[level-membership-for-cardiovascular-category]
Chapter 17 Ventricular Septal Defect
In 1879, Henri Roger wrote, “A developmental defect of the heart occurs from which cyanosis does not ensue in spite of the fact that a communication exists between the cavities of the two ventricles and in spite of the fact that admixture of venous blood and arterial blood occurs. This congenital defect, which is even compatible with a long life, is a simple one. It comprises a defect in the interventricular septum.”1
Roger went on to say that at necropsy, “The ventricular walls show no alteration, but in the upper portion of the interventricular septum beneath the mitral valve is an orifice which establishes a communication between the two ventricles.”1 And still further, “Among the congenital defects of the heart compatible with life and perhaps a long one, one of the most frequent which I have encountered … is the communication between the two ventricles because of failure of occlusion of the interventricular septum.”1
Ventricular septal defects remain the most common congenital malformation of the heart, occurring in 50% of children with congenital heart disease.2 Detection has increased dramatically with advances in imaging and with the facility of echocardiographic diagnosis in neonates, with spontaneous closure and detection of small defects in the muscular trabecular septum taken into account.3,4 A comprehensive review estimated the median incidence rate of ventricular septal defects to be 2,829 per million live births5; the defects are found to be the most common gross morphologic congenital malformation of the heart and circulation in adults, except for the bicuspid aortic valve.3,6–8
Although Roger believed that “this congenital defect … is a simple one,”1 there is still no uniform consensus on how best to characterize and classify the diverse types of defects to which the ventricular septum is subject. The ventricular septum is a complex nonplanar partition whose components are best defined according to anatomic landmarks on the right side of the septal surface (Figure 17-1).9–12 Defects are classified according to their relationship to the membranous, inlet, trabecular, and infundibular septum.2,12 The membranous septum is divided by the tricuspid annulus into ventriculoatrial and interventricular components that abut the three major segments of the muscular septum: namely, the inlet septum, which is lightly trabeculated; the trabecular septum, which is, as the name implies, heavily trabeculated; and the infundibular septum, which is nontrabeculated. The inlet septum is limited by the tensor attachments of the tricuspid valve and is so named according to its location. The trabecular septum lies between the inlet septum and the infundibular septum (see Figure 17-1). Atrioventricular conduction tissue penetrates the ventriculoatrial portion of the membranous septum. The His bundle and bundle branches run beneath the deficient interventricular component of the membranous septum close to the free edge of the ventricular septal defect.10,13
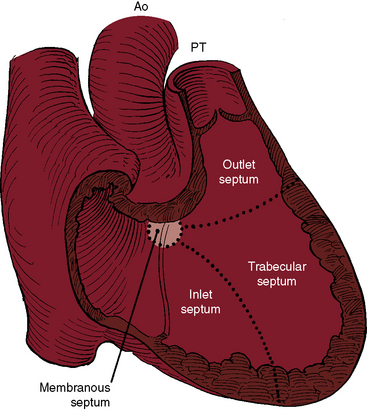
Approximately 80% of ventricular septal defects are perimembranous. The prefix peri underscores extension into adjacent portions of the inlet, trabecular, and infundubular septum. Large perimembranous defects encroach upon all three portions of the muscular septum (Figure 17-2A). Muscular ventricular septal defects are prevalent in neonates, with estimates as high as 53 of every 1000 live births. Approximately 90% of the defects close spontaneously within the first 10 months of life.
The most common types of muscular defects lie within the trabecular septum.10,14,15 These malformations are more apparent on the left septal surface and vary from small to large, from single to multiple (Figure 17-3), to a honeycombed or Swiss cheese–like structure with sieve-like fenestrations, to tortuous sinusoidal tracks threaded among septal trabeculae without through-and-through perforations.10,15 Sieve-like fenestrations or multiple small muscular defects have the net functional effect of a single large defect.
Isolated defects in the inlet septum represent approximately 8% of ventricular septal defects at surgery.16 An inlet ventricular septal defect that is bordered entirely by myocardium differs from an inlet defect that involves the basal portion of the septum near the crux where it is partially bordered by bridging atrioventricular valve tissue (see Chapter 15).10–12
The infundibular septum is represented by a small portion of muscle interposed between the outflow components of the left and right ventricles.17 A sleeve of subpulmonary infundibulum supports the leaflets of the pulmonary valve and separates the right ventricular outflow tract from the surface of the heart rather than from the left ventricular outflow track. Ventricular septal defects in the infundibular septum are also called supracristal, subpulmonary, subarterial, or doubly committed and account for approximately 5% to 7% of ventricular septal defects in North America and Western Europe, but for approximately 30% in Asian patients.14,16,17 Infundibular septal defects can be entirely muscular or can be partially rimmed by semilunar valve tissue (subarterial). Defects are considered doubly committed subarterial when little or no muscle is interposed between the outflow components of the left and right ventricles and when there is absence of the septal component of the subpulmonary infundibulum, so the aortic and pulmonary leaflets are in fibrous continuity.10,12,14,16,17 Doubly committed subarterial defects lie immediately beneath the valves of both arterial trunks, so the left and right coronary cusps of the aortic valve tend to prolapse into the outflow tract of the right ventricle (see section on Ventricular Septal Defect with Aortic Regurgitation). Much less commonly, a pulmonary cusp prolapses through the defect.18
Atrioventricular septal malalignment necessarily involves an inlet ventricular septal defect and is usually accompanied by straddling of the tensor apparatus of an atrioventricular valve that inserts (straddles) onto both sides of the ventricular septum.19 So-called Eisenmenger’s malalignment is represented by a perimembranous ventricular septal defect with anterior deviation of the infundibular septum.20 Not relevant to this chapter is malalignment between infundibular and trabecular septum that is associated with Fallot’s tetralogy (see Chapter 18) or much less commonly with coarctation of the aorta (see Chapter 8).
Henri Roger did not anticipate spontaneous closure. “The pathologic state of the heart existing before birth and consisting of an arrest of development is not susceptible to favorable changes, either by spontaneous evolution or by medical or surgical intervention.”1 However, in 1918, two reports speculated that ventricular septal defects might undergo spontaneous closure.21,22 One report was entitled “The Possibility of a Loud Congenital Heart Murmur Disappearing When a Child Grows Up.”21 The other report carried the title “Can the Clinical Manifestations of Congenital Heart Disease Disappear with the General Growth and Development of thePatient?”22 However, four decades elapsed before spontaneous closure was firmly documented.23–29 In 1960, Paul Wood wrote, “In any large series of geriatric necropsies … atrial septal defect is always well represented, but where’s the maladie de Roger? Assuming it does not provide immortality, it must either close spontaneously in middle life or have long since run its mortal course.”30
A defect may remain anatomically open but functionally closed (i.e., with absent or negligible shunt; Figures 17-2B and 17-4). The tendency for ventricular septal defects, especially perimembranous and trabecular muscular defects, to decrease in size finds an ultimate expression in complete closure24,31–33 that has been called the therapeutics of nature—the invisible sutures of spontaneous closure.34 The incidence rate of spontaneous closure varies considerably depending on the population under study, the method of diagnostic investigation, the type of defect, and whether or not the defect is solitary35 and has been estimated at 50% to 75% for restrictive perimembranous and trabecular muscular defects observed from birth.29,36,37 The incidence rate for trabecular muscular defects35 is reportedly equal to or somewhat higher than that for perimembranous defects. Moderately restrictive and nonrestrictive defects also close spontaneously, but the probability is comparatively low, with an incidence rate estimated at 5% to 10%.23–25,35,38,39 Most defects that are destined to close do so within the first year of life,35 with approximately 60% closing before 3 years of age and 90% closing before age 8 years.36,37 However, spontaneous closure also occurs in older children and young adults40 and has been documented at age 23 years,26 between 26 years and 33 years of age,41 and at age 46 years. Multiple muscular trabecular ventricular septal defects have a strong tendency to close, a conclusion in accord with the observation that multiple defects are three times more prevalent in neonates than after 1 year of age.42 In preterm infants, ventricular septal defects are about twice as frequent as in full-term infants, but the rate of spontaneous closure is the same,35 which calls into question the notion that defects reflect incomplete ventricular septation. Spontaneously closed defects in the muscular trabecular septum are funnel-shaped with a sealed orifice on the right ventricular aspect and residual patency on the left ventricular aspect, with endocardial proliferation in the lumen of the funnel and hypertrophy around the exit. Muscular defects represented by “Swiss cheese” fenestrations do not close spontaneously. Defects in the inlet septum seldom decrease in size43 but are occasionally occluded by bridging atrioventricular valve tissue (see Figure 15-87B). Infundibular defects can be closed by prolapse of the right aortic cusp.2 Perimembranous ventricular septal defects close by adherence of the septal tricuspid leaflet to the margins of the defect, less commonly by prolapse of an aortic cusp,24,31,44 and rarely by intrusion of a sinus of Valsalva aneurysm. Adherence of tricuspid leaflet tissue is seldom accompanied by tricuspid regurgitation, but there is a tendency for aneurysm formation when perimembranous defects are closed by the tricuspid valve (see Figures 17-4 and 17-31B).2,32,45–49 Ventricular septal aneurysms were described by Laennec in 1826 and are, as a rule, relatively small (see Figure 17-4). Exceptionally, however, septal aneurysms expand to considerable size, and a giant aneurysm of the membranous septum revealed itself as a mediastinal mass.50 A blind septal aneurysm occasionally represents a sui generis congenital malformation of the interventricular portion of the membranous septum rather than a sequel to spontaneous closure of a perimembranous defect.51 Familial congenital septal aneurysms have been reported, although rarely.52 With notable exceptions, aneurysms are well tolerated and have been discovered incidentally at necropsy in patients beyond the eighth decade. Complications of septal aneurysms include infective endocarditis, conduction disturbances, intraaneurysmal thrombosis with systemic embolism, aortic or tricuspid regurgitation, and obstruction to right ventricular outflow (see Figure 18-11).51 Blind congenital aneurysms may spontaneously perforate, establishing a left-to-right shunt into either the right ventricle or right atrium.51
The hemodynamic classification of ventricular septal defects is relatively straightforward because physiologic consequences depend essentially on the size of the defect and the pulmonary vascular resistance.53 These two variables change with time, and the physiologic and clinical manifestations change accordingly. A defect in the ventricular septum, even if spontaneously closed, has been postulated to constitute an abnormality that might adversely affect left ventricular systolic function.54 This postulation is relevant to impaired function or left ventricular failure in an occasional patient with a restrictive defect and a negligible shunt.54
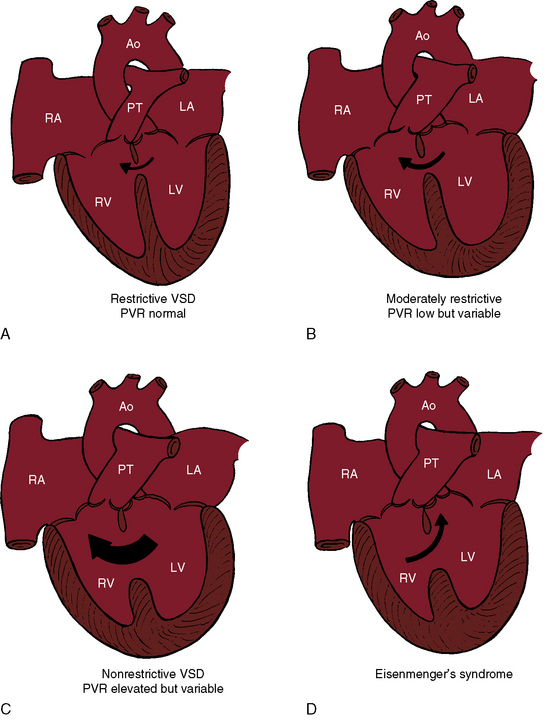
Ventricular septal defects fall into four anatomic/physiologic categories (Figure 17-5): (1) restrictive defects with normal right ventricular and pulmonary artery systolic pressure and normal pulmonary vascular resistance; (2) moderately restrictive defects with elevated right ventricular and pulmonary artery systolic pressures and low but variable pulmonary vascular resistance; (3) nonrestrictive defects with identical right ventricular and left ventricular systolic pressure and elevated but subsystemic pulmonary vascular resistance that is variable; and (4) nonrestrictive defects with identical right and left ventricular systolic pressures, suprasystemic pulmonary vascular resistance, and a right-to-left shunt—Eisenmenger’s syndrome.
Henri Roger’s account of the physiology of a restrictive ventricular septal defect with normal pulmonary vascular resistance still applies1:
Restrictive ventricular septal defects—maladie de Roger—cause little or no functional derangement because the shunt is small and the pressure and resistance in the pulmonary circulation are normal (see Figure 17-5A). A restrictive defect represents a site of obligatory resistance between the left and right ventricles, thus limiting (restricting) the magnitude of the left-to-right shunt (see previous) and precluding delivery of left ventricular systolic pressure into the right ventricle and pulmonary trunk. Shunt flow is systolic. Minor diastolic flow is generated by the normal differences in left and right ventricular distensibility and end diastolic pressure.
A moderately restrictive ventricular septal defect is characterized by right ventricular systolic pressure that is above normal but less than systemic and by low but variable pulmonary vascular resistance that rarely progresses. The left ventricle adapts to a moderate increase in volume, and the right ventricle adapts to a moderate increase in pressure. Instant-to-instant right-to-left shunting occurs because left ventricular systolic pressure falls more rapidly than right ventricular systolic pressure. However, left ventricular systolic pressure rises more rapidly than right ventricular systolic pressure, so the small right-to-left shunts are quantitatively returned to the right ventricle with the next cardiac cycle. An increase in end-diastolic pressure in the volume-loaded left ventricle results in diastolic shunting.55 Moderately restrictive defects in the trabecular muscular septum decrease in size during ventricular contraction and, in so doing, decrease the systolic shunt.56 With isotonic exercise, systemic vascular resistance falls while pulmonary vascular resistance changes little if at all,57 so a normal increment in systemic flow is achieved without a corresponding increase in left-to-right shunt.57
With a nonrestrictive ventricular septal defect, the right and left ventricles behave physiologically as a common chamber. Peak systemic systolic pressures are identical, so the volume and direction of flow through the defect depend on the relative resistances in the pulmonary and systemic circulations. A nonrestrictive defect with elevated but variable pulmonary vascular resistance (see Figure 17-5C) imposes an excessive volume load on the left ventricle and systemic afterload on the right ventricle. A persistently large left-to-right shunt culminates in depressed systolic function of the volume overloaded left ventricle.58 A rise in pulmonary vascular resistance results in a reciprocal fall in left-to-right shunt and a comparable reduction in volume overload of the left ventricle, but systolic pressure in the two ventricles necessarily remains identical. When pulmonary vascular resistance is suprasystemic, the architecture of the pulmonary vascular bed resembles primary pulmonary hypertension (see Chapter 14) and the left-to-right shunt is replaced by a right-to-left shunt, Eisenmenger’s syndrome (Figure 17-5D).37,59–65 Right ventricle function is analogous to the function of a fetal right ventricle that is equipped to cope with systemic vascular resistance.66
The vasoreactive immature pulmonary resistance vessels play a pivotal role in regulating pulmonary blood flow in neonates with a nonrestrictive ventricular septal defect. The physiologic fall in neonatal pulmonary vascular resistance is delayed because of the interplay between shunt size and pulmonary vasoreactivity. In infants with a moderately restrictive ventricular septal defect, two additional mechanisms serve to regulate the volume and direction of the shunt and the level of pulmonary arterial pressure. The most favorable mechanism is a decrease in size of the defect. A much less common mechanism is acquired obstruction to right ventricular outflow that reduces left ventricular volume overload and protects the pulmonary vascular bed (see Chapter 18).67 When and to what extent these regulatory mechanisms exert their influence determines the hemodynamic and clinical course in patients with moderately restrictive and nonrestrictive ventricular septal defects.37,62,68,69
History
Ventricular septal defects are found in a wide range of mammals and in birds with four-chambered hearts.4 The incidence in humans is unrelated to gender, race, maternal age, or birth order.4,70 Ventricular septal defects have been reported in 3.3% of first-degree relatives of index patients.71 One third of first-degree relatives of patients with congenital heart disease have ventricular septal defects.4 Between 30% and 60% of siblings of index patients have ventricular septal defects,72 and siblings of patients with ventricular septal defects have three times the incidence of ventricular septal defects compared to the general population.4 Ventricular septal defects have been reported in identical twins,73,74 but with a high frequency of discordance.4 A parent with a spontaneously closed ventricular septal defect can have offspring with a ventricular septal defect. Birth weights are low in about 18% of full-term infants with ventricular septal defects.75 Dysmaturity is in addition to and apart from occurrence of ventricular septal defects in preterm infants. Heterotrisomy is a significant factor in ventricular septal defects with Down syndrome.76
Restrictive ventricular septal defects come to light because a systolic murmur is detected at the first well-baby examination. A murmur that is present at birth is a feature of ventricular septal defect with a left ventricular-to-right atrial communication in which the shunt exists in utero and is therefore present at birth (see subsequent).77 Very small defects with trivial early systolic shunts escape detection because the early systolic murmurs are soft and are either undetected or mistaken for a normal or innocent murmur (see section Auscultation). Spontaneous closure accounts for the striking age-related disparity in the incidence of ventricular septal defects from birth to maturity.31,35,62 The patient is left with no shunt and a functionally normal heart that harbors a morphologic abnormality (see previous), especially if spontaneous closure is followed by a septal aneurysm (see Figure 17-4).32 The defect is necessarily missed if the physical examination is performed after spontaneous closure.
