[level-membership-for-pulmolory-and-respiratory-category]CHAPTER 21
Vasopressors, inotropes, and antiarrhythmic agents
After reading this chapter, the reader will be able to:
1. Define terms that pertain to vasopressors, inotropes, and antiarrhythmic drugs.
2. List the various components that make up blood pressure
3. Compare and contrast the mechanism of action of inotropes and vasopressors
4. Describe the various drug interactions that may occur with the use of vasopressors and inotropes
5. Design an algorithm for the management of hypotension
6. Describe the normal conduction of the heart
7. Define nonpharmacologic methods of treating dysrhythmias
8. Compare and contrast the categories of the Vaughan Williams classification system
9. Define the mechanism of action of digoxin
10. List all the dysrhythmias associated with cardiac arrest
11. Design an algorithm that may be used in the management of ventricular fibrillation and pulseless ventricular tachycardia
12. Design an algorithm that may be used in the management of torsades de pointes
13. Describe the proper dosing technique of intravenous magnesium therapy in the management of torsades de pointes
14. List the routes of administering medications during cardiac arrest
Group of cardiac medications that are classified according to mechanism of action; in some instances, they may have multiple mechanisms of action. The most common classification system of antiarrhythmics is the Vaughan Williams classification system, which is divided into four distinct categories and a miscellaneous section.
Irregular (faster or slower) heartbeat; the term arrhythmia is used more frequently than dysrhythmia.
Link between atrial depolarization and ventricular depolarization.
Presence of carbon dioxide aids in the release and delivery of oxygen from hemoglobin.
Amount of blood that is pumped out of the heart per unit of time.
Endogenous products that are secreted into the bloodstream and travel to nerve endings to stimulate an excitatory response.
Agent affecting the rate of contraction of the heart.
Diastolic blood pressure (DBP)
Lowest pressure reached prior to ventricular ejection.
An agent that influences the conduction of electrical impulses. A positive dromotropic agent enhances the conduction of electrical impulses to the heart.
Agent affecting the strength of muscular contraction.
Pressure that drives blood into the tissues averaged over the entire cardiac cycle.
Enzyme responsible for the breakdown of cyclic adenosine 3′,5′-monophosphate (cAMP).
Episode of ventricular fibrillation, pulseless ventricular tachycardia, pulseless electrical activity, or asystole leading to loss of life.
Peak pressure reached during ventricular ejection.
Overly rapid heartbeat, usually defined as greater than 100 beats/min in adults.
Agent causing dilation of blood vessels.
Agent causing contraction of capillaries and arteries.
Cardiac condition in which normal ventricular contractions are replaced by coarse or fine, rapid movements of the ventricular muscle.
Overview of cardiovascular system
The cardiovascular system regulates blood flow to the various regions of the body. Blood flow generally travels via a pressure gradient, shifting from areas of higher pressure to lower pressure. The central nervous system (CNS) relays electrical impulses through sensory receptors found systemically within the vasculature, affecting vascular tone and causing shunting of blood to and from various organ systems within the body. Vascular tone is regulated via the sympathetic nervous system and the circulation of neurotransmitters and hormones, such as epinephrine, vasopressin, and angiotensin. Several factors exert an effect on vascular tone as a response to tissue perfusion and circulatory volume. Hypotension is commonly present in patients with autonomic dysfunction and shock. Shock is a life-threatening medical emergency characterized by organ hypoperfusion leading to decreased delivery of oxygen and nutrients to tissues throughout the body. There are six types of shock and their effect on hemodynamic parameters can be seen in Table 21-1.
TABLE 21-1
Hemodynamic Changes in Various Shock States
| HEMODYNAMIC PARAMETER | HYPOVOLEMIC/HEMORRHAGIC | NEUROGENIC | CARDIOGENIC | SEPTIC/DISTRIBUTIVE |
| HR | ↑ | ↔ | ↔/↑ | ↑ |
| MAP | ↓ | ↑/↓ | ↑ | ↓ |
| CVP (5-12 mm Hg) | ↓ | ↓ | ↑ | ↓ |
| PCWP (10-12 mm Hg) | ↓ | ↓ | ↑ | ↓ |
| CO (5-7 L/min) | ↓ | ↔/↓ | ↓ | ↑ |
| SVR (80-1440 dyn•sec•cm−5) | ↑ | ↓ | ↑ | ↓ |

Factors affecting blood pressure
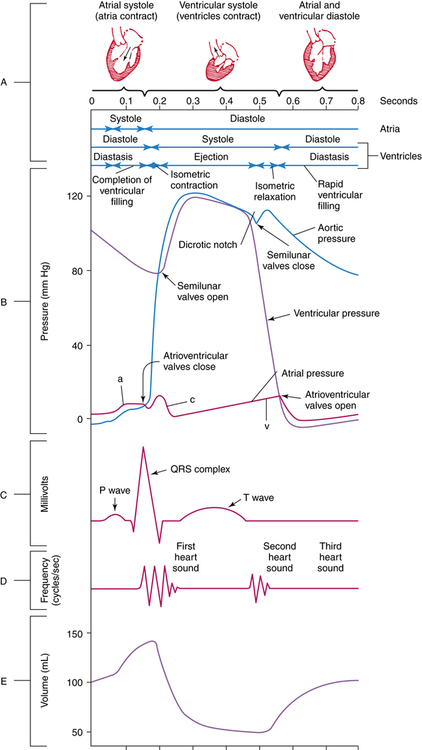
Typical measurement of blood pressure is relative to a recurring cardiac cycle of atrial and ventricular contractions and relaxations (Figure 21-1). The cycle is divided into the systolic phase and the diastolic phase. The systolic phase is the portion in which ventricular contraction occurs, resulting in ejection of blood through the aorta. Conversely, diastole is the period of ventricular relaxation and blood filling. Systolic blood pressure (SBP) is the peak pressure reached during ventricular ejection, and diastolic blood pressure (DBP) is the lowest pressure reached right before ventricular ejection. Arterial pressure is typically recorded as SBP/DBP, for example, 120/80 mm Hg. Mean arterial pressure (MAP) refers to the pressure that drives blood into the tissues averaged over the entire cardiac cycle. Because the cardiac cycle is pulsatile rather than continuous and because two-thirds of the normal cardiac cycle is spent in diastole, MAP is not the arithmetic mean of the SBP and DBP. MAP is defined as the product of cardiac output (CO) and systemic vascular resistance (SVR), as follows:
< ?xml:namespace prefix = "mml" />
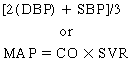
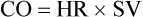
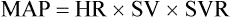
The use of therapies such as fluids, vasopressors, and inotropes to maintain cardiovascular stability is directed toward altering each of these components, as seen in Table 21-2. The various vasopressors currently on the market have different affinities for the various receptors located within the body and exert different effects on the hemodynamic parameters, as seen in Table 21-3. Vasopressors and inotropes are not always first-line therapy; on the contrary, fluids are the mainstays for improving hypotensive episodes. Vasopressors and inotropes have considerable side effects, and certain medications interact with various vasopressors and inotropes, leading to alterations in hemodynamic parameters as seen in Table 21-4.
