Ventricular Tachycardia in Patients With Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM) is a disorder characterized by left or biventricular dilatation and impaired systolic function (Figure 85-1), frequently resulting in congestive heart failure (CHF). Valvular heart disease, excess ethanol ingestion, hypertension, pregnancy, and infections are common underlying etiologic factors. In idiopathic dilated cardiomyopathy, by definition an etiology has not been determined, although genetic, autoimmune, viral, and metabolic causes have been implicated. A common feature of DCM regardless of the underlying cause is a propensity to ventricular arrhythmias and sudden death. This chapter focuses on incidence and survival, mechanisms of ventricular arrhythmogenesis, predictors of sudden death, and management of ventricular arrhythmias in DCM. For the purpose of this chapter, DCM refers to idiopathic dilated cardiomyopathy, particularly excluding cardiomyopathies related to coronary artery disease, unless otherwise stated.
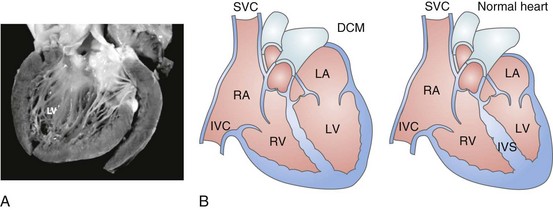
Figure 85-1 Dilated cardiomyopathy. IVC, Inferior vena cava; IVS, intraventricular septum; LA, left atria; LV, left ventricle; RA, right atria; RV, right ventricle; SVC, superior vena cava.
Incidence and Survival
Congestive heart failure afflicts approximately 5.8 million individuals in the United States.1 The incidence rises steadily with age; there is a prevalence in men that increases from 8 per 1000 at 50 to 59 years old, to 66 per 1000 at 80 to 89 years old. CHF is anticipated to be an even larger societal burden as the population ages.2 Mortality rates after a diagnosis of CHF have steadily improved since the 1980s with the widespread use of neurohormonal medical therapy, such as angiotensin-converting enzyme (ACE) inhibitors and β-blockers, and with interventions such as implantable cardioverter defibrillators (ICDs) and biventricular (BiV) pacing. However, the overall mortality from CHF remains high in the face of the rising incidence of disease with an overall adjusted survival inclusive of both ischemic and nonischemic cardiomyopathy of 48% at 5 years.3 Survival rates in DCM are principally determined by the severity of symptoms and the degree of left ventricular (LV) dysfunction in the cohort being examined.4 It is difficult to estimate the survival in dilated cardiomyopathy alone, because establishing the etiology of heart failure from population-based studies is difficult and varies widely depending on diagnostic criteria, case ascertainment, and geographic location. However, DCM accounts for a substantial proportion of the population of patients with heart failure, with only 24% of women and 32% of men having any documented coronary disease among a cohort of patients with heart failure in Olmsted County, Minnesota.3 After accounting for other contributors such as valvular, ischemic, and hypertensive heart disease, another large cohort estimated DCM to account for 32% of cases.5 After accounting for comorbidities, the prognosis of DCM compared with ischemic cardiomyopathy appears to be slightly improved.
Genetics
Approximately 40% of patients with DCM have a familial contribution to their disease, most of which appears to be inherited in an autosomal dominant fashion; however, X-linked, autosomal recessive, and mitochondrial inheritance are also seen. Advances have been made in the last 20 years in the identification of candidate genes responsible for DCM (Figure 85-2). Mutations in more than 40 candidate genes have been reported in patients with DCM, and it is reported that up to 40% of patients with DCM may have inherited this disorder.6 The more common autosomal dominant form of familial DCM also has two main forms: “pure” DCM and DCM associated with cardiac conduction system disease. Mitochondrial inheritance is seen most often in childhood forms of familial DCM, whereas X-linked and autosomal recessive forms seem to be distributed evenly between childhood and adult forms of disease. In the case of X-linked forms of DCM, two disorders have been well characterized: X-linked cardiomyopathy, which appears in adolescents and young adults, and Barth syndrome, which is most frequently identified in infancy.
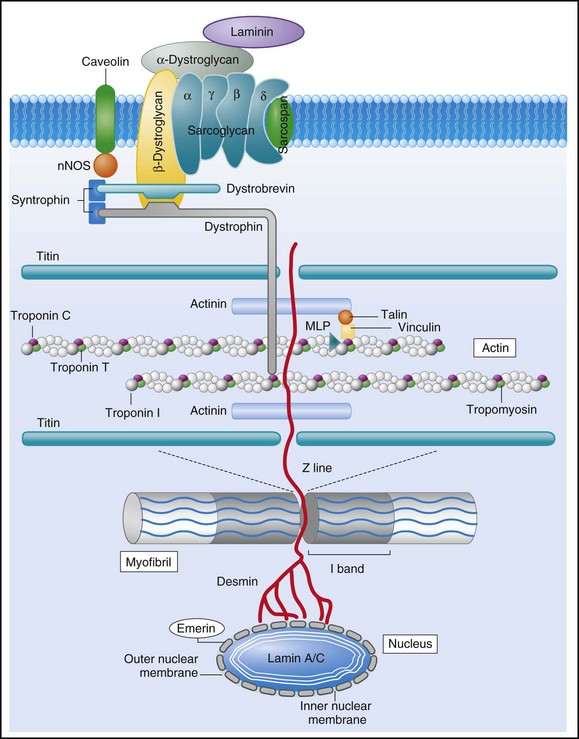
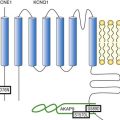
Figure 85-2 Schematic of proteins implicated in the pathogenesis of dilated cardiomyopathy. Identified genes that result in dilated cardiomyopathy include the cytoskeletal protein-encoding genes b- and d-sarcoglycan in the sarcolemma, dystrophin, and the intermediate filament protein-encoding genes desmin and laminin A/C. Sarcomeric protein-encoding genes actin, b-myosin heavy chain, a-tropomyosin, and cardiac troponin T cause either dilated cardiomyopathy or hypertrophic cardiomyopathy, whereas cardiac troponin I, titin, and the myosin light chains cause hypertrophic cardiomyopathy. Mutations in dystrobrevin cause the intermediate phenotype, left ventricular noncompaction. MLP, Muscle LIM protein; nNOS, neuronal nitric oxide.
In the case of pure DCM, linkage analysis has identified a number of candidate genes in which mutations have subsequently been found, including those that code for actin (chromosome 15q14),7 desmin (2q35),8 β-sarcoglycan (4q12),9 δ-sarcoglycan (5q33),10 cardiac troponin T (1q32), β-myosin heavy chain (chromosome 14q11),11 and α-tropomyosin (15q2).12 So far, DCM with conduction system disease candidate genes have included lamin A/C (1q21.2-3)13 and the sodium channel gene SCN5A (3p22–25).14 Recent data have shown increased susceptibility to arrhythmic events in patients with DCM and lamin A/C–related mutations.15 First described in 1987 as a form of DCM occurring in teenage boys and men in their early 20s, with rapid progression from CHF to death or transplantation, X-linked cardiomyopathy is distinguished by increased amounts of serum creatine kinase muscle isoforms, a sign of underlying skeletal muscle disease.16 Female carriers tend to develop mild to moderate DCM in their 50s, and the disease is slowly progressive. Towbin et al.17 identified a disease-causing gene that codes for dystrophin, a cytoskeletal protein. The relationship between individual genotypes and arrhythmogenicity is poorly understood. As more putative genes have been identified, genotyping has become possible; however, this has not become an important tool in determining the risk for ventricular arrhythmias or in the management of affected individuals. As with all proposals for genetic testing, certain caveats apply, such as relating to the possibility of uncertain results (either a negative screening result in someone at risk for ventricular arrhythmias or a positive result in the identification of a mutation of unclear significance for that particular individual).
