[level-membership-for-cardiovascular-category]
Chapter 18 Ventricular Septal Defect with Pulmonary Stenosis
Etienne-Louis Arthur Fallot’s classic publication, L’Anatomie Pathologique de La Maladie Bleue (Figure 18-1A), appeared in 1888 in an obscure journal published in Marseille where Fallot lived throughout his life.1 The malformation reported by Fallot was originally described 1671 by Niels Stensen,2 better known by his Latinized name, Nicholas Steno, who was equally distinguished as anatomist, geologist, and theologian.3,4 Steno wrote, “When I opened the right ventricle … the probe that was passed forward and upward along the interventricular septum entered directly into the aorta just as readily as the probe passed from left ventricle into aorta. The same aortic canal … was common to both ventricles. Thus, the aorta receives blood from both ventricles at the same time … as it partly straddles the right ventricle”4 (Figure 18-1B). In 1872, 16 years before Fallot’s publication, Sir Thomas Watson5 wrote, “The septum between the ventricles was imperfect in its upper part; and the aorta belonged as much to one ventricle as to the other. The pulmonary artery would not admit a goose-quill; the walls of the right ventricle were as thick as those of the left.” The anatomic and clinical features of Fallot’s tetralogy were also described by Eduard Sandifort (1777),4,6 William Hunter (1784),7 James Hope (1839),8 and Thomas Peacock (1866).9 Fallot made an anatomic diagnosis at the bedside, was proven right at postmortem, and coined the term tetralogy.10 He said, “This malformation consists of a true anatomo-pathologic type represented by the following tetralogy: 1) stenosis of the pulmonary artery; 2) interventricular communication; 3) deviation of the origin of the aorta to the right; 4) hypertrophy, almost always concentric, of the right ventricle.”10 Fallot requested that no eulogy be published after his death, but the tetralogy that bears his name remains one of the most familiar eponyms in cardiovascular medicine. The incidence rate is estimated at 1 in 3600 live births.11,12
The four salient anatomic components of Fallot’s tetralogy result from a specific morphogenetic abnormality: malalignment of the infundibular septum.13 In the normal heart, division of the fetal conotruncus culminates in alignment of the infundibular septum with the muscular trabecular septum. In Fallot’s tetralogy, the infundibular septum deviates anteriorly and cephalad and is therefore not aligned with the trabecular septum, creating a ventricular septal defect at the site of malalignment. The deviation of the infundibular septum encroaches on the right ventricular outflow tract and causes infundibular stenosis and a biventricular (overriding) aorta (Figure 18-2).13 The degree of override and the size of the biventricular aorta are determined chiefly by the degree of malalignment, but aortic size is also influenced by an inherent medial abnormality.14 The nonrestrictive malaligned ventricular septal defect accounts for systemic systolic pressure in the right ventricle and concentric right ventricular hypertrophy.
Malaligned ventricular septal defects are located in the perimembranous septum with extension into the infundibular septum.13 Atrioventricular conduction is normal. The crest of the muscular trabecular septum forms the floor of the defect, which is roofed by the valve of the overriding aorta, setting the stage for aortic regurgitation. Subarterial ventricular septal defects, which are more frequent among Asians, are understandably accompanied by aortic regurgitation because a supporting infundibulum is absent.15 Rarely, the ventricular septal defect is part of an atrioventricular septal defect (see Chapter 15).16–18 Muscular ventricular septal defects sometimes coexist but usually close spontaneously in the first year of life. Occasionally, a nonrestrictive malaligned defect is reduced in size by intrusion of accessory or excessive tricuspid valve tissue (see Figure 18-18) that is fixed to the edges of the defect by short chordae tendineae or tethered by long chordae, which permit wide excursions through the defect.19
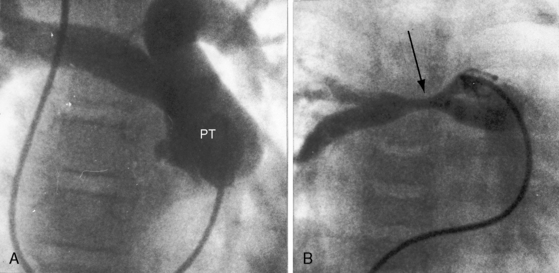
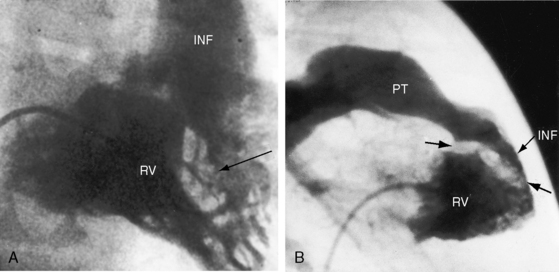
Malalignment of the infundibular septum is the essential but not the only cause of obstruction to right ventricular outflow,13 which also results from hypertrophy of the septoparietal trabeculations, the trabecula septomarginalis, and the infundibular septum (Figure 18-3A). Anterior and cephalad malalignment of the infundibular septum can narrow the entire right ventricular outflow tract (see Figure 18-3).13,20 The pulmonary valve is frequently stenotic and bicuspid (see Figure 18-3B),21 and less frequently unicommissural unicuspid.22 Occasionally, the main site of obstruction is a hypoplastic pulmonary annulus or the ostium of the infundibulum (Figure 18-4).23 The pulmonary trunk, its bifurcation, and its right and left branches may be segmentally or diffusely hypoplastic (Figure 18-5B).23 Rarely, the pulmonary arteries cross as they proceed to their respective lungs.24 Even more rarely, the tetralogy is associated with the scimitar syndrome.25
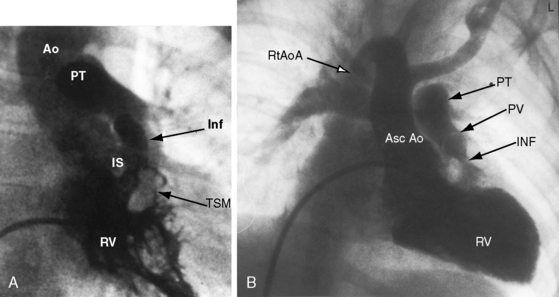
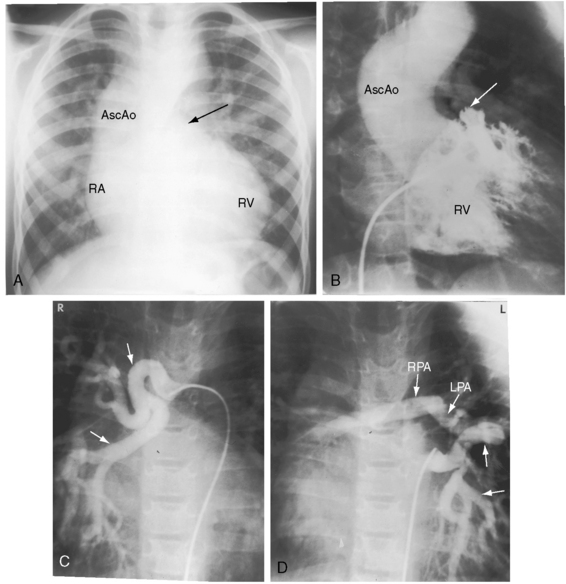
Pulmonary atresia with Fallot’s tetralogy is the ultimate expression of severity.26 The right ventricle terminates blindly against an atretic pulmonary valve or against imperforate muscle (Figures 18-6A and 18-7B). The pulmonary trunk is either a vestigial cord or a hypoplastic funnel-shaped channel that widens as it approaches the bifurcation. The proximal pulmonary arteries are hypoplastic (Figure 18-7D) and may be discontinuous.26 The entire right ventricular output enters the systemic circulation via the nonrestrictive malaligned ventricular septal defect (see Figures 18-6A and 18-7B). The biventricular aorta is dilated (see Figures 18-6A and 18-7A, B) and often continues as a right aortic arch (Figure 18-8A). The lungs are perfused by systemic-to-pulmonary arterial collaterals (see Figures 18-6B and 18-7C, D), on which survival depends (see subsequent).27,28 Exceptionally, the pulmonary circulation is supplied primarily, if not exclusively, by a long, narrow sigmoid-shaped ductus arteriosus (see Figure 18-38B) that is structurally a muscular systemic artery similar to a systemic arterial collateral. This ductal structure is appropriate for intrauterine flow, which is directed from the aorta into the pulmonary artery.
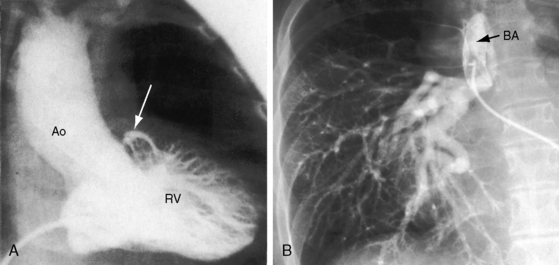
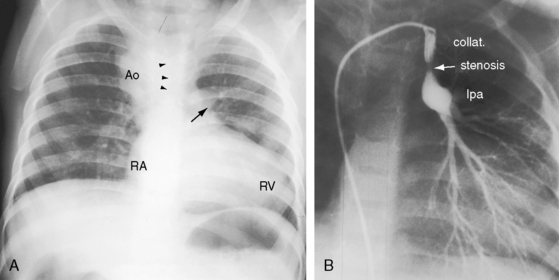
One of the most characteristic features of Fallot’s tetralogy with pulmonary atresia is a pulmonary circulation supplied entirely by collateral arteries that serve both a nutritive function and a respiratory function (gas exchange). The three types of arterial blood supply to the lungs include systemic arterial collaterals; the distinctive ductus arteriosus, which is a muscular systemic artery (see previous); and small diffuse pleural arterial plexuses.27Systemic arterial collaterals are classified according to their origins as: (1) bronchial, which originate where their name indicates and anastomose to pulmonary arteries within the lung (see Figure 18-6B)28,29; (2) direct systemic arterial collaterals, which originate from the descending aorta, enter the hilum, and then assume the structure and distribution of intrapulmonary arteries (see Figure 18-7C, D)28; and (3) indirect systemic arterial collaterals, which originate from the internal mammary, innominate, and subclavian arteries and anastomose to proximal pulmonary arteries outside the lung (see Figure 18-8B).28 Bronchial arterial collaterals are characterized by intrapulmonary anastomoses, direct arterial collaterals by hilar anatomoses, and indirect arterial collaterals by extrapulmonary anatomoses.28 Thus, systemic arterial collaterals anatomose with pulmonary arteries in three locations: (1) intrapulmonary; (2) extrapulmonary; and (3) hilar.28 All three major types of collaterals are present when Fallot’s tetralogy occurs with pulmonary atresia, but only bronchial collaterals are present when the tetralogy occurs with pulmonary stenosis, irrespective of severity.28 About 10% of arterial collaterals originate from coronary arteries.30–32 The pulmonary circulation is effective in gas exchange regardless of the type of systemic arterial collateral.
Direct aortic to pulmonary collaterals originate from intersegmental branches of the dorsal aorta during the third and fourth weeks of gestation.28,29Bronchial arterial collaterals develop in the ninth gestational week after the paired intersegmental arteries have been resorbed28,29 and do not coexist with direct aortic collaterals.28Indirect collaterals arise later in gestation and therefore coexist with bronchial collaterals but not with direct aortic collaterals.28 A particular collateral artery supplies a particular segment of lung, but duplicate blood supplies occasionally occur. A single type of collateral usually predominates in a given patient.28 Lung growth and survival depend on the size and patency of the collateral arteries. Diminished pulmonary blood flow adversely effects the growth of peripheral pulmonary arteries.28
Systemic arterial collaterals have a strong tendency to harbor intimal cushions (proliferations) that serve as sites of potential segmental stenosis (see Figure 18-8B).28 In the absence of these obstructing cushions, large collateral arteries transmit systemic arterial pressure into the pulmonary vascular bed, resulting in morphologic changes analogous to pulmonary vascular disease (see Chapter 14).28,33 Although stenotic sites protect the intrapulmonary resistance vessels from systemic pressure, regional pulmonary blood flow is compromised.
In 1947, Taussig observed that the ductus arteriosus was not structurally normal in Fallot’s tetralogy with pulmonary atresia.34,35 The normal fetal ductus functions as a conduit for right ventricular flow into pulmonary trunk, a function that cannot be served when pulmonary atresia diverts the entire right ventricular output into the aorta via the nonrestrictive malaligned ventricular septal defect. Not surprisingly, the ductus is malformed or absent when pulmonary atresia exists from early fetal life. Absence of a ductus indicates that normal intrauterine ductal function was usurped, rendering the ductus superfluous. If a ductus is present, it is represented by a long narrow branch of the aorta that carries systemic arterial blood from the aorta into the pulmonary trunk.27 This ductus is narrow because it delivers blood only to the lungs, which represents no more than 5% to 10% of the combined ventricular output. The ductus is long because it first runs distally, diverging from the aortic arch, and then turns back to join the proximal left pulmonary artery.
Aortic regurgitation in Fallot’s tetralogy occurs because the malaligned ventricular septal defect is partially roofed by the aortic valve.13,36 Herniation of aortic cusps is more frequent with an isolated subarterial ventricular septal defects than with Fallot’s tetralogy, a difference ascribed to dissimilar flow patterns and their impact on the aortic valve. In cyanotic Fallot’s tetralogy, the aortic valve is not subjected to turbulent flow because the left ventricle ejects directly into the aorta without generating a left-to-right jet. Nevertheless, there is an age-related increase in aortic regurgitation,37 in part as a result of progressive aortic root dilation associated with an inherent medial abnormality.14,38,39 Aortic regurgitation causes volume overload of both ventricles because the aorta is biventricular.37 The incompetent aortic valve is susceptible to infective endocarditis, which can suddenly and catastrophically augment the degree of biventricular regurgitation (see section The History).37 A right aortic arch is a feature of Fallot’s tetralogy.40 Its incidence rate increases as the severity of right ventricular outflow obstruction increases (see Figure 18-3) and reaches approximately 25% with pulmonary atresia (see Figures 18-6A and 18-8A).
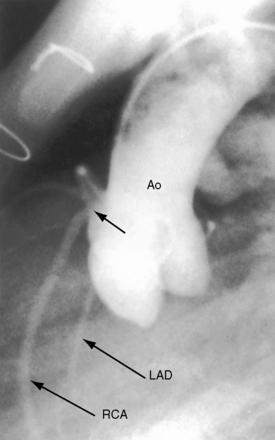
Anomalous origin and distribution of coronary arteries are common (see Chapter 32)30,41,42 and may be of no functional importance but are of considerable surgical importance. The incidence of coronary artery anomalies is influenced by aortopulmonary rotation43 and is higher when the aortic root is anterior to or side-by-side the pulmonary trunk. The most common anomalies are origin of a conus artery or the left anterior descending artery from the right coronary artery or from the right sinus of Valsalva (Figure 18-9).41 Origin of a single coronary artery from the right sinus of Valsalva is less common. Relatively frequent are fistulous communications between coronary arteries and the pulmonary artery30,44 or the right atrium and between coronary arteries and bronchial arteries.32 Rarely, the left anterior descending coronary artery originates from the pulmonary artery,45 or the left coronary artery is intramural.46
Etienne-Louis Fallot recognized that “… at times, there is an additional entirely accessory defect, namely, patency of the foramen ovale.”10 An atrial septal defect occasionally coexists with Fallot’s tetralogy, but the term pentalogy is longer used. Rarely, the tetralogy is associated with total anomalous pulmonary venous connection and an atrial septal defect.47
The combination of right ventricular outflow obstruction and ventricular septal defect is not confined to Fallot’s tetralogy. Pulmonary valve stenosis occasionally occurs with an isolated perimembranous ventricular septal defect (Figure 18-10). The degree of stenosis varies from trivial to severe, the size of the ventricular septal defect varies from small to nonrestrictive, and right ventricular systolic pressure varies from normal to suprasystemic. Other examples of the combination include pulmonary valve stenosis with a muscular ventricular septal defect,48 obstruction to right ventricular outflow caused by protrusion of a large ventricular septal aneurysm into the right ventricular outflow tract (Figure 18-11), and double-chambered right ventricle with a perimembranous49–51 or a malaligned ventricular septal defect (Figure 18-12).20,52 Sir Arthur Keith in his 1909 Hunterian lecture focused on obstructing right ventricular muscular bundles, which consist of an hypertrophied moderator band, an hypertrophied trabecula, or a fibromuscular diaphragm,53 with obstruction ranging from nil to severe to complete.50,54 A ventricular septal defect that communicates with the proximal high-pressure compartment results in a right-to-left shunt.50
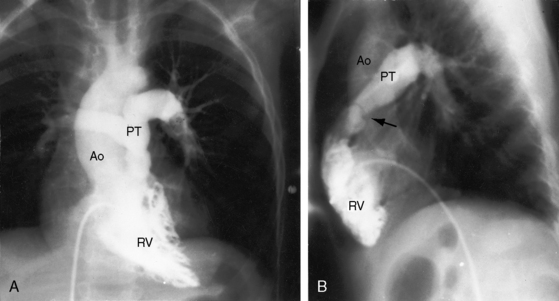
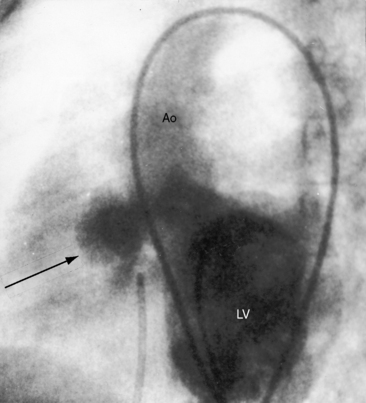
Taussig55 described two infants with a loud holosystolic murmur at the lower left sternal border and increased pulmonary blood flow, prompting the diagnosis of ventricular septal defect. Several years later, both patients were cyanotic with pulmonary stenotic murmurs and decreased pulmonary blood flow, appropriate for the diagnosis to Fallot’s tetralogy. Gasul formalized the notion that progressive infundibular obstruction sometimes occurs with ventricular septal defect (Figure 18-13), and confirmatory reports soon appeared.56–58 The acquired obstruction usually results from hypertrophy of right ventricular muscle bundles, and only rarely from a malaligned infundibular septum.57
The physiologic consequences of Fallot’s tetralogy depend essentially on two variables: the degree of obstruction to right ventricular outflow and, to a lesser extent, systemic vascular resistance. The magnitude and direction of the shunt are determined by the resistance at the site of pulmonary stenosis relative to systemic vascular resistance. When pulmonary stenosis offers lesser resistance, the shunt is left-to-right. When the resistances are equal, the shunt is balanced. When right ventricular outflow resistance exceeds systemic resistance, the shunt is right-to-left. The amount of aortic override is not the issue, although the degree of override tends to coincide with the degree of right ventricular outflow obstruction, which is the issue. When right ventricular blood preferentially flows into the aorta, pulmonary blood flow falls reciprocally, so the left side of the heart is underfilled.59,60 The ultimate expression of right ventricular outflow obstruction is pulmonary atresia, which commits the entire right ventricular output to the aorta. Pulmonary blood flow then depends on systemic arterial collaterals (see previous), which provide the lungs with normal or increased flow, so cyanosis can be mild or even absent.61 Unobstructed flow through arterial collaterals sets the stage for pulmonary vascular disease. Stenoses at intimal cushions in arterial collaterals (see previous) protect the pulmonary vascular bed, but at the price of reduced pulmonary blood flow.