TABLE 21-2
Cardiac Drugs: Dosing, Pharmacokinetics, and Hemodynamic Effects
| PHARMACOKINETICS | HEMODYNAMIC EFFECTS | ||||||||
| AGENT | DOSE RANGE | ONSET (min) | DURATION | HALF-LIFE | HR | MAP | PCWP | SVR | CO |
| Amrinone | 0.75 mg/kg bolus, then 2.5-15 μg/kg/min | 5-10 | 0.5-2 hr | 4.8-8.3 hr | ↓ | 0-↓ | 0-↓ | 0-↓ | ↓-0-↑ |
| Dobutamine (Dobutrex) | 2-20 μg/kg/min | 1-2 | 10-15 min | 2 min | 0* | 0-↓ | ↓ | ↓ | ↑ |
| Dopamine (Inotropin) | 1-5 μg/kg/min | 5 | <10 min | 2 min | 0 | 0 | 0 | 0 | 0 |
| 5-15 μg/kg/min | 5 | <10 min | 2 min | ↑ | 0-↓ | 0-↑ | ↑ | ↑ | |
| >15 μg/kg/min | 5 | <10 min | 2 min | ↑ | 0-↓ | 0-↑ | ↑ | ↑ | |
| Epinephrine (Adrenalin) | 0.01-0.1 μg/kg/min | 1 | 3-5 min | 3-5 min | ↑ | ↑ | 0-↓ | ↓†-↑* | ↑ |
| 0.1 μg/kg/min | ↑↑ | ↑↑ | ↑ | ↑↑ | ↑↑ | ||||
| Norepinephrine (Levophed) | 0.5-30 μg/min | 1-3 | 5-10 min | 1-2 min | 0-↑ | ↑↑↑ | ↑↑ | ↑↑↑ | 0-↓ |
| Phenylephrine (Neo-Synephrine) | 0.5-5 μg/kg/min | 10-15 | 1-3 hr | 2-3 hr | ↓ | ↑ | ↑ | ↑ | ↓ |
| Milrinone (Primacor) | 50 μg/kg bolus, then 0.375-0.75 μg/kg/min | 90 | 3-5 hr | 2.3 hr | 0-↑ | ↓ | ↓ | ↓ | ↑ |
| Vasopressin (Pitressin) | 0.04 U/min | 30-60 | 30-60 min | 10-20 min | 0 | ↑ | ↑ | ↑ | ↓? |
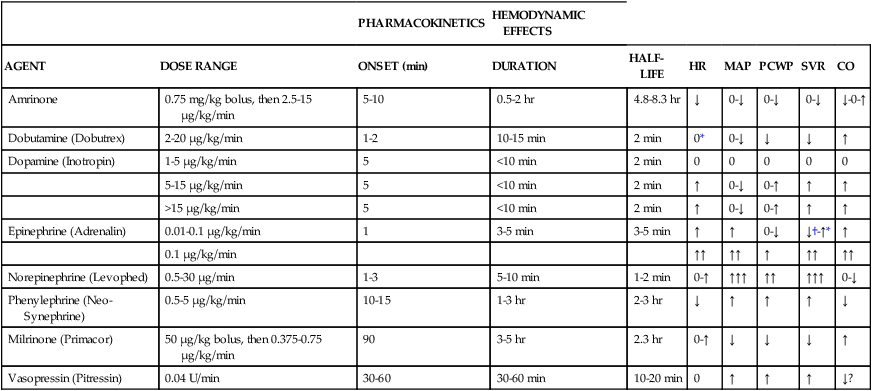
TABLE 21-3
Inotropes and Vasopressors: Receptor Affinity
| DRUG | α | β1 | β2 | DA |
| Dopamine (Inotropin) | + to +++* | +++* | + | 0/+ |
| Dobutamine (Dobutrex) | 0 to +* | 0 to +* | + | 0 |
| Epinephrine (Adrenalin) | +++* | +++ | ++* | 0 |
| Isoproterenol (Isuprel) | 0 | +++ | +++ | 0 |
| Norepinephrine (Levophed) | +++ | ++ | ++ | 0 |
| Phenylephrine (Neo-Synephrine) | +++ | 0 | 0 | 0 |
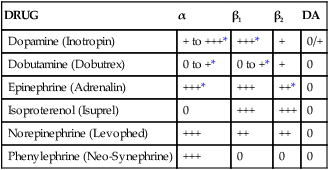
DA, Dopamine; 0, no effect; +, slight effect; ++, moderate effect; +++, pronounced effect.
TABLE 21-4
| PRECIPITANT DRUG* | EFFECT | OBJECT DRUG* | COMMENTS |
| Dobutamine, Isoproterenol, Norepinephrine | |||
| Bretylium | ↑ | Dobutamine, isoproterenol, norepinephrine | Concomitant use may potentiate effects of vasopressors, causing arrhythmias |
| Halogenated hydrocarbon anesthetics | |||
| Guanethidine | May increase pressor response, causing severe hypertension | ||
| Oxytocic drugs | |||
| Tricyclic antidepressants | |||
| Phenylephrine | |||
| Bretylium | ↑ | Phenylephrine | Concomitant use may potentiate effects of vasopressors |
| Guanethidine | |||
| Halogenated hydrocarbon anesthetics | |||
| Oxytocic drugs | |||
| Tricyclic antidepressants | ↔ | Tricyclic antidepressants may increase effects of phenylephrine | |
| Dopamine | |||
| Dopamine | ↓ | Guanethidine | Antihypertensive effects of guanethidine may be reversed |
| Phenytoin | Concomitant use may lead to seizures, severe hypotension, and bradycardia | ||
| Tricyclic antidepressants | Dopamine | Tricyclic antidepressants may increase effects of dopamine | |
| Halogenated hydrocarbon anesthetics | ↑ | Dopamine | May sensitize myocardium to actions of vasopressors, causing arrhythmia |
| MAOIs | Dopamine is metabolized by MAOIs. MAOIs increase pressor response to dopamine by 6-fold to 20-fold | ||
| Oxytocic drugs | Concomitant use may cause severe hypertension | ||
| Epinephrine | |||
| Cardiac glycosides | ↑ | Epinephrine | May sensitize myocardium to actions of vasopressors, causing arrhythmia |
| Halogenated hydrocarbon anesthetics | |||
| Levothyroxine antihistamines (chlorpheniramine, diphenhydramine) | |||
| MAOIs | Concomitant use may cause severe hypertension | ||
| Methyldopa | |||
| Oxytocic drugs | |||
| Reserpine | |||
| Sympathomimetics | |||
| Tricyclic antidepressants | |||
| β blockers | |||
| α blockers | ↓ | Epinephrine | Vasoconstricting and hypertensive effects of pressor may be reversed |
| Chlorpromazine | |||
| Diuretics | |||