Pathophysiology
Arrhythmogenesis
No single mechanism is responsible for ventricular arrhythmias in DCM; rather, multiple factors clearly contribute (Box 85-1). At autopsy, extensive subendocardial scarring in the LV has been described in 33% of patients, and multiple patchy areas of replacement fibrosis have been described in 57% of patients with DCM.18 These autopsy data correspond well with findings observed during voltage mapping of the LV and can be observed on a cardiac magnetic resonance imaging (MRI) examination (Figure 85-3). It is apparent that these areas can act as sites for reentry, one of the commonest mechanisms for ventricular tachycardia (VT) and sudden cardiac death (SCD) in patients with DCM. Ischemia, through smaller coronary artery thrombi or emboli and electrolyte imbalances (especially hypokalemia and hypomagnesemia), can also play a role in arrhythmogenesis in DCM and in other patients at risk for VT and SCD. Alterations in ventricular mechanics and geometry can predispose to reentrant arrhythmias through variable wall tension and stretch-dependent shortening of ventricular refractoriness, or by resulting in abnormal automaticity or triggered activity. Increased circulating catecholamines can cause arrhythmias indirectly by driving potassium intracellularly or directly through triggered activity. Finally, antiarrhythmic drugs themselves can exert a proarrhythmic effect in patients with DCM.
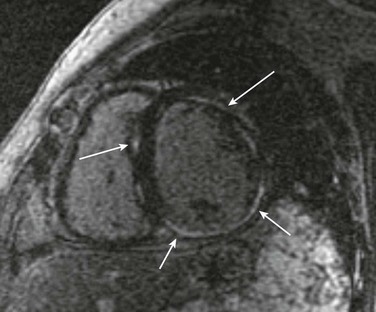
Figure 85-3 A magnetic resonance image for a patient with dilated cardiomyopathy (DCM). This delayed gadolinium enhancement image illustrates patchy areas of fibrosis at the base of the LV in the mid myocardium, with no coronary distribution typical of a DCM. Arrows indicate areas of delayed enhancement corresponding to scar.
Mechanisms of Ventricular Tachycardia
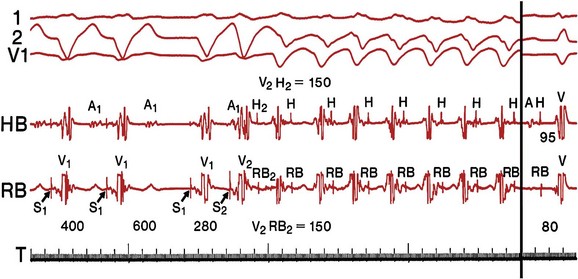
Although other mechanisms of VT are common in patients with DCM, bundle branch reentry VT (BBR-VT) is perhaps the most characteristic (Figure 85-4). It has been observed that 45% of all patients who had BBR-VT induced had underlying DCM. Bundle branch reentry produces VT through a macroreentrant circuit involving the His-Purkinje system, usually with antegrade conduction over the right bundle branch and retrograde conduction over the left bundle branch. In one series, it was the mechanism responsible for VT in up to 6% of all patients and in up to 41% of patients with DCM.19 BBR-VT is usually rapid, with a mean cycle length of 280 ms. Not surprisingly, syncope occurs in the majority of such patients. Degeneration to ventricular fibrillation can also occur. Baseline electrocardiography typically shows a nonspecific intraventricular conduction delay or left bundle branch block (LBBB) morphology. H-V intervals recorded during sinus rhythm are characteristically prolonged. Tachycardias induced by right ventricular (RV) stimulation typically show a LBBB morphology. A short-to-long change in cycle length before the premature extrastimulus has been reported to be particularly successful at initiating BBR-VT. In patients with DCM coming for an electrophysiological study with a view to catheter ablation and who have an LBBB morphology VT induced, it is important to initially exclude bundle branch reentry as the underlying mechanism. Myocardial VT in this setting can be distinguished from bundle branch reentry by certain characteristics. First, a LBBB-QRS morphology with the presence of a His signal before the ventricular signal with an H-V interval that is equal to or longer than that in sinus rhythm due to rate-related reduction in conduction velocity in the RBB. Second, the finding that preceding H-H intervals predict the following V-V intervals. BBR-VT is an important arrhythmia to diagnose because catheter ablation of the right bundle branch is curative of this arrhythmia. Some patients will require pacing after right bundle branch ablation, particularly those with a significant left bundle branch conduction delay or complete LBBB at baseline. Because of the severity of LV dysfunction, a dual-chamber or BiV ICD is usually indicated in such patients.
Insights from Electroanatomic Mapping
Electroanatomic mapping has continued to afford new insights into the basis of structural abnormalities in the genesis of ventricular arrhythmias in DCM through the creation of activation and voltage maps of the ventricles using endocardial and epicardial approaches. Mapping is typically performed during sinus rhythm or atrial fibrillation, such that ventricular activation is uniform during the mapping procedure. During mapping, attention is directed toward ensuring good catheter contact with the myocardium. Peak-to-peak electrogram voltages less than 0.5 mV are generally associated with the presence of myocardial “scar.” Voltages less than 1.5mV are generally associated with the presence of diseased myocardium and are typically found at regions near scarred myocardium. However, unlike in patients with cardiomyopathies resulting from prior myocardial infarcts, such as of low and intermediate voltages, are typically distributed in a nonregional manner. As a result, the areas of interest in patients with DCM may be found in a more diffuse and patchy distribution. Therefore, when creating voltage maps in patients with DCM, care must be taken to interrogate the entire ventricular surface. In addition, a high-density map leads to a better definition of regions, which can create a substrate for reentrant arrhythmias. While being generated, these voltage maps are displayed in a color-coded manner on the electroanatomic map (EAM), so that all areas of viable myocardium are given the same color code and the scar can be differentiated further into different zones according to the local voltage at multiple points within the scar. In patients with DCM, there is a propensity for regions of low voltage to be located at the basal LV, particularly at the level of the mitral valve. During mapping of the substrate, other electrograms of interest that might be important in the genesis of arrhythmias may be tagged on the EAM. These include fractionated electrograms that could represent regions of slow conduction, late (>10 ms after the terminal deflection of the surface QRS) and double potentials (electrograms with a wide isoelectric interval between them), which can represent regions of slow conduction or electrical block. This approach facilitates the definition of the myocardial substrate and putative regions of interest. Soejima et al.20 characterized VT in DCM using electroanatomic mapping and found macroreentrant VT to be the dominant mechanism. A critical isthmus was found in 12 patients during endocardial mapping and in 7 patients during epicardial mapping. All patients who had endocardial mapping had at least one area of scar, with an average of two scars per patient. More than half of the 32 scars extended to a valve annulus, most commonly the mitral annulus. The total area of endocardial scar per patient was approximately 16 cm2 (see Figure 85-5).
