Chapter 11
Venous Physiology
Lori L. Pounds, Lois A. Killewich
Anatomic Considerations
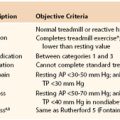
The understanding of venous anatomy has a rich history dating to Hippocrates (460-377 BC). In the field of phlebology, knowledge of anatomy is essential for evaluation and formation of a treatment plan. Venous anatomy is much more highly variable than arterial anatomy; moreover, the nomenclature is infused with duplicate and misleading names, which has led to the development of inappropriate treatment plans for patients. In 2001, an international interdisciplinary committee, under guidance of the presidents of the International Union of Phlebology and the International Federation of Anatomical Associations, formulated a consensus statement to update the Terminologia Anatomica (the official anatomic nomenclature).1 These changes, summarized in Table 11-1, represent an agreement to satisfy anatomists and clinicians, thus providing better care for the patient with a venous disorder.
Table 11-1
Changes in Nomenclature for the Superficial and Deep Veins of the Leg Based on the 2001 Conference
Venous Systems of the Extremities
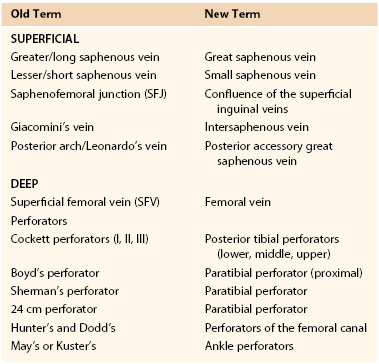
The upper and lower extremity venous systems each contain three components: the superficial, deep, and perforating veins (Fig. 11-1).
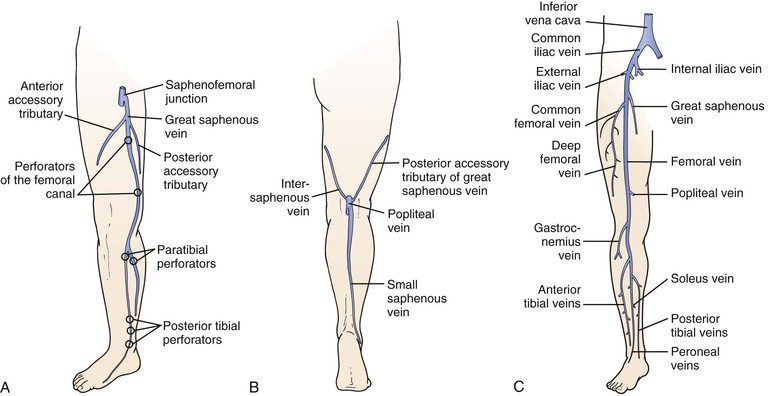
Figure 11-1 Anatomy of the veins of the lower extremity. A, Superficial veins and perforators, anterior view. B, Superficial veins, posterior view. C, Deep veins, anterior view.
Lower Extremity Veins
Superficial Veins.
The superficial veins are located above the muscular fascia. In the legs, a subcomponent, the saphenous system, is present. The saphenous compartment2,3 is bounded superficially by a hyperechoic saphenous fascia and rests on the muscular fascia (the “Egyptian eye” seen on duplex imaging; Fig. 11-2). It contains the main great (formerly “greater”) saphenous trunk, accompanying small arteries, and the saphenous nerve below the knee. The saphenous fascia has been referred to in the past as the superficial fascia, the Colles or Scarpa fascia, or the subcutaneous pseudofascia. Since the consensus update, these terms are no longer recommended. The remaining superficial compartment below the dermis contains the accessory saphenous tributaries (anterior and posterior), which ascend parallel to the great saphenous vein. It also contains the communicating veins, a complex and variable system that connects with other veins in the same superficial compartment. An example would be the intersaphenous vein (formerly known as the vein of Giacomini), which connects the great and small (formerly “lesser”) saphenous veins. The reticular venous plexus and subpapillary venous plexus are also included in this area (Fig. 11-3).
Figure 11-2 Transverse B-mode ultrasound image of the “Egyptian eye.” The pupil is the great saphenous vein; the eyelids are the superficial and deep layers of fascia surrounding the saphenous subcompartment.
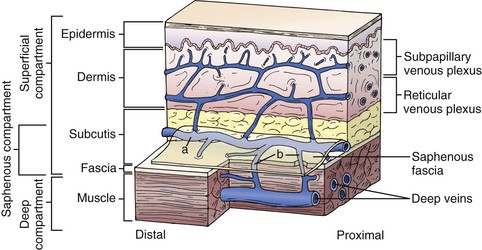
Figure 11-3 Relationship between the fascia and veins of the lower extremity. The fascia covers the muscle and separates the deep compartment from the superficial compartment. Superficial veins (a) drain the subpapillary and reticular venous plexuses and they are connected to deep veins through perforating veins (b). The saphenous fascia invests the saphenous vein. The saphenous compartment is a subcompartment of the superficial compartment. (From Mozes G, et al: New discoveries in anatomy and new terminology of leg veins: clinical implications. Vasc Endovasc Surg 38:367-374, 2004.)
Deep Veins.
The deep veins are located in the fascial muscle compartments, follow the same course as the arteries, and for the most part, have the same names. A significant change in the new terminology is that the superficial femoral vein is now named the femoral vein. Originally named because it was the venous equivalent of the superficial femoral artery, the name produced confusion for clinicians when reported that it was acutely thrombosed. Some would erroneously conclude that because it was the “superficial” femoral vein, the thrombus did not constitute deep venous thrombosis (DVT), and anticoagulation was not indicated. With the new terminology, reporting that the “femoral vein” is thrombosed should lead the practitioner to prompt initiation of anticoagulants, and possibly, thrombolytic therapy.
Perforating Veins.
There is a rich network of perforating veins that traverses the muscular fascia between the deep and superficial veins. These veins direct flow from the superficial to the deep systems for return to the heart via the calf muscle pump and a series of one-way valves. In general, the lower extremities have a series of large perforators located at 6-cm intervals from the base of the heel to the upper part of the thigh. Their names have been revised in the consensus document, and are also found in Table 11-1. The perforators may arise from the great saphenous or an accessory trunk (an indirect perforator) or connect a reticular vein directly to the deep system (a direct perforator). In the upper extremities, there are few perforating veins, which are found mostly in the arms connecting the basilic and brachial veins.
Upper Extremity Veins
Veins of the upper extremities do not function against gravity in the same fashion as the lower extremity veins do, and hence, do not contain as many valves. As in the lower extremities, deep veins accompany the arteries and have the same names. There are two main trunks of superficial veins: the cephalic and basilic veins. The cephalic vein courses on the radial (ventral) side of the forearm and on the lateral aspect of the biceps to the deltopectoral groove to join the axillary vein. The basilic vein is located on the ulnar (ventral) side of the forearm and traverses posterior to the elbow to join the brachial vein in the midportion of the arm. With the arms extended, the basilic vein is closer to the body, and the cephalic is closer to the head. There is a series of accessory ascending veins that parallel the main trunks and a series of communicating veins between the deep and superficial systems.
Venous Valves
Venous valves prevent retrograde flow of blood in a proximal-to-distal direction. They are found throughout the body, but the highest concentration is located in the lower extremities. Both superficial and deep veins contain valves. Anatomic studies have confirmed that they are present in all sizes of veins down to the level of venules.4 In general, the number of valves is higher in the more distal portions of the extremities and decreases proximally. Venous valves are most commonly bicuspid and can form sinusoidal dilatations, presumably from locally reversed flow.
Lower Extremity Venous Valves
The prevalence of valves in the iliofemoral venous system is low. Miles et al5 documented the location and extent in 70 necropsy specimens. They found no valves in the inferior vena cava and observed a mean of 1.2 valves in the iliofemoral system on the right and 0.97 valves on the left, with greater numbers in the common femoral and external iliac veins and fewer in the common iliac veins. Interestingly, no valves were found in the left common iliac vein in the area where it is crossed anteriorly by the right common iliac artery. The paucity of valves in the common iliac veins has been invoked as a possible reason why thrombolysis for iliofemoral venous thrombosis reduces the risk of developing postthrombotic syndrome. Obstruction, rather than valvular incompetence or destruction, is considered the primary cause for development of the postthrombotic syndrome following iliofemoral thrombosis.6
Moore et al7 reviewed the literature to document the number and location of venous valves in the femoro-popliteal segments in normal subjects. They found that valves were frequently present in the common femoral, femoral, and popliteal veins. A common femoral vein valve was observed in approximately 75% of cases, consistently located at the level of the inguinal ligament. The femoral vein contained one to four valves, and 90% of subjects had at least two valves. The most common location was just distal to the junction of the femoral and profunda femoris veins. The popliteal vein most commonly contained two valves, with one located just below the adductor hiatus and a second in the distal segment.