| Epinephrine | Guanethidine | Epinephrine may antagonize effects of guanethidine, resulting in decreased antihypertensive effects | |
| Digoxin | |||
| Amiodarone | ↑ | Digoxin | Amiodarone may increase digoxin blood level; reduce digoxin dose by 50% |
| β blockers | Combination may cause advanced or complete heart block | ||
| Calcium channel blockers | |||
| Calcium | Rapid administration of intravenous calcium may result in fatal arrhythmias | ||
| Succinylcholine | Succinylcholine may cause sudden extrusion of K+ from muscle cells, leading to arrhythmias | ||
| Sympathomimetics | Combination may cause increased risk of cardiac arrhythmias | ||
| Thiazide and loop diuretics | Diuretic-induced electrolyte disturbances may predispose to digitalis toxicity | ||
| Thyroid hormones | ↓ | Digoxin | Thyroid hormones may reduce digoxin blood levels; hypothyroid patients may require higher dose of digoxin |
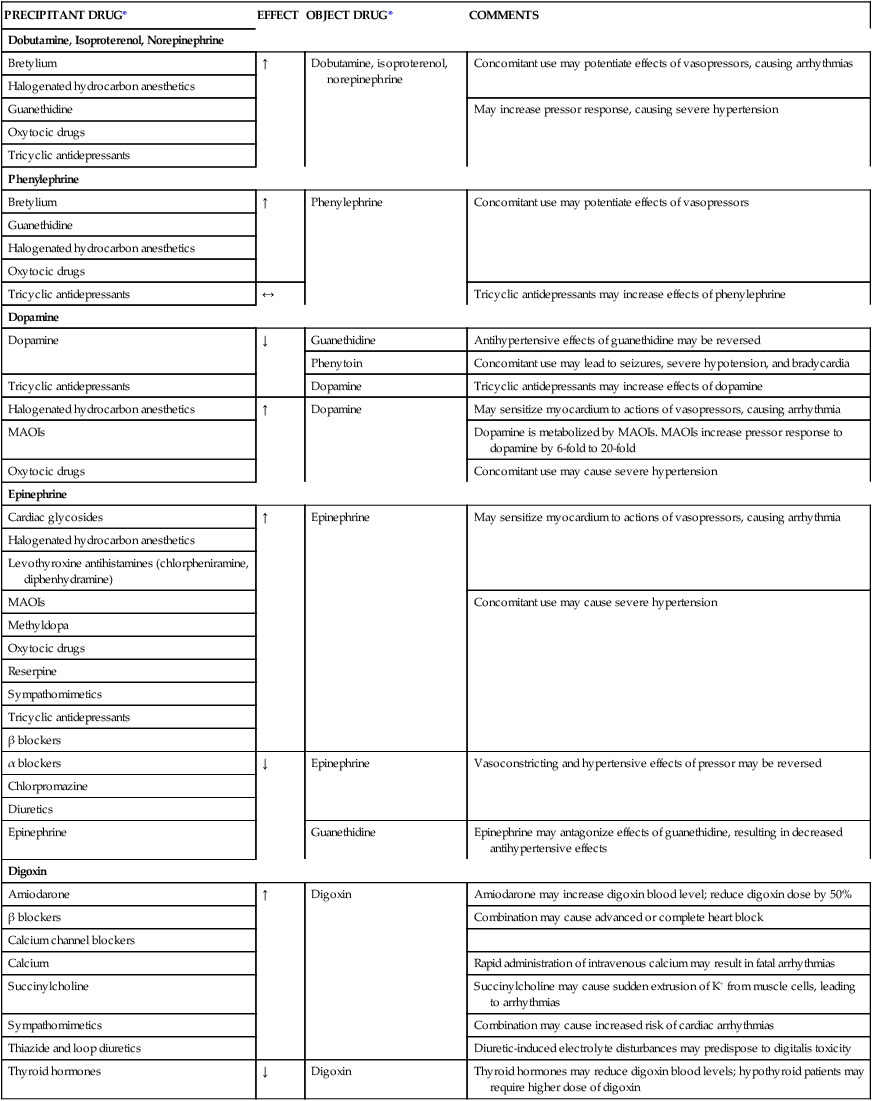
MAOIs, Monoamine oxidase inhibitors.
*Precipitant drug refers to the drug that causes the interaction; object drug refers to the drug affected by the interaction. ↑, Object drug increased; ↓, object drug decreased; ↔, object drug unaffected.
Agents used in the management of shock
Catecholamines
Norepinephrine (levophed) and epinephrine (adrenalin chloride)
Norepinephrine (Levophed) and epinephrine (Adrenalin Chloride) are endogenous catecholamines that are secreted by the adrenal medulla. Epinephrine is ultimately synthesized by the catalytic actions of tyrosine hydroxylase, which converts the amino acid tyrosine to levodopa and subsequently to dopamine, norepinephrine, and epinephrine.1,2 These neurotransmitters travel to sympathetic nerve endings, where they are released to stimulate other nerve fibers and to stimulate an excitatory response. Norepinephrine and epinephrine stimulate α receptors on the vasculature and β receptors, also located within the vasculature as well as in the myocardium. α receptors within the vasculature cause vasoconstriction, whereas β receptors cause vasodilation. Most vascular beds within the body contain β receptors; however, they are outnumbered by α receptors, so any epinephrine and norepinephrine stimulation of β receptors is negligible or of no effect, yielding a net response of vasoconstriction. In addition, β receptors more densely populate the myocardium compared with α receptors, leading to a net effect of tachycardia.3,4
Isoproterenol (isuprel)
Isoproterenol (Isuprel) is a synthetic catecholamine used for the treatment of symptomatic bradycardia or torsades de pointes. Isoproterenol works solely as an agonist of β receptors. By stimulating β1-adrenergic receptors, it exerts pronounced inotropic and chronotropic effects. By stimulating β2-adrenergic receptors, it leads to smooth muscle relaxation of the bronchi, skeletal muscle, vasculature, and gastrointestinal tract. Venous return to the heart is also increased by vasodilation of the venous bed. The use of isoproterenol is limited because of its pronounced stimulatory effect on the heart rate.4
Vasopressin
The 2008 practice guidelines5 published by the Society of Critical Care Medicine do not recommend vasopressin as the initial vasopressor of choice or as a lone agent. Precaution stems from the fact that vasopressin infusion may decrease splanchnic blood flow. Other settings in which vasopressin may be used include diabetes insipidus at doses of 5 to 10 U given intramuscularly or subcutaneously and repeated two or three times per day. Vasopressin has also been used to treat esophageal bleeding at doses up to 2 U/min. Caution is also needed when treating conditions other than shock; myocardial ischemia may ensue as a result of the potent vasoconstrictive properties at higher doses.