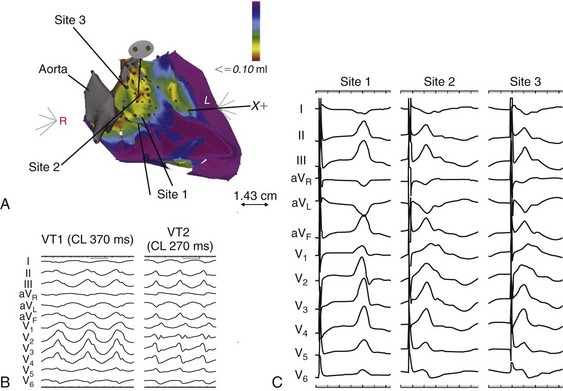
Figure 85-5 A, Voltage map of the endocardial surface of the left ventricle (LV). An extensive low-voltage area is present in the LV outflow area. B, Two morphologies of ventricular tachycardia (VT). C, Pace mapping at sites 1, 2, and 3 labeled in (A) is shown. A good pace match for VT1 is observed at sites 2 and 3. A long S-QRS delay (200 ms), consistent with slow conduction away from the pacing site, is observed during pace mapping at site 1. A series of radiofrequency lesions (red circles) across this region from the aortic annulus to the dense scar above the mitral annulus abolished both VTs. (From Soejima K, Stevenson WG, Sapp JL, et al: Endocardial and epicardial radiofrequency ablation of ventricular tachycardia associated with dilated cardiomyopathy: the importance of low-voltage scars. J Am Coll Cardiol 43:1834–1842, 2004.)
Mechanisms of Sudden Death
With the advent of telemetry and Holter monitoring, VT, previously thought to be a relatively rare condition, was noted in 50% to 60% of patients with DCM and was estimated to be responsible for 8% to 50% of deaths.4 Of patients with all-cause CHF, approximately 50% have been reported to die suddenly. An approximately equal number succumb to progressive ventricular dysfunction with subsequent mortality being related to progressive pump failure. Although the likelihood of death from progressive pump failure rather than sudden arrhythmic death increases with the severity of heart failure symptoms, the absolute likelihood of sudden death (presumed arrhythmic death) increases with New York Heart Association (NYHA) class. In the absence of an implanted device or telemetry, distinguishing sudden death from death caused by pump failure can be difficult.
Despite their common occurrence, ventricular arrhythmias are not the only cause of sudden death in patients with DCM. Luu et al.21 reviewed 21 episodes of cardiac arrest in 20 hospitalized, monitored patients with end-stage heart failure caused by ischemic or nonischemic cardiomyopathy. Primary ventricular tachyarrhythmias occurred in 38% of arrests. Bradycardia or electromechanical dissociation was the initial event in 62% of the patients. First- and second-degree atrioventricular (AV) block have been identified as markers of poor prognosis in patients with DCM. Others have also noted that ischemia, secondary to acute coronary artery thrombi or emboli, can lead to sudden death in a small number of patients. Pulmonary emboli and electrolyte imbalances are other significant precipitants of sudden death in patients with heart failure.21,22
Imaging
A more detailed examination of the presence, extent, and transmurality of myocardial scar is possible with cardiac magnetic resonance imaging (Figure 85-5). As previously observed on autopsy findings, it has been observed with MRI that the regions and depth of scar in patients with DCM differ from other forms of cardiomyopathies. As discussed previously, the location of the scar, as defined by late gadolinium enhancement (LGE), has a propensity for the basal regions. Transmurality is less commonly observed than in patients with ischemia as an underlying cause for their cardiomyopathy and subepicardial and midmyocardial scar is frequently observed in patients with DCM. Initially described by McCrohon et al.23 who found longitudinal striae of midwall enhancement consistent with fibrosis in 28% of patients with DCM, MRI can be used to quantitate the extent of scar and define its location. Nazarian et al.24 demonstrated a propensity for scar in the basal midmyocardial layers of the LV in patients with DCM and found that the amount of nontransmural scar (26% to 75% wall thickness), as defined by LGE, was an independent predictor of the presence of inducible VT during programmed ventricular stimulation. In several cases, the location of scar could be correlated with the origin of the clinical VT (see Figure 85-5). In a series of 141 patients studied by Hombach et al.25 predictors of poor clinical outcomes (cardiac death) included a longer QRS duration (>110 ms), comorbid diabetes mellitus, and the presence of LGE on MRI .
Predictors of Mortality and Ventricular Arrhythmias
Clinical Predictors of Mortality
The severity of LV dysfunction is a powerful predictor of mortality in patients with DCM and CHF. A reduced LV ejection fraction (LVEF) has been shown to correlate strongly with poor survival. In one study including patients with both ischemic and nonischemic cardiomyopathy, with a mean follow-up of 37 months, mortality was substantial in all LVEF groups (range, LVEF ≤ 15%, 51.7%; LVEF > 55%, 23.5%). Among patients with LVEF ≤ 45%, mortality decreased in a near linear fashion across successively higher LVEF groups (LVEF < 15%, 51.7%; LVEF 36% to 45%, 25.6%; P < .0001).26 Other studies have also shown LVEF to be a powerful predictor of mortality in a population, excluding ischemic cardiomyopathy.27 Information on LVEF, as well as other data on cardiac function and structure, can be obtained noninvasively from an echocardiogram. Other invasively obtained indices of LV and RV function including pulmonary capillary wedge pressure, cardiac index, stroke work index, stroke volume, and right atrial pressure also predict mortality in patients with DCM.4
Given the subjective nature of the data, clinical observations and heart failure classification are not as robust predictors of mortality as the more objective indices of LV function. Nevertheless, both higher NYHA classification and the presence of a third heart sound correlate with mortality in DCM and other causes of CHF.28 In DCM, the incidence of sudden death is significantly greater in patients with a history of syncope.29 Laboratory values found to be predictive of mortality include low serum sodium and increased plasma norepinephrine, renin, and both atrial and brain natriuretic peptide levels. Most studies that have included patients with ischemic and nonischemic cardiomyopathy have demonstrated a marginally worse prognosis in patients with coronary artery disease; however, this has not been universally observed.
Electrophysiological Predictors of Arrhythmias and Death
Electrocardiogram
The presence of LBBB and first- and second-degree AV block have been associated with poorer outcomes in DCM. Data from the Vesnarinone Study Trial (VEST) have confirmed a significant association between the degree of QRS prolongation and mortality (Figure 85-6). It has also been observed that the development of atrial fibrillation appears to portend a poorer prognosis, although the reason for this remains controversial. The emergence of atrial fibrillation can contribute to worsening cardiac performance and precipitate acute heart failure, particularly if ventricular rates are poorly controlled. Some patients can have ventricular arrhythmias precipitated by uncontrolled supraventricular arrhythmias. In others, atrial fibrillation may simply be a marker of progressive cardiac dysfunction and not the primary cause of this progression. In one large study, 19% of 390 patients with advanced heart failure,30 including 191 patients with DCM, developed paroxysmal or persistent atrial fibrillation. One-year actuarial survival of patients with atrial fibrillation compared with patients in sinus rhythm was 52% versus 71% (P = .001); however, this association did not remain significant when corrected for an increased pulmonary wedge pressure. Atrial fibrillation can also reduce survival by increasing thromboembolic events or ventricular arrhythmias, the latter through long-short cycle lengths increasing the dispersion of refractoriness in the His-Purkinje system and ventricles.
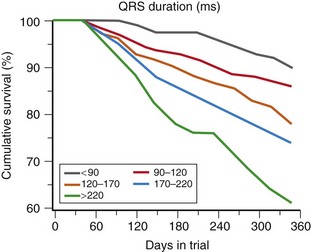
Figure 85-6 Cumulative survival as a function of QRS duration on 12-lead electrocardiogram at trial entry in the VEST study. (Adapted from Venkateshwar K, Gottipaty SP, Krelis FL, et al: The resting electrocardiogram provides an inexpensive marker of prognosis in patients with chronic congestive heart failure. JACC 33[2]:145A [abstract 847-4], 1999.)
