Chapter 12
Venous Pathology
Peter K. Henke
Veins are complex organs, much like arteries, and well suited to their physiologic purpose. Venous diseases are common in the general population and are influenced by genetics, environment, and acquired conditions (Fig. 12-1). Understanding the basic physiologic and molecular responses to venous injury is essential for designing effective and safe therapies.
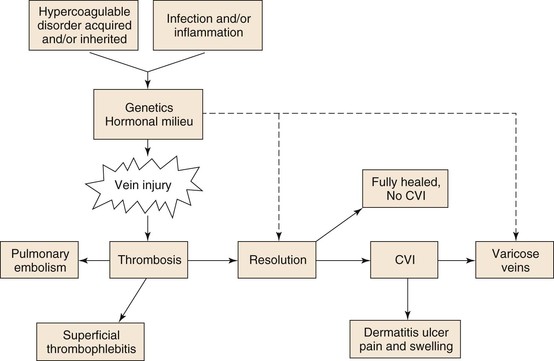
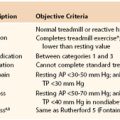
Figure 12-1 Hypercoagulable, acquired, or inherited disorders as well as infection, inflammation, and stasis, in conjunction with the patient’s physiologic milieu and genetics, affect vein wall injury. This injury may lead to thrombosis, vein injury, or varicose veins. Of those patients in whom thrombosis develops, a pulmonary embolism may occur or thrombosis resolves with no sequela. More commonly however, chronic venous insufficiency (CVI) develops, which may potentiate varicosities as well as lead to postthrombotic vein injury and dermatitis with ulcers, pain, or swelling.
Basic Considerations
Venous Cellular and Developmental Concepts
The physiology of veins has become better studied over the last several decades. Veins are not passive conduits but rather have regional and anatomically specific functions that contribute in diverse ways to organism homeostasis. Numerous differences exist between arteries and veins, including the physical structure of the vein wall, the presence of valves, the fact that veins carry deoxygenated blood, and the magnitude of shear and wall stresses (Table 12-1).
Table 12-1
Artery and Vein Comparisons
| Artery | Vein | |
| Embryology | Branchial arches | Cardinal veins |
| Cellular markers | EphB2, delta-like ligand 4, AIK-1, EPAS-1 | EphB4, NRP2, COUP-TF II |
| Hemostasis mechanism | Platelet-mediated, extrinsic pathway, exposed subendothelial collagen | Tissue factor– and fibrin-mediated, extrinsic pathway, cell adhesion molecules, von Willebrand factor –platelet |
| Response to injury | Plaque formation; occlusion | Thrombosis; fibrosis |
| Vasoregulation mechanisms | Adrenergic > endothelin Nitric oxide |
Endothelin > adrenergic Nitric oxide |
| Anticoagulant mechanisms | Thrombomodulin, tissue plasminogen activator, tissue factor pathway inhibitor | Thrombomodulin, endothelial protein C receptor , urinary plasminogen activator |
The embryonic vascular primordia of both arteries and veins are progenitor hematopoietic stem cells that arise in the yolk sac. These stem cells, called “blood islands,” separate into peripheral cells that differentiate into formed vascular structures. The initial angiogenic capillaries, which are essentially endothelial tubes, recruit mesenchymal stem cells that become smooth muscle cells, secrete matrix, and are followed later by adventitial fibroblasts.
Research now suggests that vascular morphogenesis is primarily related to morphologic changes in endothelial stem cells that depend on vascular endothelial growth factor (VEGF), the NOTCH signaling pathway, and the transforming growth factorβ (TGF-β)/bone morphogenic protein (BMP) signaling pathways.1 Angiogenic sprouting is dependent on VEGF, whereas arterial and vein identity depends on NOTCH signaling. Not surprisingly, arteries and veins express different embryologic endothelial molecular markers. For example, venous endothelium–specific genes include EPHB4, neurophilin-2, and COUP-TFII.2 These signaling pathways may mediate venous and arterial development. For instance, suppression of NOTCH signaling by COUP-TFII (chicken ovalbumin upstream promoter transcription factor II) is essential for vein identity.3 The pathways of vascular morphogenesis have also become better delineated in zebrafish and other basic models. Disruption of the bone morphogenic protein–chordin pathway results in severe experimental venous malformations in experimental models, depending on where anatomically the inhibition occurs.4
Although embryonic vasculogenesis may differ from adult vasculogenesis, interaction between the endothelium and the basement membrane matrix is critical for certain processes, including endothelial cell migration, proliferation, and survival. Laminin, integrins, and glycoproteins are all important components that direct intracellular signaling.5 How these processes specifically relate to venous development has not been as well studied but is probably similar to the relationship in arteriogenesis.
The macroembryology of veins is well described and is more variable than arterial development. The embryologic origin of the venous system is the cardinal veins, which form the main venous drainage system in the embryo, consisting of the anterior and posterior segments. Other venous structures are formed between the fifth and seventh weeks of embryogenesis, including the supracardial and sacrocardinal veins. Because there are multiple embryologic trunks, numerous anomalies can develop from the pairing of these basic venous structures and contribute to the variability of venous anatomy in individuals.
The histologic structure of veins is similar to that of all vessels, consisting of an intima (the endothelium with basement membrane), a thin medial layer composed of smooth muscle cells and a surrounding matrix, and an adventitia. In normal veins, the muscle layers are composed of smooth muscle cells, which are spindle shaped and have a contractile phenotype.6 These cells lie in close proximity to one another in parallel arrays and are surrounded by bundles of regularly arranged collagen fibers.
The vein wall endothelium separates the intravascular and extravascular spaces and has a regulated and selective function.7 Endothelial gap junctions, although nonfenestrated, regulate the diapedesis of leukocytes as well as efflux of plasma and macromolecules, which has been best studied on the high endothelial venules. Vein wall endothelial cells are shorter and broader than arterial wall cells and are not aligned to flow, in contrast to arterial endothelium.2
Venous Biomechanics
The biomechanics of veins suggest excellent adaptation for their function. Veins allow a very large volume capacitance, and tonal regulation, to rapidly redistribute overall blood volume. Approximately, 60% to 80% of circulating blood is stored in the venules and the systemic veins at any given time. The function of the blood capacitance system, via vasoregulation, is to maintain the filling pressure of the heart as well as to compensate for orthostatic changes. The physiology of venous blood flow in the limb related to the calf muscle pump and other actions is detailed in Chapter 11.
Everyday activities and changes in body position cause large changes in venous pressure. The average venous pressure at the foot is approximately 100 mm Hg in a person 5 feet 10 inches tall and weighing 75 kg. This pressure drops significantly with ambulation and while the person is recumbent. The venous valves are endothelium-lined folds of tunica intima that allow directional flow and contribute to this pressure reduction as well as maintaining prograde blood flow. To accommodate pressure and volume changes, veins undergo complex alterations in shape, depending on the blood volume, resistance, and the amount of blood flow within the system. Less vascular resistance occurs with a circular shape than an elliptical shape, and thus as venous volume increases, resistance to flow lessens.
Unlike arteries, large veins lack an extensive elastic lamella (composed of elastin) but exhibit marked distensibility. Veins have a much smaller ratio of wall thickness to radius and higher incremental distensibility in the low-pressure range then arteries do, thus indicating that the elastic modulus of veins can greatly exceed the stress modulus of arteries. As a result, veins have a high breaking pressure, nearly four atmospheres.8 Much of the stress-bearing function of the vein wall may depend on its smooth muscle cell and elastin content, in contrast to the abundance of collagen in the arterial wall. Indeed, vein wall compliance is decreased after experimental venous thrombosis (VT) injury, which correlates with its increased collagen content,9 and disrupted elastin, as measured histologically.10
Venous Vasoregulation
Whereas the elastic properties of the vein wall provide passive tone, active vasoregulation is provided by smooth muscle cells in the medial layer mediated through sympathetic nerves, as well as vasoactive circulating mediators, as in arteries. Central responses include changes in hormones, body temperature, blood volume, physiologic stress, and other conditions. Typical adrenergic agonists are noradrenalin and epinephrine, whereas acetylcholine causes both constriction and relaxation. Locally, vasodilation and vasoconstriction are mediated by endothelium-derived relaxing factor, namely, nitric oxide (NO) and the constricting agent endothelin-1. Stimulators for NO include muscarinic activation, thrombin, and α2-adrenergic agents. Interestingly, the venous endothelium synthesizes less NO and more endothelin-1 than the endothelium of arteries.11 However, the endothelium is a major regulator of venous tone, through similar mechanisms that regulate arterial tone.12 The vein wall also produces vasoactive prostanoids, both intraluminal and extraluminal, that alter tone. For example, prostacyclin synthesis in the vein wall is stimulated by numerous substances and by mechanical stretching and promotes venodilation. Other direct and indirect vasoregulators include angiotensin II, bradykinin, histamine, serotonin, and vasoactive intestinal peptide.
The local physiologic environment also affects venous tone, particularly hypoxemia and the local pH. For example, decreased pH, elevated PCO2, and elevated lactate values correlate with decreased contractility in isolated vein ring experiments.13 Age-related declines in vasodilatory and vasoconstrictor responses occur and may contribute to the demographics of venous disease, which is much more common with advancing age.14
Blood flow rates can also affect venous tone independently of the pressure. Mechanically, vasoregulation occurs with pulsatile blood flow and increased endothelial shear stress,15 which induces synthesis of various substances. In a manner similar to that in arteries, the Na, K, and Ca pumps are also important at the cellular level of venous regulation. Interestingly, the local ionic environment of vein walls may contribute to development of essential hypertension, with greater amounts of sodium ions in juxtaposition with the vein walls in hypertensive patients than in nonhypertensive patients.16
Acute Deep Venous Thrombosis
Venous thromboembolism (VTE) is a significant health care problem in this country, with an estimated 900,000 cases of deep venous thrombosis (DVT) and pulmonary embolism (PE), causing approximately 300,000 deaths yearly.17 For the past 150 years, understanding the pathogenesis of VTE has centered on Virchow’s triad of stasis, changes in the vessel wall (now recognized as injury), and thrombogenic changes in the blood. Stasis is probably permissive, and not a direct cause, whereas systemic infection and systemic inflammation may be more causal than previously thought.18,19
Venous Thrombosis Pathways
Hemostasis is typically initiated by damage to the vessel wall and disruption of the endothelium, although it may be initiated in the absence of vessel wall damage, particularly in venous thrombosis.20 Vessel wall damage simultaneously results in release of tissue factor (TF), a cell membrane protein, from injured cells and the circulation, with subsequent activation of the extrinsic pathway of the coagulation cascade. These two events are critical to the activation and acceleration of thrombosis. Tissues also vary with regard to their susceptibility to thrombosis, and the local organ mechanisms may be somewhat different. For example, hemostasis in cardiac muscle may be more dependent on the extrinsic pathway for thrombosis, whereas skeletal muscle may be more dependent on the intrinsic pathway for thrombosis.21
Platelet activation and the formation of an effective hemostatic “platelet plug” is a primary thrombotic event. Two platelet activation routes are thought to exist physiologically.22 Without direct vessel damage, platelet activation may occur via TF de-encryption and activation by protein disulfide isomerase, with factor VIIa generation and activation of platelets. Alternatively, subendothelial collagen may directly bind to glycoprotein (GP) VI and von Willebrand factor (vWF), leading to platelet capture and activation.