Upper Extremity Venous Valves
The location and number of valves in the subclavian and axillary veins was recently investigated by Celepci and Brenner8 in 59 limbs of 30 cadavers. These investigators found a terminal valve in the subclavian vein just distal to the venous angle (junction of the subclavian and internal jugular veins) in 100% of cases. In most cases, no additional valves were found in the subclavian vein. Three-quarters of axillary vein specimens possessed at least two valves, with the remainder possessing up to four valves. All valves in the axillary veins were found in the distal half.
Venous Wall Structure and Composition
Vein walls are composed of three layers—the intima, media, and adventitia. The intima is a single layer of cells resting on a thin layer of connective tissue. Valves are lined on both sides with a layer of intima over a thin connective tissue skeleton. Adjacent to the intima is the internal elastic lamina, which is more developed in larger veins and may be absent in small veins. The media consists of smooth muscle cells and connective tissue, such as collagen. In the great saphenous vein, it is thick and has a great capacity for muscular contraction. This conveys protection from dilatation and varicosity formation. In contrast, tributaries of the great saphenous vein have little media and are prone to the formation of varicosities. In the deep system, the content of collagen also varies greatly; calf veins have a large amount, which provides great wall strength and resistance to varicosity formation. The central veins have fewer smooth muscle cells and an increasing amount of connective tissue. The adventitia is not well differentiated from the media and contains loose connective tissue, vasa vasorum, and adrenergic nerve fibers.2
Normal Venous Hemodynamics
The venous system has two important functions—return of blood to the heart from the capillary bed and maintenance of cardiovascular hemostasis through changes in capacitance.9 In addition, these functions contribute to the peripheral pumping mechanism, which assists the heart in transporting blood during exercise and participates in the thermoregulatory system of the body.
Venous return is accomplished through the interactions of pressure gradients, muscle pumps, and valves. Pressure gradients dominate in the supine position, but in the upright position, gravity is counteracted by an efficient system of muscle pumps and valves.10–13 Unlike arteries, the volume of blood carried in a vein fluctuates considerably without concomitant changes in venous pressure. This large capacitance maintains cardiovascular stability.
Pressure-Flow Relationships and Venous Return
Throughout the human body, the pumping action of the heart causes movement of blood through both arteries and veins. The pressure generated by cardiac pumping is termed dynamic pressure. Under normal conditions in the supine position, blood flow is determined by dynamic pressure gradients, with arterial pressure being higher than venous pressure. The majority of dynamic pressure is dissipated in the arterial system before it reaches the capillary bed. At the venous end of the capillary bed, it ranges from 12 to 18 mm Hg. Atrial pressure averages 4 to 7 mm Hg under normal conditions. Hence, blood flows along this gradient and is returned to the heart.
In the upright position, venous flow in the lower extremities is dominated by the effects of hydrostatic pressure, which is derived from the weight of the column of blood below the right atrium (Fig. 11-4). A small effect of dynamic pressure is also present. Hydrostatic pressure is determined by the density of blood and the acceleration of gravity, and is expressed as a constant multiplier (0.77 mm Hg/cm) of the vertical distance in centimeters below the atrium.9 Return of blood from the dependent lower extremity to the heart requires overcoming the effects of hydrostatic pressure, which is accomplished by muscle pumps working in concert with venous valves. The leg muscle pumps, of which the calf pump is the most important, generate high pressure during muscle contraction, which propels venous blood toward the heart. During relaxation, valves close, and blood is prevented from refluxing down the leg and breaking up the hydrostatic pressure column.10,11 The negative pressure generated by valve closure also draws blood from the superficial to the deep systems via perforating veins, thus further enhancing return of blood to the heart.14 In the upright position under normal conditions, venous pressure measured on the dorsum of the foot ranges from approximately 90 to 120 mm Hg, depending on the individual’s height.
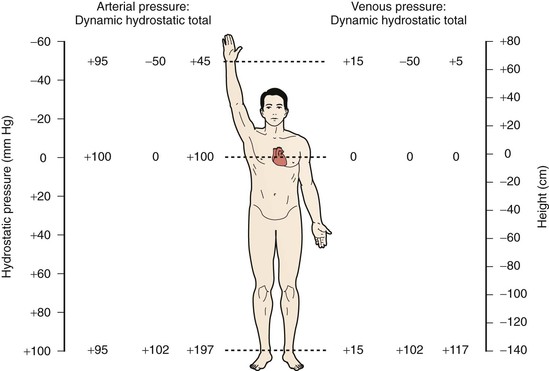
Figure 11-4 Effect of upright position on venous and arterial pressure. Pressure at the right atrial level is 0. In the supine position, hydrostatic pressure is essentially 0, so total intravascular pressure closely approximates dynamic pressure.
Upper extremity venous flow is governed by dynamic pressure gradients in a similar fashion to lower extremity flow. When the upper extremity is dependent (similar to the lower extremity flow in the upright position), pressure is governed not only by the distance below the atria but also by the distance between the atria and the first rib. This occurs because anatomy dictates that blood draining from the arm must travel vertically to the level of the first rib. When the arm is held above the atria in the upright posture, the upper extremity, head, and neck veins collapse.9
Venous Compliance and Capacitance
In addition to returning blood to the heart, veins play a significant role in maintaining cardiovascular hemostasis by storing large volumes of blood and by their ability to change shape and maintain pressure despite relatively large changes in volume.15 The venous system at times contains as much as 75% of the systemic blood volume. As such, reflex changes in vasomotor tone, together with passive distention or recoil of veins, provide mechanisms by which blood volume can be rapidly redistributed to compensate for loss of volume in conditions such as hemorrhage. For example, because of changes in venous capacitance, a healthy individual can tolerate an acute decrease in blood volume of up to 10% without serious cardiovascular consequences.16 A healthy individual can also compensate for large increases in total body fluid by using changes in venous volume and capacitance.
A complete discussion of the relationship between venous capacitance and cardiac output is beyond the scope of this chapter, and the reader is referred to several excellent reviews.17–19 A brief summary follows.
The relationship between pressure and volume at a given level of smooth muscle tone in the venous system is termed capacitance. Compliance is the change in blood volume that occurs for each unit of change in transmural pressure in a segment of vein; to put it another way, it is the slope of the capacitance curve.15,17–19
Venous capacitance is governed by the collapsible nature of the venous wall. A low volume of blood in a vein results in an elliptical shape with nearly coapted walls and low pressure (Fig. 11-5).9,19 A large increase in volume can occur with only a minimal increase in pressure. Eventually, the system is overwhelmed, and the vein dilates, which results in an increase in pressure per unit of volume. The difference between intraluminal pressure acting to expand a vein and tissue pressure acting to collapse the vein is termed transmural pressure. An increase in venous transmural pressure from 0 to 15 mm Hg results in an increase in venous volume of up to 250%. This corresponds to a change in shape from elliptical to circular (see Fig. 11-5).20 However, once the vein is circular, much higher pressure is required to stretch the venous wall to add additional volume. At arterial pressures, veins become as stiff as arteries.21–23
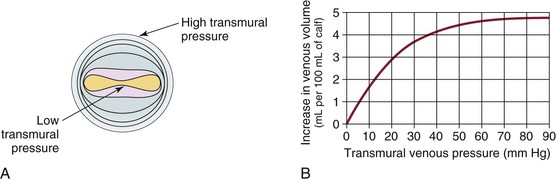
Figure 11-5 A, Cross-section of a venous lumen at various transmural pressures. At lower pressures the vein is elliptical, whereas at high pressures it is circular. B, Relationship of venous volume to transmural pressure. At low pressures, veins are compliant and change shape easily to accommodate large increases in volume. At high pressures, they become stiff and cannot accommodate large changes in volume. (A, Adapted from Moreno AH, et al: Mechanics of distention of dog veins and other thin-walled tubular structures. Circ Res 27:1069, 1970.)