Spontaneous Ventricular Arrhythmias
Multiform premature ventricular complexes, ventricular couplets, and nonsustained VT (NSVT) are nearly ubiquitous in patients with dilated cardiomyopathy and in some cases can represent a cause of the left ventricular dysfunction.31 The significance of NSVT is difficult to assess given its high prevalence among patients with DCM. There have been conflicting reports of the significance of NSVT on overall mortality and sudden death in well-treated patients with DCM after efforts to control for other variables.32
Programmed Electrical Stimulation
The inducibility of VT is much less predictive of arrhythmia recurrence and sudden death in patients with DCM than in patients with coronary artery disease (Table 85-1). Analysis of pooled data shows that, of patients with inducible VT suppressed by drug therapy, 28% who initially had VT experienced recurrence, whereas 43% with cardiac arrest later died suddenly.33 Failure to induce sustained monomorphic VT during programmed electrical stimulation is associated with high rates of arrhythmia recurrence and sudden death, although at lower incidence rates than in patients with inducible VT. Therefore, programmed ventricular stimulation should not be used for risk stratification in patients with DCM.
Table 85-1
Programmed Stimulation in Patients with Dilated Cardiomyopathy and No History of Sustained Ventricular Arrhythmias
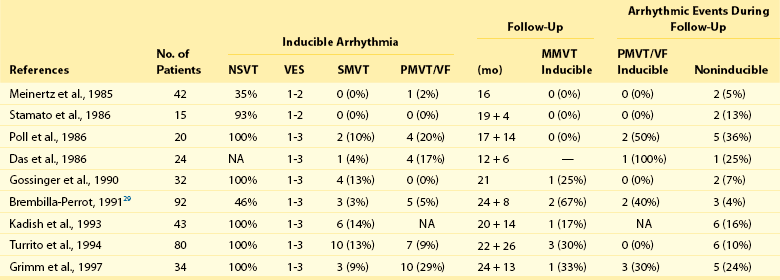
(From Grimm W, Hoffmann J, Menz V, et al.: Programmed ventricular stimulation for arrhythmia risk prediction in patients with idiopathic dilated cardiomyopathy and nonsustained ventricular tachycardia. J Am Coll Cardiol 32:739–745, 1998.)
Noninvasive Electrocardiograph Predictors
A number of noninvasive electrocardiograph (ECG) predictors have been studied in small series as predictors of clinical and inducible VT as well as sudden death in patients with DCM. In a large trial, Grimm et al.34 examined the predictive value of echocardiography, 24-hour Holter monitoring, signal averaged ECG (SAECG), heart rate variability, QT dispersion, microvolt T wave alternans, and baroreflex sensitivity in 343 patients with DCM. After an average follow-up of 52 months, 13% of patients had a sustained ventricular arrhythmia or died suddenly. The relative risk of VT and SCD was increased 2.3-fold for each 10% reduction in EF and 1.7-fold when VT was present on a Holter monitor. The relative risk of VT and SCD was reduced by 40% when patients were treated with β-blockers. However, none of the other five noninvasive predictors proved useful in predicting VT or SCD. In the asymptomatic patient with DCM, LVEF and the presence of VT on a Holter monitor remain the most powerful and clinically useful predictors of risk for sudden cardiac death. Data on the presence and extent of myocardial fibrosis on cardiac MRI and subsequent fatal or nonfatal ventricular arrhythmias in DCM have also been reported.25
Drug Therapy
Vasodilator Therapy
Vasodilators improve survival in patients with CHF by lessening the progression of LV dysfunction. Neurohormonal blockade with ACE inhibitors and angiotensin receptor blockers, in contrast to other vasodilators, could be unique in reducing risk for sudden death in such patients. In the Veterans Heart Failure Trial II,35 the reduction in all-cause mortality rates (18% vs. 25%) with enalapril compared with hydralazine-isosorbide dinitrate was attributed to a reduction in the incidence of sudden death (37% vs. 46% of the total mortality) in the enalapril group. Ventricular tachycardia was less frequent at 3 months, and less new VT developed at 1 and 2 years in the enalapril group. In the Evaluation of Losartan in the Elderly (ELITE) Trial,36 an angiotensin II receptor antagonist (losartan) was shown to reduce overall mortality rates by 46% and, from within total mortality, sudden death by 64% compared with captopril in an older group of patients with symptomatic heart failure, and LV systolic dysfunction. These results were not confirmed in the subsequent ELITE II trial,37 which showed no difference in mortality rates, total or sudden, between an ACE inhibitor and angiotensin II receptor blocker.
Adrenergic Receptor Blocking Agents
In 1975, Waagstein et al.38 first reported an improvement in ventricular function and exercise tolerance in seven patients with DCM treated with β-receptor blocking agents. Since then, several studies have shown symptomatic improvement in patients with nonischemic dilated cardiomyopathies treated with β-blockers. Since that time, β-blockers have clearly shown mortality benefit in patients with both ischemic and nonischemic cardiomyopathy. A metaanalysis of more than 10,000 patients, more than 4000 of which had nonischemic cardiomyopathy pooled from 22 studies, revealed substantially reduced mortality at 1 and 2 years (odds ratios, 0.65 and 0.72).39 The rationale for β-blocker therapy in patients with dilated cardiomyopathies is to counteract the deleterious effects of heightened sympathetic tone and circulating catecholamines, including cytosolic calcium overload mediated through ryanodine receptor hyperphosphorylation and decreased synthesis of contractile proteins (Figure 85-7).
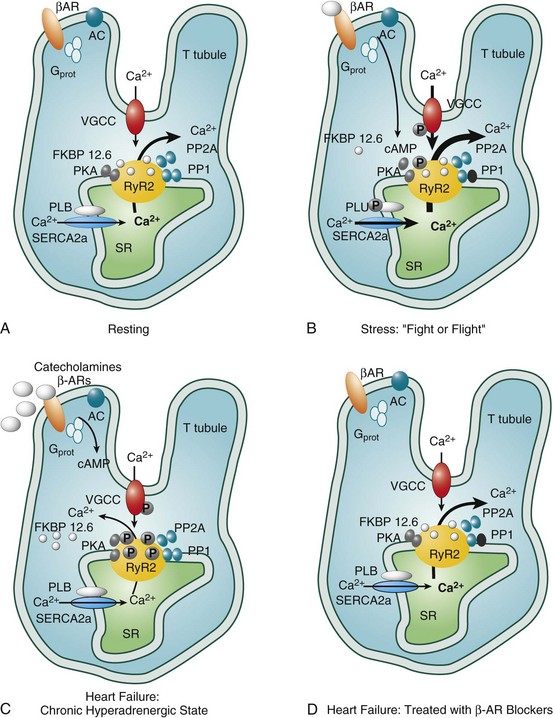
Figure 85-7 Catecholamine-induced polymorphic ventricular tachycardia (PMVT). Constant adrenergic stimulation through β-adrenergic receptors (β-AR) causes a positive shift in ryanodine receptor 2 (RyR2) kinetics and Ca2+ leaks back into the cytosol in diastole causing early afterdepolarizations (EADs) and premature ventricular contractions (PVCs). These Ca2+ leaks can be reduced by β-blockade through inhibition of adenylate cyclase (AC)–mediated cyclic adenosine monophosphate (cAMP) production and less RyR2 activation. Identified mutations include: P2328S, Q4201R, and V4653F in the calsequestrin 2 gene (Calsq2). Gprot, G proteins; PLB, phospholamban; SR, sarcoplasmic reticulum; VGCC, voltage-gated Ca2+ channel.
