by adding glucocorticoids to the treatment regimen or by using Burow’s solution (aluminum acetate in water). Antibiotics are most effective when given topically. Otic mixtures provide adequate pathogen coverage; these preparations usually combine neomycin with polymyxin, with or without glucocorticoids. Systemic antimicrobial agents typically are reserved for severe disease or infections in immunocompromised hosts.
Chronic otitis externa is caused primarily by repeated local irritation, most commonly arising from persistent drainage from a chronic middle-ear infection. Other causes of repeated irritation, such as insertion of cotton swabs or other foreign objects into the ear canal, can lead to this condition, as can rare chronic infections such as syphilis, tuberculosis, and leprosy. Chronic otitis externa typically presents as erythematous, scaling dermatitis in which the predominant symptom is pruritus rather than pain; this condition must be differentiated from several others that produce a similar clinical picture, such as atopic dermatitis, seborrheic dermatitis, psoriasis, and dermatomycosis. Therapy consists of identifying and treating or removing the offending process, although successful resolution is frequently difficult.
Invasive otitis externa, also known as malignant or necrotizing otitis externa, is an aggressive and potentially life-threatening disease that occurs predominantly in elderly diabetic patients and other immunocompromised persons. The disease begins in the external canal as a soft tissue infection that progresses slowly over weeks to months and often is difficult to distinguish from a severe case of chronic otitis externa because of the presence of purulent otorrhea and an erythematous swollen ear and external canal. Severe, deep-seated otalgia, frequently out of proportion to findings on examination, is often noted and can help differentiate invasive from chronic otitis externa. The characteristic finding on examination is granulation tissue in the posteroinferior wall of the external canal, near the junction of bone and cartilage. If left unchecked, the infection can migrate to the base of the skull (resulting in skull-base osteomyelitis) and onward to the meninges and brain, with a high mortality rate. Cranial nerve involvement is seen occasionally, with the facial nerve usually affected first and most often. Thrombosis of the sigmoid sinus can occur if the infection extends to the area. CT, which can reveal osseous erosion of the temporal bone and skull base, can be used to help determine the extent of disease, as can gallium and technetium-99 scintigraphy studies. P. aeruginosa is by far the most common offender, although S. aureus, Staphylococcus epidermidis, Aspergillus, Actinomyces, and some gram-negative bacteria also have been associated with this disease. In all cases, the external ear canal should be cleansed and a biopsy specimen of the granulation tissue within the canal (or of deeper tissues) obtained for culture of the offending organism. IV antibiotic therapy should be given for a prolonged course (6–8 weeks) and directed specifically toward the recovered pathogen. For P. aeruginosa, the regimen typically includes an antipseudomonal penicillin or cephalosporin (e.g., piperacillin or cefepime), often with an aminoglycoside or a fluoroquinolone, the latter of which can even be administered orally given its excellent bioavailability. In addition, antibiotic drops containing an agent active against Pseudomonas (e.g., ciprofloxacin) are usually prescribed and are combined with glucocorticoids to reduce inflammation. Cases of invasive Pseudomonas otitis externa recognized in the early stages can sometimes be treated with oral and otic fluoroquinolones alone, albeit with close follow-up. Extensive surgical debridement, once an important component of the treatment approach, is now rarely indicated.
In necrotizing otitis externa, recurrence is documented up to 20% of the time. Aggressive glycemic control in diabetics is important not only for effective treatment but also for prevention of recurrence. The role of hyperbaric oxygen has not been clearly established.
INFECTIONS OF MIDDLE-EAR STRUCTURES
Otitis media is an inflammatory condition of the middle ear that results from dysfunction of the eustachian tube in association with a number of illnesses, including URIs and chronic rhinosinusitis. The inflammatory response in these conditions leads to the development of a sterile transudate within the middle ear and mastoid cavities. Infection may occur if bacteria or viruses from the nasopharynx contaminate this fluid, producing an acute (or sometimes chronic) illness.