Neural Regulation of Venous Capacitance and Compliance
Changes in venous capacitance are modulated both passively, by distention and recoil, and actively, by changes in the smooth muscle activity of vein walls. Smooth muscle activity is, in turn, controlled by the sympathetic nervous system.19–21 Passive transfer of blood out of an organ takes place when blood flow through the organ decreases. This occurs because the decrease in pressure across the resistance to venous flow is reduced, which follows from Ohm’s law applied to hydraulics (ΔP = F × R). An example is that splanchnic blood volume decreases to compensate for blood pooling in skin veins when body temperature rises during exercise.19
Active changes in capacitance occur in response to stimulation of the sympathetic nervous system. Studies performed in dogs by Brooksby and Donald showed that stimulation of the sympathetic system resulted in expulsion of approximately 70 mL of blood from the splanchnic circulation.24 Of this, the investigators determined that reduced flow across the splanchnic bed produced passive recoil of the venous system, which accounted for 45 mL of the blood expelled. The remaining 25 mL was expelled actively by stimulation of α-adrenergic receptors of the sympathetic nervous system.24
Arterial baroreceptors such as the carotid sinus also influence capacitance through modulation of the sympathetic nervous system. Decreased arterial blood pressure is sensed by the carotid sinus and results in secretion of catecholamines, which, in turn, increase smooth muscle activity in small veins and venules. Constriction of these vessels (reduced capacitance) causes venous blood to be transferred from the periphery to the heart, thereby boosting cardiac output.23
Effects of Respiration
Respiratory Effects on Lower Extremity Venous Return
Respiration modulates venous return from the legs through alterations in venous capacitance and collapsibility related to movement of the diaphragm and changes in intrathoracic and intra-abdominal pressure. Sumner and Zierler25 developed a model of this relationship as shown in Figures 11-6 and 11-7. In this model, the pressure driving blood toward the heart (15 cm H2O) is represented by an elevated fluid reservoir. A collapsible tube, representing the inferior vena cava, passes through a closed container with a pressure of 5 cm H2O, representing the abdominal cavity. The end of the tube, which represents the right atrium, is open to the atmosphere, and therefore, has an outflow pressure of 0 cm H2O (see Fig. 11-6).
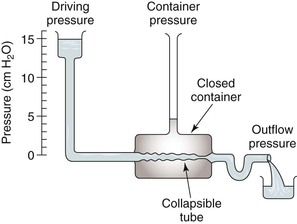
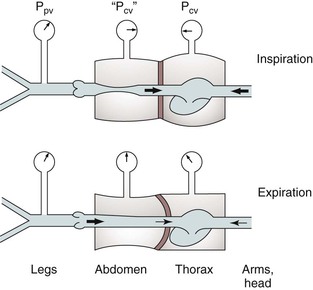
Figure 11-7 Effects of respiration on venous return. Pcv, Central venous pressure; Ppv, peripheral venous pressure. (From Sumner DS: In Bergan JJ, Yao JST, editors: Surgery of the veins, Orlando, FL, 1985, Grune & Stratton; pp 3-23.)
For blood to drain from the elevated fluid reservoir (the periphery) through the closed container (the abdominal cavity) to the heart, the pressure in the collapsible tube (the inferior vena cava) must rise until it exceeds that in the closed container, which allows the tube to open (increase in venous capacitance associated with a change in shape from elliptical to circular). This gradient is equal to 15 − 5 cm H2O or 10 cm H2O, rather than the gradient across the entire system, which is 1 to 15 cm H2O. Elevation of the driving pressure will increase the gradient, and hence, flow, but changes in outflow pressure have no effect unless the pressure rises above that in the closed container, in which case the gradient and flow will be decreased. Increasing pressure in the closed container will also decrease the gradient and flow. These relationships derive from the low pressure and “collapsibility” of veins, which means that flow cannot occur until the inferior vena cava, represented by the collapsible tube, achieves a transmural pressure greater than that in the abdominal cavity.
When inspiration occurs, the diaphragm descends, and intra-abdominal pressure is increased (see Fig. 11-7). This results in a decrease in the gradient, and venous return from the lower extremity is reduced. The increase is frequently large enough to reduce the gradient to 0, and venous flow ceases. This situation results in the well-described phenomenon of venous phasicity with respiration. Conditions that markedly increase venous pressure in the lower extremities augment the gradient to an extent that the effects of intra-abdominal pressure are overcome, and venous flow becomes continuous. This occurs in the setting of lower extremity venous thrombosis.
Respiratory Effects on Upper Extremity Venous Return
In the upper extremities (see Fig. 11-7), intrathoracic pressure, which is representative of outflow pressure in the model, is generally lower than extrathoracic, or driving, pressure. Hence, the gradient supports return of blood to the heart, and flow is less dependent on respiration. Conditions that alter this dynamic, such as congestive heart failure or pulmonary hypertension, cause central venous pressure to be elevated significantly above peripheral pressure, and the peripheral flow pattern is then derived from right heart function. This causes pulsatile flow patterns in veins of both the upper and lower extremities.
Calf Muscle Pump
Return of venous blood to the heart is facilitated by the action of muscle pumps in the legs, which occur in the thigh, in the calf, and in conjunction with the venous plexus on the plantar aspect of the foot.10,11,13,26 These pumps forcefully expel blood from the intramuscular and surrounding veins and propel it toward the heart (Fig. 11-8A). Ludbrook11 was the first to show the importance of these pumps and also noted that the calf pump appeared to be more important than the thigh pump. During vigorous exercise involving both the calf and thigh, 65% of the blood that entered the calf was expelled, whereas only 15% of the blood entering the thigh was expelled. At the same time, Stegall26 demonstrated that up to 30% of the energy required to circulate blood during vigorous exercise could be furnished by the muscle pumps. More recent evidence14 suggests that the calf muscle pump generates up to 200 mm Hg during muscular contraction and expels 40% to 60% of the venous volume of the calf (100 to 150 mL).
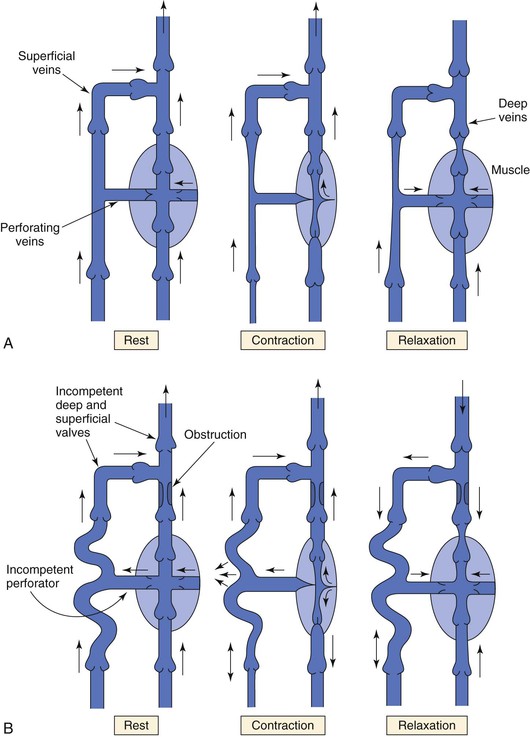
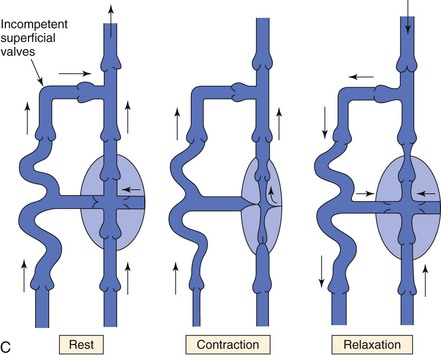
Figure 11-8 A, Dynamics of venous flow in response to calf muscle contraction in a normal limb. B, Dynamics of venous flow in response to calf muscle contraction in a postthrombotic limb with residual deep venous obstruction, incompetent valves, and secondary varicose veins. Blood flow in perforating veins is bidirectional. C, Dynamics of venous flow in response to calf muscle contraction in a limb with primary varicose veins. (From Sumner DS: Venous dynamics—varicosities. Clin Obstet Gynecol 24:743-760, 1981.)
