5 Ventricular Septal Defect
I. CASE
A. Fetal echocardiography findings
1. Fetal echocardiography reveals situs solitus of the atria, levocardia, left aortic arch, and normal cardiothoracic ratio (0.3).
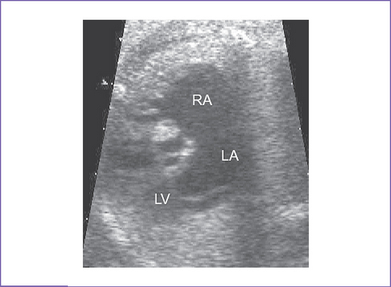
2. The four-chamber view is normal.
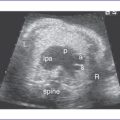
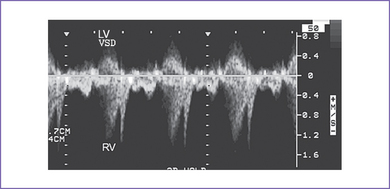
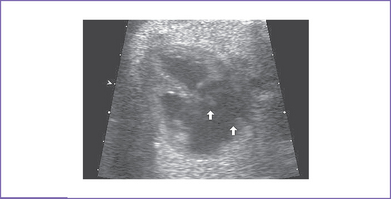
3. There is a moderate (5 mm) perimembranous ventricular septal defect (VSD) (subaortic), with bidirectional shunting and velocity of 1 m/s (Fig. 5-1).
4. Outflow assessment shows normally related great arteries with normal valve morphology and size. The pulmonary artery is anterior to and to the left of the aorta.
5. Both the aortic arch and ductal arch are of equal size and have normal antegrade flow.
6. There is a nonrestricted right-to-left shunt across the foramen ovale; however, no aneurysm or atrial septal tissue could be seen across the fossa ovalis.
7. The pulmonary venous drainage is normal.
8. The RV and left Tei indices (myocardial performance index) were normal. There are no signs of heart failure.
B. Cardiovascular profile score
The cardiovascular profile score is 10/10. The arterial and venous flow patterns are normal.
D. Fetal management and counseling
a. Because of the possibility of aneuploidy, amniocentesis was offered but was declined.
b. Abnormal karyotype with a single VSD is common in a fetus with a trisomy and otherwise structurally normal heart.
a. Serial antenatal fetal echocardiography studies are performed at 6- to 8-week intervals to assess the size of the defect and compare it with the aortic root size serially.
b. At each visit, a renewed search is made for possible developing associated cardiac lesions, such as coarctation of the aorta.
c. In an outlet defect, pulmonary stenosis can appear later in pregnancy, as evidenced by a pulmonary artery smaller than the aorta, thus evolving into tetralogy of Fallot or pulmonary valve stenosis.
d. The size of the ascending aorta and transverse aortic arch is monitored.
e. Size and function of both ventricles are monitored. The size of the ventricles should be compared.
f. The size of the aortic valve annulus is monitored; check for subaortic stenosis.
g. The size and direction of flow across the foramen ovale should be checked to ensure the right-to-left flow is normal in the fetus.
h. The patency of the ductus arteriosus must be ensured.
i. Development of heart failure would not be expected in this condition.
j. Patient management options must be re-evaluated in view of any evolving lesions or new documentation of extracardiac findings that might alter the outcome.
E. Delivery
1. If the fetus remains well compensated and the lesion is isolated, delivery can be normal at term in a secondary care center.
2. Once the baby is delivered, an echocardiogram should be performed to determine the size, number, and hemodynamics of the VSD(s) and to exclude additional pathology. This could be done within the first 2 weeks of delivery if the third-trimester fetal echocardiogram suggests no critical heart disease.
F. Neonatal management
With normal appearance and no other malformations, the baby should go home with the mother.
a. Heart failure with a VSD often does not manifest for several weeks after birth.
b. If the size of the arch is questionable, the baby should be managed as a neonate with possible coarctation (see Chapter 14).
c. Antifailure medication in the form of oral digoxin and furosemide can be started in the hospital, if surgery is likely, or at the first sign of congestive heart failure (see Chapter 30). Use of angiotensin-converting enzyme (ACE) inhibition to reduce the systemic vascular resistance out of proportion to the pulmonary vascular resistance can also assist in managing the heart failure by reducing the net shunting or  p/
p/ s (ratio of pulmonary flow to systemic flow).
s (ratio of pulmonary flow to systemic flow).
d. If the VSD is large and there is evidence of significant shunting, a high-calorie formula may be needed to maintain growth, given the high metabolic rate such infants can have.
