[level-membership-for-surgery-category]Chapter 7
Ischemia-Reperfusion
George P. Casale, Iraklis I. Pipinos
Based on a chapter in the seventh edition by Robert S. Crawford and Michael T. Watkins
The purpose of this chapter is to discuss the pathophysiology of severe acute ischemia, followed by acute reperfusion in the limbs and organs of the body. Ischemia-reperfusion (I/R) is a complex pathologic process involving intracellular and extracellular pathways that result in metabolic, thrombotic, and inflammatory changes in the affected tissues. A devastating component of I/R injury is the paradoxical increase in tissue damage associated with restitution of blood flow to ischemic tissues.
Haimovici1 described the development of myonephropathic syndrome in a few patients who underwent lower extremity revascularization after acute ischemia in the late 1950s. This report provided one of the first published clinical observations of limb I/R. These patients experienced ongoing lower extremity muscle necrosis and myoglobin-induced renal failure in the presence of palpable pulses. Cerra et al.2 completed an experimental animal and human clinical correlation of reperfusion in 1975. Histologic assessment of canine myocardium showed that restoration of blood flow was associated with the development of subendothelial hemorrhagic necrosis. Moreover, evaluation of the medical records and autopsy tissue from patients who died after aortic valve replacement provided evidence of subendothelial hemorrhagic necrosis in individuals subjected to more than 70 minutes of cardiopulmonary bypass. These findings implicated reperfusion injury as a cause of death in patients who underwent cardiopulmonary bypass for heart operations.
I/R injury has clinical relevance because it affects the outcome of patients after cardiac and vascular surgery, organ transplantation, plastic surgical reconstruction, and recovery from traumatic injury. The cellular damage that occurs as a consequence of I/R injury has been studied intensively in humans, experimental animals, and cell culture systems. To understand and characterize I/R injury from a biochemical perspective, it is useful to discuss this process in terms of its two components—ischemic injury and reperfusion injury.
Components of Ischemia-Reperfusion Injury
Injury During Ischemia
In the ischemic phase of I/R injury, the predominant mechanisms of injury result from tissue hypoxia or anoxia and stasis in the microcirculation (Fig. 7-1). The degree to which tissue from various organs is capable of tolerating ischemia varies widely and depends on each tissue’s baseline metabolic demand. Ischemia of human skeletal muscle under normothermic conditions is tolerated for more than 2 hours, whereas histologic evidence of ischemic injury develops in the jejunum after only 30 minutes of ischemia.3
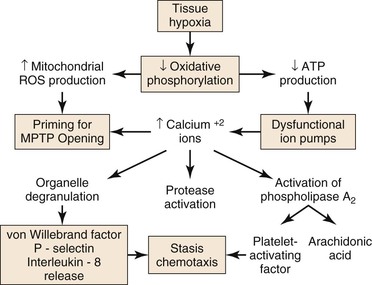
Figure 7-1 Injury during ischemia. Decreased oxygen supply activates a complex cascade of metabolic, inflammatory, and prothrombotic pathways and sets the conditions for MPTP opening during reperfusion. ATP, adenosine triphosphate; MPTP, mitochondrial membrane permeability transition pore; ROS, reactive oxygen species.
During ischemia, mitochondria deprived of O2 can no longer produce adenosine triphospate (ATP) by oxidative phosphorylation; consequently, cellular (ATP) falls rapidly with a concomitant increase in adenosine diphosphate (ADP), adenosine mononphosphate (AMP), and Pi. Glycolysis is stimulated, but is unable to produce sufficient ATP to meet the needs of the cell. The intracellular pH decreases with accumulation of lactic acid, progressively inhibiting glycolysis and activating the Na+/H+ antiporter that pumps H+ out of the cell. Because of ATP deficiency, the entering Na+ ions are not pumped out of the cell by Na+/K+ ATPase. High intracellular Na+ inhibits the Na+/Ca2+ antiporter, producing high intracellular Ca2+, which, in turn, activates degradative enzymes, including phospholipase A2, and proteases, such as calpains. In addition, ATP deficiency impairs ATP-dependent repair processes.4 Consequently, prolonged ischemia will lead to cell membrane damage and necrotic cell death.
As ischemia progresses, reactive oxygen species (ROS) are produced by and accumulate within the mitochondria.4,5 Increased mitochondrial matrix ROS together with increased cytosolic Ca2+, elevated Pi, and depletion of adenine nucleotides are most of the conditions needed for opening of the mitochondrial membrane permeability transition pore (MPTP), which causes necrotic cell death.6,7 The data indicate that the increase of mitochondrial ROS during ischemia primes the mitochondrial MPTP for opening, and that the magnitude of ROS damage to the mitochondria is the most important factor that determines the duration of opening of the MPTP, and therefore, the extent of tissue damage.4 Pore opening, however, is inhibited by the low intracellular pH during ischemia, but occurs during reperfusion when the pH increases, as is discussed in “Injury During Reperfusion” section in this chapter.
Tissue hypoxia leads to mobilization of neutrophils into the interstitium, where they have both beneficial and deleterious effects on tissue during reperfusion.8 Migration of neutrophils and macrophages to sites of inflammation is dependent on hypoxia-adaptive pathways.9,10 Activated neutrophils release the soluble mediators glutamate11 and adenine nucleotides (in the form of ATP or AMP)12,13 during ischemia that are converted to adenosine at the vascular endothelial surface. Adenosine protects the function of the microvascular endothelial barrier by re-establishing endothelial cell-cell contact after neutrophil transmigration. Transcellular metabolism (neutrophils provide ATP as a substrate for enzymes located on the endothelial membrane) and signaling are enhanced by hypoxia-induced transcriptional increases in functional endothelial surface apyrase (CD39), 5′-ectonucleotidase (CD73), and adenosine receptors (AdoRA2B).13,14 Polymorphonuclear neutrophils have a deleterious effect on local tissue by releasing factors that can disrupt the endothelial barrier. Activation of neutrophils by β2 integrins stimulates neutrophils to release soluble factors that induce endothelial cytoskeletal rearrangement, gap formation, and increased permeability. One neutrophil-derived permeabilizing factor is heparin-binding protein (HBP), also known as azurocidin or CAP37. HBP induces Ca2+-dependent cytoskeletal changes in endothelial cells and triggers macromolecular leakage in vivo.15
Injury During Reperfusion
Reperfusion injury represents the complex response to tissue injury when blood flow is restored after ischemia (Fig. 7-2). There are metabolic, thrombotic, and inflammatory components of reperfusion injury. The degree to which reperfusion either restores tissue integrity or exacerbates ischemic injury depends primarily on the duration of the ischemia. It is paradoxical that moderate ischemia followed by reperfusion may cause more fulminant postischemic tissue injury than seen with ischemia alone. However, without reperfusion, the loss of function in the brain, gut, heart, or limb may have a more catastrophic outcome than if perfusion is not restored.
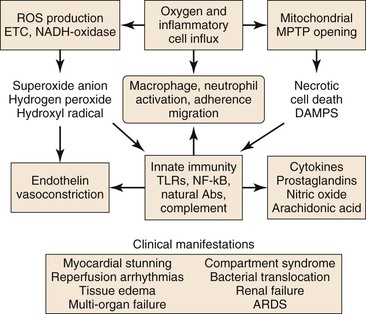
Figure 7-2 Injury during reperfusion. The return of oxygen, influx of inflammatory cells, and washout of metabolites contribute to cell death and an inflammatory, prothrombotic milieu that exacerbates tissue injury. Abs, Antibodies; ARDS, acute respiratory distress syndrome; DAMPS, danger-associated molecular patterns; METC, mitochondrial electron transport chain; MPTP, mitochondrial membrane permeability transition pore; NADH, reduced nicotinamide adenine dinucleotide; NF-κB, nuclear factor kappa B; ROS, reactive oxygen species; TLRs, toll-like receptors.
Reperfusion is characterized by a burst of ROS production (primarily by the mitochondrial electron transport chain), increased Ca2+ uptake into the mitochondrial matrix, and a shift of the intracellular pH from acidic toward neutral.6 Mitochondrial oxidative damage leading to programmed cell death is the major cause of tissue loss produced by reperfusion.16 The two forms of programmed cell death during reperfusion are apoptosis and necrosis.
Increased ROS generated during both ischemia and reperfusion can initiate apoptosis and necrosis, cell death programs mediated through the mitochondria. The BCL-2 family of proteins is central to regulating mitochondrial apoptosis. Antiapoptotic members, including BCL-2 and BCL-XL, act by blocking proapoptotic members, including BAX and BAK. The latter initiate apoptosis by increasing the permeability of the outer mitochondrial membrane, causing the release of proapoptotic proteins, such as cytochrome c.17 BH3-only proteins, proapoptotic BCL-2 family members, can initiate apoptosis by directly activating BAX and BAK or by blocking BCL-2 and BCL-XL.17 ROS initiate mitochondrial apoptosis by activating BH3-only proteins or by triggering opening of the mitochondrial MPTP, causing release of cytochrome c.7 Alternatively, apoptosis may occur via ROS-dependent lipid peroxidation of cardiolipin, a mitochondria-specific phospholipid of the inner membrane, which binds strongly to cytochrome c. The latter dissociates from peroxidized cardiolipin and is released from the inner membrane.18 The evidence, however, suggests that ROS-dependent necrosis, rather than apoptosis, is the main mechanism of cell death caused by I/R injury.6,7,19
Programmed necrotic cell death may be mediated by signaling via a tumor necrosis factor (TNF) death receptor20–22 or by direct opening of the mitochondrial MPTP.16,17 Both processes require calcium and oxidative stress.16,17 TNF receptor 1 (TNFR1) signaling for necrotic cell death begins with a conformational change caused by binding of TNF to the receptor. This conformational change promotes binding of cytosolic TNF receptor-associated death domain (TRADD) to a cytosolic domain of the receptor, initiating formation of a complex consisting of receptor-interacting protein 1 (RIP1) kinase, TNF receptor-associated factor 2/5 (TRAF2/5), cellular inhibitor of apoptosis 1 (cIAP1) and cellular inhibitor of apoptosis 2 (cIAP2). The TNFR1 complex is internalized and produces secondary cytosolic complexes, including the necrosome. The latter comprises Fas-associated death domain (FADD) protein, inactivated caspase 8, RIP1, and receptor-interacting protein 3 (RIP3). Activation of necrotic signaling requires phosphorylation of RIP1 and RIP3 and is critically dependent on ROS production by the mitochondrial electron transport system.21 The ultimate effector in this pathway is not yet established; however, the present data suggest that signaling leads to opening of the mitochondrial MPTP.20
Direct opening of the mitochondrial MPTP is central to tissue injury caused by ischemia and reperfusion.16,17,19 The mitochondrial MPTP is a nonselective protein channel spanning the inner and outer mitochondrial membranes at points of contact. When open, the pore permits free passage of water and solutes less than 1.5 kDa, causing osmotic damage to the mitochondria and uncoupling mitochondrial oxidative phosphorylation, the major source of cellular ATP, by dissipation of the pH gradient and the electrical potential across the inner mitochondrial membrane.4,6 Mitochondrial damage and collapse of oxidative phosphorylation promote disruption of metabolism and ionic homeostasis initiated before pore opening during ischemia, and thereby, promote and expand necrotic cell death. The composition of mitochondrial MPTP is not yet solved, but the voltage-dependent anion channel (VDAC), adenine nucleotide translocase (ANT), and mitochondrial phosphate carrier (PiC) appear to be constituents.6 Cyclophilin D (CyP-D), a prolyl isomerase in the mitochondrial matrix, is an essential component of the mitochondrial MPTP and the central regulator of pore opening.16 Pore opening depends on mitochondrial calcium concentration (Ca2+ m) but is relatively insensitive to Ca2+ m per se, occurring at very high Ca2+ m. The sensitivity of pore opening to Ca2+ m is greatly enhanced by oxidative stress caused by increased mitochondrial ROS production during ischemia and a surge of mitochondrial ROS production during reperfusion.6 Oxidative stress promotes binding of CyP-D to an ANT/PiC dimer, facilitating a Ca2+-dependent conformational change in these proteins and opening of the mitochondrial MPTP. The evidence supports activation of CyP-D via binding of p53, which accumulates in the mitochondrial matrix in response to oxidative stress.16 Low intracellular pH that occurs during ischemia inhibits pore opening; however, during reperfusion, when blood flow is restored, the intracellular pH increases, and the mitochondrial MPTP opens.6
Reperfusion of ischemic tissue produces a burst of ROS production by the mitochondrial ETC and by parenchymal and endothelial cell reduced nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase and xanthine oxidase, neutrophils and macrophages, and necrotic cell death with opening of the MPTP.23 These processes lead to release of normally sequestered cellular proteins and protein fragments in addition to extracellular and cell surface accumulation of oxidatively modified proteins and lipids. Some of these structures have molecular configurations recognized by Toll-like receptors (TLR) of the innate immune system as “danger-associated molecular patterns” (DAMPS),24 and a growing body of work supports TLR-mediated inflammation as central to I/R injury.25 Reperfusion-associated DAMPS include heat shock protein 60 (HSP60), a ligand for TLR4; high-mobility group protein B1 (HMGB1), a ligand for TLR2, 4, and 9; low-molecular-weight hyaluronic acid, a ligand for TLR2 and 4; the extra domain A of fibronectin (EDA), a ligand for TLR2 and TLR4; and cardiac myosin, a ligand for TLR2 and 8.26 Ligand binding to TLRs present on parenchymal cells, endothelial cells, and circulating leukocytes activates TLR signaling pathways that lead to apoptosis or to nuclear factor kappa B (NF-κB) signaling.26 Inflammation induced by TLR-mediated NF-κB signaling is a significant contributor to I/R injury.25 NF-κB is a transcription factor present in the cytosol in an inactive complex with inhibitor of NF-κB (I-κB). Signaling via TLRs leads to removal and proteolysis of I-κB, permitting NF-κB to translocate to the nucleus, where it initiates transcription of proinflammatory genes, including those that code for TNF-α, interleukin-1β (IL-1β), and IL-6, as well as intercellular adhesion molecule-1 (ICAM-1), all of which recruit neutrophils and macrophages to the damaged tissue.23,25,27 In addition, NF-κB signaling may be initiated by binding of TNF to the TNFR1 receptor.21
Activation of innate immunity in response to DAMPS, during reperfusion, is not limited to their recognition by TLRs, but also occurs in response to their recognition by natural antibodies.27 These antibodies belong predominantly to the immunoglobulin-M (IgM) class, are secreted by B-1 cells (a specialized subset of B-cells) and are germline encoded.24 Many of these antibodies recognize epitopes formed by oxidative modification of lipids and proteins (e.g., during reperfusion). Some recognize normally sequestered self-antigen, for example, non-muscle myosin heavy chain II released as a consequence of cell injury during both ischemia and reperfusion.28 Reperfusion elicits an inflammatory response dependent on activation of complement by natural IgM antibodies.28 Binding of IgM natural antibodies to target molecules on the cell surface or in the extracellular environment activates the complement system, causing production of cleavage products, including complement component C3b, which tags cells and other targets for removal by macrophages, and C5a, a fragment chemotactic for neutrophils.27 Alternatively, the complement system may be activated in the absence of antibody by mannose-binding lectin (MBL) bound to cell surface carbohydrates.29 Complement activation, detected as tissue-bound C3b and MBL, occurs during ischemia and increases during reperfusion.30
A variety of inflammatory mediators are delivered to reperfused tissue by blood-borne cells (macrophages, lymphocytes, neutrophils, mast cells, platelets).31,32 Noncellular elements, such as the complement system, ROS, nitric oxide (NO), and proinflammatory and anti-inflammatory cytokines are also believed to modulate the complex scenario of reperfusion injury. The presence of O2, washout of lactatic acid, development of tissue edema, and influx of numerous cell types during reperfusion produce a complex environment in which numerous mediators contribute to inflammation and tissue injury. The cellular and molecular components of the I/R response will be discussed individually.