Irrespective of the degree of right ventricular outflow obstruction, right ventricular systolic pressure cannot exceed systemic because the ventricular septal defect is nonrestrictive. Accordingly, a systemic ceiling is placed on the pressure overload that pulmonary stenosis can impose on the right ventricle. When pulmonary stenosis is severe, right ventricular pressure overload is determined by systemic vascular resistance. Increased resistance associated with systemic hypertension or, less commonly, with acquired calcific aortic stenosis (see Figure 18-33B) improves pulmonary blood flow but increases right ventricular afterload. Aortic regurgitation imposes volume load on the already pressure-overloaded right ventricle.
In addition to concentric hypertrophy of the right ventricle, certain morphologic changes are secondary to the physiologic derangements of Fallot’s tetralogy. Tricuspid leaflets develop fibrous thickening because right ventricular systolic pressure is systemic, but the thickened leaflets are seldom incompetent. The right ventricle ejects against systemic resistance without an increase in filling pressure, so right atrial pressure remains normal, and overall systolic function of the hypertrophied right ventricle remains normal (Figure 18-14B, C; see subsequent).60 The underfilled left ventricle tends to be reduced in size with reduced stroke volume.59,60 Low pressure and low flow in the pulmonary circulation alter the small muscular arteries and arterioles and cause thinning of the media with interruption of elastic tissue and widespread thromboses.62–64
When severe pulmonary stenosis occurs with a restrictive ventricular septal defect, right ventricular systolic pressure exceeds systemic and the hypertrophied right ventricle dilates and fails. A physiologically analogous state exists when accessory tricuspid leaflet tissue partially occludes the malaligned ventricular septal defect (see Figure 18-18).19
History
Gender distribution in Fallot’s tetralogy is approximately equal. The malformation recurs in families and has been reported in siblings,65–67 including triplets,68 and in parents and offspring.67,69 Two brothers with DiGeorge syndrome had Fallot’s tetralogy. Birth weight tends to be lower than normal, and growth and development are generally retarded. Hereditary cardiovascular defects in Keeshond dogs include typical Fallot’s tetralogy and isolated ventricular septal defect with pulmonary stenosis.70 Familial tetralogy has been associated with mutation of the jagged 1 gene,71 with NKX2.5 mutations,72 and with chromosome 22q11.2 deletion.73
The clinical course in early infancy is often benign. Mild to moderate neonatal cyanosis tends to increase, but cyanosis may be delayed for months and is coupled with increased oxygen requirements of the growing infant rather than with progressive obstruction to right ventricular outflow.74 Patients seldom remain acyanotic after the first few years of life, and by 5 to 8 years of age, most children are conspicuously cyanotic, with cyanosis closely coupled to the severity of pulmonary stenosis. Infants with Fallot’s tetralogy and pulmonary atresia are mildly cyanotic or acyanotic when collateral flow is abundant.61
In an analysis of survival patterns based on 566 necropsy cases of Fallot’s tetralogy, two thirds of patients reached their first birthday, approximately half reached age 3 years, and approximately a quarter completed the first decade of life.75,76 The attrition rate was then 6.4% per year with 11% alive at age 20 years, 6% at age 30 years, and 3% at age 40 years.75,76 Nevertheless, Fallot’s tetralogy remains the most common cyanotic congenital heart disease after 4 years of age and constitutes a large proportion of adults with cyanotic congenital heart disease. Fallot recognized this tendency when he wrote, “We have seen from our observations that cyanosis, especially in the adult, is the result of a small number of cardiac malformations well determined. One of these cardiac malformations is much more frequent than others…,” namely, the tetralogy to which he referred.10 Fallot’s oldest patient was 36 years of age.10 Survivals between the fifth and seventh decades are uncommon but not rare.12,77–79 A 64-year-old woman with the tetralogy was diagnosed in 1895 by G.A. Gibson, best known for his description of the continuous murmur of patent ductus arteriosus. In 1929, White and Sprague80 published an account of the American composer Henry F. Gilbert who lived a productive life to age 60 years. Another patient played cricket and football as a schoolboy and survived to age 62 years. Patients without repair have lived to age 75 years,81 78 years,82 84 years,83 and 86 years.84 In Fallot’s tetralogy with pulmonary atresia, life expectancy without surgery is as low as 50% in 1 year and 8% in 10 years,75 but adequate collateral blood flow occasionally permits survival into adolescence and adulthood.18,37,78,85,86 One such patient lived to age 54 years,78 and another lived to age 55 years, despite acquired calcific aortic stenosis and regurgitation (see Figure 18-33B.)
Pregnancy is poorly tolerated in females who reach childbearing age.87 The gestational fall in systemic vascular resistance increases the right-to-left shunt, and labile systemic vascular resistance during labor and delivery results in abrupt oscillations in hypoxemia. Fetal wastage is high,23 and live born infants are dysmature.
The neonatal right ventricle is well equipped to eject against systemic vascular resistance because the nonrestrictive ventricular septal defect permits decompression into the aorta.60 The right ventricle has been analyzed in terms of its inlet, apical trabecular, and outlet portions.88 The apical trabecular portion took up the greatest portion of the overload, the outlet portion had decreased ejecting force, and the inlet portion was not significantly affected.88 Right ventricular failure is uncommon.89 However, biventricular failure in the first few weeks of life accompanies pulmonary atresia with excessive flow through large systemic arterial collaterals.61 Accessory tricuspid leaflet tissue that partially occludes the ventricular septal defect results in suprasystemic right ventricular systolic pressure and the right ventricular failure.19,89 Absence of the pulmonary valve (see subsequent section) results in volume overload of the pressure-overloaded right ventricle.90 Systemic hypertension increases left and right ventricular afterload and can induce right ventricular or biventricular failure.89,91 Acquired calcific stenosis of the biventricular aortic valve imposes increased afterload on both the right and the left ventricles (see Figure 18-33B). Regurgitation of a biventricular aortic valve sets the stage for right ventricular failure by imposing volume overload on the already pressure-overloaded right ventricle.37 Infective endocarditis on an incompetent aortic valve can result in catastrophic acute severe biventricular aortic regurgitation.
Isotonic exercise is accompanied by a fall in systemic vascular resistance in the face of fixed obstruction to right ventricular outflow, increasing venoarterial mixing and significantly influencing the dynamics of O2 uptake and ventilation.92,93 Exercise-induced hypoxemia and increased carbon dioxide content stimulate the respiratory center and the carotid body, provoking hyperventilation that is subjectively perceived as dyspnea.92,93
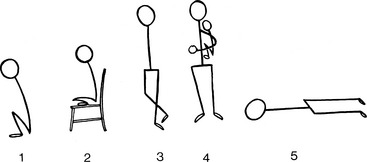
Hypoxic spells, variously called paroxysmal hyperpnea, syncopal attacks, hypoxic or hypercyanotic spells, are dramatic and alarming features of Fallot’s tetralogy.94–96 A typical spell begins with a progressive increase in the rate and depth of breathing and culminates in paroxysmal hyperpnea, deepening cyanosis, limpness, syncope, and occasionally convulsions, cerebrovascular accidents, and death.95,96 Electroencephalographic abnormalities during an hypoxic spell are similar to those of hypoxic episodes of other causes.97 Peak incidence is between the second and sixth month of life, with an occasional spell as early as the first month but comparatively few spells after age 2 years, and only rarely in adults.98 Spells in infants are typically initiated by the stress of feeding, crying, or a bowel movement, particularly after awakening from a long deep sleep.94,95 However, attacks sometimes occur without an apparent precipitating cause, especially in deeply cyanotic infants, although spells are not necessarily related to the degree of cyanosis.95 Spells were originally attributed to infundibular contraction caused by sympathetic stimulation, which was believed to divert right ventricular blood into the aorta,96 but occurrence in patients with pulmonary atresia argued against this theory. It is now believed that vulnerable respiratory control mechanisms, which are especially sensitive after prolonged deep sleep, react to the sudden increase in cardiac output provoked by feeding, crying, or straining, by initiating the following vicious cycle.94,95 As heart rate and cardiac output increase, venous return increases in the face of fixed obstruction to right ventricular outflow, so the right-to-left shunt increases. Infundibular contraction reinforces this pattern but does not initiate it. The increased right-to-left shunt causes a fall in systemic arterial pO2 and pH and a rise in pCO2, a blood gas composition to which a sleep-sensitive respiratory center and carotid body overreact, provoking hyperpnoea, which in turn further increases the cardiac output and perpetuates the cycle. Supraventricular tachycardia and rapid atrial pacing initiate spells by inducing infundibular narrowing, which increases the right-to-left shunt.99 Five mechanisms are therefore involved in the pathogenesis of Fallot spells: (1) an acceleration in heart rate; (2) an increase in cardiac output and venous return; (3) an increase in right-to-left shunt; (4) vulnerable respiratory control centers; and (5) infundibular contraction. Manual compression of the abdominal aorta can abort a spell by decreasing cardiac output and venous return.100 Squatting for relief of dyspnea is a time-honored hallmark of Fallot’stetralogy (Figure 18-15).101,102 In 1784, William Hunter7 made the following observations on the effects of posture: “Any hurry upon his spirits or brisk motion of his body would generally occasion a fit. And for some of the last years of his life he found out by his own observations that when the fit was coming upon him, he would escape it altogether, or at least take considerably from its violence or duration by instantly lying down upon the carpet on his left side, and remaining immovable in that position for about 10 minutes. I saw the experiments made with success.”

Figure 18-15 Typical squatting postures assumed effortlessly in two children with Fallot’s tetralogy.
Taussig described the preference for certain postures other than squatting, namely, the knee-chest position, lying down, or sitting with legs drawn underneath (Figure 18-16).103 Parents may hold their breathless infant upright with its legs flexed against its abdomen (see Figure 18-16, panel 4). Young adults cross their legs during quiet standing or sitting, a relatively ineffective variation. Habitual squatters assume the position effortlessly (see Figure 18-15). The mechanisms by which squatting exerts its beneficial effects are as follows.101,102,104,105 (1) Quiet standing after exercise-induced peripheral vasodilation predisposes to orthostatic hypotension and faintness, a tendency that is exaggerated in hypoxemic patients. Squatting counteracts orthostatic hypotension and diminishes or prevents postexertion orthostatic faintness.102,104 (2) Squatting increases systemic vascular resistance, diverts right ventricular blood into the pulmonary circulation, and increases the amount of oxygenated blood entering the left side of the heart.102,104,106 The left ventricle delivers the larger volume of oxygenated blood into the systemic circulation, so systemic arterial pO2 and pH increase and pCO2 decreases, blunting the stimulus to the respiratory center and carotid body and relieving hyperventilatory dyspnea.104 The effect of squatting on systemic venous return is an even more effective means by which hyperventilatory dyspnea is relieved.104 (3) Isotonic leg exercise reduces the oxygen saturation of venous effluent returning to the heart from the lower extremities. Squatting mechanically curtails lower extremity venous return, decreases the volume of unsaturated venous blood delivered to the heart, and increases the oxygen saturation of right ventricular blood. (4) Right ventricular blood shunted into systemic circulation has a higher oxygen content and pH and a lower pCO2 content.104 (5) The higher pO2 and pH and the lower pCO2 reduce the stimulus to the respiratory center and carotid body and reduce the hyperventilatory dyspnea.
Recurrent hypoxic spells sometimes lead to brain damage and mental retardation. Cerebral venous sinus thromboses and small occult thromboses may become manifest after prolonged hypoxic spells. Hypernasal resonance or nasal speech (velopharyngeal insufficiency) may develop after repeated or prolonged spells because nasal resonance is compromised by improper approximation of the velum (soft palate) and the pharyngeal walls, a disturbance that has been ascribed in part to central nervous system damage caused by hypoxic spells.107
Brain abscess and cerebral embolism add to the list of central nervous system complications.108–110 Iron-deficient erythrocytosis in patients less than 4 years of age increases the risk of cerebral venous sinus thrombosis.110 Wheezing and stridor have been attributed to tracheal compression by an enlarged aorta.14,111 A stenotic pulmonary valve112 and an incompetent aortic valve113 are substrates for infective endocarditis.
The physiologic consequences and clinical course of a nonrestrictive ventricular septal defect are favorably influenced by mild to moderate acquired obstruction to right ventricular outflow (see previous57 and Chapter 17). The clinical picture initially resembles an isolated nonrestrictive ventricular septal defect with large left-to-right shunt (see Figure 18-13A.) With the development of right ventricular outflow obstruction, excessive pulmonary blood flow and volume overload of the left ventricle are curtailed,56,58,74 symptoms related to the left-to-right shunt diminish, and physical development improves. Obstruction to right ventricular outflow may progress sufficiently to reverse the shunt, resulting in late onset cyanosis (see Figure 18-13B).
When a restrictive ventricular septal defect is accompanied by severe pulmonary valve stenosis, the clinical picture resembles isolated pulmonary stenosis with intact ventricular septum (see Chapter 11). A restrictive ventricular septal defect with mild pulmonary stenosis is associated with a conspicuous murmur and few or no symptoms but with the risk of infective endocarditis.
Physical Appearance
Patients with cyanotic Fallot’s tetralogy are as a rule physically underdeveloped,114 and infants with excessive collateral arterial blood flow accompanying pulmonary atresia have development of congestive heart failure and the catabolic effects of poor physical development.
Cyanosis varies from absent to severe and is symmetrically distributed. John Hunter described such a patient: “I was consulted about a young gentleman’s health. From his infancy, every considerable exertion produced a seeming tendency to suffocation and a change from the scarlet tinge to the modena or purple.”115 Cyanosis may become manifest only after crying, feeding, or exercise when the accompanying stress increases venous return to the obstructed right ventricle and augments the right-to-left shunt. When there is a history of squatting or an analogous posture, it is useful to have the patient or parent illustrate the posture, so it can be witnessed by the examiner (see Figures 18-15 and 18-16).
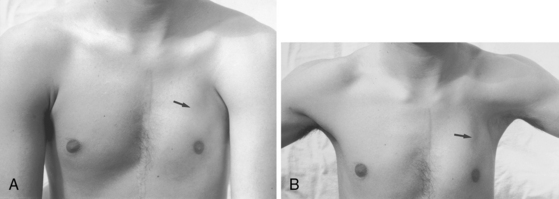
Fallot’s tetralogy is associated with a number of distinctive phenotypes116: CATCH 22 monosomy 22q11.2,117,118 Down trisomy 21,16,17 velocardiofacial (Shprintzen-Goldberg) syndrome,119 Goldenhar’s syndrome120 (oculo-auriculo-vertebral dysplasia), absent thumb and first metacarpal,121 absence of a pectoralis major muscle (congenital pectoral dysplasia or Poland’s syndrome; Figure 18-17),1 syndactyly, brachydactyly with hypoplasia of the ipsilateral hand,122 underdevelopment of the left arm secondary to an isolated left subclavian artery, and hypoplsia of a hand.123
Arterial Pulse
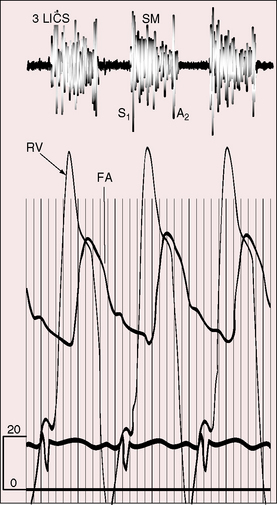
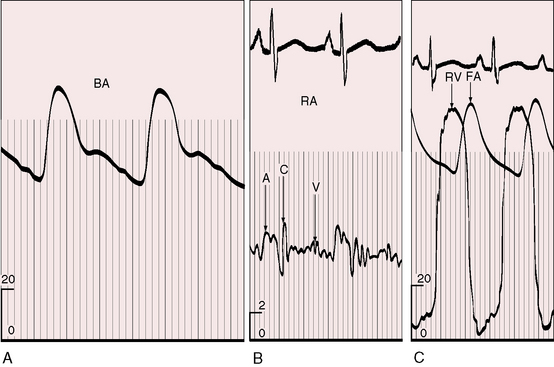
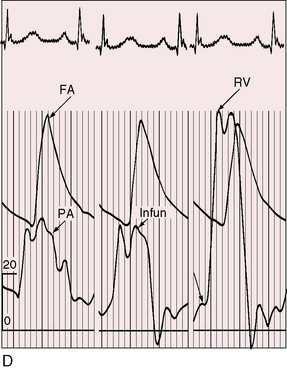
The arterial pulse is normal, irrespective of the severity of pulmonary stenosis (see Figure 18-14C, D). When the shunt is balanced, the left ventricle maintains a normal stroke volume, and when there is severe pulmonary stenosis or atresia, a reduced left ventricular stroke volume is supplemented by right ventricular blood ejected directly into the aorta.60 A brisk arterial pulse with wide pulse pressure is reserved for large systemic arterial collateral flow61 or aortic regurgitation.