Platelet interactions and activation are mediated by vWF, whose receptor is GPIb, and via GPIIb/IIIa to fibrin.23Activation of platelets leads to the release of the prothrombotic contents of platelet granules, which contain receptors for coagulation factors Va and VIIIa. In addition, platelet activation also leads to the elaboration of arachidonic acid metabolites such as thromboxane A2, further promoting platelet aggregation (as well as vasoconstriction). Changes in platelet shape result in exposure of negatively charged procoagulant phospholipids normally located within the inner leaflet of the platelet membrane.24 Platelets also release microparticles (MPs), rich in TF and other procoagulants, which accelerate and concentrate the thrombus generation. Interestingly, circulating TF may be more important in venous thrombosis than in arterial thrombosis.20,25
The extrinsic pathway begins with activation of factor VII, by complexing TF with factor VII. The TF-VIIa complex then activates factors IX and X to IXa and Xa, respectively, in the presence of calcium. Feedback amplification occurs with factors VIIa, IXa, and Xa, all of which are capable of activating VII to VIIa, especially when bound to TF.26 Factor Xa is also capable of activating factor V to Va. Factors Xa, Va, and II (prothrombin) form on the platelet phospholipid surface in the presence of Ca2+ and initiate the prothrombinase complex, which catalyzes the formation of thrombin from prothrombin. Thrombin feedback amplifies the system by activating not only factor V to Va but also factors VIII (normally circulating bound to vWF) to VIIIa and XI to XIa. After activation, factor VIIIa dissociates from vWF and assembles with factors IXa and X on the platelet surface in the presence of Ca2+ to form a complex called the Xase complex, which catalyzes the activation of factor X to Xa.
Thrombin (factor II) is central to coagulation through its action of cleavage and release of fibrinopeptide A (FPA) from the α chain of fibrinogen and fibrinopeptide B (FPB) from the β chain of fibrinogen. This causes fibrin monomer polymerization and cross-linking, which stabilizes the thrombus and the initial platelet plug. Thrombin also activates factor XIII to XIIIa, which catalyzes the cross-linking of fibrin as well as that of other plasma proteins, such as fibronectin and α2-antitrypsin, resulting in their incorporation into the clot and increasing resistance to thrombolysis.27 In addition, factor XIIIa activates platelets as well as factors V and VIII, further amplifying thrombin production.
Coagulation can be activated through the intrinsic pathway with activation of factor XI to XIa, which subsequently converts factor IX to IXa and promotes formation of the Xase complex and ultimately thrombin. Another mechanism by which this occurs in vitro is through the contact activation system, whereby factor XII (Hageman factor) is activated to XIIa when complexed to prekallikrein and high-molecular-weight kininogen (HMWK) on a negatively charged surface; factor XIIa then activates factor XI to XIa. Both thrombin and factor XIa are also capable of activating factor XI.28 The physiologic importance of the intrinsic pathway is not completely clear and is probably not as physiologically important in the venous system as in the arterial system.
Natural Anticoagulants
Several interrelated processes localize thrombotic activity to sites of vascular injury. First, antithrombin (AT) is a central anticoagulant protein that binds to thrombin and interferes with coagulation by three major mechanisms: (1) inhibition of thrombin prevents removal of fibrinopeptides A and B from fibrinogen, limiting fibrin formation; (2) thrombin becomes unavailable for activation of factors V and VIII, thus slowing the coagulation cascade; and (3) thrombin-mediated platelet activation and aggregation are inhibited. In the presence of heparin, the accelerated inhibition of thrombin by antithrombin results in systemic anticoagulation. Antithrombin has been shown to directly inhibit factors VIIa, IXa, Xa, XIa, and XIIa. Thus, patients with a genetic deficiency of antithrombin are at much higher risk for development of VTE than the normal population is.
A second natural anticoagulant is activated protein C (APC), which is produced on the surface of intact endothelium when thrombin binds to its receptor, thrombomodulin, and endothelial protein C receptor (EPCR). The thrombin-thrombomodulin complex inhibits the actions of thrombin and also activates protein C to APC. APC, in the presence of its cofactor protein S, inactivates factors Va and VIIIa, therefore reducing Xase and prothrombinase activity.29
The third innate anticoagulant is tissue factor pathway inhibitor (TFPI). This protein binds the TF-VIIa complex, thus inhibiting the activation of factor X to Xa and formation of the prothrombinase complex. Interestingly, factor IX activation is not inhibited. Finally, heparin cofactor II is another inhibitor of thrombin whose action is in the extravascular compartment. The activity of heparin cofactor II is augmented by glycosaminoglycans, including both heparin and dermatan sulfate, but its deficiency is not associated with increased VTE risk.30
Physiologic Thrombolysis
Physiologic clot formation is balanced by controlled thrombolysis to prevent pathologic intravascular thrombosis. The central fibrinolytic enzyme is plasmin, a serine protease generated by the proteolytic cleavage of the proenzyme plasminogen. Its main substrates include fibrin, fibrinogen, and other coagulation factors. Plasmin also interferes with vWF-mediated platelet adhesion by proteolysis of GPIb.31
Activation of plasminogen occurs by several mechanisms. In the presence of thrombin, vascular endothelial cells produce and release tissue plasminogen activator (tPA) as well as α2-antiplasmin, a natural inhibitor of excess fibrin-bound plasmin. As clot is formed, plasminogen, tPA, and α2-antiplasmin become incorporated into it. In contrast to free circulating tPA, fibrin-bound tPA is an efficient activator of plasminogen.
A second endogenous activator of plasminogen is through the urinary plasminogen activator (uPA), also produced by endothelial cells but with less affinity for fibrin. Activation of uPA in vivo is not completely understood. However, it is hypothesized that plasmin in small amounts (produced through tPA) activates uPA, leading to further plasminogen activation and amplification of fibrinolysis.32
The third mechanism of plasminogen activation involves factors of the contact activation system; activated forms of factor XII, kallikrein, and factor XI can each independently convert plasminogen to plasmin. These activated factors may also catalyze the release of bradykinin from high-molecular-weight kininogen, which further augments tPA secretion. Finally, APC has been found to proteolytically inactivate plasminogen activator inhibitor type 1 (PAI-1), an inhibitor of plasmin activators that is released by endothelial cells in the presence of thrombin.33
The degradation of fibrin polymers by plasmin ultimately results in the creation of fragment E and two molecules of fragment D, which are released as a covalently linked dimer (D-dimer).34 Detection of D-dimer in the circulation is a marker for ongoing thrombus metabolism and has been shown to accurately predict ongoing risk of recurrent VTE.35
Interestingly, the resting state of the fibrinolytic system within the vein wall is lower in the area of the valvular cusps.36 In comparison with other anatomic locations, the deep veins of the lower limb have the lowest fibrinolytic activity in soleal sinuses as well as in the popliteal and femoral vein regions. This observation underlies a popular hypothesis as to why DVT most commonly originates in the lower limb. However, no in vivo real-time imaging studies have ever shown how and where DVT forms.
Plasminogen Inhibitors and Thrombosis
Activation of plasminogen provides localized proteolytic activity,37–39 and in plasma, PAI-1 is the primary inhibitor of plasminogen activators. It is secreted in an active form from liver and endothelial cells and is stabilized by binding to vitronectin (and inhibits thrombin in this form). PAI-1 is stored in the alpha-granules of quiescent platelets.40 PAI-1 levels are elevated by hyperlipidemia, and PAI-1 elevation appears to synergize with factor V Leiden genetic abnormalities.
Studies on the role of elevated PAI-1 in venous thrombosis have been contradictory,41,42 although it is plausible that elevated PAI-1 could suppress fibrinolysis and increase thrombosis potential. In humans, genetic polymorphisms correlate with increased risk of VTE. The highest levels of PAI-1 have been noted in those individuals carrying the 4G/4G polymorphism. Studies have found an eightfold higher risk for VTE in patients with the 4G allele in combination with other thrombophilic markers,43 and a 4.5-fold higher risk for pulmonary embolism in patients with 4G/4G polymorphism and protein S deficiency.44
Endothelium and Hemostasis
Most of the thrombosis-thrombolysis processes occur in juxtaposition to the endothelium, and hence the endothelium is one of the pivotal regulators of homeostasis (Fig. 12-2). Under normal conditions, endothelial cells maintain a vasodilatory and local fibrinolytic state in which coagulation, platelet adhesion, and activation are suppressed. A nonthrombogenic endothelial surface is maintained by a number of mechanisms, including (1) endothelial production of thrombomodulin and subsequent activation of protein C; (2) endothelial expression of heparan sulfate and dermatin sulfate, which accelerate antithrombin and heparin cofactor II activity; (3) constitutive expression of TFPI; and (4) local production of tPA and uPA. In addition, the production of NO and prostacyclin by the endothelium inhibits the adhesion and activation of leukocytes and produces vasodilation.45 TF production is also inhibited by NO.46
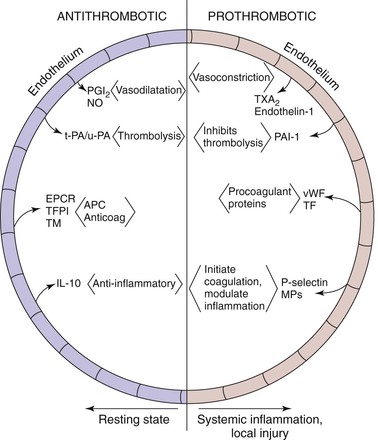
Figure 12-2 The careful balance between an antithrombotic and a prothrombotic milieu exists primarily at the endothelial level. Antithrombotic mediators include prostacyclin (PGI2) and nitric oxide (NO). Local thrombolysis is conferred by tissue (tPA) and urinary (uPA) plasminogen activators. Endothelial receptor for protein-C (EPCR), tissue factor pathway inhibitor (TFPI), and thrombomodulin (TM) inhibit thrombosis. Lastly, interleukin-10 (IL-10) is an anti-inflammatory cytokine. On the prothrombotic side, thromboxane (TXA2) and endothelin-1 promote vasoconstriction. Plasminogen activator inhibitor-1 (PAI-1) inhibits thrombolysis, and von Willebrand factor (vWF) and tissue factor (TF) are both procoagulant proteins. Lastly, P-selectin and microparticles (MPs) initiate coagulation and also modulate inflammation.