a. The majority of VSDs (>50%) close spontaneously, usually in the first year of life. Only a small percentage of VSDs require surgery.
b. Surgery is confined to patients with elevation of pulmonary artery pressure, significant heart failure, failure to thrive, and evolution of additional pathology such as subaortic obstruction, aortic valve prolapse with regurgitation, and right ventricular outflow tract (RVOT) obstruction.
c. The surgical procedure of choice is primary open heart patch closure of the VSD and atrial septal defect (ASD) through the right atrium (RA) on bypass. Most patients are hospitalized 3 to 10 days after surgery for VSD and ASD closure
d. The risks of surgery are low: 1% to 2% for mortality or a complication such as complete heart block.
G. Follow-up
1. Subacute bacterial endocarditis (SBE) prophylaxis should be continued as long as any VSD is present and for 6 months after successful closure of the defect. ASD is not an indication for SBE prophylaxis.
2. Yearly visits to the cardiologist are indicated to detect late problems with arrhythmia such as sinus bradycardia from sinus node dysfunction or atrial tachycardia. A new heart murmur could indicate development of subaortic stenosis.
I. Outcome of this case
1. The baby was delivered at term weighing 3.0 kg. Apgar scores were good: 8 and 9 at 1 and 5 minutes, respectively. Pulse oximeter reading was 98% in room air.
2. Echocardiogram confirmed the diagnosis of a large perimembranous VSD (Fig. 5-2), a small apical muscular VSD (Fig. 5-3), and a moderate-sized secundum ASD.
3. Medical therapy with digoxin, furosemide, and captopril was given for the first 8 weeks after birth.
4. Due to poor weight gain, the baby was referred to a dietician.
5. Surgery was performed at 2 months of age with division of the patent ductus arteriosus, patch closure of the secundum ASD, and patch closure of the VSD. Postoperatively, a large pericardial effusion developed that required pericardiocentesis.
6. The patient was discharged on PO digoxin and furosemide at 2 weeks after surgery.
7. At the 2-week follow-up visit in the cardiologist’s office, the baby was well and medications were stopped. SBE prophylaxis was continued for 6 months and then discontinued. He was seen yearly thereafter.
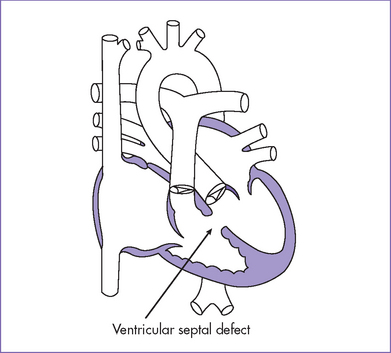
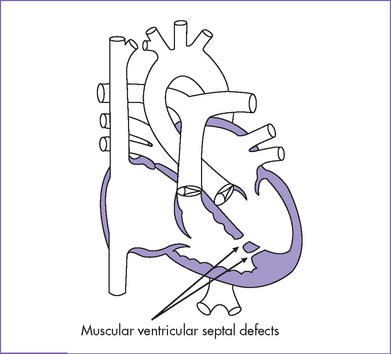
II. YOUR HANDY REFERENCE
A. Ventricular septal defect
a. VSDs are the commonest form of congenital heart disease detected in infancy, accounting for more than 30% of the total cases of congenital heart disease. The incidence is 2 in 1000 live births.
b. The spectrum of VSDs seen in prenatal life is very different from that manifesting postnatally. Isolated VSDs constituted only 6% of a large series of congenital heart diseases identified prenatally.
c. Perimembranous and small VSDs (the most common type postnatally) are rarely seen prenatally, particularly in isolation, and the moderate and large defects predominate.
a. VSDs are the most common type of congenital heart malformation to be overlooked in the fetus. Their detection prenatally depends on the image quality, the size and site of the defect, and the color Doppler capabilities of the ultrasound machine. A large VSD may be seen only on imaging.
b. Most overlooked defects tend to be small.
c. VSDs are commonly associated with other cardiac defects as part of more complex disease.
d. VSDs can vary in location and are divided into inlet, outlet, perimembranous, and muscular defects.
e. There have been some reports of VSDs closing spontaneously in prenatal life.