An accurate estimate of the right ventricular outflow gradient requires little more than the bedside determination of blood pressure. Right ventricular systolic pressure is systemic (see Figure 18-14C, D) so the stenotic gradient is the difference between the cuff brachial arterial systolic pressure and an estimated pulmonary arterial systolic pressure of 15 to 25 mm Hg, depending on age and the severity of pulmonary stenosis. The estimate is further refined by taking into account that pulmonary arterial pressure is lowest when cyanosis is severe and normal when cyanosis is mild.
Jugular Venous Pulse
The neonatal right ventricle has an inherent capacity to eject against systemic resistance without extra help from its atrium. In Fallot’s tetralogy, resistance to right ventricular discharge is at, but not above, systemic because the nonrestrictive ventricular septal defect permits decompression into the aorta (see Figure 18-14C, D.) The right ventricle maintains its neonatal capacity to eject at systemic resistance without increasing its filling pressure. Accordingly, the right atrium is not required to increase its contractile force, so the jugular venous pulse is normal in height and wave form (see Figure 18-14B).60 If accessory tricuspid leaflet tissue partially occludes the ventricular septal defect, right ventricular systolic pressure exceeds systemic (Figure 18-18) and the jugular A wave becomes prominent. With systemic hypertension, the right ventricle contracts from an increased end-diastolic fiber length induced by forceful right atrial contraction, which is reflected in the jugular venous pulse as an increase in the A wave. Acquired stenosis of the biventricular aortic valve has a similar effect on the right ventricle and right atrium. A minor feature of the jugular venous pulse is related to a persistent left superior vena cava.124 The left jugular pulse is then more prominent than the right.124,125
Precordial Movement and Palpation
In 1839, James Hope8 described an “increase of pulsation at the inferior part of the sternum” as a sign of right ventricular hypertrophy. The right ventricle in Fallot’s tetralogy ejects at systemic pressure with little or no increase in its force of contraction. Accordingly, the accompanying precordial impulse is gentle, analogous to the impulse of a normal neonatal right ventricle. The right ventricular impulse is relegated to the fourth and fifth left intercostal spaces and subzyphoid area because the stenosis is infundibular (see Figure 18-14D.) A left ventricular impulse is conspicuous by its absence because the left ventricle is underfilled. Abundant flow through large systemic arterial collaterals augments left ventricular filling, but even then, a left ventricular impulse is seldom palpated. Subinfundibular stenosis or double-chambered right ventricle (see Figure 18-12) relegates the right ventricular impulse to the fifth left intercostal space and subxyphoid area.
A dilated right aortic arch (see Figure 18-6) reveals itself by an impulse at the right sternoclavicular junction. The aortic component of the second heart sound is often palpable in the second left intercostal space because a hypoplastic or atretic anterior pulmonary trunk is all that guards the enlarged aortic root. Systolic thrills do not originate at sites of severe stenosis because right ventricular blood flow is diverted from the pulmonary trunk into the aorta. In acyanotic Fallot’s tetralogy, the lesser degree of obstruction permits sufficient flow across the stenotic site to generate a thrill.
Auscultation
The physiology of Fallot’s tetralogy is nicely reflected in the accompanying auscultatory signs. Ejection sounds originate in a dilated aorta (see Figures 18-6A and 18-7A) and are therefore important auscultatory signs of severe pulmonary stenosis or atresia (Figures 18-19 through 18-23).74,86,112 The aortic ejection sound is maximal at the upper right sternal border, but when loud, it is heard along the left sternal border and toward the apex. The ejection sound may selectively decrease with inspiration (see Figure 18-19), even though it originates in the aortic root rather than in the pulmonary valve (see Chapter 11). Pulmonary ejection sounds are absent because the stenotic bicuspid pulmonary valve is not sufficiently mobile (see Figure 18-3).
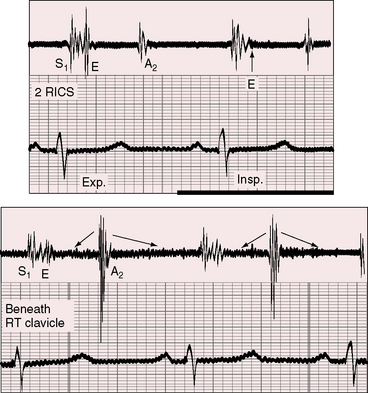
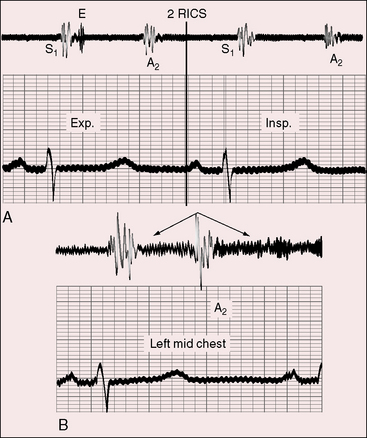
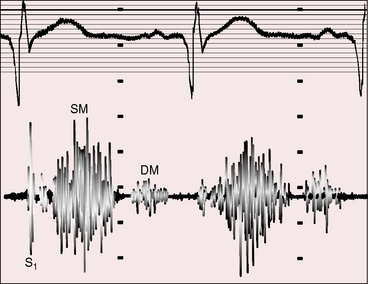
Nearly 50 years before Fallot’s report, James Hope8 wrote: “A loud superficial murmur with the first heart sound in the third left intercostal space may proceed from a contraction of the pulmonary orifice or from an opening out of the right ventricle into the left ventricle, or from both these lesions conjoined. When these lesions coincide with cyanosis, the double lesion is almost positive and an increase of pulsation at the inferior part of the sternum, indicative of right ventricular hypertrophy is a corroborative circumstance.” The murmur that Hope described referred to cyanotic Fallot’s tetralogy and originated at the site of stenosis rather than across the ventricular septal defect (Figure 18-24).126 The murmur is maximal in the third left intercostal space because the stenosis is infundibular. Subinfundibular stenosis results in a lower location of the murmur.51 The duration and configuration of the systolic murmur are determined by the balance between resistance at the site of stenosis and resistance in the systemic vascular bed. Changes in this balance are reflected in changes in the length and loudness of the systolic murmur. These auscultatory signs are closely coupled with the severity of pulmonary stenosis (see Figure 18-20).112 A holosystolic murmur extending up to the aortic component of the second heart sound reflects a left-to-right shunt across the ventricular septal defect (see Figure 18-20B). As pulmonary stenosis increases, the shunt murmur becomes decrescendo, diminishing and ending before the aortic component of the second sound (see Figure 18-20C). When the shunt is balanced, the ventricular septal defect is silent and the previously obscured pulmonary stenotic murmur emerges (see Figure 18-20D). A further increase in the degree of pulmonary stenosis diverts right ventricular blood into the aorta and away from the pulmonary trunk, so the pulmonary stenotic murmur becomes shorter and softer (Figure 18-20E). Severe pulmonary stenosis is reflected in an even shorter and softer systolic murmur and by an ejection sound that is generated in the dilated aortic root (see Figure 18-20F). Pulmonary atresia abolishes right ventricular-to-pulmonary arterial flow and abolishes the pulmonary stenotic murmur. An aortic ejection sound may be followed by no murmur at all (see Figure 18-20G) or a trivial midsystolic murmur into the dilated aorta.
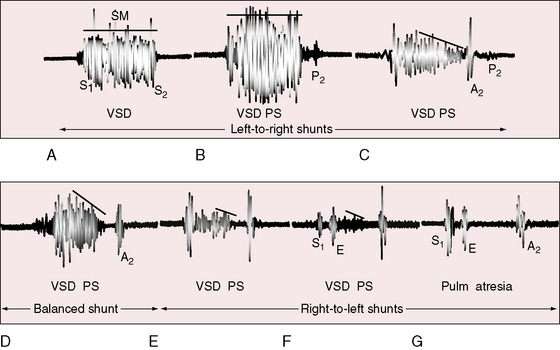
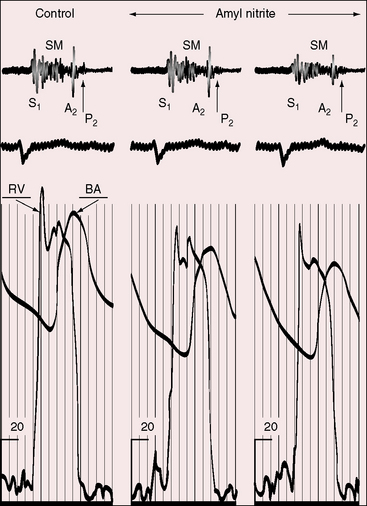
During hypoxic spells, pulmonary arterial blood flow sharply declines, the pulmonary stenotic murmur shortens and softens, and with loss of consciousness, the murmur disappears.96,112 Vasoactive drugs induce analogous changes by altering systemic vascular resistance.112,127 Pressor agents increase systemic vascular resistance and increase the resistance to right ventricular discharge into the aorta.91 The right-to-left shunt decreases, right ventricular blood is diverted into the pulmonary artery, and the pulmonary stenotic murmur becomes louder and longer. Amyl nitrite has the opposite effect, inducing a decrease in systemic vascular resistance, a decrease in resistance to right ventricular discharge into the aorta, and a decrease in flow into the pulmonary trunk, with softening and shortening of the stenotic murmur (Figure 18-25).
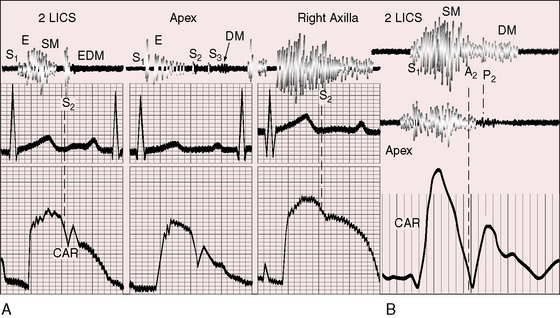
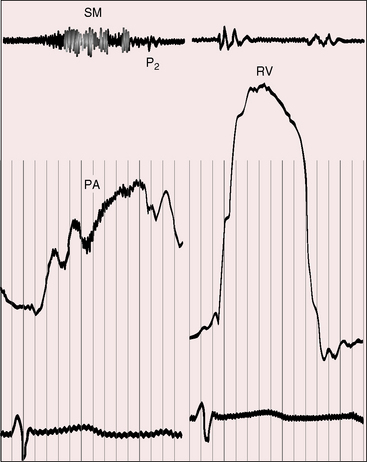
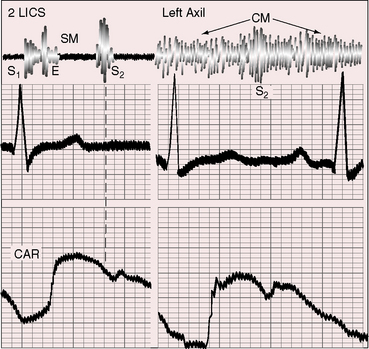
Continuous murmurs are auscultatory signs of pulmonary atresia and occur in more than 80% of such patients (see Figures 18-19 and 18-21 through 18-23). Continuous murmurs originate in direct and indirect systemic arterial collaterals (see previous) and therefore do not occur in Fallot’s tetralogy with pulmonary stenosis in which collaterals are confined to bronchial arteries (see Figure 18-6B).28 The intensity of continuous murmurs ranges from grade 3/6 to soft and easily overlooked, especially at nonprecordial sites. Continuous murmurs are heard beneath the clavicles, in the back, to the right and left of the sternum, and in the right and left axillae (see Figures 18-21 through 18-23). Thoracic locations vary from patient to patient, and from time to time, in the same patient.86,128 Continuous murmurs may peak before and after the second heart sound (see Figures 18-21 and 18-23) or may be more prominent in systolic, as with other arterial continuous murmurs (see Figure 18-22), creating the mistaken impression of a long intracardiac systolic murmur, especially if the murmur is located at the left sternal edge.
Diastolic murmurs are caused by aortic regurgitation, or much less frequently, by absent pulmonary valve (see subsequent section).112 The aortic regurgitant murmur is typically high-frequency decrescendo, beginning with the prominent single aortic component of the second heart sound (see Figure 18-22A). Continuous murmurs from collateral circulation obscure the murmur of aortic regurgitation. A brisk arterial pulse may result from abundant collateral flow rather than aortic regurgitant flow. However, conspicuous cyanosis with a brisk arterial pulse favors aortic regurgitation because arterial collaterals large enough to cause bounding pulses are also large enough to increase pulmonary arterial blood flow and minimize the cyanosis. Large collateral arteries that deliver abundant pulmonary blood flow occasionally cause a mitral mid-diastolic murmur (see Figure 18-22A). Rarely, a bicuspid pulmonary valve is incompetent, generating a delayed medium-frequency diastolic murmur of low-pressure pulmonary regurgitation (see Figure 18-22B).
The pulmonary component of the second heart sound is soft or absent because right ventricular blood preferentially enters the aorta, so pulmonary blood flow and artery pressure are abnormally low. A delay in the pulmonary component is the result of the relatively long interval required for high right ventricular systolic pressure to fall below the low pulmonary arterial diastolic incisura and to delayed relaxation of the infundibulum that contributes to late pulmonary valve closure by supporting the column of blood in the pulmonary trunk after the right ventricle has begun to relax.112,129
With mild or absent cyanosis, a soft delayed pulmonary component can be detected (see Figures 18-20C and 18-24).129,130 When cyanosis is marked, the pulmonary component is typically inaudible, and with pulmonary atresia, there can be no pulmonary component because there is no functional pulmonary valve. Amyl nitrite inhalation reduces pulmonary arterial flow still further, so an audible pulmonary second sound disappears (see Figure 18-25).127,131 Inaudibility also results from thickened bicuspid pulmonary leaflets that preclude brisk closing excursions. In 1866, Thomas Peacock9 wrote: “The aorta is unusually large and from the powerful reaction on the valves during diastole of the heart, a loud ringing second sound is heard on listening at the upper part of the sternum.” Peacock’s loud ringing second sound was the loud aortic component (Figures 18-23 and 18-26).
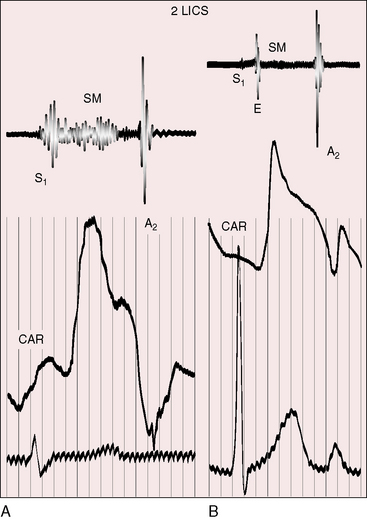
Fourth heart sounds are exceptional because the force of right atrial contraction is not increased. Third heart sounds rarely occur on either side of the heart because right ventricular failure is uncommon and left ventricular filling is reduced. With systemic hypertension, right ventricular or biventricular failure may be accompanied by third and fourth heart sounds. With pulmonary atresia and large systemic to pulmonary arterial collaterals, increased blood flow across the mitral valve sometimes generates a left ventricular third sound and a short mid-diastolic murmur (see Figure 18-22).
Auscultatory signs in severe pulmonary valve stenosis with a restrictive perimembranous ventricular septal defect are similar to if not identical to the auscultatory signs of severe isolated pulmonary stenosis. Suprasystemic right ventricular systolic pressure abolishes the left-to-right shunt through the ventricular septal defect, so a long pulmonary stenotic murmur exists alone.132 A loud long pulmonary stenotic murmur is also heard when accessory tricuspid leaflet tissue partially occludes the ventricular septal defect (see Figure 18-18).132
Electrocardiogram
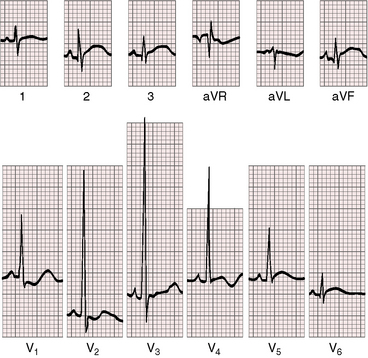
The electrocardiogram tends to be a reliable reflection of the physiology of Fallot’s tetralogy.133,134 Because the force of right atrial contraction is not increased and the right atrium is not enlarged, P waves are often normal in young patients and are seldom increased in amplitude but may be peaked (Figures 18-27 and 18-28). P wave duration tends to be short (see Figure 18-27) because the underfilled and relatively small left atrium writes the terminal force.135–137 The PR interval is normal because the conduction system is normal.