Infective endocarditis is a tangible risk in restrictive ventricular septal defects, but rarely occurs before eruption of the second teeth.62,67,78–80 Roger aptly stated, “Prevention of complications is by means of hygiene.”1 Infective endocarditis is usually located on the septal tricuspid leaflet at site of the jet impact. Muscular defects have a low incidence of infective endocarditis because the jet is dissipated within the right ventricular cavity without striking the septal tricuspid leaflet.15 Occasionally, the site of infective endocarditis is on an aneurysm of the ventricular septum,81 and rarely, vegetations at the base of the septal tricuspid leaflet cause perforation and a left ventricular-to-right atrial shunt, an acquired Gerbode defect.82
Longevity is likely to be normal, as Roger stated:
Several subjects whom I have been able to observe, I have attended for periods of five, twelve and fifteen years; these children have grown like others, not one has died prematurely. I have occasionally visited as physician a woman whose children I attended from early ages. She had always been in excellent health and had never complained of cardiac difficulties. On auscultation I was greatly surprised to hear a murmur. I asked her if physicians had ever found anything wrong with her heart, and she told me that Guersant the Elder (the famous pediatrician who was my first master in infantile pathology) had recognized in her a few days after birth a cardiac malformation. This woman has now passed her fiftieth year; her health continues to be perfect and she is the mother of four children. In spite of the existence of an uncomplicated defect in the interventricular septum, patients may attain or even surpass the average span of human life.1
Moderately restrictive ventricular septal defects with low but variable pulmonary vascular resistance (Figure 17-5B) escape detection in the newborn nursery because the delayed fall in neonatal pulmonary vascular resistance delays the onset of the shunt and the onset of the murmur. Congestive heart failure within the first few months of life is in response to a large left-to-right shunt that is established after the fall in pulmonary vascular resistance. Parents report that their infant fatigues and coughs while feeding, sweats excessively, is restless when recumbent, and sleeps poorly. Parents may detect a thrill when they touch their infant’s chest and may detect a hyperactive precordium when holding the infant next to their chest. Symptomatic improvement is related to a spontaneous decrease in size of the defect, a favorable trend that may culminate in spontaneous closure (Figure 17-6; see previous).
Patients with moderately restrictive defects and moderate left-to-right shunts tolerate isotonic exercise because the exercise-induced increment in systemic blood flow is achieved without a corresponding increase in shunt flow (see previous).57 However, persistence of a significant shunt incurs the risk of infective endocarditis, and protracted volume overload of the left ventricle incurs the risk of congestive heart failure.67 Only an occasional patient achieves adulthood (Figure 17-7), with one reaching age 65 years (Figure 17-8B) and another reaching age 79 years.83
Nonrestrictive ventricular septal defects with elevated but variable pulmonary vascular resistance (see Figure 17-5C) present in infancy with congestive heart failure and with little prospect that the defect will decrease in size. Poor growth and development, labored breathing, frequent lower respiratory infections, difficulty feeding, and excessive diaphoresis are typical.37,67,84 Dyspnea and irritability are especially pronounced when the infant is supine and improve when the baby is held upright or is placed in an infant seat. Feeding patterns are also typical. A hungry infant awakens from a fretful sleep and feeds vigorously only to stop short of satisfaction because of dyspnea, falls asleep again, exhausted by the effort, and awakens with renewed hunger and repeats the frustrating cycle. Regulation of the large left-to-right shunt with amelioration of symptoms is almost always the result of a rise in pulmonary vascular resistance.25,38,62,68 A minority of these patients undergo relatively little fall in neonatal pulmonary vascular resistance, so the shunts are small and the early clinical course is deceptively benign. When pulmonary resistance exceeds systemic, the shunt is reversed, the patient is cyanotic,37,60,64 and the condition that Maude Abbott called Eisenmenger’s complex exists.85 Victor Eisenmenger published his account in 1897 in a paper entitled “Congenital Defects of the Ventricular Septum.”86,87 Paul Wood, in his landmark publication of 1958, introduced the term Eisenmenger’s syndrome (Figure 17-9), which he defined as pulmonary hypertension with reversed shunt.64 Wood quoted Eisenmenger:
Cyanotic congenital heart disease, represented here by Eisenmenger’s syndrome, is a multisystem systemic disorder that involves red cell mass, hemostasis, the central nervous system, bilirubin kinetics, the systemic vascular bed, the coronary circulation, the myocardium, uric acid clearance, the kidneys, respiration, the digits, the long bones, and gynecologic endocrinology.88,89 Morbidity and mortality take into account contemporary medical management of the multisystem disorders.88–90 Longevity is about one to two decades longer than in early reports.63,89,91 Right ventricular failure is uncommon66 unless acquired systemic hypertension imposes disproportionate afterload.
Erythrocytosis is an adaptive response to the decrease in tissue oxygenation—arterial hypoxemia—that stimulates renal release of erythropoietin, which in turn stimulates an increase in the number of red blood cells. Equilibrium conditions are established at elevated hematocrit levels. Erythrocytosis is a desirable adaptive response designed to offset the hypoxemic deficit in tissue oxygenation and does not incur a risk of stroke from cerebral arterial thrombosis irrespective of hematocrit level, iron stores, or cerebral symptoms of hyperviscosity.88 However, in cyanotic children younger than age 4 years, iron-deficient erythrocytosis incurs the risk of thrombosis of intracranial venous sinuses.88 The right-to-left shunts of cyanotic congenital heart disease are pathways for paradoxical emboli that cause transient ischemic attacks and strokes.
Hemostatic abnormalities have been recognized for more than 50 years in patients with cyanotic congenital heart disease.88 Platelet counts are in the low range of normal, and many clotting factor disorders have been identified, most recently acquired abnormalities of the von Willebrand’s factor.92 A mucocutaneous bleeding tendency is characterized by easy bruising, gingival bleeding, epistaxis, menorrhagia, and increased risk of traumatic bleeding. Pulmonary hemorrhage can be external (hemoptysis) or intrapulmonary, is the most serious type of bleeding, and varies from mild and occasional to copious, recurrent, massive, and fatal.64,88,93 Massive intrapulmonary hemorrhage is a common cause of sudden death in Eisenmenger’s syndrome.89 Victor Eisenmenger’s patient “died more or less suddenly … following a large hemoptysis.”64
Bilirubin is formed from the breakdown of heme, a process that is excessive in cyanotic congenital heart disease and that coincides with a substantial increase in the amount of unconjugated bilirubin in the bile.88 Calcium bilirubinate gallstones develop because unconjugated bilirubin is virtually water insoluble at physiologic pH.88
Systemic vascular dilation is the result of endothelial-derived nitric oxide and prostaglandins that are released in response to the increase in shear stress inherent in the erythrocytotic perfusate. Increased arteriolar dilation and tissue vascularity contribute to the bleeding tendency (Figure 17-10).88 Syncope can be caused by the heat-induced vasodilation of hot humid weather or by a hot shower or bath, especially when standing.
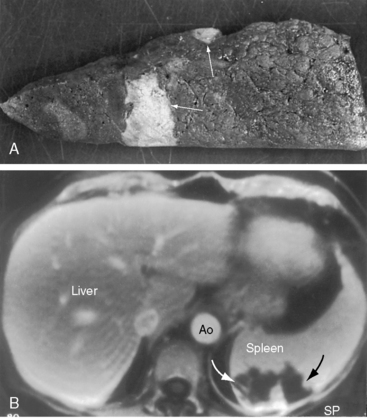
The extramural coronary arteries are dilated, elongated, and tortuous, even aneurysmal (coronary ectasia).88 Vasodilator nitric oxide and prostaglandins are released from coronary artery endothelium by the increased shear stress of erythrocytosis. Dilation of the coronary arteries is accompanied by an increase in extramural coronary artery blood flow that does not encroach on coronary vascular reserve because of remodeling of the myocardial microvascular circulation.94
Hyperuricemia results from decreased renal clearance and increased production of urate. Elevated plasma uric acid levels are common in cyanotic adults and are secondary to enhanced urate reabsorption that results from renal hypoperfusion reinforced by a high filtration fraction.88 Acute gouty arthritis is uncommon but not rare.
Renal involvement is represented by abnormalities of both structure and function.88,89 Decreased clearance of urate is a functional abnormality, but the resulting hyperuricemia in turn exerts little or no deleterious effect on renal function.88,89 Proteinuria is a functional abnormality caused by increased glomerular hydraulic pressure in response to the high viscosity of erythrocytotic blood entering the afferent glomerular arteriole in concert with obligatory ultrafiltration from afferent to afferent arterioles.
Two distinct structural (morphologic) glomerular abnormalities are found in cyanotic adults.89,95 The vascular abnormality is characterized by dilation of hilar arterioles and glomerular capillaries because of intraglomerular release of nitric oxide.95 The nonvascular abnormality is characterized by an increase in glomerular cellularity in response to platelet-derived growth factor and transforming growth factor beta in the cytoplasm of systemic venous megakaryocytes that are shunted into the systemic arterial circulation and lodge in glomerular tufts.95
Cardiorespiratory responses to isotonic exercise in cyanotic congenital heart disease significantly influence the dynamics of oxygen uptake and ventilation.88,96 Prolonged onset and recovery of oxygen uptake kinetics incur large oxygen deficits that result in hypoxemia even with low levels of exercise, which suggests that patients with right-to-left shunts rely heavily on anaerobic metabolism. Hyperventilation, which is subjectively perceived as dyspnea, is present at rest and increases excessively during exercise because augmentation of the right-to-left shunt induced by exercise is accompanied by an increase in systemic arterial carbon dioxide and a decrease in pH that stimulate the respiratory center and carotid bodies.88,96
Clubbing of the digits and hypertrophic osteoarthropathy share a common pathogenesis.88 Systemic venous megakaryocytes are shunted into the systemic arterial circulation (see previous) and impact in the digits and subperiostium. Platelet-derived growth factor and transforming growth factor beta are released from megakaryocytic cytoplasm and promote cell proliferation, protein synthesis, connective tissue formation, and deposition of extracellular matrix that are responsible for clubbing and hypertrophic osteoarthropathy.88
Pregnancy poses an excessive risk for women with Eisenmenger’s syndrome.87 The maternal mortality rate exceeds 50%, and there is an independent excessive risk of fetal wastage because of uterine hypoxemia associated with cyanosis.62,89,97,98
Sudden death often results from massive intrapulmonary hemorrhage,89 less commonly from rupture (dissection) of a dilated hypertensive pulmonary trunk,65,99 and rarely from ventricular tachycardia. A cerebral abscess in early life can result in a seizure disorder in adulthood, or the abscess can originate in adulthood.64,88
Physical Appearance
The abnormalities in physical appearance are the result of cyanosis caused by the reversed shunt in Eisenmenger’s syndrome, the catabolic effects of congestive heart failure in infants (poor growth and development, frailty, and cachexia),62 and Harrison’s grooves caused by the thoracic retractions of chronic dyspnea (Figure 17-11). Growth and development are also influenced by intrauterine and genetic factors and by low birth weight.75 Infants and young patients with nonrestrictive ventricular septal defects and balanced shunts may become cyanotic only after crying or exercise.100 Doubly committed subarterial ventricular septal defects are more common among Asians (see previous).17 The physical appearance of trisomy 18 Down syndrome (see Figures 19-5 and 20-11) coincides with the presence of an inlet ventricular septal defect (see Figures 15-90 and 15-91).101–104 In addition, a relationship exists between ventricular septal defect and physical appearance in trisomy 13; trisomy 8 and 9 mosaic; rare aberrations, such as 5p (cri du chat) and 13a- and 18q-105; Holt-Oram syndrome29; Cornelia de Lange’s syndrome72; Klippel-Feil syndrome54; cardiofacial syndrome106; and fetal alcohol syndrome.107
Arterial Pulse
Moderately restrictive defects with relatively low pulmonary vascular resistance are associated with a brisk arterial pulse because of vigorous ejection from a normal volume-loaded left ventricle. A diminished arterial pulse with pulsus alternans accompanies nonrestrictive defects with large left-to-right shunts and congestive heart failure. The arterial pulse in Eisenmenger’s syndrome is normal because systemic stroke volume is maintained (Figure 17-12).64
Jugular Venous Pulse
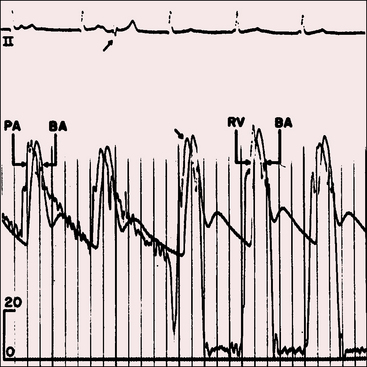
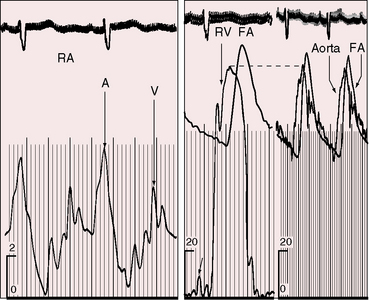
Moderately restrictive and nonrestrictive ventricular septal defects with congestive heart failure are accompanied by an elevated mean jugular venous pressure and an increase in A and V waves. However, the jugular venous pulse in Eisenmenger’s syndrome is normal or nearly so, with a small dominant A wave (Figure 17-13). Large A waves are exceptional because right ventricular systolic pressure does not exceed systemic. Systemic afterload exists from birth, so the right ventricle requires little extra help from its atrium.
Precordial Movement and Palpation
Roger stated that when a ventricular septal defect is restrictive, “it coincides with no other sign of organic disease with the exception of the harsh thrill which accompanies it.”1 The thrill is maximal in the third or fourth left intercostal space at the left sternal border. A subarterial ventricular septal defect directs its shunt into the pulmonary trunk, so the accompanying thrill is maximal in the second or first and second left intercostal space with radiation upward, to the left, and into the neck.108
Auscultation
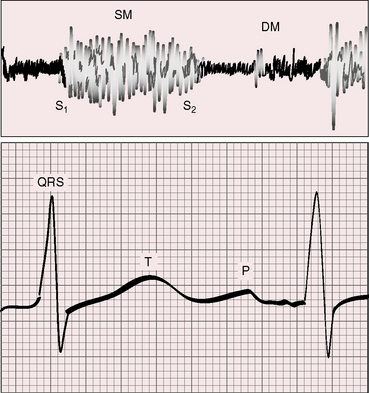
A very restrictive defect in the perimembranous or trabecular septum (see Figure 17-2B) is accompanied by a soft, localized, high-frequency, decrescendo, early systolic murmur (Figure 17-14) that is easily missed in restless infants. Early systolic timing indicates that the shunt is early systole because small perimembranous or muscular defects decrease in size or obliterate in late systole.109 The early systolic murmur lengthens during a premature ventricular beat because reduced left ventricular contractility fails to close the defect in late systole.78 Conversely, vigorous left ventricular contraction during a postpremature beat closes the defect early and shortens the murmur still further. During the evolution of spontaneous closure, a holosystolic murmur becomes early systolic before disappearing altogether (see Figure 17-18).23
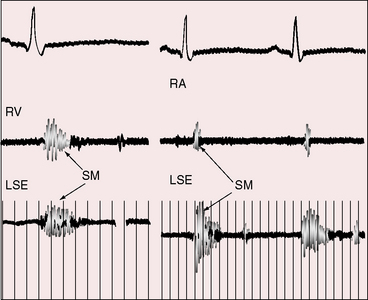
Moderately restrictive ventricular septal defects with pulmonary vascular resistance that is below systemic are accompanied by a loud harsh holosystolic murmur (see Figure 17-16) that is sometimes accentuated in midsystolic because of superimposition of a pulmonary flow murmur. The murmur of a restrictive perimembranous ventricular septal defect is also holosystolic, high-frequency, grade 4/6 or louder, and maximal in the third and fourth intercostal spaces at the left sternal border (Figures 17-15 and 17-16). These precordial sites are topographically appropriate for the intracardiac location of the murmur (see Figure 17-15).110
The typical holosystolic murmur (see Figures 17-15 and 17-16) corresponds to the holosystolic pressure difference across the defect. Roger recognized this mechanism when he described the murmur as “extending right through systole covering both heart sounds…” and “accompanied by no other physical signs save only the purring thrill.”1,30 When tricuspid chordae tendineae traverse the defect, the murmur is musical, assuming the pitch of an Aeolian harp. Muscular trabecular ventricular septal defect murmurs are maximal at the same precordial sites as murmurs of perimembranous defects.