Leukocytes
In contrast to neutrophil activation during ischemia (discussed previously), neutrophil activation during reperfusion is characterized by a logarithmic increase in neutrophil chemotaxis, neutrophil–endothelial cell adhesion, and transmigration. In contrast to the slow transendothelial migration of resident neutrophils in the static ischemic environment, during reperfusion, flow modulates the interaction between neutrophils and endothelium. Transendothelial migration during reperfusion occurs in sequential steps: rolling, adherence, and transendothelial migration.9 The first step is initiated by increases in the surface expression of endothelial P-selectin (CD62P); CD62P interacts with its leukocyte ligand P-selectin glycoprotein ligand 1 (PSGL-1), which results in leukocyte rolling. Firm neutrophil adherence is modulated by interaction of the β2 integrins CD11a/CD18 and CD11b/CD18 with endothelial ICAM-1. Transmigration of leukocytes is facilitated by platelet–endothelial cell adhesion molecule-1 (PECAM-1), which is constitutively expressed along endothelial cell junctions. Once inside the interstitium, activated leukocytes release toxic ROS, proteases, and elastases.31 These substances contribute to increased microvascular permeability, edema, thrombosis, and parenchymal cell death.32,33 Neutrophil accumulation in the extravascular compartment is facilitated by chemotactic agents, including complement components C3a and C5a, chemokines such as IL-8, DAMPs, and leukotriene B4 produced by arachidonic acid metabolism.34 The release of chemotactic agents creates a gradient that attracts neutrophils from the intravascular space toward the interstitium.34–36
Cytokines-Chemokines/Antagonists
Multiple growth factors and cytokines are present at sites of inflammation, and each can influence the nature of the inflammatory response.37 There is a constant interaction between proinflammatory and anti-inflammatory signals in the vascular wall (Box 7-1).
Liver and skeletal muscle I/R have been correlated with increased levels of local and systemic cytokines that are believed to modulate local38–40 and systemic injury.41,42 TNF41,43 and macrophage inflammatory protein-2 (MIP-2) levels are markedly increased in hind limbs exposed to acute limb I/R.43,44 Direct cytokine antagonists (receptor antagonists and anticytokine antibodies),45 metabolic rescue agents,43,46 and anti-inflammatory molecules47–51 have been shown to ameliorate skeletal muscle injury and decrease tissue levels of proinflammatory cytokines when administered to animals during limb I/R. Recent literature suggests that administration of an IL-1 receptor antagonist attenuates spinal cord I/R injury by reducing neuronal necrosis and apoptosis.52 Administration of this same IL-1 receptor antagonist has also been shown to ameliorate renal I/R injury in animal experiments.53 Because this receptor antagonist is clinically used to treat rheumatoid arthritis, it may soon be used in clinical trials of I/R injury in humans. A word of caution regarding inhibition of cytokines or their receptors comes from the work of Shireman et al,54 who showed that chronic deficiency of monocyte chemoattractant protein-1 resulted in a decrease in the inflammatory response, as expected, but also inhibited skeletal muscle regeneration after chronic limb ischemia.
Mitochondrial Injury
The mitochondria are a dynamically growing and shrinking network of double-membrane organelles that comprise the primary energy-generating system of eukaryotic cells. Energy is generated by coupling ATP production with oxidative transfer of electrons from reduced substrates to molecular O2 (i.e., oxidative phosphorylation).55 This process of energy conversion is mediated by the mitochondrial electron transport chain that consists of five multiunit protein complexes tightly configured within the mitochondrial inner membrane.56 The largest of these is Complex I, which consists of 46 protein subunits and 9 redox centers, 1 of which is flavin mononucleotide (FMN) located at the terminus of the hydrophilic arm extending into the mitochondrial matrix. High-energy electrons from reduced nicotinamide adenine dinucleotide (NADH) enter the ETC by reducing the FMN of Complex I. The electrons pass through Complex I to its coenzyme Q (CoQ) reductase site located in the inner membrane and where CoQ is reduced. The latter shuttles the electrons to Complex III, which couples oxidation of reduced CoQ with reduction of (passage of the electrons to) cytochrome c located in the intermembrane space. Finally, the electrons are passed to Complex IV, which couples oxidation of cytochrome c with reduction of molecular O2 to water. Complex II can also shuttle electrons to CoQ by coupling conversion of succinate to fumarate with reduction of flavin adenine dinucleotide (FAD), which is present at the mitochondrial matrix side of the complex. Importantly, passage of two electrons through Complexes I, III, and IV is coupled with transfer of protons from the mitochondrial matrix across the inner membrane to the intermembrane space (i.e., a total of eight protons per electron pair).56 This process generates a pH gradient and a membrane potential used by Complex V to generate ATP from ADP and Pi.
Approximately 90% of inhaled O2 is consumed by mitochondria and reduced to water to produce energy. Under optimal physiologic conditions, 2% to 4% of the O2 consumed by mitochondria can capture single electrons that leak along the complexes of the electron transport chain generating the ROS superoxide anion (O•−).57 Oxidative stress caused by excess accumulation of ROS is held in check by mitochondrial superoxide dismutase, which converts it to H2O2 (another ROS) and catalase, which converts the latter to H2O and O2.57 During tissue ischemia, when O2 tension in the mitochondria is reduced (hypoxia), ROS generation paradoxically is increased, overwhelming oxidative defense enzymes and causing an excessive accumulation of ROS (oxidative stress).5 This happens because under tissue hypoxia with reduced availability of O2 as the final electron acceptor of the ETC, the NADH/NAD+ ratio in the mitochondrial matrix increases, thereby saturating the FMN of Complex I with electrons. Saturated FMN transfers electrons to available O2 in the matrix, producing O•−.57 This FMN-dependent process is amplified by (1) the accumulation of reduced CoQ generated by both Complex I and II under the condition of low electron flow, and (2) the presence of a pH gradient across the inner membrane. Under these conditions, present during ischemia, electrons back up through Complex I to its FMN, where electrons are passed to O2, generating O•−.57 With the onset of reperfusion, O2 returns to meet the conditions of increased NADH/NAD+, accumulated reduced CoQ, electron-saturated FMN, and an incapacitated electron transport chain. The surge of O2 thus provides a sink for flow of electrons from reduced FMN leading to a burst of O•−. The duration of this burst, and therefore, the extent of acute reperfusion damage are dependent on the duration and extent of ROS accumulation in the preceding ischemic phase of I/R. The evidence supports Complex I as the main source of this reperfusion burst of ROS generation.4
Complement
The complement system has long been recognized as an important mediator in innate immune defense and inflammation.58 Complement can be activated by any of three pathways—the antibody-dependent classical pathway, the alternative pathway, or the MBL-associated serine protease pathway. The classical pathway is initiated when IgM or IgG antigen-antibody complexes bind to C1, the first component of complement. Activation of the alternative pathway is triggered by microbial surfaces and complex polysaccharides, and results in the generation of C3. The lectin pathway is initiated by binding of the MBL protein to mannose and glucosamine residues on bacterial cell walls. Activation of these proteins results in cleavage of complement factors 4 and 2, followed by subsequent activation of C3.
There is considerable evidence to support a role of the complement system in hind limb I/R injury. In a model of rodent hind limb I/R, there was direct correlation between increased complement deposition, tissue injury, and increased glycolysis.59 Complement receptor 2–deficient (CR2−/−) mice exhibit deficient IgM secretion and are protected against I/R injury by defective B cells.60 IgM-secreting B-cells clones were studied from CR2 knockout mice and wild-type mice. Only one clone was found to restore injury to previously protected mice, thus suggesting that a single clone of self-reactive IgM can initiate complement-dependent I/R injury.61 In a follow-up study, peptides that bound the IgM clone were found to block injury in wild-type mice, a novel observation providing a potential therapeutic venue.62 Complement inhibition has been demonstrated to improve skeletal muscle contractility after I/R.63 C5-deficient mice have been shown to exhibit decreased lung vascular permeability, lung myeloperoxidase levels, and serum alanine aminotransferase levels, along with less skeletal muscle injury after reperfusion.64 Because the complement system is well described, and numerous inhibitors are available for each pathway, a beneficial role for these inhibitors in the brain, intestine, liver, and myocardium has been described.58
Nitric Oxide
NO is known to regulate vascular tone and to have potent anti-inflammatory and antithrombotic properties at the endothelial surface. It is also known to have concentration-dependent toxic effects on tissue and organ systems.65,66 The primary physiologic effect of NO results from stimulation of the activity of guanylate cyclase, accumulation of intracellular cyclic guanosine monophosphate, and a consequent decrease in intracellular calcium that results in relaxation of smooth muscle, decreased cardiac contractility, and reduced platelet and inflammatory cell activation. NO is produced from L-arginine by oxidation of one or two equivalent guanido nitrogens by NO synthase (NOS) in a reaction that requires O2, and the cofactors NADPH and tetrahydrobiopterin. There are at least three distinct genes for NOS; neuronal NOS (nNOS or NOS-1) and endothelial NOS (eNOS or NOS-3) are dependent on micromolar concentrations of calcium for activity and are constitutively expressed. The inducible isoform, iNOS (or NOS-2), is calcium independent and widely expressed in a variety of cells after exposure to specific stimuli. iNOS is often referred to as the high-output source of NO because it can be strongly induced by proinflammatory stimuli. However, the enzyme does not produce NO at a rate substantially greater than does eNOS or nNOS; rather, more of the protein can be transiently induced and activated at normal levels of calcium.67,68
One of the first observations implicating NO as a marker of ischemic injury was made by Beckman.65 His laboratory went on to propose that the production of NO and O2 radicals by neurons when the ischemic or hypoxic brain is reperfused might contribute to cerebral injury. Ischemia is believed to depolarize neuronal membranes and cause synaptic discharge of the excitatory neurotransmitter glutamate, which, in turn, opens the voltage-dependent, N-methyl-d-aspartate–specific glutamate receptor/ionophore and allows calcium to accumulate in the neuron. Calcium then activates an O2-dependent nNOS that oxidizes arginine to produce NO when O2 is readmitted to the brain by reperfusion. NO reacts with the O2 radical superoxide (O•−), also produced by reperfusion, to form peroxynitrite (ONOO−). ONOO− has been shown to interact directly with proteins (i.e., the mitochondrial electron transport chain, prostaglandin synthase) and amino acids (oxidizes critical cysteine residues, leading to deactivation of proteins), and triggers membrane lipid peroxidation, thereby altering membrane permeability.69 ONOO− is a powerful oxidant capable of nitrating phenolic moieties, such as tyrosine or tyrosine residues in proteins, and increases after traumatic brain injury.70 This powerful molecule also activates mitochondrial-dependent apoptosis, poly (ADP-ribose) polymerase (PARP),69 and NF-κB.71 Alternatively, some in vitro studies have shown that ONOO− may inhibit the generation of NF-κB.72,73 ONOO− is believed to diffuse for several micrometers before decomposing to form the powerful and cytotoxic oxidants hydroxyl radical and nitrogen dioxide. Beckman’s hypothesis is consistent with available evidence on the protective action of glutamate antagonists and O2 radical scavengers in limiting cerebral infarction after focal ischemia.66 Furthermore, ONOO− decomposition catalysts are known to limit reperfusion injury in a variety of organ systems.74
There are also anti-inflammatory effects of NO that deserve mention. NO donors have been shown to ameliorate tissue injury in in vivo and ex vivo models of intestinal, neuronal, and hepatic I/R.75–77 NO is known to inhibit activation of NF-κB,78,79 which regulates a wide variety of proinflammatory cytokines and adhesion molecules. NO alone is an important antiatherosclerotic autacoid with antiaggregatory effects on platelets and with antioxidant, anti-inflammatory, and antiproliferative effects on the vasculature.80 NO inhibits vascular permeability in the postischemic intestinal microcirculation, probably by preventing endothelial cell contraction and gap formation.81 Endogenous NO also inhibits neutrophil adhesion to endothelium.82 Finally, NO regulates the activation of platelets by decreasing aggregation and adhesion responses.83 The duration of ischemia, magnitude of the injury, and specific organ bed will determine whether NO has a protective or deleterious effect. In contrast to the deleterious effects of NO when synthesized in large amounts by inflammatory cells via iNOS, the protective effects of NO appear to be related primarily to endogenous sources of constitutively synthesized NO generated by eNOS or nNOS or exogenous pharmacologic doses of NO donors.84
Endothelin
Endothelin (ET), a 21-amino acid peptide with potent vasoconstrictor properties,85 is produced mainly in endothelial cells, yet it is present throughout the entire cardiovascular system. Shear stress, growth factors, vasoactive hormones, and I/R are major stimuli for its release. Three distinct human ET-related genes have been cloned: “classic” ET-1, ET-2, and ET-3.86 ET-1 is the most predominant isoform in blood vessels and is synthesized by endothelial cells. ET-1 is produced as a precursor peptide that is cleaved by a converting enzyme into its active form. ET plays a major role in the regulation of vascular tone through its potent vasoconstrictive actions. ET acts on two different types of G-protein–coupled receptors: ET-A and ET-B. ET-1, the most potent vasoconstrictor, mediates its effects mainly through ET-A receptors present on vascular smooth muscle cells.87 ET-B receptors mediate both vasodilatory effects of ET through prostacyclin and NO (ET-B1) and vasoconstrictive effects on vascular smooth muscle (ET-B2).88,89
Increased production, upregulation of its receptors, enhanced sensitivity, and potentiation of the action of serotonin and norepinephrine all potentiate ET’s actions during I/R.90,91 Bosentan, a mixed ET-A/ET-B endothelin receptor antagonist, was shown to exert a marked tissue protective effect, as assessed by histologic evaluation in a rat model of myocardial I/R. This effect was probably the result of antioxidant protection derived from preservation of levels of the antioxidant enzymes superoxide dismutase and catalase in the treated animals.92 McMurdo et al93 showed that the vasoconstrictive effects of ET-1 in the coronary vasculature of the rabbit were primarily mediated by the ET-A receptor. However, FR139317, an ET-A receptor antagonist, failed to decrease infarct size after the induction of ischemia and reperfusion in that same model.93 A more contemporary assessment of the action of BQ-123, a selective ET-A receptor antagonist, revealed decreased myocardial levels of lipid peroxidation; an increase in NO, glutathione, catalase, and superoxide dismutase activity; and a decrease in the ratio of infarcted area to area at risk compared with I/R alone.94 Further evidence has proved that ET-A antagonism is useful in I/R. The ET-A antagonists ABT-627, S-0139, BSF208075, and LU135252 all show significant tissue protection in models of I/R.95–98
Adenosine
Adenosine is an endogenous nucleoside produced in part by degradation of ATP. Formation of adenosine follows the metabolism of ATP into its by-products (ADP, AMP, and cyclic AMP [cAMP]). These by-products increase during ischemia partly as a result of the inability of mitochondria to rephosphorylate ATP.99 Depletion of the adenosine pool occurs during reperfusion because of washout and conversion of adenosine first to inosine and then to xanthine through the action of xanthine oxidase.100 Adenosine acts as a hormone and mediates its effects through the actions of four G-protein–coupled receptors (A1, A2A, A2B, A3). These receptors are expressed widely and differentially in most cells and vascular beds of the body, including cells involved in the immune response. Specific agonists and antagonists and knockout mice have elucidated the role of each receptor in the cardiovascular system.101 The majority of data on the beneficial effects of adenosine treatment of I/R have involved the myocardium.