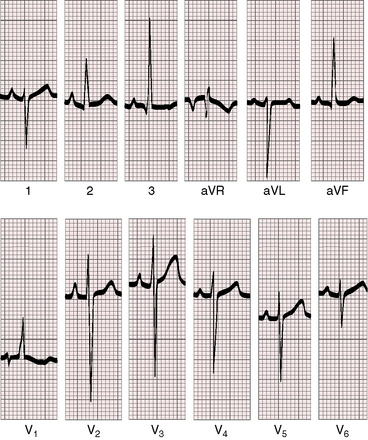
The QRS axis is the same as that of a healthy newborn (see Figure 18-27).134,138 However, the axis and direction of ventricular depolarization do not change as the neonate matures because the functional demands on the right ventricle do not change (see Figures 18-27 and 18-28).134,138 The QRS complex is neither notched or slurred, and the duration is normal,133,134 but peripheral conduction delay occurs in an occasional adult.139 Left axis deviation with counterclockwise depolarization is reserved for Fallot’s tetralogy with an atrioventricular septal defect (see Chapter 15).140
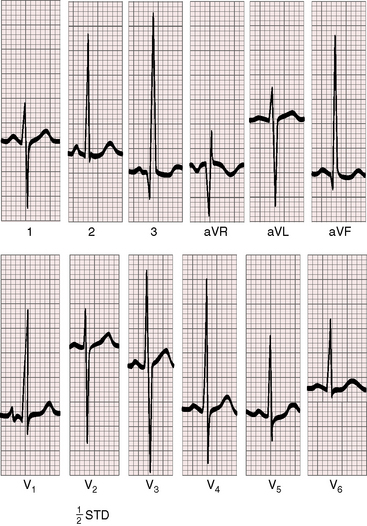
Right ventricular hypertrophy is characterized by a tall monophasic R wave confined to lead V1, with an abrupt change to an rS pattern in V2 (Figures 18-27, 18-28, and 18-29).133,134,136,138 The presence and depth of Q waves and the amplitude of R waves in leads V5-6 are sensitive signs of the magnitude of pulmonary blood flow and of left ventricular filling. Reduced pulmonary flow with an underfilled left ventricle is accompanied by rS patterns in leads V2-6 (see Figure 18-27). A balanced shunt is accompanied by small q waves and well-developed R waves in lead V5-6 (see Figure 18-28). A left-to-right shunt is accompanied by deeper left precordial Q waves and relatively tall R waves (see Figure 18-29).
Right precordial T waves are upright or inverted with almost equal frequency (see Figures 18-27 through 18-29). The deeply inverted right precordial T waves that characterize severe pulmonary stenosis with an intact ventricular septum seldom occur because right ventricular systolic pressure seldom exceeds systemic.
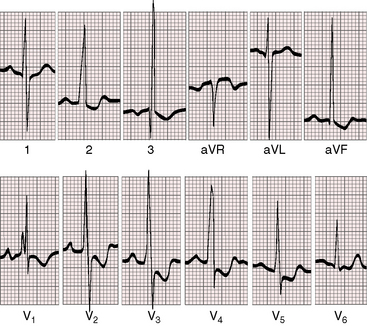
With pulmonary atresia and an abundant collateral arterial circulation, P waves are broad and bifid because of increased flow into the left atrium. Q waves with well-developed R waves appear in leads V5-6 because of increased flow into the left ventricle. ST segment and T wave abnormalities may be found in midprecordial leads (Figure 18-30).
Acquired obstruction to right ventricular outflow with a nonrestrictive ventricular septal defect results in an electrocardiogram that initially resembles isolated ventricular septal defect of equivalent size. Right ventricular systolic pressure remains unchanged, but pulmonary blood flow and left ventricular volume necessarily decrease. As outflow obstruction increases, left precordial qR complexes are less prominent and tall right precordial R waves persist.74
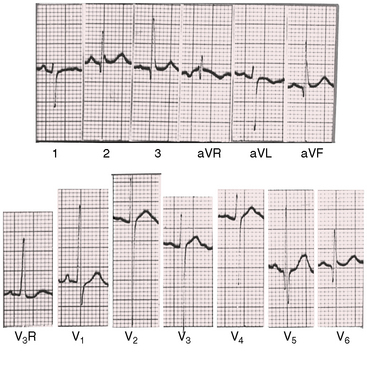
In double-chambered right ventricle, the increase in right ventricular pressure and mass are confined to the hypertensive proximal compartment, so precordial leads display a normal QRS progression from V1-6 (Figure 18-31).49,141 An upright T wave in lead V3R may be the only electrocardiographic sign of right ventricular hypertrophy.49
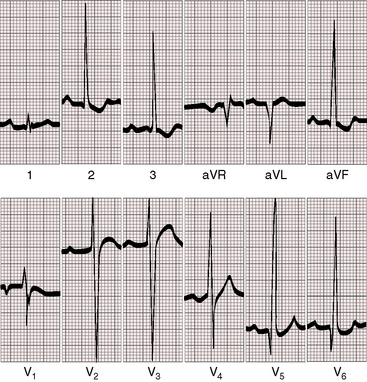
The electrocardiogram of a restrictive ventricular septal defect with severe pulmonary valve stenosis is indistinguishable from isolated pulmonary stenosis with intact ventricular septum.132Restrictive ventricular septal defects with mild pulmonary stenosis are associated with modest signs of right ventricular hypertrophy manifested by a relatively vertical QRS axis, an rSr′ pattern in lead V1, and persistent upright right precordial T waves.
X-Ray
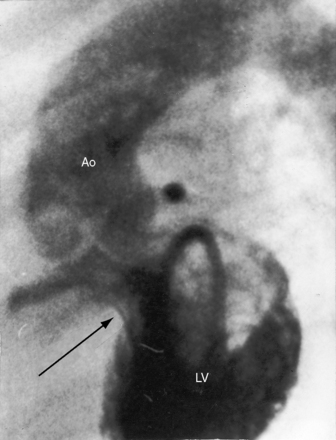
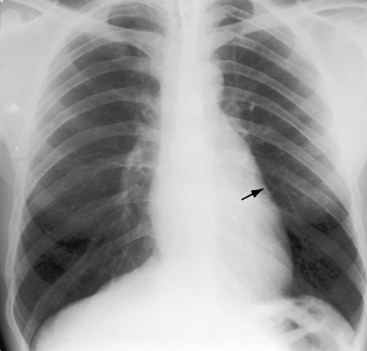
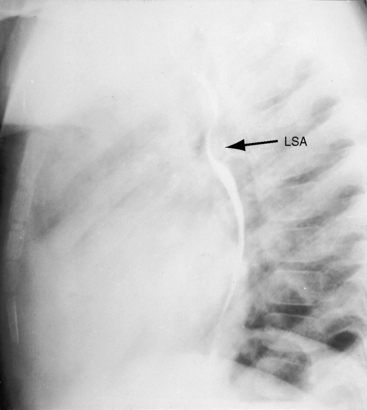
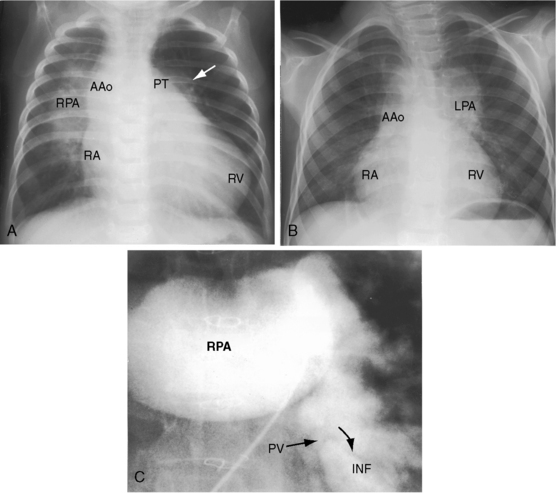
In cyanotic Fallot’s tetralogy, pulmonary vascularity is reduced and the middle and outer thirds of the lung fields display a paucity of vascular markings because of a reduction in size of intrapulmonary arteries and veins (Figure 18-32).142 The right lung is occasionally less vascular than the left.143 In pulmonary atresia, there is a lacy reticular pattern without the normal diminution in vessel caliber toward the periphery because systemic arterial collaterals anastomose with segmental or lobar intrapulmonary arteries (Figure 18-33A).133,144 When systemic arterial collaterals or bronchial collaterals anastomose with hilar or extrapulmonary arteries, intrapulmonary branching is normal (see Figures 18-6A, 18-7C, D, and 18-8B). The patterns of collateral arterial circulation are not uniform, with some areas oligemic and others normal or hypervascular (Figure 18-34). Systemic collateral arteries rarely cause rib notching because they do not run in intercostal grooves.145
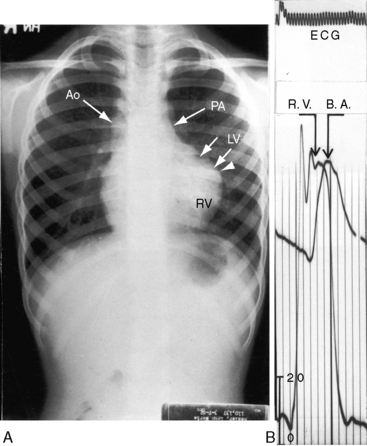
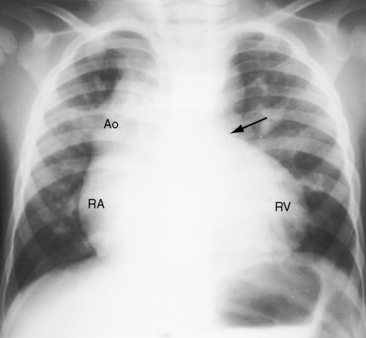
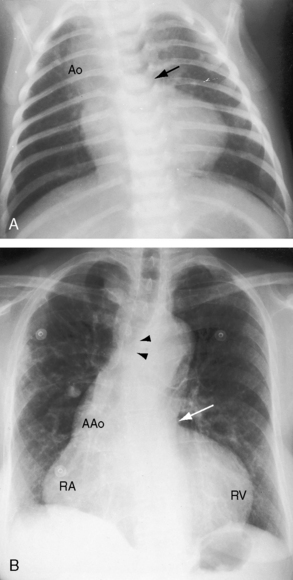
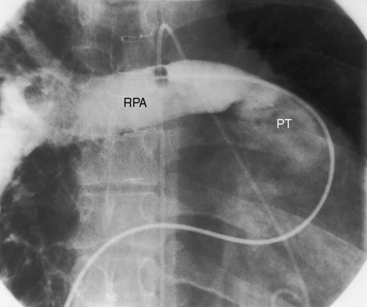
Figure 18-34 X-ray from the 3-year-old boy with Fallot’s tetralogy and pulmonary atresia (angiograms in Figure 18-7). Pulmonary vascularity is not homogeneous because systemic arterial collateral flow is not uniform (see Figure 18-7). The ascending aorta (Ao) is dilated, and the main pulmonary artery segment is concave (arrow). A prominent right atrium (RA) occupies the lower right cardiac border. The apex-forming right ventricle (RV) is not boot-shaped because the left ventricle was well-developed from adequate filling from systemic arterial collaterals.
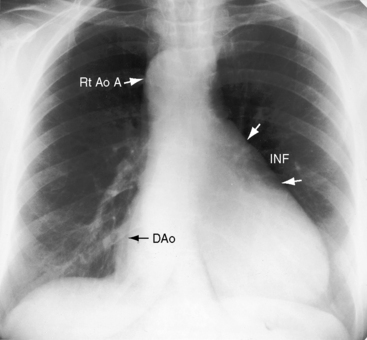
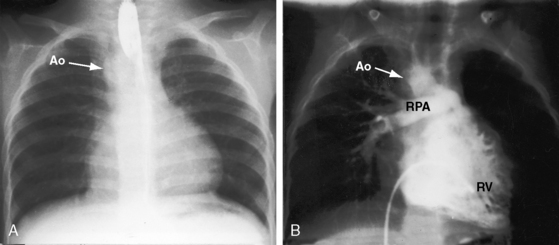
The concave main pulmonary artery segment of pulmonary atresia stands out in bold contrast to the dilated right aortic arch (see Figures 18-32 and 18-33A).86 The size of the ascending aorta tends to vary inversely with the size of the pulmonary trunk (see Figure 18-3), culminating in the conspicuously dilated ascending aorta of pulmonary atresia (see Figure 18-33). However, dilation of the ascending aorta is also determined by inherent medial abnormalities.14,38,39,146 There is a right aortic arch in 20% to 30% of patients with Fallot’s tetralogy, with the highest incidence rate in pulmonary atresia (see Figures 18-6A and 18-33A).40,86 A right arch is typically accompanied by a right descending aorta, which runs as a fine line parallel to the vertebral column (Figures 18-33 and 18-36). A right aortic arch indents the right side of the trachea (see Figure 18-8A), and a left aortic arch indents the left side of the trachea (see Figure 18-33B). Occasionally, there is a double aortic arch.147 A dilated anomalous left subclavian artery may pass behind the esophagus and cause localized posterior indentation (Figure 18-35).148 A left superior vena cava casts its shadow at the left thoracic inlet.149
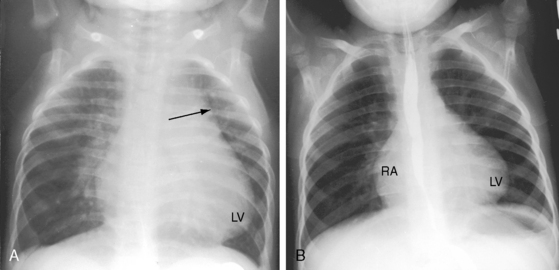
The size of the heart in cyanotic Fallot’s tetralogy is normal (see Figure 18-32).103 The right atrium and right ventricle cope with systemic resistance without dilating, and the left atrium and left ventricle are underfilled and are therefore small. In pulmonary atresia, cardiac size tends to be larger in response to flow through systemic arterial collaterals (see Figures 18-33 and 18-34).86
The configuration of the heart has long been a subject of interest in Fallot’s tetralogy because of the distinctive boot-shaped or coeur en sabot appearance (see Figure 18-32).103,150 The configuration has also been likened to a golf club wood or driver, especially when a right aortic arch and a concave pulmonary artery segment accentuate the configuration of the left cardiac border (see Figures 18-6A and 18-8A). The boot shape results from the combination of a small underfilled left ventricle that lies above a horizontal ventricular septum, inferior to which is a concentrically hypertrophied but nondilated right ventricle (see Figure 18-32).150 The coeur en sabot is uncommon in neonates because intrauterine left ventricular volume is normal. In pulmonary atresia, the boot shape may be present at birth because of a reduction in pulmonary blood flow and left ventricular volume caused by atresia of the fetal ductus arteriosus (see Figure 18-33A). In acyanotic Fallot’s tetralogy or with pulmonary at resia and large systemic arterial collaterals, a well-formed convex apex is the result of normal if not increased left ventricular filling (see Figure 18-34). When right ventricular outflow obstruction is caused by stenosis of the ostium of the infundibulum, a localized indentation marks the site of obstruction, above which the infundibular chamber is slightly convex (see Figures 18-4 and 18-36).
A nonrestrictive ventricular septal defect with acquired obstruction to right ventricular outflow exhibits an initial increase in pulmonary vascularity, with enlargement of the left ventricle, left atrium, and pulmonary trunk (see Figure 18-13A). Progressive obstruction results in a decline in pulmonary arterial blood flow and in normalization of the size of the left atrium and left ventricle (see Figure 18-13B).74 When a nonrestrictive perimembranous ventricular septal defect occurs with pulmonary valve stenosis, the radiologic picture is determined by the degree of pulmonary stenosis. The size of the ascending is normal, the pulmonary trunk is dilated, and the ventricular septum is vertical rather than horizontal (see Figure 18-10). When a malaligned ventricular septal defect is partially occluded by tricuspid leaflet tissue, the right ventricle may fail and dilate. When a restrictive perimembranous ventricular septal defect occurs with mild pulmonary valve stenosis, the x-ray resembles isolated pulmonary valve stenosis.
Echocardiogram
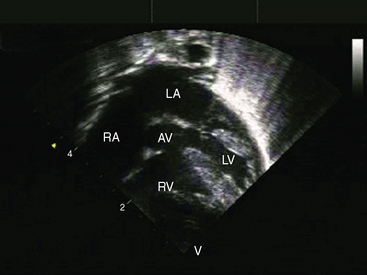
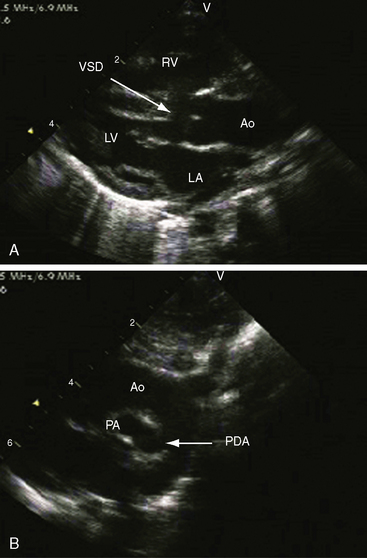
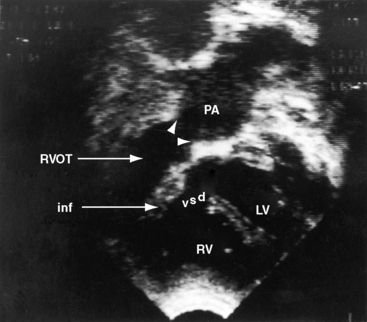
Echocardiography with color flow imaging and Doppler interrogation provides morphologic and physiologic assessment of Fallot’s tetralogy and its variations.151,152 Thymic aplasia or hypoplasia identifies patients with DiGeorge syndrome.153 Continuous wave Doppler scan establishes the outflow gradient and the combination of valvular and infundibular pulmonary stenosis (Figure 18-37). The malaligned ventricular septal defect is recognized by anterior and cephalad deviation of the infundibular septum and a biventricular aorta (Figures 18-38 and 18-39). The pulmonary trunk and its proximal branches can be visualized (see Figure 18-39). A right aortic arch is imaged from suprasternal notch and subclavicular windows, and the presence and degree of aortic regurgitation can be established with color flow. Echocardiography identifies tricuspid leaflet tissue partially occluding the malaligned ventricular septal defect, and color flow imaging with continuous-wave Doppler interrogation establishes the degree of restriction across the partially obstructed defect. Pulmonary atresia is diagnosed when pulmonary valve echoes are absent and when color flow imaging and continuous wave Doppler scan detect no flow across the right ventricular outflow tract (see Figure 18-38A). The distinctive narrow serpentine elongated arterialized ductus arteriosus can be identified (see Figure 18-38B).