Inotropic agents
Dobutamine
Dobutamine is indicated for the short-term treatment of decompensated heart failure secondary to depressed contractility. Dobutamine is a synthetic catecholamine that is chemically related to dopamine; however, in contrast to dopamine, it is not metabolized to norepinephrine, and it does not stimulate dopamine receptors.4 Its pharmacologic actions are due to the effects of its racemic components. The (R)-isomer is responsible for its activity on the β1 and β2 receptors, causing predominant positive inotropic and chronotropic effects and vasodilatory effects, respectively. This combination of effects enhances cardiac output and stroke volume. The (S)-isomer is responsible for its activity on the α1 receptors, causing vasoconstriction.1,6 The vasodilatory β2-adrenergic effects counterbalance the vasoconstrictive α1 effects, leading to minor changes in systemic vascular resistance usually seen at lower doses. With increasing doses, the β2-vasodilatory actions predominate over the α1-vasoconstrictive effect, causing a decrease in systemic and pulmonary vascular resistance. The decline in systemic and pulmonary vascular resistance may also be secondary to enhanced cardiac output.
As an inotropic agent, dobutamine has adverse cardiac effects, which include arrhythmias, increase in myocardial oxygen consumption and demand, tachycardia, and hypotension. A limiting factor when dobutamine is used for more than 72 hours is tachyphylaxis; this may be due to a downregulation of β1 receptors and may be overcome by increasing the dose. In patients with sulfite sensitivity, allergic reactions such as anaphylaxis or life-threatening asthmatic episodes may occur because dobutamine formulations contain sulfites.4
Phosphodiesterase inhibitors: inamrinone and milrinone
Milrinone is the phosphodiesterase inhibitor most commonly used in practice today because it has a shorter half-life than inamrinone and is less likely to cause thrombocytopenia. It undergoes renal elimination with an elimination half-life of 1 to 3 hours in patients with normal renal function; steady-state concentrations are reached in 4 to 6 hours if initiated without a loading dose. The risk of hypotension occurring is higher when a loading dose is given. Milrinone may be given as an initial intravenous (IV) bolus dose of 50 μg/kg administered slowly over 10 minutes followed by continuous infusion at a rate of 0.375 to 0.75 μg/kg/min and titrated to effect. Dosage adjustment should be made in patients with severe cardiac failure or renal impairment because of the considerable reduction in clearance.1,4,6
Cardiac glycosides: digoxin (lanoxin)
In contrast, digitalis toxicity may cause hyperkalemia by its inhibitory actions on the Na+,K+-ATPase pump. Digoxin toxicity may manifest as serious life-threatening ventricular arrhythmias, including premature ventricular contractions, AV junctional rhythm, bigeminal rhythm, and second-degree AV blockade. Bradycardia may also occur early on in the setting of digoxin toxicity. The initial symptoms of digitalis toxicity are nausea, vomiting, anorexia, and abdominal pain. These symptoms may be due to a direct effect on the gastrointestinal tract or result from CNS stimulation of the chemoreceptor trigger zone. Other rare but possible neuropsychiatric effects may manifest as disorientation and hallucination, especially in elderly patients, and visual disturbances such as yellow-green halos. Digoxin immune Fab is the antidote used to facilitate the speedy elimination of digoxin from the body. Digoxin immune Fab is indicated in the setting of life-threatening toxicity such as ventricular arrhythmias, bradyrhythmias, ingestion of greater than 10 mg in adults or 4 mg in children, a steady state level greater than 10 ng/mL, progressive elevation of potassium, or a potassium level greater than 5 mEq/L.7
Electrophysiology of myocardium
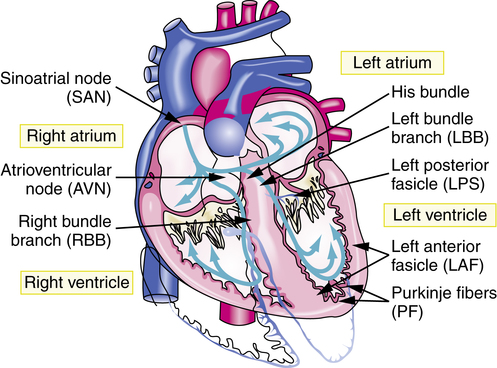
Electrical activity is initiated by an innate pacemaker located at the sinoatrial (SA) node. Electrical potential exists across the cell membrane, and it changes in response to transmembrane movement of Na+, K+, Ca2+, and Cl− ions. These ions mediate the process of myocardial contraction and relaxation. When an electrical stimulus is evoked from the SA node, it generates an action potential (AP). Once generated, the AP produces a local current, which evokes further APs along the myocardium. An AP elicits myocardial depolarization or contraction. The link between atrial depolarization and ventricular depolarization is a portion of the conduction system called the atrioventricular (AV) node. The AV node slows down the electrical impulse to ensure that atrial excitation is completed before ventricular excitation. After leaving the AV node, the impulse travels to the wall between the two ventricles via the conducting system fibers known as the bundle of His. From the bundle of His, the cardiac conduction system bifurcates into three main bundle branches: the right bundle and two left bundles. These bundle branches form a conduction network, referred to as Purkinje fibers (Figure 21-2). The conduction system innervates the myocardium and causes changes in membrane polarization of the muscle fiber.8
An AP (Figure 21-3) can be divided into the following five different phases:
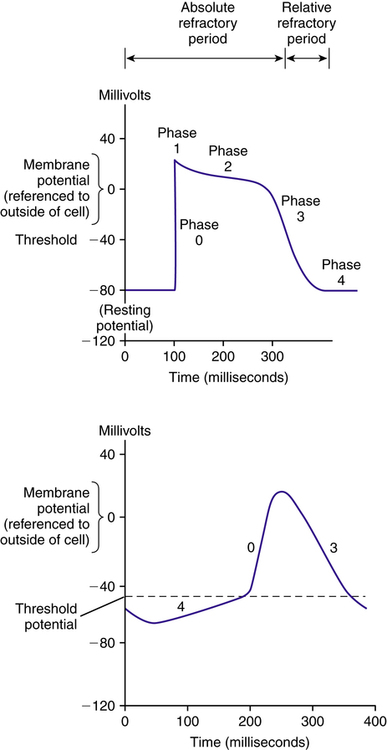
Phase 0: Initial rapid depolarization of myocardial tissues secondary to an abrupt transmembrane influx of sodium through “fast” sodium channels
Phase 1: Fast sodium channels are inactivated; this, coupled with the movement of K+ and Cl− ions, leads to a transient net outward current and the beginning of repolarization
Phase 2: “Plateau” phase, maintained by a balance between calcium influx and potassium efflux
Phase 3: Calcium channels close, but membrane remains permeable to potassium, resulting in cellular repolarization
Phase 4: Cell returns to its “resting” state; the resting membrane potential is reached through gradual depolarization related to a constant sodium influx balanced by a decreasing efflux of potassium
Ablation with radiofrequency current
Catheter ablation is very effective when atrial fibrillation (AF) is due to a single primary circuit. The procedure involves inserting a catheter into a blood vessel in the groin or the neck and guiding it toward the heart. When the tip of the catheter is placed against the part of the heart causing the arrhythmia, radiofrequency electrical current is applied through the catheter to produce a small burn about 6 to 8 mm in diameter. Patients should be adequately anticoagulated at least 1 month before the ablation procedure to prevent the formation of thrombi in the atria. The procedure carries a success rate in maintaining sinus rhythm over the next year of 30% to 90%.9
Implantable cardioverter-defibrillators
• Cardiac arrest caused by pulseless ventricular tachycardia (VT) or ventricular fibrillation (VF) not due to a transient or reversible cause
• Syncope of undetermined origin with clinically relevant, electrophysiologically inducible sustained VT or VF when drug therapy is ineffective, not tolerated, or not preferred
• Nonsustained VT in patients with coronary artery disease, before myocardial infarction, left ventricular dysfunction, and electrophysiologically inducible VT or VF not suppressed by class I antiarrhythmics
Of patients with ICDs, 40% to 70% require antiarrhythmic drug therapy, which puts them at risk for drug-ICD interactions.10
Pharmacology of antiarrhythmics
Antiarrhythmics are classified according to their mechanisms of action. In some instances, these drugs may manifest multiple mechanisms of action. The most common classification system of antiarrhythmics is the Vaughan Williams classification system, which divides antiarrhythmics into classes: I (IA, IB, IC), II, III, IV, and a miscellaneous class. See Table 21-5 for the detailed pharmacology of antiarrhythmics, and see Table 21-6 for their pharmacokinetic parameters.