The largest trials of β-blockers in CHF included mainly patients with ischemic cardiomyopathy. Carvedilol, a nonselective β-blocker that is also an α-receptor antagonist and antioxidant was associated with a remarkable 65% reduction in mortality rates among 1094 patients with class III or IV heart failure and an ejection fraction of less than 0.35 who could tolerate the drug and were randomized to carvedilol or placebo in addition to conventional therapy.40 The Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure randomized 3991 patients with an ejection fraction of 0.40 or less and NYHA class II-IV heart failure to metoprolol or placebo in addition to conventional therapy. Overall mortality rate was reduced 34% at 21 months among the patients treated with metoprolol, with significant reductions noted in patients with and without coronary disease (40% and 30%, respectively). There was a 49% reduction in heart failure–related deaths and a 41% reduction in sudden cardiac deaths.41 In the Cardiac Insufficiency Bisoprolol Study II (CIBIS II) trial, 2647 patients with NYHA III or IV symptoms and an ejection fraction less than 0.35 were randomized to bisoprolol or placebo in addition to an ACE inhibitor and diuretic. There was a highly significant 34% reduction in total mortality rate at 1.3 years mainly because of a 44% reduction in the rate of sudden cardiac death, which led to the trial being halted prematurely.42 The striking similarities in the outcomes of these three trials indicate that the large reductions in mortality rates observed with β-adrenergic blockade argue strongly for a class effect and add to the weight of evidence supporting the role of ACE inhibition and β-receptor blockade in all patients with CHF unless a specific contraindication exists.
Antiarrhythmic Drug Therapy
Antiarrhythmic drugs can be used for the treatment of VT or atrial arrhythmias in DCM. They have also been used in an attempt to reduce mortality in DCM in those without clinically evident arrhythmias. Unfortunately, most conventional antiarrhythmic agents can exacerbate LV dysfunction, particularly class IA and IC agents (e.g., disopyramide and flecainide). Life-threatening complications, including CHF and proarrhythmia, occur far more frequently in patients with impaired ventricular function. Amiodarone, which is also a vasodilator with antiadrenergic effects, is the most frequently used antiarrhythmic drug in DCM. Neri et al.43 found that amiodarone significantly reduced the incidence of complex ventricular arrhythmias and sudden death among 65 patients with DCM and VT. Side effects developed in slightly more than half of the patients, making it necessary to terminate therapy in 9.8%. In another study, among 232 patients with DCM being evaluated for cardiac transplantation, treatment with amiodarone was not found to prolong survival, although patients were not randomized.44 Similar to conventional antiarrhythmic agents, the efficacy and tolerance of amiodarone decreases with deteriorating ventricular function. In the largest trial to date, Sudden Cardiac Death Heart Failure Trial (SCD-HeFT),45 2521 patients with an LVEF less than 0.35 with CAD (52%) and nonischemic DCM (48%) with NYHA II-III CHF, but no arrhythmia requiring amiodarone therapy, were randomized to placebo, amiodarone, or an ICD. In this large primary prevention trial, there was no mortality difference between amiodarone-treated and placebo-treated patients with nonischemic cardiomyopathy. Based on this trial and other observations, it has been recommended that amiodarone not be used routinely in patients with DCM unless a specific, well-defined arrhythmia indication exists. When used to treat VT in the setting of DCM, antiarrhythmic drug therapy is most commonly used as an adjunct to ICD therapy.
Device Therapy
Implantable Cardioverter-Defibrillators
For patients with DCM who have survived a cardiac arrest or who have had spontaneous sustained VT, implantation of an ICD is indicated. Currently, guidelines also recommend ICD implantation for DCM patients with an LVEF less than or equal to 35% with NYHA class II or III heart failure (class I indication); for those with DCM and a history of syncope with documented significant LV dysfunction, implantation of an ICD is deemed reasonable (class IIA indication).46 For primary prevention, a number of trials have examined the role of prophylactic ICDs in the treatment of DCM.
In the Defibrillators in Non-Ischemic Cardiomyopathy Evaluation Trial, Kadish et al.47 randomized 458 subjects with DCM and an LVEF less than 36% to standard medical therapy or ICD therapy. More than 90% were already on best medical therapy with a β-blocker and an ACE inhibitor or ARB. Mean LVEF was 21%, and 90% of patients had nonsustained VT. Symptom class was NYHA class I in 22%, class II in 57%, and class III in 21%. The primary endpoint was total mortality. After 2 years, 14.1% of the standard therapy group had died and 7.9% of the ICD therapy group had died, resulting in a 6.2% absolute and 35% relative risk reduction. This difference did not reach statistical significance (P = .08) likely because of insufficient sample size and a lower than expected death rate in the standard therapy group. Arrhythmic mortality, a secondary endpoint, was reduced by 80% (P = .006) in the ICD therapy group. Mortality reduction was greater in NYHA class III than in class II patients.
In the Amiodarone versus Implantable Cardiovert-Defibrillator Trial, Strickberger et al.46 randomized 103 patients with DCM, NSVT, and an LVEF less than 35% to receive either amiodarone or an ICD. Mortality at 1 and 2 years was similar (10% vs. 4% and 12% vs. 13%, respectively; P = not significant). There was no difference in quality of life between groups.48
In the SCD-HeFT, Bardy et al.45 randomized 2521 patients with both coronary disease and DCM with class II or III CHF and LVEF less than 35% to receive placebo, weight-adjusted amiodarone, or an ICD. The ICD implanted in this trial was a single chamber device with “shock only” therapies programmed. The device was not programmed to deliver antitachycardia pacing. At 5 years of follow-up, absolute and relative mortality reductions in the ICD group compared with placebo were 7.2% and 23%, respectively (P = .007; Figure 85-8). An interesting subgroup analysis, which must be interpreted with caution, suggested that the mortality benefit was limited to those with class II rather than class III heart failure. Although the overall result in favoring ICD implantation was positive, the P value was .06 for the relative risk reduction in mortality in patients with nonischemic cardiomyopathy. Nonetheless, these trials show similar significant reductions in total mortality among patients with DCM who receive an ICD rather than standard medical therapy or the addition of amiodarone, and support the use of prophylactic ICD therapy in patients with DCM and severely reduced LV function in the absence of NYHA class IV CHF. This recommendation is supported by current guidelines on the use of ICDs for primary prevention of sudden death in DCM.46
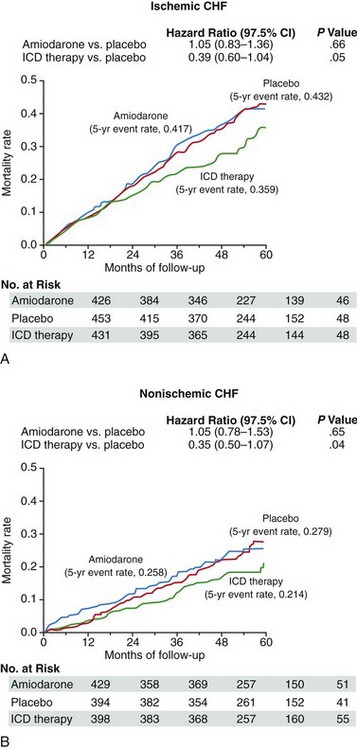
Figure 85-8 Kaplan-Meier analysis of freedom from death among patients with ischemic and nonischemic dilated cardiomyopathy randomized to placebo, implantable cardiac defibrillators (ICDs), or amiodarone in the Sudden Cardiac Death Heart Failure Trial. Mortality reduction with ICD therapy compared with placebo was similar in the ischemic and nonischemic groups. Amiodarone failed to reduce mortality in either group. (From Bardy GH, Lee KL, Mark DB et al: Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N Engl J Med 2005;352:225–337.)