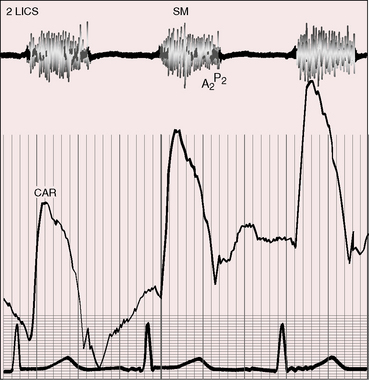
The shunt in a subarterial ventricular septal defect is directly into the pulmonary trunk, so the accompanying murmur and thrill are maximal in the second or first and second left intercostal space (Figure 17-17) with radiation upward toward the left clavicle into the suprasternal notch and into the left side of the neck (see Figure 17-17).108
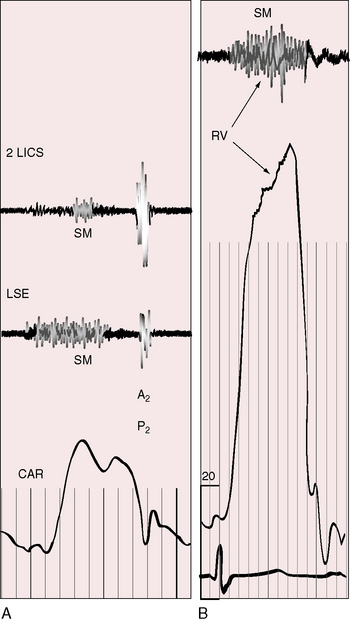
A decrease in size or spontaneous closure of a perimembranous ventricular septal defect is sometimes accompanied by a septal aneurysm111 that may harbor a small residual left-to-right shunt (see Figure 17-4). Otherwise, the aneurysm resembles a blind congenital membranous septal aneurysm (see previous).44 Septal aneurysms are usually occult, but occasionally there are auscultatory clues. The aneurysm may contain one or more small perforations (see Figure 17-4) that generate a late systolic murmur, or a holosystolic murmur with late systolic accentuation, because stretching of the aneurysmal pouch accentuates late systolic patency of small perforations.112–114 Murmurs have also been attributed to the entry of blood into a sealed blind aneurysmal pouch during ventricular systole. Systolic tensing of the aneurysm sometimes produces a midsystolic click that introduces a late systolic murmur.113 The aneurysm occasionally protrudes into the right ventricular outflow tract outflow tract (see Figure 17-4), causing a midsystolic murmur in the second left intercostal space. A septal aneurysm may cause tricuspid regurgitation115 with the murmur identified during active respiration (Carvallo’s sign).116
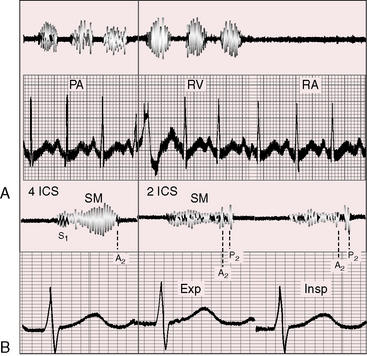
The prominent holosystolic murmur of a moderately restrictive ventricular septal defect usually buries both components of the second sound, rendering the pulmonary component audible only during inspiration as it emerges from the murmur (see Figure 17-17). Analysis of the second heart sound benefits from exaggerated splitting (see Figure 17-15B).117 Attention has been called to wide splitting with subarterial defects (see Figure 17-17).108
Moderately restrictive or nonrestrictive ventricular septal defects with large left-to-right shunts are associated with short mid-diastolic murmurs at the apex because of torrential flow across the mitral valve (see Figure 17-16)118 as originally described by Laubry and Pezzi in 1921.119 These murmurs are often preceded by a third heart sound, especially in the presence of left ventricular failure. Intracardiac phonocardiograms confirm the presence of mid-diastolic murmurs within the inflow tract of the left ventricle just beyond the mitral valve.110
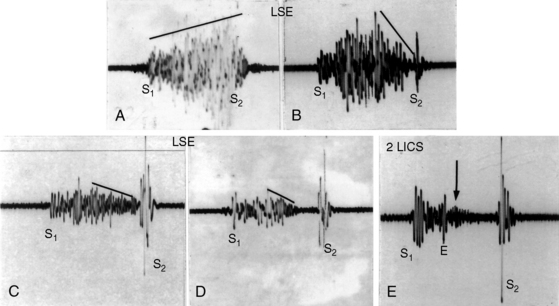
Nonrestrictive ventricular septal defects with a progressive increase in pulmonary vascular resistance illustrate the effects of pulmonary vascular disease on the length and loudness of the accompanying systolic murmur.120 As pulmonary resistance approaches systemic, the holosystolic murmur shortens and softens (Figures 17-18C, 17-18D, and 17-19), and its shape becomes decrescendo (see Figures 17-18 and 17-19). The murmur is early systolic (see Figure 17-18D) before disappearing altogether as the shunt is reversed (see Figure 17-18E).64,120
Auscultatory analysis of the second heart sound benefits from an increase in intensity of the pulmonary component and from a rise in pulmonary vascular resistance that softens the murmur in latter systole.120 As pulmonary resistance increases, the degree of splitting decreases (see Figure 17-19),120 so the second sound is single in Eisenmenger’s syndrome (see Figure 17-18E).64,120,121 The two components fuse because the respective descending limbs of the left and right ventricular pressure pulses cross the aortic and pulmonary arterial pressure pulses simultaneously. Because the aortic and pulmonary arterial dicrotic notches are synchronous, the two components of the second sound are synchronous.
When suprasystemic pulmonary vascular resistance abolishes the left-to-right shunt, the auscultatory signs are those of pure pulmonary hypertension (see Figure 17-18E and Chapter 14).120 Flow into the dilated hypertensive pulmonary trunk causes a pulmonary ejection sound that introduces a short, soft, midsystolic murmur.120 A high-frequency Graham Steell murmur issues from a loud pulmonary component of the second heart sound and is the only diastolic murmur when the shunt is reversed. The mid-diastolic murmur across the mitral valve disappears as pulmonary blood flow decreases. Rarely, the murmur of pulmonary regurgitation is caused by prolapse of a pulmonary cusp through a subarterial ventricular septal defect.18
Electrocardiogram
The electrocardiogram is an important indication of the physiologic derangements of ventricular septal defects, but not of their anatomic location. The scalar tracing is influenced by the size of the defect, the size of the left-to-right shunt, and the pulmonary vascular resistance (see Figure 17-5; i.e., by the presence and degree of volume overload of the left ventricle and the presence and degree of pressure overload of the right ventricle).
A restrictive ventricular septal defect with normal pulmonary artery pressure is associated with a normal electrocardiogram and an occasional rsr prime pattern in lead V1.122,123 When a restrictive perimembranous defect is accompanied by a septal aneurysm, there is an increase in the incidence of rhythm and conduction disturbances, especially atrial fibrillation, paroxysmal atrial tachycardia, junctional rhythm, atrial flutter, and complete heart block.51
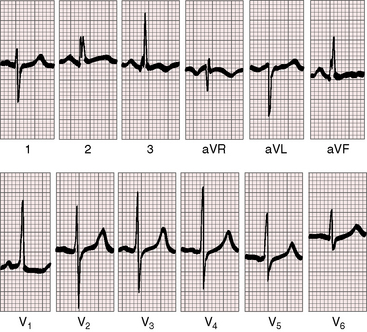
Moderately restrictive ventricular septal defects with a large left-to-right shunt are associated with broad notched left atrial P waves in lead 1 and 2 and with a broad deep P terminal force in lead V1 (Figure 17-20).122 The QRS axis is normal (see Figure 17-20), although left axis deviation occurs in about 5% of restrictive or moderately restrictive perimembranous defects (Figure 17-21).124–126 Inlet defects are associated with left axis deviation when they are a component of an atrioventricular septal defect (see Chapter 15).127 In the presence of ventricular septal aneurysms, the incidence rate of left axis deviation increases; it has been found in as many as 40% of patients with multiple ventricular septal defects. Volume overload of the left ventricle is reflected in tall R waves and tall peaked T waves in leads 2, 3, and aVF and in prominent Q waves, tall R waves, and tall peaked T waves in leads V5-6 (see Figure 17-20).123,128
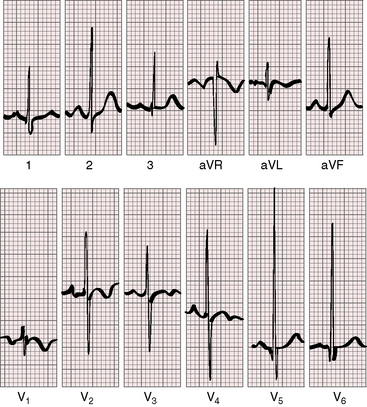
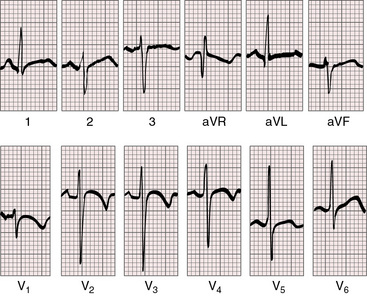
A nonrestrictive ventricular septal defect with a large left-to-right shunt exhibits right atrial or combined right and left atrial P wave abnormalities, especially in lead 2 and in leads V1-2 (Figure 17-22). The QRS axis shifts moderately to the right (Figures 17-22 and 17-23). Biventricular hypertrophy is reflected in the increased R wave amplitude in lead V1, the deep Q waves, tall R waves, and tall peaked T waves in leads V5-6 and in large equidiphasic RS complexes (the Katz-Wachtel pattern) in midprecordial leads (see Figures 17-22 and 17-23).122,129 Infants with a nonrestrictive ventricular septal defect and large left-to-right shunts occasionally exhibit marked right axis deviation and pure or relatively pure right ventricular hypertrophy, except for large equidiphasic complexes in one or more midprecordial leads (Figure 17-24).
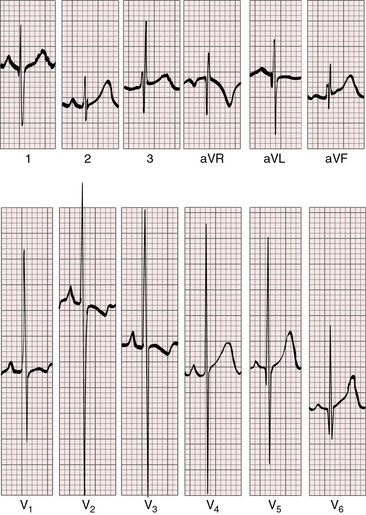
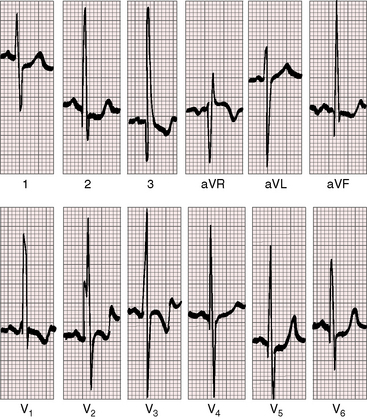
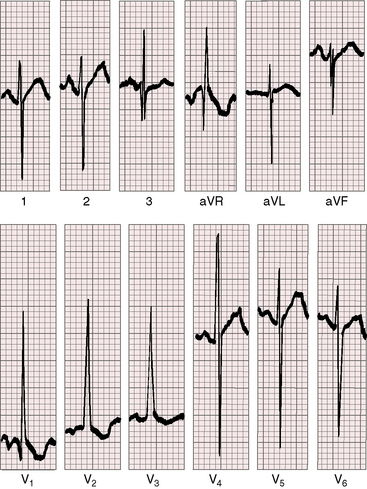
In Eisenmenger’s syndrome, P waves are often normal (Figure 17-25) in younger patients and moderately peaked in older patients. Right axis deviation is moderate. Lead V1 exhibits a tall monophasic R wave (see Figure 17-25) that is occasionally notched on its upstroke and followed by a small s wave. Prominent S waves appear in left precordial leads, but combined ventricular hypertrophy is lacking (see Figure 17-25).
X-Ray
Vaquez and Bordet130 described the radiologic signs of ventricular septal defect in 1913. Small defects that were moderately restrictive at birth show residual signs of an initially larger left-to-right shunt, especially an increase in size of the left ventricle and dilation of the pulmonary trunk (Figures 17-6B and 17-26).
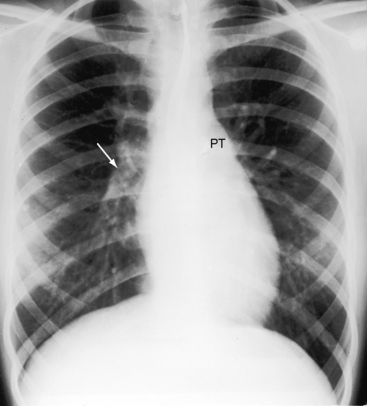
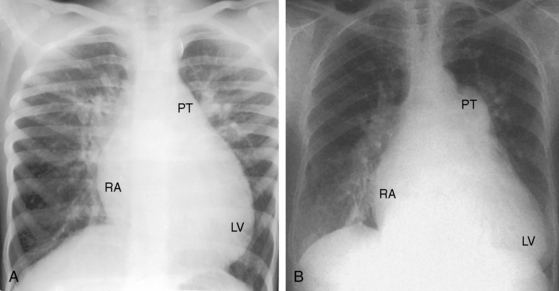
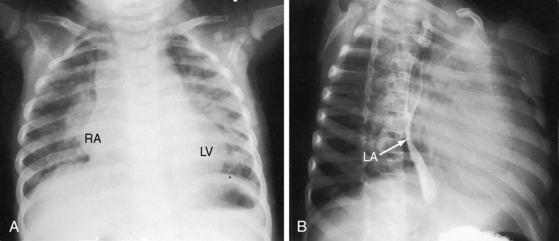
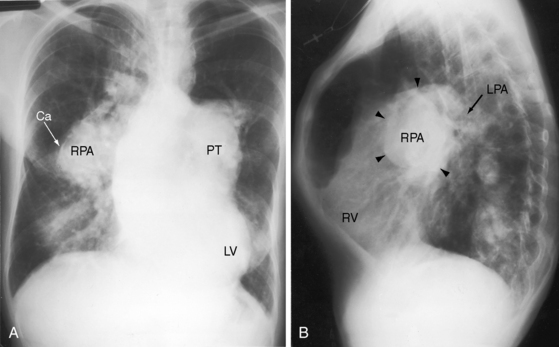
Moderately restrictive ventricular septal defects with low but variable pulmonary vascular resistance (see Figure 17-5B) exhibit radiologic signs appropriate for the magnitude of the left-to-right shunt and the presence and degree of pulmonary hypertension. When pulmonary vascular resistance is relatively low, increased pulmonary arterial vascularity and pulmonary venous congestion coexist (Figures 17-6A and 17-27). Large shunts in infants are accompanied by hyperinflated lungs with flat hemidiaphragms.131 Right atrial dilation accompanies congestive heart failure (see Figures 17-6A and 17-27). Enlargement of the left atrium is best recognized in the lateral projection (see Figure 17-27B) and can be more impressive than enlargement of the left ventricle. An increase in size of the pulmonary trunk and its branches reflects the magnitude and chronicity of pulmonary arterial blood flow and the level of pulmonary arterial pressure (see Figures 17-7 and 17-8).132 The size of the ascending aorta is not increased because the left-to-right shunt is intracardiac and therefore does not traverse the aortic root (see Figure 17-8). In the lateral projection, the lower sternum is apt to protrude anteriorly whereas the upper sternum is relatively straight.
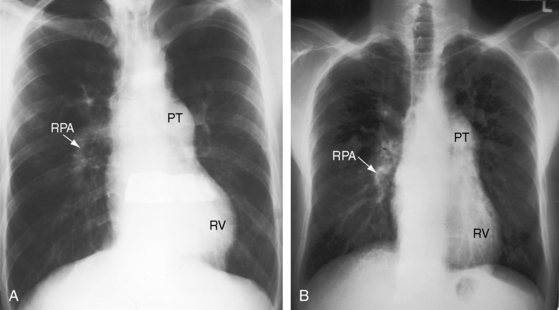
Nonrestrictive ventricular septal defects with elevated but variable pulmonary vascular resistance present in infancy with congestive heart failure, pulmonary venous congestion, and enlargement of all four chambers. The x-rays resemble those of moderately restrictive defects with large left-to-right shunts (see Figures 17-6A and 17-7). In the exceptional adult, the pulmonary trunk and its branches are aneurysmal (Figure 17-28). As pulmonary vascular resistance rises, the left-to-right shunt falls reciprocally, congestive heart failure is ameliorated, and the heart size decreases, but enlargement of the pulmonary trunk and its branches persists (see Figure 17-6B) When the shunt is reversed by suprasystemic pulmonary vascular resistance, Eisenmenger’s syndrome (see Figure 17-5D), the lung fields are oligemic; right atrial, left atrial, and left ventricular sizes are normal; and the hypertrophied but nondilated right ventricle occupies the apex (see Figure 17-9). Except for enlargement of the pulmonary trunk and its branches and a convex apical right ventricle, the cardiac silhouette is deceptively unimpressive (see Figure 17-9).
Echocardiogram
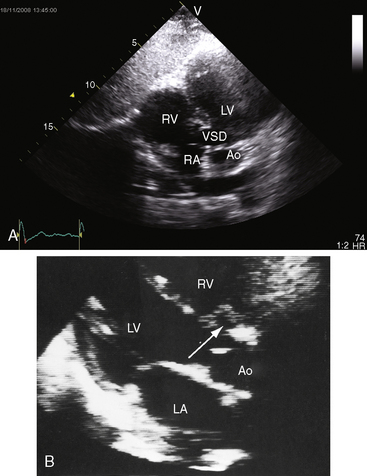
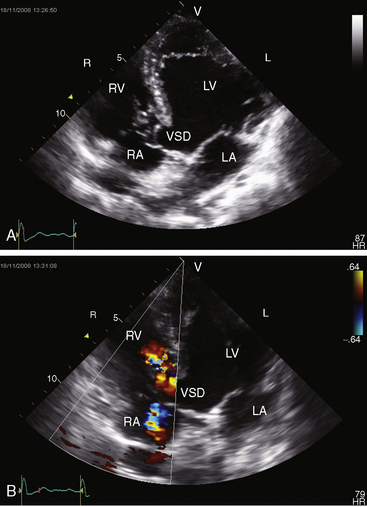
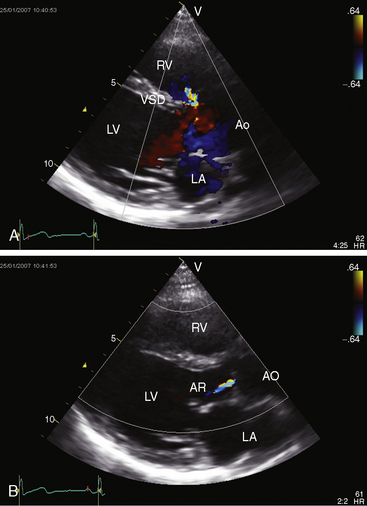
Tranthoracic and tranesophageal echocardiography with color flow imaging and Doppler interrogation establish the presence, location, and physiologic consequences of ventricular septal defects (Figure 17-29 and Video 17-1).133–135 Shunt flow can be examined in the fetus with color Doppler M-mode scan.136 Three-dimensional echocardiography improves assessment137,138 and has proven accurate in quantifying shunts.2 Small multiple defects in the muscular septum,139 spontaneous closure of perimembranous or muscular defects, and septal aneurysms140 can be identified, and Swiss cheese defects in the trabecular muscular septum can be recognized with color flow imaging. Right ventricular systolic pressure and pulmonary artery systolic and diastolic pressure can be estimated (Figure 17-30).