After endothelial injury a prothrombotic and proinflammatory state of vasoconstriction is supported by the endothelial surface. Release of platelet-activating factor (PAF) and endothelin-1 promotes vasoconstriction,47 whereas production of vWF, TF, PAI-1, and factor V augments thrombosis. Indeed, vWF is expressed to a greater extent on the endothelium of veins than on the endothelium of arteries, and tPA is less commonly expressed in venous endothelium.2 Systemic inflammatory insults such as conferred by tumor necrosis factor-α may cause endothelial activation and result in increased surface expression of cell adhesion molecules (CAMs) such as P-selectin, E-selectin, and intracellular CAM (ICAM), thereby promoting the adhesion and activation of leukocytes as well as platelets.7
Inflammation and Thrombosis
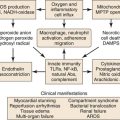
The relationship between thrombosis and inflammation was first suggested in the early 1970s.48 Inflammation increases TF, membrane phospholipids, fibrinogen, and the reactivity of platelets while decreasing thrombomodulin and inhibiting fibrinolysis (Fig. 12-3).49
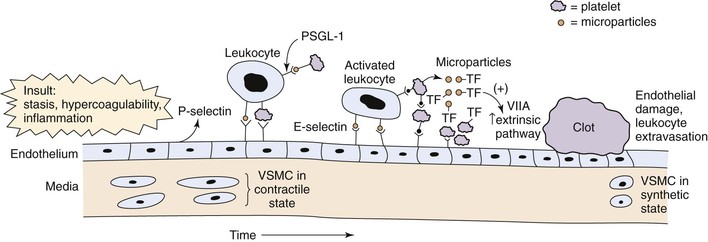
Figure 12-3 Acute venous thrombosis most commonly occurs in the lower extremity veins. Insults such as stasis, hypercoagulability, and inflammation result in damage to the endothelium and/or upregulation of P-selectin. Leukocytes bind via P-selectin glycoprotein ligand (PSGL-1) to P-selectin as well as platelets. This step leads to activated leukocytes, which then more firmly bind via E-selectin and release inflammatory mediators. Platelets also bind to leukocytes, endothelium, and other platelets and release microparticles that express tissue factor (TF). Tissue factor activates the extrinsic pathway by activation of factor VIIA (VIIA). A clot develops, and endothelial damage with further leukocyte adherence, extravasation, and inflammation occur. In the media, the normal state of the vascular smooth muscle cells (VSMCs) is contractile. Once activated and in the inflammatory state, the cells may become synthetic with production of profibrotic mediators and collagen.
Microparticles are involved in the initiation and amplification of thrombosis. They are small (<1 µm) phospholipid vesicles shed from platelets, leukocytes, and endothelial cells in a calcium-dependent fashion.50–52 Microparticles lack DNA and RNA, but subpopulations of microparticles rich in TF and phosphatidylserine have been identified.53,54 Fusion of microparticles with activated platelets results in decryption of TF and the initiation of thrombosis.55 Indeed, several circulating markers of inflammation once thought to be soluble are actually carried by microparticles.56–58 These vesicles have direct exogenous procoagulant activity, as shown by normalization of tail bleeding times in hemophilic mice.57 Moreover, microparticles shed from platelets express PAI-1, and these microparticles add to the growing thrombus via interactions between P-selectin and its receptor, P-selectin glycoprotein ligand-1 (PSGL-1). In this manner, platelet microparticles not only are prothrombotic but also inhibit fibrinolysis, facilitating thrombus growth.59
In veins, CAMs and microparticles interface in promoting thrombosis and inflammation. CAMs allow leukocyte transmigration, and selectins (P- and E-selectin) are integrally involved in venous thrombosis. P-selectin is upregulated in the vein wall as early as 6 hours after thrombus induction, and E-selectin has been found upregulated at later time points.60 Both venous stasis and hypoxia result in the upregulation of P-selectin, which localizes prothrombotic microparticles to the area of stasis and promotes the development of DVT.61–63 PSGL-1 is expressed on leukocytes and platelets as well as on their derived microparticles. Indeed, microparticles coexpressing TF and leukocyte markers have been shown to accumulate in growing thrombi in a P-selectin/PSGL-1–dependent fashion.64–66 Furthermore, P-selectin/PSGL-1 interaction stimulates the production of thrombogenic microparticles from leukocytes, along with platelets and endothelial cells.67
Although it was long thought to be a bystander in venous thrombosis unlike in the arterial system, the platelet is now thought to play a critical role.68 First, in stasis and nonstasis experimental murine venous thrombosis, genetic deletion of vWF was associated with significantly reduced size of venous thrombi that was not restored with recombinant factor VIII.69 Intravital microscopy also showed direct association of leukocytes and platelets in a growing acute thrombus. Consistently, platelets, via GPIb2, may promote venous thrombosis by colocalizing leukocytes and coagulation factors at the site of injury or stasis in the vein.70
Thrombus Resolution and Vein Wall Remodeling
Regardless of the location and extent of acute DVT, the resolution process is complex71–73 (Table 12-2 and Fig. 12-4). In humans, the natural fibrinolytic mechanisms break down the thrombus over time and at variable rates.74,75 Resolution of experimental venous thrombus resembles wound healing, involving profibrotic growth factors, collagen deposition, and activation of matrix metalloproteinase (MMP). The fact that leukocytes invade the thrombus in a specific sequence suggests their importance in normal thrombus resolution.76,77 Real-time in vivo imaging has also shown that leukocyte concentration and MMP activity colocalize to areas of recanalization.48,78,79
Table 12-2
Experimental Venous Thrombosis Resolution Characteristics
| Acute (<8 Days) | Chronic (>8 Days) | |
| Effector leukocyte | Neutrophil | Monocyte |
| Chemokines/cytokine | IL-8, IL-1β, IL-6 | Monocyte chemotactic protein-1 Secondary lymphoid chemokine |
| Growth factors | — | Vascular endothelial growth factor Basic fibroblast growth factor, transforming growth factor–β |
| Neovascularization of clot | Minimal | Yes von Willebrand factor–positive channels, hypoxia-inducible factor-1α |
| Matrix remodeling | MMP-9, elastase? | Urinary plasminogen activator–plasmin MMP-2, plasminogen activator inhibitor-1–vironectin |
| Vein wall, collagen type | Collagen III | Collagen I |
IL, Interleukin; MMP, matrix metalloproteinase.
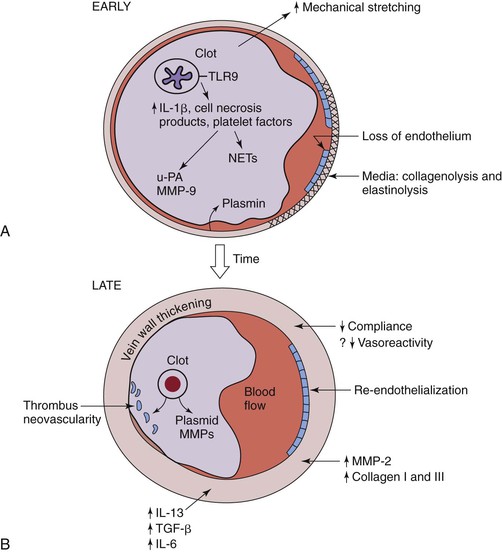
Figure 12-4 Hypothesized thrombosis resolution, based on small rodent models, is shown. A, Early thrombus resolution involves a large clot releasing interlukin-1β (IL-1β), cell necrosis products, and platelet factors that drive neutrophil influx, with release of plasminogen activators, such as urinary platelet activator (uPA). Concurrently, matrix metalloproteinase-9 (MMP-9) is released, and plasmin is upregulated. This allows for early thrombolysis. At the same time, dying polymorphonuclear neutrophils (PMNs) may contribute to thrombosis with release of neutrophil extracellular traps (NETs). Concurrently, loss of endothelium exposes the subendothelial matrix proteins that may further potentiate thrombosis. In the media, collagenolysis and elastinolysis occur in addition to direct mechanical stretch. B, Later (usually after 8 days), vein wall medial thickening occurs with decreased compliance and, possibly, decreased vasoreactivity. Reendothelization commences but is incomplete until a much later time point (>14 days). Thrombus neovascularity and cellularity of the thrombus are associated with resolution, which is a monocyte/macrophage-driven process. Within the vein wall, matrix turnover occurs with increased MMP-2 expression, as well as collagen I and collagen III production. IL-13 and transforming growth factor-β (TGF-β) are two profibrotic growth factors that may be involved with late vein wall remodeling.