The calf muscle pump includes the soleal and gastrocnemius muscles, their intramuscular venous sinusoids, and the superficial and deep veins. The soleal and gastrocnemius muscle sinusoids constitute the major “bellows” of the calf pump. Even in the standing position, contraction of the calf muscles produces enough pressure to eject blood and propel it toward the heart. Intramuscular veins are also affected because of the strong fascia investing the muscles.
Muscle contraction produces higher pressure in the deep venous system than in the superficial system, but blood is prevented from refluxing in a superficial direction by the system of venous valves within the perforating veins that connect the two systems (see Fig. 11-8A).27 When the calf muscles relax, the soleal and gastrocnemius sinusoids are refilled by capillary inflow from the distal deep veins, and to a lesser extent, by flow in a superficial-to-deep direction through the perforators.26–28
Ambulatory Venous Pressure
In the erect posture, venous pressure measured on the dorsum of the foot is slightly higher than arm pressure, depending on height. Walking, which produces contractions of the calf muscle pump, reduces venous pressure at the foot level quickly (within 7 to 12 steps) to a mean of 22 mm Hg.29,30 “Tiptoe” maneuvers, consisting of standing ankle plantar flexion or heel raise, produce the same effects (Fig. 11-9A). Ambulatory venous pressure measurements have been used to quantify changes in venous function associated with chronic venous insufficiency and the postthrombotic syndrome.31 This is accomplished by insertion of a 21-gauge needle into a dorsal foot vein. After exercise is discontinued, a mean of 31 seconds is required to restore hydrostatic pressure via inflow from the capillary bed32 (see Fig. 11-9A).
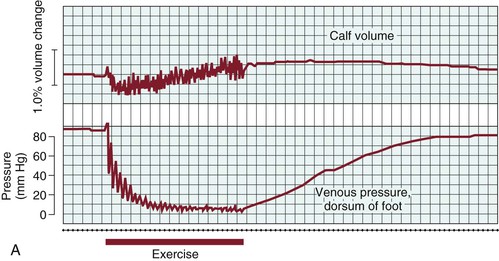
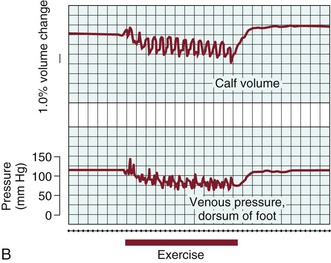
Figure 11-9 A, Effects of exercise on calf volume and venous pressure measured on the dorsum of the foot in a normal limb. B, Effects of exercise on calf volume and venous pressure measured on the dorsum of the foot in a patient with postthrombotic syndrome and venous hypertension. (From Strandness DE, Jr, et al: Hemodynamics for surgeons, New York, NY, 1975, Grune & Stratton.)
Flow through the capillary beds of exercising muscles is facilitated by reductions in ambulatory venous pressure. Pressure in peripheral arteries consists of the sum of hydrostatic pressure and the pressure generated by the left ventricle. As an example, the pressure at the arteriolar end of a calf muscle capillary would be
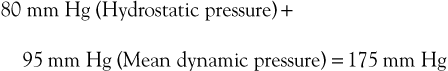
Venous pressure in the erect posture would be
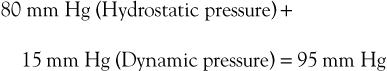
The pressure gradient across the bed is thus
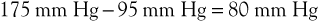
Upon exercising, with resultant calf muscle contraction, venous pressure is decreased to a mean of 22 mm Hg (see previous discussion). The pressure gradient would then be
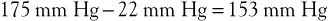
This higher pressure gradient facilitates muscle perfusion.
Venous Pathophysiology
Venous Obstruction
Venous obstruction secondary to DVT and superficial thrombophlebitis results in elevations of peripheral venous pressure. Sumner and Zierler25 described the relationship between peripheral venous pressure (Ppv), central venous pressure (Pcv), flow through the area of concern (Qv), and the hemodynamic resistance of the intervening veins (Rv):
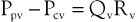
or
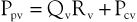
Normal central venous pressure is lower than intra-abdominal pressure; therefore, in the supine position, Pcv is a reflection of intra-abdominal pressure. When venous obstruction occurs, peripheral venous resistance (Rv) increases, which results in an increase in Ppv. The degree of increase is a function of the location, length, and number of obstructed venous segments, as well as the presence of collateral venous channels. In the case of a localized, distal thrombosis, elevations in venous pressure are minimal.33 In a classic paper, Husni et al34 demonstrated that the mean venous pressure in the limb of a supine patient with an acute thrombosis was 17 mm Hg. The increase was approximately 2.5 times that found in a normal limb. In the erect position, foot pressures were equivalent in normal subjects and those with thrombosis. This occurs because the major contributor to foot pressure measurements is hydrostatic pressure, and the increases in Ppv associated with increased Rv are small in comparison. Ambulation results in significant decreases in Ppv in a normal person. However, in the case of acute thrombosis, only minimal changes in Ppv occur.
Acute venous obstruction also results in changes in venous outflow. Such changes have been measured with strain gauge plethysmography. Briefly, the technique uses changes in resistance through a conductor tube to measure changes in calf volume via a strain gauge attached around the calf. A pneumatic cuff is placed around the thigh and inflated to 50 mm Hg until the recorded tracing reaches a plateau. It is then deflated rapidly, and changes in volume are recorded. The increase in limb volume that occurs with cuff inflation is defined as the venous volume, and the tangent to the steepest portion of the venous outflow curve after cuff deflation is defined as the maximal venous outflow (MVO). MVO is usually recorded over the interval between 0.5 and 2.0 seconds after cuff deflation to obtain the most accurate measurement.35–37
Barnes et al35,38 measured MVO in normal volunteers and patients with acute DVT. MVO in normal volunteers was 45 ± 18 mL/100 mL/min. In patients with DVT, MVO ranged from 11 to 20 mL/100 mL/min.
MVO is also decreased in patients with upper extremity DVT. One group found that MVO was 162 ± 52 mL/100 mL/min in normal volunteers versus 79 ± 30 mL/100 mL/min in patients with acute DVT.39
Valvular Incompetence
Valvular incompetence occurs in all categories of extremity veins, including the deep, superficial, and perforator systems. It can occur in anatomically normal valves that do not coapt or may follow an event such as a DVT, in which case the valve cusps are most often destroyed. Regardless of the mechanism of valvular incompetence, it is intimately associated with the venous hypertension (see Fig. 11-8B and C).9,40–42
Valvular incompetence in which the valve cusps are present in some form, but do not coapt normally, most frequently occurs in the superficial and perforator systems in the setting of varicosities (see Fig. 11-8C).9,40,41 Originally, it was believed that valvular incompetence was the primary cause of venous varicosities; more recent data suggest that incompetent valves result from changes in the vein wall leading to dilatation of the veins.43–45 This is discussed more fully under “Varicose Veins.”
Animal Models
Venous valve failure has been studied in an animal model of venous hypertension using the Wistar rat.46–48 In this model, venous hypertension was produced by creation of an arteriovenous fistula between the femoral artery and femoral vein. Femoral venous pressure increased to approximately 90 mm Hg compared with 9 mm Hg in the contralateral normal femoral vein. Valves were immediately stretched by the increased pressure; reflux developed by 2 days and continued increasing up to 42 days.47 The development of reflux was accompanied by changes in valve morphology, including increased diameter of the valvular annulus and shortening of annular height. By 42 days, most of the valves disappeared. Changes and ultimate loss of valves were associated with increased inflammation, manifested as infiltration of granulocytes, monocytes, macrophages, and lymphocytes, and expression of matrix metalloproteinases (MMPs), in particular MMP-2 and MMP-9. The investigators concluded that when valves were subjected to prolonged venous hypertension, inflammation, valve remodeling and loss, and reflux occur.48
The same authors have also explored the effects of shear stress on valve function and reflux.48 In normal conditions, valve function is based on pressure rather than flow.49 Valve closure occurs with minimal reflux. Flow through the valve consists of a proximally directed jet and vertical flow into the sinus behind the valve cusp. The vertical flow ensures that stasis does not occur in the area behind the cusp, and shear stress is maintained. When the pressure associated with flow behind the cusp exceeds anterograde flow through the valve, closure occurs.