The complex shunt flow across a ventricular septal defect was first characterized with angiocardiography141 but is now characterized with echocardiography.139 Isovolumetric contraction is accompanied by left-to-right shunting, with flow patterns during ventricular ejection depending in part on the size of the ventricular septal defect (see Figure 17-30). In nonrestrictive defects, the direction of shunt flow is determined by the relative resistances in the pulmonary and systemic vascular beds. Isovolumetric relaxation coincides with right-to-left flow. Two patterns have been identified during ventricular diastole: transient right-to-left flow at the time of mitral valve opening followed by left-to-right flow from mid-diastole to the time of mitral valve closure.
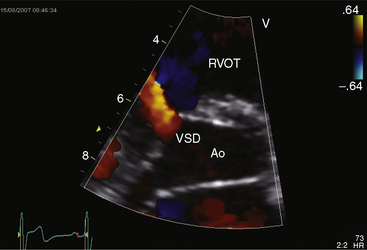
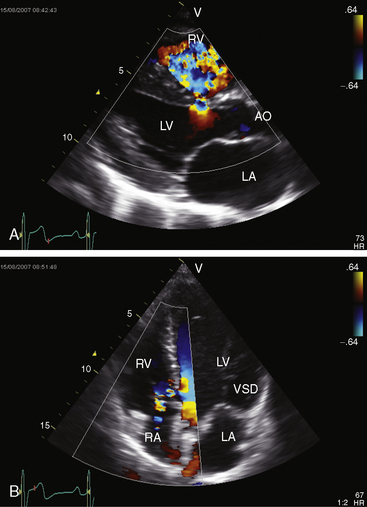
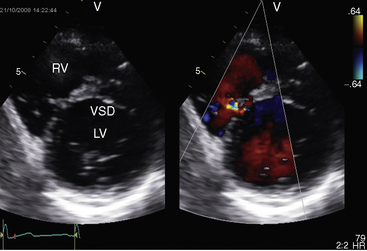
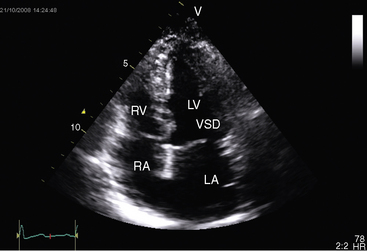
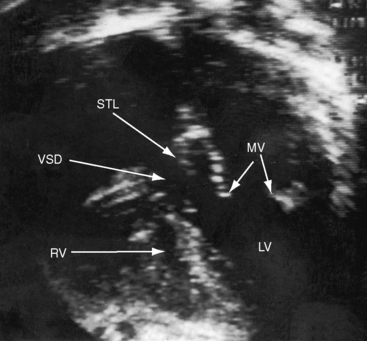
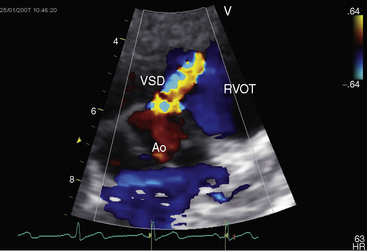
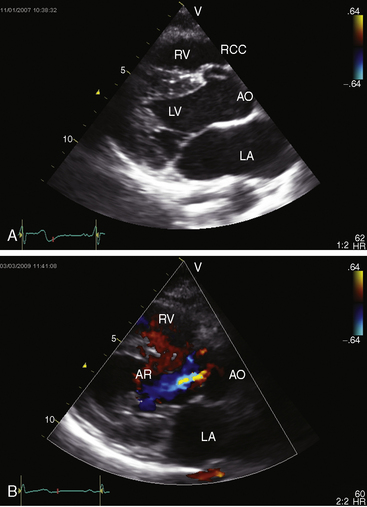
Echocardiography with color flow imaging identifies the ventricular septal defect in the perimembranous septum (Figure 17-31 and Video 17-2), in the muscular septum (Figures 17-32 and 17-33 and Videos 17-3 and 17-4), in the infundibular septum, or in the inlet septum (Figure 17-34). Two-dimensional imaging identifies a septal aneurysm (Figures 17-31B and 17-35A and Video 17-5A), and color flow imaging determines whether or not the aneurysm harbors a residual shunt (Figure 17-35B and Video 17-5B). Aneurysm formation warrants color flow interrogation of the tricuspid valve for the presence of regurgitation.115 Inlet ventricular septal defects can be accompanied by atrioventricular valve straddling (see Figure 17-34). A subarterial defect is associated with flow directed toward the pulmonary trunk (Figure 17-36), with continuity of aortic and pulmonary valves in the roof of the defect43,133 and with aortic regurgitation from prolapse of the right or noncoronary cusp (see Figure 17-45 and Videos 17-6A and 17-6B).
Ventricular septal defect with aortic regurgitation
In 1921, Laubry and Pezzi119 published a postmortem account of ventricular septal defect with an incompetent aortic valve. Eleven years later, Laubry, Routier, and Soulie142 described two patients with a ventricular septal defect, a loud aortic diastolic murmur, a wide pulse pressure, and at necropsy, prolapsed aortic cusps. The prevalence of aortic regurgitation with ventricular septal defect depends largely on the relative prevalence of perimembranous versus doubly committed subarterial defects.17,143,144 Perimembranous defects are more common in whites, with an incidence rate of aortic regurgitation of approximately 5% to 8%.143,144 Subarterial defects are more common in Asians, with an incidence rate of aortic regurgitation of approximately 30%.17,144
Perimembranous defects are located immediately beneath the commissure between the right coronary cusp and the noncoronary cusp.143 Aortic regurgitation is associated with sagging or herniation of the right coronary cusp or the right coronary cusp and the noncoronary cusp (Figures 17-37 and 17-38).32,144 Herniation sometimes protrudes into the right ventricular outflow tract and causes obstruction145 or protrudes through the ventricular septal defect and reduces the left-to-right shunt.143
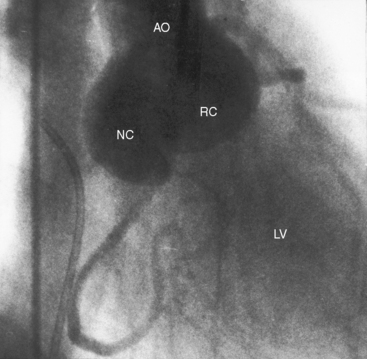
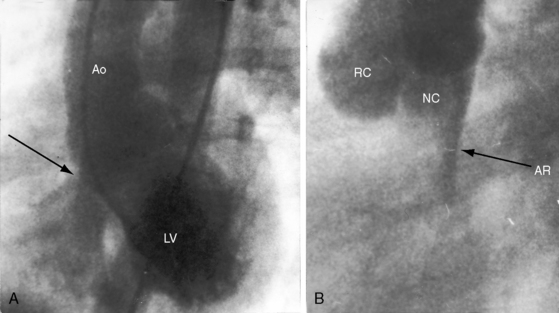
The inherent morphologic basis for aortic regurgitation has been assigned to the right coronary/noncoronary commissure where a structural abnormality is aggravated if not caused by the high-velocity jet accompanying the ventricular septal defect shunt.145,146 This is in accord with observations that aortic regurgitation develops years after birth147 and that when the high-velocity jet is absent as in Eisenmenger’s syndrome, aortic regurgitation is virtually unknown.143
The typical sequence begins with the basic structural abnormality responsible for faulty aortic leaflet apposition, followed by sagging of the noncoronary cusp and right coronary cusp (see Figures 17-37 and 17-38).145,146 Once the cusps fall below the normal closure line, the aortic valve becomes incompetent, the impact of the regurgitant jet provokes leaflet elongation and thickening, and aortic regurgitation begets aortic regurgitation.146
Ventricular septal defects in the infundibular septum lie immediately beneath the aortic and pulmonary valves that constitute a contiguous roof over the defect, which is therefore doubly committed and subarterial.17 The aortic and pulmonary valves are in fibrous continuity because the infundibular septum, including the septal portion of the subpulmonary infundibulum, is deficient.17 The aortic valve lacks support, so the right and noncoronary sinuses move into the right ventricular outflow tract, causing aortic regurgitation and occasionally a subpulmonary gradient.12,143,145
The physiologic consequences of aortic regurgitation with a ventricular septal defect are borne by the left ventricle, which receives the sum of the regurgitant volume from the aorta and the shunt volume from the ventricular septal defect. Aortic regurgitation begins insidiously and progresses gradually unless infective endocarditis supervenes (see section The History). The left-to-right shunt is larger for a given degree of incompetence when regurgitation is associated with a subarterial defect, which does not decrease in size unless partially occluded by the prolapsed right coronary cusp. Conversely, a perimembranous ventricular defect tends to get smaller as the degree of aortic regurgitation increases, so aortic regurgitant volume rather than shunt volume becomes the major contributor to left ventricular volume overload.143 The shunt can then be trivial or even absent in the presence of severe aortic regurgitation (Figure 17-39).143
History
When aortic regurgitation coexists with a ventricular septal defect, the male:female ratio is as high as 2:1, in contrast to the equal gender distribution when a ventricular septal defect occurs in isolation (see previous).143,144 Echocardiography and aortography in perimembranous and subarterial ventricular septal defects detect herniation of the right and noncoronary cusps before the onset of regurgitation,17 which seldom begins before age 2 years or after age 10 years and peaks between 5 and 9 years of age.143,147
The diagnosis can sometimes be suspected based on the history. The typical murmur of ventricular septal defect is known for years, more likely in a male. Between age 5 years and 9 years, an additional murmur is discovered. The patient gradually becomes aware of neck pulsations, chest pain, diaphoresis, and vigorous precordial movement, especially at night when lying in the left lateral recumbent position. Infective endocarditis, to which the incompetent aortic valve is highly susceptible, dramatically accelerates regurgitant flow,144 and the occasional coexistence of a bicuspid aortic valve increases susceptibility to infection.148
Arterial Pulse
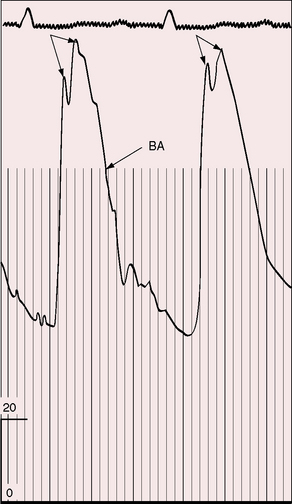
In gradually progressive chronic severe aortic regurgitation, the arterial pulse is the classic bounding Corrigan water hammer with a wide pulse pressure, a brisk rate of rise, bisferiens peaks (see Figure 17-39), pistol shot sounds over the femoral arteries, and capillary pulsations in the nail beds (see Chapter 7).143 Taussig said of one patient, “The head shook with each cardiac impulse and the pulsations of the dorsalis pedis were readily felt through the bed clothes.”149
In acute severe aortic regurgitation, the arterial pulse differs considerably from the previous description.150 The left ventricle cannot adapt to the sudden increase in volume, so contractility is depressed, the velocity of ejection declines, stroke volume and peak systolic pressure cannot increase, and end diastolic pressure rises steeply. Accordingly, the pulse pressure is not increased, and the rate of rise is not brisk.
Auscultation
The murmurs of ventricular septal defect with aortic regurgitation are not continuous (see Figure 17-41B) but are contiguous holosystolic and early diastolic (Figures 17-40 and 17-41A).116,143 The systolic portion of the murmur is typical of ventricular septal defect (see Figures 17-40 and 17-41A),110 and the diastolic portion is typical of chronic aortic regurgitation (see Figures 17-40 and 17-41A).150 These contiguous murmurs do not peak around the second heart sound and therefore differ from the continuous murmur of a patent ductus arteriosus that envelops the second heart sound and peaks around it (see Figure 17-41B and Chapter 20).

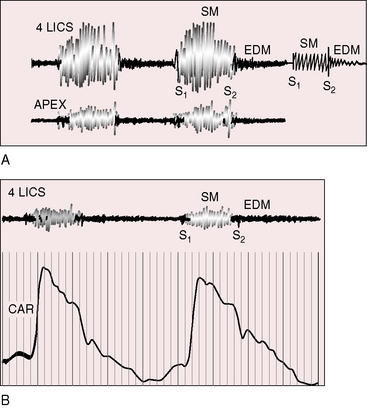
Figure 17-40 A, Tracings from an 8-year-old boy with a moderately restrictive perimembranous ventricular septal defect, a 2 to 1 left-to-right shunt, and mild aortic regurgitation. A prominent holosystolic murmur (SM) is followed by a soft, high-frequency early diastolic murmur of aortic regurgitation (EDM). The holosystolic and early diastolic murmurs are schematically illustrated on the right of the same strip (also see Figure 17-41A). At the apex, the holosystolic murmur remained prominent, but the aortic regurgitation murmur was absent. B, Tracings from a 17-year-old boy with a restrictive perimembranous ventricular septal defect, a 1.3 to 1 shunt, and moderate aortic regurgitation (brachial arterial pressure, 135/50 mm Hg). In the fourth left intercostal space (4 LICS), there was the high-frequency holosystolic murmur of a restrictive ventricular septal defect and a soft early diastolic murmur (EDM) of aortic regurgitation. (CAR = carotid pulse.)
The systolic and diastolic portions of the continuous murmur of patent ductus are both maximal in the first or second left intercostal space, the systolic portion of the murmur of ventricular septal defect is loudest in the third or fourth intercostal space, and the diastolic aortic regurgitation portion is best heard at the mid to lower left sternal border (see Figure 17-40). Uncertainty arises in the third left intercostal space where the systolic and diastolic murmurs are both prominent and sometimes create the mistaken impression of a continuous murmur. Distinctive eddy sounds punctuate the ductus murmur.116
The distinction between the murmur of aortic regurgitation and a Graham Steell murmur is difficult if not impossible on purely auscultatory grounds.116 However, aortic regurgitation rarely occurs with the Eisenmenger’s syndrome (see previous), so a high-frequency early diastolic murmur in that contest is likely to be Graham Steell.120 Conversely, in the presence of a restrictive ventricular septal defect, an early diastolic murmur is likely to be aortic regurgitation, especially if the patient is male and certainly if the systemic arterial pulse is water hammer. When the shunt is small and aortic regurgitation is severe, an apical mid-diastolic murmur is an Austin Flint rumble and not increased shunt flow across the mitral valve.143
Electrocardiogram
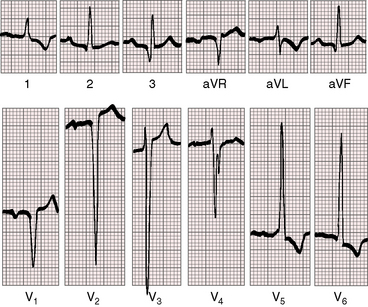
The scalar tracing is useful in calling attention to the left ventricular hypertrophy of aortic regurgitation unexplained by a restrictive ventricular septal defect (Figure 17-42) (i.e., the electrocardiogram resembles aortic regurgitation rather than ventricular septal defect). Deep Q waves, tall R waves, deeply inverted T waves, and coved ST segments appear in left precordial leads (Figure 17-42). When a subarterial ventricular septal defect coexists with moderate aortic regurgitation and a moderate left-to-right shunt, these distinctions are less clear.
X-Ray
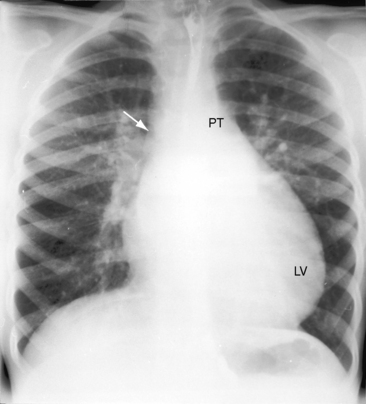
A key point in the x-ray is the contrast between appreciable enlargement of the left ventricle in the context of no more than a modest left-to-right shunt (Figure 17-43).143 The x-ray resembles severe aortic regurgitation rather than ventricular septal defect (see Figure 17-43). The ascending aorta is prominent and pulsates vigorously on fluoroscopy. When the ventricular septal defect is subarterial, the aortic root is inconspicuous despite appreciable regurgitant flow because the ventricular septal defect remains large enough to divert a sizable fraction of left ventricular stroke volume into the pulmonary circulation (Figure 17-44). Whether the ventricular septal defect is perimembranous or subarterial, the onset of aortic regurgitation is insidious, so the x-ray initially reflects the left-to-right shunt. As time goes on, the balance shifts as described previously.