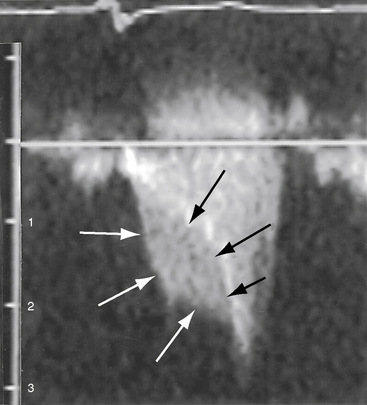
Obstructing or nonobstructing subinfundibular muscle bundles of a double-chambered right ventricle can be identified, the degree of obstruction can be established with color flow and Doppler interrogation (Figure 18-40 and Videos 18-1A and 18-1B), and a coexisting perimembranous ventricular septal defect can be detected.154
Fallot’s tetralogy with absent pulmonary valve
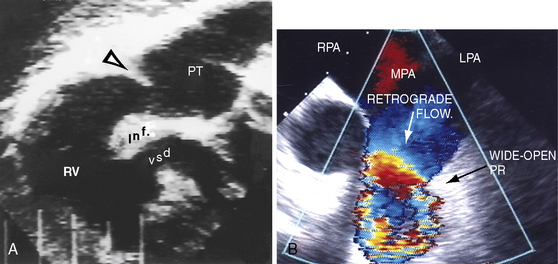
In 1847, Chevers155 described the combination of absent pulmonary valve, ventricular septal defect, annular stenosis, and dilation of the pulmonary trunk and its branches. The malformation was confirmed in 1908 by Royer and Wilson156 and reconfirmed in 1927 by Kurtz, Sprague, and White.157 The incidence rate of absent pulmonary valve with Fallot’s tetralogy ranges from 2.4% to 6.3%.158 Pulmonary valve tissue is lacking completely or consists of rudimentary remnants of avascular myxomatous connective tissue. Rarely, absence of the pulmonary valve occurs with absence of a pulmonary artery (see subsequent section)159,160 and with systemic to pulmonary artery collaterals.161 Obstruction to right ventricular outflow resides at the narrow pulmonary annulus, not at the malaligned infundibular septum.159 The pulmonary trunk, especially its proximal branches, dilates massively (Figure 18-41) together with the infundibulum (see Figure 18-41C).90,159,162
The malformation can be recognized in the fetus.163 Regurgitant volume in utero is returned to the pulmonary trunk during each right ventricular systole, distending the pulmonary trunk and its branches, more so when egress is curtailed by agenesis of the ductus arteriosus,163 even though decompression is achieved via the nonrestrictive ventricular septal defect.163Diastolic collapse of the central pulmonary arteries occurs after each systole, and diastolic flow is accelerated by elastic recoil of the proximal pulmonary arteries.163 Medial abnormalities in the walls of the dilated proximal pulmonary arteries have been attributed to abnormal flow patterns in utero, but inherent medial abnormalities contribute materially to the massive dilation (Figures 18-41 and 18-42).14,164 Morphometric studies reveal a bizarre pattern of abnormal hilar branching in addition to abnormalities of the proximal pulmonary artery.165 Tufts of pulmonary arteries are entwined among compressed intrapulmonary bronchi. Compression of small bronchi by abnormal branching patterns of intrapulmonary arteries together with compression of the trachea and bronchi by dilated proximal pulmonary arteries are major complications of the malformation.159,166 An important feature of the experimental fetal rat model of congenitally absent pulmonary valve is the degree of bronchial deformity that suggests an inherent bronchial abnormality as an essential part of the syndrome.167
The physiologic consequences of Fallot’s tetralogy with absent pulmonary valve are borne by the right ventricle, which is subjected to the massive volume overload of severe pulmonary regurgitation in addition to the resistance to discharge incurred by annular obstruction.160 The doubly beset right ventricle dilates and fails, its filling pressure rises and equilibrates with pulmonary arterial diastolic pressure, and right atrial pressure rises in parallel. Right ventricular failure and tracheobronchial compression are responsible for the high morbidity and mortality rates.168
History
The malformation causes respiratory distress (tracheobronchial obstruction) and right ventricular failure soon after birth, but an occasional patient experiences infancy with surprisingly few symptoms.159 The symptoms occasionally improve during the course of tracheobronchial maturation, although emphysema, atelectasis, and pulmonary infection are common.159 Early cyanosis is the result of respiratory distress, and a right-to-left shunt diminishes with age because a fall in pulmonary vascular resistance decreases pulmonary regurgitant flow, decreases volume overload of the pressure-overloaded right ventricle, and decreases the right-to-left shunt.
Arterial Pulse, Jugular Venous Pulse, and Precordial Palpation
Right ventricular failure curtails aortic flow and reduces the systemic arterial pulse. The jugular venous pulse is elevated in parallel with the elevated right ventricular filling pressure. The A wave remains dominant until tricuspid regurgitation obliterates the X descent and increases the crest of the V wave. The right ventricular impulse is especially dynamic because of the combined effects of volume and pressure overload. A dilated infundibulum (see Figure 18-41C) is readily palpated in the third left intercostal space, and a dilated pulsatile pulmonary trunk is palpated in the second left intercostal space (see Figure 18-41A). A systolic thrill is common because augmented right ventricular stroke volume is ejected rapidly across a hypoplastic pulmonary annulus. A diastolic thrill of pulmonary regurgitation is common because diastolic flow is accelerated by recoil of the dilated proximal pulmonary arteries.
Auscultation
The wheezing and stertorous breathing of tracheobronchial compression compromise auscultation. A pulmonary ejection sound is absent because the pulmonary valve is absent. The pulmonary component of the second heart sound is necessarily absent because the pulmonary valve mechanism is absent.90 The aortic component of the second sound is muted by anterior interposition of the dilated pulmonary trunk. A midsystolic murmur is maximal in the second left intercostal space and is loud, harsh, and long because a large right ventricular stroke volume is ejected across a narrow annulus into a dilated pulmonary trunk (see Figure 18-42).169 The pulmonary regurgitant murmur is analogous to the diastolic murmur of isolated severe congenital pulmonary valve regurgitation (see Chapter 12).112,169 A distinct gap exists between the aortic component of the second heart sound and the onset of the diastolic murmur, which coincides with the delayed timing of the absent pulmonary closure sound. The diastolic murmur is usually grade 3/6 and is impure and often harsh, ending well before the subsequent first heart sound (see Figure 18-42). The crescendo portion of the diastolic murmur may be comparatively long because a delayed fall in pressure is associated with impaired infundibular relaxation and increased diastolic stiffness.169 The combination of a long, loud, harsh systolic murmur followed by a shorter harsh diastolic murmur creates the auscultatory impression of sawing wood.
Electrocardiogram
The P wave is usually peaked and tall (Figure 18-43). The QRS axis is rightward. However, the chief electrocardiographic distinction between Fallot’s tetralogy with absent pulmonary valve and classic cyanotic Fallot’s tetralogy is the precordial lead pattern of right ventricular hypertrophy. When the pulmonary valve is absent, the tall monophasic R wave in lead V1 extends to adjacent precordial leads (see Figure 18-43), in contrast to Fallot’s tetralogy in which the tall right precordial R wave is confined to lead V1 (see Figure 18-27).
X-Ray
Radiologic features of Fallot’s tetralogy with absent pulmonary valve are striking.90 The pulmonary trunk and proximal branches dilate massively (see Figures 18-41 and 18-42). Infundibular dilation (see Figure 18-41C) projects leftward as a hump-shaped shadow.90 A conspicuously dilated right ventricle occupies the apex, and an enlarged right atrium forms the right lower cardiac silhouette (see Figure 18-41A, B). Pulmonary vascularity is normal rather than decreased, although assessment is compromised by emphysema, hyperinflation, and atelectasis. Rarely, the right or left pulmonary artery arises directly from the aorta; lung vascularity is greater on the ipsilateral side.158
Echocardiogram
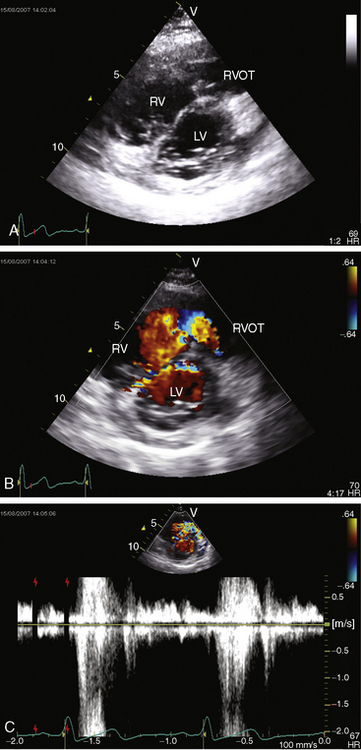
Echocardiography with color flow imaging and Doppler interrogation establishes the diagnosis and hemodynamics consequences of Fallot’s tetralogy with absent pulmonary valve.163 The malaligned ventricular septal defect is nonrestrictive (Figures 18-44 and 18-45). An echodense ridge projects into the lumen at the expected site of the absent pulmonary valve (see Figures 18-44 and 18-45A). The infundibulum, pulmonary trunk, and proximal branches are dilated, but the annulus is obstructed (see Figures 18-44 and 18-45A). Color flow imaging and Doppler interrogation characterize the pulmonary regurgitation and the flow patterns across the absent pulmonary valve and the narrow pulmonary annulus (Figures 18-45B).
Fallot’s Tetralogy with Absence of a Pulmonary Artery
Congenital absence of a pulmonary artery almost always involves the left pulmonary artery143,170 and only rarely occurs in isolation (Figures 18-46 and 18-47).139 Congenital absence of a pulmonary artery differs from anomalous origin of a pulmonary artery from the ascending aorta, a malformation that also occurs with the tetralogy.171 When the left pulmonary artery is absent, the murmur of pulmonary stenosis radiates into the right pulmonary artery and right upper chest.170 The x-ray is diagnostically useful because the left hemithorax is small, the left hemidiaphragm is elevated, the left lung is hypovascular (see Figure 18-46A),143,170 and the aortic arch is on the right (see Figure 18-46).172,173 When the left pulmonary artery originates from the ascending aorta, the ipsilateral lung is relatively hypervascular. When the right pulmonary artery is absent, the blood supply to the ipsilateral lung is usually derived from the coronary arteries.174
1 Acierno L.J. Etienne-Louis Fallot: is it his tetralogy? Clin Cardiol. 1999;22:321-322.
2 Steno N. Anatomicus regij hafniensis embryo monstro affinis paisiis dissectus. Acta Med et Philosophia, Hafminsia. 1671;1:200.
3 Maas V. Nicolai Steno. Opera Philosophica. 1910;2:49.
4 Willius F.A. An unusually early description of the so-called tetralogy of Fallot. Mayo Clin Proc. 1948;23:316-320.
5 Watson T. Lectures on the principles and practice of physic. Philadelphia: Henry C- Lea; 1872.
6 Sandifort E. Observationes Anatomico-pathological. Lugduni Batavorum: Eyk and D. Vygh; 1777.
7 Hunter W. Three cases of malformation of the heart. Medical Observations and Inquiries by a Society of Physicians in London. 1784;6:291.
8 Hope J. Diseases of the heart, 3rd ed. London: John Churchill & Sons; 1839.
9 Peacock T. On malformations of the human heart. London: J. Churchill & Sons; 1866.
10 Fallot A. Contribution à. l’anatomie pathologique de la maladie bleue (cyanose cardiaque). Mars Med. 1888;25:77-93.
11 Apitz C., Webb G.D., Redington A.N. Tetralogy of Fallot. Lancet. 2009;374:1462-1471.
12 Shinebourne E.A., Babu-Narayan S.V., Carvalho J.S. Tetralogy of Fallot: from fetus to adult. Heart. 2006;92:1353-1359.
13 Soto B., Pacifico A.D., Ceballos R., Bargeron L.M.Jr. Tetralogy of Fallot: an angiographic-pathologic correlative study. Circulation. 1981;64:558-566.
14 Niwa K., Perloff J.K., Bhuta S.M., et al. Structural abnormalities of great arterial walls in congenital heart disease: light and electron microscopic analyses. Circulation. 2001;103:393-400.
15 Ando M., Takahashi Y., Kikuchi T., Tatsuno K. Tetralogy of Fallot with subarterial ventricular septal defect. Ann Thorac Surg. 2003;76:1059-1064. discussion 1064–1055
16 Tandon R., Moller J.H., Edwards J.E. Tetralogy of Fallot associated with persistent common atrioventricular canal (endocardial cushion defect). Br Heart J. 1974;36:197-206.
17 Uretzky G., Puga F.J., Danielson G.K., et al. Complete atrioventricular canal associated with tetralogy of Fallot. Morphologic and surgical considerations. J Thorac Cardiovasc Surg. 1984;87:756-766.
18 Okamura T., Nagase Y., Matsumoto Y., Park I.-S., Mitsui F., Shibairi M. Complete atrioventricular canal and tetralogy of Fallot with pulmonary atresia. Ann Thorac Surg. 2004;78:e69-e71.
19 Faggian G., Frescura C., Thiene G., Bortolotti U., Mazzucco A., Anderson R.H. Accessory tricuspid valve tissue causing obstruction of the ventricular septal defect in tetralogy of Fallot. Br Heart J. 1983;49:324-327.
20 Daliento L., Grisolia E.F., Frescura C., Thiene G. Anomalous muscle bundle of the sub-pulmonary outflow in tetralogy of Fallot. Int J Cardiol. 1984;6:547-550.
21 Nair V., Thangaroopan M., Cunningham K.S., et al. A bicuspid pulmonary valve associated with tetralogy of fallot. J Card Surg. 2006;21:185-187.
22 Altrichter P.M., Olson L.J., Edwards W.D., Puga F.J., Danielson G.K. Surgical pathology of the pulmonary valve: a study of 116 cases spanning 15 years. Mayo Clin Proc. 1989;64:1352-1360.
23 Sharma S.N., Sharma S., Shrivastava S., Rajani M., Tandon R. Pulmonary arterial anatomy in tetralogy of Fallot. Int J Cardiol. 1989;25:33-37.
24 Chaturvedi R., Mikailian H., Freedom R.M. Crossed pulmonary arteries in tetralogy of Fallot. Cardiol Young. 2005;15:537.
25 Azhari N., Al-Fadley F., Bulbul Z.R. Tetralogy of Fallot associated with scimitar syndrome. Cardiol Young. 2000;10:70-72.
26 Edwards J.E., Mcgoon D.C. Absence of anatomic origin from heart of pulmonary arterial supply. Circulation. 1973;47:393-398.
27 Liao P.K., Edwards W.D., Julsrud P.R., Puga F.J., Danielson G.K., Feldt R.H. Pulmonary blood supply in patients with pulmonary atresia and ventricular septal defect. J Am Coll Cardiol. 1985;6:1343-1350.
28 Rabinovitch M., Herrera-Deleon V., Castaneda A.R., Reid L. Growth and development of the pulmonary vascular bed in patients with tetralogy of Fallot with or without pulmonary atresia. Circulation. 1981;64:1234-1249.
29 Deruiter M.C., Gittenberger-De Groot A.C., Poelmann R.E., Vaniperen L., Mentink M.M. Development of the pharyngeal arch system related to the pulmonary and bronchial vessels in the avian embryo. With a concept on systemic-pulmonary collateral artery formation. Circulation. 1993;87:1306-1319.
30 Wu L., Liu F. Anomalous origin of the pulmonary arteries from the left coronary artery in tetralogy of Fallot with pulmonary atresia. Cardiol Young. 2009;19:620-621.
31 Amin Z., Mcelhinney D.B., Reddy V.M., Moore P., Hanley F.L., Teitel D.F. Coronary to pulmonary artery collaterals in patients with pulmonary atresia and ventricular septal defect. Ann Thorac Surg. 2000;70:119-123.
32 Pahl E., Fong L., Anderson R.H., Park S.C., Zuberbuhler J.R. Fistulous communications between a solitary coronary artery and the pulmonary arteries as the primary source of pulmonary blood supply in tetralogy of Fallot with pulmonary valve atresia. Am J Cardiol. 1989;63:140-143.
33 Andriko J.A., Robinowitz M., Moore J., Virmani R. Necrotizing arteritis in uncorrected tetralogy of Fallot with pulmonary artery. Pediatr Cardiol. 1992;13:233-236.
34 Berry T.E., Muster A.J., Paul M.H. Transient neonatal tricuspid regurgitation: possible relation with premature closure of the ductus arteriosus. J Am Coll Cardiol. 1983;2:1178-1182.
35 Taussig H.B. Clinical and pathological findings in cases of truncus arteriosus in infancy. Am J Med. 1947;2:26-34.
36 Glancy D.L., Morrow A.G., Roberts W. Malformations of the aortic valve in patients with the tetralogy of Fallot. Am Heart J. 1968;76:755-759.
37 Marelli A.J., Perloff J.K., Child J.S., Laks H. Pulmonary atresia with ventricular septal defect in adults. Circulation. 1994;89:243-251.