Adenosine has a multitude of physiologic effects, which makes it an ideal treatment of I/R. Adenosine promotes preservation of vascular flow both through endogenous vasodilator properties and by opposing the action of vasoconstrictor substances.102,103 It can inhibit multiple cell types involved in immunity, including neutrophils,101 and can enhance recovery of the depleted intracellular nucleotide pool.104 Finally, adenosine can reduce the production of O2-derived free radicals and reduce intracellular calcium through the opening of receptor-activated KATP channels.105,106 Multiple animal experiments and human clinical trials have established adenosine as a viable therapy for myocardial I/R. We refer the reader to two excellent reviews on this subject100,107 and to a discussion of the two most important human studies documenting the effectiveness of adenosine in reducing infarct size in patients presenting with anterior wall myocardial infarction (MI)108,109 presented in the Clinical Trials section of this chapter.
The protective effects of adenosine have been demonstrated in other organ systems. In a mouse model of skeletal muscle I/R, the role of adenosine receptors was elucidated by using specific agonists. The results showed that the A3 receptor could induce potent cytoprotection of skeletal muscle through phospholipase C receptor signaling.110 A recent study on the role of ATL-146e, an adenosine A2 receptor agonist, showed improved hind limb function at 48 hours after reperfusion in a rabbit model of spinal I/R injury.111 Activation of the adenosine A2A receptor with the selective agonist DWH-146e also resulted in protection in a model of renal I/R injury, most likely by reducing neutrophil infiltration and suppressing the molecules involved in neutrophil rolling and adhesion (P-selectin and ICAM-1).112 Finally, a large amount of data is also available to show the beneficial effects of adenosine in models of hepatic I/R.113
Adrenomedullin
Adrenomedullin (AM) is a peptide with powerful vasodilatory properties that was originally isolated from human pheochromocytoma.114 AM binds to a specific AM-binding protein-1 (AMBP-1) receptor in human plasma, which has been identified as complement factor H.115 Besides its vasodilatory effects, mediated by increasing both cAMP and endothelium-derived NO, AM inhibits the alternative complement pathway through AMBP-1 binding and can modulate neutrophil and monocyte function.116–118 During gut I/R, AM levels increase significantly after reperfusion, but not with ischemia alone, whereas levels of AMBP-1 decrease with ischemia and remain depressed during reperfusion. Administration of AM/AMBP-1 after I/R dramatically downregulates inflammatory cytokines (TNF-α, IL-1β, IL-6), attenuates tissue injury (as manifested by decreased levels of alanine and aspartate aminotransferase and lactate), and improves survival.119 Studies in AM knockout mice have delineated the protective effects of AM in renal I/R. Taken together, results from these studies show that eNOS, which is stimulated by AM, may play a protective role, but the role of iNOS remains to be delineated in renal I/R.120 Similar findings (attenuation of inflammatory cytokines and improved NO) have also been found to be responsible for attenuating injury in a hepatic model of I/R.121
Heme Oxygenase System and Carbon Monoxide
In humans, heme oxygenase (HO) opens up the heme ring and releases equimolar quantities of three biologically active molecules: biliverdin IXα, reduced iron, and carbon monoxide (CO).122 Three different isoforms of HO catalyze the degradation of heme. HO-1 is also known as HSP32. HO-1 is the only isoform that is inducible. Maines et al123 first described induction of HO-1 during ischemia and reperfusion in a model of kidney I/R, the activity of which goes from half that of HO-2 at baseline to 10-fold more than its counterpart after induction. HO-1 expression is increased in various models of hypoxia and exogenous stress mediated by a variety of oxidant-sensitive regulatory molecules.124
The protective actions of HO-1 induction are manifold. O2-derived free radical damage is known to be a major component of the pathophysiology of I/R injury. Heme production has been shown to accumulate in excess of its needs on resumption of the circulation in I/R.125 First, the heme molecule is an effective catalyst for the production of powerful oxidants (O•−), and H2O2).126 Induction of HO-1 decreases the levels of heme itself, hence protecting against free radical damage–mediated cellular injury and the induction of further inflammatory signals. Second, metabolism of the heme molecule generates biliverdin, and subsequently, bilirubin, both of which have been shown to exhibit strong antioxidant activity.127 The third product of heme metabolism is CO, which provides protection against I/R at many cellular levels. Like NO, CO has been recognized as a vasodilator substance through its activation of soluble guanylate cyclase.128 In a model of lung ischemia and reperfusion injury, CO has been shown to inhibit apoptosis and protect cultured cells through a p38 mitogen-activated protein kinase (MAPK) and caspase-3–mediated mechanism.129 In a model of transplant-induced I/R injury, CO provided anti-inflammatory protection by suppressing the early expression of TNF-α and IL-6. Possible mechanisms for this protection include downregulation of the MEK/ERK1/2 signaling pathway.130 Furthermore, working with a mouse hind limb I/R injury model, Patel et al have demonstrated that inhaled CO can protect skeletal muscle from structural injury and energy depletion.131 Finally, through its regulatory role on platelet–vessel wall interactions, CO has also been shown to decrease platelet aggregation.132 Although the idea of using CO as a therapeutic agent might be complicated by its well-known potential toxic effects, the ability to reliably deliver it and also to measure its levels in humans makes it an attractive agent for the treatment of I/R-mediated injury.
Anticoagulants and Ischemia-Reperfusion
Following restitution of vascular flow by either open surgical or endovascular means, anticoagulants have been the mainstay of clinical therapy after acute vascular occlusion. In clinical practice, unfractionated heparin, and recently, low-molecular-weight heparin are the agents most widely used to prevent extension of arterial thrombus, and hence, improve vascular flow in the affected organ. Experimental studies in animal models have indicated that unfractionated heparin may modulate endothelial cell permeability and pH.133 Recent experimental studies of the effect of enoxaparin administered soon after the onset of hind limb ischemia revealed a significant decrease in markers of local thrombosis but no significant salvage of skeletal muscle.134 In contrast to the findings noted in ischemic skeletal muscle, some data suggest that enoxaparin administered 30 hours after the onset of traumatic brain injury in a rodent model decreases tissue edema and infarct size.135 This finding may indicate that ischemic brain tissue may benefit from treatment with low-molecular-weight heparin when administered after an acute injury. More recently, attention has focused on new antithrombotic agents, tested previously in models of sepsis and shock, such as activated protein C (APC)136 and antithrombin III,137 for their potential role in the treatment of I/R injury. APC’s mode of action depends on its initial binding to thrombomodulin, whereupon activated thrombomodulin then decreases the activity of thrombin. In experimental models of renal reperfusion injury, APC prevented histologic changes and the inflammation associated with I/R injury.138 In rodents subjected to bilateral lower extremity I/R, antithrombin III decreased remote lung and local skeletal muscle injury, but did not alter the biochemical profile of tissue inflammation.139
Hypothermia
Hypothermia is known to reduce cellular metabolism and thus decrease demand for O2 and nutrients during times of stress. Thus, hypothermia has been shown to decrease skeletal muscle injury in isolated gracilis muscle140 and spinal cord dysfunction141,142 in response to I/R injury. In vascular and cardiovascular surgery, moderate hypothermia must be used cautiously to protect tissue yet avoid devastating coagulopathy. To this end, localized spinal cord cooling has been used by Black et al,143 Cambria et al,144 Davison et al,145 and El-Wahsh146 to ameliorate spinal cord injury. In clinical applications of organ transplantation, explanted organs are routinely stored under hypothermic conditions to decrease basal metabolic rates.146 Total body hypothermia combined with total circulatory arrest has been useful in replacement of the great vessels.147–149 New technology has now made systemic hypothermia feasible in patients with acute ischemic events. Current endovascular systems can provide rapid and safe systemic cooling and have been evaluated in patients with acute MI in the COOL MI (Hypothermia as an Adjunctive Therapy to PCI in Patients with Acute Myocardial Infarction) and ICE-IT (Intravascular Cooling Adjunctive to Percutaneous Coronary Intervention) trials. Results from both of these trials have been presented in scientific meetings, but have not been published yet in peer-reviewed journals. Initial conclusions from the available data include that mild systemic hypothermia is safe and well tolerated during coronary intervention in patients presenting with acute MI, and that although neither of the two trials met its primary end point, which was a reduction of infarct size, patients with anterior infarction, who were successfully cooled to the target temperature of ≤35° C, appear to have a significant reduction in infarct size.
Neurogenic Inflammation
Recent studies have shown that vagal nerve stimulation inhibits cytokine synthesis induced by endotoxin by activating the cholinergic anti-inflammatory pathway.150–156 Vagal stimulation has also been shown to decrease myocardial I/R injury by decreasing tissue injury and inflammation.153,154 In a rodent model of suprarenal aortic occlusion, vagal nerve stimulation has been found to decrease cytokine synthesis in serum and pulmonary/visceral tissue and shock during reperfusion.150 Further studies have shown that the α7 subunit-containing nicotinic acetylcholine receptor (α7nAChR) is an essential component in the vagus nerve–based “cholinergic anti-inflammatory pathway” that regulates levels of TNF, high-mobility group protein B1 (HMGB1), and other cytokines during inflammation. Examination of the effects of cholinergic stimulation with the agonists nicotine or GTS-21 given before or after bilateral renal I/R injury in rats revealed that pretreatment with either agonist significantly attenuated the renal dysfunction and tubular necrosis induced by renal ischemia. Similarly, TNF-α expression and leukocyte infiltration of the kidney were markedly reduced after treatment with cholinergic agonists.157
Clinical Manifestations of Ischemia-Reperfusion Injury
The following discussions provide a brief overview of some clinical examples of I/R injury.
Compartment Syndrome
Acute limb compartment syndrome is often associated with acute penetrating arterial or blunt/crush injury, followed by reperfusion.158 Isolated arterial and venous injuries can give rise to lower extremity compartment syndrome.159 In the setting of combined injury, the combination of edema and ischemia may increase the need to perform fasciotomy. Iliofemoral thrombosis causing phlegmasia cerulea dolens can produce compartment syndrome. Embolectomy, thrombolysis, and bypass surgery can cause compartment syndrome at an incidence of up to 21%. In this setting, the condition is known as postischemic compartment syndrome and is caused by revascularization of the leg after a prolonged period of ischemia. The pathophysiology of compartment syndrome is believed to be due to a reduction in the arteriovenous pressure gradient. Specifically, as edema and intracompartmental pressure increase, the veins are compressed, and venous pressure increases. The increase in venous pressure decreases the arteriovenous gradient, thereby resulting in diminished or absent local perfusion. Reduced venous drainage as a result of increased interstitial pressure exacerbates the development of edema, such that arteriolar compression develops and leads to muscle and nerve ischemia. Sensory changes in the nerves, manifested by clinical complaints of paresthesias and hyperesthesia, may develop after only 30 minutes of ischemia. Irreversible nerve damage begins after 12 to 24 hours of total ischemia. Failure to decompress all fascial compartments usually leads to muscle ischemia, rhabdomyolysis, and renal insufficiency. Treatment is directed toward improving tissue perfusion pressure, usually by expediently performing fasciotomy.160,161 A high index of suspicion is required to make this diagnosis, particularly in sedated or unconscious patients.
No–Reflow Phenomenon
One of the most formidable advanced consequences of I/R injury is the “no-reflow phenomenon,” whereby, despite restoration of blood flow to the muscular arteries, occlusion of the microvasculature results in tissue ischemia and anoxic injury. In 1968, Ames et al162 described the no-reflow phenomenon in the brain, and May et al163 reported the same phenomenon 10 years later in skin flaps. In the no-reflow state, the flow in the reperfused tissue is disturbed because of downstream microembolization of platelets, neutrophils, thrombus, and atheroma particles, as well as endothelial cell dysfunction and severe microvascular damage. Clinically, this syndrome is associated with tissue hypoperfusion in the absence of a significant obstruction in the named arteries, and in its advanced forms, it can result in loss of the affected organ or limb.
Myocardial Stunning/Arrhythmias
Myocardial stunning is defined as reversible, transient myocardial dysfunction persisting after reperfusion despite the absence of irreversible damage.164,165 This kind of injury may require inotropic support. A multitude of conditions contribute to myocardial stunning, including decreased ATP resynthesis, coronary microvascular spasm/plugging, and cytotoxic injury secondary to reactive O2 metabolites or increased intracellular calcium release and uptake.166,167 Reperfusion arrhythmias may cause sudden death after surgical revascularization or coronary thrombolysis. These arrhythmias may be due in part to rapid and sudden changes in ion concentrations within ischemic tissue on reperfusion.168–170 Staged gradual reflow or transient acid reperfusion may reduce the incidence of malignant arrhythmias.171
Central Nervous System
Reperfusion injury of the central nervous system (CNS) may contribute to the morbidity and mortality of stroke, head trauma, cerebrovascular surgery, and deep hypothermic circulatory arrest. Disruption of the blood-brain barrier leading to increased vascular permeability results in cerebral edema, increased intracranial pressure, and leukocyte migration into brain tissue.11 As mentioned earlier in this chapter, leukocytes release proteases, ROS, and lipid mediators that result in irreversible tissue damage. The clinical manifestations of CNS I/R injury usually coincide with severe changes in sensory, motor, or cognitive function, and often lead to death.