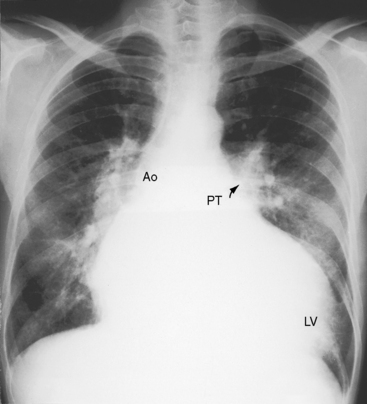
Figure 17-43 X-ray from a 21-year-old man with a restrictive perimembranous ventricular septal defect, a left-to-right shunt of 1.4 to 1, and severe aortic regurgitation. The electrocardiogram is shown in Figure 17-42. The pulmonary trunk (PT) only mildly convex, but the relatively prominent pulmonary vascularity implies that the shunt had previously been larger and the ventricular septal defect had decreased in size. The large left ventricle (LV) and the dilated ascending aorta (Ao) are the result of the aortic regurgitation, not the left-to-right shunt of the ventricular septal defect.
Echocardiogram
Transthoracic and transesophageal echocardiography with Doppler interrogation and color flow imaging identify the ventricular septal defect as perimembranous or subarterial, identify prolapse of the right and noncoronary cusps, and establish the presence and degree of aortic regurgitation (Figure 17-45 and Videos 17-6A and 17-6B).2,17,151 Prolapse of the right coronary artery can be distinguished from an aneurysm of the membranous septum. Color flow imaging identifies flow across a perimembranous ventricular septal defect (Figure 17-46A) and establishes the presence and degree of aortic regurgitation (see Figures 17-45B and 17-46B). A subarterial ventricular septal defect is recognized by its proximity to the pulmonary and aortic valves, whose sinuses are contiguous without interposition of an infundibular septum. The systolic jet through the subarterial ventricular septal defect is directed into the pulmonary trunk (see Figure 17-36). Echocardiography with color flow imaging identifies prolapse of the right coronary cusp or noncoronary aortic cusp before the onset of aortic regurgitation.17 Doppler interrogation determines the presence and degree of right ventricular outflow obstruction caused by prolapse of an elongated right coronary cusp through the ventricular septal defect.
Left ventricular to right atrial communication
In 1838, Thurnman152 described a left ventricular–to–right atrial communication in which a cleft tricuspid septal leaflet adhered to a ventricular septal defect. Two decades later, Hillier153 reported on “Congenital Malformation of the Heart; Perforation of the Septum Ventriculorum, Establishing a Communication between the Left Ventricle and the Right Atrium.” Similar if not identical reports appeared from Merkel in 1869 and from Preicz in 1890.154 The 1958 publication by Gerbode and associates155 stimulated renewed interest in the malformation that is sometimes called the Gerbode defect.
A perimembranous ventricular septal defect is an obligatory component of the two types of left ventricular–to–right atrial communications illustrated in Figure 17-47.77,155–158 The first and more common type is characterized by a defect in the intraventricular portion of the membranous septum that opens into the right ventricle and then communicates with the right atrium through an anatomic deficiency in the septal tricuspid leaflet, which is tethered to the crest of the ventricular septum (see Figure 17-47A). The deficiency in the septal tricuspid leaflet takes the form of perforations, clefts, and widening or absence of the commissure between the anterior and septal tricuspid leaflets.157 A septal aneurysm is almost always present.159,160 The ventriculoatrial communication originally described by Thurnman was this first type.152 In the second and much less common type, the defect is in the atrioventricular portion of the membranous septum, so left ventricular blood enters the right atrium directly (see Figure 17-47E). This type of communication differs structurally from the atrioventricular septal defect discussed in Chapter 15.
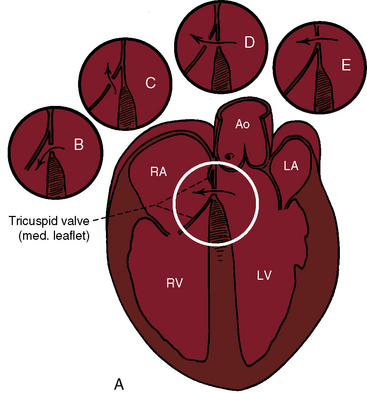
Figure 17-47 Schematic illustrations of the two varieties of left ventricular–to–right atrial communications originally proposed by Perry, Burchell, and Edwards156 in 1949. The variety depicted in (A), the large sketch, consists of a defect in the intraventricular portion of the membranous septum with tethering of the septal tricuspid leaflet to the crest of the ventricular septal defect. (RA/LA = right atrium/left atrium; RV/LV = right ventricle/left ventricle.) Circular insert (E) depicts the variety in which a defect in the atrioventricular portion of the membranous septum creates a direct communication from left ventricle into right atrium (arrow). Inserts B, C, and D are variations of a subanular defect in the membranous septum.
(With permission from Perry EL, Burchell HB, Edwards JE. Congenital communication between left ventricle and right atrium. Proc Staff Meet Mayo Clin 1949;24:198.)
The physiologic consequences of left ventricular to right atrial communications depend on the size of the defect and on the pulmonary vascular resistance, as is the case with other types of ventricular septal defects. Shunt flow begins in utero because of the obligatory difference in systolic pressure between the left ventricle and right atrium.77 The right atrium accepts the shunt with little or no elevation of pressure because right atrial distensibility and volume increase appropriately and because the ventricular septal defect is usually restrictive,77,155 a feature that also accounts for normal or near-normal pulmonary artery pressure. Left ventricular blood that is received by the right atrium during ventricular systole (Figure 17-48) enters the right ventricle during the next diastole, establishing right ventricular volume overload. The shunt volume then finds its way through the pulmonary circulation, so volume overload of the left side of the heart coexists.
History
Gender distribution is equal or with slight female preponderance.77,161 One patient with a left ventricular–to–right atrial communication had a sibling with an isolated perimembranous ventricular septal defect.160 The clinical course is, for all practical purposes, indistinguishable from that described previously for restrictive or moderately restrictive perimembranous ventricular septal defects. However, an important difference is the chronology of the murmur because onset of the shunt and therefore the onset of the murmur do not depend on the neonatal fall in pulmonary vascular resistance. The shunt is present in utero (see previous) because of the obligatory differences in systolic pressure between left ventricle and right atrium, so the murmur generated by the shunt is necessarily present at birth.77 The lesion is susceptible to infective endocarditis as with other perimembranous ventricular septal defects.162 Occasionally, infective endocarditis is responsible for creating an acquired Gerbode defect (see previous).82
Jugular Venous Pulse
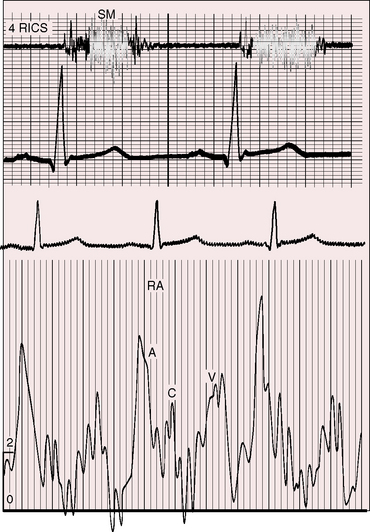
Because right atrial volume and distensibility increase in proportion to the shunt,77,155 large V waves are seldom present and the mean jugular venous pressure is seldom elevated (Figure 17-49), even though the left ventricle ejects into the right atrium (see previous).155
Precordial Movement and Palpation
The left-to-right shunt received by the right atrium then enters the right ventricle, which is therefore volume overloaded. The right ventricular impulse is disproportionately prominent compared with ventricular septal defects of comparable size in other locations and is analogous to the volume-overloaded right ventricular impulse of an ostium secundum atrial septal defect (see Chapter 15). However, the apex is occupied by a volume-overloaded left ventricle.
Auscultation
The quality, configuration, and intensity of the systolic murmur generated by a left ventricular–to–right atrial communication are indistinguishable from murmurs of other ventricular septal defects of comparable size.155 The topographic location of the murmur is its only distinguishing feature. Because the shunt is received by a right atrium that underlies the right lower sternal border (see Figure 17-49), the systolic murmur is recorded within the right atrial cavity (Figure 17-50)163 and is located in a relatively rightward thoracic position (see Figure 17-49).164 Equal volumes of blood traverse the tricuspid and mitral valves, so mid-diastolic murmurs are generated across both atrioventeicular valves, depending on shunt volume.
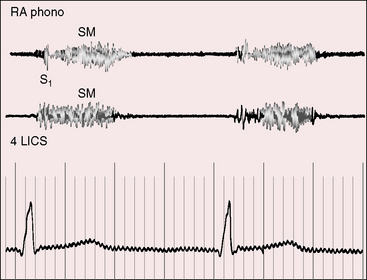
Figure 17-50 Tracings from the 17-year-old boy referred to in Figure 17-49. Intracardiac phonocardiogram within the right atrial cavity (RA pHONO) recorded a holosystolic murmur (SM), and the thoracic wall phonocardiogram in the fourth left intercostal space (4 LICS) recorded an identical murmur.
The intensity, splitting, and respiratory movement of the second heart sound are the same as in comparable isolated perimembranous ventricular septal defects. The split widens normally during inspiration because systemic venous return to the right side of the heart increases without a reciprocal decline in left-to-right shunt. The systolic shunt that is responsible for volume overload of the right ventricle also traverses the left ventricle, so both ventricles handle equal volumes that are ejected against different resistances. Low pulmonary vascular resistance and increased pulmonary capacitance delay the pulmonary component of the second sound. The split is persistent but moves normally with respiration because an intact atrial septum prevents a reciprocal decline in left-to-right shunt that accompanies an ostium secundum atrial septal defect (see Chapter 15). Normal intensity of the pulmonary component is a useful sign because it indicates that right ventricular enlargement is not the result of pulmonary hypertension but instead of volume overload.
Electrocardiogram
There is an age-related increase in the incidence of supraventricular tachycardia, atrial flutter, and atrial fibrillation.77,165 Tall peaked right atrial P waves date from infancy because the right atrium is the early recipient of the left-to-right shunt. PR interval prolongation reflects right atrial enlargement, and biatrial P wave abnormalities are common because both atria handle an equivalent increase in volume (Figure 17-51).77,166
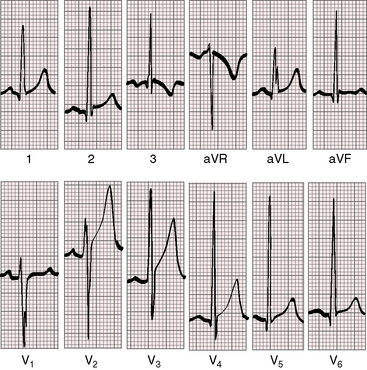
Figure 17-51 Electrocardiogram from the 17-year-old boy referred to in Figure 17-49. The tracing resembles an isolated ventricular septal defect. Volume overload of the left ventricle is represented by prominent Q waves, tall R waves, and peaked T waves in leads V5-6. Slight peaking of the P wave in lead V2 is the only suggestion of a right atrial abnormality.
An rSr prime in lead V1 and a terminal r wave in aVR and V3R are electrocardiographic signs of right ventricular volume overload. Prominent left precordial Q waves, tall R waves, and upright T waves are signs of coexisting left ventricular volume overload (see Figure 17-51). A useful electrocardiographic sign is the combination of tall peaked right atrial P waves with left ventricular hypertrophy.77,155
X-Ray
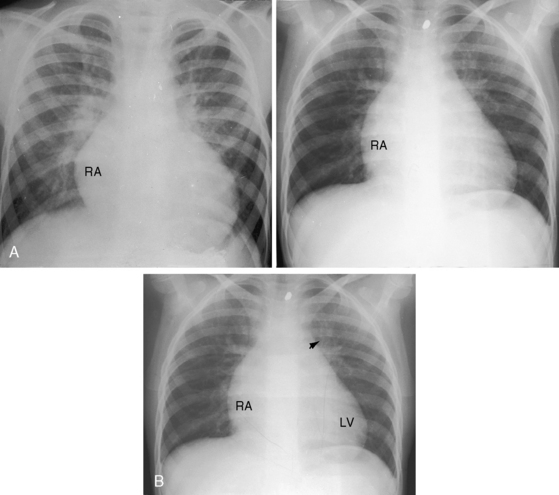
The most distinctive feature in the x-ray is disproportionate right atrial enlargement (Figure 17-52).155,161,165 The cardiac silhouette occasionally has a ball-like shape with the right side of the ball formed by the large right atrium and the left side of the ball formed by dilation of the right ventricular infundibulum and the left ventricle (see Figure 17-52A).77,165
Echocardiogram
Echocardiography identifies the perimemranous ventricular septal defect, and color flow imaging identifies the flow pattern from left ventricle into right atrium.133,135 The relationship of the ventricular septal defect to the tricuspid valve can be established, and a ventricular septal aneurysm can be identified.
1 Roger H.. Clinical researches on the congenital communication of the two sides of the heart by failure of occlusion of the interventricular septum. Bull, de l’Acad. de Méd. 8:1074,1879. Willius E.A., Keys T.E., editors. Classics of Cardiology. Malabar: Floida Robert E. Krieger Publishing Company, 1983.
2 Minette M.S., Sahn D.J. Ventricular septal defects. Circulation. 2006;114:2190-2197.
3 Martin G.R., Perry L.W., Ferencz C. Increased prevalence of ventricular septal defect: epidemic or improved diagnosis. Pediatrics. 1989;83:200-203.
4 Newman T.B. Etiology of ventricular septal defects: an epidemiologic approach. Pediatrics. 1985;76:741-749.
5 Hoffman J.I., Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890-1900.
6 Ferencz C., Rubin J.D., Mccarter R.J., et al. Congenital heart disease: prevalence at livebirth. The Baltimore-Washington Infant Study. Am J Epidemiol. 1985;121:31-36.
7 Fyler D.C. Report of the New England Regional Infant Cardiac Program. Pediatrics. 1980;65:375-461.
8 Hoffman J.I., Christianson R. Congenital heart disease in a cohort of 19,502 births with long-term follow-up. Am J Cardiol. 1978;42:641-647.
9 Mcdaniel N.L. Ventricular and atrial septal defects. Pediatr Rev. 2001;22:265-270.
10 Anderson R.H., Wilcox B.R. The surgical anatomy of ventricular septal defect. J Card Surg. 1992;7:17-35.
11 Baker E.J., Leung M.P., Anderson R.H., Fischer D.R., Zuberbuhler J.R. The cross sectional anatomy of ventricular septal defects: a reappraisal. Br Heart J. 1988;59:339-351.
12 Soto B., Becker A.E., Moulaert A.J., Lie J.T., Anderson R.H. Classification of ventricular septal defects. Br Heart J. 1980;43:332-343.
13 Titus J.L., Daugherty G.W., Edwards J.E. Anatomy of the atrioventricular conduction system in ventricular septal defect. Circulation. 1963;28:72-81.
14 Becu L.M., Burchell H.B., Dushane J.W., Edwards J.E., Fontana R.S., Kirklin J.W. Anatomic and pathologic studies in ventricular septal defect. Circulation. 1956;14:349-364.
15 Saab N.G., Burchell H.B., Dushane J.W., Titus J.L. Muscular ventricular septal defects. Am J Cardiol. 1966;18:713-723.
16 Lincoln C., Jamieson S., Joseph M., Shinebourne E., Anderson R.H. Transatrial repair of ventricular septal defects with reference to their anatomic classification. J Thorac Cardiovasc Surg. 1977;74:183-190.
17 Griffin M.L., Sullivan I.D., Anderson R.H., Macartney F.J. Doubly committed subarterial ventricular septal defect: new morphological criteria with echocardiographic and angiocardiographic correlation. Br Heart J. 1988;59:474-479.
18 Gould L., Lyon A.F. Prolapse of the pulmonic valve through a ventricular septal defect. Am J Cardiol. 1966;18:127-131.
19 Wenink A.C., Gittenberger-De Groot A.C. Straddling mitral and tricuspid valves: morphologic differences and developmental backgrounds. Am J Cardiol. 1982;49:1959-1971.
20 Zielinsky P., Rossi M., Haertel J.C., Vitola D., Lucchese F.A., Rodrigues R. Subaortic fibrous ridge and ventricular septal defect: role of septal malalignment. Circulation. 1987;75:1124-1129.
21 French H. Possibility of loud congenital heart murmur disappearing when child grows up. Guys Hospital Reports. 1918;32:87.
22 Weber F.P. Can the clinical manifestation of congenital heart disease disappear with the general growth and development of the patient? British Journal of Children’s Diseases. 1918;15:113.
23 Evans J.R., Rowe R.D., Keith J.D. Spontaneous closure of ventricular septal defects. Circulation. 1960;22:1044-1054.
24 Glancy D.L., Roberts W.C. Complete spontaneous closure of ventricular septal defect: necropsy study of five subjects. Am J Med. 1967;43:846-853.
25 Moore D., Vlad P., Lambert E.C. Spontaneous closure of ventricular septal defect following cardiac failure in infancy. J Pediatr. 1965;66:712-721.
26 Schott G.D. Documentation of spontaneous functional closure of a ventricular septal defect during adult life. Br Heart J. 1973;35:1214-1216.
27 Simmons R.L., Moller J.H., Edwards J.E. Anatomic evidence for spontaneous closure of ventricular septal defect. Circulation. 1966;34:38-45.
28 Wade G., Wright J.P. Spontaneous closure of ventricular septal defects. Lancet. 1963;1:737-740.
29 Krovetz L.J. Spontaneous closure of ventricular septal defect. Am J Cardiol. 1998;81:100-101.
30 Wood P.H. Diseases of the heart and circulation. Philadelphia: Lippincott; 1956.
31 Anderson R.H., Lenox C.C., Zuberbuhler J.R. Mechanisms of closure of perimembranous ventricular septal defect. Am J Cardiol. 1983;52:341-345.
32 Eroglu A.G., Oztunc F., Saltik L., Bakari S., Dedeoglu S., Ahunbay G. Evolution of ventricular septal defect with special reference to spontaneous closure rate, subaortic ridge and aortic valve prolapse. Pediatr Cardiol. 2003;24:31-35.