The first cell type in the thrombus is the polymorphonuclear neutrophil (PMN). Although PMNs may cause vein wall injury, they contribute to both early thrombus amplification70 and early thrombus resolution. The role of PMNs in promoting early DVT by release of neutrophil extracellular traps (NETs) has been shown in primate and murine models of thrombosis.80–82 Indeed, inflammatory activation of PMNs causes degranulation of nucleic DNA, which is complexed with antimicrobial peptides and allows platelets and coagulation factors to assemble in juxtaposition to the vein wall. Consistently, DNAse treatment can directly accelerate venous thrombosis resolution. Conversely, in a rat model of stasis DVT, neutropenia was associated with larger thrombi at 2 and 7 days and was correlated with increased thrombus fibrosis and significantly lower thrombus levels of both uPA and MMP-9.83–85
The monocyte is likely the most important cell for later DVT resolution.76 Monocyte influx into the thrombus peaks at day 8 after thrombogenesis and correlates with elevations of monocyte chemotactic protein-1 (MCP-1), one of the primary CC chemokines that direct monocyte chemotaxis and activation;86 MCP-1 has also been associated with DVT resolution.87 Targeted deletion of CC receptor-2 (CCR-2 KO) in the mouse model of stasis thrombosis is associated with late impairment of thrombus resolution, probably via impaired interferon-γ–inducible proteinase activity, which may be independent of monocytes.88
The local venous environment is by definition hypoxic. A major angiogenic growth factor is hypoxia-inducible factor-1α (HIF-Iα). Experimental data in a stasis model of venous thrombosis suggests thrombosis stimulates increased vein wall HIF-Iα, and that by exogenous stimulation of HIF-Iα expression, thrombus recanalization was increased and associated with accelerated resolution of venous thrombi.89 Moreover, adenoviral transfection of the downstream proangiogenic growth factor VEGF in a stasis rat model of DVT was associated with increased thrombus recanalization and smaller venous thrombi. This was also correlated with higher numbers of thrombus monocytes.90 Thus, thrombus resolution is in part dependent on neovascularization. The effect of mechanical stretch, such as by a venous thrombus, may also stimulate HIF-Iα as well as MMP-2 and MMP-9, all of which lead to reduced vein contractility.91
As the thrombus resolves, a number of proinflammatory factors are released into the local environment, including interleukin-1β (IL-1β) and tumor necrosis factor-α.77 The cellular sources of these different mediators have not been specifically defined but probably include leukocytes and smooth muscle–like cells within the resolving thrombus and adjacent vein wall. Leukocyte kinetics in the vein wall after DVT are similar to what is observed in the thrombus, with an early influx of PMNs followed by monocytes. On the basis of experimental studies, elastinolysis and collagenolysis seem to occur early, as measured by an increase in vein wall stiffness, persisting through 14 days, and are accompanied by elevated MMP-2 and MMP-9 activities.9,10
Data linking inflammation to fibrosis demonstrate that inhibition of the inflammatory response can decrease vein wall fibrosis. In a rat model of stasis DVT, treated with either low-molecular-weight heparin (LMWH) or an oral inhibitor to P-selectin 2 days after establishment of thrombosis, inhibition of P-selectin significantly decreased vein wall injury (independent of thrombus size), as measured by vein wall tensiometry (stiffness), intimal thickness score, IL-13 levels, MCP-1 levels, and platelet-derived growth factor-β (PDGF-β) levels.92 The mechanism accounting for this protective effect is not yet known but probably does not involve leukocyte blockade, because no differences in influx of monocytes into the vein wall were observed.
Loss of venous endothelium likely also contributes to the vein wall fibrosis as well as the predisposition to recurrent thrombosis. An experimental model of DVT showed lower expression of homeostatic endothelial gene products, such as NO and thrombomodulin, than in controls, which correlated with loss of vWF positive cell luminal staining.93 Other investigators have found that prolonged venous stasis is associated with decreased plasminogen activators, probably related to loss of endothelium.94
Associated with the early biomechanical injury from DVT is an elevation of profibrotic mediators, including TGF-β, IL-13, IL-6, and MCP-1.10,92,95 These mediators are present within the vein wall and thrombus and may drive the fibrotic response. Although exogenous MCP-1 may hasten DVT resolution, it promotes organ fibrosis in vivo. The profibrotic growth factor TGF-β is also present in the thrombus and is activated with normal thrombolysis.96 TGF-β may be a key mechanism promoting vein wall fibrosis. Late fibrosis has been observed in our mouse model of DVT, which demonstrated a significant increase in vein wall collagen after stasis thrombogenesis.9,97 Correlating with this increase in fibrosis is an increase in collagen I and III gene expression, as well as increases in MMP-2 and MMP-9 gene expression and activity. Genetic deletion of MMP-2 or MMP-2/9 is associated with decreased vein wall fibrosis, possibly by modulating vein wall elastin/collagen metabolism as well as monocyte influx.97a Moreover, the thrombus size itself does not drive the vein wall injury response; rather, the mechanism of thrombosis seems more important.10,98
Thus, early vein wall injury is associated with active matrix remodeling that seems to promote late fibrosis, not unlike many organ responses to inflammation “burnout.”6 Indeed, precedent exists in humans that the monocytic activation state may predict long-term DVT resolution.99 The specific mechanisms and strategies to reverse this process are being actively investigated.
Chronic Venous Insufficiency
Limb manifestations of CVI are easy to recognize: variable swelling, varicosities, hyperpigmentation, stasis dermatitis, ulceration, and pain (Figure 12-5). The symptoms are widespread in the general population and are not always directly related to the magnitude of venous insufficiency (e.g., degree of reflux). However, the most severe CVI symptoms are related to venous reflux with an obstructed venous segment.100,101
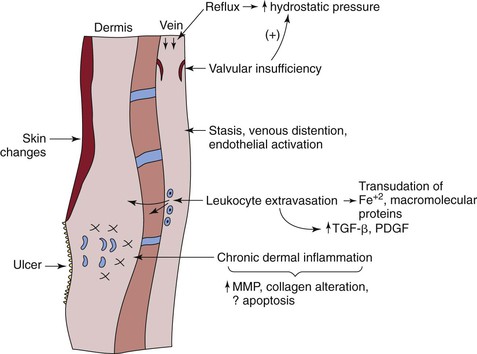
Figure 12-5 The pathogenesis of chronic venous insufficiency (CVI) pathogenesis is complex. Primarily, CVI is caused by reflux, which increases hydrostatic pressure in the vein and is transmitted to the subcutaneous dermis and skin. This process occurs with both primary and secondary valvular insufficiency. Reflux also potentiates blood flow stasis, with vein distention and endothelial activation, followed by leukocyte extravasation and transudative macromolecules and iron. Chronic dermal inflammation occurs with increased matrix metalloproteinases (MMPs), collagen alteration, and possibly apoptosis. A venous ulcer is the most severe manifestation of CVI. FE+2, Ferrous iron; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor-β.
The best way to categorize CVI is with the CEAP (clinical, etiologic, anatomic, pathologic) classification. This system is discussed in detail in Chapter 53; in brief, it is a standardized system to grade CVI on the basis of symptoms, etiology, anatomy, and pathophysiology.102 The higher the class, the more severe the symptoms and the patient’s disability.19 For clarity of discussion, this section separately focuses on the pathology of class 2 to 3 and class 4 to 6 CVI, although there are many common factors and the pathology clearly represents a spectrum.
Mechanisms of chronic vein injury with primary and secondary valve damage have been reasonably well described (Table 12-3). The common mechanism is venous reflux between the superficial and deep systems, either at the site of perforators or through other deep and superficial system connections, which accounts for the increased venous hydrostatic pressure transmitted to the superficial veins and tissues. An obstructed vein segment worsens this process. Experimentally, venous hypertension activates leukocytes,103 although the activation is probably a local phenomenon.104 The process of vein wall fibrosis plays a contributing role in valvular damage, which then worsens the hydrostatic pressure regulation and consequently promotes greater venous hypertension with the upright position. The molecular and cellular events seem to point to adaptive responses to injury as well as abnormal healing.
Table 12-3
Characteristics of Venous Insufficiency

bFGF, Basic fibroblast growth factor; MMP, matrix metalloproteinase; TGF-β, transforming growth factor-β.
Historical Perspective and General Background
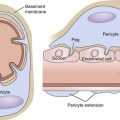
Several important theories have been postulated regarding the etiology and pathophysiology of CVI. In 1917, John Homans105 produced a clinical treatise on the diagnosis and the management of patients with CVI and coined the term “post thrombotic syndrome.” Dr. Alfred Blalock106 put forth the hypothesis that local hypoxia precipitated CVI. Local tissue hypoxia and alterations in nutrient blood flow were proposed as an underlying etiology by Browse and Burnand.107 Their important study directly demonstrated the effect of venous hypertension on the venous microcirculation, and they observed histologically that in large capillaries, pericapillary fibrin deposition, which they called the “fibrin cuff,” occurred (Fig. 12-6). Dr. P.D. Coleridge Smith108 proposed that leukocyte trapping in slow-flow and distended venous segments may underlie much of CVI development. Burnand et al109 also showed a positive correlation between the inability of the calf pump to reduce foot vein pressure during exercise and the number of capillaries in the skin.
Varicose Veins (CEAP Class 2 to 3 Disease)
Varicose veins (VVs) have been described since before the Common Era and are obvious on the lower limbs of many people. The fact that varicose veins primarily affect the lower limb directly implicates the upright nature of humans—specifically, the effect of hydrostatic pressure on the pathophysiology of such veins. The relationship between body weight and extent of varicose veins and symptoms is variable. Limb symptoms are generally local and consist of pruritus and swelling. Occasionally, varicose veins can erode and bleed. Conversely, most such veins do not thrombose, despite relatively slow blood flow through these torturous structures, and this fact underlies the natural anticoagulant nature of venous endothelium, even in structurally abnormal vessels.
The initial anatomic location of varicose veins is typically in the great and small saphenous distributions and their tributaries in the superficial system.110 Related risk factors are multiple, including primary etiologies associated with pregnancy, prolonged standing, female gender, and, rarely, congenital absence of valves.111 In addition, varicosities may develop as a result of prior DVT or trauma.
Studies support a genetic predisposition to varicose veins.112 In a prospective study of 402 subjects, the risk of development of varicose veins was 90% if both parents were affected, 25% for males and 62% for females if one parent was affected, and 20% if neither parent was affected.113 These data suggest an autosomal dominant with variable penetrance mode of genetic transmission.
Studies are generally lacking in relation to early vein wall changes associated with varicose veins, because most histologic studies are limited to end-stage surgical specimens. In varicose veins, the orderly appearance of the medial layer is replaced by an intense and disorganized deposition of collagen that separates the closely apposed muscle cells. Smooth muscle cells appear elliptical and are likely a secretory phenotype, and both TGF-β and basic fibroblast growth factor (bFGF) have been documented to be significantly increased in hypertrophic segments of varicose veins.114 The underlying mechanism for these histologic changes is unknown, but the inciting event of increased hydrostatic pressure or an intrinsic genetic defect is probably primary.
Active vein wall remodeling is consistently observed in specimens of varicose veins with abnormal matrix collagen metabolism.115 Quantitatively, higher collagen content and lower elastin content in varicose veins have been measured in human samples, suggesting an imbalance in connective tissue matrix regulation and turnover.116 Specific alteration in the type of collagen may also contribute to vessel weakening, with an observed increase in tissue water and collagen type I content in comparison with normal saphenous veins.117
Conversely, collagen types III and V levels were lower than in normal veins, and less type III collagen is associated with decreased elasticity. Similarly, in a separate study, varicose veins had increased type I and decreased type III collagen gene expression.118 Comparison of smooth muscle cells from varicose veins with controls has shown matrix dysregulation119 as well as regional differentiation as measured by antigen markers in the cells from varicose veins.120 Interestingly, a similar collagen dysregulation pattern has also been observed in patients with varicose veins after dermal biopsies,121 suggesting an intrinsic genetic abnormal response to injury.