Gastrointestinal System
Gastrointestinal I/R injury is characterized by a decrease in intestinal barrier function. Under normal physiologic conditions, the gastrointestinal mucosa separates the bacteria of the bowel lumen from the vascular space and the interstitium of the bowel wall. After I/R, there is an increase in intestinal permeability that allows translocation of bacteria from the bowel lumen into the portal and systemic circulation.87,172,173 Activation of complement and circulating leukocytes by translocated bacteria may eventually lead to development of the systemic inflammatory response after gastrointestinal I/R injury. The relatively unpredictable nature of gastrointestinal I/R injury has led to widespread adoption of a policy to consider a second-look operation within 24 hours of acute mesenteric or celiac revascularization.174–176
Multiorgan Dysfunction Syndrome
Remote organ injury after I/R is a leading cause of death, with mortality often linked to the number of failed organ systems. The pulmonary system is the most frequently injured organ in patients with multiple-organ dysfunction syndrome,177 and onset of the syndrome is commonly preceded by the development of acute respiratory insufficiency 24 to 72 hours after the initiating ischemic event.178,179 Respiratory failure is often followed by hepatic, renal, gastrointestinal, myocardial, and CNS dysfunction. In its terminal stages, multiorgan dysfunction is characterized by dysfunction of the coagulation and immune systems, which leads to thrombosis, disseminated intravascular coagulation, and immunocompromise.
Clinical Trials
The principal therapy in patients who sustain acute ischemic events in one of their organs or limbs is prompt reperfusion by mechanical and/or pharmacologic (thrombolytic) intervention. To date, there have been very few human studies providing evidence that a pharmacologic agent, administered as an adjunct to prompt reperfusion, can actually reduce I/R injury.
Coronary Interventions
Two of the most encouraging trials are the AMISTAD-I108 (Acute Myocardial Infarction Study of Adenosine Trial) and AMISTAD-II studies,109 which have provided evidence that adenosine reduces infarct size in patients presenting with anterior MI when given in conjunction with thrombolysis or primary angioplasty therapy. Specifically, a 3-hour infusion of adenosine at 70 μg/kg/min produced a significantly lower infarct size (11% vs 27% in controls as measured by technetium-99m sestamibi tomography; P = .023), and this finding correlated with fewer adverse clinical events. As previously discussed, adenosine has direct protective effects on organ parenchymal cells; vasodilatation of native arteries, but also anti-inflammatory effects that may inhibit neutrophil adhesion and migration into the myocardium; cytokine release from mononuclear cells; release of O2 radicals; and antiplatelet effects that may have a role in maintaining infarct artery patency.
Several studies of experimental animal models of acute MI have also documented the effectiveness of ischemic conditioning (applications of brief periods of ischemia) in reducing infarct size. When ischemic conditioning is applied before the I/R episode, it is called ischemic preconditioning and is applicable mostly during operations for the performance of either a heart180 or leg bypass procedure.181 Ischemic conditioning can also be applied at the time of reperfusion (ischemic postconditioning), making it applicable for patients who are being treated for an acute ischemic event. Postconditioning can be applied either as inflation and deflation of the angioplasty balloon after reopening the occluded (usually coronary) artery or as inflation and deflation of a blood pressure cuff around an unaffected arm or leg (remote conditioning). Several small clinical studies have demonstrated that postconditioning by coronary angioplasty protects the human heart during acute MI.182 The initial promise presented by these studies has lead to postconditioning being currently evaluated in nine ongoing clinical studies, including the DANAMI-3 (DANish Study of Optimal Acute Treatment of Patients With ST-elevation Myocardial Infarction) with 2000 anticipated patient enrollments. Clinicians hope that these ongoing studies either alone or in combination will provide data that will have adequate statistical power to detect the cardioprotective properties of postconditioning.183
Other treatments targeting the biochemical pathways that are activated during IR, including the production of ROS, cytosolic and mitochondrial Ca2+-overload, complement and neutrophil activation, endothelial dysfunction, and shift in metabolism and substrate use, have been investigated for patients with acute MI with mostly disappointing results.184,185 The most encouraging exception among these works is a pilot study evaluating the cardioprotective effects of cyclosporine, which in addition to its well-known immunosuppressive properties, is a potent inhibitor of MPTP.186 In a randomized trial of 58 patients, the investigators reported that administration of the medication immediately before primary coronary intervention reduced the size of the infarct (measured using surrogate biomarkers and cardiac magnetic resonance imaging) by as much as 40%. This study has provided the preliminary data for a larger European trial, does Cyclosporine ImpRove Clinical oUtcome in ST elevation (CIRCUS), which aims to confirm the beneficial effects of cyclosporine in a multicenter, randomized, placebo-controlled, double-blinded fashion.
Transplantation Interventions
In the field of organ transplantation, the strategies used to improve organ preservation and minimize I/R injury have not changed very much in the last 10 years, with cold preservation being the mainstay procedure. More recently, additives to the cold storage solution, including the p38 MAPK inhibitor FR 167653 (with anti-inflammatory properties) and the colloid polyethylene glycol, which appear to reduce ATP depletion and calcium accumulation within the cells, have been successfully used to reduce I/R damage after transplantation.187 In transplantation, the benefit of ischemic preconditioning of the donor is still under evaluation, with most studies not showing a clinical benefit with respect to graft or graft-recipient survival.188 In the direction of treatments applied to the donor before harvesting of the organs, a study of pretreatment of donors with low-dose dopamine demonstrated a reduction of the need for dialysis after kidney transplantation.189 The investigators believe that the mechanism of protection from dopamine involves direct interaction with the cellular membrane (related to the dihydroxy-phenolic ring structure of the dopamine molecule), which is capable of protecting endothelial cells from oxidative stress during cold storage reperfusion.
Lower Extremity Interventions
Therapeutic intervention for acute lower extremity arterial occlusion has centered on open surgical intervention or thrombolysis. Several prospective, randomized trials were designed to compare thrombolytic therapy with direct operative intervention190–193 in a post hoc scenario (i.e., after the onset of ischemia). The STILE (Surgery vs. Thrombolysis for Ischemia of the Lower Extremity) trial showed that patients with peripheral artery occlusion and ischemia of less than 14 days’ duration who were treated with catheter-directed urokinase or alteplase had significantly lower amputation rates, shorter hospital stays, and fewer subsequent surgical interventions than those initially treated with surgery did.190 These benefits were achieved at the cost of a greater risk of bleeding complications than in patients treated by surgery alone. In contrast, for patients with ischemia lasting longer than 14 days, surgery resulted in significantly less ongoing or recurrent ischemia than fibrinolysis did. In the TOPAS trial (Thrombolysis or Peripheral Arterial Surgery), intra-arterial infusion of urokinase reduced the need for open surgical procedures and yielded similar rates of amputation and death despite a higher frequency of hemorrhagic complications.193 Even with these major milestones in the management of acute arterial occlusion, high rates of limb loss and mortality continue to be documented in this complex patient population.191
A cell-free extracorporeal perfusion system used to minimize both local and systemic injury has been evaluated in patients with severe limb ischemia (sensorimotor loss, rest pain/gangrene).194 In this post hoc treatment study, reperfusion of the ischemic limb was achieved with hypertonic, hyperoncotic perfusate containing antioxidants delivered through the arterial tree with initial venous drainage. Rapid lactate washout and decreased neutrophil activation, serum levels of F2-isoprostanes (marker of free radical–mediated systemic lipid peroxidation), and mortality were observed in patients treated with the study perfusion method versus controls. In this small study, modification of limb perfusion in patients with severe limb ischemia via a simple and rapid (15-20 minutes) method provided beneficial systemic effects.
Another post hoc therapeutic intervention, controlled reperfusion using a simple blood bag perfusion system, has been found to reduce reperfusion injury and facilitate return of normal function.195 Patients with severe, acute lower limb ischemia were allocated to treatment with either embolectomy alone or surgical embolectomy plus controlled reperfusion via a simplified perfusion system. Although the duration of ischemia was greater in the patients who underwent controlled reperfusion, there were no deaths or amputations in this treatment group. In contrast, in patients who had shorter periods of ischemia treated by embolectomy alone, there were three in-hospital deaths and two major amputations. This study strongly suggests that the results of conventional embolectomy for acute, severe lower limb ischemia can be improved by controlled reperfusion.
The state of our knowledge for the treatment/prevention of I/R injury in patients undergoing vascular operations is quite limited. Almost all of the clinical work thus far has focused on prevention of I/R after the performance of major vascular operations, mostly aortic repairs for aneurysmal and occlusive disease. The two main interventions evaluated in small pilot studies are ischemic preconditioning and administration of antioxidants. A recent review comparing the outcomes from vascular and endovascular surgical procedures with and without the use of remote ischemic preconditioning demonstrated that perioperative mortality was not clearly different between the two groups, whereas MIs were reduced in the remote ischemic preconditioning group, and unplanned critical care admissions tended to increase in the remote ischemic preconditioning group. The authors concluded that based on current evidence from small pilot trials, there are too few data to be able to say whether remote ischemic preconditioning has any beneficial or harmful effects and that the safety of this technique needs to be confirmed in adequately powered randomized trials.181
A similar review compared the effects of antioxidant treatment on a variety of ROS- and inflammation-related markers in patients who underwent vascular and endovascular surgical procedures. The authors demonstrated that antioxidant supplementation might reduce oxidative stress and activation of inflammatory pathways. The correlation of this finding to clinical outcomes in these patients was evaluated in only one study, which demonstrated an association between plasma malondialdehyde (one of the most frequently used biomarkers of lipid peroxidation and oxidative stress) levels and the need for transfer to the intensive care unit during vascular operations. The authors concluded that their data pointed to a potential therapeutic advantage associated with antioxidant supplementation in patients undergoing vascular operations.196
The Future of Reperfusion Therapy
The first line of therapy for patients with acute nontraumatic limb ischemia is the institution of anticoagulant therapy, usually with heparin. The goal of anticoagulation is to avoid propagation of a clot as the patient is being evaluated, resuscitated, and prepared for intervention, be it open surgical- or catheter-based therapy. Despite the plethora of experimental data indicating a metabolic, inflammatory, and thrombotic component to I/R197,198 that starts locally in the ischemic limb before reperfusion, conventional post hoc clinical trials have not instituted metabolic rescue or anti-inflammatory treatments before reperfusion. In addition, most clinical trials have used therapeutic agents that have a specific action on a single inflammatory, metabolic, or procoagulant pathway despite the overwhelming evidence that multiple pathways are induced by I/R.
To date, the most successful treatment scenario for reperfusion injury has been achieved in the transplant arena, where a donated organ becomes ischemic in a planned fashion so that steps can be taken to institute therapy before the onset of ischemia. This ideal therapeutic window is not available for patients with acute vascular catastrophes; despite the differences in the therapeutic window, an important lesson can be gleaned from the experiences of our transplant colleagues. Early delivery of an agent capable of addressing the metabolic, inflammatory, and thrombotic components of I/R is necessary. In the setting of a vascular catastrophe, a pleiotropic agent (a compound having more than one effect) must be started at high doses as soon as the diagnosis of limb ischemia becomes clear. Transplant surgeons have used a pleiotropic agent, the University of Wisconsin solution, with great success179; this solution involves the use of a multimodal therapeutic approach, thereby addressing the problem of I/R on metabolic, thrombotic, and anti-inflammatory levels simultaneously. It is likely that a similar approach will be needed to adequately address vascular reperfusion injury reliably in complex patient populations with a variety of risk factors.
Acknowledgment
The authors acknowledge funding from NIH grant R01AG034995, from the Charles and Mary Heider Fund for Excellence in Vascular Surgery, and from the Department of Surgery, University of Nebraska Medical Center.
Selected Key References
Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011;8(5):292–300.
This paper provides an excellent review of innate immune activation through Toll-like receptors and a therapeutic perspective on “danger-associated molecular patterns as attractive targets for intervention”.
Blaisdell F. The pathophysiology of skeletal muscle ischemia and the reperfusion syndrome: a review. Cardiovasc Surg. 2002;10:620–630.
This paper is one of the best overall reviews on the topic of skeletal muscle ischemia-reperfusion injury. This paper contains extensive historical, clinical, and pathologic details.
Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125(7):1241–1252.
This is a detailed review of the consequence of mitochondrial injury during acute and chronic ischemia-reperfusion.
Dirksen MT, Laarman GJ, Simoons ML, Duncker DJ. Reperfusion injury in humans: a review of clinical trials on reperfusion injury inhibitory strategies. Cardiovasc Res. 2007;74(3):343–355.
This paper provides an excellent review of clinical trials on reperfusion injury inhibitory strategies.
Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nat Med. 2011;17(11):1391–1401.
This paper provides an excellent review of the cellular and molecular mechanisms of innate and adaptive immune responses activated during ischemia/reperfusion and their contributions to tissue injury.
Frank A, Bonney M, Bonney S, Weitzel L, Koeppen M, Eckle T. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Semin Cardiothorac Vasc Anesth. 2012;16(3):123–132.
This paper provides an excellent review of clinical trials on reperfusion injury inhibitory strategies.
Gourgiotis S, Villias C, Germanos S, Foukas A, Ridolfini MP. Acute limb compartment syndrome: a review. J Surg Educ. 2007;64:178–186.
This is an extensive detailed review of the lower extremity compartment syndrome associated with arterial, venous, and nonvascular causes.
Halestrap AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans. 2010;38(4):841–860.
This paper is a detailed review of the physiological and clinical significance of the mitochondria and their mitochondrial permeability transition pore in ischemia-reperfusion injury.
Jaeschke H, Woolbright BL. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transplant Rev (Orlando). 2012;26(2):103–114.
This review centers on sources and mediators of oxidative stress during hepatic ischemia/reperfusion and the status of current antioxidant interventions.
Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111(9):1208–1221.
This is an excellent review highlighting the molecular mechanisms of the participation of mitochondria in cell death and mitophagy during ischemia/reperfusion injury.
McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312:159–163.
This paper was one of the first important studies to implicate a definitive role for oxygen-derived free radicals in postischemic human tissue injury.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13.
This paper is a detailed review of the characterization of the reactive oxygen species and their implication to the pathophysiology of human disease.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Haimovici H. Arterial embolism with acute massive ischemic myopathy and myoglobinuria: evaluation of a hitherto unreported syndrome with report of two cases. Surgery. 1960;47:739–747.
2. Cerra FB, et al. Hemorrhagic infarction: a reperfusion injury following prolonged myocardial ischemic anoxia. Surgery. 1975;78:95–104.