In cases in which shear stress is low or flow is turbulent, which occurs in the setting of valve cusps that do not meet, inflammatory mediators are released and free radicals generated. Although the mechanisms are not fully understood, these investigators have suggested that the inflammatory processes are instrumental in producing the changes in venous valves that result in destruction and incompetence.46–48
Human Studies
In humans, damaged valves have been observed in patients undergoing saphenectomy by preoperative duplex scanning and intraoperative angioscopy.50 Four types of valvular abnormalities have been described: type I (elongated and atrophic cusps), type II (expanded and depressed commissures with cusp changes), type III (cusps with other deformities), and type IV (absent valves). In a study of 153 limbs in 116 patients, Yamaki et al50 observed roughly 22% with type I, 37% with type II, 16% with type III, and 26% with type IV. Sales et al51 showed that patients undergoing saphenectomy for venous insufficiency had significantly fewer valves in their saphenous veins (mean, 2.3 ± 0.83) than did a control group in which a normal saphenous vein was used for infrainguinal revascularization (mean, 4.8 ± 2.0).
After an episode of superficial thrombophlebitis or DVT, valves are frequently destroyed and venous hypertension ensues. The clinical syndrome associated with these events is secondary venous disease, sometimes referred to as the postthrombotic syndrome.42 Bauer, a Swedish phlebologist, was the first to recognize that although recanalization occurs after an episode of DVT, the valves were destroyed or rendered incompetent.52
Valvular Incompetence and Secondary Chronic Venous Disease
Numerous studies have produced evidence showing the relationship between incompetent valves and the syndromes of secondary chronic venous disease. Although deep venous incompetence was originally thought to be the primary mechanism leading to the development of chronic disease, most investigators now believe that both superficial reflux and deep reflux contribute to these diseases.9,42,53–57 In addition, residual obstruction is often present.53,57,58 The contribution of valvular incompetence to the syndromes of chronic disease has been studied by two methods, measurement of the venous filling index (VFI) by air plethysmography and measurement of valve function by duplex scanning.
Studies Using Air Plethysmography.
Air plethysmography, introduced by Christopoulos and Nicolaides in the 1980s,59,60 is a noninvasive method that uses changes in leg volume as a means to quantify changes in venous volume. Changes in leg volume are measured by wrapping a plastic sleeve around the calf connected to a pressure transducer. The test is begun with the subject in the supine position and the leg elevated; changes in volume are then recorded during a series of movements in which the subject rises to a standing non–weight-bearing position, performs one tiptoe maneuver to eject blood from the calf followed by 10 tiptoe maneuvers, and then resumes the erect non–weight-bearing position to allow the veins to refill (Fig. 11-10). In regard to valvular incompetence, the most meaningful measurement is VFI, which is the rate of refilling of the limb veins in milliliters per second. It is an estimation of the degree of overall valvular incompetence in the limb and is calculated by dividing 90% of the increase in venous volume that occurs on standing by the time to reach 90% of venous volume. Christopoulos and Nicolaides reported values for VFI in normal subjects (<1.7 mL/sec) and those with superficial incompetence (2 to 30 mL/sec) and deep incompetence (7 to 28 mL/sec).59 Other investigators have shown that VFI values >7 mL/sec are associated with a high incidence of venous ulceration.61
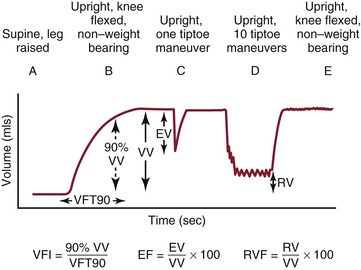
Figure 11-10 Limb volume changes recorded during the standard sequence of postural changes and tiptoe exercise performed with calibrated air plethysmography. A, After calibration, the patient lies supine with 45-degree passive elevation of the leg together with slight knee flexion and external rotation until maximum venous filling occurs. B, The patient stands up with the leg under examination flexed and non–weight bearing. C, The patient performs a single tiptoe exercise with both legs and returns to non–weight bearing on the examined leg. D, After reaching the second complete filling plateau, the patient performs 10 consecutive tiptoe exercises. E, The patient returns to the non–weight-bearing position. EF, Ejection fraction; EV, ejected volume; RV, residual volume; RVF, residual volume fraction; VFI, venous filling index; VFT, venous filling time; VV, functional venous volume. (Courtesy Cynthia Burnham, MD.)
Studies Using Duplex Ultrasound Scanning.
The location of incompetent valves has been evaluated by duplex scanning with measurement of valve closure times (VCTs), a technique first described by van Bemmelen et al.62 VCT, or the time to “abrupt cessation of reverse flow,” is measured by duplex scanning after rapid deflation of a distal cuff in the upright position. van Bemmelen et al62 reported that a VCT of >0.5 second constituted pathologic reflux. This was later modified by Labropoulos et al,63 who found that reflux was present in the superficial and deep calf veins when the VCT was greater than 0.5 second, but that reflux was present in the femoropopliteal veins when the value was greater than 1 second.63
Welch et al55 measured VCTs in 69 limbs of patients with chronic venous disease. They found that the incidence of deep venous reflux increased with the severity of disease and that it correlated with ulcer formation. Myers et al.54 showed that combined deep and superficial reflux was present in 34% of 776 limbs with lipodermatosclerosis and in 48% with previous ulceration.
Perforator Incompetence
The role of perforating vein incompetence in the development of secondary chronic venous disease is less well understood, although it has been suggested that perforating veins become major collaterals in the presence of deep venous obstruction or incompetence (or both).9 Perforator incompetence occurs both in the presence and in the absence of deep incompetence.64 It was originally suggested that perforator incompetence transmitted high venous pressure to the skin and contributed to dermal hypoxia and ulceration,65 but in a trial of perforator ligation performed by subfascial endoscopic perforator surgery, the rate of ulcer recurrence at 1 year was 30%.66 These results led investigators to conclude that perforator incompetence was less important in the development of chronic venous disease. Nevertheless, perforator incompetence occurs in up to 66% of limbs with deep reflux and secondary venous disease.54
Venous Hypertension
Venous hypertension is the underlying pathophysiology of venous disorders. This phenomenon was recognized almost 40 years ago in a study of elastic compression. Husni et al34 measured venous pressure at the foot level in normal volunteers and in patients with varicose veins, acute DVT, and chronic venous disease. They found that pressure in the supine position and during quiet standing was not significantly elevated in patients with various manifestations of DVT compared with controls, but that pressure increased dramatically during ambulation (see Fig. 11-9B). In patients with acute DVT or chronic disease, pressure was increased threefold (90 ± 18 mm Hg vs 35 ± 9 mm Hg) in comparison to controls. In the normal lower extremity, calf muscle contraction with ambulation ejects blood from the extremity, and when the muscles relax, valve closure prevents blood from refluxing distally. In the presence of incompetent valves, residual obstruction, or both, blood refluxes distally during muscle relaxation, and the hydrostatic column is rapidly re-established. Peripheral venous pressure rises rapidly between contractions, and hypertension is established.
Although the mechanism by which ambulatory venous hypertension leads to the skin changes associated with chronic venous disease is not fully understood, it was recognized long ago that ulcer formation and pressure are linearly related. Nicolaides and Zukowski67 reported ambulatory venous pressure in patients with varicose veins (25-70 mm Hg), deep valvular incompetence (55-85 mm Hg), and deep valvular incompetence combined with proximal occlusion (60-110 mm Hg) versus normal individuals (15-30 mm Hg). They also reported the incidence of venous ulceration in relation to pressure (Table 11-2). In limbs with ambulatory venous pressure greater than 80 mm Hg, the incidence of ulceration was approximately 80%, whereas in patients with pressure less than 40 mm Hg, ulcers rarely developed.68
Table 11-2
Relationship between Ambulatory Venous Pressure and the Incidence of Venous Ulceration
| Ambulatory Venous Pressure (mm Hg) | Incidence of Venous Ulceration (%) |
| ≤45 | 0 |
| 45-49 | 5 |
| 50-59 | 15 |
| 60-69 | 50 |
| 70-79 | 75 |
| ≥80 | 80 |
Data derived from Nicolaides AN: Noninvasive assessment of primary and secondary varicose veins. In Bernstein EF, editor: Noninvasive diagnostic techniques in vascular disease, ed 2, St Louis, MO, 1982, CV Mosby, pp 575-586.