Acute Otitis Media Acute otitis media results when pathogens from the nasopharynx are introduced into the inflammatory fluid collected in the middle ear (e.g., by nose blowing during a URI). Pathogenic proliferation in this space leads to the development of the typical signs and symptoms of acute middle-ear infection. The diagnosis of acute otitis media requires the demonstration of fluid in the middle ear (with tympanic membrane [TM] immobility) and the accompanying signs or symptoms of local or systemic illness (Table 44-2).
|
GUIDELINES FOR THE DIAGNOSIS AND TREATMENT OF ACUTE OTITIS MEDIA |
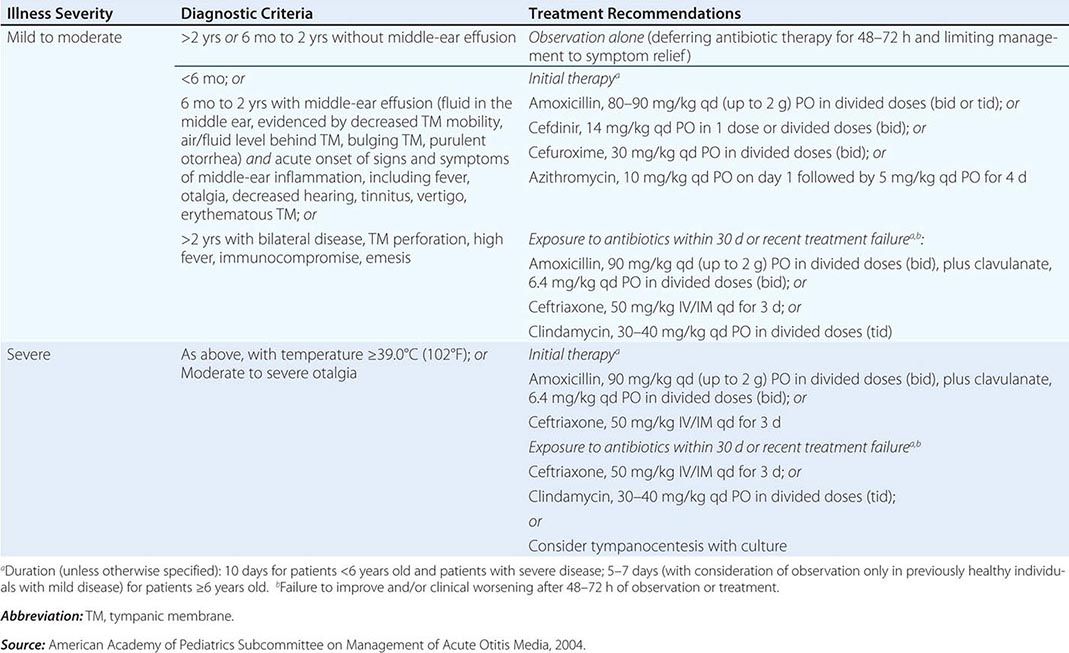
ETIOLOGY Acute otitis media typically follows a viral URI. The causative viruses (most commonly RSV, influenza virus, rhinovirus, and enterovirus) can themselves cause subsequent acute otitis media; more often, they predispose the patient to bacterial otitis media. Studies using tympanocentesis have consistently found S. pneumoniae to be the most important bacterial cause, isolated in up to 35% of cases. H. influenzae (nontypable strains) and M. catarrhalis also are common bacterial causes of acute otitis media, and concern is increasing with MRSA as an emerging etiologic agent. Viruses, such as those mentioned above, have been recovered either alone or with bacteria in 17–40% of cases.
CLINICAL MANIFESTATIONS Fluid in the middle ear is typically demonstrated or confirmed with pneumatic otoscopy. In the absence of fluid, the TM moves visibly with the application of positive and negative pressure, but this movement is dampened when fluid is present. With bacterial infection, the TM can also be erythematous, bulging, or retracted and occasionally can perforate spontaneously. The signs and symptoms accompanying infection can be local or systemic, including otalgia, otorrhea, diminished hearing, and fever. Erythema of the TM is often evident but is nonspecific as it frequently is seen in association with inflammation of the upper respiratory mucosa. Other signs and symptoms occasionally reported include vertigo, nystagmus, and tinnitus.