3. Associated syndromes and extracardiac anomalies.
a. When a VSD is detected, a complete sequential analysis of the heart is mandatory.
b. An isolated VSD is rarely associated with situs anomalies.
c. VSD(s) are commonly found as part of complex cardiac anomalies, some of which are not obvious when the study is performed during early pregnancy.
d. VSD can be part of RVOT obstruction, such as tetralogy of Fallot.
e. VSD can be associated with left heart obstructions, including aortic valve stenosis, coarctation of the aorta, and interrupted aortic arch.
f. Extracardiac anomalies associated with a VSD (>40%) include trisomies 21, 13, and 18. This rate is higher than expected from postnatal series. This may be related to the selection of patients referred for fetal echocardiography as well as spontaneous fetal loss in chromosomally abnormal fetuses that could not be included in postnatal series.
g. The 22q 11 chromosome deletion in association with a VSD has been reported.
h. Aortic coarctation occurring in isolation or with VSDs or other forms of left heart obstruction may be associated with chromosomal abnormalities including Turner’s syndrome, trisomy 18, and deletion of chromosome 22q11 (especially in the presence of a malalignment VSD).
i. Chromosomal anomalies were found in 25% to 50% of cases of isolated VSD in one fetal series.
4. Clues to fetal sonographic diagnosis.
a. Two-dimensional echocardiogram, four-chamber view.
d. The velocity of shunting across a ventricular septal defect is usually low (0.4-0.7 m/s) and does not occur throughout the cardiac cycle.
e. Apical muscular septal defects—even moderate to large, single or multiple—can be missed in the fetus. This may be due to the imaging difficulty in this area and due to the heavily trabeculated right ventricle.
a. In the fetus, the right and left ventricular pressures are equal or similar. Small dynamic differences cause systolic left-to-right and diastolic right-to-left shunting through a large VSD. After birth, the pulmonary artery pressure drops, and the shunt through the VSD increases in magnitude. Pulmonary resistance continues to fall postnatally, reaching its nadir at 3 to 4 months of age, when the worst symptoms of CHF can be expected.
b. If the VSD is smaller than the aorta, a gradient can develop between the LV and RV over time, resulting in a lower pulmonary than aortic pressure.
c. In addition to perimembranous VSDs (those that touch the tricuspid valve), there are muscular (apical, mid, or outlet) and subarterial (or outlet or supracristal) VSDs. Muscular VSD is likely to close spontaneously over time. Subarterial defects can affect the aortic valve and cause regurgitation secondary to cusp prolapse.
d. Important associated defects with VSD are coarctation of the aorta, ASD, multiple VSDs, patent ductus arteriosus, aortic stenosis, mitral valve anomalies, pulmonary stenosis, anomalous muscle bundle of the RV, and anomalies of pulmonary venous return.
a. The size of a large perimembranous VSD noted early in gestation tends to remain the same relative to the aorta.
b. Many small VSDs close spontaneously before birth, especially those in the muscular septum.
7. Perinatal management (for VSDs not diagnosed in the fetus).
a. After birth, the clinical presentation of VSD is variable, but the baby is usually acyanotic.
b. If cyanosis is present at birth (oxygen saturation <90%), then obstruction to pulmonary blood flow should be suspected as in tetralogy of Fallot (see Chapter 7). Pulmonary hypertension may be another cause. Cardiology referral is mandatory.
c. Surgical repair of VSD without significant other disease is usually performed for the following indications:
d. The incidence of closure is 0% to 2%, compared to 1% in older infants, children, or adolescents. In the presence of a large, hemodynamically significant VSD, the repair (VSD closure) is usually performed in one stage.
e. Additional cardiac lesions that increase the need for surgical intervention include subaortic obstruction, aortic valve prolapse with insufficiency, and evolving RVOT obstruction.
f. Although surgical closure is currently the standard of practice, there is growing experience in device closure at catheterization, particularly with smaller defects that are confined to the membranous or muscular–trabecular septum away from the aortic and mitral valves.
g. Long-term survival following repair of isolated VSD is 86% to 93% at 12 to 15 years following repair.
![]() B.Abnormalities of the atrial septum
B.Abnormalities of the atrial septum
a. An ASD occurs in about 1 in 1500 live births. It is a common component of complex disease, but only isolated defects are discussed here. It is usually sporadic, but it can occur as part of genetic syndromes such as Holt–Oram syndrome (autosomal dominant).
c. There are essentially four types of ASD, with different mechanisms underlying their formation.
c. In the absence of atrial isomerism or complete left ventricular outflow obstruction, the prognosis for all forms of atrial septal defect is good.