Calf Muscle Pump Dysfunction
Dysfunction of the calf muscle pump, quantified as ejection fraction (EF) and residual volume fraction (RVF) by air plethysmography, is present in patients with chronic venous disease. Araki et al69 measured these parameters in 69 limbs of 55 patients with no ulceration, healed ulceration, or active ulceration. They found that EF (P = .0002) and RVF (P = .0006) were significantly reduced in patients with active ulceration in comparison to the other groups. In a second study, the same group reported that ankle range of motion was also significantly reduced in patients with active ulceration and correlated with reduced EF and RVF.70 In an earlier study, Christopoulos et al60 proposed that low EF was the primary cause of venous ulceration in limbs with minimal valvular incompetence. Padberg et al71 later performed a randomized trial of 6 months of exercise and found that improving calf muscle strength resulted in normalization of EF values in patients with chronic venous disease.
Clinical Syndromes
A complete discussion of the pathophysiology of clinical syndromes associated with abnormalities in venous physiology can be found in Chapters 49 to 65. What follows is a brief discussion.
Deep Venous Thrombosis
Acute DVT produces obstruction of the outflow of venous blood from the extremity. As the thrombosis becomes chronic, organization occurs with recanalization, valve incompetence, partial obstruction, and collateral formation. Ambulatory venous hypertension is the result.
An important clinical result of venous hypertension is the concomitant increase in mean capillary pressure, which leads to the formation of edema. According to the Starling equation72,73
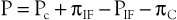
where P equals the net pressure driving fluid out of the capillary, Pc equals the capillary pressure, πIF equals the osmotic pressure of interstitial fluid, PIF equals the interstitial fluid pressure, and πC equals the osmotic pressure of blood.
Under normal conditions, fluid escaping from the arteriolar end of a capillary is returned to the circulation at the venular end, and any excess is removed by the lymphatics. The equation remains balanced. Although Pc rises considerably when a person stands, thus producing a larger pressure gradient across the capillary wall and promoting escape of fluid, even minimal amounts of calf muscle activity reduce the pressure quickly and decrease edema formation.
In the case of acute DVT, the Starling equation is upset by the increase in Pc, and edema develops. Edema formation appears to be proportional to the degree of pressure elevation. In an early study, edema was found in 70% of lower limbs with popliteal or calf vein thrombosis, 86% of limbs with femoral thrombosis, and 100% of limbs with iliofemoral thrombosis.74
Massive iliofemoral venous thrombosis can produce phlegmasia cerulea dolens, a condition in which enormous increases in venous pressure (reported as 16 to 17 times normal values within 6 hours) produce equally large increases in tissue pressure and edema formation.75 Elevated tissue pressure, in turn, can produce systemic circulatory collapse and gangrene of the affected extremity.
Chronic Venous Insufficiency
Postthrombotic Syndrome
The term postthrombotic syndrome refers to the symptoms of chronic lower extremity pain, edema, hyperpigmentation, and stasis ulceration occurring after an episode of lower extremity DVT.9,42 Postthrombotic syndrome occurs as a result of ambulatory venous hypertension, which, in turn, results from venous outflow obstruction, valvular incompetence, and calf muscle pump dysfunction. The results of a number of studies have demonstrated that the majority of deep vein thrombi lyse by 3 to 6 months after an acute episode of DVT.53,76–78 Killewich et al53 studied 21 patients with acute lower extremity DVT by duplex scanning and found that the thrombi were fully lysed in 53% of the study group by 90 days after initial evaluation. However, in 15% of cases, some residual obstruction was still present at 270 days. Similar findings were observed by van Ramshorst et al76 and more recently by Killewich et al77 and Meissner et al.78 Valvular incompetence is present in virtually all cases of postthrombotic syndrome. Incompetence has been identified in both deep and superficial veins, as well as in veins originally involved by the DVT and those that were not involved. Caps et al79 demonstrated that at 20 months after the original episode of DVT, 28% of major lower extremity venous segments contained incompetent valves. These investigators also found that incompetence was present in the great saphenous vein, a superficial vein, in 53% of cases of isolated DVT. Valvular incompetence has been shown as early as 7 days after the development of DVT80; in other studies, additional noninvolved venous segments were still developing reflux 7 years after the original DVT.81 Originally, it was believed that deep venous incompetence was the main contributor, but more recent data suggest that a significant number of patients experience both deep and superficial incompetence.
Although the relationship between ambulatory venous hypertension and the physiologic changes of outflow obstruction, valvular incompetence, and calf muscle pump dysfunction is reasonably well understood, how these phenomena lead to the clinical syndrome is still unclear. The preponderance of current evidence suggests that reduced skin oxygenation, originally thought to be the primary underlying mechanism, is not significant.37 Most investigators believe that white cell “trapping” occurs and produces neutrophil and monocyte activation with resultant endothelial injury. This causes lipodermatosclerosis and stimulates skin fibroblast growth, which produces fibrous tissue. Stimulation of skin capillary proliferation by vascular endothelial growth factor results in fragility and susceptibility of the skin to ulceration.82
Venous Claudication
Venous claudication is defined as “bursting” thigh pain and “tightness” that develops during exercise in patients with iliofemoral venous thrombosis.83 Peripheral arterial disease is not a factor. The pathophysiology of venous claudication is related to the high outflow resistance associated with venous collaterals. Venous volume increases, but proximal venous flow is unable to increase because of the fixed resistance of the collaterals, which results in the clinical syndrome.83 Delis et al84 observed a reduced EF (37%), high VFI (3.8 mL/sec), and increased RVF (45%) measured by air plethysmography in 39 patients with a history of iliofemoral venous thrombosis.
Superficial Venous Disease
Varicose Veins.
The etiology of varicose veins is not fully understood. It was originally believed that varicose veins develop as a result of valvular incompetence and venous hypertension, beginning at the saphenofemoral or saphenopopliteal junction and descending distally.85 More recent evidence suggests that varicose veins result from weakness of the vein wall.44 Using ultrasound and analysis of surgical specimens, valvular incompetence has been observed simultaneously in multiple discontinuous venous segments.86
The histologic changes that have been observed in varicose saphenous veins include reduced elastin content and degradation of the elastic lamella secondary to increases in MMP-9, as well as increased collagen with changes in the relative amounts of collagen types I and III. Endothelial dysfunction has been observed, in particular with a decrease in constrictor mediators such as noradrenaline, which may predispose the vein wall to dilatation. An upregulation in the nitric oxide/cyclic guanosine monophosphate system also contributes to potential vasodilatation.44
Factors that have been proposed to contribute to these observed ultrastructural alterations include hypoxia, changes in apoptosis, energy metabolism, and lysosomal activity.85
Pregnancy
The venous changes associated with pregnancy occur as a result of both hormonal and mechanical phenomena. By the third trimester, the enlarged uterus compresses the inferior vena cava and iliac veins,87,88 and thereby, decreases venous return to the heart, which, in turn, reduces cardiac output and produces hypotension.89 Pregnancy does not “cause” varicose veins, but the increased pressure and venous distention exaggerate predisposing factors.90 Population studies have found that only 12% of women with varicose veins have never been pregnant.91 Pregnancy-induced alterations in venous physiology and “secondary” varicose veins can become progressively worse during gestation, and in many cases, regress in the postpartum period. Hormonal factors are believed to play the most important role, because varicose veins will develop in as many as 80% of patients during the first trimester, often within 2 to 3 weeks of gestation, when the uterus is nominally enlarged. Progesterone is necessary for successful pregnancy because it inhibits smooth muscle contractility and provides stability for the pregnant uterus. A negative effect of increased progesterone levels is inhibition of smooth muscle contractility in the venous wall, which allows dilatation of subcutaneous veins. This is also seen in nonpregnant females on the first day of a menstrual period; the effects of progesterone-stimulated venous dilatation may cause the sudden formation of a cluster of varicosities or a telangiectatic blemish from a valve that is acutely incompetent.92
These hormonal influences, together with the generalized increase in total blood volume and progressive outflow obstruction in an individual who has a hereditary predilection, can result in a high prevalence of varicose vein formation in pregnancy.93,94 Graduated compression stockings can minimize dilatation of the superficial vessels and maintain valvular competence. The most ideal are 20 to 30 mm Hg graduated pantyhose.95
Selected Key References
Bergan JJ, Schmid-Schonbein GW, Coleridge-Smith PD, Nicolaides AN, Boisseau MR, Eklof B. Chronic venous disease. N Engl J Med. 2006;355:488–498.