38 Chowdhury U.K., Mishra A.K., Ray R., Kalaivani M., Reddy S.M., Venugopal P. Histopathologic changes in ascending aorta and risk factors related to histopathologic conditions and aortic dilatation in patients with tetralogy of Fallot. J Thorac Cardiovasc Surg. 2008;135:69-77.
39 Niwa K., Siu S.C., Webb G.D., Gatzoulis M.A. Progressive aortic root dilatation in adults late after repair of tetralogy of Fallot. Circulation. 2002;106:1374-1378.
40 Hastreiter A.R., D’cruz I.A., Cantez T., Namin E.P., Licata R. Right-sided aorta. I. Occurrence of right aortic arch in various types of congenital heart disease. II. Right aortic arch, right descending aorta, and associated anomalies. Br Heart J. 1966;28:722-739.
41 Fellows K.E., Freed M.D., Keane J.F., Praagh R., Bernhard W.F., Castaneda A.C. Results of routine preoperative coronary angiography in tetralogy of Fallot. Circulation. 1975;51:561-566.
42 Gupta D., Saxena A., Kothari S.S., et al. Detection of coronary artery anomalies in tetralogy of Fallot using a specific angiographic protocol. Am J Cardiol. 2001;87:241-244. A249
43 Chiu I.S., Wu C.S., Wang J.K., et al. Influence of aortopulmonary rotation on the anomalous coronary artery pattern in tetralogy of fallot. Am J Cardiol. 2000;85:780-784. A789
44 Talwar S., Sharma P., Gulati G.S., Kothari S.S., Choudhary S.K. Tetralogy of fallot with coronary artery to pulmonary artery fistula and unusual coronary pattern: missed diagnosis. J Card Surg. 2009;24:752-755.
45 Yamaguchi M., Tsukube T., Hosokawa Y., Ohashi H., Oshima Y. Pulmonary origin of left anterior descending coronary artery in tetralogy of Fallot. Ann Thorac Surg. 1991;52:310-312.
46 Gandhi S.K., Pigula F.A., Siewers R.D. Intramural left coronary artery associated with right ventricular outflow tract obstruction. J Thorac Cardiovasc Surg. 2003;125:729-730.
47 Talwar S., Choudhary S.K., Shivaprasad M.B., et al. Tetralogy of Fallot with total anomalous pulmonary venous drainage. Ann Thorac Surg. 2008;86:1937-1940.
48 Sautter R.D., Emanuel D.A., Doege K.H. Association of pulmonary valvular stenosis and muscular ventricular septal defect. Report of a case in a patient aged 75 years. Am J Cardiol. 1965;16:743-745.
49 Goitein K.J., Neches W.H., Park S.C., Mathews R.A., Lenox C.C., Zuberbuhler J.R. Electrocardiogram in double chamber right ventricle. Am J Cardiol. 1980;45:604-608.
50 Hartmann A.F.Jr, Tsifutis A.A., Arvidssonh D., Goldring D. The two-chambered right ventricle. Report of nine cases. Circulation. 1962;26:279-287.
51 Perloff J.K., Ronan J.A.Jr, De Leon A.C.Jr. Ventricular septal defect with the “two-chambered right ventricle”. Am J Cardiol. 1965;16:894-900.
52 Wang J.K., Wu M.H., Chang C.I., et al. Malalignment-type ventricular septal defect in double-chambered right ventricle. Am J Cardiol. 1996;77:839-842.
53 Bashour T.T., Kabbani S., Sandouk A., Cheng T.O. Double-chambered right ventricle due to fibromuscular diaphragm. Am Heart J. 1984;107:792-794.
54 Hindle W.V.Jr, Engle M.A., Hagstrom J.W. Anomalous right ventricular muscles: a clinicopathologic study. Am J Cardiol. 1968;21:487-495.
55 Taussig H.B. Left to right shunts in infancy. In: Lam C.R., editor. Henry Ford Hospital International Symposium on Cardiovascular Surgery. Philadelphia: W. B. Saunders Company, 1955.
56 Maron B.J., Ferrans V.J., White R.I.Jr. Unusual evolution of acquired infundibular stenosis in patients with ventricular septal defect. Clinical and morphologic observations. Circulation. 1973;48:1092-1103.
57 Pongiglione G., Freedom R.M., Cook D., Rowe R.D. Mechanism of acquired right ventricular outflow tract obstruction in patients with ventricular septal defect: an angiocardiographic study. Am J Cardiol. 1982;50:776-780.
58 Shepherd R.L., Glancy D.L., Jaffe R.B., Perloff J.K., Epstein S.E. Acquired subvalvular right ventricular outflow obstruction in patients with ventricular septal defect. Am J Med. 1972;53:446-455.
59 Fukuda J., Izumi T., Matsukawa T., Eguchi S. Development of left ventricular muscle in tetralogy of Fallot. Jpn Circ J. 1984;48:465-473.
60 Jarmakani J.M., Graham T.P.Jr, Canent R.V.Jr, Jewett P.H. Left heart function in children with tetralogy of Fallot before and after palliative or corrective surgery. Circulation. 1972;46:478-490.
61 Danilowicz D., Ross J.Jr. Pulmonary atresia with cyanosis. Report of two cases with ventricular septal defect and increased pulmonary blood flow. Br Heart J. 1971;33:138-141.
62 Best P.V., Heath D. Pulmonary thrombosis in cyanotic congenital heart disease without pulmonary hypertension. J Pathol Bacteriol. 1958;75:281-291.
63 Ferencz C. The pulmonary vascular bed in tetralogy of Fallot. I. Changes associated with pulmonic stenosis. Bull Johns Hopkins Hosp. 1960;106:81-99.
64 Rich A.R. A hitherto unrecognized tendency to the development of widespread pulmonary vascular obstruction in patients with congenital pulmonary stenosis (tetralogy of Fallot). Bull Johns Hopkins Hosp. 1948;82:389-401.
65 Der Kaloustian V.M., Ratl H., Malouf J., et al. Tetralogy of Fallot with pulmonary atresia in siblings. Am J Med Genet. 1985;21:119-122.
66 Pankau R., Siekmeyer W., Stoffregen R. Tetralogy of Fallot in three sibs. Am J Med Genet. 1990;37:532-533.
67 Zellers T.M., Driscoll D.J., Michels V.V. Prevalence of significant congenital heart defects in children of parents with Fallot’s tetralogy. Am J Cardiol. 1990;65:523-526.
68 Cassidy S.C., Allen H.D. Tetralogy of Fallot in triplet siblings. Am J Cardiol. 1991;67:1442-1444.
69 Dichiara J.A., Pieroni D.R., Gingell R.L., Bannerman R.M., Vlad P. Familial pulmonary atresia. Its occurrence with a ventricular septal defect. Am J Dis Child. 1980;134:506-508.
70 Patterson D.F., Pyle R.L., Van Mierop L., Melbin J., Olson M. Hereditary defects of the conotruncal septum in Keeshond dogs: pathologic and genetic studies. Am J Cardiol. 1974;34:187-205.
71 Eldadah Z.A., Hamosh A., Biery N.J., et al. Familial Tetralogy of Fallot caused by mutation in the jagged1 gene. Hum Mol Genet. 2001;10:163-169.
72 Goldmuntz E., Geiger E., Benson D.W. NKX2.5 mutations in patients with tetralogy of fallot. Circulation. 2001;104:2565-2568.
73 Momma K. Coarctation with tetralogy and pulmonary astresia. Cardiol Young. 2001;11:478.
74 Lendrum B., Agustsson M., Arcilla R., Gasul B. Natural history of patients with” acyanotic” tetralogy of Fallot [abstract]. Circulation. 1961;24:979.
75 Bertranou E.G., Blackstone E.H., Hazelrig J.B., Turner M.E., Kirklin J.W. Life expectancy without surgery in tetralogy of Fallot. Am J Cardiol. 1978;42:458-466.
76 Rygg I.H., Olesen K., Boesen I. The life history of tetralogy of Fallot. Dan Med Bull. 1971;18(suppl 2):25-30.
77 Bain G.O. Tetralogy of fallot: survival to seventieth year; report of a case. AMA Arch Pathol. 1954;58:176-179.
78 Smitherman T.C., Nimetz A.A., Friedlich A.L. Pulmonary atresia with ventricular septal defect: report of the oldest known surviving case. Chest. 1975;67:603-606.
79 Huang M.-H., Hu H., Plata E.T., Yahia A.M., Goldfarb A.L., Izzo J.L.Jr. Exceptional survival of a patient with large ventricular septal defect, bidirectional shunt, and severe pulmonary valve stenosis. J Am Soc Echocardiogr. 2002;15:665-667.
80 White P.D., Sprague H.B. The Tetralogy of fallot. report of a case in a noted musician, who lived to his sixtieth year. J Am Med Assoc. 1929;92:787-791.
81 Stanescu C.M., Branidou K. A case of 75-year-old survivor of unrepaired tetralogy of Fallot and quadricuspid aortic valve. Eur J Echocardiogr. 2008;9:167-170.
82 Chandrasekaran B., Wilde P., Mccrea W.A. Tetralogy of fallot in a 78-year-old man. N Engl J Med. 2007;357:1160-1161.
83 Gerlis L.M., Ho S.Y., Sheppard M.N. Longevity in the setting of tetralogy of Fallot: survival to the 84th year. Cardiol Young. 2004;14:664-666.
84 Alonso A., Downey B.C., Kuvin J.T. Uncorrected tetralogy of Fallot in an 86-year-old patient. Am J Geriatr Cardiol. 2007;16:38-41.
85 Garcia R., Cargill J.W., Drake E.H. Pseudotruncus arteriosus. Report of the oldest surviving patient. Am Heart J. 1969;78:537-540.
86 Lafargue R.T., Vogel J.H., Pryor R., Blount S.G.Jr. Pseudotruncus arteriosus. A review of 21 cases with observations on oldest reported case. Am J Cardiol. 1967;19:239-246.
87 Perloff J.K. Pregnancy in congenital heart disease. In Perloff J.K., Child J.S., Aboulhosn J., editors: Congenital heart disease in adults, 3rd ed, Philadelphia: WB Saunders, 2009.
88 Bodhey N.K., Beerbaum P., Sarikouch S., et al. Functional analysis of the components of the right ventricle in the setting of tetralogy of Fallot. Circulation. 2008;1:141-147. Cardiovascular imaging
89 Chesler E., Joffe H.S., Beck W., Schrire V. Tetralogy of Fallot and heart failure. Am Heart J. 1971;81:321-326.
90 Ruttenberg H.D., Carey L.S., Adams P., Adams P.Jr, Edwards J.E. Absence of the pulmonary valve in the tetralogy of Fallot. Am J Roentgenol Radium Ther Nucl Med. 1964;91:500-510.
91 Benge W., White C.W. Systemic hypertension complicating tetralogy of Fallot: effects of antihypertensive therapy. Am J Cardiol. 1978;42:294-298.
92 Sietsema K.E., Cooper D.M., Perloff J.K., et al. Dynamics of oxygen uptake during exercise in adults with cyanotic congenital heart disease. Circulation. 1986;73:1137-1144.
93 Sietsema K.E., Cooper D.M., Perloff J.K., et al. Control of ventilation during exercise in patients with central venous-to-systemic arterial shunts. J Appl Physiol. 1988;64:234-242.
94 Guntheroth W.G., Morgan B.C., Mullins G.L. Physiologic studies of paroxysmal hyperpnea in cyanotic congenital heart disease. Circulation. 1965;31:70-76.
95 Morgan B.C., Guntheroth W.G., Bloom R.S., Fyler D.C. A clinical profile of paroxysmal hyperpnea in cyanotic congenital heart disease. Circulation. 1965;31:66-69.
96 Wood P. Attacks of deeper cyanosis and loss of consciousness (syncope) in Fallot’s tetralogy. Br Heart J. 1958;20:282-286.
97 Daniels S.R., Bates S.R., Kaplan S. EEG monitoring during paroxysmal hyperpnea of tetralogy of Fallot: an epileptic or hypoxic phenomenon? J Child Neurol. 1987;2:98-100.
98 Weng Y.-M., Chang Y.-C., Chiu T.-F., Weng C.-S. Tet spell in an adult. Am J Emerg Med. 2009;27:130-e135. e133
99 King S.B., Franch R.H. Production of increased right-to-left shunting by rapid heart rates in patients with tetralogy of Fallot. Circulation. 1971;44:265-271.
100 Van Roekens C.N., Zuckerberg A.L. Emergency management of hypercyanotic crises in tetralogy of Fallot. Ann Emerg Med. 1995;25:256-258.
101 Brotmacher L. Haemodynamic effects of squatting during recovery from exertion. Br Heart J. 1957;19:567-573.
102 O’donnell T.V., Mc I.M. The circulatory effects of squatting. Am Heart J. 1962;64:347-356.
103 Taussig H.B. Congenital malformations of the heart. New York: The Commonwealth Fund; 1947.
104 Guntheroth W.G., Mortan B.C., Mullins G.L., Baum D. Venous return with knee-chest position and squatting in tetralogy of Fallot. Am Heart J. 1968;75:313-318.
105 Lurie P.R. Postural effects in tetralogy of Fallot. Am J Med. 1953;15:297-306.
106 Sharpey-Schafer E.P. Effects of squatting on the normal and failing circulation. Br Med J. 1956;1:1072-1074.
107 Laskin R.L., Salazer R., Witzel M.A., Rose V. Velopharyngeal insufficiency in tetralogy of Fallot: a report of four cases. Pediatr Cardiol. 1983;4:41-44.
108 Berthrong M., Sabiston D.C.Jr. Cerebral lesions in congenital heart disease, a review of autopsies on 162 cases. Bull Johns Hopkins Hosp. 1951;89:384-406.
109 Martelle R.R., Linde L.M. Cerebrovascular accidents with tetralogy of Fallot. Am J Dis Child. 1961;101:206-209.
110 Perloff J.K., Marelli A. Neurological and psychosocial disorders in adults with congenital heart disease. Heart Dis Stroke. 1992;1:218-224.
111 Capitanio M.A., Wolfson B.J., Faerber E.N., Williams J.L., Balsara R.K. Obstruction of the airway by the aorta: an observation in infants with congenital heart disease. AJR Am J Roentgenol. 1983;140:675-679.
112 Vogelpoel L., Schrire V. Auscultatory and phonocardiographic assessment of Fallot’s tetralogy. Circulation. 1960;22:73.
113 Emanuel R., Somerville J., Prusty S., Ross D.N. Aortic regurgitation from infective endocarditis in Fallot’s tetralogy and pulmonary atresia. Br Heart J. 1975;37:365-370.
114 Baum D., Stern M.P. Adipose hypocellularity in cyanotic congenital heart disease. Circulation. 1977;55:916-920.
115 Qvist G. John Hunter 1728–1793. London: William Heinemann Medical Books Limited; 1981.
116 Kinouchi A., Mori K., Ando M., Takao A. Facial appearance of patients with conotruncal anomalies. Pediatr Jap. 1976;17:84-87.
117 Hofbeck M., Rauch A., Buheitel G., et al. Monosomy 22q11 in patients with pulmonary atresia, ventricular septal defect, and major aortopulmonary collateral arteries. Heart. 1998;79:180-185.
118 Momma K., Kondo C., Ando M., Matsuoka R., Takao A. Tetralogy of Fallot associated with chromosome 22q11 deletion. Am J Cardiol. 1995;76:618-621.
119 Pauliks L.B., Chan K.-C., Lorts A., Elias E.R., Cayre R.O., Valdes-Cruz L.M. Shprintzen-Goldberg syndrome with tetralogy of fallot and subvalvar aortic stenosis. J Ultrasound Med. 2005;24:703-706.
120 Greenwood R.D., Rosenthal A., Sommer A., Wolff G., Craenen J. Cardiovascular malformations in oculoauriculovertebral dysplasia (Goldenhar syndrome). J Pediatr. 1974;85:816-818.
121 Sajeev C.G., Fassaludeen M., Venugopal K. Tetralogy of Fallot with absent thumb and first metacarpal. Int J Cardiol. 2005;102:349-350.
122 Mace J.W., Kaplan J.M., Schanberger J.E., Gotlin R.W. Poland’s syndrome. Report of seven cases and review of the literature. Clin Pediatr (Phila). 1972;11:98-102.
123 Carnero Alcazar M., Marianeschi S., Ruiz Alonso E., Garcia Torres E., Comas J.V. Left arm underdevelopment secondary to an isolated left subclavian artery in tetralogy of Fallot. Ann Thorac Surg. 2010;89:637-639.
124 Horwitz S., Esquivel J., Attie F., Lupi E., Espino-Vela J. Clinical diagnosis of persistent left superior vena cava by observation of jugular pulses. Am Heart J. 1973;86:759-763.
125 Colman A.L. Diagnosis of left superior vena cava by clinical inspection, a new physical sign. Am Heart J. 1967;73:115-120.
126 Feruglio G.A., Gunton R.W. Intracardiac phonocardiography in ventricular septal defect. Circulation. 1960;21:49-58.
127 Vogelpoel L., Schrire V., Nellen M., Swanepoel A. The use of amyl nitrite in the differentiation of Fallot’s tetralogy and pulmonary stenosis with intact ventricular septum. Am Heart J. 1959;57:803-819.
128 Campeau L., Gilbert G., Aerichide N. Absence of the pulmonary valve. Report of two cases associated with other congenital lesions. Am J Cardiol. 1961;8:113-124.
129 Bousvaros G.A. Pulmonary second sound in the tetralogy of Fallot. Am Heart J. 1961;61:570-571.
130 Tofler O.B. The pulmonary component of the second heart sound in Fallot’s tetralogy. Br Heart J. 1963;25:509-513.
131 Perloff J.K., Calvin J., Deleon A.C., Bowen P. Systemic hemodynamic effects of amyl nitrite in normal man. Am Heart J. 1963;66:460-469.