Recurrent Acute Otitis Media Recurrent acute otitis media (more than three episodes within 6 months or four episodes within 12 months) generally is due to relapse or reinfection, although data indicate that the majority of early recurrences are new infections. In general, the same pathogens responsible for acute otitis media cause recurrent disease; even so, the recommended treatment consists of antibiotics active against β-lactamase-producing organisms. Antibiotic prophylaxis (e.g., with trimethoprim-sulfamethoxazole [TMP-SMX] or amoxicillin) can reduce recurrences in patients with recurrent acute otitis media by an average of one episode per year, but this benefit is small compared with the high likelihood of colonization with antibiotic-resistant pathogens. Other approaches, including placement of tympanostomy tubes, adenoidectomy, and tonsillectomy plus adenoidectomy, are of questionable overall value in light of the relatively small benefit compared with the potential for complications.
Serous Otitis Media In serous otitis media (otitis media with effusion), fluid is present in the middle ear for an extended period in the absence of signs and symptoms of infection. In general, acute effusions are self-limited; most resolve in 2–4 weeks. In some cases, however (in particular after an episode of acute otitis media), effusions can persist for months. These chronic effusions are often associated with significant hearing loss in the affected ear. The great majority of cases of otitis media with effusion resolve spontaneously within 3 months without antibiotic therapy. Antibiotic therapy or myringotomy with insertion of tympanostomy tubes typically is reserved for patients in whom bilateral effusion (1) has persisted for at least 3 months and (2) is associated with significant bilateral hearing loss. With this conservative approach and the application of strict diagnostic criteria for acute otitis media and otitis media with effusion, it is estimated that 6–8 million courses of antibiotics could be avoided each year in the United States.
Chronic Otitis Media Chronic suppurative otitis media is characterized by persistent or recurrent purulent otorrhea in the setting of TM perforation. Usually, there is also some degree of conductive hearing loss. This condition can be categorized as active or inactive. Inactive disease is characterized by a central perforation of the TM, which allows drainage of purulent fluid from the middle ear. When the perforation is more peripheral, squamous epithelium from the auditory canal may invade the middle ear through the perforation, forming a mass of keratinaceous debris (cholesteatoma) at the site of invasion. This mass can enlarge and has the potential to erode bone and promote further infection, which can lead to meningitis, brain abscess, or paralysis of cranial nerve VII. Treatment of chronic active otitis media is surgical; mastoidectomy, myringoplasty, and tympanoplasty can be performed as outpatient surgical procedures, with an overall success rate of ~80%. Chronic inactive otitis media is more difficult to cure, usually requiring repeated courses of topical antibiotic drops during periods of drainage. Systemic antibiotics may offer better cure rates, but their role in the treatment of this condition remains unclear.
Mastoiditis Acute mastoiditis was relatively common among children before the introduction of antibiotics. Because the mastoid air cells connect with the middle ear, the process of fluid collection and infection is usually the same in the mastoid as in the middle ear. Early and frequent treatment of acute otitis media is most likely the reason that the incidence of acute mastoiditis has declined to only 1.2–2.0 cases per 100,000 person-years in countries with high prescribing rates for acute otitis media.
![]() In countries such as the Netherlands, where antibiotics are used sparingly for acute otitis media, the incidence rate of acute mastoiditis is roughly twice that in countries like the United States. However, neighboring Denmark has a rate of acute mastoiditis similar to that in the Netherlands but an antibiotic-prescribing rate for acute otitis media more similar to that in the United States.
In countries such as the Netherlands, where antibiotics are used sparingly for acute otitis media, the incidence rate of acute mastoiditis is roughly twice that in countries like the United States. However, neighboring Denmark has a rate of acute mastoiditis similar to that in the Netherlands but an antibiotic-prescribing rate for acute otitis media more similar to that in the United States.