3. Clues to fetal sonographic diagnosis. Normally, the foramen ovale defect occupies about the middle one third of the atrial septum.
a. A true secundum atrial defect is larger than normal for gestation (0.3 mm at 20 weeks to 0.8 mm at term).
b. Atrial defect is larger than the aorta.
c. There is a defect in an abnormal position in the atrial septum.
d. There is a restrictive foramen ovale (obstruction at the foramen itself or at the passage around the flap).
e. Chiari malformation is seen in the right atrium at the site of the eustachian valve. This is more commonly seen with tricuspid valve anomalies such as tricuspid atresia.
4. Associated syndromes and extracardiac anomalies: Secundum ASD can be associated with upper limb anomalies as in Holt–Oram syndrome or with other genetic disorders.
a. Secundum ASD occurs when the septum primum fails to cover the oval fossa or when there are fenestrations in the septum primum. There may be one or more communications.
b. A primum ASD involves the lower part of the atrial septum, which is in continuity with the AV valves and is part of the spectrum of AV septal defect.
c. A sinus venosus ASD is located in the posterosuperior or posteroinferior portion of the atrial septum.
d. A coronary sinus ASD is rare and occurs when the atriosinus venosus fold fails to form. It consists of a communication between the atria at the site of the absent coronary sinus with persistence of a left superior vena cava (SVC) directly draining to the LA. This defect is almost always associated with a persistent left SVC opening into the LA.
6. Fetal management and counseling.
a. In cases of isolated atrial defect, delivery should be at term and does not require a tertiary care center. In case of restrictive foramen ovale, it may be necessary to deliver early in the presence of fetal hydrops.
b. ASDs are usually closed electively between 1 and 5 years of age using either a transcatheter device (as in secundum ASD) or open heart surgery (sinus venosus or coronary sinus type defect). Such patients are usually asymptomatic, at least in the neonatal period.
c. In a few fetuses, spontaneous prenatal closure of a small VSD as part of a complete AV septal defect has resulted in a partial AV septal defect. If the fetus has a primum defect with coarctation or hypoplastic left heart syndrome, delivery should be in a tertiary care center.
7. Risk of recurrence: For a secundum atrial septal defect in a baby with one affected sibling and no other family history, the recurrence risk is 2.5%.
III. TAKE-HOME MESSAGE
A. Diagnosis
1. Perimembranous and small VSDs (the most common type postnatally) are rarely seen prenatally, particularly in isolation, and the moderate and large defects predominate.
2. A small VSD could be missed in fetal life. This should be mentioned during fetal counseling.
3. Chromosomal anomalies are common with VSD.
4. Secundum ASD occurs when the flap valve fails to seal the foramen after birth, so it is almost impossible to predict this before birth unless the foramen ovale defect is complete (see Fig. 5-6). Therefore, the prenatal diagnosis of an isolated secundum ASD is rare.
5. Primum ASD can be part of complex congenital heart defects and is associated with situs anomalies such as left atrial isomerism. Isolated secundum ASD is rarely associated with extracardiac anomalies or genetic disorders.
6. Neither sinus venosus defect nor coronary sinus ASDs have been reported in utero.
7. A dilated coronary sinus could be mistaken for a primum septal defect. A left superior vena cava is usually the cause of the enlarged coronary sinus and will be found in other projections. In this setting, both AV valves insert at the ventricular septum at different levels.
Allan LD, Sharland GK, Milburn A, et al. Prospective diagnosis of 1,006 consecutive cases of congenital heart disease in the fetus. J Am Coll Cardiol. 1994;23(6):1452-1458.
Ferenz C: Epidemiology of Congenital Heart Disease: The Baltimore–Washington Infant Study, 1981-1989. Perspectives in Pediatric Cardiology, . Armonk, NY;Futura: 1993;vol 4.
Huhta JC, Edwards WD, Danielson GK, Feldt RH. Abnormalities of the tricuspid valve in complete transposition of the great arteries with ventricular septal defect. J Thorac Cardiovasc Surg. 1982;83(4):569-576.
Murphy DJJr, Ludomirsky A, Huhta JC. Continuous-wave Doppler in children with ventricular septal defect: Noninvasive estimation of interventricular pressure gradient. Am J Cardiol. 1986;57(6):428-432.
Phillipos EZ, Robertson MA, Still KD. The echocardiographic assessment of the human fetal foramen ovale. J Am Soc Echocardiogr. 1994;7(3 Pt 1):257-263.
Sharif DS, Huhta JC, Marantz P, et al. Two-dimensional echocardiographic determination of ventricular septal defect size: Correlation with autopsy. Am Heart J. 1989;117(6):1333-1336.