33 Garne E. Atrial and ventricular septal defects – epidemiology and spontaneous closure. J Matern Fetal Neonatal Med. 2006;19:271-276.
34 Perloff J.K. Therapeutics of nature–the invisible sutures of “spontaneous closure”. Am Heart J. 1971;82:581-585.
35 Moe D.G., Guntheroth W.G. Spontaneous closure of uncomplicated ventricular septal defect. Am J Cardiol. 1987;60:674-678.
36 Alpert B.S., Mellits E.D., Rowe R.D. Spontaneous closure of small ventricular septal defects: probability rates in the first five years of life. Am J Dis Child. 1973;125:194-196.
37 Collins G., Calder L., Rose V., Kidd L., Keith J. Ventricular septal defect: clinical and hemodynamic changes in the first five years of life. Am Heart J. 1972;84:695-705.
38 Diehl A.M., Kittle C.F., Crockett J.E. Spontaneous complete closure of a high-flow, high-pressure ventricular septal defect. J Lancet. 1961;81:572-576.
39 Nadas A.S., Scott L.P., Hauck A.J., Rudolph A.M. Spontaneous functional closing of ventricular septal defects. N Engl J Med. 1961;264:309-316.
40 Suzuki H., Lucas R.V.Jr. Spontaneous closure of ventricular septal defects. Anatomic evidence in three patients. Arch Pathol. 1967;84:31-36.
41 Wise J.R.Jr, Wilson W.S. Angiographic documentation of spontaneous closure of ventricular septal defect in an adult. Chest. 1979;75:90-93.
42 Fellows K.E., Westerman G.R., Keane J.F. Angiocardiography of multiple ventricular septal defects in infancy. Circulation. 1982;66:1094-1099.
43 Shirali G.S., Smith E.O., Geva T. Quantitation of echocardiographic predictors of outcome in infants with isolated ventricular septal defect. Am Heart J. 1995;130:1228-1235.
44 Chesler E., Korns M.E., Edwards J.E. Anomalies of the tricuspid valve, including pouches, resembling aneurysms of the membranous ventricular septum. Am J Cardiol. 1968;21:661-668.
45 Beerman L.B., Park S.C., Fischer D.R., et al. Ventricular septal defect associated with aneurysm of the membranous septum. J Am Coll Cardiol. 1985;5:118-123.
46 Hamby R.I., Raia F., Apiado O. Aneurysm of the pars membranacea. Report of three adult cases and a review of the literature. Am Heart J. 1970;79:688-699.
47 Misra K.P., Hildner F.J., Cohen L.S., Narula O.S., Samet P. Aneurysm of the membranous ventricular septum. A mechanism for spontaneous closure of ventricular septal defect. N Engl J Med. 1970;283:58-61.
48 Ramaciotti C., Keren A., Silverman N.H. Importance of (perimembranous) ventricular septal aneurysm in the natural history of isolated perimembranous ventricular septal defect. Am J Cardiol. 1986;57:268-272.
49 Varghese P.J., Izukawa T., Celermajer J., Simon A., Rowe R.D. Aneurysm of the membranous ventricular septum: A method of spontaneous closure of small ventricular septal defect. Am J Cardiol. 1969;24:531-536.
50 Sethia B., Cotter L. Giant aneurysm of membranous septum. Unusual cause of mediastinal mass. Br Heart J. 1981;46:107-109.
51 Thery C., Lekieffre J., Dupuis C. Atrioventricular block secondary to a congenital aneurysm of the membranous septum. Histological examination of conduction system. Br Heart J. 1975;37:1097-1100.
52 Chen M.R., Rigby M.L., Redington A.N. Familial aneurysms of the interventricular septum. Br Heart J. 1991;65:104-106.
53 Rudolph A.M. The changes in the circulation after birth. Their importance in congenital heart disease. Circulation. 1970;41:343-359.
54 Nora J.J., Cohen M., Maxwell G.M. Klippel-Feil syndrome with congenital heart disease. Am J Dis Child. 1961;102:110.
55 Graham T.P.Jr, Atwood G.F., Boucek R.J.Jr, Cordell D., Boerth R.C. Right ventricular volume characteristics in ventricular septal defect. Circulation. 1976;54:800-804.
56 Herbert W.H. Hydrogen-detected ventricular septal defects. Br Heart J. 1969;31:766-769.
57 Bendien C., Bossina K.K., Buurma A.E., et al. Hemodynamic effects of dynamic exercise in children and adolescents with moderate-to-small ventricular septal defects. Circulation. 1984;70:929-934.
58 Jarmakani J.M., Graham T.P.Jr, Canent R.V.Jr. Left ventricular contractile state in children with successfully corrected ventricular septal defect. Circulation. 1972;45:I102-110.
59 Friedli B., Kidd B.S., Mustard W.T., Keith J.D. Ventricular septal defect with increased pulmonary vascular resistance. Late results of surgical closure. Am J Cardiol. 1974;33:403-409.
60 Haworth S.G. Pulmonary vascular disease in ventricular septal defect: structural and functional correlations in lung biopsies from 85 patients, with outcome of intracardiac repair. J Pathol. 1987;152:157-168.
61 Wagenvoort C.A., Neufeld H.N., Dushane J.W., Edwards J.E. The pulmonary arterial tree in ventricular septal defect. A quantitative study of anatomic features in fetuses, infants. and children. Circulation. 1961;23:740-748.
62 Walker W.J., Garcia-Gonzalez E., Hall R.J., et al. Interventricular septal defect: Analysis of 415 catheterized cases, ninety with serial hemodynamic studies. Circulation. 1965;31:54-65.
63 Warnes C.A., Boger J.E., Roberts W.C. Eisenmenger ventricular septal defect with prolonged survival. Am J Cardiol. 1984;54:460-462.
64 Wood P. The Eisenmenger syndrome, or pulmonary hypertension with reversed central shunt. Br Med J. 1958;2(701–709):755-762.
65 Shuhaiber J.H. Eisenmenger’s syndrome and pulmonary-artery dissection. N Engl J Med. 2007;357:718. author reply 718–719
66 Hopkins W.E., Waggoner A.D. Severe pulmonary hypertension without right ventricular failure: the unique hearts of patients with Eisenmenger syndrome. Am J Cardiol. 2002;89:34-38.
67 Van Hare G.F., Soffer L.J., Sivakoff M.C., Liebman J. Twenty-five-year experience with ventricular septal defect in infants and children. Am Heart J. 1987;114:606-614.
68 Bliss H.A., Moffat J.E. Hemodymaic events during the development of cyanosis and heart failure in a patient with large ventricular septal defect. Circulation. 1964;30:101-105.
69 Lucas R.V.Jr., Adams P.Jr, Anderson R.C., Meyne N.G., Lillehei C.W., Varco R.L. The natural history of isolated ventricular septal defect. A serial physiologic study. Circulation. 1961;24:1372-1387.
70 Weidman W.H., Blount S.G.Jr., Dushane J.W., Gersony W.M., Hayes C.J., Nadas A.S. Clinical course in ventricular septal defect. Circulation. 1977;56:I56-69.
71 Czeizel A., Meszaros M. Two family studies of children with ventricular septal defect. Eur J Pediatr. 1981;136:81-85.
72 Nora J.J., Nora A.H. The genetic contribution to congenital heart disease. In: Nora J.J., Takao A., editors. Congenital Heart Disease Mount Kisco. New York: Futura Publishing Company, 1984.
73 Blair E., Herman R., Cowley R.A. Interventricular septal defect in identical twins. J Thorac Cardiovasc Surg. 1965;50:197-201.
74 Rubenstein H.J., Weaver K.H. Monozygotic twins concordant for ventricular septal defects. Case report and review of the literature of congenital heart disease in monozygotic twins. Am J Cardiol. 1965;15:386-390.
75 Levy R.J., Rosenthal A., Miettinen O.S., Nadas A.S. Determinants of growth in patients with ventricular septal defect. Circulation. 1978;57:793-797.
76 Baptista M.J., Fairbrother U.L., Howard C.M., et al. Heterotrisomy, a significant contributing factor to ventricular septal defect associated with Down syndrome? Hum Genet. 2000;107:476-482.
77 Riemenschneider T.A., Moss A.J. Left ventricular-right atrial communication. Am J Cardiol. 1967;19:710-718.
78 Mudd J.G., Aykent Y., Willman V.L., Hanlon C.R., Fagan L.F. The natural and postoperative history of 252 patients with proved ventricular septal defects. Am J Med. 1965;39:946-951.
79 Shah P., Singh W.S., Rose V., Keith J.D. Incidence of bacterial endocarditis in ventricular septal defects. Circulation. 1966;34:127-131.
80 Zakrzewski T., Keith J. Bacterial endocarditis in infants and children. J Pediatr. 1965;67:1179.
81 Walpot J., Peerenboom P., Van Wylick A., Klazen C. Aneurysm of the membranous septum with ventricular septal defect and infective endocarditis. Eur J Echocardiogr. 2004;5:391-393.
82 Matt P., Winkler B., Gutmann M., Eckstein F. Acquired Gerbode defect after endocarditis. Eur J Cardiothorac Surg. 2009;36:402.
83 Fontana R.S., Edwards J.E. Congenital Cardiac Disease. Philadelphia: W.B. Saunders Company; 1962.
84 Alter B.P., Czapek E.E., Rowe R.D. Sweating in congenital heart disease. Pediatrics. 1968;41:123-129.
85 Abbott M.E. Congenital Heart Disease. New York: Thomas Nelson & Sons; 1932.
86 Eisenmenger V. Die angeborenen Defecte der Kammerscheidewand des Herzens. Z Klin Med. 32(suppl 1), 1897.
87 Kumar R.K., Sandoval J. Advanced pulmonary vascular disease: the Eisenmenger syndrome. Cardiol Young. 2009;19:622-626.
88 Perloff J.K., Rosove M.H., Sietsema K.E., Territo M.C. Cyanotic congenital heart disease: A multisystem systemic disorder. In Perloff J.K., Child J.S., Aboulhosn J., editors: Congenital Heart Disease in Adults, 3rd ed, Philadelphia: W.B. Saunders, 2009.
89 Niwa K., Perloff J.K., Kaplan S., Child J.S., Miner P.D. Eisenmenger syndrome in adults: ventricular septal defect, truncus arteriosus, univentricular heart. J Am Coll Cardiol. 1999;34:223-232.
90 Territo M.C., Rosove M.H. Cyanotic congenital heart disease: hematologic management. J Am Coll Cardiol. 1991;18:320-322.
91 Rosove M.H., Perloff J.K., Hocking W.G., Child J.S., Canobbio M.M., Skorton D.J. Chronic hypoxaemia and decompensated erythrocytosis in cyanotic congenital heart disease. Lancet. 1986;2:313-315.
92 Terito M., Perloff J., Rosove M., Moake J., Runge A. Acquired von Willebrand factor abnormalities in adults with congenital heart disease. Clin Appl Thromb Hemost. 1998;4:257.
93 Clarkson P.M., Frye R.L., Dushane J.W., Burchell H.B., Wood E.H., Weidman W.H. Prognosis for patients with ventricular septal defect and severe pulmonary vascular obstructive disease. Circulation. 1968;38:129-135.
94 Brunken R.C., Perloff J.K., Czernin J., et al. Coronary blood flow and myocardial perfusion reserve in adults with cyanotic congenital heart disease. Am J Physiol. 2005:H1798-H1806.
95 Tutar H.E., Atalay S., Turkay S., Imamoglu A. QRS axis in isolated perimembranous ventricular septal defect and influences of morphological factors on QRS axis. J Electrocardiol. 2001;34:197-203.
96 Sietsema K.E., Cooper D.M., Perloff J.K., et al. Dynamics of oxygen uptake during exercise in adults with cyanotic congenital heart disease. Circulation. 1986;73:1137-1144.
97 Perloff J.K. Pregnancy in congenital heart disease: The mother and the fetus. In Perloff J.K., Child J.S., Aboulhosn J., editors: Congenital Heart Disease in Adults, 3rd ed, Philadelphia: WB Saunders, 2009.
98 Pitts J.A., Crosby W.M., Basta L.L. Eisenmenger’s syndrome in pregnancy: does heparin prophylaxis improve the maternal mortality rate? Am Heart J. 1977;93:321-326.
99 Niwa K., Perloff J.K., Bhuta S.M., et al. Structural abnormalities of great arterial walls in congenital heart disease: light and electron microscopic analyses. Circulation. 2001;103:393-400.
100 Albers H.J., Carroll S.E., Coles J.C. Spontaneous closure of a membranous ventricular septal defect. Necrospy finding with clinical application. Br Med J. 1962;2:1162-1163.
101 Balderston S.M., Shaffer E.M., Washington R.L., Sondheimer H.M. Congenital polyvalvular disease in trisomy 18: echocardiographic diagnosis. Pediatr Cardiol. 1990;11:138-142.
102 Matsuoka R., Misugi K., Goto A., Gilbert E.F., Ando M. Congenital heart anomalies in the trisomy 18 syndrome, with reference to congenital polyvalvular disease. Am J Med Genet. 1983;14:657-668.
103 Moene R.J., Sobotka-Plojhar M., Oppenheimer-Dekker A., Lindhout D. Ventricular septal defect with overriding aorta in trisomy-18. Eur J Pediatr. 1988;147:556-557.
104 Van Praagh S., Truman T., Firpo A., et al. Cardiac malformations in trisomy-18: a study of 41 postmortem cases. J Am Coll Cardiol. 1989;13:1586-1597.
105 Lin A.E., Perloff J.K. Upper limb malformations associated with congenital heart disease. Am J Cardiol. 1985;55:1576-1583.
106 Cayler G.G. Cardiofacial syndrome. Congenital heart disease and facial weakness, a hitherto unrecognized association. Arch Dis Child. 1969;44:69-75.
107 Steeg C.N., Woolf P. Cardiovascular malformations in the fetal alcohol syndrome. Am Heart J. 1979;98:635-637.
108 Farru O., Duffau G., Rodriguez R. Auscultatory and phonocardiographic characteristics of supracristal ventricular septal defect. Br Heart J. 1971;33:238-245.
109 Moncada R., Bicoff J.P., Arcilla R.A., Agustsson M.H., Lendrum B.L., Gasul B.M. Retrograde left ventricular angiocardiography in ventricular septal defect. Am J Cardiol. 1963;11:436-446.
110 Feruglio G.A., Gunton R.W. Intracardiac phonocardiography in ventricular septal defect. Circulation. 1960;21:49-58.
111 Carr M., Kearney D.L., Eidem B.W. Congenital aneurysm of the muscular interventricular septum. J Am Soc Echocardiogr. 2008;21:1282.e1281-1282.e1286.
112 Linhart J.W., Razi B. Late systolic murmur: a clue to the diagnosis of aneurysm of the membranous ventricular septum. Chest. 1971;60:283-286.
113 Pickering D., Keith J.D. Systolic clicks with ventricular septal defects. A sign of aneurysm of ventricular septum? Br Heart J. 1971;33:538-539.
114 Pieroni D.R., Bell B.B., Krovetz L.J., Varghese P.J., Rowe R.D. Auscultatory recognition of aneurysm of the membranous ventricular septum associated with small ventricular septal defect. Circulation. 1971;44:733-739.
115 Winslow T.M., Redberg R.F., Foster E., Schiller N.B. Transesophageal echocardiographic detection of abnormalities of the tricuspid valve in adults associated with spontaneous closure of perimembranous ventricular septal defect. Am J Cardiol. 1992;70:967-969.
116 Perloff J.K. Physical Examination of the Heart and Circulation, 4th ed. Shelton, Connecticut: People’s Medical Publishing House; 2009.
117 Harris C., Wise J.Jr., Oakley C.M. “Fixed” splitting of the second heart sound in ventricular septal defect. Br Heart J. 1971;33:428-431.
118 Nadas A.S., Ellison R.C. Phonocardiographic analysis of diastolic flow murmurs in secundum atrial septal defect and ventricular septal defect. Br Heart J. 1967;29:684-688.
119 Laubry C., Pezzi C. Traits des Maladies Congenitales du Coeur. Paris: J. B. Bailliere et Fits; 1921.
120 Perloff J.K. Auscultatory and phonocardiographic manifestations of pulmonary hypertension. Prog Cardiovasc Dis. 1967;9:303-340.
121 Sutton G., Harris A., Leatham A. Second heart sound in pulmonary hypertension. Br Heart J. 1968;30:743-756.
122 Toscano-Barboza E., Dushane J.W. Ventricular septal defect: correlation of electrocardiographic and hemodynamic findings in 60 proved cases. Am J Cardiol. 1959;3:721-732.
123 Vince D.J., Keith J.D. The electrocardiogram in ventricular septal defect. Circulation. 1961;23:225-240.
124 Backman H. Influence of structural and functional features of ventricular septal defect on frontal plane QRS axis of the electrocardiogram. Br Heart J. 1972;34:274-283.
125 Farru-Albohaire O., Arcil G., Hernandez I. An association between left axis deviation and an aneurysmal defect in children with a perimembranous ventricular septal defect. Br Heart J. 1990;64:146-150.
126 Shaw N.J., Godman M.J., Hayes A., Sutherland G.R. Superior QRS axis in ventricular septal defect. Br Heart J. 1989;62:281-283.
127 Neufeld H.N., Titus J.L., Dushane J.W., Burchell H.B., Edwards J.E. Isolated ventricular septal defect of the persistent common atrioventricular canal type. Circulation. 1961;23:685-696.
128 Witham A.C., Mcdaniel J.S. Electrocardiogram, vectorcardiogram, and hemodynamics in ventricular septal defect. Am Heart J. 1970;79:335-346.