The observed pathology suggests a net effect of matrix deposition. One mechanism for these changes may be local upregulation of MMPs and fibrinolytic activity within the microenvironment.122 The upstream regulators of MMPs are in part, the plasmin system. Urinary PA levels are three to five times higher than in controls, as assessed from media of specimen organ culture from varicose veins.123 Interestingly, there is no difference in tPA activity or PAI-1 levels. Investigators have found that TIMP-1 and MMP-1 protein levels are higher at the saphenofemoral junction in patients with varicose veins and that MMP-2 activity is lower in normal controls.124 Similarly, high TIMP-1 activity and low MMP-2 activity have been observed in varicose vein segments, with a threefold significant difference in comparison with normal controls.125 Overall differences in MMP-9 protein have also been identified, and it is likely, with the inflammatory influx, that MMP-9 is released primarily from PMNs but may be less important than MMP-2.126,127
This disordered vein structure also correlates with altered vasoreactivity. The contractual response of varicose and normal great saphenous rings to various alpha-adrenergic and non–alpha-adrenergic receptors has shown decreased contractility as compared with controls.128 This lower contractility may be due to repeated overdistention or impaired contractility related to persistent vein wall tension. However, it is a segmental response and likely adaptive.129 Receptor downregulation may also play a role. For example, feedback inhibition of ETA receptor secondary to increased endothelin-1 is also postulated to mediate the lower vasoreactivity content in the walls of varicose veins.130 The ratio of prostacyclin to thromboxane A2 is also lower, suggesting an increased basal contractile state.131 Finally, one investigation has suggested that MMP-2 may act to dilate the vein by direct hyperpolarization effects, via a Ca+2 channel mechanism.132
Pathophysiology of Stasis Dermatitis and Dermal Fibrosis (CEAP Class 4 to 6 Disease)
Stasis venous dermatitis is a disease of chronic dermal inflammation due to persistent and sustained cutaneous and dermal injury secondary to venous hypertension. The primary injury may be extravasation of macromolecules and red blood cell products into the dermal interstitium, which creates a secondary inflammatory response, and multiple pathophysiologic hypotheses have been proposed.133 The clinical appearance is that of brawny induration, skin thickening, swelling, and tissue breakdown with ulceration in the gaiter regions. Alteration in extracellular matrix is clear on histologic assessment, with dermal space extracellular disorganized collagen deposition and perivascular tissue cuffs.
The role of the common growth factor TGF-β1, which is secreted by activated endothelial cells, fibroblasts, and platelets and stimulates matrix protein production,134,135 has been studied in relation to venous ulcer development. TGF-β is an inducer of TIMP-1 and collagen production and inhibits MMP activity and gene expression.135 The local upregulation of TGF-β1 thus favors net collagen and matrix accumulation and is supported by histologic and clinical analysis, although gene upregulation may occur at an earlier stage than protein production.136 Whether TGF-β is acting directly or is associated with this process has not been substantiated in human studies to date. Several other growth factors are elevated in the dermis of patients with CVI, including platelet-derived growth factors α and β and VEGF.137 These various molecules are found within the capillaries surrounding fibroblasts and inflammatory cells in patients with venous skin changes.
Further studies have suggested that endothelial activation results from venous hypertension.138 A significant rise in plasma levels of endothelial leukocyte adhesion molecule-1 (ELAM-1), ICAM-1, and vascular cell adhesion molecule-1 were observed in patients with venous hypertension, indirectly suggesting a role of these molecules in the pathogenesis.139 Similarly, increased endothelial expression of ICAM-1, vascular cell adhesion molecule-1, and leukocyte function-associated antigen-1 (LFA-1) has been documented in patients with dermal disease.140
Several studies suggest conflicting data with regard to whether MMP-2, MMP-9, and TIMP-1 levels are higher or unaltered in patients with venous ulcers. This uncertainty may have to do with the measurement of MMP gene, protein, or activity expression as well as with the ulcer stage and patient characteristics that may not be controlled for. Another important proteinase is elastase, primarily derived from PMNs. Higher levels of plasma elastase were found in patients with venous ulceration than in those with uncomplicated varicose veins, perhaps reflecting active degranulation in those with ulcers.141 However, this evidence is only indirect, and whether proteinases are directly responsible for venous ulcer development is not known.
Consistent with the tissue inflammation, leukocytes are markedly elevated in the gaiter region in association with the venous ulcers.142 This finding correlates with preceding elevation of IL-1α and ICAM-1 in the tissue of lipodermatosclerotic skin.143 The type and distribution of leukocytes in patients with CEAP class 5/6 CVI have been studied histologically.144 Numbers of mast cells and macrophages were two to four times greater around arterioles and postcapillary venules in patients with CEAP class 4/5 CVI than in controls. Similarly, increased macrophages are demonstrated around arterioles and postcapillary venules, although fibroblasts are the most common cell observed in both gaiter and thigh biopsy specimens. It is likely that these leukocytes regulate the tissue remodeling that results in dermal fibrosis.
Sluggish venous blood flow related to increased hydrostatic pressure leads to hypoxia and PMN activation, with degranulation of mediators and proteinases that cause endothelial damage. Skin hypoxia also occurs on the gaiter areas of limbs with severe venous disease and is significantly different from controls, oxygen tension (tcPO2) differing by more than 20 mm.145 Although leukocyte trapping within the capillaries is probably not the sole cause, it is likely that leukocytes become activated, transmigrate into the vein walls, and mediate some of the observed damage. Consistently, findings in punch biopsy specimens of ulcers suggest that leukocytes play a role in the dermal manifestations of CVI.142 For example, leukocytes, macrophages, and mast cells have all been observed in immunohistochemical and electron microscopic examinations of affected tissue.144
The dermal fibroblast may also be dysfunctional and allow perpetuation of the disease. Decreased motility, in part mediated by the microenvironment, plays a role in the impaired healing process of ulcer tissue.146 Comparison of venous ulcer fibroblasts with control fibroblasts with stimulation with TGF-β suggested that there are differences in collagen production.147 With stimulation, collagen production was increased 60% in controls, whereas the venous ulcer fibroblasts were unresponsive. This finding may be related to an end-stage process, overstimulation of the ulcer fibroblast, or an intrinsically dysfunctional fibroblast. Consistently, the proliferative responses of fibroblasts from patients with CVI to TGF-1β correlated with disease severity.148 Fibroblasts in patients with CEAP class 2 or 3 CVI retained agonist induce proliferative capacity, whereas those from patients with class 4 or 5 CVI showed diminished agonist-induced proliferation. Fibroblasts from patients with class 6 CVI and active ulcers did not proliferate with TGF-1β, suggesting that these ulcer fibroblasts are refractory to stimulation and may contribute to the inability to promote healing. Histologically, these fibroblasts appear morphologically similar to fibroblasts undergoing cellular senescence, and therefore may be pro-apoptotic from repeated injury. Another study showed similar impairment of dermal wound fibroblast proliferation response to both basic fibroblast growth factor and EGF.149
An interesting but unanswered question is which patients with similar degrees of reflux and hydrostatic pressures from CVI are more predisposed to development of venous ulcers. Data now suggest that iron metabolism and ulcer development are interrelated. Although commonly thought of as a consequence of all the other mechanisms of CVI, the iron deposition itself may cause tissue damage.150 More convincingly, the risk of ulcer development among patients with class 4 to 6 CVI was sevenfold higher in those with the C282Y genotype, a mutation related to iron processing.151
Taken together, venous insufficiency and secondary cutaneous manifestations suggest that active tissue remodeling occurs, likely via multiple mechanisms and in different stages. Whether the growth factors, cytokines, and proteinases are directly responsible or secondary to other factors of the disease has not been definitively answered. Regardless, the significant benefit of compression in ulcer healing suggests these factors may play a secondary role. In one study the benefit of compression therapy and how it may alter histologic and biochemical features was shown after 4 weeks.152 Complete epithelialization was frequent, the hemosiderin and red blood cell extravasation products had decreased, and fibrin cuffs were reversed. However, a single “silver bullet” approach to therapy is unlikely to be successful. Rather, identifying patients at risk for CVI with biomarkers (via genomics and proteomics) may be the best immediate translational approach.
Venous Aneurysms
The etiology of venous aneurysms is poorly understood, and little clinical series data regarding their pathogenesis is available in the literature. Not surprisingly, no genetic studies related to any underlying connective tissue or matrix enzyme abnormalities have been performed. Venous aneurysms are more common in South America than in North America or Europe. The primary clinical risk associated with venous aneurysms is stasis, and VTE may develop in up to 71% of those with venous aneurysms.153 Few symptoms are associated with venous aneurysms, and the main indication for surgical management of such a lesion is the fact it may be a nidus for thrombus and contribute to VTE. Venous aneurysms are not really at risk for rupture per se.
Venous aneurysms may be associated with either an acquired condition, such as an arteriovenous fistula or primary, or may be related to venous distention. Case reports have shown them to be present in jugular veins, the vena cava, axillary-subclavian veins, femoral veins, and popliteal veins.153,154 The overall primary pathologic etiology has not been determined. It is likely they are flow-related phenomena in one setting and a local degenerative process in other settings.
In contrast to thrombotic injury, characterized as an occlusive fibrotic process, the flow dynamics in a venous aneurysm direct the vein in “outward” remodeling. Histologic analysis shows vein wall fragmentation with elastin degeneration.154 Pathologically the wall is thin, in which elastin fragmentation alternates with smooth muscle cell attenuation and fibrous tissue deposition, thus suggesting a typical response to vascular injury.155
Thrombophlebitis
Thrombophlebitis may be sterile or associated with a bacterial infection, and may be a marker for DVT.156 Most often it involves the superficial venous system, such as the great saphenous or cephalic vein, and occurs after placement of an intravenous catheter or other superficial trauma. Symptoms include pain, redness, swelling, and tenderness to palpation, often with a cord present in the affected limb. A systemic febrile response is usually absent in noninfected cases, whereas bacterial thrombophlebitis may be a source of postoperative fever. Occasionally the latter may be associated with bacteremia and a septic picture.