Biventricular Pacing
Between 30% and 50% of patients with DCM have intraventricular conduction delays resulting in a QRS duration greater than 120 ms, most commonly with an LBBB pattern. Delayed activation of the LV free wall can result in dyssynchronous free wall contraction relative to the septum with resulting reduction in LV stroke volume and cardiac output and increased functional mitral regurgitation. In addition, delayed activation can result in subsequent delayed relaxation with the consequence that passive filling of the LV can be nearly simultaneous with atrial contraction, particularly at faster heart rates. It has been shown that earlier activation of the LV free wall by pacing (1) improves systolic function by shortening the duration of mechanical systole and increasing dP/dt, (2) improves diastolic function by prolonging diastolic filling time, and (3) reduces presystolic mitral regurgitation by earlier activation of the lateral papillary muscle, without the potential adverse effects associated with inotropic drugs. The results of prospective randomized controlled trials published to date have consistently demonstrated an improvement in acute hemodynamics, LV dP/dt, LV ejection fraction, NYHA class, and 6-minute walk duration, as well as a reduction in hospitalization for heart failure and cost effectiveness, even in minimally symptomatic patients,49 after implantation of BiV pacemakers. In addition, among those patients randomized to BiV ICDs, there was a reduction in the frequency of ICD shocks, number of therapies required, and number of episodes of nonsustained VT.
Large trials assessing the effects of BiV pacing on mortality have included patients with DCM. The CARE-HF study50 followed 813 patients (46% with DCM) who had NYHA class III or IV heart failure despite receiving standard pharmacologic therapy, a left ventricular ejection fraction of 35% or less, and a QRS interval duration of at least 120 ms on the resting electrocardiogram. Patients with a QRS interval of 120 to 149 ms were required to meet two of three additional criteria for dyssynchrony: an aortic preejection delay of more than 140 ms, an interventricular mechanical delay of more than 40 ms, or delayed activation of the posterolateral left ventricular wall. After 2.5 years of follow-up, the investigators found a 36% reduction in mortality from 30% to 20% and a 37% reduction in death or hospitalization from 55% to 39% among those randomized to BiV pacing. Compared with medical therapy, cardiac resynchronization reduced the interventricular mechanical delay, the end-systolic volume index, and the area of the mitral regurgitant jet; it also increased the left ventricular ejection fraction and improved symptoms and quality of life (P < .01 for all comparisons).
In the Comparison of Medical Therapy, Pacing, and Defibrillation in Heart Failure Trial,51 1520 patients with NYHA class III-IV heart failure, QRS duration longer than 120 ms, PR interval longer than 150 ms, and LV ejection fraction less than 0.35 were randomized in a 1 : 2 : 2 manner to optimal medical therapy, BiV pacing alone, or BiV-ICD therapy. Of note, 44% of the patients had nonischemic cardiomyopathy. BiV pacing and BiV-ICD therapy reduced the combined end-point of mortality and hospitalization by 34% and 40%, respectively, compared with optimal medical therapy alone; mortality was reduced by 24% and 36%, respectively, with the larger reduction in the BiV-ICD group reaching statistical significance. Recently, the benefit of cardiac resynchonization therapy (CRT) has been demonstrated among asymptomatic and minimally symptomatic patients with LBBB and decreased ejection fraction,52 resulting in an expansion of the guidelines in the recommendation for CRT implant to include a class I indication for NYHA class II with LBBB with QRS duration of 150 ms or longer and the addition of a class IIb recommendation for patients who have LVEF of 30% or less, ischemic etiology of HF, sinus rhythm, LBBB with a QRS duration of 150 ms or longer, and NYHA class I symptoms. NYHA class IV heart failure has been considered a contraindication to ICD implantation (except in those patients awaiting cardiac transplant) because of the high likelihood of death caused by pump failure 1 year. Therefore, BiV pacemakers would seem to be the most appropriate therapy in patients with severe (NYHA class IV) heart failure who have fixed contraindications for cardiac transplant, whereas BiV ICDs might be more appropriate for patients with DCM, NYHA class II-III CHF, and advanced LV dysfunction; however, this approach might need to be revised in cases in which BiV pacing results in marked improvement in heart failure symptoms to NYHA class II-III such that an ICD would also be indicated. Although it has been reported that up to 30% of patients fail to respond BiV pacing, a more aggressive and multiple-specialty follow-up and echo-guided device programming approach has shown to lead to better outcomes in patients undergoing BiV device outcome compared with a non-multidisciplinary approach.53
Because of the increasing volume of patients with implanted BiV-ICDs and availability of close prospective follow-up in some centers, further information regarding predictors of ventricular arrhythmias can be gleaned. In a recent study of 269 patients (46% with nonischemic cardiomyopathy) who had undergone BiV-ICD implantation for standard indications, the 4-year incidence of appropriate device therapy was 36%. An observation was made that those with an LV end-systolic diameter greater than 61 mm had an incidence of ventricular arrhythmias of 51% at 3 years; for those with a less dilated LV, the corresponding figure was 26% (P = .001). In patients with a less dilated LV (<61 mm), multivariate predictors of appropriate therapy were absence of β-blocker therapy, severely impaired left ventricular ejection fraction (LVEF < 20%), and a history of sustained ventricular arrhythmia.54
Ventricular Assist Devices
Ventricular assist devices constitute an important advance in the management of patients with end-stage heart failure. Previously used solely as an in-hospital bridge to transplant, many patients have now been discharged from hospital while awaiting heart transplantation. Such home-bridging to transplantation has been studied as part of clinical trials of the HeartMate (Thoratec, Pleasanton, CA) and AbioCor (AbioMed, Danvers, MA)55 and other vented electrical implantable devices. There are many reports of patients with assist devices who tolerate ventricular arrhythmias, including a prolonged period of ventricular fibrillation, remarkably well because cardiac output is maintained by the assist device rather than the left ventricle. However, patients with poor RV function and others with ventricular fibrillation can develop significant hemodynamic compromise with a solely left ventricular assist device. Currently, the use of these devices is limited by their cost, the risk for infection related to the percutaneous power or drive cable, and the risk for systemic thromboembolism. However, their increasing use as destination therapy or a bridge to recovery rather than as a bridge to transplant is an indication of their success in the treatment of end-stage DCM.
Catheter Ablation
Although ICDs have reduced the likelihood of arrhythmic death, they do not prevent the recurrence of symptomatic VT. Pharmacologic control of VT is successful in approximately 40% of cases, leaving a significant proportion of patients with symptomatic VT or ICD shocks. Previous experience with catheter ablation of VT in DCM had been limited and was associated with lower success rates than VT ablation in the setting of coronary disease and post–myocardial infarction. However, the advent of electroanatomic mapping systems, irrigated tip ablation catheters and epicardial mapping and ablation has been associated with significant improvement in success rates. With the wider acceptance and performance of ablation for patients with DCM and VT, more of these procedures are performed annually. Guidelines for patient selection have been published.56 Of importance for patients with DCM, catheter ablation is recommended for symptomatic sustained monomorphic VT that has been unresponsive to antiarrhythmic medications (or when these are believed to be contraindicated), for the treatment of incessant sustained monomorphic VT or VT storm that is not due to reversible cause, and for bundle branch reentrant VT.