3. Olding M, et al. Ischemic tolerance of canine jejunal flaps. Plast Reconstr Surg. 1994;94:167–173.
4. Halestrap AP, et al. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta. 2009;1787(11):1402–1415.
5. Clanton TL. Hypoxia-induced reactive oxygen species formation in skeletal muscle. J Appl Physiol. 2007;102(6):2379–2388.
6. Halestrap AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans. 2010;38(4):841–860.
7. Loor G, et al. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim Biophys Acta. 2011;1813(7):1382–1394.
8. Kong T, et al. Leukocyte adhesion during hypoxia is mediated by HIF-1–dependent induction of beta2 integrin gene expression. Proc Natl Acad Sci U S A. 2004;101:10440–10445.
9. Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314.
10. Tamura DY, et al. Acute hypoxemia in humans enhances the neutrophil inflammatory response. Shock. 2002;17:269–273.
11. Collard CD, et al. Neutrophil-derived glutamate regulates vascular endothelial barrier function. J Biol Chem. 2002;277:14801–14811.
12. Eltzschig HK, et al. Vascular ischaemia and reperfusion injury. Br Med Bull. 2004;70:71–86.
13. Eltzschig HK, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–3992.
14. Eltzschig HK, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796.
15. Gautam N, et al. Heparin-binding protein (HBP/CAP37): a missing link in neutrophil-evoked alteration of vascular permeability. Nat Med. 2001;7:1123–1127.
16. Vaseva AV, et al. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149(7):1536–1548.
17. Kubli DA, et al. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111(9):1208–1221.
18. Nomura K, et al. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J. 2000;351(Pt 1):183–193.
19. Jaeschke H, et al. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transplant Rev (Orlando). 2012;26(2):103–114.
20. Morgan MJ, et al. Reactive oxygen species in TNFalpha-induced signaling and cell death. Mol Cells. 2010;30(1):1–12.
21. Vandenabeele P, et al. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3(115).
22. Narayan N, et al. The NAD-dependent deacetylase SIRT2 is required for programmed necrosis. Nature. 2012;492(7428):199–204.
23. Marchant DJ, et al. Inflammation in myocardial diseases. Circ Res. 2012;110(1):126–144.
24. Miller YI, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108(2):235–248.
25. Ha T, et al. Toll-like receptors: new players in myocardial ischemia/reperfusion injury. Antioxid Redox Signal. 2011;15(7):1875–1893.
26. Arslan F, et al. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011;8(5):292–300.
27. Eltzschig HK, et al. Ischemia and reperfusion—from mechanism to translation. Nat Med. 2011;17(11):1391–1401.
28. Zhang M, et al. Natural antibody mediated innate autoimmune response. Mol Immunol. 2007;44(1-3):103–110.
29. Zhang M, et al. Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. J Immunol. 2006;177(7):4727–4734.
30. Collard CD, et al. Complement activation after oxidative stress: role of the lectin complement pathway. Am J Pathol. 2000;156(5):1549–1556.
31. Belkin M, et al. The role of leukocytes in the pathophysiology of skeletal muscle ischemic injury. J Vasc Surg. 1989;10:14–18.
32. Carden DL, et al. Pathophysiology of ischaemia-reperfusion injury. J Pathol. 2000;190:255–266.
33. Alexander JS, et al. Inflammatory mediators induce sequestration of VE-cadherin in cultured human endothelial cells. Inflammation. 2000;24:99–113.
34. Phillipson M, et al. The neutrophil in vascular inflammation. Nat Med. 2011;17(11):1381–1390.
35. Albadawi H, et al. Human microvascular endothelial synthesis of interleukin-8 during in vitro ischemia and reperfusion. J Cell Biochem. 2007;100:412–420.
36. Colgan SP, et al. Epithelial exposure to hypoxia modulates neutrophil transepithelial migration. J Exp Med. 1996;184:1003–1015.
37. Tedgui A, et al. Anti-inflammatory mechanisms in the vascular wall. Circ Res. 2001;88:877–887.
38. Lentsch AB, et al. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and KC. Hepatology. 1998;27:1172–1177.
39. Conrad MF, et al. Local inflammatory and thrombotic responses differ in a murine model of partial and complete hindlimb ischemia/reperfusion. Surgery. 2005;138:375–381.
40. Hua HT, et al. CXC chemokine expression and synthesis in skeletal muscle during ischemia/reperfusion. J Vasc Surg. 2005;42:337–343.
41. Seekamp A, et al. Requirements for tumor necrosis factor-alpha and interleukin-1 in limb ischemia/reperfusion injury and associated lung injury. Am J Pathol. 1993;143:453–463.
42. Colletti LM, et al. Lung and liver injury following hepatic ischemia/reperfusion in the rat is increased by exogenous lipopolysaccharide which also increases hepatic TNF production in vivo and in vitro. Shock. 2001;16:312–319.
43. Wakai A, et al. Inosine attenuates tourniquet-induced skeletal muscle reperfusion injury. J Surg Res. 2001;99:311–315.
44. Kang J, et al. Apolipoprotein E-/- have delayed skeletal muscle healing after hind limb ischemia-reperfusion injury. J Vasc Surg. 2008;48:701–708.
45. Seekamp A, et al. Ischemia-reperfusion injury. Agents Actions Suppl. 1993;41:137–152.
46. Crawford RS, et al. Postischemic treatment with ethyl pyruvate prevents adenosine triphosphate depletion, ameliorates inflammation, and decreases thrombosis in a murine model of hind-limb ischemia and reperfusion. J Trauma. 2011;70(1):103–110.
47. Stone DH, et al. Effect of PJ34 on spinal cord tissue viability and gene expression in a murine model of thoracic aortic reperfusion injury. Vasc Endovascular Surg. 2009;43(5):444–451.
48. Crawford RS, et al. Postischemic poly (ADP-ribose) polymerase (PARP) inhibition reduces ischemia reperfusion injury in a hind-limb ischemia model. Surgery. 2010;148(1):110–118.
49. Murphy AD, et al. Poloxamer 188 protects against ischemia-reperfusion injury in a murine hind-limb model. Plast Reconstr Surg. 2010;125(6):1651–1660.
50. Conrad MF, et al. Local administration of the poly ADP-ribose polymerase (PARP) inhibitor, PJ34 during hindlimb ischemia modulates skeletal muscle reperfusion injury. J Surg Res. 2006;135:233–237.
51. Hua HT, et al. Polyadenosine diphosphate-ribose polymerase inhibition modulates skeletal muscle injury following ischemia reperfusion. Arch Surg. 2005;140:344–351.
53. Rusai K, et al. Administration of interleukin-1 receptor antagonist ameliorates renal ischemia-reperfusion injury. Transpl Int. 2008;21:572–580.
54. Shireman PK, et al. MCP-1 deficiency causes altered inflammation with impaired skeletal muscle regeneration. J Leukoc Biol. 2007;81:775–785.
55. Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125(7):1241–1252.
56. Lenaz G, et al. Structural and functional organization of the mitochondrial respiratory chain: a dynamic super-assembly. Int J Biochem Cell Biol. 2009;41(10):1750–1772.
57. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13.
58. Arumugam TV, et al. Complement mediators in ischemia-reperfusion injury. Clin Chim Acta. 2006;374:33–45.
59. Wong MS, et al. Hindlimb ischemia-reperfusion increases complement deposition and glycolysis. J Surg Res. 1999;85:130–135.
60. Fleming SD, et al. Mice deficient in complement receptors 1 and 2 lack a tissue injury-inducing subset of the natural antibody repertoire. J Immunol. 2002;169:2126–2133.
61. Austen WG, et al. Murine hindlimb reperfusion injury can be initiated by a self-reactive monoclonal IgM. Surgery. 2004;136:401–406.
62. Chan RK, et al. Attenuation of skeletal muscle reperfusion injury with intravenous 12 amino acid peptides that bind to pathogenic IgM. Surgery. 2006;139:236–243.
63. Toomayan C, et al. C1-esterase inhibitor and a novel peptide inhibitor improve contractile function in reperfused skeletal muscle. Mircosurgery. 2003;23:561–567.
64. Kyriakides C, et al. Skeletal muscle reperfusion injury is mediated by neutrophils and the complement membrane attack complex. Am J Physiol Cell Physiol. 1999;277:C1263–C1268.
65. Beckman JS. Ischaemic injury mediator. Nature. 1990;345:27–28.
66. Beckman JS. The double-edged role of nitric oxide in brain function and superoxide-mediated injury. J Dev Physiol. 1991;15:53–59.
67. Nathan C. Nitric oxide and biopterin: a study in Chiaroscuro. J Clin Invest. 1994;93:1875–1876.
68. Nathan C, et al. Regulation of biosynthesis of nitric oxide. J Biol Chem. 1994;269:13725–13728.
69. Pacher P, et al. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424.
70. Avila MA, et al. l-Arginine decreases fluid-percussion injury–induced neuronal nitrotyrosine immunoreactivity in rats. J Cereb Blood Flow Metab. 2008;28:1733–1741.
71. Matata BM, et al. Peroxynitrite is an essential component of cytokines production mechanism in human monocytes through modulation of nuclear factor-kappa B DNA binding activity. J Biol Chem. 2002;277:2330–2335.
72. Park SW, et al. Tyrosine nitration on p65: a novel mechanism to rapidly inactivate nuclear factor-κB. Mol Cell Proteomics. 2005;4:300–309.
73. Park SW, et al. Regulation of c-myc gene by nitric oxide via inactivating NF-kappa B complex in P19 mouse embryonal carcinoma cells. J Biol Chem. 2003;278:29776–29782.
74. Rabkin SW, et al. Metalloporphrins as a therapeutic drug class against peroxynitrite in cardiovascular diseases involving ischemic reperfusion injury. Eur J Pharmacol. 2008;586:1–8.
75. Greco R, et al. Neuroprotective effect of nitroglycerin in a rodent model of ischemic stroke: evaluation of Bcl-2 expression. Int Rev Neurobiol. 2007;82:423–435.
76. Katsumi H, et al. Prevention of hepatic ischemia/reperfusion injury by prolonged delivery of nitric oxide to the circulating blood in mice. Transplantation. 2008;85:264–269.
77. Namazi H. Sodium nitroprusside as a nitric oxide donor in a rat intestinal ischemia reperfusion model: a novel molecular mechanism. Clinics. 2008;63:405.
78. De Caterina R, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–68.
79. Peng HB, et al. Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J Biol Chem. 1995;270:14214–14219.
80. Rubbo H, et al. Nitric oxide, peroxynitrite and lipoxygenase in atherogenesis: mechanistic insights. Toxicology. 2005;208:305–317.
81. Kubes P. Polymorphonuclear leukocyte–endothelium interactions: a role for pro-inflammatory and anti-inflammatory molecules. Can J Physiol Pharmacol. 1993;71:88–97.
82. Kubes P, et al. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A. 1991;88:4651–4655.
83. Radomski MW, et al. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet. 1987;2:1057–1058.
84. Tuor UI, et al. Dexamethasone prevents cerebral infarction without affecting cerebral blood flow in neonatal rats. Stroke. 1993;24:452–457.
85. Yanagisawa M, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415.
86. Inoue A, et al. The human endothelin family: three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc Natl Acad Sci U S A. 1989;86:2863–2867.
87. Mallick IH, et al. Ischemia-reperfusion injury of the intestine and protective strategies against injury. Dig Dis Sci. 2004;49:1359–1377.
88. Clozel M, et al. The endothelin ETB receptor mediates both vasodilation and vasoconstriction in vivo. Biochem Biophys Res Commun. 1992;186:867–873.
89. Haynes WG, et al. Endothelium-dependent modulation of responses to endothelin-I in human veins. Clin Sci (Lond). 1993;84:427–433.
90. Sargent CA, et al. Role of endothelin receptor subtype B (ET-B) in myocardial ischemia. Life Sci. 1994;55:1833–1844.
91. Henrion D, et al. Potentiation of norepinephrine-induced contractions by endothelin-1 in the rabbit aorta. Hypertension. 1993;22:78–83.
92. Gupta SK, et al. Bosentan, the mixed ETA-ETB endothelin receptor antagonist, attenuated oxidative stress after experimental myocardial ischemia and reperfusion. Mol Cell Biochem. 2005;275:67–74.
93. McMurdo L, et al. The effects of the endothelin ETA receptor antagonist, FR 139317, on infarct size in a rabbit model of acute myocardial ischaemia and reperfusion. Br J Pharmacol. 1994;112:75–80.
94. Ozdemir R, et al. Selective endothelin A (ETA) receptor antagonist (BQ-123) reduces both myocardial infarct size and oxidant injury. Toxicology. 2006;219:142–149.
95. Kuro T, et al. Selective antagonism of the ETA receptor, but not the ETB receptor, is protective against ischemic acute renal failure in rats. Jpn J Pharmacol. 2000;82:307–316.
96. Matsuo Y, et al. Protective effect of endothelin type A receptor antagonist on brain edema and injury after transient middle cerebral artery occlusion in rats. Stroke. 2001;32:2143–2148.
97. Uhlmann D, et al. Improvement of postischemic hepatic microcirculation after endothelin A receptor blockade—endothelin antagonism influences platelet-endothelium interactions. J Gastrointest Surg. 2005;9:187–197.
98. Andrasi TB, et al. ET(A) receptor blockade protects the small intestine against ischaemia/reperfusion injury in dogs via an enhancement of antioxidant defences. Clin Sci (Lond). 2002;103(Suppl 48):59S–63S.
99. Jennings RB, et al. Total ischemia in dog hearts, in vitro. 1. Comparison of high energy phosphate production, utilization, and depletion, and of adenine nucleotide catabolism in total ischemia in vitro vs. severe ischemia in vivo. Circ Res. 1981;49:892–900.
100. Forman MB, et al. Role of adenosine as adjunctive therapy in acute myocardial infarction. Cardiovasc Drug Rev. 2006;24:116–147.
101. Yaar R, et al. Animal models for the study of adenosine receptor function. J Cell Physiol. 2005;202:9–20.
102. Berne RM. The role of adenosine in the regulation of coronary blood flow. Circ Res. 1980;47:807–813.
104. Pasque MK, et al. Ribose-enhanced myocardial recovery following ischemia in the isolated working rat heart. J Thorac Cardiovasc Surg. 1982;83:390–398.
105. Gross GJ, et al. Role of ATP dependent potassium channels in myocardial ischaemia. Cardiovasc Res. 1992;26:1011–1016.
106. Cronstein BN, et al. Adenosine; a physiologic modulator of superoxide anion generation by human neutrophils. Adenosine acts via an A2 receptor on human neutrophils. J Immunol. 1985;135:1366–1371.
107. Quintana M, et al. Pharmacological prevention of reperfusion injury in acute myocardial infarction. A potential role for adenosine as a therapeutic agent. Am J Cardiovasc Drugs. 2004;4:159–167.