132 Padmanabhan J., Varghese P.J., Lloyd S., Haller J.A.Jr. Tetralogy of Fallot with suprasystemic pressure in the right ventricle. A case report and review of the literature. Am Heart J. 1971;82:805-811.
133 Bender S.R., Dreifus L.S., Downing D. Anatomic and electrocardiographic correlation of Fallot’s tetralogy: a study of 100 proved cases. Am J Cardiol. 1961;7:475-480.
134 Depasquale N.P., Burch G.E. The electrocardiogram, vectorcardiogram, and ventricular gradient in the tetralogy of Fallot. Circulation. 1961;24:94-109.
135 Macruz R., Perloff J.K., Case R.B. A method for the electrocardiographic recognition of atrial enlargement. Circulation. 1958;17:882-889.
136 Coelho E., De P., et al. Tetralogy of Fallot. Angiocardiographic, electrocardiographic, vectorcardiographic and hemodynamic studies of the Fallot-type complex. Am J Cardiol. 1961;7:538-564.
137 Lev M. The architecture of the conduction system in congenital heart disease. II. Tetralogy of Fallot. AMA Arch Pathol. 1959;67:572-587.
138 Pileggi F., Bocanegra J., Tranchesi J., et al. The electrocardiogram in tetralogy of Fallot: a study of 142 cases. Am Heart J. 1960;59:667-680.
139 Abraham K.A., Cherian G., Rao V.D., Sukumar I.P., Krishnaswami S., John S. Tetralogy of Fallot in adults. A report on 147 patients. Am J Med. 1979;66:811-816.
140 Feldt R.H., Dushane J.W., Titus J.L. The anatomy of the atrioventricular conduction system in ventricular septal defect and tetralogy of fallot: correlations with the electrocardiogram and vectorcardiogram. Circulation. 1966;34:774-782.
141 Gale G.E., Heimann K.W., Barlow J.B. Double-chambered right ventricle. A report of five cases. Br Heart J. 1969;31:291-298.
142 Hislop A., Reid L. Structural changes in the pulmonary arteries and veins in tetralogy of Fallot. Br Heart J. 1973;35:1178-1183.
143 Wilson W.J., Amplatz K. Unequal vascularity in tetralogy of Fallot. Am J Roentgenol Radium Ther Nucl Med. 1967;100:318-321.
144 Campbell M., Gardner F. Radiological features of enlarged bronchial arteries. Br Heart J. 1950;12:183-200.
145 Wong H.O., Ang A.H. Unilateral rib notching in Fallot’s tetralogy due to systemic-pulmonary collateral vessels. Br Heart J. 1973;35:226-228.
146 Tan J.L., Davlouros P.A., Mccarthy K.P., Gatzoulis M.A., Ho S.Y. Intrinsic histological abnormalities of aortic root and ascending aorta in tetralogy of Fallot: evidence of causative mechanism for aortic dilatation and aortopathy. Circulation. 2005;112:961-968.
147 Emmel M., Schmidt B., Schickendantz S. Double aortic arch in a patient with Fallot’s tetralogy. Cardiol Young. 2005;15:52-53.
148 Velasquez G., Nath P.H., Castaneda-Zuniga W.R., Amplatz K., Formanek A. Aberrant left subclavian artery in tetralogy of Fallot. Am J Cardiol. 1980;45:811-818.
149 Fraser R.S., Dvorkin J., Rossall R.E., Eidem R. Left superior vena cava: a review of associated congenital heart lesions, catheterization data and roentgenologic findings. Am J Med. 1961;31:711-716.
150 Haider E.A. The boot-shaped heart sign. Radiology. 2008;246:328-329.
151 Dadlani G.H., John J.B., Cohen M.S. Echocardiography in tetralogy of Fallot. Cardiol Young. 2008;18(suppl 3):22-28.
152 Moon-Grady A.J., Tacy T.A., Brook M.M., Hanley F.L., Silverman N.H. Value of clinical and echocardiographic features in predicting outcome in the fetus, infant, and child with tetralogy of Fallot with absent pulmonary valve complex. Am J Cardiol. 2002;89:1280-1285.
153 Moran A.M., Colan S.D., Mayer J.E.Jr, Van Der Velde M.E. Echocardiographic identification of thymic hypoplasia in tetralogy of fallot/tetralogy pulmonary atresia. Am J Cardiol. 1999;84:1268-1271. A1269
154 Von Doenhoff L.J., Nanda N.C. Obstruction within the right ventricular body: two-dimensional echocardiographic features. Am J Cardiol. 1983;51:1498-1501.
155 Chevers N. Rétrécissement congenital de l’orifice pulmonaire. Arch Gen Med Fourth Series. 1847;15:488.
156 Royer B.E., Wilson J.D. Incomplete heterotaxy with unusual heart malformations. Arch Pediatr. 1908;25:881-896.
157 Kurtz C.M., Sprague H.B., White P.D. Congenital heart disease. Interventricular septal defects with associated anomalies in a series of three cases examined postmortem, and a living patient fifty-eight years old with cyanosis and clubbing of the fingers. Am Heart J. 1927;3:77-90.
158 Calder A.L., Brandt P.W., Barratt-Boyes B.G., Neutze J.M. Variant of tetralogy of fallot with absent pulmonary valve leaflets and origin of one pulmonary artery from the ascending aorta. Am J Cardiol. 1980;46:106-116.
159 Buendia A., Attie F., Ovseyevitz J., et al. Congenital absence of pulmonary valve leaflets. Br Heart J. 1983;50:31-41.
160 Jekel L., Benatar A., Bennink G.B., Woolley S.R., Van De Wal H.J. Tetralogy of Fallot with absent pulmonary valve. A continuing challenge. Scand Cardiovasc J. 1998;32:213-217.
161 Siwik E.S., Preminger T.J., Patel C.R. Association of systemic to pulmonary collateral arteries with tetralogy of Fallot and absent pulmonary valve syndrome. Am J Cardiol. 1996;77:547-549.
162 Kirshbom P.M., Kogon B.E. Tetralogy of Fallot with absent pulmonary valve syndrome. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2004;7:65-71.
163 Fouron J.C., Sahn D.J., Bender R., et al. Prenatal diagnosis and circulatory characteristics in tetralogy of Fallot with absent pulmonary valve. Am J Cardiol. 1989;64:547-549.
164 Bedard E., Mccarthy K.P., Dimopoulos K., Giannakoulas G., Gatzoulis M.A., Ho S.Y. Structural abnormalities of the pulmonary trunk in tetralogy of Fallot and potential clinical implications: a morphological study. J Am Coll Cardiol. 2009;54:1883-1890.
165 Milanesi O., Talenti E., Pellegrino P.A., Thiene G. Abnormal pulmonary artery branching in tetralogy of Fallot with “absent” pulmonary valve. Int J Cardiol. 1984;6:375-380.
166 Hiraishi S., Bargeron L.M., Isabel-Jones J.B., Emmanouilides G.C., Friedman W.F., Jarmakani J.M. Ventricular and pulmonary artery volumes in patients with absent pulmonary valve. Factors affecting the natural course. Circulation. 1983;67:183-190.
167 Momma K., Ando M., Takao A. Fetal cardiac morphology of tetralogy of Fallot with absent pulmonary valve in the rat. Circulation. 1990;82:1343-1351.
168 Donofrio M.T., Jacobs M.L., Rychik J. Tetralogy of Fallot with absent pulmonary valve: echocardiographic morphometric features of the right-sided structures and their relationship to presentation and outcome. J Am Soc Echocardiogr. 1997;10:556-561.
169 Fontana M.E., Wooley C.F. The murmur of pulmonic regurgitation in tetralogy of Fallot with absent pulmonic valve. Circulation. 1978;57:986-990.
170 Nadas A.S., Rosenbaum H.D., Wittenborg M.H., Rudolph A.M. Tetralogy of Fallot with unilateral pulmonary atresia. A clinically diagnosable and surgically significant variant. Circulation. 1953;8:328-336.
171 Morgan J.R. Left pulmonary artery from ascending aorta in tetralogy of Fallot. Circulation. 1972;45:653-657.
172 Barrett O.J., Walker W. Tetralogy of Fallot with absent left pulmonary artery: report of a case with anomalous development of the right hilar vasculature and non-functioning right lung. Am Heart J. 1958;55:357.
173 Pool P.E., Vogel J.H., Blount S.G.Jr. Congenital unilateral absence of a pulmonary artery. The importance of flow in pulmonary hypertension. Am J Cardiol. 1962;10:706-732.
174 Gupta K., Livesay J.J., Lufschanowski R. Absent right pulmonary artery with coronary collaterals supplying the affected lung. Circulation. 2001;104:E12-E13.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 18 Ventricular Septal Defect with Pulmonary Stenosis
Etienne-Louis Arthur Fallot’s classic publication, L’Anatomie Pathologique de La Maladie Bleue (Figure 18-1A), appeared in 1888 in an obscure journal published in Marseille where Fallot lived throughout his life.1 The malformation reported by Fallot was originally described 1671 by Niels Stensen,2 better known by his Latinized name, Nicholas Steno, who was equally distinguished as anatomist, geologist, and theologian.3,4 Steno wrote, “When I opened the right ventricle … the probe that was passed forward and upward along the interventricular septum entered directly into the aorta just as readily as the probe passed from left ventricle into aorta. The same aortic canal … was common to both ventricles. Thus, the aorta receives blood from both ventricles at the same time … as it partly straddles the right ventricle”4 (Figure 18-1B). In 1872, 16 years before Fallot’s publication, Sir Thomas Watson5 wrote, “The septum between the ventricles was imperfect in its upper part; and the aorta belonged as much to one ventricle as to the other. The pulmonary artery would not admit a goose-quill; the walls of the right ventricle were as thick as those of the left.” The anatomic and clinical features of Fallot’s tetralogy were also described by Eduard Sandifort (1777),4,6 William Hunter (1784),7 James Hope (1839),8 and Thomas Peacock (1866).9 Fallot made an anatomic diagnosis at the bedside, was proven right at postmortem, and coined the term tetralogy.10 He said, “This malformation consists of a true anatomo-pathologic type represented by the following tetralogy: 1) stenosis of the pulmonary artery; 2) interventricular communication; 3) deviation of the origin of the aorta to the right; 4) hypertrophy, almost always concentric, of the right ventricle.”10 Fallot requested that no eulogy be published after his death, but the tetralogy that bears his name remains one of the most familiar eponyms in cardiovascular medicine. The incidence rate is estimated at 1 in 3600 live births.11,12
The four salient anatomic components of Fallot’s tetralogy result from a specific morphogenetic abnormality: malalignment of the infundibular septum.13 In the normal heart, division of the fetal conotruncus culminates in alignment of the infundibular septum with the muscular trabecular septum. In Fallot’s tetralogy, the infundibular septum deviates anteriorly and cephalad and is therefore not aligned with the trabecular septum, creating a ventricular septal defect at the site of malalignment. The deviation of the infundibular septum encroaches on the right ventricular outflow tract and causes infundibular stenosis and a biventricular (overriding) aorta (Figure 18-2).13 The degree of override and the size of the biventricular aorta are determined chiefly by the degree of malalignment, but aortic size is also influenced by an inherent medial abnormality.14 The nonrestrictive malaligned ventricular septal defect accounts for systemic systolic pressure in the right ventricle and concentric right ventricular hypertrophy.
Malaligned ventricular septal defects are located in the perimembranous septum with extension into the infundibular septum.13 Atrioventricular conduction is normal. The crest of the muscular trabecular septum forms the floor of the defect, which is roofed by the valve of the overriding aorta, setting the stage for aortic regurgitation. Subarterial ventricular septal defects, which are more frequent among Asians, are understandably accompanied by aortic regurgitation because a supporting infundibulum is absent.15 Rarely, the ventricular septal defect is part of an atrioventricular septal defect (see Chapter 15).16–18 Muscular ventricular septal defects sometimes coexist but usually close spontaneously in the first year of life. Occasionally, a nonrestrictive malaligned defect is reduced in size by intrusion of accessory or excessive tricuspid valve tissue (see Figure 18-18) that is fixed to the edges of the defect by short chordae tendineae or tethered by long chordae, which permit wide excursions through the defect.19
Malalignment of the infundibular septum is the essential but not the only cause of obstruction to right ventricular outflow,13 which also results from hypertrophy of the septoparietal trabeculations, the trabecula septomarginalis, and the infundibular septum (Figure 18-3A). Anterior and cephalad malalignment of the infundibular septum can narrow the entire right ventricular outflow tract (see Figure 18-3).13,20 The pulmonary valve is frequently stenotic and bicuspid (see Figure 18-3B),21 and less frequently unicommissural unicuspid.22 Occasionally, the main site of obstruction is a hypoplastic pulmonary annulus or the ostium of the infundibulum (Figure 18-4).23 The pulmonary trunk, its bifurcation, and its right and left branches may be segmentally or diffusely hypoplastic (Figure 18-5B).23 Rarely, the pulmonary arteries cross as they proceed to their respective lungs.24 Even more rarely, the tetralogy is associated with the scimitar syndrome.25
Pulmonary atresia with Fallot’s tetralogy is the ultimate expression of severity.26 The right ventricle terminates blindly against an atretic pulmonary valve or against imperforate muscle (Figures 18-6A and 18-7B). The pulmonary trunk is either a vestigial cord or a hypoplastic funnel-shaped channel that widens as it approaches the bifurcation. The proximal pulmonary arteries are hypoplastic (Figure 18-7D) and may be discontinuous.26 The entire right ventricular output enters the systemic circulation via the nonrestrictive malaligned ventricular septal defect (see Figures 18-6A and 18-7B). The biventricular aorta is dilated (see Figures 18-6A and 18-7A, B) and often continues as a right aortic arch (Figure 18-8A). The lungs are perfused by systemic-to-pulmonary arterial collaterals (see Figures 18-6B and 18-7C, D), on which survival depends (see subsequent).27,28 Exceptionally, the pulmonary circulation is supplied primarily, if not exclusively, by a long, narrow sigmoid-shaped ductus arteriosus (see Figure 18-38B) that is structurally a muscular systemic artery similar to a systemic arterial collateral. This ductal structure is appropriate for intrauterine flow, which is directed from the aorta into the pulmonary artery.
One of the most characteristic features of Fallot’s tetralogy with pulmonary atresia is a pulmonary circulation supplied entirely by collateral arteries that serve both a nutritive function and a respiratory function (gas exchange). The three types of arterial blood supply to the lungs include systemic arterial collaterals; the distinctive ductus arteriosus, which is a muscular systemic artery (see previous); and small diffuse pleural arterial plexuses.27Systemic arterial collaterals are classified according to their origins as: (1) bronchial, which originate where their name indicates and anastomose to pulmonary arteries within the lung (see Figure 18-6B)28,29; (2) direct systemic arterial collaterals, which originate from the descending aorta, enter the hilum, and then assume the structure and distribution of intrapulmonary arteries (see Figure 18-7C, D)28; and (3) indirect systemic arterial collaterals, which originate from the internal mammary, innominate, and subclavian arteries and anastomose to proximal pulmonary arteries outside the lung (see Figure 18-8B).28 Bronchial arterial collaterals are characterized by intrapulmonary anastomoses, direct arterial collaterals by hilar anatomoses, and indirect arterial collaterals by extrapulmonary anatomoses.28 Thus, systemic arterial collaterals anatomose with pulmonary arteries in three locations: (1) intrapulmonary; (2) extrapulmonary; and (3) hilar.28 All three major types of collaterals are present when Fallot’s tetralogy occurs with pulmonary atresia, but only bronchial collaterals are present when the tetralogy occurs with pulmonary stenosis, irrespective of severity.28 About 10% of arterial collaterals originate from coronary arteries.30–32 The pulmonary circulation is effective in gas exchange regardless of the type of systemic arterial collateral.
Direct aortic to pulmonary collaterals originate from intersegmental branches of the dorsal aorta during the third and fourth weeks of gestation.28,29Bronchial arterial collaterals develop in the ninth gestational week after the paired intersegmental arteries have been resorbed28,29 and do not coexist with direct aortic collaterals.28Indirect collaterals arise later in gestation and therefore coexist with bronchial collaterals but not with direct aortic collaterals.28 A particular collateral artery supplies a particular segment of lung, but duplicate blood supplies occasionally occur. A single type of collateral usually predominates in a given patient.28 Lung growth and survival depend on the size and patency of the collateral arteries. Diminished pulmonary blood flow adversely effects the growth of peripheral pulmonary arteries.28
Systemic arterial collaterals have a strong tendency to harbor intimal cushions (proliferations) that serve as sites of potential segmental stenosis (see Figure 18-8B).28 In the absence of these obstructing cushions, large collateral arteries transmit systemic arterial pressure into the pulmonary vascular bed, resulting in morphologic changes analogous to pulmonary vascular disease (see Chapter 14).28,33 Although stenotic sites protect the intrapulmonary resistance vessels from systemic pressure, regional pulmonary blood flow is compromised.
In 1947, Taussig observed that the ductus arteriosus was not structurally normal in Fallot’s tetralogy with pulmonary atresia.34,35 The normal fetal ductus functions as a conduit for right ventricular flow into pulmonary trunk, a function that cannot be served when pulmonary atresia diverts the entire right ventricular output into the aorta via the nonrestrictive malaligned ventricular septal defect. Not surprisingly, the ductus is malformed or absent when pulmonary atresia exists from early fetal life. Absence of a ductus indicates that normal intrauterine ductal function was usurped, rendering the ductus superfluous. If a ductus is present, it is represented by a long narrow branch of the aorta that carries systemic arterial blood from the aorta into the pulmonary trunk.27 This ductus is narrow because it delivers blood only to the lungs, which represents no more than 5% to 10% of the combined ventricular output. The ductus is long because it first runs distally, diverging from the aortic arch, and then turns back to join the proximal left pulmonary artery.