In typical acute mastoiditis, purulent exudate collects in the mastoid air cells (Fig. 44-1), producing pressure that may result in erosion of the surrounding bone and formation of abscess-like cavities that are usually evident on CT. Patients typically present with pain, erythema, and swelling of the mastoid process along with displacement of the pinna, usually in conjunction with the typical signs and symptoms of acute middle-ear infection. Rarely, patients can develop severe complications if the infection tracks under the periosteum of the temporal bone to cause a subperiosteal abscess, erodes through the mastoid tip to cause a deep neck abscess, or extends posteriorly to cause septic thrombosis of the lateral sinus.
FIGURE 44-1 Acute mastoiditis. Axial CT image shows an acute fluid collection within the mastoid air cells on the left.
Purulent fluid should be cultured whenever possible to help guide antimicrobial therapy. Initial empirical therapy usually is directed against the typical organisms associated with acute otitis media, such as S. pneumoniae, H. influenzae, and M. catarrhalis. Patients with more severe or prolonged courses of illness should be treated for infection with S. aureus and gram-negative bacilli (including Pseudomonas). Broad empirical therapy should be narrowed once culture results become available. Most patients can be treated conservatively with IV antibiotics; surgery (cortical mastoidectomy) is reserved for complicated cases and those in which conservative treatment has failed.
INFECTIONS OF THE PHARYNX AND ORAL CAVITY
Oropharyngeal infections range from mild, self-limited viral illnesses to serious, life-threatening bacterial infections. The most common presenting symptom is sore throat—one of the most common reasons for ambulatory care visits by both adults and children. Although sore throat is a symptom in many noninfectious illnesses as well, the overwhelming majority of patients with a new sore throat have acute pharyngitis of viral or bacterial etiology.
ACUTE PHARYNGITIS
Millions of visits to primary care providers each year are for sore throat; the majority of cases of acute pharyngitis are caused by typical respiratory viruses. The most important source of concern is infection with group A β-hemolytic Streptococcus (S. pyogenes) that is associated with acute glomerulonephritis and acute rheumatic fever. The risk of rheumatic fever can be reduced by timely penicillin therapy.
Etiology A wide variety of organisms cause acute pharyngitis. The relative importance of the different pathogens can only be estimated, since a significant proportion of cases (~30%) have no identified cause. Together, respiratory viruses are the most common identifiable cause of acute pharyngitis, with rhinoviruses and coronaviruses accounting for large proportions of cases (~20% and at least 5%, respectively). Influenza virus, parainfluenza virus, and adenovirus also account for a measurable share of cases, with the former two more seasonal and the latter as part of the more clinically severe syndrome of pharyngoconjunctival fever. Other important but less common viral causes include herpes simplex virus (HSV) types 1 and 2, coxsackievirus A, cytomegalovirus (CMV), and Epstein-Barr virus (EBV). Acute HIV infection can present as acute pharyngitis and should be considered in at-risk populations.
Acute bacterial pharyngitis is typically caused by S. pyogenes, which accounts for ~5–15% of all cases of acute pharyngitis in adults; rates vary with the season and with utilization of the health care system. Group A streptococcal pharyngitis is primarily a disease of children 5–15 years of age; it is uncommon among children <3 years old, as is rheumatic fever. Streptococci of groups C and G account for a minority of cases, although these serogroups are nonrheumatogenic. Fusobacterium necrophorum has been increasingly recognized as a cause of pharyngitis in adolescents and young adults and is isolated nearly as often as group A streptococci. This organism is important because of the rare but life-threatening Lemierre’s disease, which is generally associated with F. necrophorum and is usually preceded by pharyngitis (see “Oral Infections,” below). The remaining bacterial causes of acute pharyngitis are seen infrequently (<1% of cases each) but should be considered in appropriate exposure groups because of the severity of illness if left untreated; these etiologic agents include Neisseria gonorrhoeae, Corynebacterium diphtheriae, Corynebacterium ulcerans, Yersinia enterocolitica, and Treponema pallidum (in secondary syphilis). Anaerobic bacteria also can cause acute pharyngitis (Vincent’s angina) and can contribute to more serious polymicrobial infections, such as peritonsillar or retropharyngeal abscesses (see below). Atypical organisms such as M. pneumoniae and C. pneumoniae have been recovered from patients with acute pharyngitis; whether these agents are commensals or causes of acute infection is debatable.