Thorough review of current evidence about the development of postthrombotic syndrome in an animal model of venous hypertension.
Caps MT, Manzo RA, Bergelin RO, Meissner MH, Strandness DE Jr. Venous valvular reflux in veins not involved at the time of acute deep vein thrombosis. J Vasc Surg. 1995;22:524–531.
Classic paper addressing the development of valvular incompetence in patients with postthrombotic syndrome from the investigators at the University of Washington. This article also provides a good reference list for other publications from the same group.
Meissner MH, Eklof B, Smith PC, Dalsing MC, DePalma RG, Gloviczki P, Moneta G, Neglen P, O’Donnell T, Partsch H, Raju S. Secondary chronic venous disorders. J Vasc Surg. 2007;46:S68–S83.
Recent comprehensive review on secondary venous disorders.
Meissner MH, Gloviczki P, Bergan J, Kistner RL, Morrison N, Pannier F, Pappas PJ, Rabe E, Raju S, Villavicencio JL. Primary chronic venous disorders. J Vasc Surg. 2007;46:S54–S67.
Thorough, recently published review.
Meissner MH, Moneta G, Burnand K, Gloviczki P, Lohr MJ, Lurie F, Mattos MA, McLafferty RB, Mozes G, Rutherford RB, Padberg F, Sumner DS. The hemodynamics and diagnosis of venous disease. J Vasc Surg. 2007;46:4S–24S.
Another thorough, recently published review of the hemodynamics and diagnosis of venous disease.
Nicolaides AN, Hussein MK, Szendro G, Christopoulos D, Vasdekis S, Clarke H. The relationship of venous ulceration with ambulatory venous pressure measurements. J Vasc Surg. 1993;17:414–419.
Demonstrates the importance of ambulatory venous pressure in ulcer formation.
Strandness DE Jr, Sumner DS. Hemodynamics for surgeons. Grune & Stratton: New York; 1975.
Although this book is out of print, and in some places, out of date, it still remains the “bible” of both arterial and venous hemodynamics.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Caggiati A, et al. Nomenclature of the veins of the lower limb: an international interdisciplinary consensus statement. J Vasc Surg. 2002;36:416–422.
2. Caggiati A. Fascial relationships of the long saphenous vein. Circulation. 1999;100:2547–2549.
3. Caggiati A. Fascial relationships of the short saphenous vein. J Vasc Surg. 2001;34:241–246.
4. Caggiati A, et al. Valves in small veins and venules. Eur J Vasc Endovasc Surg. 2006;32:447–452.
5. Miles RN, et al. Some surgical implication of the anatomy of the cava-iliofemoral system. Ann Surg. 1973;177:740–747.
6. Akesson H, et al. Venous function assessed during a 5 year period after acute ilio-femoral venous thrombosis treated with anticoagulation. Eur J Vasc Surg. 1990;4:43–48.
7. Moore, et al. Number and location of venous valves within the popliteal and femoral veins-a review of the literature. J Anat. 2011;219:439–443.
8. Celepci, et al. Position of valves within the subclavian and axillary veins. J Vasc Surg. 2011;54:70S–76S.
9. Meissner MH, et al. The hemodynamics and diagnosis of venous disease. J Vasc Surg. 2007;46:4S–24S.
10. Alimi YS, et al. Venous pump of the calf: a study of venous and muscular pressures. J Vasc Surg. 1994;20:728–735.
11. Ludbrook J. The musculovenous pumps of the human lower limb. Am Heart J. 1966;71:635–641.
12. Pollack AA, et al. Venous pressure in the saphenous vein at the ankle in man during exercise and changes in posture. J Appl Physiol. 1949;1:649–662.
13. White JV, et al. Venous outflow of the leg: anatomy and physiologic mechanism of the plantar venous plexus. J Vasc Surg. 1996;24:819–824.
14. Burnand KG. The physiology and hemodynamics of chronic venous insufficiency of the lower limb. Gloviczki P, Yao JST. Handbook of venous disorders. Guidelines of the American Venous Forum. ed 2. Arnold: London; 2001:49–57.
15. Rothe CF. Physiology of venous return. Arch Intern Med. 1986;146:977–982.
16. Shepherd JT. Role of the veins in the circulation. Circulation. 1966;33:484–491.
17. Tyberg JV. How changes in venous capacitance modulate cardiac output. Pflugers Arch Eur J Physiol. 2002;445:10–17.
18. Rothe CF. Mean circulatory filling pressure: its meaning and measurement. J Appl Physiol. 1993;74:499–509.
19. Rothe CF. Reflex control of veins and vascular capacitance. Physiol Rev. 1983;63:1281–1342.
20. Moreno AH, et al. Mechanics of distension of dog veins and other thin-walled tubular structures. Circ Res. 1970;27:1069–1080.
21. Attinger EO. Wall properties of veins. IEEE Trans Biomed Eng. 1969;16:253–261.
22. Lye CR, et al. The transcutaneous measurement of the elastic properties of the human saphenous vein femoropopliteal bypass graft. Surg Gynecol Obstet. 1975;141:891–895.
23. Strandness DE Jr, et al. Hemodynamics for surgeons. Grune & Stratton: New York; 1975.
24. Brooksby GA, et al. Release of blood from the splanchnic circulation in dogs. Circ Res. 1972;31:105–118.
25. Sumner DS, et al. Vascular physiology: essential hemodynamic principles. Rutherford RB. Vascular surgery. ed 6. WB Saunders: Philadelphia; 2005:75–123.
26. Stegall HR. Muscle pumping in the dependent leg. Circ Res. 1966;19:180–190.
27. Arnoldi CC, et al. Venous blood pressures in the lower limb at rest and during exercise in patients with idiopathic dysfunction of the venous pump of the calf. Act Chir Scand. 1969;135:601–609.
28. Almen T, et al. Serial phlebography of the normal leg during muscular contraction and relaxation. Acta Radiol. 1962;57:264–272.
29. Hjelmstedt A. The pressure in the veins of the dorsum of the foot in quiet standing and during exercise in limbs without signs of venous disorder. Acta Chir Scand. 1968;134:235–244.
30. Hojensgard IC, et al. Static and dynamic pressures in the superficial and deep veins of the lower extremity in man. Acta Physiol Scand. 1952;27:49–67.
31. Criado E, et al. Physiological assessment of the venous system. Rutherford RB. Vascular surgery. ed 3. WB Saunders: Philadelphia; 1989:175–177.
32. Nicolaides AN, et al. The relationship of venous ulceration with ambulatory venous pressure measurements. J Vasc Surg. 1993;17:414–419.
33. Ellwood RA, et al. Pedal venous pressure: correlation with presence and site of deep-venous abnormalities. Radiology. 1979;131:73–74.
34. Husni EA, et al. Elastic support of the lower limbs in hospital patients—a critical study. JAMA. 1970;214:1456–1462.
35. Barnes RW, et al. Noninvasive quantitation of maximum venous outflow in acute thrombophlebitis. Surgery. 1972;72:971–979.
36. Cooke ED, et al. Thrombosis: preclinical diagnosis by deep vein thermography. Br J Surg. 1974;61:971–978.
37. Hallbook T, et al. Strain gauge plethysmography and phlebography in diagnosis of deep venous thrombosis. Acta Chir Scand. 1971;137:37–52.