It is unclear whether there is a specific genetic predisposition to this disease, but clinical factors and environment likely play the biggest roles. A special case of thrombophlebitis is a migratory thrombophlebitis, which was described by Trousseau in the 1800s and is specifically linked to patients with pancreatic cancer. Mondor’s disease is superficial thrombophlebitis of the veins in the breast tissue, particularly in the upper outer quadrant.
Sterile phlebitis may occur in varicosities, as well as after stripping or endovenous ablation. An experimental study of sterile thrombophlebitis showed a typical sequence of early PMN attachment followed by transmigration and inflammatory changes.157 Approximately 25% of the time, thrombosis occurs before symptoms. The histologic pathology is an acute vasculitis with marked thickening, inflammatory cells, and fibrin deposition.158 Later resolution involves fibrotic changes and recanalization of the venous segment at variable rates over weeks to months. One study has shown that anticoagulation for 45 days significantly decreased proximal venous thrombus propagation by approximately 85%.159
The role of bacterial infection in directly promoting thrombosis has been described in various clinical settings.160 The pathophysiology of infectious thrombophlebitis is similar to that of any closed-space infection, and bacterial antigens and proteins may directly stimulate venous thrombosis and the extrinsic pathway. Multiple organisms may be cultured from infectious thrombophlebitis, including both gram-positive and gram-negative species.
Interface of Arterial Thrombosis and Venous Thrombosis
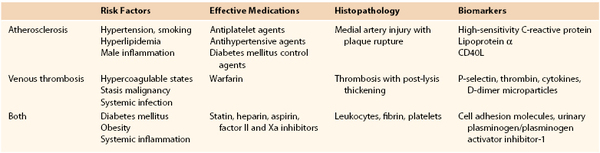
Several interesting epidemiologic and basic science studies have suggested a common interface between risk for venous thrombosis and arterial vascular pathology (Table 12-4). Dysfunctional endothelium contributes to atherosclerosis and may also contribute to clinical VTE. Conversely, systemic vascular inflammation may affect both arteries and veins,161 and the differences are manifested as the typical clinical pathology of arterial and venous occlusions.
Epidemiologic studies support risks common to these two vascular diseases. The concept that VTE and atherosclerosis may have common risk factors has been assessed by a metaanalysis. Compared with control subjects, the risk of VTE was 2.33 for obesity, 1.5 for hypertension, 1.4 for diabetes, 1.2 for smoking, and 1.2 for hypercholesterolemia.162 Interestingly, mean high-density lipoprotein cholesterol levels were lower in patients with VTE than in controls. Conversely, patients with spontaneous VTE had a 1.6-fold measured incidence of symptomatic atherosclerosis.163 Clinical data also suggest that aspirin may decrease the rate of recurrent VTE164 as well as the number of arterial atheroembolic events in at-risk patients with VTE.165
Logically, any proendothelial agent will affect both arterial and venous systems. In addition to the endothelium-protective effects of statins, these agents may increase thrombomodulin expression and enhance APC activity, thereby tipping the balance of endothelium toward a more antiinflammatory and anticoagulant state. Registry cohort patient data suggests a 22% lower rate in patients with symptomatic VTE undergoing statin therapy, than in those not receiving such therapy.166 Reports are forthcoming that 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) may be associated with decreased DVT incidence. Glynn and colleagues,167 in a multicenter trial, showed that incident VTE was significantly reduced by the use of a statin agent over 3 years of follow-up.
Experimentally, hyperlipidemic mice (in which the APOE gene has been deleted) have impaired thrombus resolution associated with significantly reduced plasmin activity, fewer monocytes, and reduced MCP-1 levels.97 Whether this finding directly translates to human pathophysiology is not clear. Statin treatment of mice with stasis DVT is associated with smaller venous thrombi, but the effect is time dependent.168 Statins also inhibit monocyte TF expression, possibly accounting for their antithrombotic effect.25
Much remains to be uncovered about the mechanisms and treatment of venous disease, but consideration of the similarities and differences between venous and arterial pathology will likely lead to novel therapies (Fig. 12-7).
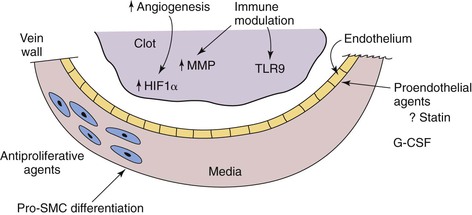
Figure 12-7 The potential future therapies for venous disease are speculative but promising, on the basis of experimental and epidemiologic studies. Proendothelial agents, such as statins, may decrease the risk of venous thrombosis or increase vein wall healing. Antiproliferative agents and pro–smooth muscle cell (SMC) differentiation agents may one day reverse the vein wall thickening and fibrosis that occur after deep venous thrombosis. Immune modulation with thrombus resolution—perhaps via an increase in local matrix metalloproteinase (MMP) activity, or through priming and driving thrombolysis mechanisms via Toll-like receptor 9 (TLR9)—is a potentially attractive mechanism by which to accelerate thrombus resolution without anticoagulant risk. G-CSF, Granulocyte colony stimulating factor; HIF1α, hypoxia-inducible factor 1α.
Selected Key References
Bergan JJ, Schmid-Schönbein GW, Smith PD, Nicolaides AN, Boisseau MR, Eklof B. Chronic venous disease. N Engl J Med. 2006;355:488–498.
Comprehensive review of CVI by several of the world’s experts.
Browse NL, Burnand KG. The cause of venous ulceration. Lancet. 1982;2(8292):243–245.
A classic treatise on venous ulcer pathophysiology by two of the early investigators.
Henke PK, Mitsuya M, Luke CE, Elfline MA, Baldwin JF, Deatrick KB, Diaz JA, Sood V, Upchurch GR, Wakefield TW, Hogaboam C, Kunkel SL. Toll-like receptor 9 signaling is critical for early experimental deep vein thrombosis resolution. Arterioscler Thromb Vasc Biol. 2011;31:43–49.
An experimental study that suggests that immunomodulation of DVT is possible.
Monos E, Bérczi V, Nádasy G. Local control of veins: biomechanical, metabolic, and humoral aspects. Physiol Rev. 1995;75:611–666.
An extremely comprehensive review of venous physiology.
Singh I, Burnand KG, Collins M, Luttun A, Collen D, Boelhouwer B, Smith A. Failure of thrombus to resolve in urokinase-type plasminogen activator gene-knockout mice: rescue by normal bone marrow-derived cells. Circulation. 2003;107:869–875.
An elegant study that suggests the central role of uPA in mediating later DVT resolution.
von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Timiceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–835.
This comprehensive experimental study shows the critical early interactions of leukocytes, their functions and the coagulation system including the platelet in experimental venous thrombosis. It also confirms the role of innate immunity in venous thrombosis.
Wakefield TW, Strieter RM, Wilke CA, Kadell AM, Wrobleski SK, Burdick MD, Schmidt R, Kunkel SL, Greenfield LJ. Venous thrombosis-associated inflammation and attenuation with neutralizing antibodies to cytokines and adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15:258–268.
One of the first studies to follow up on the concept that venous thrombosis is an inflammatory disease.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Holderfield MT, et al. Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-β in vascular morphogenesis. Circ Res. 2008;102:637–652.
2. Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158–173.
3. You LR, et al. Suppression of notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature. 2005;435:98–104.
4. Delot EC, et al. Abnormal venous and arterial patterning in chordin mutants. Dev Dyn. 2007;236:2586–2593.
5. Davis GE, et al. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97:1093–1107.
6. Deroo S, et al. The vessel wall: a forgotten player in post thrombotic syndrome. Thromb Haemost. 2010;104:681–692.
7. Eriksson EE, et al. Powerful inflammatory properties of large vein endothelium in vivo. Arterioscler Thromb Vasc Biol. 2005;25:723–728.
8. Archie JP Jr, et al. Saphenous vein rupture pressure, rupture stress, and carotid endarterectomy vein patch reconstruction. Surgery. 1990;107:389–396.
9. Deatrick KB, et al. Vein wall remodeling after deep vein thrombosis involves matrix metalloproteinases and late fibrosis in a mouse model. J Vasc Surg. 2005;42:140–148.
10. Henke PK, et al. Fibrotic injury after experimental deep vein thrombosis is determined by the mechanism of thrombogenesis. Thromb Haemost. 2007;98:1045–1055.
11. De Mey JG, et al. Heterogeneous behavior of the canine arterial and venous wall: importance of the endothelium. Circ Res. 1982;51:439–447.
12. Raffetto JD, et al. Endothelium-dependent nitric oxide and hyperpolarization-mediated venous relaxation pathways in rat inferior vena cava. J Vasc Surg. 2012;55:1716–1725.
13. Vanhoutte PM, et al. Endothelium-dependent contractions. Blood Vessels. 1991;28:74–83.
14. Hiremath AN, et al. Comparison of age-related changes in prostaglandin E1 and beta-adrenergic responsiveness of vascular smooth muscle in adult males. J Gerontol. 1989;44:M13–M17.
15. Bevan JA, et al. Comparable sensitivity of flow contraction and relaxation to Na reduction may reflect flow-sensor characteristics. Am J Physiol. 1992;263:H182–H187.
16. Simon G. Increased vascular wall sodium in hypertension: where is it, how does it get there and what does it do there? Clin Sci (Lond). 1990;78:533–540.
17. Heit JA, et al. Estimated annual number of incident and recurrent, non-fatal venous thromboembolism (VTE) events in the US. Blood. 2005;106.
18. Gangireddy C, et al. Risk factors and clinical impact of postoperative symptomatic venous thromboembolism. J Vasc Surg. 2007;45:335–341.
19. Meissner MH, et al. Acute venous disease: venous thrombosis and venous trauma. J Vasc Surg. 2007;46(Suppl S):25S–53S.
20. Mackman N, et al. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1687–1693.
21. Mackman N. Tissue-specific hemostasis in mice. Arterioscler Thromb Vasc Biol. 2005;25:2273–2281.
22. Furie B, et al. Mechanisms of thrombus formation. N Engl J Med. 2008;359:938–949.
23. Savage B, et al. Selective recognition of adhesive sites in surface-bound fibrinogen by glycoprotein IIb-IIIa on nonactivated platelets. J Biol Chem. 1991;266:11227–11233.
24. Ferguson JJ, et al. Fundamentals of coagulation and glycoprotein IIb/IIIa receptor inhibition. Eur Heart J. 1998;19(Suppl D):D3–D9.