Clinical Manifestations Although the signs and symptoms accompanying acute pharyngitis are not reliable predictors of the etiologic agent, the clinical presentation occasionally suggests one etiology over another. Acute pharyngitis due to respiratory viruses such as rhinovirus or coronavirus usually is not severe and typically is associated with a constellation of coryzal symptoms better characterized as nonspecific URI. Findings on physical examination are uncommon; fever is rare, and tender cervical adenopathy and pharyngeal exudates are not seen. In contrast, acute pharyngitis from influenza virus can be severe and is much more likely to be associated with fever as well as with myalgias, headache, and cough. The presentation of pharyngoconjunctival fever due to adenovirus infection is similar. Since pharyngeal exudate may be present on examination, this condition can be difficult to differentiate from streptococcal pharyngitis. However, adenoviral pharyngitis is distinguished by the presence of conjunctivitis in one-third to one-half of patients. Acute pharyngitis from primary HSV infection can also mimic streptococcal pharyngitis in some cases, with pharyngeal inflammation and exudate, but the presence of vesicles and shallow ulcers on the palate can help differentiate the two diseases. This HSV syndrome is distinct from pharyngitis caused by coxsackievirus (herpangina), which is associated with small vesicles that develop on the soft palate and uvula and then rupture to form shallow white ulcers. Acute exudative pharyngitis coupled with fever, fatigue, generalized lymphadenopathy, and (on occasion) splenomegaly is characteristic of infectious mononucleosis due to EBV or CMV. Acute primary infection with HIV is frequently associated with fever and acute pharyngitis as well as with myalgias, arthralgias, malaise, and occasionally a nonpruritic maculopapular rash, which may be followed by lymphadenopathy and mucosal ulcerations without exudate.
The clinical features of acute pharyngitis caused by streptococci of groups A, C, and G are similar, ranging from a relatively mild illness without many accompanying symptoms to clinically severe cases with profound pharyngeal pain, fever, chills, and abdominal pain. A hyperemic pharyngeal membrane with tonsillar hypertrophy and exudate is usually seen, along with tender anterior cervical adenopathy. Coryzal manifestations, including cough, are typically absent; when present, they suggest a viral etiology. Strains of S. pyogenes that generate erythrogenic toxin can also produce scarlet fever characterized by an erythematous rash and strawberry tongue. The other types of acute bacterial pharyngitis (e.g., gonococcal, diphtherial, and yersinial) often present as exudative pharyngitis with or without other clinical features. Their etiologies are often suggested only by the clinical history.
Diagnosis The primary goal of diagnostic testing is to separate acute streptococcal pharyngitis from pharyngitis of other etiologies (particularly viral) so that antibiotics can be prescribed more efficiently for patients in whom they may be beneficial. The most appropriate standard for the diagnosis of streptococcal pharyngitis, however, has not been established definitively. Throat swab culture is generally regarded as the most appropriate but cannot distinguish between infection and colonization and requires 24–48 h to yield results that vary with technique and culture conditions. Rapid antigen-detection tests offer good specificity (>90%) but lower sensitivity when implemented in routine practice. The sensitivity has also been shown to vary across the clinical spectrum of disease (65–90%). Several clinical prediction systems (Fig. 44-2) can increase the sensitivity of rapid antigen-detection tests to >90% in controlled settings. Since the sensitivities achieved in routine clinical practice are often lower, several medical and professional societies continue to recommend that all negative rapid antigen-detection tests in children be confirmed by a throat culture to limit transmission and complications of illness caused by group A streptococci. The Centers for Disease Control and Prevention, the Infectious Diseases Society of America, and the American Academy of Family Physicians do not recommend backup culture when adults have negative results from a highly sensitive rapid antigen-detection test, however, because of the lower prevalence and smaller benefit in this age group.