38. Barnes RW, et al. Detection of deep vein thrombosis with an automatic electrically calibrated strain gauge plethysmograph. Surgery. 1977;82:219–223.
39. Zufferey P, et al. Assessment of acute and old deep venous thrombosis in upper extremity by venous strain gauge plethysmography. Vasa. 1992;21:263–267.
40. Raju S, et al. Venous valve station changes in “primary” and post-thrombotic reflux: an analysis of 149 cases. Ann Vasc Surg. 2000;14:193–199.
41. Labropoulos N, et al. Superficial venous insufficiency: correlation of anatomic extent of reflux with clinical symptoms and signs. J Vasc Surg. 1994;20:953–958.
42. Meissner MH, et al. Secondary chronic venous disorders. J Vasc Surg. 2007;46:S68–S83.
43. Clarke H, et al. Role of venous elasticity in the development of varicose veins. Br J Surg. 1989;76:577–580.
44. Somers P, et al. The histopathology of varicose vein disease. Angiology. 2006;57:546–555.
45. Raffetto J, et al. Mechanisms of varicose vein formation: valve dysfunction and wall dilation. Phlebology. 2008;23:85–98.
46. Takase S, et al. Hypertension-induced venous valve remodeling. J Vasc Surg. 2004;39:1329–1334.
47. Pascarella L, et al. An animal model of venous hypertension: the role of inflammation in venous valve failure. J Vasc Surg. 2005;41:303–311.
48. Bergan JJ, et al. Chronic venous disease. N Engl J Med. 2006;355:488–498.
49. Qui Y, et al. Fluid dynamics of venous valve closure. Ann Biomed Eng. 1995;23:750–759.
50. Yamaki T, et al. Preoperative duplex-derived parameters and angioscopic evidence of valvular incompetence associated with superficial venous insufficiency. J Endovasc Ther. 2002;9:229–233.
51. Sales CM, et al. The valvular apparatus in venous insufficiency: a problem of quantity? Ann Vasc Surg. 1998;12:153–155.
52. Bauer G. The etiology of leg ulcers and their treatment by resection of the popliteal vein. J Int Chir. 1948;8:937–967.
53. Killewich LA, et al. Spontaneous lysis of deep venous thrombi: rate and outcome. J Vasc Surg. 1989;9:89–97.
54. Myers KA, et al. Duplex ultrasonography scanning for chronic venous disease: patterns of venous reflux. J Vasc Surg. 1995;21:605–612.
55. Welch HJ, et al. Duplex assessment of venous reflux and chronic venous insufficiency: the significance of deep venous reflux. J Vasc Surg. 1996;24:755–762.
56. O’Shaughnessy A, et al. The patterns and distribution of residual abnormalities between the individual proximal venous segments after an acute deep vein thrombosis. J Vasc Surg. 2001;33:379–384.
58. Neglen P, et al. Venous outflow obstruction: an underestimated contributor to chronic venous disease. J Vasc Surg. 2003;38:879–885.
59. Christopoulos DG, et al. Air-plethysmography and the effect of elastic compression on venous hemodynamics of the leg. J Vasc Surg. 1987;5:148–159.
60. Christopoulos D, et al. Pathogenesis of venous ulceration in relation to the calf muscle pump function. Surgery. 1989;106:829–835.
61. Criado E, et al. The role of air-plethysmography in the diagnosis of chronic venous insufficiency. J Vasc Surg. 1998;27:660–670.
62. van Bemmelen PS, et al. The mechanism of venous valve closure: its relationship to the velocity of reverse flow. Arch Surg. 1990;125:617–619.
63. Labropoulos N, et al. Definition of venous reflux in lower extremity veins. J Vasc Surg. 2003;38:793–798.
64. Delis KT, et al. In situ hemodynamics of perforating veins in chronic venous insufficiency. J Vasc Surg. 2001;33:773–782.
65. Browse NL, et al. The cause of venous ulceration. Lancet. 1982;2:243–245.
66. Gloviczki P, et al. Mid-term results of endoscopic perforator vein interruption for chronic venous insufficiency: lessons learned from the North American subfascial endoscopic perforator surgery registry. J Vasc Surg. 1999;29:489–502.
67. Nicolaides AN, et al. The value of dynamic venous pressure measurements. World J Surg. 1986;10:919–924.
68. Nicolaides AN. Noninvasive assessment of primary and secondary varicose veins. Bernstein EF. Noninvasive diagnostic techniques in vascular disease. ed 2. CV Mosby: St Louis; 1982:575–586.
69. Araki CT, et al. The significance of calf muscle pump function in venous ulceration. J Vasc Surg. 1994;20:872–877.
70. Back TL, et al. Limited range of motion is a significant factor in venous ulceration. J Vasc Surg. 1995;22:519–523.
71. Padberg FT, et al. Structured exercise improves calf muscle pump function in chronic venous insufficiency: a randomized trial. J Vasc Surg. 2004;39:79–87.
72. Starling EH. On the absorption of fluids from the connective tissue spaces. J Physiol. 1896;19:312–326.
73. Taylor AE. Capillary fluid filtration. Starling forces and lymph flow. Circ Res. 1981;49:557–575.
74. DeWeese JA, et al. Phlebographic patterns of acute deep venous thrombosis of the leg. Surgery. 1963;53:99–108.
75. Snyder MA, et al. Hemodynamics of phlegmasia cerulea dolens. Surg Gynecol Obstet. 1967;125:342–346.
76. van Ramshorst B, et al. Thrombus regression in deep venous thrombosis: quantification of spontaneous thrombolysis with duplex scanning. Circulation. 1992;86:414–419.
77. Killewich LA, et al. Regression of proximal deep venous thrombosis is associated with fibrinolytic enhancement. J Vasc Surg. 1997;26:861–868.
78. Meissner JH, et al. Coagulation, fibrinolysis, and recanalization after acute deep venous thrombosis. J Vasc Surg. 2002;35:278–285.
79. Caps MT, et al. Venous valvular reflux in veins not involved at the time of acute deep vein thrombosis. J Vasc Surg. 1995;22:524–531.
80. Markel A, et al. Valvular reflux after deep vein thrombosis: incidence and time of occurrence. J Vasc Surg. 1992;15:377–382.
81. van Haarst EP, et al. The development of duplex scanning. Eur J Vasc Endovasc Surg. 1996;12:295–299.
82. Coleridge Smith P. The causes of skin damage and leg ulceration in chronic venous disease. Lower extremity Wounds. 2006;5:160–168.
83. Killewich LA, et al. Pathophysiology of venous claudication. J Vasc Surg. 1984;1:507–511.
84. Delis KT, et al. Venous claudication in iliofemoral thrombosis: long-term effects on venous hemodynamics, clinical status, and quality of life. Ann Surg. 2004;239:118–126.
85. Meissner MH, et al. Primary chronic venous disorders. J Vasc Surg. 2007;46:S54–S67.
86. Labropoulos N, et al. Where does venous reflux start? J Vasc Surg. 1997;26:736–742.
87. Kerr MG, et al. Studies of the inferior vena cava in late pregnancy. BMJ. 1964;1:532–533.
88. Sumner DS. Venous dynamics—varicosities. Clin Obstet Gynecol. 1981;24:743–760.
89. Ueland K. Pregnancy and cardiovascular disease. Med Clin North Am. 1977;61:17–41.
90. Barwin BN, et al. Venous distensibility during pregnancy determined by graded venous congestion. Am J Obstet Gynecol. 1976;125:921–923.
91. Henry M, et al. The incidence of varicose veins in. Ireland. Phlebology. 1989;4:133–136.
92. Bergan JJ. Causes of venous varicosities and telangiectasias: implications for treatment. J Vasc Biol Med. 1995;85:1101–1106.
93. Evans CJ, et al. Epidemiology of varicose veins: a review. Int Angiol. 1994;13:263–270.
94. Boivin P, et al. Pregnancy-induced changes in lower extremity superficial veins: an ultrasound scan study. J Vasc Surg. 2000;32:570–574.
95. Sustrell C, et al. The effects of long-term graduated compression treatment on venous function during pregnancy. Phlebology. 1995;10:165–168.