108. Mahaffey KW, et al. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol. 1999;34(6):1711–1720.
109. Ross AM, et al. a randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). [AMISTAD-II investigators] J Am Coll Cardiol. 2005;45(11):1775–1780.
110. Zheng J, et al. Protective roles of adenosine A1, A2A, and A3 receptors in skeletal muscle ischemia and reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;293:H3685–H3691.
111. Reece TB, et al. Early adenosine receptor activation ameliorates spinal cord reperfusion injury. J Cardiovasc Med (Hagerstown). 2008;9:363–367.
112. Okusa MD, et al. A(2A) adenosine receptor–mediated inhibition of renal injury and neutrophil adhesion. Am J Physiol Renal Physiol. 2000;279:F809–F818.
113. Koti RS, et al. Protection of the liver by ischemic preconditioning: a review of mechanisms and clinical applications. Dig Surg. 2003;20:383–396.
114. Kitamura K, et al. Adrenomedullin: a novel hypotensive peptide isolated from human pheochromocytoma. Biochem Biophys Res Commun. 1993;192:553–560.
115. Pio R, et al. Complement factor H is a serum-binding protein for adrenomedullin, and the resulting complex modulates the bioactivities of both partners. J Biol Chem. 2001;276:12292–12300.
116. Shimekake Y, et al. Adrenomedullin stimulates two signal transduction pathways, cAMP accumulation and Ca2+ mobilization, in bovine aortic endothelial cells. J Biol Chem. 1995;270:4412–4417.
117. Harrison RA, et al. The physiological breakdown of the third component of human complement. Mol Immunol. 1980;17:9–20.
118. DiScipio RG, et al. Human polymorphonuclear leukocytes adhere to complement factor H through an interaction that involves alphaMbeta2 (CD11b/CD18). J Immunol. 1998;160:4057–4066.
119. Carrizo GJ, et al. Adrenomedullin and adrenomedullin-binding protein-1 downregulate inflammatory cytokines and attenuate tissue injury after gut ischemia-reperfusion. Surgery. 2007;141:245–253.
120. Lien YH, et al. Pathogenesis of renal ischemia/reperfusion injury: lessons from knockout mice. Life Sci. 2003;74:543–552.
121. Kerem M, et al. Effect of adrenomedullin on hepatic damage in hepatic ischemia reperfusion injury in rats. Liver Int. 2008.
122. Tenhunen R, et al. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci U S A. 1968;61:748–755.
123. Maines MD, et al. Induction of kidney heme oxygenase-1 (HSP32) mRNA and protein by ischemia/reperfusion: possible role of heme as both promotor of tissue damage and regulator of HSP32. J Pharmacol Exp Ther. 1993;264:457–462.
124. Ferrandiz ML, et al. Inducers of heme oxygenase-1. Curr Pharm Des. 2008;14:473–486.
125. Ryter SW, et al. Carbon monoxide and bilirubin: potential therapies for pulmonary/vascular injury and disease. Am J Respir Cell Mol Biol. 2007;36:175–182.
126. Misra HP, et al. The generation of superoxide radical during the autoxidation of hemoglobin. J Biol Chem. 1972;247:6960–6962.
127. Hayashi S, et al. Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress: role of bilirubin generated by the enzyme. Circ Res. 1999;85:663–671.
128. Christova T, et al. Heme oxygenase–carbon monoxide signalling pathway as a physiological regulator of vascular smooth muscle cells. Acta Physiol Pharmacol Bulg. 2000;25:9–17.
129. Zhang X, et al. Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen–activated protein kinase pathway and involves caspase 3. J Biol Chem. 2003;278:1248–1258.
130. Kaizu T, et al. Protection of transplant-induced hepatic ischemia/reperfusion injury with carbon monoxide via MEK/ERK1/2 pathway downregulation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G236–G244.
131. Patel R, et al. Inhalation of carbon monoxide reduces skeletal muscle injury after hind limb ischemia-reperfusion injury in mice. Am J Surg. 2012;203(4):488–495.
132. Morse D, et al. Carbon monoxide–dependent signaling. Crit Care Med. 2002;30:S12–S17.
133. Hobson RW 2nd, et al. Heparinization reduces endothelial permeability and hydrogen ion accumulation in a canine skeletal muscle ischemia-reperfusion model. J Vasc Surg. 1988;7:585–591.
134. Abbruzzese TA, et al. Enoxaparin does not ameliorate limb ischemia-reperfusion injury. J Surg Res. 2008;147:260–266.
135. Stutzmann JM, et al. Neuroprotective profile of enoxaparin, a low molecular weight heparin, in in vivo models of cerebral ischemia or traumatic brain injury in rats: a review. CNS Drug Rev. 2002;8:1–30.
136. Levi M, et al. Beyond sepsis: activated protein C and ischemia-reperfusion injury. Crit Care Med. 2004;32:S309–S312.
137. Roemisch J, et al. A new look at the actions of a serine protease inhibitor. Blood Coagul Fibrinolysis. 2002;13:657–670.
138. Laszik Z, et al. Lack of suppressed renal thrombomodulin expression in a septic rat model with glomerular thrombotic microangiopathy. Lab Invest. 1994;70:862–867.
139. Duru S, et al. Antithrombin III pretreatment reduces neutrophil recruitment into the lung and skeletal muscle tissues in the rat model of bilateral lower limb ischemia and reperfusion: a pilot study. Acta Anaesthesiol Scand. 2005;49:1142–1148.
140. Wright JG, et al. Regional hypothermia protects against ischemia-reperfusion injury in isolated canine gracilis muscle. J Trauma. 1988;28:1026–1031.
141. Kang J, et al. The effects of systemic hypothermia on a murine model of thoracic aortic ischemia reperfusion. J Vasc Surg. 2010;52(2):435–443.
142. Motoyoshi N, et al. Establishment of a local cooling model against spinal cord ischemia representing prolonged induction of heat shock protein. J Thorac Cardiovasc Surg. 2001;122:351–357.
143. Black JH, et al. Regional hypothermia with epidural cooling for prevention of spinal cord ischemic complications after thoracoabdominal aortic surgery. Semin Thorac Cardiovasc Surg. 2003;15:345–352.
144. Cambria RP, et al. Epidural cooling for spinal cord protection during thoracoabdominal aneurysm repair: a five-year experience. J Vasc Surg. 2000;31:1093–1102.
145. Davison JK, et al. Epidural cooling for regional spinal cord hypothermia during thoracoabdominal aneurysm repair. J Vasc Surg. 1994;20:304–310.
146. El-Wahsh M. Liver graft preservation: an overview. Hepatobiliary Pancreat Dis Int. 2007;6:12–16.
147. Coselli JS, et al. Hypothermic circulatory arrest: safety and efficacy in the operative treatment of descending and thoracoabdominal aortic aneurysms. Ann Thorac Surg. 2008;85:956–963.
148. Kazui T, et al. The safety of moderate hypothermic circulatory arrest with selective cerebral perfusion. J Thorac Cardiovasc Surg. 2008;135:715.
149. Khaladj N, et al. Hypothermic circulatory arrest with selective antegrade cerebral perfusion in ascending aortic and aortic arch surgery: a risk factor analysis for adverse outcome in 501 patients. J Thorac Cardiovasc Surg. 2008;135:908–914.
151. Czura CJ, et al. Neural inhibition of inflammation: the cholinergic anti-inflammatory pathway. J Endotoxin Res. 2003;9:409–413.
152. Wang X, et al. Inhibition of factor Xa reduces ischemic brain damage after thromboembolic stroke in rats. Stroke. 2003;34:468–474.
153. Mioni C, et al. Activation of an efferent cholinergic pathway produces strong protection against myocardial ischemia/reperfusion injury in rats. Crit Care Med. 2005;33:2621–2628.
154. Uemura K, et al. Efferent vagal nerve stimulation induces tissue inhibitor of metalloproteinase-1 in myocardial ischemia-reperfusion injury in rabbit. Am J Physiol Heart Circ Physiol. 2007;293:H2254–H2261.
155. Borovikova LV, et al. Role of vagus nerve signaling in CNI-1493–mediated suppression of acute inflammation. Auton Neurosci. 2000;85:141–147.
156. Borovikova LV, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462.
157. Yeboah MM, et al. Cholinergic agonists attenuate renal ischemia-reperfusion injury in rats. Kidney Int. 2008;74:62–69.
158. Gourgiotis S, et al. Acute limb compartment syndrome: a review. J Surg Educ. 2007;64:178–186.
159. Tiwari A, et al. Acute compartment syndromes. Br J Surg. 2002;89:397–412.
160. Heemskerk J, et al. Acute compartment syndrome of the lower leg: retrospective study on prevalence, technique, and outcome of fasciotomies. World J Surg. 2003;27:744–747.
161. Rush DS, et al. Does open fasciotomy contribute to morbidity and mortality after acute lower extremity ischemia and revascularization? J Vasc Surg. 1989;10:343–350.
162. Ames A 3rd, et al. Cerebral ischemia. II. The no-reflow phenomenon. Am J Pathol. 1968;52:437–453.
163. May JW Jr, et al. The no-reflow phenomenon in experimental free flaps. Plast Reconstr Surg. 1978;61:256–267.
164. Laky D, et al. Myocardial stunning. Morphological studies in acute experimental ischemia and intraoperatory myocardial biopsies. Rom J Morphol Embryol. 2008;49:153–158.
165. Page B, et al. Persistent regional downregulation in mitochondrial enzymes and upregulation of stress proteins in swine with chronic hibernating myocardium. Circ Res. 2008;102:103–112.
166. Li C, et al. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol. 2002;282:C227–C241.
167. Maxwell SR, et al. Reperfusion injury: a review of the pathophysiology, clinical manifestations and therapeutic options. Int J Cardiol. 1997;58:95–117.
168. Forman MB, et al. Importance of tissue perfusion in ST segment elevation myocardial infarction patients undergoing reperfusion strategies: role of adenosine. Clin Cardiol. 2007;30:583–585.
169. Pereda D, et al. Elective cardiac surgery using Celsior or St. Thomas No. 2 solution: a prospective, single-center, randomized pilot study. Eur J Cardiothorac Surg. 2007;32:501–506.
170. Richling N, et al. Thrombolytic therapy vs primary percutaneous intervention after ventricular fibrillation cardiac arrest due to acute ST-segment elevation myocardial infarction and its effect on outcome. Am J Emerg Med. 2007;25:545–550.
171. Yamazaki S, et al. Effects of staged versus sudden reperfusion after acute coronary occlusion in the dog. J Am Coll Cardiol. 1986;7:564–572.
172. Cerqueira NF, et al. Pathophysiology of mesenteric ischemia/reperfusion: a review. Acta Cir Bras. 2005;20:336–343.
173. Oldenburg WA, et al. Acute mesenteric ischemia: a clinical review. Arch Intern Med. 2004;164:1054–1062.
174. Kaminsky O, et al. Does a second-look operation improve survival in patients with peritonitis due to acute mesenteric ischemia? A five-year retrospective experience. World J Surg. 2005;29:645–648.
175. Park WM, et al. Contemporary management of acute mesenteric ischemia: factors associated with survival. J Vasc Surg. 2002;35:445–452.
176. Yanar H, et al. Planned second-look laparoscopy in the management of acute mesenteric ischemia. World J Gastroenterol. 2007;13:3350–3353.
177. Tang PS, et al. Acute lung injury and cell death: how many ways can cells die? Am J Physiol Lung Cell Mol Physiol. 2008;294:L632–L641.
178. Landis C. Why the inflammatory response is important to the cardiac surgical patient. J Extra Corpor Technol. 2007;39:281–284.
179. Menger MD, et al. Pathomechanisms of ischemia-reperfusion injury as the basis for novel preventive strategies: is it time for the introduction of pleiotropic compounds? Transplant Proc. 2007;39:485–488.
180. D’Ascenzo F, et al. Remote ischaemic preconditioning in coronary artery bypass surgery: a meta-analysis. Heart. 2012;98(17):1267–1271.
181. Desai M, et al. Remote ischaemic preconditioning versus no remote ischaemic preconditioning for vascular and endovascular surgical procedures. Cochrane Database Syst Rev. 2011;(12).
182. Hausenloy DJ. Cardioprotection techniques: preconditioning, postconditioning and remote conditioning (basic science). Curr Pharm Des. 2012;19:4544–4563.
183. Frank A, et al. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Semin Cardiothorac Vasc Anesth. 2012;16(3):123–132.
184. Dirksen MT, et al. Reperfusion injury in humans: a review of clinical trials on reperfusion injury inhibitory strategies. Cardiovasc Res. 2007;74(3):343–355.
185. Steffens S, et al. The inflammatory response as a target to reduce myocardial ischaemia and reperfusion injury. Thromb Haemost. 2009;102(2):240–247.
186. Piot C, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359(5):473–481.
187. Lutz J, et al. Anti-inflammatory treatment strategies for ischemia/reperfusion injury in transplantation. J Inflamm (Lond). 2010;7:27.
188. de Rougemont O, et al. Preconditioning, organ preservation, and postconditioning to prevent ischemia-reperfusion injury to the liver. Liver Transpl. 2009;15(10):1172–1182.
189. Schnuelle P, et al. Effects of donor pretreatment with dopamine on graft function after kidney transplantation: a randomized controlled trial. JAMA. 2009;302(10):1067–1075.
190. Graor R, et al. Results of a prospective randomized trial evaluating surgery vs thrombolysis for ischemia of the lower extremities. Ann Surg. 1994;220:251–268.
191. Ouriel K, et al. A comparison of thrombolytic therapy with operative revascularization in the initial treatment of acute peripheral arterial ischemia. J Vasc Surg. 1994;19:1021–1030.
192. Ouriel K, et al. Thrombolysis or peripheral arterial surgery: phase I results. TOPAS Investigators. J Vasc Surg. 1996;23:64–73.
193. Ouriel K, et al. A comparison of recombinant urokinase with vascular surgery as initial treatment for acute arterial occlusion of the legs. Thrombolysis or Peripheral Arterial Surgery (TOPAS) Investigators. N Engl J Med. 1998;339:1105–1111.
194. Walker PM, et al. Lower limb ischemia: phase 1 results of salvage perfusion. J Surg Res. 1999;84:193–198.
195. Wilhelm MP, et al. Controlled reperfusion using a simplified perfusion system preserves function after acute and persistent limb ischemia: a preliminary study. J Vasc Surg. 2005;42:690–694.
196. Aivatidi C, et al. Oxidative stress during abdominal aortic aneurysm repair—biomarkers and antioxidant’s protective effect: a review. Eur Rev Med Pharmacol Sci. 2011;15(3):245–252.
197. Blaisdell FW. The pathophysiology of skeletal muscle ischemia and the reperfusion syndrome: a review. Cardiovasc Surg. 2002;10:620–630.