Aortic regurgitation in Fallot’s tetralogy occurs because the malaligned ventricular septal defect is partially roofed by the aortic valve.13,36 Herniation of aortic cusps is more frequent with an isolated subarterial ventricular septal defects than with Fallot’s tetralogy, a difference ascribed to dissimilar flow patterns and their impact on the aortic valve. In cyanotic Fallot’s tetralogy, the aortic valve is not subjected to turbulent flow because the left ventricle ejects directly into the aorta without generating a left-to-right jet. Nevertheless, there is an age-related increase in aortic regurgitation,37 in part as a result of progressive aortic root dilation associated with an inherent medial abnormality.14,38,39 Aortic regurgitation causes volume overload of both ventricles because the aorta is biventricular.37 The incompetent aortic valve is susceptible to infective endocarditis, which can suddenly and catastrophically augment the degree of biventricular regurgitation (see section The History).37 A right aortic arch is a feature of Fallot’s tetralogy.40 Its incidence rate increases as the severity of right ventricular outflow obstruction increases (see Figure 18-3) and reaches approximately 25% with pulmonary atresia (see Figures 18-6A and 18-8A).
Anomalous origin and distribution of coronary arteries are common (see Chapter 32)30,41,42 and may be of no functional importance but are of considerable surgical importance. The incidence of coronary artery anomalies is influenced by aortopulmonary rotation43 and is higher when the aortic root is anterior to or side-by-side the pulmonary trunk. The most common anomalies are origin of a conus artery or the left anterior descending artery from the right coronary artery or from the right sinus of Valsalva (Figure 18-9).41 Origin of a single coronary artery from the right sinus of Valsalva is less common. Relatively frequent are fistulous communications between coronary arteries and the pulmonary artery30,44 or the right atrium and between coronary arteries and bronchial arteries.32 Rarely, the left anterior descending coronary artery originates from the pulmonary artery,45 or the left coronary artery is intramural.46
Etienne-Louis Fallot recognized that “… at times, there is an additional entirely accessory defect, namely, patency of the foramen ovale.”10 An atrial septal defect occasionally coexists with Fallot’s tetralogy, but the term pentalogy is longer used. Rarely, the tetralogy is associated with total anomalous pulmonary venous connection and an atrial septal defect.47
The combination of right ventricular outflow obstruction and ventricular septal defect is not confined to Fallot’s tetralogy. Pulmonary valve stenosis occasionally occurs with an isolated perimembranous ventricular septal defect (Figure 18-10). The degree of stenosis varies from trivial to severe, the size of the ventricular septal defect varies from small to nonrestrictive, and right ventricular systolic pressure varies from normal to suprasystemic. Other examples of the combination include pulmonary valve stenosis with a muscular ventricular septal defect,48 obstruction to right ventricular outflow caused by protrusion of a large ventricular septal aneurysm into the right ventricular outflow tract (Figure 18-11), and double-chambered right ventricle with a perimembranous49–51 or a malaligned ventricular septal defect (Figure 18-12).20,52 Sir Arthur Keith in his 1909 Hunterian lecture focused on obstructing right ventricular muscular bundles, which consist of an hypertrophied moderator band, an hypertrophied trabecula, or a fibromuscular diaphragm,53 with obstruction ranging from nil to severe to complete.50,54 A ventricular septal defect that communicates with the proximal high-pressure compartment results in a right-to-left shunt.50
Taussig55 described two infants with a loud holosystolic murmur at the lower left sternal border and increased pulmonary blood flow, prompting the diagnosis of ventricular septal defect. Several years later, both patients were cyanotic with pulmonary stenotic murmurs and decreased pulmonary blood flow, appropriate for the diagnosis to Fallot’s tetralogy. Gasul formalized the notion that progressive infundibular obstruction sometimes occurs with ventricular septal defect (Figure 18-13), and confirmatory reports soon appeared.56–58 The acquired obstruction usually results from hypertrophy of right ventricular muscle bundles, and only rarely from a malaligned infundibular septum.57
The physiologic consequences of Fallot’s tetralogy depend essentially on two variables: the degree of obstruction to right ventricular outflow and, to a lesser extent, systemic vascular resistance. The magnitude and direction of the shunt are determined by the resistance at the site of pulmonary stenosis relative to systemic vascular resistance. When pulmonary stenosis offers lesser resistance, the shunt is left-to-right. When the resistances are equal, the shunt is balanced. When right ventricular outflow resistance exceeds systemic resistance, the shunt is right-to-left. The amount of aortic override is not the issue, although the degree of override tends to coincide with the degree of right ventricular outflow obstruction, which is the issue. When right ventricular blood preferentially flows into the aorta, pulmonary blood flow falls reciprocally, so the left side of the heart is underfilled.59,60 The ultimate expression of right ventricular outflow obstruction is pulmonary atresia, which commits the entire right ventricular output to the aorta. Pulmonary blood flow then depends on systemic arterial collaterals (see previous), which provide the lungs with normal or increased flow, so cyanosis can be mild or even absent.61 Unobstructed flow through arterial collaterals sets the stage for pulmonary vascular disease. Stenoses at intimal cushions in arterial collaterals (see previous) protect the pulmonary vascular bed, but at the price of reduced pulmonary blood flow.
Irrespective of the degree of right ventricular outflow obstruction, right ventricular systolic pressure cannot exceed systemic because the ventricular septal defect is nonrestrictive. Accordingly, a systemic ceiling is placed on the pressure overload that pulmonary stenosis can impose on the right ventricle. When pulmonary stenosis is severe, right ventricular pressure overload is determined by systemic vascular resistance. Increased resistance associated with systemic hypertension or, less commonly, with acquired calcific aortic stenosis (see Figure 18-33B) improves pulmonary blood flow but increases right ventricular afterload. Aortic regurgitation imposes volume load on the already pressure-overloaded right ventricle.
In addition to concentric hypertrophy of the right ventricle, certain morphologic changes are secondary to the physiologic derangements of Fallot’s tetralogy. Tricuspid leaflets develop fibrous thickening because right ventricular systolic pressure is systemic, but the thickened leaflets are seldom incompetent. The right ventricle ejects against systemic resistance without an increase in filling pressure, so right atrial pressure remains normal, and overall systolic function of the hypertrophied right ventricle remains normal (Figure 18-14B, C; see subsequent).60 The underfilled left ventricle tends to be reduced in size with reduced stroke volume.59,60 Low pressure and low flow in the pulmonary circulation alter the small muscular arteries and arterioles and cause thinning of the media with interruption of elastic tissue and widespread thromboses.62–64
When severe pulmonary stenosis occurs with a restrictive ventricular septal defect, right ventricular systolic pressure exceeds systemic and the hypertrophied right ventricle dilates and fails. A physiologically analogous state exists when accessory tricuspid leaflet tissue partially occludes the malaligned ventricular septal defect (see Figure 18-18).19
History
Gender distribution in Fallot’s tetralogy is approximately equal. The malformation recurs in families and has been reported in siblings,65–67 including triplets,68 and in parents and offspring.67,69 Two brothers with DiGeorge syndrome had Fallot’s tetralogy. Birth weight tends to be lower than normal, and growth and development are generally retarded. Hereditary cardiovascular defects in Keeshond dogs include typical Fallot’s tetralogy and isolated ventricular septal defect with pulmonary stenosis.70 Familial tetralogy has been associated with mutation of the jagged 1 gene,71 with NKX2.5 mutations,72 and with chromosome 22q11.2 deletion.73
The clinical course in early infancy is often benign. Mild to moderate neonatal cyanosis tends to increase, but cyanosis may be delayed for months and is coupled with increased oxygen requirements of the growing infant rather than with progressive obstruction to right ventricular outflow.74 Patients seldom remain acyanotic after the first few years of life, and by 5 to 8 years of age, most children are conspicuously cyanotic, with cyanosis closely coupled to the severity of pulmonary stenosis. Infants with Fallot’s tetralogy and pulmonary atresia are mildly cyanotic or acyanotic when collateral flow is abundant.61
In an analysis of survival patterns based on 566 necropsy cases of Fallot’s tetralogy, two thirds of patients reached their first birthday, approximately half reached age 3 years, and approximately a quarter completed the first decade of life.75,76 The attrition rate was then 6.4% per year with 11% alive at age 20 years, 6% at age 30 years, and 3% at age 40 years.75,76 Nevertheless, Fallot’s tetralogy remains the most common cyanotic congenital heart disease after 4 years of age and constitutes a large proportion of adults with cyanotic congenital heart disease. Fallot recognized this tendency when he wrote, “We have seen from our observations that cyanosis, especially in the adult, is the result of a small number of cardiac malformations well determined. One of these cardiac malformations is much more frequent than others…,” namely, the tetralogy to which he referred.10 Fallot’s oldest patient was 36 years of age.10 Survivals between the fifth and seventh decades are uncommon but not rare.12,77–79 A 64-year-old woman with the tetralogy was diagnosed in 1895 by G.A. Gibson, best known for his description of the continuous murmur of patent ductus arteriosus. In 1929, White and Sprague80 published an account of the American composer Henry F. Gilbert who lived a productive life to age 60 years. Another patient played cricket and football as a schoolboy and survived to age 62 years. Patients without repair have lived to age 75 years,81 78 years,82 84 years,83 and 86 years.84 In Fallot’s tetralogy with pulmonary atresia, life expectancy without surgery is as low as 50% in 1 year and 8% in 10 years,75 but adequate collateral blood flow occasionally permits survival into adolescence and adulthood.18,37,78,85,86 One such patient lived to age 54 years,78 and another lived to age 55 years, despite acquired calcific aortic stenosis and regurgitation (see Figure 18-33B.)
Pregnancy is poorly tolerated in females who reach childbearing age.87 The gestational fall in systemic vascular resistance increases the right-to-left shunt, and labile systemic vascular resistance during labor and delivery results in abrupt oscillations in hypoxemia. Fetal wastage is high,23 and live born infants are dysmature.
The neonatal right ventricle is well equipped to eject against systemic vascular resistance because the nonrestrictive ventricular septal defect permits decompression into the aorta.60 The right ventricle has been analyzed in terms of its inlet, apical trabecular, and outlet portions.88 The apical trabecular portion took up the greatest portion of the overload, the outlet portion had decreased ejecting force, and the inlet portion was not significantly affected.88 Right ventricular failure is uncommon.89 However, biventricular failure in the first few weeks of life accompanies pulmonary atresia with excessive flow through large systemic arterial collaterals.61 Accessory tricuspid leaflet tissue that partially occludes the ventricular septal defect results in suprasystemic right ventricular systolic pressure and the right ventricular failure.19,89 Absence of the pulmonary valve (see subsequent section) results in volume overload of the pressure-overloaded right ventricle.90 Systemic hypertension increases left and right ventricular afterload and can induce right ventricular or biventricular failure.89,91 Acquired calcific stenosis of the biventricular aortic valve imposes increased afterload on both the right and the left ventricles (see Figure 18-33B). Regurgitation of a biventricular aortic valve sets the stage for right ventricular failure by imposing volume overload on the already pressure-overloaded right ventricle.37 Infective endocarditis on an incompetent aortic valve can result in catastrophic acute severe biventricular aortic regurgitation.
Isotonic exercise is accompanied by a fall in systemic vascular resistance in the face of fixed obstruction to right ventricular outflow, increasing venoarterial mixing and significantly influencing the dynamics of O2 uptake and ventilation.92,93 Exercise-induced hypoxemia and increased carbon dioxide content stimulate the respiratory center and the carotid body, provoking hyperventilation that is subjectively perceived as dyspnea.92,93
Hypoxic spells, variously called paroxysmal hyperpnea, syncopal attacks, hypoxic or hypercyanotic spells, are dramatic and alarming features of Fallot’s tetralogy.94–96 A typical spell begins with a progressive increase in the rate and depth of breathing and culminates in paroxysmal hyperpnea, deepening cyanosis, limpness, syncope, and occasionally convulsions, cerebrovascular accidents, and death.95,96 Electroencephalographic abnormalities during an hypoxic spell are similar to those of hypoxic episodes of other causes.97 Peak incidence is between the second and sixth month of life, with an occasional spell as early as the first month but comparatively few spells after age 2 years, and only rarely in adults.98 Spells in infants are typically initiated by the stress of feeding, crying, or a bowel movement, particularly after awakening from a long deep sleep.94,95 However, attacks sometimes occur without an apparent precipitating cause, especially in deeply cyanotic infants, although spells are not necessarily related to the degree of cyanosis.95 Spells were originally attributed to infundibular contraction caused by sympathetic stimulation, which was believed to divert right ventricular blood into the aorta,96 but occurrence in patients with pulmonary atresia argued against this theory. It is now believed that vulnerable respiratory control mechanisms, which are especially sensitive after prolonged deep sleep, react to the sudden increase in cardiac output provoked by feeding, crying, or straining, by initiating the following vicious cycle.94,95 As heart rate and cardiac output increase, venous return increases in the face of fixed obstruction to right ventricular outflow, so the right-to-left shunt increases. Infundibular contraction reinforces this pattern but does not initiate it. The increased right-to-left shunt causes a fall in systemic arterial pO2 and pH and a rise in pCO2, a blood gas composition to which a sleep-sensitive respiratory center and carotid body overreact, provoking hyperpnoea, which in turn further increases the cardiac output and perpetuates the cycle. Supraventricular tachycardia and rapid atrial pacing initiate spells by inducing infundibular narrowing, which increases the right-to-left shunt.99 Five mechanisms are therefore involved in the pathogenesis of Fallot spells: (1) an acceleration in heart rate; (2) an increase in cardiac output and venous return; (3) an increase in right-to-left shunt; (4) vulnerable respiratory control centers; and (5) infundibular contraction. Manual compression of the abdominal aorta can abort a spell by decreasing cardiac output and venous return.100 Squatting for relief of dyspnea is a time-honored hallmark of Fallot’stetralogy (Figure 18-15).101,102 In 1784, William Hunter7 made the following observations on the effects of posture: “Any hurry upon his spirits or brisk motion of his body would generally occasion a fit. And for some of the last years of his life he found out by his own observations that when the fit was coming upon him, he would escape it altogether, or at least take considerably from its violence or duration by instantly lying down upon the carpet on his left side, and remaining immovable in that position for about 10 minutes. I saw the experiments made with success.”
Figure 18-15 Typical squatting postures assumed effortlessly in two children with Fallot’s tetralogy.
Taussig described the preference for certain postures other than squatting, namely, the knee-chest position, lying down, or sitting with legs drawn underneath (Figure 18-16).103 Parents may hold their breathless infant upright with its legs flexed against its abdomen (see Figure 18-16, panel 4). Young adults cross their legs during quiet standing or sitting, a relatively ineffective variation. Habitual squatters assume the position effortlessly (see Figure 18-15). The mechanisms by which squatting exerts its beneficial effects are as follows.101,102,104,105 (1) Quiet standing after exercise-induced peripheral vasodilation predisposes to orthostatic hypotension and faintness, a tendency that is exaggerated in hypoxemic patients. Squatting counteracts orthostatic hypotension and diminishes or prevents postexertion orthostatic faintness.102,104 (2) Squatting increases systemic vascular resistance, diverts right ventricular blood into the pulmonary circulation, and increases the amount of oxygenated blood entering the left side of the heart.102,104,106 The left ventricle delivers the larger volume of oxygenated blood into the systemic circulation, so systemic arterial pO2 and pH increase and pCO2 decreases, blunting the stimulus to the respiratory center and carotid body and relieving hyperventilatory dyspnea.104 The effect of squatting on systemic venous return is an even more effective means by which hyperventilatory dyspnea is relieved.104 (3) Isotonic leg exercise reduces the oxygen saturation of venous effluent returning to the heart from the lower extremities. Squatting mechanically curtails lower extremity venous return, decreases the volume of unsaturated venous blood delivered to the heart, and increases the oxygen saturation of right ventricular blood. (4) Right ventricular blood shunted into systemic circulation has a higher oxygen content and pH and a lower pCO2 content.104 (5) The higher pO2 and pH and the lower pCO2 reduce the stimulus to the respiratory center and carotid body and reduce the hyperventilatory dyspnea.
Recurrent hypoxic spells sometimes lead to brain damage and mental retardation. Cerebral venous sinus thromboses and small occult thromboses may become manifest after prolonged hypoxic spells. Hypernasal resonance or nasal speech (velopharyngeal insufficiency) may develop after repeated or prolonged spells because nasal resonance is compromised by improper approximation of the velum (soft palate) and the pharyngeal walls, a disturbance that has been ascribed in part to central nervous system damage caused by hypoxic spells.107
Brain abscess and cerebral embolism add to the list of central nervous system complications.108–110 Iron-deficient erythrocytosis in patients less than 4 years of age increases the risk of cerebral venous sinus thrombosis.110 Wheezing and stridor have been attributed to tracheal compression by an enlarged aorta.14,111 A stenotic pulmonary valve112 and an incompetent aortic valve113 are substrates for infective endocarditis.
The physiologic consequences and clinical course of a nonrestrictive ventricular septal defect are favorably influenced by mild to moderate acquired obstruction to right ventricular outflow (see previous57 and Chapter 17). The clinical picture initially resembles an isolated nonrestrictive ventricular septal defect with large left-to-right shunt (see Figure 18-13A.) With the development of right ventricular outflow obstruction, excessive pulmonary blood flow and volume overload of the left ventricle are curtailed,56,58,74 symptoms related to the left-to-right shunt diminish, and physical development improves. Obstruction to right ventricular outflow may progress sufficiently to reverse the shunt, resulting in late onset cyanosis (see Figure 18-13B).
[/not-level-membership-for-cardiovascular-category]