TABLE 21-5
Pharmacology of Antiarrhythmics
| CLASS/MOA | ION BLOCK | DRUG | QRS | Q–Tc | INDICATIONS | DOSES | ROUTE |
| IA/↓ phase 0, ↑ AP | Sodium (intermediate) | Moricizine* | ↑ | 0 | VA | 600-900 mg/day in three divided doses | PO |
| Quinidine | ↑ | ↑ | AF/AFL/VA | Quinidine sulfate, 200-600 mg q 4-12 hr; quinidine gluconate, AF/AFL cardioversion and VA, 324-648 mg q 8-12 hr | PO | ||
| Quinidine gluconate, AF/AFL cardioversion and VA, 10 mg/min infusion up to 400 mg | IV | ||||||
| Procainamide | ↑ | ↑ | VA | 40-50 kg, 2 g/day; 60-70 kg, 3 g/day; 80-90 kg, 4 g/day; >100 kg, 5 g/day | IV | ||
| Disopyramide | ↑ | ↑ | VA | 400-800 mg/day in divided doses, IR divided q 6 hr, CR divided q 12 hr | PO | ||
| IB/↓ phase 0 slightly; shorten AP | Sodium (fast on/off) | Lidocaine | 0 | 0-↓ | VA | 50-100 mg (may repeat in 5 min) up to 300 mg in any 1-hr period; maintenance 1-4 mg/min | IV |
| VT | 1-1.5 mg/kg; may repeat at 0.5-0.75 mg/kg q 5-10 min (maximum 3 mg/kg) | ||||||
| Mexiletine | 0 | 0 | VA | 200-400 mg q 8 hr | PO | ||
| 150-250 mg over 10 min, then 250 mg over 30-60 min, then 250 mg over 2.5 hr, then 500 mg over 8 hr; maintenance 250-500 mg q 12 hr | IV | ||||||
| Tocainide | 0 | 0-↓ | VA | 400 mg q 8 hr, then 1200-1800 mg/day divided q 8 hr (maximum 2400 mg/day) | PO | ||
| Phenytoin | 0 | ↓ | VA | 4 mg/kg q 6 hr for 1 day, then 5-6 mg/kg/day divided q 12 hr | PO | ||
| 15 mg/kg over 1 hr (or target level of 15-20 μg/ml) | IV | ||||||
| IC/Marked ↓ of phase 0; affect repolarization | Sodium (slow on/off) | Flecainide | ↑↑ | 0-↑ | AF/AFL/PSVT | 50 mg q 12 hr; ↑ by 100 mg q 4 days (maximum 300 mg/day) | PO |
| VA/VT | 200-400 mg/day | ||||||
| Propafenone | ↑ | 0-↑ | AF | 225 mg q 12 hr (SR) | PO | ||
| AFL | 325-425 mg q 12 hr (IR) | ||||||
| PSVT | 150 mg q 12 hr (IR) | ||||||
| AF/AFL/PSVT/VA | AF, 225 mg (SR) q 12 hr, ↑ to 325-425 mg q 12 hr; AFL/PSVT/VA, 150-300 mg (IR) q 8 hr | ||||||
| II/↓ phase 4 (depolarization) | Calcium (indirect) | Propranolol | 0 | 0-↓ | AF/AFL/PSVT/PVC | Loading dose, 0.5-1 mg q 2 min (up to 0.1-0.15 mg/kg); maintenance dose, 0.04 mg/kg/min | IV |
| Maintenance dose, 10-120 mg three times daily | PO | ||||||
| Esmolol | 0 | 0-↓ | Loading dose, 0.5 mg/kg over 1 min; maintenance dose, 50-300 μg/kg/min (bolus between dose increases) | IV | |||
| Acebutolol | 0 | 0-↓ | Initial, 200 mg twice a day; maintenance, 600-1200 mg/day (in two or three divided doses) | PO | |||