FIGURE 44-2 Algorithm for the diagnosis and treatment of acute pharyngitis.
Cultures and rapid diagnostic tests for other causes of acute pharyngitis, such as influenza virus, adenovirus, HSV, EBV, CMV, and M. pneumoniae, are available in many locations and can be used when suspected. The diagnosis of acute EBV infection depends primarily on the detection of antibodies to the virus with a heterophile agglutination assay (monospot slide test) or enzyme-linked immunosorbent assay. Testing for HIV RNA or antigen (p24) should be performed when acute primary HIV infection is suspected. If other bacterial causes are suspected (particularly N. gonorrhoeae, C. diphtheriae, or Y. enterocolitica), specific cultures should be requested since these organisms may be missed on routine throat swab culture.
TREATMENTPHARYNGITIS
Antibiotic treatment of pharyngitis due to S. pyogenes confers numerous benefits, including a decrease in the risk of rheumatic fever, the primary focus of treatment. The magnitude of this benefit is fairly small, since rheumatic fever is now a rare disease, even among untreated patients. Nevertheless, when therapy is started within 48 h of illness onset, symptom duration is decreased modestly. An additional benefit of therapy is the potential to reduce the transmission of streptococcal pharyngitis, particularly in areas of overcrowding or close contact. Antibiotic therapy for acute pharyngitis is therefore recommended in cases in which S. pyogenes is confirmed as the etiologic agent by rapid antigen-detection test or throat swab culture. Otherwise, antibiotics should be given in routine cases only when another bacterial cause has been identified. Effective therapy for streptococcal pharyngitis consists of either a single dose of IM benzathine penicillin or a full 10-day course of oral penicillin (Fig. 44-2).
![]() Azithromycin can be used in place of penicillin, although resistance to azithromycin among S. pyogenes strains in some parts of the world (particularly Europe) can prohibit the use of this drug. Newer (and more expensive) antibiotics also are active against streptococci but offer no greater efficacy than the agents mentioned above. Testing for cure is unnecessary and may reveal only chronic colonization. There is no evidence to support antibiotic treatment of group C or G streptococcal pharyngitis or pharyngitis in which mycoplasmas or chlamydiae have been recovered. Cultures can be of benefit because F. necrophorum, an increasingly common cause of bacterial pharyngitis in young adults, is not covered by macrolide therapy. Long-term penicillin prophylaxis (benzathine penicillin G, 1.2 million units IM every 3–4 weeks; or penicillin VK, 250 mg PO bid) is indicated for patients at risk of recurrent rheumatic fever.
Azithromycin can be used in place of penicillin, although resistance to azithromycin among S. pyogenes strains in some parts of the world (particularly Europe) can prohibit the use of this drug. Newer (and more expensive) antibiotics also are active against streptococci but offer no greater efficacy than the agents mentioned above. Testing for cure is unnecessary and may reveal only chronic colonization. There is no evidence to support antibiotic treatment of group C or G streptococcal pharyngitis or pharyngitis in which mycoplasmas or chlamydiae have been recovered. Cultures can be of benefit because F. necrophorum, an increasingly common cause of bacterial pharyngitis in young adults, is not covered by macrolide therapy. Long-term penicillin prophylaxis (benzathine penicillin G, 1.2 million units IM every 3–4 weeks; or penicillin VK, 250 mg PO bid) is indicated for patients at risk of recurrent rheumatic fever.
Treatment of viral pharyngitis is entirely symptom based except in infection with influenza virus or HSV. For influenza, the armamentarium includes the adamantanes amantadine and rimantadine and the neuraminidase inhibitors oseltamivir and zanamivir. Administration of all these agents needs to be started within 48 h of symptom onset to reduce illness duration meaningfully. Among these agents, only oseltamivir and zanamivir are active against both influenza A and influenza B and therefore can be used when local patterns of infection and antiviral resistance are unknown. Oropharyngeal HSV infection sometimes responds to treatment with antiviral agents such as acyclovir, although these drugs are often reserved for immunosuppressed patients.