Preprocedural planning is extremely important when a catheter ablation procedure is planned in a patient with DCM. Preprocedural imaging should be undertaken to assess the degree of LV dysfunction and associated valvular abnormalities, and to exclude the presence of an LV thrombus, which would be a contraindication to endocardial mapping. Baseline ECGs and any available ECGs of the ventricular tachycardia should be reviewed, as well as device-based electrograms. Endocardial mapping can be performed via a retrograde aortic approach or an antegrade approach following atrial transseptal puncture. Mapping of the epicardial surface is generally straightforward after gaining access to the epicardium following an epicardial puncture as described by Sosa et al.57 The ablation of VT in DCM is usually performed with one or more of a number of mapping modalities, including entrainment mapping, pace-mapping, and electroanatomically guided voltage, and in some cases activation sequence mapping (i.e., EAM).58 The use of integrated mapping systems that permit merging of three-dimensional CT or MRI images with real-time electroanatomic mapping has proved helpful to some operators (Figure 85-9). Similarly, integration of the EAM with a real-time acquisition of intracardiac echocardiographic images can be helpful in mapping larger LV cavities. Mapping of both the endocardial and, if necessary, epicardial surfaces of the ventricle permits the creation of a representation of the ventricular myocardium that can be merged in real time with a previously obtained CT or MRI or with ultrasound images obtained using intracardiac echocardiography. Activation and voltage maps can be simultaneously created to guide localization of a VT circuit (see Figure 85-4). The resolution of these EAMs is sufficient to permit mapping of the chamber and guide targeted ablation, with minimization of fluoroscopy times. Voltage mapping using electroanatomic mapping systems allows rapid identification of the presence of scar, particularly on the endocardium. The presence of low voltage on the epicardium can represent either local scar or epicardial fat. As discussed previously, these mapping systems can be extremely useful in identifying reentrant circuits that might be responsible for a patient’s clinically observed VT. EAMs thus enable mapping and guidance for the selection of ablation targets, linear lesion sets, or both. It has been demonstrated using entrainment mapping that myocardial reentry is the dominant mechanism responsible for VT in patients with DCM in whom BBR-VT has been ruled out. In hemodynamically stable patients in whom entrainment mapping can be performed, a critical reentry isthmus can usually be identified. As many scar-related VT circuits in patients with DCM extend to the level of the mitral valve, many operators design lesion sets involving not only a defined critical isthmus but also continuing the lesion set to the mitral valve annulus, a site of fixed anatomical block. The availability of hemodynamic support devices can now facilitate the mapping of rapid VTs that previously would not have been mappable because of hemodynamic instability. Failure to ablate VT from the epicardium or endocardium can be due to a midmyocardial source or critical isthmus, or inability to reach an epicardial source because of the presence of epicardial fat. The use of cryoablation on the epicardial surface has been reported to be successful in such circumstances.59 Novel catheter designs including needle-tipped catheters are under development to help address the problem of intramural targets. Irrigated tip catheters have also proved useful in both increasing lesion depth toward the midmyocardial layers where myocardial fibrosis has been frequently identified in DCM patients, as well as reducing the likelihood of endocardial charring and thrombus formation. The increasing use of percutaneous epicardial catheter ablation is a result of the moderate success rates observed with endocardial approaches alone. In patients with DCM, approximately one third of VTs are ablated epicardially rather than endocardially. In patients with DCM, challenges of epicardial catheter ablation include the need to avoid the epicardial coronary vasculature to prevent arterial injury during ablation and the presence of epicardial fat, which can prevent access to target sites.
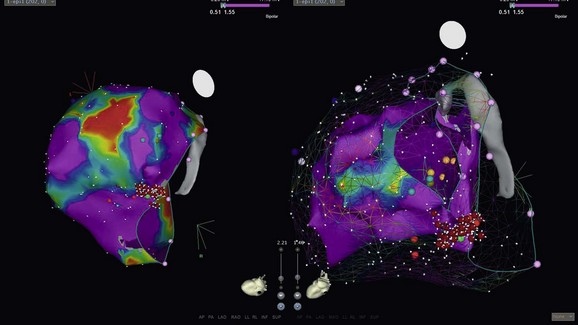
Figure 85-9 CARTO (Biosense-Webster, Diamond Bar, CA) electroanatomical voltage maps of the left ventricle in a patient with DCM. Epicardial map on left showing more low voltages and “scar” than an endocardial map on the right (voltages set at 0.5 to 1.5 mV.) The VT was successfully ablated; the region of exit was at the basal level at the mitral valve. Such propensity of epicardial low voltages (“scar”) and perimitral mechanism of VT is not uncommon in patients with DCM.
References
1. Lloyd-Jones, D, Adams, RJ, Brown, TM, et al. Heart disease and stroke statistics–2010 update: a report from the American Heart Association. Circulation. 2010; 121:e46–e215.
2. Ho, KK, Pinsky, JL, Kannel, WB, et al. The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol. 1993; 22:6A–13A.
3. Roger, VL, Weston, SA, Redfield, MM, et al. Trends in heart failure incidence and survival in a community-based population. JAMA. 2004; 292:344–350.
4. Tamburro, P, Wilber, D. Sudden death in idiopathic dilated cardiomyopathy. Am Heart J. 1992; 124:1035–1045.
5. Baldasseroni, S, Opasich, C, Gorini, M, et al. Left bundle-branch block is associated with increased 1-year sudden and total mortality rate in 5517 outpatients with congestive heart failure: a report from the Italian network on congestive heart failure. Am Heart J. 2002; 143:398–405.
6. Lakdawala, NK, Winterfield, JR, Funke, BH. Dilated cardiomyopathy. Circulation Arrhythm Electrophysiol. 2013; 6:228–237.
7. Olson, TM, Michels, VV, Thibodeau, SN, et al. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science. 1998; 280:750–752.
8. Li, D, Tapscoft, T, Gonzalez, O, et al. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation. 1999; 100:461–464.
9. Barresi, R, Di Blasi, C, Negri, T, et al. Disruption of heart sarcoglycan complex and severe cardiomyopathy caused by beta sarcoglycan mutations. J Med Genet. 2000; 37:102–107.
10. Tsubata, S, Bowles, KR, Vatta, M, et al. Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest. 2000; 106:655–662.
11. Kamisago, M, Sharma, SD, DePalma, SR, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000; 343:1688–1696.
12. Olson, TM, Kishimoto, NY, Whitby, FG, et al. Mutations that alter the surface charge of alpha-tropomyosin are associated with dilated cardiomyopathy. J Mol Cell Cardiol. 2001; 33:723–732.
13. Brodsky, GL, Muntoni, F, Miocic, S, et al. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation. 2000; 101:473–476.
14. Olson, TM, Keating, MT. Mapping a cardiomyopathy locus to chromosome 3p22-p25. J Clin Invest. 1996; 97:528–532.
15. van Rijsingen, IA, Arbustini, E, Elliott, PM, et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol. 2012; 59:493–500.
16. Berko, BA, Swift, M. X-linked dilated cardiomyopathy. N Engl J Med. 1987; 316:1186–1191.
17. Towbin, JA, Hejtmancik, JF, Brink, P, et al. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993; 87:1854–1865.
18. Roberts, WC, Siegel, RJ, McManus, BM. Idiopathic dilated cardiomyopathy: analysis of 152 necropsy patients. Am J Cardiol. 1987; 60:1340–1355.
19. Caceres, J, Jazayeri, M, McKinnie, J, et al. Sustained bundle branch reentry as a mechanism of clinical tachycardia. Circulation. 1989; 79:256–270.
20. Soejima, K, Stevenson, WG, Sapp, JL, et al. Endocardial and epicardial radiofrequency ablation of ventricular tachycardia associated with dilated cardiomyopathy: the importance of low-voltage scars. J Am Coll Cardiol. 2004; 43:1834–1842.
21. Luu, M, Stevenson, WG, Stevenson, LW, et al. Diverse mechanisms of unexpected cardiac arrest in advanced heart failure. Circulation. 1989; 80:1675–1680.
22. Leier, CV. The cardiomyopathies: mortality, sudden death, and ventricular arrhythmias. Cardiovasc Clin. 1992; 22:275–306.