| Metoprolol | 0 | 0-↓ | Initial, 2.5-5 mg q 2-5 min (up to 15 mg over 10-15 min) | IV | |||
| Maintenance dose, 25-100 mg twice a day | PO | ||||||
| Atenolol | 0 | 0-↓ | 0.5 mg/min in aliquots of 2.5 mg with 10-min interval between aliquots (maximum single dose 10 mg) | IV | |||
| Initial, 50-100 mg daily | PO | ||||||
| Nadolol | 0 | 0-↓ | 0.01-0.05 mg/kg at 1 mg/min (maximum cumulative dose 10 mg) | IV | |||
| 60-160 mg/day in single or divided doses | PO | ||||||
| III/↑ phase 3 (repolarization) | Potassium | Amiodarone | ↑ | ↑↑ | VA | 800-1600 mg for 1-3 wk, then 600-800 mg for 1 mo, then 400-600 mg daily | PO |
| 150-300 mg bolus, then 1 mg/min for 6 hr, then 0.5 mg/min for 18 hr | IV | ||||||
| Dronedarone | ↑ | ↑↑ | AF/AFL | 400 mg twice a day with meals | PO | ||
| Bretylium | 0 | 0 | VA | Loading dose, 5-10 mg/kg bolus, may repeat to a maximum of 30 mg/kg, then 1-2 mg/min or 5-10 mg/kg over 8 min q 6 hr | IV | ||
| Dofetilide | AF/AFL | Q–Tc ≤440 msec, 500 μg twice a day 2-3 hr after first dose if Q–Tc increases >15% or >500 msec, ↓ dose to 250 μg twice a day | PO | ||||
| Sotalol | 0 | ↑↑ | AF/AFL | CrCl >60 mL/min, 160 mg/day; CrCl 40-60 mL/min, 80 mg/day; titrate to Q–Tc <520 msec (maximum 320 mg/day) | PO | ||
| VA | 80 mg twice a day, ↑ at 40-80 mg q 2-3 days (maximum 480-640 mg/day) | ||||||
| Ibutilide | 0 | ↑↑ | AF/AFL | ≥60 kg, 1 mg; <60 kg, 0.1 mg/kg over 10 min (may repeat once) | IV | ||
| IV/↓ phase 4, ↑ phases 1 and 2 | Calcium | Verapamil | 0 | 0 | SVT | IR, 240-320 mg/day in three or four divided doses; up to 480 mg/day in three or four divided doses for patients not on digoxin therapy | PO |
| 0.075-0.15 mg/kg over 2 min; may give 10 mg after 30 min if no response | IV | ||||||
| Diltiazem | 0 | ↓ | PSVT | 0.25 mg/kg over 2 min; if no response, may give 0.35 mg/kg after 15 min; maintenance, 5-10 mg/hr; ↑ in 5-mg/hr increments up to 15 mg/hr for up to 24 hr | IV | ||
| ↑ phase 4 ↓ AP | Na+,K+ pump | Digoxin | 0 | ↓ | SVT | 8-12 μg/kg | IV |
| ↓ conduction time; interrupts reentry through AV node | Adenosine receptor | Adenosine | 0 | 0 | SVT | 6 mg over 1-2 sec; ↑ to 12 mg q 1-2 min as needed for two doses (maximal single dose 12 mg) | IV |
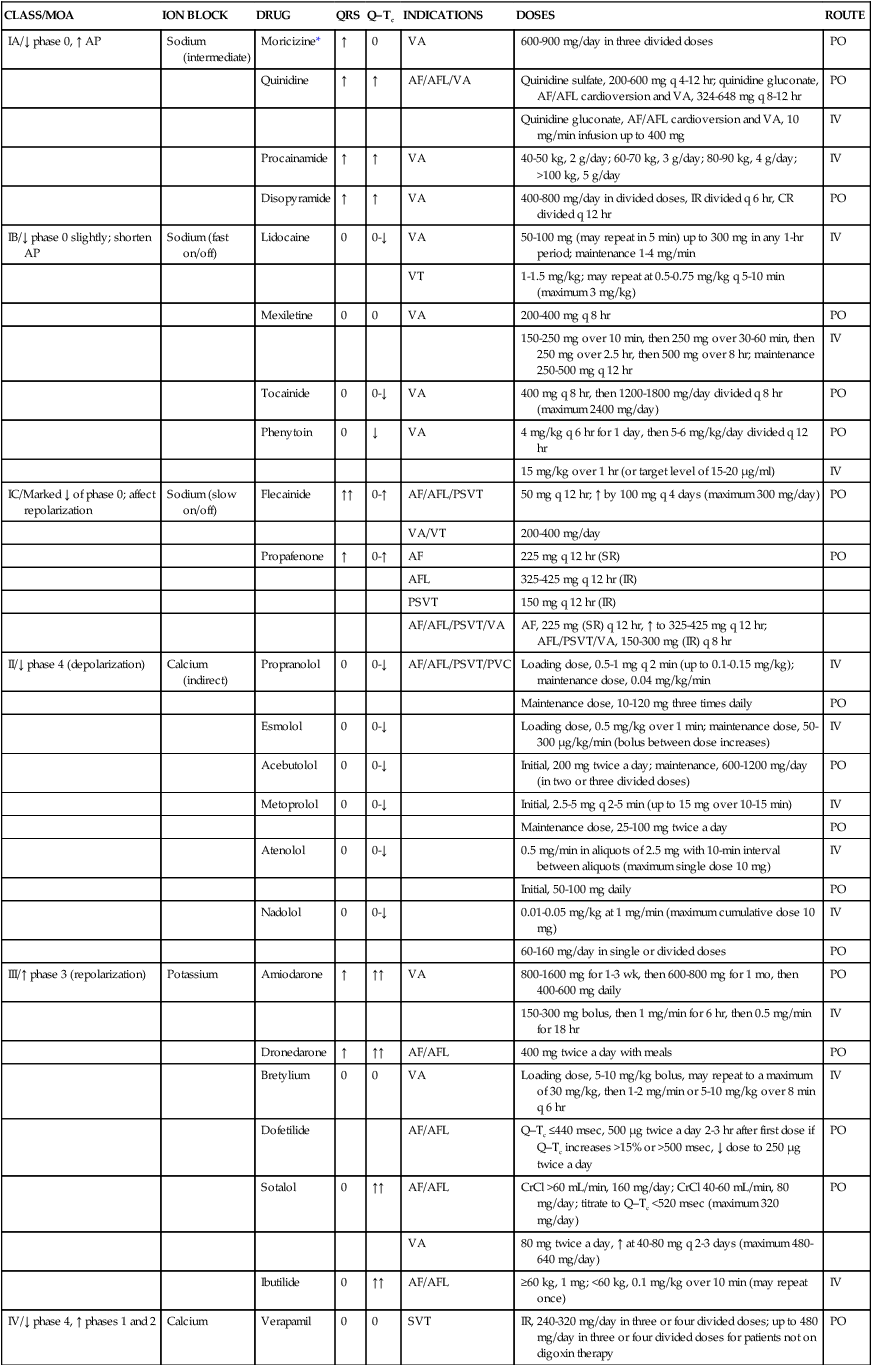
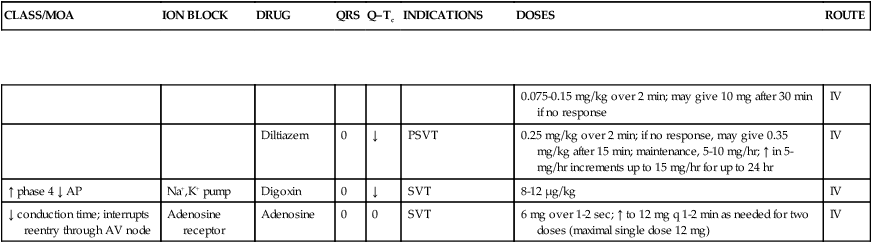
*Moricizine does not belong to any subclass (IA, IB, or IC) of antiarrhythmic but does have some properties of each.