Complications Although rheumatic fever is the best-known complication of acute streptococcal pharyngitis, the risk of its following acute infection remains quite low. Other complications include acute glomerulonephritis and numerous suppurative conditions, such as peritonsillar abscess (quinsy), otitis media, mastoiditis, sinusitis, bacteremia, and pneumonia—all of which occur at low rates. Although antibiotic treatment of acute streptococcal pharyngitis can prevent the development of rheumatic fever, there is no evidence that it can prevent acute glomerulonephritis. Some evidence supports antibiotic use to prevent the suppurative complications of streptococcal pharyngitis, particularly peritonsillar abscess, which can also involve oral anaerobes such as Fusobacterium. Abscesses usually are accompanied by severe pharyngeal pain, dysphagia, fever, and dehydration; in addition, medial displacement of the tonsil and lateral displacement of the uvula are often evident on examination. Although early use of IV antibiotics (e.g., clindamycin, penicillin G with metronidazole) may obviate the need for surgical drainage in some cases, treatment typically involves needle aspiration or incision and drainage.
ORAL INFECTIONS
Aside from periodontal disease such as gingivitis, infections of the oral cavity most commonly involve HSV or Candida species. In addition to causing painful cold sores on the lips, HSV can infect the tongue and buccal mucosa, causing the formation of irritating vesicles. Although topical antiviral agents (e.g., acyclovir and penciclovir) can be used externally for cold sores, oral or IV acyclovir is often needed for primary infections, extensive oral infections, and infections in immunocompromised patients. Oropharyngeal candidiasis (thrush) is caused by a variety of Candida species, most often C. albicans. Thrush occurs predominantly in neonates, immunocompromised patients (especially those with AIDS), and recipients of prolonged antibiotic or glucocorticoid therapy. In addition to sore throat, patients often report a burning tongue, and physical examination reveals friable white or gray plaques on the gingiva, tongue, and oral mucosa. Treatment, which usually consists of an oral antifungal suspension (nystatin or clotrimazole) or oral fluconazole, is typically successful. In the uncommon cases of fluconazole-refractory thrush that are seen in some patients with HIV/AIDS, other therapeutic options include oral formulations of itraconazole or voriconazole as well as an IV echinocandin (caspofungin, micafungin, or anidulafungin) or amphotericin B deoxycholate, if needed. In these cases, therapy based on culture and susceptibility test results is ideal.
Vincent’s angina, also known as acute necrotizing ulcerative gingivitis or trench mouth, is a unique and dramatic form of gingivitis characterized by painful, inflamed gingiva with ulcerations of the interdental papillae that bleed easily. Since oral anaerobes are the cause, patients typically have halitosis and frequently present with fever, malaise, and lymphadenopathy. Treatment consists of debridement and oral administration of penicillin plus metronidazole, with clindamycin or doxycycline alone as an alternative.
Ludwig’s angina is a rapidly progressive, potentially fulminant form of cellulitis that involves the bilateral sublingual and submandibular spaces and that typically originates from an infected or recently extracted tooth, most commonly the lower second and third molars. Improved dental care has reduced the incidence of this disorder substantially. Infection in these areas leads to dysphagia, odynophagia, and “woody” edema in the sublingual region, forcing the tongue up and back with the potential for airway obstruction. Fever, dysarthria, and drooling also may be noted, and patients may speak in a “hot potato” voice. Intubation or tracheostomy may be necessary to secure the airway, as asphyxiation is the most common cause of death. Patients should be monitored closely and treated promptly with IV antibiotics directed against streptococci and oral anaerobes. Recommended agents include ampicillin/sulbactam, clindamycin, or high-dose penicillin plus metronidazole.