25. Mackman N. New insights into the mechanisms of venous thrombosis. J Clin Invest. 2012;122:2331–2336.
26. Dahlback B. Blood coagulation. Lancet. 2000;355:1627–1632.
27. Davie EW, et al. The coagulation cascade: initiation, maintenance, and regulation. Biochemistry. 1991;30:10363–10370.
28. Naito K, et al. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J Biol Chem. 1991;266:7353–7358.
29. Marlar RA, et al. Mechanism of action of human activated protein C, a thrombin-dependent anticoagulant enzyme. Blood. 1982;59:1067–1072.
30. Corral J, et al. Homozygous deficiency of heparin cofactor II: relevance of p17 glutamate residue in serpins, relationship with conformational diseases, and role in thrombosis. Circulation. 2004;110:1303–1307.
31. Adelman B, et al. Plasmin effect on platelet glycoprotein Ib-von Willebrand factor interactions. Blood. 1985;65:32–40.
32. Sidelmann JJ, et al. Fibrin clot formation and lysis: basic mechanisms. Semin Thromb Hemost. 2000;26:605–618.
33. Esmon CT. The regulation of natural anticoagulant pathways. Science. 1987;235:1348–1352.
34. Hassouna HI. Laboratory evaluation of hemostatic disorders. Hematol Oncol Clin North Am. 1993;7:1161–1249.
35. Palareti G, et al. D-dimer testing to determine the duration of anticoagulation therapy. N Engl J Med. 2006;355:1780–1789.
36. Ljungner H, et al. Decreased fibrinolytic activity in the bottom of human vein valve pockets. Vasa. 1983;12:333–336.
37. Dano K, et al. Plasminogen activators, tissue degradation, and cancer. Adv Cancer Res. 1985;44:139–266.
38. Vassalli JD, et al. The plasminogen activator/plasmin system. J Clin Invest. 1991;88:1067–1072.
39. Kohler HP, et al. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. 2000;342:1792–1801.
40. Booth NA, et al. Plasminogen activator inhibitor (PAI-1) in plasma and platelets. Br J Haematol. 1988;70:327–333.
41. Schulman S, et al. The significance of hypofibrinolysis for the risk of recurrence of venous thromboembolism. Duration of anticoagulation (DURAC) trial study group. Thromb Haemost. 1996;75:607–611.
42. Crowther MA, et al. Fibrinolytic variables in patients with recurrent venous thrombosis: a prospective cohort study. Thromb Haemost. 2001;85:390–394.
43. Segui R, et al. PAI-1 promoter 4g/5g genotype as an additional risk factor for venous thrombosis in subjects with genetic thrombophilic defects. Br J Haematol. 2000;111:122–128.
44. Zoller B, et al. A common 4g allele in the promoter of the plasminogen activator inhibitor-1 (PAI-1) gene as a risk factor for pulmonary embolism and arterial thrombosis in hereditary protein S deficiency. Thromb Haemost. 1998;79:802–807.
45. Becker MD, et al. Reduced leukocyte migration, but normal rolling and arrest, in interleukin-8 receptor homologue knockout mice. Invest Ophthalmol Vis Sci. 2000;41:1812–1817.
46. Yang Y, et al. Regulation of tissue factor expression in human microvascular endothelial cells by nitric oxide. Circulation. 2000;101:2144–2148.
47. Gross PL, et al. The endothelium and thrombosis. Semin Thromb Hemost. 2000;26:463–478.
48. Stewart GJ. Neutrophils and deep venous thrombosis. Haemostasis. 1993;23(Suppl 1):127–140.
49. Esmon CT. Inflammation and thrombosis. J Thromb Haemost. 2003;1:1343–1348.
50. Gilbert GE, et al. Platelet-derived microparticles express high affinity receptors for factor VIII. J Biol Chem. 1991;266:17261–17268.
51. Mesri M, et al. Endothelial cell activation by leukocyte microparticles. J Immunol. 1998;161:4382–4387.
52. Sabatier F, et al. Interaction of endothelial microparticles with monocytic cells in vitro induces tissue factor-dependent procoagulant activity. Blood. 2002;99:3962–3970.
53. Falati S, et al. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med. 2003;197:1585–1598.
55. Osterud B. The role of platelets in decrypting monocyte tissue factor. Semin Hematol. 2001;38:2–5.
56. Ahn ER, et al. Differences of soluble CD40l in sera and plasma: implications on CD40l assay as a marker of thrombotic risk. Thromb Res. 2004;114:143–148.
57. Hrachovinova I, et al. Interaction of P-selectin and PSGL-1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nat Med. 2003;9:1020–1025.
58. Andre P, et al. Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc Natl Acad Sci U S A. 2000;97:13835–13840.
59. Podor TJ, et al. Vimentin exposed on activated platelets and platelet microparticles localizes vitronectin and plasminogen activator inhibitor complexes on their surface. J Biol Chem. 2002;277:7529–7539.
60. Myers D Jr, et al. Selectins influence thrombosis in a mouse model of experimental deep venous thrombosis. J Surg Res. 2002;108:212–221.
61. Myers DD, et al. P-selectin and leukocyte microparticles are associated with venous thrombogenesis. J Vasc Surg. 2003;38:1075–1089.
62. Myers DD, et al. Inflammation-dependent thrombosis. Front Biosci. 2005;10:2750–2757.
63. Polgar J, et al. The P-selectin, tissue factor, coagulation triad. J Thromb Haemost. 2005;3:1590–1596.
64. Siddiqui FA, et al. The presence and release of tissue factor from human platelets. Platelets. 2002;13:247–253.
65. Giesen PL, et al. Blood-borne tissue factor: another view of thrombosis. Proc Natl Acad Sci U S A. 1999;96:2311–2315.
66. Celi A, et al. P-selectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci U S A. 1994;91:8767–8771.
67. Jy W, et al. Platelet factor 3 in plasma fractions: its relation to microparticle size and thromboses. Thromb Res. 1995;80:471–482.
68. Reitsma PH, et al. Mechanistic view of risk factors for venous thromboembolism. Arterioscler Thromb Vasc Biol. 2012;32:563–568.
69. Brill A, et al. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117:1400–1407.
70. von Bruhl ML, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–835.
71. Day SM, et al. Macrovascular thrombosis is driven by tissue factor derived primarily from the blood vessel wall. Blood. 2005;105:192–198.
72. Henn V, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591–594.
73. Horrevoets AJ. Plasminogen activator inhibitor 1 (PAI-1): in vitro activities and clinical relevance. Br J Haematol. 2004;125:12–23.
74. Meissner MH, et al. Deep venous insufficiency: the relationship between lysis and subsequent reflux. J Vasc Surg. 1993;18:596–605.
75. Killewich LA, et al. Regression of proximal deep venous thrombosis is associated with fibrinolytic enhancement. J Vasc Surg. 1997;26:861–868.
76. Saha P, et al. Leukocytes and the natural history of deep vein thrombosis: current concepts and future directions. Arterioscler Thromb Vasc Biol. 2011;31:506–512.
77. Wakefield TW, et al. Venous thrombosis-associated inflammation and attenuation with neutralizing antibodies to cytokines and adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15:258–268.
78. Ripplinger CM, et al. Inflammation modulates murine venous thrombosis resolution in vivo: assessment by multimodal fluorescence molecular imaging. Arterioscler Thromb Vasc Biol. 2012;32:2616–2624.
79. Varma MR, et al. Neutropenia impairs venous thrombosis resolution in the rat. J Vasc Surg. 2003;38:1090–1098.
80. Fuchs TA, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–15885.
81. Fuchs TA, et al. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:1777–1783.
82. Brill A, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10:136–144.
83. Varma MR, et al. Neutropenia impairs venous thrombosis resolution in the rat. J Vasc Surg. 2003;38:1090–1098.
84. Henke PK, et al. Neutrophils modulate post-thrombotic vein wall remodeling but not thrombus neovascularization. Thromb Haemost. 2006;95:272–281.
85. Varma MR, et al. Deep vein thrombosis resolution is not accelerated with increased neovascularization. J Vasc Surg. 2004;40:536–542.
86. Hogaboam CM, et al. Novel roles for chemokines and fibroblasts in interstitial fibrosis. Kidney Int. 1998;54:2152–2159.
87. Humphries J, et al. Monocyte chemotactic protein-1 (MCP-1) accelerates the organization and resolution of venous thrombi. J Vasc Surg. 1999;30:894–899.
88. Henke PK, et al. Targeted deletion of CCR2 impairs deep vein thrombosis resolution in a mouse model. J Immunol. 2006;177:3388–3397.
89. Evans CE, et al. Upregulation of hypoxia-inducible factor 1 alpha in local vein wall is associated with enhanced venous thrombus resolution. Thromb Res. 2011;128:346–351.
90. Modarai B, et al. Adenovirus-mediated VEGF gene therapy enhances venous thrombus recanalization and resolution. Arterioscler Thromb Vasc Biol. 2008;28:1753–1759.
91. Lim CS, et al. Prolonged mechanical stretch is associated with upregulation of hypoxia-inducible factors and reduced contraction in rat inferior vena cava. J Vasc Surg. 2011;53:764–773.
92. Myers DD Jr, et al. Treatment with an oral small molecule inhibitor of P selectin (PSI-697) decreases vein wall injury in a rat stenosis model of venous thrombosis. J Vasc Surg. 2006;44:625–632.
93. Moaveni DK, et al. Vein wall re-endothelialization after deep vein thrombosis is improved with low-molecular-weight heparin. J Vasc Surg. 2008;47:616–624.
94. Stenberg B, et al. Effect of venous stasis on vessel wall fibrinolysis. Thromb Haemost. 1984;51:240–242.
95. Wojcik BM, et al. Interleukin-6: a potential target for post-thrombotic syndrome. Ann Vasc Surg. 2011;25:229–239.
96. Grainger DJ, et al. Release and activation of platelet latent TGFB in blood clots during dissolution with plasmin. Nature Med. 1995;1:932–937.
97. Diaz JA, et al. Impaired fibrinolytic system in APOE gene-deleted mice with hyperlipidemia augments deep vein thrombosis. J Vasc Surg. 2012;55:815–822.
97a. [Deatrick et al: JVS; In press] Deatrick KB, et al. The effect of matrix metalloproteinase 2/9 deletion in experimental post-thrombotic vein wall remodeling. J Vasc Surg. 2013.
98. Baldwin JF, et al. The role of urokinase plasminogen activator and plasmin activator inhibitor-1 on vein wall remodeling in experimental deep vein thrombosis. J Vasc Surg. 2012;56:1089–1097.