TABLE 21-6
Antiarrhythmics: Pharmacokinetics and Adverse Reactions
| PHARMACOKINETICS | ||||||
| DRUG | ONSET (PO) (hr) | DURATION (hr) | HALF-LIFE (hr) | THERAPEUTIC RANGE (μg/mL) | TOXIC LEVEL (μg/mL) | ADVERSE REACTIONS |
| Moricizine (Ethmozine) | 2 | 10-24 | 1.5-3.5 | NA | NA |
All class I agents: Negative inotropic effect, infranodal conduction block Quinidine: N/V/D, cinchonism (tinnitus, blurred vision, dizziness, tremor) Procainamide: N/V/D (30%), bitter taste, rash, hepatitis, mental depression, psychosis Disopyramide: Anticholinergic effects (dry mouth, blurred vision, urinary retention), hypoglycemia, cholestatic jaundice, agranulocytosis Class IB: Muscle twitch, seizures, proarrhythmia, dyspnea Phenytoin: Hypotension, gingival hyperplasia, antiepileptic hypersensitivity syndrome Class IC: Ventricular proarrhythmia Propafenone: Dyspnea (2%), worsening of asthma, metallic taste |
All: Sinus bradycardia; torsades de pointes, heart failure exacerbation
Amiodarone: GI (25%), ocular (10%), CNS, hepatic (40%-55%), dermatologic (15%), hypothyroidism/hyperthyroidism (4%)
Dronedarone: N/V/D, asthenia, elevated serum creatinine (51%)
Dofetilide: Headache (11%), chest pain (10%), dizziness (8%), dyspnea (6%)
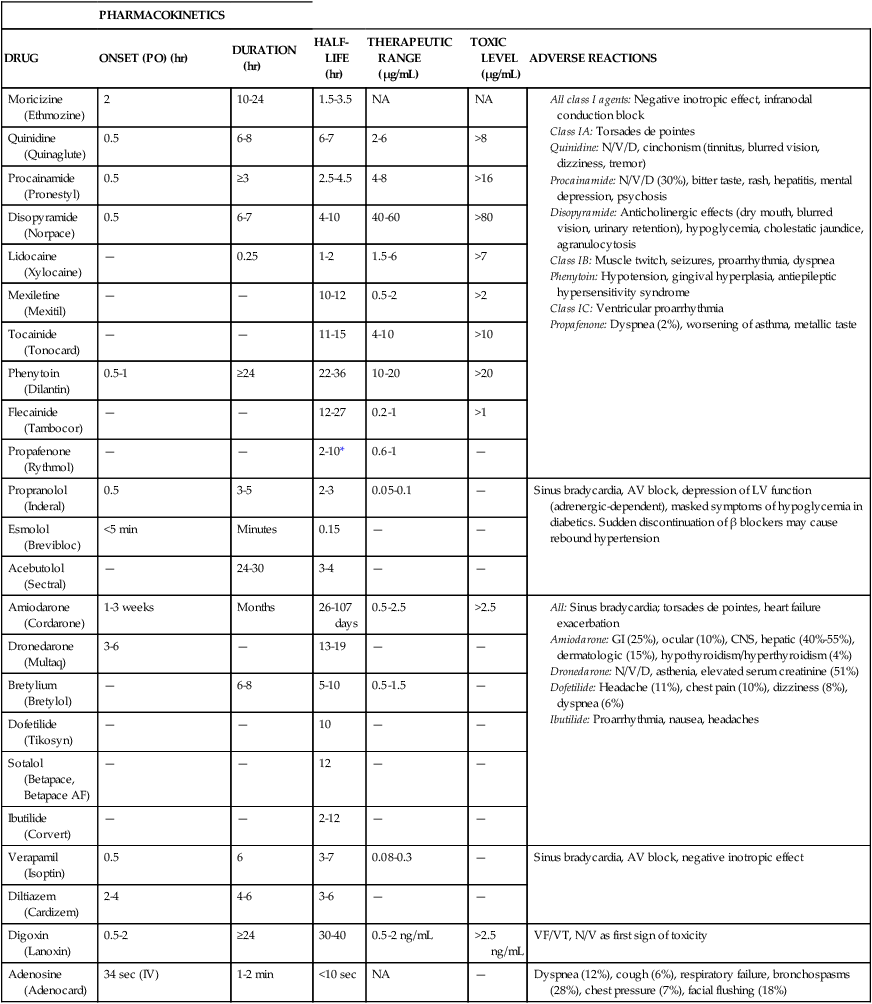
*Half-life is 6-36 hours in patients who are poor metabolizers of propafenone (i.e., patients with low-activity CYP2D6 isozyme).
Class IA
Quinidine
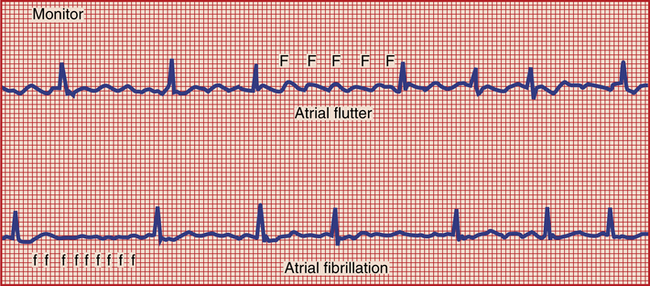
Quinidine, although less commonly used, is efficacious in the treatment of atrial fibrillation/flutter (AF/AFL) (Figure 21-4). The effects of quinidine on the AV node are bimodal. At lower concentrations, quinidine has antivagal properties, enhancing AV nodal conduction. At higher concentrations, the AV nodal conduction is slowed down. Because of difficulty in predicting response to quinidine, it is important to initiate a rate-controlling agent first. Quinidine should be used with caution in patients with preexisting asthma, muscle weakness, or infection with fever because hypersensitivity reactions to this medication may be masked by these conditions. Overdosage of quinidine has produced respiratory depression or distress, apnea, diarrhea and vomiting, seizures, hypotension, syncope, and electrocardiogram (ECG) changes.11
Procainamide
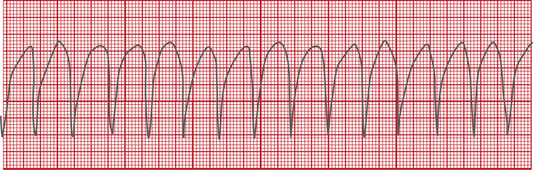
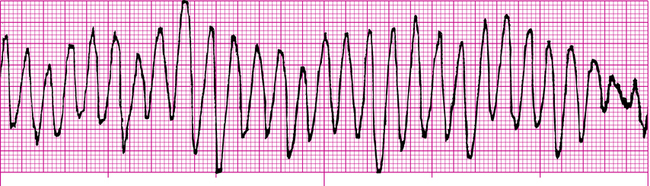
Procainamide is available only as an IV formulation in the United States and is indicated for the treatment of ventricular tachycardia (VT) (Figure 21-5) that is life-threatening; because of its proarrhythmic effects, including torsades de pointes (Figure 21-6), the use of this agent for lesser arrhythmias is not recommended. In addition, procainamide has the potential to produce serious hematologic disorders, particularly leukopenia and agranulocytosis; it is used only when benefits outweigh the risks. Procainamide has also been used to convert AF/AFL to sinus rhythm. It is necessary to monitor levels of both procainamide and its active metabolite N-acetyl procainamide (NAPA) for efficacy and toxicity. An adverse effect unique to procainamide is the development of lupus erythematosus–like syndrome, which can manifest with pleural or abdominal pain, myalgias, arthralgias, pleural effusion, pericarditis, fever, chills, and skin lesions. Lupus erythematosus–like syndrome occurs in 30% of patients after prolonged administration of procainamide, especially in slow acetylators, who are at risk of accumulating the hydroxylamine metabolite responsible for the pathogenesis of this syndrome. If the lupoid syndrome does not resolve with discontinuation of procainamide, treatment with corticosteroids may be warranted.4
Disopyramide
Disopyramide is indicated for the treatment of life-threatening VT; it is also used for the treatment of paroxysmal supraventricular tachycardia (PSVT). Treatment with disopyramide should be initiated in the hospital. Patients with AF/AFL must receive digoxin therapy to achieve an adequate serum digoxin level before administration of disopyramide to ensure there is no further elevation of ventricular rate. Potassium should be corrected before initiation of therapy because the drug may be ineffective in patients with hypokalemia, and its toxic effects may be enhanced in hyperkalemia. Disopyramide may cause or aggravate congestive heart failure or episodes of hypotension because of its negative inotropic properties. Overdose with disopyramide may be followed by apnea, loss of consciousness, cardiac arrhythmias, and loss of spontaneous respirations requiring mechanical ventilation or other vigorous treatment modalities. This agent has limited use because of its anticholinergic side effects, including dry mouth, difficulty in urination, dizziness, tachycardia, hyperthermia, and blurred vision.4
Class IB
Lidocaine
Lidocaine is used frequently to treat ventricular arrhythmia (VA) occurring during cardiac surgery or after an acute myocardial infarction. After administering IV bolus doses (owing to its short half-life of approximately 1.5 to 2 hours), continuous infusion is necessary to maintain sinus rhythm. Lidocaine is metabolized extensively in the liver to two toxic metabolites, monoethylglycinexylidide and glycinexylidide; these metabolites display antiarrhythmogenic properties but are also highly prone to seizure activity. Patients need to be monitored vigilantly for signs of seizure, such as tremors.11 Other CNS side effects associated with lidocaine are insomnia, drowsiness, ataxia, agitation, and dysarthria. Caution should also be exercised in patients with hepatic failure or congestive heart failure because the rate of drug clearance is significantly reduced in either condition. Lidocaine infusions lasting longer than 24 hours may prolong the half-life of lidocaine to approximately 3 hours leading to a greater risk of lidocaine accumulation and toxicity. In the setting of lidocaine infusion longer than 24 hours, the infusion rate should be reduced by approximately 50%. Lidocaine has also been implicated in causing respiratory depression and arrest.4