198. Blaisdell FW, et al. Management of acute lower extremity arterial ischemia due to embolism and thrombosis. Surgery. 1978;84:822–834.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]Chapter 7
Ischemia-Reperfusion
George P. Casale, Iraklis I. Pipinos
Based on a chapter in the seventh edition by Robert S. Crawford and Michael T. Watkins
The purpose of this chapter is to discuss the pathophysiology of severe acute ischemia, followed by acute reperfusion in the limbs and organs of the body. Ischemia-reperfusion (I/R) is a complex pathologic process involving intracellular and extracellular pathways that result in metabolic, thrombotic, and inflammatory changes in the affected tissues. A devastating component of I/R injury is the paradoxical increase in tissue damage associated with restitution of blood flow to ischemic tissues.
Haimovici1 described the development of myonephropathic syndrome in a few patients who underwent lower extremity revascularization after acute ischemia in the late 1950s. This report provided one of the first published clinical observations of limb I/R. These patients experienced ongoing lower extremity muscle necrosis and myoglobin-induced renal failure in the presence of palpable pulses. Cerra et al.2 completed an experimental animal and human clinical correlation of reperfusion in 1975. Histologic assessment of canine myocardium showed that restoration of blood flow was associated with the development of subendothelial hemorrhagic necrosis. Moreover, evaluation of the medical records and autopsy tissue from patients who died after aortic valve replacement provided evidence of subendothelial hemorrhagic necrosis in individuals subjected to more than 70 minutes of cardiopulmonary bypass. These findings implicated reperfusion injury as a cause of death in patients who underwent cardiopulmonary bypass for heart operations.
I/R injury has clinical relevance because it affects the outcome of patients after cardiac and vascular surgery, organ transplantation, plastic surgical reconstruction, and recovery from traumatic injury. The cellular damage that occurs as a consequence of I/R injury has been studied intensively in humans, experimental animals, and cell culture systems. To understand and characterize I/R injury from a biochemical perspective, it is useful to discuss this process in terms of its two components—ischemic injury and reperfusion injury.
Components of Ischemia-Reperfusion Injury
Injury During Ischemia
In the ischemic phase of I/R injury, the predominant mechanisms of injury result from tissue hypoxia or anoxia and stasis in the microcirculation (Fig. 7-1). The degree to which tissue from various organs is capable of tolerating ischemia varies widely and depends on each tissue’s baseline metabolic demand. Ischemia of human skeletal muscle under normothermic conditions is tolerated for more than 2 hours, whereas histologic evidence of ischemic injury develops in the jejunum after only 30 minutes of ischemia.3
Figure 7-1 Injury during ischemia. Decreased oxygen supply activates a complex cascade of metabolic, inflammatory, and prothrombotic pathways and sets the conditions for MPTP opening during reperfusion. ATP, adenosine triphosphate; MPTP, mitochondrial membrane permeability transition pore; ROS, reactive oxygen species.
During ischemia, mitochondria deprived of O2 can no longer produce adenosine triphospate (ATP) by oxidative phosphorylation; consequently, cellular (ATP) falls rapidly with a concomitant increase in adenosine diphosphate (ADP), adenosine mononphosphate (AMP), and Pi. Glycolysis is stimulated, but is unable to produce sufficient ATP to meet the needs of the cell. The intracellular pH decreases with accumulation of lactic acid, progressively inhibiting glycolysis and activating the Na+/H+ antiporter that pumps H+ out of the cell. Because of ATP deficiency, the entering Na+ ions are not pumped out of the cell by Na+/K+ ATPase. High intracellular Na+ inhibits the Na+/Ca2+ antiporter, producing high intracellular Ca2+, which, in turn, activates degradative enzymes, including phospholipase A2, and proteases, such as calpains. In addition, ATP deficiency impairs ATP-dependent repair processes.4 Consequently, prolonged ischemia will lead to cell membrane damage and necrotic cell death.
As ischemia progresses, reactive oxygen species (ROS) are produced by and accumulate within the mitochondria.4,5 Increased mitochondrial matrix ROS together with increased cytosolic Ca2+, elevated Pi, and depletion of adenine nucleotides are most of the conditions needed for opening of the mitochondrial membrane permeability transition pore (MPTP), which causes necrotic cell death.6,7 The data indicate that the increase of mitochondrial ROS during ischemia primes the mitochondrial MPTP for opening, and that the magnitude of ROS damage to the mitochondria is the most important factor that determines the duration of opening of the MPTP, and therefore, the extent of tissue damage.4 Pore opening, however, is inhibited by the low intracellular pH during ischemia, but occurs during reperfusion when the pH increases, as is discussed in “Injury During Reperfusion” section in this chapter.
Tissue hypoxia leads to mobilization of neutrophils into the interstitium, where they have both beneficial and deleterious effects on tissue during reperfusion.8 Migration of neutrophils and macrophages to sites of inflammation is dependent on hypoxia-adaptive pathways.9,10 Activated neutrophils release the soluble mediators glutamate11 and adenine nucleotides (in the form of ATP or AMP)12,13 during ischemia that are converted to adenosine at the vascular endothelial surface. Adenosine protects the function of the microvascular endothelial barrier by re-establishing endothelial cell-cell contact after neutrophil transmigration. Transcellular metabolism (neutrophils provide ATP as a substrate for enzymes located on the endothelial membrane) and signaling are enhanced by hypoxia-induced transcriptional increases in functional endothelial surface apyrase (CD39), 5′-ectonucleotidase (CD73), and adenosine receptors (AdoRA2B).13,14 Polymorphonuclear neutrophils have a deleterious effect on local tissue by releasing factors that can disrupt the endothelial barrier. Activation of neutrophils by β2 integrins stimulates neutrophils to release soluble factors that induce endothelial cytoskeletal rearrangement, gap formation, and increased permeability. One neutrophil-derived permeabilizing factor is heparin-binding protein (HBP), also known as azurocidin or CAP37. HBP induces Ca2+-dependent cytoskeletal changes in endothelial cells and triggers macromolecular leakage in vivo.15
Injury During Reperfusion
Reperfusion injury represents the complex response to tissue injury when blood flow is restored after ischemia (Fig. 7-2). There are metabolic, thrombotic, and inflammatory components of reperfusion injury. The degree to which reperfusion either restores tissue integrity or exacerbates ischemic injury depends primarily on the duration of the ischemia. It is paradoxical that moderate ischemia followed by reperfusion may cause more fulminant postischemic tissue injury than seen with ischemia alone. However, without reperfusion, the loss of function in the brain, gut, heart, or limb may have a more catastrophic outcome than if perfusion is not restored.
Figure 7-2 Injury during reperfusion. The return of oxygen, influx of inflammatory cells, and washout of metabolites contribute to cell death and an inflammatory, prothrombotic milieu that exacerbates tissue injury. Abs, Antibodies; ARDS, acute respiratory distress syndrome; DAMPS, danger-associated molecular patterns; METC, mitochondrial electron transport chain; MPTP, mitochondrial membrane permeability transition pore; NADH, reduced nicotinamide adenine dinucleotide; NF-κB, nuclear factor kappa B; ROS, reactive oxygen species; TLRs, toll-like receptors.
Reperfusion is characterized by a burst of ROS production (primarily by the mitochondrial electron transport chain), increased Ca2+ uptake into the mitochondrial matrix, and a shift of the intracellular pH from acidic toward neutral.6 Mitochondrial oxidative damage leading to programmed cell death is the major cause of tissue loss produced by reperfusion.16 The two forms of programmed cell death during reperfusion are apoptosis and necrosis.
Increased ROS generated during both ischemia and reperfusion can initiate apoptosis and necrosis, cell death programs mediated through the mitochondria. The BCL-2 family of proteins is central to regulating mitochondrial apoptosis. Antiapoptotic members, including BCL-2 and BCL-XL, act by blocking proapoptotic members, including BAX and BAK. The latter initiate apoptosis by increasing the permeability of the outer mitochondrial membrane, causing the release of proapoptotic proteins, such as cytochrome c.17 BH3-only proteins, proapoptotic BCL-2 family members, can initiate apoptosis by directly activating BAX and BAK or by blocking BCL-2 and BCL-XL.17 ROS initiate mitochondrial apoptosis by activating BH3-only proteins or by triggering opening of the mitochondrial MPTP, causing release of cytochrome c.7 Alternatively, apoptosis may occur via ROS-dependent lipid peroxidation of cardiolipin, a mitochondria-specific phospholipid of the inner membrane, which binds strongly to cytochrome c. The latter dissociates from peroxidized cardiolipin and is released from the inner membrane.18 The evidence, however, suggests that ROS-dependent necrosis, rather than apoptosis, is the main mechanism of cell death caused by I/R injury.6,7,19
Programmed necrotic cell death may be mediated by signaling via a tumor necrosis factor (TNF) death receptor20–22 or by direct opening of the mitochondrial MPTP.16,17 Both processes require calcium and oxidative stress.16,17 TNF receptor 1 (TNFR1) signaling for necrotic cell death begins with a conformational change caused by binding of TNF to the receptor. This conformational change promotes binding of cytosolic TNF receptor-associated death domain (TRADD) to a cytosolic domain of the receptor, initiating formation of a complex consisting of receptor-interacting protein 1 (RIP1) kinase, TNF receptor-associated factor 2/5 (TRAF2/5), cellular inhibitor of apoptosis 1 (cIAP1) and cellular inhibitor of apoptosis 2 (cIAP2). The TNFR1 complex is internalized and produces secondary cytosolic complexes, including the necrosome. The latter comprises Fas-associated death domain (FADD) protein, inactivated caspase 8, RIP1, and receptor-interacting protein 3 (RIP3). Activation of necrotic signaling requires phosphorylation of RIP1 and RIP3 and is critically dependent on ROS production by the mitochondrial electron transport system.21 The ultimate effector in this pathway is not yet established; however, the present data suggest that signaling leads to opening of the mitochondrial MPTP.20
Direct opening of the mitochondrial MPTP is central to tissue injury caused by ischemia and reperfusion.16,17,19 The mitochondrial MPTP is a nonselective protein channel spanning the inner and outer mitochondrial membranes at points of contact. When open, the pore permits free passage of water and solutes less than 1.5 kDa, causing osmotic damage to the mitochondria and uncoupling mitochondrial oxidative phosphorylation, the major source of cellular ATP, by dissipation of the pH gradient and the electrical potential across the inner mitochondrial membrane.4,6 Mitochondrial damage and collapse of oxidative phosphorylation promote disruption of metabolism and ionic homeostasis initiated before pore opening during ischemia, and thereby, promote and expand necrotic cell death. The composition of mitochondrial MPTP is not yet solved, but the voltage-dependent anion channel (VDAC), adenine nucleotide translocase (ANT), and mitochondrial phosphate carrier (PiC) appear to be constituents.6 Cyclophilin D (CyP-D), a prolyl isomerase in the mitochondrial matrix, is an essential component of the mitochondrial MPTP and the central regulator of pore opening.16 Pore opening depends on mitochondrial calcium concentration (Ca2+ m) but is relatively insensitive to Ca2+ m per se, occurring at very high Ca2+ m. The sensitivity of pore opening to Ca2+ m is greatly enhanced by oxidative stress caused by increased mitochondrial ROS production during ischemia and a surge of mitochondrial ROS production during reperfusion.6 Oxidative stress promotes binding of CyP-D to an ANT/PiC dimer, facilitating a Ca2+-dependent conformational change in these proteins and opening of the mitochondrial MPTP. The evidence supports activation of CyP-D via binding of p53, which accumulates in the mitochondrial matrix in response to oxidative stress.16 Low intracellular pH that occurs during ischemia inhibits pore opening; however, during reperfusion, when blood flow is restored, the intracellular pH increases, and the mitochondrial MPTP opens.6
Reperfusion of ischemic tissue produces a burst of ROS production by the mitochondrial ETC and by parenchymal and endothelial cell reduced nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase and xanthine oxidase, neutrophils and macrophages, and necrotic cell death with opening of the MPTP.23 These processes lead to release of normally sequestered cellular proteins and protein fragments in addition to extracellular and cell surface accumulation of oxidatively modified proteins and lipids. Some of these structures have molecular configurations recognized by Toll-like receptors (TLR) of the innate immune system as “danger-associated molecular patterns” (DAMPS),24 and a growing body of work supports TLR-mediated inflammation as central to I/R injury.25 Reperfusion-associated DAMPS include heat shock protein 60 (HSP60), a ligand for TLR4; high-mobility group protein B1 (HMGB1), a ligand for TLR2, 4, and 9; low-molecular-weight hyaluronic acid, a ligand for TLR2 and 4; the extra domain A of fibronectin (EDA), a ligand for TLR2 and TLR4; and cardiac myosin, a ligand for TLR2 and 8.26 Ligand binding to TLRs present on parenchymal cells, endothelial cells, and circulating leukocytes activates TLR signaling pathways that lead to apoptosis or to nuclear factor kappa B (NF-κB) signaling.26 Inflammation induced by TLR-mediated NF-κB signaling is a significant contributor to I/R injury.25 NF-κB is a transcription factor present in the cytosol in an inactive complex with inhibitor of NF-κB (I-κB). Signaling via TLRs leads to removal and proteolysis of I-κB, permitting NF-κB to translocate to the nucleus, where it initiates transcription of proinflammatory genes, including those that code for TNF-α, interleukin-1β (IL-1β), and IL-6, as well as intercellular adhesion molecule-1 (ICAM-1), all of which recruit neutrophils and macrophages to the damaged tissue.23,25,27 In addition, NF-κB signaling may be initiated by binding of TNF to the TNFR1 receptor.21
Activation of innate immunity in response to DAMPS, during reperfusion, is not limited to their recognition by TLRs, but also occurs in response to their recognition by natural antibodies.27 These antibodies belong predominantly to the immunoglobulin-M (IgM) class, are secreted by B-1 cells (a specialized subset of B-cells) and are germline encoded.24 Many of these antibodies recognize epitopes formed by oxidative modification of lipids and proteins (e.g., during reperfusion). Some recognize normally sequestered self-antigen, for example, non-muscle myosin heavy chain II released as a consequence of cell injury during both ischemia and reperfusion.28 Reperfusion elicits an inflammatory response dependent on activation of complement by natural IgM antibodies.28 Binding of IgM natural antibodies to target molecules on the cell surface or in the extracellular environment activates the complement system, causing production of cleavage products, including complement component C3b, which tags cells and other targets for removal by macrophages, and C5a, a fragment chemotactic for neutrophils.27 Alternatively, the complement system may be activated in the absence of antibody by mannose-binding lectin (MBL) bound to cell surface carbohydrates.29 Complement activation, detected as tissue-bound C3b and MBL, occurs during ischemia and increases during reperfusion.30
A variety of inflammatory mediators are delivered to reperfused tissue by blood-borne cells (macrophages, lymphocytes, neutrophils, mast cells, platelets).31,32 Noncellular elements, such as the complement system, ROS, nitric oxide (NO), and proinflammatory and anti-inflammatory cytokines are also believed to modulate the complex scenario of reperfusion injury. The presence of O2, washout of lactatic acid, development of tissue edema, and influx of numerous cell types during reperfusion produce a complex environment in which numerous mediators contribute to inflammation and tissue injury. The cellular and molecular components of the I/R response will be discussed individually.