129 Katz L.N., Wachtel H. Diphasic QRS type of electrocardiogram in congenital heart disease. Am Heart J. 1937;13:202.
130 Vaquez H., Bordet E. Le Coeur et l’Aorte, Etudes Radiographiques. Paris: J. B. Bailliere et Fils; 1913.
131 Davies H., Williams J., Wood P. Lung stiffness in states of abnormal pulmonary blood flow and pressure. Br Heart J. 1962;24:129-138.
132 Jarmakani J.M., Graham T.P.Jr, Benson D.W.Jr, Canent R.V.Jr, Greenfield J.C.Jr. In vivo pressure-radius relationships of the pulmonary artery in children with congenital heart disease. Circulation. 1971;43:585-592.
133 Helmcke F., De Souza A., Nanda N.C., et al. Two-dimensional and color Doppler assessment of ventricular septal defect of congenital origin. Am J Cardiol. 1989;63:1112-1116.
134 Sutherland G.R., Godman M.J., Smallhorn J.F., Guiterras P., Anderson R.H., Hunter S. Ventricular septal defects. Two dimensional echocardiographic and morphological correlations. Br Heart J. 1982;47:316-328.
135 Child J. Transthoracic and transesophageal echocardiographic imaging: Anatomic and homodynamic assessment. In Perloff J., Child J., editors: Congenital Heart Disease in Adults, 2nd ed, Phialdelphia: WB Saunders Co, 1998.
136 Lethor J.P., Marcon F., De Moor M., King M.E. Physiology of ventricular septal defect shunt flow in the fetus examined by color Doppler M-mode. Circulation. 2000;101:E93.
137 Ishii M., Hashino K., Eto G., et al. Quantitative assessment of severity of ventricular septal defect by three-dimensional reconstruction of color Doppler-imaged vena contracta and flow convergence region. Circulation. 2001;103:664-669.
138 Dall’agata A., Cromme-Dijkhuis A.H., Meijboom F.J., et al. Three-dimensional echocardiography enhances the assessment of ventricular septal defect. Am J Cardiol. 1999;83:1576-1579. A1578
139 Sutherland G.R., Fraser A.G. Colour flow mapping in cardiology: indications and limitations. Br Med Bull. 1989;45:1076-1091.
140 Dipchand A.I., Boutin C. Left ventricular septal aneurysm. Circulation. 1998;98:1697.
141 Levin A.R., Spach M.S., Canent R.V.Jr, et al. Intracardiac pressure-flow dynamics in isolated ventricular septal defects. Circulation. 1967;35:430-441.
142 Laubry C., Routier D., Soulie P. Les souffles de la maladie de Roger. Revue de Medécine. 1933;50:439.
143 Nadas A.S., Thilenius O.G., Lafarge C.G., Hauck A.J. Ventricular septal defect with aortic regurgitation: Medical and pathologic aspects. Circulation. 1964;29:862-873.
144 Rhodes L.A., Keane J.F., Keane J.P., et al. Long follow-up (to 43 years) of ventricular septal defect with audible aortic regurgitation. Am J Cardiol. 1990;66:340-345.
145 Van Praagh R., Mcnamara J.J. Anatomic types of ventricular septal defect with aortic insufficiency. Diagnostic and surgical considerations. Am Heart J. 1968;75:604-619.
146 Tatsuno K., Konno S., Ando M., Sakakibara S. Pathogenetic mechanisms of prolapsing aortic valve and aortic regurgitation associated with ventricular septal defect. Anatomical, angiographic, and surgical considerations. Circulation. 1973;48:1028-1037.
147 Halloran K.H., Talner N.S., Browne M.J. A study of ventricular septal sefect associated with aortic insufficiency. Am Heart J. 1965;69:320-326.
148 Hor K.N., Border W.L., Cripe L.H., Benson D.W., Hinton R.B. The presence of bicuspid aortic valve does not predict ventricular septal defect type. Am J Med Genet Part A. 2008;146A:3202-3205.
149 Taussig H.B., Semans J.H. Severe aortic insufficiency in association with a congenital malformation of the heart of the Eisenmenger type. Bull Johns Hopkins Hosp. 1940;66:157.
150 Morganroth J., Perloff J.K., Zeldis S.M., Dunkman W.B. Acute severe aortic regurgitation. Pathophysiology, clinical recognition, and management. Ann Intern Med. 1977;87:223-232.
151 Nishimura R.A., Miller F.A.Jr, Callahan M.J., Benassi R.C., Seward J.B., Tajik A.J. Doppler echocardiography: theory, instrumentation, technique, and application. Mayo Clinic Proceedings. 1985;60:321-343.
152 Thurman J. On aneurysms of the heart. Trans Med Soc Lond. 1838;21:187.
153 Hillier T. Congenital malformation of the heart; perforation of the septum ventriculorum, establishing a communication between the left ventricle and the right atrium. Trans Pathol Soc Lond. 1859;10:110.
154 Preicz H. Beitrage zur Lehre von den angeborenen Herzanomalien. Beitr Pathol Anat. 1890;7:234.
155 Gerbode F., Hultgren H., Melrose D., Osborn J. Syndrome of left ventricular-right atrial shunt; successful surgical repair of defect in five cases, with observation of bradycardia on closure. Ann Surg. 1958;148:433-446.
156 Perry E.L., Burchell H.B., Edwards J.E. Congenital communication between the left ventricle and the right atrium; co-existing ventricular septal defect and double tricuspid orifice. Mayo Clinic Proceedings. 1949;24:198-206.
157 Rosenquist G.C., Sweeney L.J. Normal variations in tricuspid valve attachments to the membranous ventricular septum: a clue to the etiology of left ventricle-to-right atrial communication. Am Heart J. 1975;89:186-188.
158 Stahlman M., Kaplan S., Helmsworth J.A., Clark L.C., Scott H.W.Jr. Syndrome of left ventricular-right atrial shunt resulting from high interventricular septal defect associated with defective septal leaflet of the tricuspid valve. Circulation. 1955;12:813-818.
159 Burrows P.E., Fellows K.E., Keane J.F. Cineangiography of the perimembranous ventricular septal defect with left ventricular-right atrial shunt. J Am Coll Cardiol. 1983;1:1129-1134.
160 Grenadier E., Shem-Tov A., Motro M., Palant A. Echocardiographic diagnosis of left ventricular-right atrial communication. Am Heart J. 1983;106:407-409.
161 Edwards J., Carey L.S., Neufeld H.N., Lester R.G. Congenital Heart Disease. Philadelphia: WB. Saunders Company; 1965.
162 Yacoub M.H., Mansur A., Towers M., Westbury H. Bacterial endocarditis complicating left ventricle to right atrium communication. Br J Dis Chest. 1972;66:78-82.
163 Bouchard F., Wolff F., Kalmanson D. Left ventricle-right atrium communication: diagnosis by catheterization and intracardiac phonocardiography. Arch Mal Coeur Vaiss. 1961;54:1310-1325.
164 Sakakibara S., Konno S. Left ventricular-right atrial communication. Ann Surg. 1963;158:93-99.
165 Levy M., Lillehei C.W. Left ventricular-right atrial canal. Ten cases treated surgically. Am J Cardiol. 1962;10:623-633.
166 Macruz R., Perloff J.K., Case R.B. A method for the electrocardiographic recognition of atrial enlargement. Circulation. 1958;17:882-889.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 17 Ventricular Septal Defect
In 1879, Henri Roger wrote, “A developmental defect of the heart occurs from which cyanosis does not ensue in spite of the fact that a communication exists between the cavities of the two ventricles and in spite of the fact that admixture of venous blood and arterial blood occurs. This congenital defect, which is even compatible with a long life, is a simple one. It comprises a defect in the interventricular septum.”1
Roger went on to say that at necropsy, “The ventricular walls show no alteration, but in the upper portion of the interventricular septum beneath the mitral valve is an orifice which establishes a communication between the two ventricles.”1 And still further, “Among the congenital defects of the heart compatible with life and perhaps a long one, one of the most frequent which I have encountered … is the communication between the two ventricles because of failure of occlusion of the interventricular septum.”1
Ventricular septal defects remain the most common congenital malformation of the heart, occurring in 50% of children with congenital heart disease.2 Detection has increased dramatically with advances in imaging and with the facility of echocardiographic diagnosis in neonates, with spontaneous closure and detection of small defects in the muscular trabecular septum taken into account.3,4 A comprehensive review estimated the median incidence rate of ventricular septal defects to be 2,829 per million live births5; the defects are found to be the most common gross morphologic congenital malformation of the heart and circulation in adults, except for the bicuspid aortic valve.3,6–8
Although Roger believed that “this congenital defect … is a simple one,”1 there is still no uniform consensus on how best to characterize and classify the diverse types of defects to which the ventricular septum is subject. The ventricular septum is a complex nonplanar partition whose components are best defined according to anatomic landmarks on the right side of the septal surface (Figure 17-1).9–12 Defects are classified according to their relationship to the membranous, inlet, trabecular, and infundibular septum.2,12 The membranous septum is divided by the tricuspid annulus into ventriculoatrial and interventricular components that abut the three major segments of the muscular septum: namely, the inlet septum, which is lightly trabeculated; the trabecular septum, which is, as the name implies, heavily trabeculated; and the infundibular septum, which is nontrabeculated. The inlet septum is limited by the tensor attachments of the tricuspid valve and is so named according to its location. The trabecular septum lies between the inlet septum and the infundibular septum (see Figure 17-1). Atrioventricular conduction tissue penetrates the ventriculoatrial portion of the membranous septum. The His bundle and bundle branches run beneath the deficient interventricular component of the membranous septum close to the free edge of the ventricular septal defect.10,13
Approximately 80% of ventricular septal defects are perimembranous. The prefix peri underscores extension into adjacent portions of the inlet, trabecular, and infundubular septum. Large perimembranous defects encroach upon all three portions of the muscular septum (Figure 17-2A). Muscular ventricular septal defects are prevalent in neonates, with estimates as high as 53 of every 1000 live births. Approximately 90% of the defects close spontaneously within the first 10 months of life.
The most common types of muscular defects lie within the trabecular septum.10,14,15 These malformations are more apparent on the left septal surface and vary from small to large, from single to multiple (Figure 17-3), to a honeycombed or Swiss cheese–like structure with sieve-like fenestrations, to tortuous sinusoidal tracks threaded among septal trabeculae without through-and-through perforations.10,15 Sieve-like fenestrations or multiple small muscular defects have the net functional effect of a single large defect.
Isolated defects in the inlet septum represent approximately 8% of ventricular septal defects at surgery.16 An inlet ventricular septal defect that is bordered entirely by myocardium differs from an inlet defect that involves the basal portion of the septum near the crux where it is partially bordered by bridging atrioventricular valve tissue (see Chapter 15).10–12
The infundibular septum is represented by a small portion of muscle interposed between the outflow components of the left and right ventricles.17 A sleeve of subpulmonary infundibulum supports the leaflets of the pulmonary valve and separates the right ventricular outflow tract from the surface of the heart rather than from the left ventricular outflow track. Ventricular septal defects in the infundibular septum are also called supracristal, subpulmonary, subarterial, or doubly committed and account for approximately 5% to 7% of ventricular septal defects in North America and Western Europe, but for approximately 30% in Asian patients.14,16,17 Infundibular septal defects can be entirely muscular or can be partially rimmed by semilunar valve tissue (subarterial). Defects are considered doubly committed subarterial when little or no muscle is interposed between the outflow components of the left and right ventricles and when there is absence of the septal component of the subpulmonary infundibulum, so the aortic and pulmonary leaflets are in fibrous continuity.10,12,14,16,17 Doubly committed subarterial defects lie immediately beneath the valves of both arterial trunks, so the left and right coronary cusps of the aortic valve tend to prolapse into the outflow tract of the right ventricle (see section on Ventricular Septal Defect with Aortic Regurgitation). Much less commonly, a pulmonary cusp prolapses through the defect.18
Atrioventricular septal malalignment necessarily involves an inlet ventricular septal defect and is usually accompanied by straddling of the tensor apparatus of an atrioventricular valve that inserts (straddles) onto both sides of the ventricular septum.19 So-called Eisenmenger’s malalignment is represented by a perimembranous ventricular septal defect with anterior deviation of the infundibular septum.20 Not relevant to this chapter is malalignment between infundibular and trabecular septum that is associated with Fallot’s tetralogy (see Chapter 18) or much less commonly with coarctation of the aorta (see Chapter 8).
Henri Roger did not anticipate spontaneous closure. “The pathologic state of the heart existing before birth and consisting of an arrest of development is not susceptible to favorable changes, either by spontaneous evolution or by medical or surgical intervention.”1 However, in 1918, two reports speculated that ventricular septal defects might undergo spontaneous closure.21,22 One report was entitled “The Possibility of a Loud Congenital Heart Murmur Disappearing When a Child Grows Up.”21 The other report carried the title “Can the Clinical Manifestations of Congenital Heart Disease Disappear with the General Growth and Development of thePatient?”22 However, four decades elapsed before spontaneous closure was firmly documented.23–29 In 1960, Paul Wood wrote, “In any large series of geriatric necropsies … atrial septal defect is always well represented, but where’s the maladie de Roger? Assuming it does not provide immortality, it must either close spontaneously in middle life or have long since run its mortal course.”30
A defect may remain anatomically open but functionally closed (i.e., with absent or negligible shunt; Figures 17-2B and 17-4). The tendency for ventricular septal defects, especially perimembranous and trabecular muscular defects, to decrease in size finds an ultimate expression in complete closure24,31–33 that has been called the therapeutics of nature—the invisible sutures of spontaneous closure.34 The incidence rate of spontaneous closure varies considerably depending on the population under study, the method of diagnostic investigation, the type of defect, and whether or not the defect is solitary35 and has been estimated at 50% to 75% for restrictive perimembranous and trabecular muscular defects observed from birth.29,36,37 The incidence rate for trabecular muscular defects35 is reportedly equal to or somewhat higher than that for perimembranous defects. Moderately restrictive and nonrestrictive defects also close spontaneously, but the probability is comparatively low, with an incidence rate estimated at 5% to 10%.23–25,35,38,39 Most defects that are destined to close do so within the first year of life,35 with approximately 60% closing before 3 years of age and 90% closing before age 8 years.36,37 However, spontaneous closure also occurs in older children and young adults40 and has been documented at age 23 years,26 between 26 years and 33 years of age,41 and at age 46 years. Multiple muscular trabecular ventricular septal defects have a strong tendency to close, a conclusion in accord with the observation that multiple defects are three times more prevalent in neonates than after 1 year of age.42 In preterm infants, ventricular septal defects are about twice as frequent as in full-term infants, but the rate of spontaneous closure is the same,35 which calls into question the notion that defects reflect incomplete ventricular septation. Spontaneously closed defects in the muscular trabecular septum are funnel-shaped with a sealed orifice on the right ventricular aspect and residual patency on the left ventricular aspect, with endocardial proliferation in the lumen of the funnel and hypertrophy around the exit. Muscular defects represented by “Swiss cheese” fenestrations do not close spontaneously. Defects in the inlet septum seldom decrease in size43 but are occasionally occluded by bridging atrioventricular valve tissue (see Figure 15-87B). Infundibular defects can be closed by prolapse of the right aortic cusp.2 Perimembranous ventricular septal defects close by adherence of the septal tricuspid leaflet to the margins of the defect, less commonly by prolapse of an aortic cusp,24,31,44 and rarely by intrusion of a sinus of Valsalva aneurysm. Adherence of tricuspid leaflet tissue is seldom accompanied by tricuspid regurgitation, but there is a tendency for aneurysm formation when perimembranous defects are closed by the tricuspid valve (see Figures 17-4 and 17-31B).2,32,45–49 Ventricular septal aneurysms were described by Laennec in 1826 and are, as a rule, relatively small (see Figure 17-4). Exceptionally, however, septal aneurysms expand to considerable size, and a giant aneurysm of the membranous septum revealed itself as a mediastinal mass.50 A blind septal aneurysm occasionally represents a sui generis congenital malformation of the interventricular portion of the membranous septum rather than a sequel to spontaneous closure of a perimembranous defect.51 Familial congenital septal aneurysms have been reported, although rarely.52 With notable exceptions, aneurysms are well tolerated and have been discovered incidentally at necropsy in patients beyond the eighth decade. Complications of septal aneurysms include infective endocarditis, conduction disturbances, intraaneurysmal thrombosis with systemic embolism, aortic or tricuspid regurgitation, and obstruction to right ventricular outflow (see Figure 18-11).51 Blind congenital aneurysms may spontaneously perforate, establishing a left-to-right shunt into either the right ventricle or right atrium.51
The hemodynamic classification of ventricular septal defects is relatively straightforward because physiologic consequences depend essentially on the size of the defect and the pulmonary vascular resistance.53 These two variables change with time, and the physiologic and clinical manifestations change accordingly. A defect in the ventricular septum, even if spontaneously closed, has been postulated to constitute an abnormality that might adversely affect left ventricular systolic function.54 This postulation is relevant to impaired function or left ventricular failure in an occasional patient with a restrictive defect and a negligible shunt.54
Ventricular septal defects fall into four anatomic/physiologic categories (Figure 17-5): (1) restrictive defects with normal right ventricular and pulmonary artery systolic pressure and normal pulmonary vascular resistance; (2) moderately restrictive defects with elevated right ventricular and pulmonary artery systolic pressures and low but variable pulmonary vascular resistance; (3) nonrestrictive defects with identical right ventricular and left ventricular systolic pressure and elevated but subsystemic pulmonary vascular resistance that is variable; and (4) nonrestrictive defects with identical right and left ventricular systolic pressures, suprasystemic pulmonary vascular resistance, and a right-to-left shunt—Eisenmenger’s syndrome.