Postanginal septicemia (Lemierre’s disease) is a rare anaerobic oropharyngeal infection caused predominantly by F. necrophorum. The illness typically starts as a sore throat (most commonly in adolescents and young adults), which may present as exudative tonsillitis or peritonsillar abscess. Infection of the deep pharyngeal tissue allows organisms to drain into the lateral pharyngeal space, which contains the carotid artery and internal jugular vein. Septic thrombophlebitis of the internal jugular vein can result, with associated pain, dysphagia, and neck swelling and stiffness. Sepsis usually occurs 3–10 days after the onset of sore throat and is often coupled with metastatic infection to the lung and other distant sites. Occasionally, the infection can extend along the carotid sheath and into the posterior mediastinum, resulting in mediastinitis, or it can erode into the carotid artery, with the early sign of repeated small bleeds into the mouth. The mortality rate from these invasive infections can be as high as 50%. Treatment consists of IV antibiotics (clindamycin or ampicillin/sulbactam) and surgical drainage of any purulent collections. The concomitant use of anticoagulants to prevent embolization remains controversial and is sometimes advised, with careful consideration of both the risks and the benefits.
INFECTIONS OF THE LARYNX AND EPIGLOTTIS
LARYNGITIS
Laryngitis is defined as any inflammatory process involving the larynx and can be caused by a variety of infectious and noninfectious processes. The vast majority of laryngitis cases seen in clinical practice in developed countries are acute. Acute laryngitis is a common syndrome caused predominantly by the same viruses responsible for many other URIs. In fact, most cases of acute laryngitis occur in the setting of a viral URI.
Etiology Nearly all major respiratory viruses have been implicated in acute viral laryngitis, including rhinovirus, influenza virus, parainfluenza virus, adenovirus, coxsackievirus, coronavirus, and RSV. Acute laryngitis can also be associated with acute bacterial respiratory infections such as those caused by group A Streptococcus or C. diphtheriae (although diphtheria has been virtually eliminated in the United States). Another bacterial pathogen thought to play a role (albeit unclear) in the pathogenesis of acute laryngitis is M. catarrhalis, which has been recovered from nasopharyngeal culture in a significant percentage of cases.
![]() Chronic laryngitis of infectious etiology is much less common in developed than in developing countries. Laryngitis due to Mycobacterium tuberculosis is often difficult to distinguish from laryngeal cancer, in part because of the frequent absence of signs, symptoms, and radiographic findings typical of pulmonary disease. Histoplasma and Blastomyces may cause laryngitis, often as a complication of systemic infection. Candida species can cause laryngitis as well, often in association with thrush or esophagitis and particularly in immunosuppressed patients. Rare cases of chronic laryngitis are due to Coccidioides and Cryptococcus.
Chronic laryngitis of infectious etiology is much less common in developed than in developing countries. Laryngitis due to Mycobacterium tuberculosis is often difficult to distinguish from laryngeal cancer, in part because of the frequent absence of signs, symptoms, and radiographic findings typical of pulmonary disease. Histoplasma and Blastomyces may cause laryngitis, often as a complication of systemic infection. Candida species can cause laryngitis as well, often in association with thrush or esophagitis and particularly in immunosuppressed patients. Rare cases of chronic laryngitis are due to Coccidioides and Cryptococcus.
Clinical Manifestations Laryngitis is characterized by hoarseness and also can be associated with reduced vocal pitch or aphonia. As acute laryngitis is caused predominantly by respiratory viruses, these symptoms usually occur in association with other symptoms and signs of URI, including rhinorrhea, nasal congestion, cough, and sore throat. Direct laryngoscopy often reveals diffuse laryngeal erythema and edema, along with vascular engorgement of the vocal folds. In addition, chronic disease (e.g., tuberculous laryngitis) often includes mucosal nodules and ulcerations visible on laryngoscopy; these lesions are sometimes mistaken for laryngeal cancer.
CROUP
The term croup actually denotes a group of diseases collectively referred to as “croup syndrome,” all of which are acute and predominantly viral respiratory illnesses characterized by marked swelling of the subglottic region of the larynx. Croup primarily affects children <6 years old. For a detailed discussion of this entity, the reader should consult a textbook of pediatric medicine.