Leukocytes
In contrast to neutrophil activation during ischemia (discussed previously), neutrophil activation during reperfusion is characterized by a logarithmic increase in neutrophil chemotaxis, neutrophil–endothelial cell adhesion, and transmigration. In contrast to the slow transendothelial migration of resident neutrophils in the static ischemic environment, during reperfusion, flow modulates the interaction between neutrophils and endothelium. Transendothelial migration during reperfusion occurs in sequential steps: rolling, adherence, and transendothelial migration.9 The first step is initiated by increases in the surface expression of endothelial P-selectin (CD62P); CD62P interacts with its leukocyte ligand P-selectin glycoprotein ligand 1 (PSGL-1), which results in leukocyte rolling. Firm neutrophil adherence is modulated by interaction of the β2 integrins CD11a/CD18 and CD11b/CD18 with endothelial ICAM-1. Transmigration of leukocytes is facilitated by platelet–endothelial cell adhesion molecule-1 (PECAM-1), which is constitutively expressed along endothelial cell junctions. Once inside the interstitium, activated leukocytes release toxic ROS, proteases, and elastases.31 These substances contribute to increased microvascular permeability, edema, thrombosis, and parenchymal cell death.32,33 Neutrophil accumulation in the extravascular compartment is facilitated by chemotactic agents, including complement components C3a and C5a, chemokines such as IL-8, DAMPs, and leukotriene B4 produced by arachidonic acid metabolism.34 The release of chemotactic agents creates a gradient that attracts neutrophils from the intravascular space toward the interstitium.34–36
Cytokines-Chemokines/Antagonists
Multiple growth factors and cytokines are present at sites of inflammation, and each can influence the nature of the inflammatory response.37 There is a constant interaction between proinflammatory and anti-inflammatory signals in the vascular wall (Box 7-1).
Liver and skeletal muscle I/R have been correlated with increased levels of local and systemic cytokines that are believed to modulate local38–40 and systemic injury.41,42 TNF41,43 and macrophage inflammatory protein-2 (MIP-2) levels are markedly increased in hind limbs exposed to acute limb I/R.43,44 Direct cytokine antagonists (receptor antagonists and anticytokine antibodies),45 metabolic rescue agents,43,46 and anti-inflammatory molecules47–51 have been shown to ameliorate skeletal muscle injury and decrease tissue levels of proinflammatory cytokines when administered to animals during limb I/R. Recent literature suggests that administration of an IL-1 receptor antagonist attenuates spinal cord I/R injury by reducing neuronal necrosis and apoptosis.52 Administration of this same IL-1 receptor antagonist has also been shown to ameliorate renal I/R injury in animal experiments.53 Because this receptor antagonist is clinically used to treat rheumatoid arthritis, it may soon be used in clinical trials of I/R injury in humans. A word of caution regarding inhibition of cytokines or their receptors comes from the work of Shireman et al,54 who showed that chronic deficiency of monocyte chemoattractant protein-1 resulted in a decrease in the inflammatory response, as expected, but also inhibited skeletal muscle regeneration after chronic limb ischemia.
Mitochondrial Injury
The mitochondria are a dynamically growing and shrinking network of double-membrane organelles that comprise the primary energy-generating system of eukaryotic cells. Energy is generated by coupling ATP production with oxidative transfer of electrons from reduced substrates to molecular O2 (i.e., oxidative phosphorylation).55 This process of energy conversion is mediated by the mitochondrial electron transport chain that consists of five multiunit protein complexes tightly configured within the mitochondrial inner membrane.56 The largest of these is Complex I, which consists of 46 protein subunits and 9 redox centers, 1 of which is flavin mononucleotide (FMN) located at the terminus of the hydrophilic arm extending into the mitochondrial matrix. High-energy electrons from reduced nicotinamide adenine dinucleotide (NADH) enter the ETC by reducing the FMN of Complex I. The electrons pass through Complex I to its coenzyme Q (CoQ) reductase site located in the inner membrane and where CoQ is reduced. The latter shuttles the electrons to Complex III, which couples oxidation of reduced CoQ with reduction of (passage of the electrons to) cytochrome c located in the intermembrane space. Finally, the electrons are passed to Complex IV, which couples oxidation of cytochrome c with reduction of molecular O2 to water. Complex II can also shuttle electrons to CoQ by coupling conversion of succinate to fumarate with reduction of flavin adenine dinucleotide (FAD), which is present at the mitochondrial matrix side of the complex. Importantly, passage of two electrons through Complexes I, III, and IV is coupled with transfer of protons from the mitochondrial matrix across the inner membrane to the intermembrane space (i.e., a total of eight protons per electron pair).56 This process generates a pH gradient and a membrane potential used by Complex V to generate ATP from ADP and Pi.
Approximately 90% of inhaled O2 is consumed by mitochondria and reduced to water to produce energy. Under optimal physiologic conditions, 2% to 4% of the O2 consumed by mitochondria can capture single electrons that leak along the complexes of the electron transport chain generating the ROS superoxide anion (O•−).57 Oxidative stress caused by excess accumulation of ROS is held in check by mitochondrial superoxide dismutase, which converts it to H2O2 (another ROS) and catalase, which converts the latter to H2O and O2.57 During tissue ischemia, when O2 tension in the mitochondria is reduced (hypoxia), ROS generation paradoxically is increased, overwhelming oxidative defense enzymes and causing an excessive accumulation of ROS (oxidative stress).5 This happens because under tissue hypoxia with reduced availability of O2 as the final electron acceptor of the ETC, the NADH/NAD+ ratio in the mitochondrial matrix increases, thereby saturating the FMN of Complex I with electrons. Saturated FMN transfers electrons to available O2 in the matrix, producing O•−.57 This FMN-dependent process is amplified by (1) the accumulation of reduced CoQ generated by both Complex I and II under the condition of low electron flow, and (2) the presence of a pH gradient across the inner membrane. Under these conditions, present during ischemia, electrons back up through Complex I to its FMN, where electrons are passed to O2, generating O•−.57 With the onset of reperfusion, O2 returns to meet the conditions of increased NADH/NAD+, accumulated reduced CoQ, electron-saturated FMN, and an incapacitated electron transport chain. The surge of O2 thus provides a sink for flow of electrons from reduced FMN leading to a burst of O•−. The duration of this burst, and therefore, the extent of acute reperfusion damage are dependent on the duration and extent of ROS accumulation in the preceding ischemic phase of I/R. The evidence supports Complex I as the main source of this reperfusion burst of ROS generation.4
Complement
The complement system has long been recognized as an important mediator in innate immune defense and inflammation.58 Complement can be activated by any of three pathways—the antibody-dependent classical pathway, the alternative pathway, or the MBL-associated serine protease pathway. The classical pathway is initiated when IgM or IgG antigen-antibody complexes bind to C1, the first component of complement. Activation of the alternative pathway is triggered by microbial surfaces and complex polysaccharides, and results in the generation of C3. The lectin pathway is initiated by binding of the MBL protein to mannose and glucosamine residues on bacterial cell walls. Activation of these proteins results in cleavage of complement factors 4 and 2, followed by subsequent activation of C3.
There is considerable evidence to support a role of the complement system in hind limb I/R injury. In a model of rodent hind limb I/R, there was direct correlation between increased complement deposition, tissue injury, and increased glycolysis.59 Complement receptor 2–deficient (CR2−/−) mice exhibit deficient IgM secretion and are protected against I/R injury by defective B cells.60 IgM-secreting B-cells clones were studied from CR2 knockout mice and wild-type mice. Only one clone was found to restore injury to previously protected mice, thus suggesting that a single clone of self-reactive IgM can initiate complement-dependent I/R injury.61 In a follow-up study, peptides that bound the IgM clone were found to block injury in wild-type mice, a novel observation providing a potential therapeutic venue.62 Complement inhibition has been demonstrated to improve skeletal muscle contractility after I/R.63 C5-deficient mice have been shown to exhibit decreased lung vascular permeability, lung myeloperoxidase levels, and serum alanine aminotransferase levels, along with less skeletal muscle injury after reperfusion.64 Because the complement system is well described, and numerous inhibitors are available for each pathway, a beneficial role for these inhibitors in the brain, intestine, liver, and myocardium has been described.58
Nitric Oxide
NO is known to regulate vascular tone and to have potent anti-inflammatory and antithrombotic properties at the endothelial surface. It is also known to have concentration-dependent toxic effects on tissue and organ systems.65,66 The primary physiologic effect of NO results from stimulation of the activity of guanylate cyclase, accumulation of intracellular cyclic guanosine monophosphate, and a consequent decrease in intracellular calcium that results in relaxation of smooth muscle, decreased cardiac contractility, and reduced platelet and inflammatory cell activation. NO is produced from L-arginine by oxidation of one or two equivalent guanido nitrogens by NO synthase (NOS) in a reaction that requires O2, and the cofactors NADPH and tetrahydrobiopterin. There are at least three distinct genes for NOS; neuronal NOS (nNOS or NOS-1) and endothelial NOS (eNOS or NOS-3) are dependent on micromolar concentrations of calcium for activity and are constitutively expressed. The inducible isoform, iNOS (or NOS-2), is calcium independent and widely expressed in a variety of cells after exposure to specific stimuli. iNOS is often referred to as the high-output source of NO because it can be strongly induced by proinflammatory stimuli. However, the enzyme does not produce NO at a rate substantially greater than does eNOS or nNOS; rather, more of the protein can be transiently induced and activated at normal levels of calcium.67,68
One of the first observations implicating NO as a marker of ischemic injury was made by Beckman.65 His laboratory went on to propose that the production of NO and O2 radicals by neurons when the ischemic or hypoxic brain is reperfused might contribute to cerebral injury. Ischemia is believed to depolarize neuronal membranes and cause synaptic discharge of the excitatory neurotransmitter glutamate, which, in turn, opens the voltage-dependent, N-methyl-d-aspartate–specific glutamate receptor/ionophore and allows calcium to accumulate in the neuron. Calcium then activates an O2-dependent nNOS that oxidizes arginine to produce NO when O2 is readmitted to the brain by reperfusion. NO reacts with the O2 radical superoxide (O•−), also produced by reperfusion, to form peroxynitrite (ONOO−). ONOO− has been shown to interact directly with proteins (i.e., the mitochondrial electron transport chain, prostaglandin synthase) and amino acids (oxidizes critical cysteine residues, leading to deactivation of proteins), and triggers membrane lipid peroxidation, thereby altering membrane permeability.69 ONOO− is a powerful oxidant capable of nitrating phenolic moieties, such as tyrosine or tyrosine residues in proteins, and increases after traumatic brain injury.70 This powerful molecule also activates mitochondrial-dependent apoptosis, poly (ADP-ribose) polymerase (PARP),69 and NF-κB.71 Alternatively, some in vitro studies have shown that ONOO− may inhibit the generation of NF-κB.72,73 ONOO− is believed to diffuse for several micrometers before decomposing to form the powerful and cytotoxic oxidants hydroxyl radical and nitrogen dioxide. Beckman’s hypothesis is consistent with available evidence on the protective action of glutamate antagonists and O2 radical scavengers in limiting cerebral infarction after focal ischemia.66 Furthermore, ONOO− decomposition catalysts are known to limit reperfusion injury in a variety of organ systems.74
There are also anti-inflammatory effects of NO that deserve mention. NO donors have been shown to ameliorate tissue injury in in vivo and ex vivo models of intestinal, neuronal, and hepatic I/R.75–77 NO is known to inhibit activation of NF-κB,78,79 which regulates a wide variety of proinflammatory cytokines and adhesion molecules. NO alone is an important antiatherosclerotic autacoid with antiaggregatory effects on platelets and with antioxidant, anti-inflammatory, and antiproliferative effects on the vasculature.80 NO inhibits vascular permeability in the postischemic intestinal microcirculation, probably by preventing endothelial cell contraction and gap formation.81 Endogenous NO also inhibits neutrophil adhesion to endothelium.82 Finally, NO regulates the activation of platelets by decreasing aggregation and adhesion responses.83 The duration of ischemia, magnitude of the injury, and specific organ bed will determine whether NO has a protective or deleterious effect. In contrast to the deleterious effects of NO when synthesized in large amounts by inflammatory cells via iNOS, the protective effects of NO appear to be related primarily to endogenous sources of constitutively synthesized NO generated by eNOS or nNOS or exogenous pharmacologic doses of NO donors.84
Endothelin
Endothelin (ET), a 21-amino acid peptide with potent vasoconstrictor properties,85 is produced mainly in endothelial cells, yet it is present throughout the entire cardiovascular system. Shear stress, growth factors, vasoactive hormones, and I/R are major stimuli for its release. Three distinct human ET-related genes have been cloned: “classic” ET-1, ET-2, and ET-3.86 ET-1 is the most predominant isoform in blood vessels and is synthesized by endothelial cells. ET-1 is produced as a precursor peptide that is cleaved by a converting enzyme into its active form. ET plays a major role in the regulation of vascular tone through its potent vasoconstrictive actions. ET acts on two different types of G-protein–coupled receptors: ET-A and ET-B. ET-1, the most potent vasoconstrictor, mediates its effects mainly through ET-A receptors present on vascular smooth muscle cells.87 ET-B receptors mediate both vasodilatory effects of ET through prostacyclin and NO (ET-B1) and vasoconstrictive effects on vascular smooth muscle (ET-B2).88,89
Increased production, upregulation of its receptors, enhanced sensitivity, and potentiation of the action of serotonin and norepinephrine all potentiate ET’s actions during I/R.90,91 Bosentan, a mixed ET-A/ET-B endothelin receptor antagonist, was shown to exert a marked tissue protective effect, as assessed by histologic evaluation in a rat model of myocardial I/R. This effect was probably the result of antioxidant protection derived from preservation of levels of the antioxidant enzymes superoxide dismutase and catalase in the treated animals.92 McMurdo et al93 showed that the vasoconstrictive effects of ET-1 in the coronary vasculature of the rabbit were primarily mediated by the ET-A receptor. However, FR139317, an ET-A receptor antagonist, failed to decrease infarct size after the induction of ischemia and reperfusion in that same model.93 A more contemporary assessment of the action of BQ-123, a selective ET-A receptor antagonist, revealed decreased myocardial levels of lipid peroxidation; an increase in NO, glutathione, catalase, and superoxide dismutase activity; and a decrease in the ratio of infarcted area to area at risk compared with I/R alone.94
[/not-level-membership-for-surgery-category]