23. McCrohon, JA, Moon, JC, Prasad, SK, et al. Differentiation of heart failure related to dilated cardiomyopathy and coronary artery disease using gadolinium-enhanced cardiovascular magnetic resonance. Circulation. 2003; 108:54–59.
24. Nazarian, S, Bluemke, DA, Lardo, AC, et al. Magnetic resonance assessment of the substrate for inducible ventricular tachycardia in nonischemic cardiomyopathy. Circulation. 2005; 112:2821–2825.
25. Hombach, V, Merkle, N, Bernhard, P, et al. Prognostic significance of cardiac magnetic resonance imaging: Update 2010. Cardiol J. 2010; 17:549–557.
26. Curtis, JP, Sokol, SI, Wang, Y, et al. The association of left ventricular ejection fraction, mortality, and cause of death in stable outpatients with heart failure. J Am Coll Cardiol. 2003; 42:736–742.
27. Juilliere, Y, Danchin, N, Briancon, S, et al. Dilated cardiomyopathy: long-term follow-up and predictors of survival. Int J Cardiol. 1988; 21:269–277.
28. Stewart, RA, McKenna, WJ, Oakley, CM. Good prognosis for dilated cardiomyopathy without severe heart failure or arrhythmia. Q J Med. 1990; 74:309–318.
29. Brembilla-Perrot, B, Donetti, J, de la Chaise, AT, et al. Diagnostic value of ventricular stimulation in patients with idiopathic dilated cardiomyopathy. Am Heart J. 1991; 121:1124–1131.
30. Middlekauff, HR, Stevenson, WG, Stevenson, LW. Prognostic significance of atrial fibrillation in advanced heart failure. A study of 390 patients. Circulation. 1991; 84:40–48.
31. Ban, JE, Park, HC, Park, JS, et al. Electrocardiographic and electrophysiological characteristics of premature ventricular complexes associated with left ventricular dysfunction in patients without structural heart disease. Europace. 2012.
32. Singh, SN, Fisher, SG, Carson, PE, et al. Prevalence and significance of nonsustained ventricular tachycardia in patients with premature ventricular contractions and heart failure treated with vasodilator therapy. Department of Veterans Affairs CHF STAT Investigators. J Am Coll Cardiol. 1998; 32:942–947.
33. Grimm, W, Hoffmann, J, Menz, V, Luck, K, et al. Programmed ventricular stimulation for arrhythmia risk prediction in patients with idiopathic dilated cardiomyopathy and nonsustained ventricular tachycardia. J Am Coll Cardiol. 1998; 32:739–745.
34. Grimm, W, Christ, M, Bach, J, et al. Noninvasive arrhythmia risk stratification in idiopathic dilated cardiomyopathy: results of the Marburg Cardiomyopathy Study. Circulation. 2003; 108:2883–2891.
35. Cohn, JN, Johnson, G, Ziesche, S, et al. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med. 1991; 325:303–310.
36. Pitt, B, Segal, R, Martinez, FA, et al. Randomised trial of losartan versus captopril in patients over 65 with heart failure (Evaluation of Losartan in the Elderly Study, ELITE). Lancet. 1997; 349:747–752.
37. Pitt, B, Poole-Wilson, PA, Segal, R, et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomised trial–the Losartan Heart Failure Survival Study ELITE II. Lancet. 2000; 355:1582–1587.
38. Waagstein, F, Hjalmarson, A, Varnauskas, E, et al. Effect of chronic beta-adrenergic receptor blockade in congestive cardiomyopathy. Br Heart J. 1975; 37:1022–1036.
39. Brophy, JM, Joseph, L, Rouleau, JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med. 2001; 134:550–560.
40. Packer, M, Bristow, MR, Cohn, JN, et al. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U. S. Carvedilol Heart Failure Study Group. N Engl J Med. 1996; 334:1349–1355.
41. M-HS Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet. 1999; 353:2001–2007.
42. C-IIa Committees. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999; 353:9–13.
43. Neri, R, Mestroni, L, Salvi, A, et al. Ventricular arrhythmias in dilated cardiomyopathy: efficacy of amiodarone. Am Heart J. 1987; 113:707–715.
44. Keogh, AM, Baron, DW, Hickie, JB. Prognostic guides in patients with idiopathic or ischemic dilated cardiomyopathy assessed for cardiac transplantation. Am J Cardiol. 1990; 65:903–908.
45. Bardy, GH, Lee, KL, Mark, DB, et al. Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N Engl J Med. 2005; 352:225–237.
46. Epstein, AE, Dimarco, JP, Ellenbogen, KA, et al. ACC/AHA/HRS 2008 guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: executive summary. Heart Rhythm. 2008; 5:934–955.
47. Kadish, A, Dyer, A, Daubert, JP, et al. Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy. N Engl J Med. 2004; 350:2151–2158.
48. Strickberger, SA, Hummel, JD, Bartlett, TG, et al. Amiodarone versus implantable cardioverter-defibrillator:randomized trial in patients with nonischemic dilated cardiomyopathy and asymptomatic nonsustained ventricular tachycardia–AMIOVIRT. J Am Coll Cardiol. 2003; 41:1707–1712.
49. Noyes, K, Veazie, P, Hall, WJ, et al. Cost-effectiveness of cardiac resynchronization therapy in the MADIT-CRT trial. J Cardiovasc Electrophysiol. 2013; 24:66–74.
50. Cleland, JG, Daubert, JC, Erdmann, E, et al. The effect of cardiac resynchronization on morbidity and mortality in heart failure. N Engl J Med. 2005; 352:1539–1549.
51. Saxon, LA, Bristow, MR, Boehmer, J, et al. Predictors of sudden cardiac death and appropriate shock in the Comparison of Medical Therapy, Pacing, and Defibrillation in Heart Failure (COMPANION) Trial. Circulation. 2006; 114:2766–2772.
52. Moss, AJ, Hall, WJ, Cannom, DS, et al. Cardiac-resynchronization therapy for the prevention of heart-failure events. N Engl J Med. 2009; 361:1329–1338.
53. Altman, RK, Parks, KA, Schlett, CL, et al. Multidisciplinary care of patients receiving cardiac resynchronization therapy is associated with improved clinical outcomes. Eur Heart J. 2012; 33:2181–2188.
54. Friedman, DJ, Altman, RK, Orencole, M, et al. Predictors of sustained ventricular arrhythmias in cardiac resynchronization therapy. Circulation Arrhythm Electrophysiol. 2012; 5:762–772.
55. Poirier, VL. The heartmate left ventricular assist system: worldwide clinical results. Eur J Cardiothorac Surg. 1997; 11(Suppl):S39–S44.
56. Aliot, EM, Stevenson, WG, Almendral-Garrote, JM, et al. EHRA/HRS Expert Consensus on Catheter Ablation of Ventricular Arrhythmias: developed in a partnership with the European Heart Rhythm Association (EHRA), a Registered Branch of the European Society of Cardiology (ESC), and the Heart Rhythm Society (HRS); in collaboration with the American College of Cardiology (ACC) and the American Heart Association (AHA). Heart Rhythm. 2009; 6:886–933.
57. Sosa, E, Scanavacca, M. Epicardial mapping and ablation techniques to control ventricular tachycardia. J Cardiovasc Electrophysiol. 2005; 16:449–452.
58. Barrett, CD, Di Biase, L, Vacca, M, et al. Ventricular tachycardia ablation—for whom, when, and how? Card Electrophysiol Clin. 2009; 1:201–211.
59. Di Biase, L, Saliba, WI, Natale, A. Successful ablation of epicardial arrhythmias with cryoenergy after failed attempts with radiofrequency energy. Heart Rhythm. 2009; 6:109–112.