99. Deatrick KB, et al. Chronic venous insufficiency: current management of varicose vein disease. Am Surg. 2010;76:125–132.
100. Delis KT, et al. Venous claudication in iliofemoral thrombosis: long-term effects on venous hemodynamics, clinical status, and quality of life. Ann Surg. 2004;239:118–126.
101. Franzeck UK, et al. On the relationship between changes in the deep veins evaluated by duplex sonography and the postthrombotic syndrome 12 years after deep vein thrombosis. Thromb Haemost. 1997;77:1109–1112.
102. Eklof B, et al. Revision of the CEAP classification for chronic venous disorders: consensus statement. J Vasc Surg. 2004;40:1248–1252.
103. Lalka SG, et al. Elevated cutaneous leukocyte concentration in a rodent model of acute venous hypertension. J Surg Res. 1998;74:59–63.
104. Pappas PJ, et al. Role of leukocyte activation in patients with venous stasis ulcers. J Surg Res. 1995;59:553–559.
105. Homans J. The etiology and treatment of varicose ulcer of the leg. Surg Gynecol Obstet. 1917;24:300–311.
106. Blalock A. Oxygen content of blood in patients with varicose veins. Arch Surg. 1929;19:898–905.
107. Browse NL, et al. The cause of venous ulceration. Lancet. 1982;2:243–245.
108. Coleridge Smith PD, et al. Causes of venous ulceration: a new hypothesis. Br Med J (Clin Res Ed). 1988;296:1726–1727.
109. Burnand KG, et al. The relationship between the number of capillaries in the skin of the venous ulcer-bearing area of the lower leg and the fall in foot vein pressure during exercise. Br J Surg. 1981;68:297–300.
110. Labropoulos N, et al. Where does venous reflux start? J Vasc Surg. 1997;26:736–742.
111. Sadick NS. Predisposing factors of varicose and telangiectatic leg veins. J Dermatol Surg Oncol. 1992;18:883–886.
112. Gundersen J, et al. Hereditary factors in venous insufficiency. Angiology. 1969;20:346–355.
113. Cornu-Thenard A, et al. Importance of the familial factor in varicose disease. Clinical study of 134 families. J Dermatol Surg Oncol. 1994;20:318–326.
114. Badier-Commander C, et al. Smooth muscle cell modulation and cytokine overproduction in varicose veins: an in situ study. J Pathol. 2001;193:398–407.
115. Niebes P. Vessel wall modification in venous pathology. Application to the study of phlebotonic drugs. Int Angiol. 1996;15:88–92.
116. Gandhi RH, et al. Analysis of the connective tissue matrix and proteolytic activity of primary varicose veins. J Vasc Surg. 1993;18:814–820.
117. Waksman Y, et al. Collagen subtype pattern in normal and varicose saphenous veins in humans. Isr J Med Sci. 1997;33:81–86.
118. Sansilvestri-Morel P, et al. Imbalance in the synthesis of collagen type I and collagen type III in smooth muscle cells derived from human varicose veins. J Vasc Res. 2001;38:560–568.
119. Sansilvestri-Morel P, et al. Abnormal deposition of extracellular matrix proteins by cultured smooth muscle cells from human varicose veins. J Vasc Res. 1998;35:115–123.
120. Porto LC, et al. Immunolabeling of type IV collagen, laminin, and alpha-smooth muscle actin cells in the intima of normal and varicose saphenous veins. Angiology. 1998;49:391–398.
121. Sansilvestri-Morel P, et al. Synthesis of collagen is dysregulated in cultured fibroblasts derived from skin of subjects with varicose veins as it is in venous smooth muscle cells. Circulation. 2002;106:479–483.
122. Somers P, et al. The histopathology of varicose vein disease. Angiology. 2006;57:546–555.
123. Shireman PK, et al. Plasminogen activator levels are influenced by location and varicosity in greater saphenous vein. J Vasc Surg. 1996;24:719–724.
124. Parra JR, et al. Tissue inhibitor of metalloproteinase-1 is increased in the saphenofemoral junction of patients with varices in the leg. J Vasc Surg. 1998;28:669–675.
125. Badier-Commander C, et al. Increased TIMP/MMP ratio in varicose veins: a possible explanation for extracellular matrix accumulation. J Pathol. 2000;192:105–112.
126. Woodside KJ, et al. Morphologic characteristics of varicose veins: possible role of metalloproteinases. J Vasc Surg. 2003;38:162–169.
127. Kosugi I, et al. Matrix metalloproteinase-9 and urokinase-type plasminogen activator in varicose veins. Ann Vasc Surg. 2003;17:234–238.
128. Rizzi A, et al. Effects of vasoactive agents in healthy and diseased human saphenous veins. J Vasc Surg. 1998;28:855–861.
129. Raffetto JD, et al. Functional adaptation of venous smooth muscle response to vasoconstriction in proximal, distal, and varix segments of varicose veins. J Vasc Surg. 2010;51:962–971.
130. Barber DA, et al. Characterization of endothelin receptors in human varicose veins. J Vasc Surg. 1997;26:61–69.
131. Nemcova S, et al. Cyclic nucleotides and production of prostanoids in human varicose veins. J Vasc Surg. 1999;30:876–883.
132. Raffetto JD, et al. Matrix metalloproteinase 2-induced venous dilation via hyperpolarization and activation of K+ channels: relevance to varicose vein formation. J Vasc Surg. 2007;45:373–380.
133. Cheatle TR, et al. The pathogenesis of skin damage in venous disease: a review. Eur J Vasc Surg. 1991;5:115–123.
134. O’Kane S, et al. Transforming growth factor beta S and wound healing. Int J Biochem Cell Biol. 1997;29:63–78.
135. Border WA, et al. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–1292.
136. Pappas PJ, et al. Dermal tissue fibrosis in patients with chronic venous insufficiency is associated with increased transforming growth factor-beta1 gene expression and protein production. J Vasc Surg. 1999;30:1129–1145.
137. Peschen M, et al. Increased expression of platelet-derived growth factor receptor alpha and beta and vascular endothelial growth factor in the skin of patients with chronic venous insufficiency. Arch Dermatol Res. 1998;290:291–297.
138. Nicolaides AN. Chronic venous disease and the leukocyte-endothelium interaction: from symptoms to ulceration. Angiology. 2005;56(Suppl 1):S11–S19.
139. Saharay M, et al. Endothelial activation in patients with chronic venous disease. Eur J Vasc Endovasc Surg. 1998;15:342–349.
140. Nicolaides AN. Investigation of chronic venous insufficiency: a consensus statement (France, March 5-9, 1997). Circulation. 2000;102:E126–E163.
141. Shields DA, et al. Plasma elastase in venous disease. Br J Surg. 1994;81:1496–1499.
142. Scott HJ, et al. Histological study of white blood cells and their association with lipodermatosclerosis and venous ulceration. Br J Surg. 1991;78:210–211.
143. Wilkinson LS, et al. Leukocytes: their role in the etiopathogenesis of skin damage in venous disease. J Vasc Surg. 1993;17:669–675.
144. Pappas PJ, et al. Morphometric assessment of the dermal microcirculation in patients with chronic venous insufficiency. J Vasc Surg. 1997;26:784–795.
145. Clyne CA, et al. Oxygen tension on the skin of the gaiter area of limbs with venous disease. Br J Surg. 1985;72:644–647.
146. Raffetto JD, et al. Changes in cellular motility and cytoskeletal actin in fibroblasts from patients with chronic venous insufficiency and in neonatal fibroblasts in the presence of chronic wound fluid. J Vasc Surg. 2001;33:1233–1241.
147. Hasan A, et al. Dermal fibroblasts from venous ulcers are unresponsive to the action of transforming growth factor-beta1. J Dermatol Sci. 1997;16:59–66.
148. Lal BK, et al. Altered proliferative responses of dermal fibroblasts to TGF-beta1 may contribute to chronic venous stasis ulcer. J Vasc Surg. 2003;37:1285–1293.
149. Stanley AC, et al. Reduced growth of dermal fibroblasts from chronic venous ulcers can be stimulated with growth factors. J Vasc Surg. 1997;26:994–999.
150. Ackerman Z, et al. Overload of iron in the skin of patients with varicose ulcers: possible contributing role of iron accumulation in progression of the disease. Arch Dermatol. 1988;124:1376–1378.
151. Zamboni P, et al. Hemochromatosis C282Y gene mutation increases the risk of venous leg ulceration. J Vasc Surg. 2005;42:309–314.
152. Herrick SE, et al. Sequential changes in histologic pattern and extracellular matrix deposition during the healing of chronic venous ulcers. Am J Pathol. 1992;141:1085–1095.
153. Calligaro KD, et al. Venous aneurysms: surgical indications and review of the literature. Surgery. 1995;117:1–6.
154. Gillespie DL, et al. Presentation and management of venous aneurysms. J Vasc Surg. 1997;26:845–852.
155. Winchester D, et al. Popliteal venous aneurysms. Surgery. 1993;114:600–607.
156. Jorgensen JO, et al. The incidence of deep venous thrombosis in patients with superficial thrombophlebitis of the lower limbs. J Vasc Surg. 1993;18:70–73.
157. Woodhouse CR. Infusion thrombophlebitis: the histological and clinical features. Ann R Coll Surg Engl. 1980;62:364–368.
158. Luis Rodriguez-Peralto J, et al. Superficial thrombophlebitis. Semin Cutan Med Surg. 2007;26:71–76.
160. Tapper H, et al. Modulation of hemostatic mechanisms in bacterial infectious diseases. Blood. 2000;96:2329–2337.
161. Shebuski RJ, et al. Role of inflammatory mediators in thrombogenesis. J Pharmacol Exp Ther. 2002;300:729–735.
162. Ageno W, et al. Cardiovascular risk factors and venous thromboembolism: a meta-analysis. Circulation. 2008;117:93–102.
163. Prandoni P, et al. Venous thromboembolism and the risk of subsequent symptomatic atherosclerosis. J Thromb Haemost. 2006;4:1891–1896.
164. Becattini C, et al. Aspirin for preventing the recurrence of venous thromboembolism. N Engl J Med. 2012;366:1959–1967.
165. Brighton TA, et al. Low-dose aspirin for preventing recurrent venous thromboembolism. N Engl J Med. 2012;367:1979–1987.
166. Ray JG, et al. Use of statins and the subsequent development of deep vein thrombosis. Arch Intern Med. 2001;161:1405–1410.
167. Glynn RJ, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med. 2009;360:1851–1861.
168. Diaz J: Unpublished data, 2011.