Henri Roger’s account of the physiology of a restrictive ventricular septal defect with normal pulmonary vascular resistance still applies1:
Restrictive ventricular septal defects—maladie de Roger—cause little or no functional derangement because the shunt is small and the pressure and resistance in the pulmonary circulation are normal (see Figure 17-5A). A restrictive defect represents a site of obligatory resistance between the left and right ventricles, thus limiting (restricting) the magnitude of the left-to-right shunt (see previous) and precluding delivery of left ventricular systolic pressure into the right ventricle and pulmonary trunk. Shunt flow is systolic. Minor diastolic flow is generated by the normal differences in left and right ventricular distensibility and end diastolic pressure.
A moderately restrictive ventricular septal defect is characterized by right ventricular systolic pressure that is above normal but less than systemic and by low but variable pulmonary vascular resistance that rarely progresses. The left ventricle adapts to a moderate increase in volume, and the right ventricle adapts to a moderate increase in pressure. Instant-to-instant right-to-left shunting occurs because left ventricular systolic pressure falls more rapidly than right ventricular systolic pressure. However, left ventricular systolic pressure rises more rapidly than right ventricular systolic pressure, so the small right-to-left shunts are quantitatively returned to the right ventricle with the next cardiac cycle. An increase in end-diastolic pressure in the volume-loaded left ventricle results in diastolic shunting.55 Moderately restrictive defects in the trabecular muscular septum decrease in size during ventricular contraction and, in so doing, decrease the systolic shunt.56 With isotonic exercise, systemic vascular resistance falls while pulmonary vascular resistance changes little if at all,57 so a normal increment in systemic flow is achieved without a corresponding increase in left-to-right shunt.57
With a nonrestrictive ventricular septal defect, the right and left ventricles behave physiologically as a common chamber. Peak systemic systolic pressures are identical, so the volume and direction of flow through the defect depend on the relative resistances in the pulmonary and systemic circulations. A nonrestrictive defect with elevated but variable pulmonary vascular resistance (see Figure 17-5C) imposes an excessive volume load on the left ventricle and systemic afterload on the right ventricle. A persistently large left-to-right shunt culminates in depressed systolic function of the volume overloaded left ventricle.58 A rise in pulmonary vascular resistance results in a reciprocal fall in left-to-right shunt and a comparable reduction in volume overload of the left ventricle, but systolic pressure in the two ventricles necessarily remains identical. When pulmonary vascular resistance is suprasystemic, the architecture of the pulmonary vascular bed resembles primary pulmonary hypertension (see Chapter 14) and the left-to-right shunt is replaced by a right-to-left shunt, Eisenmenger’s syndrome (Figure 17-5D).37,59–65 Right ventricle function is analogous to the function of a fetal right ventricle that is equipped to cope with systemic vascular resistance.66
The vasoreactive immature pulmonary resistance vessels play a pivotal role in regulating pulmonary blood flow in neonates with a nonrestrictive ventricular septal defect. The physiologic fall in neonatal pulmonary vascular resistance is delayed because of the interplay between shunt size and pulmonary vasoreactivity. In infants with a moderately restrictive ventricular septal defect, two additional mechanisms serve to regulate the volume and direction of the shunt and the level of pulmonary arterial pressure. The most favorable mechanism is a decrease in size of the defect. A much less common mechanism is acquired obstruction to right ventricular outflow that reduces left ventricular volume overload and protects the pulmonary vascular bed (see Chapter 18).67 When and to what extent these regulatory mechanisms exert their influence determines the hemodynamic and clinical course in patients with moderately restrictive and nonrestrictive ventricular septal defects.37,62,68,69
History
Ventricular septal defects are found in a wide range of mammals and in birds with four-chambered hearts.4 The incidence in humans is unrelated to gender, race, maternal age, or birth order.4,70 Ventricular septal defects have been reported in 3.3% of first-degree relatives of index patients.71 One third of first-degree relatives of patients with congenital heart disease have ventricular septal defects.4 Between 30% and 60% of siblings of index patients have ventricular septal defects,72 and siblings of patients with ventricular septal defects have three times the incidence of ventricular septal defects compared to the general population.4 Ventricular septal defects have been reported in identical twins,73,74 but with a high frequency of discordance.4 A parent with a spontaneously closed ventricular septal defect can have offspring with a ventricular septal defect. Birth weights are low in about 18% of full-term infants with ventricular septal defects.75 Dysmaturity is in addition to and apart from occurrence of ventricular septal defects in preterm infants. Heterotrisomy is a significant factor in ventricular septal defects with Down syndrome.76
Restrictive ventricular septal defects come to light because a systolic murmur is detected at the first well-baby examination. A murmur that is present at birth is a feature of ventricular septal defect with a left ventricular-to-right atrial communication in which the shunt exists in utero and is therefore present at birth (see subsequent).77 Very small defects with trivial early systolic shunts escape detection because the early systolic murmurs are soft and are either undetected or mistaken for a normal or innocent murmur (see section Auscultation). Spontaneous closure accounts for the striking age-related disparity in the incidence of ventricular septal defects from birth to maturity.31,35,62 The patient is left with no shunt and a functionally normal heart that harbors a morphologic abnormality (see previous), especially if spontaneous closure is followed by a septal aneurysm (see Figure 17-4).32 The defect is necessarily missed if the physical examination is performed after spontaneous closure.
Infective endocarditis is a tangible risk in restrictive ventricular septal defects, but rarely occurs before eruption of the second teeth.62,67,78–80 Roger aptly stated, “Prevention of complications is by means of hygiene.”1 Infective endocarditis is usually located on the septal tricuspid leaflet at site of the jet impact. Muscular defects have a low incidence of infective endocarditis because the jet is dissipated within the right ventricular cavity without striking the septal tricuspid leaflet.15 Occasionally, the site of infective endocarditis is on an aneurysm of the ventricular septum,81 and rarely, vegetations at the base of the septal tricuspid leaflet cause perforation and a left ventricular-to-right atrial shunt, an acquired Gerbode defect.82
Longevity is likely to be normal, as Roger stated:
Several subjects whom I have been able to observe, I have attended for periods of five, twelve and fifteen years; these children have grown like others, not one has died prematurely. I have occasionally visited as physician a woman whose children I attended from early ages. She had always been in excellent health and had never complained of cardiac difficulties. On auscultation I was greatly surprised to hear a murmur. I asked her if physicians had ever found anything wrong with her heart, and she told me that Guersant the Elder (the famous pediatrician who was my first master in infantile pathology) had recognized in her a few days after birth a cardiac malformation. This woman has now passed her fiftieth year; her health continues to be perfect and she is the mother of four children. In spite of the existence of an uncomplicated defect in the interventricular septum, patients may attain or even surpass the average span of human life.1
Moderately restrictive ventricular septal defects with low but variable pulmonary vascular resistance (Figure 17-5B) escape detection in the newborn nursery because the delayed fall in neonatal pulmonary vascular resistance delays the onset of the shunt and the onset of the murmur. Congestive heart failure within the first few months of life is in response to a large left-to-right shunt that is established after the fall in pulmonary vascular resistance. Parents report that their infant fatigues and coughs while feeding, sweats excessively, is restless when recumbent, and sleeps poorly. Parents may detect a thrill when they touch their infant’s chest and may detect a hyperactive precordium when holding the infant next to their chest. Symptomatic improvement is related to a spontaneous decrease in size of the defect, a favorable trend that may culminate in spontaneous closure (Figure 17-6; see previous).
Patients with moderately restrictive defects and moderate left-to-right shunts tolerate isotonic exercise because the exercise-induced increment in systemic blood flow is achieved without a corresponding increase in shunt flow (see previous).57 However, persistence of a significant shunt incurs the risk of infective endocarditis, and protracted volume overload of the left ventricle incurs the risk of congestive heart failure.67 Only an occasional patient achieves adulthood (Figure 17-7), with one reaching age 65 years (Figure 17-8B) and another reaching age 79 years.83
Nonrestrictive ventricular septal defects with elevated but variable pulmonary vascular resistance (see Figure 17-5C) present in infancy with congestive heart failure and with little prospect that the defect will decrease in size. Poor growth and development, labored breathing, frequent lower respiratory infections, difficulty feeding, and excessive diaphoresis are typical.37,67,84 Dyspnea and irritability are especially pronounced when the infant is supine and improve when the baby is held upright or is placed in an infant seat. Feeding patterns are also typical. A hungry infant awakens from a fretful sleep and feeds vigorously only to stop short of satisfaction because of dyspnea, falls asleep again, exhausted by the effort, and awakens with renewed hunger and repeats the frustrating cycle. Regulation of the large left-to-right shunt with amelioration of symptoms is almost always the result of a rise in pulmonary vascular resistance.25,38,62,68 A minority of these patients undergo relatively little fall in neonatal pulmonary vascular resistance, so the shunts are small and the early clinical course is deceptively benign. When pulmonary resistance exceeds systemic, the shunt is reversed, the patient is cyanotic,37,60,64 and the condition that Maude Abbott called Eisenmenger’s complex exists.85 Victor Eisenmenger published his account in 1897 in a paper entitled “Congenital Defects of the Ventricular Septum.”86,87 Paul Wood, in his landmark publication of 1958, introduced the term Eisenmenger’s syndrome (Figure 17-9), which he defined as pulmonary hypertension with reversed shunt.64 Wood quoted Eisenmenger:
Cyanotic congenital heart disease, represented here by Eisenmenger’s syndrome, is a multisystem systemic disorder that involves red cell mass, hemostasis, the central nervous system, bilirubin kinetics, the systemic vascular bed, the coronary circulation, the myocardium, uric acid clearance, the kidneys, respiration, the digits, the long bones, and gynecologic endocrinology.88,89 Morbidity and mortality take into account contemporary medical management of the multisystem disorders.88–90 Longevity is about one to two decades longer than in early reports.63,89,91 Right ventricular failure is uncommon66 unless acquired systemic hypertension imposes disproportionate afterload.
Erythrocytosis is an adaptive response to the decrease in tissue oxygenation—arterial hypoxemia—that stimulates renal release of erythropoietin, which in turn stimulates an increase in the number of red blood cells. Equilibrium conditions are established at elevated hematocrit levels. Erythrocytosis is a desirable adaptive response designed to offset the hypoxemic deficit in tissue oxygenation and does not incur a risk of stroke from cerebral arterial thrombosis irrespective of hematocrit level, iron stores, or cerebral symptoms of hyperviscosity.88 However, in cyanotic children younger than age 4 years, iron-deficient erythrocytosis incurs the risk of thrombosis of intracranial venous sinuses.88 The right-to-left shunts of cyanotic congenital heart disease are pathways for paradoxical emboli that cause transient ischemic attacks and strokes.
Hemostatic abnormalities have been recognized for more than 50 years in patients with cyanotic congenital heart disease.88 Platelet counts are in the low range of normal, and many clotting factor disorders have been identified, most recently acquired abnormalities of the von Willebrand’s factor.92 A mucocutaneous bleeding tendency is characterized by easy bruising, gingival bleeding, epistaxis, menorrhagia, and increased risk of traumatic bleeding. Pulmonary hemorrhage can be external (hemoptysis) or intrapulmonary, is the most serious type of bleeding, and varies from mild and occasional to copious, recurrent, massive, and fatal.64,88,93 Massive intrapulmonary hemorrhage is a common cause of sudden death in Eisenmenger’s syndrome.89 Victor Eisenmenger’s patient “died more or less suddenly … following a large hemoptysis.”64
Bilirubin is formed from the breakdown of heme, a process that is excessive in cyanotic congenital heart disease and that coincides with a substantial increase in the amount of unconjugated bilirubin in the bile.88 Calcium bilirubinate gallstones develop because unconjugated bilirubin is virtually water insoluble at physiologic pH.88
Systemic vascular dilation is the result of endothelial-derived nitric oxide and prostaglandins that are released in response to the increase in shear stress inherent in the erythrocytotic perfusate. Increased arteriolar dilation and tissue vascularity contribute to the bleeding tendency (Figure 17-10).88 Syncope can be caused by the heat-induced vasodilation of hot humid weather or by a hot shower or bath, especially when standing.
The extramural coronary arteries are dilated, elongated, and tortuous, even aneurysmal (coronary ectasia).88 Vasodilator nitric oxide and prostaglandins are released from coronary artery endothelium by the increased shear stress of erythrocytosis. Dilation of the coronary arteries is accompanied by an increase in extramural coronary artery blood flow that does not encroach on coronary vascular reserve because of remodeling of the myocardial microvascular circulation.94
Hyperuricemia results from decreased renal clearance and increased production of urate. Elevated plasma uric acid levels are common in cyanotic adults and are secondary to enhanced urate reabsorption that results from renal hypoperfusion reinforced by a high filtration fraction.88 Acute gouty arthritis is uncommon but not rare.
Renal involvement is represented by abnormalities of both structure and function.88,89 Decreased clearance of urate is a functional abnormality, but the resulting hyperuricemia in turn exerts little or no deleterious effect on renal function.88,89 Proteinuria is a functional abnormality caused by increased glomerular hydraulic pressure in response to the high viscosity of erythrocytotic blood entering the afferent glomerular arteriole in concert with obligatory ultrafiltration from afferent to afferent arterioles.
Two distinct structural (morphologic) glomerular abnormalities are found in cyanotic adults.89,95 The vascular abnormality is characterized by dilation of hilar arterioles and glomerular capillaries because of intraglomerular release of nitric oxide.95 The nonvascular abnormality is characterized by an increase in glomerular cellularity in response to platelet-derived growth factor and transforming growth factor beta in the cytoplasm of systemic venous megakaryocytes that are shunted into the systemic arterial circulation and lodge in glomerular tufts.95
Cardiorespiratory responses to isotonic exercise in cyanotic congenital heart disease significantly influence the dynamics of oxygen uptake and ventilation.88,96 Prolonged onset and recovery of oxygen uptake kinetics incur large oxygen deficits that result in hypoxemia even with low levels of exercise, which suggests that patients with right-to-left shunts rely heavily on anaerobic metabolism. Hyperventilation, which is subjectively perceived as dyspnea, is present at rest and increases excessively during exercise because augmentation of the right-to-left shunt induced by exercise is accompanied by an increase in systemic arterial carbon dioxide and a decrease in pH that stimulate the respiratory center and carotid bodies.88,96
Clubbing of the digits and hypertrophic osteoarthropathy share a common pathogenesis.88 Systemic venous megakaryocytes are shunted into the systemic arterial circulation (see previous) and impact in the digits and subperiostium. Platelet-derived growth factor and transforming growth factor beta are released from megakaryocytic cytoplasm and promote cell proliferation, protein synthesis, connective tissue formation, and deposition of extracellular matrix that are responsible for clubbing and hypertrophic osteoarthropathy.88
Pregnancy poses an excessive risk for women with Eisenmenger’s syndrome.87 The maternal mortality rate exceeds 50%, and there is an independent excessive risk of fetal wastage because of uterine hypoxemia associated with cyanosis.62,89,97,98
Sudden death often results from massive intrapulmonary hemorrhage,89 less commonly from rupture (dissection) of a dilated hypertensive pulmonary trunk,65,99 and rarely from ventricular tachycardia. A cerebral abscess in early life can result in a seizure disorder in adulthood, or the abscess can originate in adulthood.64,88
Physical Appearance
The abnormalities in physical appearance are the result of cyanosis caused by the reversed shunt in Eisenmenger’s syndrome, the catabolic effects of congestive heart failure in infants (poor growth and development, frailty, and cachexia),62 and Harrison’s grooves caused by the thoracic retractions of chronic dyspnea (Figure 17-11). Growth and development are also influenced by intrauterine and genetic factors and by low birth weight.75 Infants and young patients with nonrestrictive ventricular septal defects and balanced shunts may become cyanotic only after crying or exercise.100 Doubly committed subarterial ventricular septal defects are more common among Asians (see previous).17 The physical appearance of trisomy 18 Down syndrome (see Figures 19-5 and 20-11) coincides with the presence of an inlet ventricular septal defect (see Figures 15-90 and 15-91).101–104 In addition, a relationship exists between ventricular septal defect and physical appearance in trisomy 13; trisomy 8 and 9 mosaic; rare aberrations, such as 5p (cri du chat) and 13a- and 18q-105; Holt-Oram syndrome29; Cornelia de Lange’s syndrome72; Klippel-Feil syndrome54; cardiofacial syndrome106; and fetal alcohol syndrome.107
Arterial Pulse
Moderately restrictive defects with relatively low pulmonary vascular resistance are associated with a brisk arterial pulse because of vigorous ejection from a normal volume-loaded left ventricle. A diminished arterial pulse with pulsus alternans accompanies nonrestrictive defects with large left-to-right shunts and congestive heart failure. The arterial pulse in Eisenmenger’s syndrome is normal because systemic stroke volume is maintained (Figure 17-12).64
Jugular Venous Pulse
Moderately restrictive and nonrestrictive ventricular septal defects with congestive heart failure are accompanied by an elevated mean jugular venous pressure and an increase in A and V waves. However, the jugular venous pulse in Eisenmenger’s syndrome is normal or nearly so, with a small dominant A wave (Figure 17-13). Large A waves are exceptional because right ventricular systolic pressure does not exceed systemic. Systemic afterload exists from birth, so the right ventricle requires little extra help from its atrium.
Precordial Movement and Palpation
Roger stated that when a ventricular septal defect is restrictive, “it coincides with no other sign of organic disease with the exception of the harsh thrill which accompanies it.”1 The thrill is maximal in the third or fourth left intercostal space at the left sternal border. A subarterial ventricular septal defect directs its shunt into the pulmonary trunk, so the accompanying thrill is maximal in the second or first and second left intercostal space with radiation upward, to the left, and into the neck.108
[/not-level-membership-for-cardiovascular-category]