Mexiletine
Mexiletine has a mechanism of action similar to lidocaine and is available as an oral formulation. It is indicated for the treatment of life-threatening VAs. Because of its anesthetic properties, it is also used at lower doses to reduce neuropathic pain associated with diabetic neuropathy. In controlled trials, the most frequent adverse events were gastrointestinal disturbances (41%), tremor (12%), and lightheadedness and difficulty in coordination (more than 10%). Dyspnea and respiratory problems occurred in 5.7% of patients. Coma and respiratory arrest may occur with massive overdoses.11
Tocainide
Tocainide is the oral congener of lidocaine and is used to treat VAs and may also be used to treat myotonic dystrophy and trigeminal neuralgia. Tocainide carries a U.S. Food and Drug Administration (FDA) black box warning (so called for the black border surrounding the warning located in the manufacturer information sheet or the package insert) for causing pulmonary disorders, including pulmonary edema, fibrosing alveolitis, pneumonitis, and respiratory arrest (0.11%). These pulmonary manifestations are detectable on radiographic studies within 3 to 18 weeks of therapy. Another black box warning is for blood dyscrasias, which are not that prevalent (0.18%) but are associated with a fatality rate of up to 25%.4
Class IC
Class IC agents are generally not used mainly because of their relatively higher proarrhythmic potential. Other agents from this class have been withdrawn from the market (i.e., encainide and moricizine) because of their substantial proarrhythmic potential as shown in two landmark trials: Cardiac Arrhythmia Suppression Trial I (CAST I) and CAST II12 Class IC agents are commonly used in the management of supraventricular arrhythmias, but they have activity against ventricular arrhythmias as well.
Flecainide (tambocor)
Flecainide (Tambocor) is indicated for the prevention of paroxysmal AF/AFL associated with disabling symptoms and PSVT, including AV nodal reentrant tachycardia, AV tachycardia, other supraventricular tachycardia (SVT) in patients without structural heart disease, and sustained VT. It is efficacious in suppressing AF in 61% to 92% of patients treated. Flecainide has a long half-life, and the dose should not be increased more often than every 4 days. Flecainide was one of the antiarrhythmics studied in CAST in patients with asymptomatic non–life-threatening arrhythmias occurring 6 days to 2 years after documented myocardial infarction. Flecainide contributed to an excessive mortality or nonfatal cardiac arrest rate of 5.1% versus 2.3% for its matched placebo. Long-term oral prophylaxis with an antiarrhythmic agent poses a great risk of adverse events, and relapse rates are high. Also, flecainide elimination is affected by urinary pH, leading to either toxic or subtherapeutic levels. Alkaline pH decreases and acidic pH increases renal excretion of flecainide.4
The “pill-in-the-pocket” approach is the alternative treatment of recurrent arrhythmias, in which a pill is taken by the patient at the time of onset of palpitations. One study assessed this approach in the conversion of AF to sinus rhythm with class IC agents, using either flecainide or propafenone as a single oral dose to convert patients to sinus rhythm out of hospital. Flecainide was shown to be equally effective for pill-in-the-pocket treatment of recurrent AF, with a 94% efficacy rate.13
Propafenone (rythmol)
Propafenone (Rythmol) seems to be comparable to other antiarrhythmics in preventing PSVT and maintaining sinus rhythm after successful cardioversion. It is considered a first-line agent for conversion of recent-onset (less than 48 hours) AF, with efficacy rates of 60% to 90%. Therapy is 15% to 30% less effective in patients manifesting symptoms of AF for more than 48 hours. Propafenone displays nonselective β-blocking activity, and it generally should not be used to treat patients with asthma or bronchospastic disease because β-blocking properties may inhibit bronchodilation. The highest concentrations of the drug are found in the lungs (10-fold higher than in the heart muscles or liver and 24-fold higher than in the kidneys).11
Class III
Amiodarone (cordarone)
Amiodarone-induced pulmonary toxicity warrants substantial concern when treating patients with arrhythmias. The main caveat associated with amiodarone is its distinctive side-effect profile.5 Baseline parameters that must be obtained before starting therapy, along with incidences of various side effects, are presented in Table 21-7. Pulmonary toxicity is quite common, as evidenced by cough and by local or diffuse infiltrates on chest radiographs, and occurs at a rate of up to 20%. Amiodarone-induced pulmonary toxicity is managed best by discontinuation or by corticosteroid therapy; in some cases, fatalities of approximately 10% have been reported 11 In addition, amiodarone is probably regarded as one of the most potent inhibitors of the cytochrome P450 (CYP450) 3A4 isoenzyme system, and it inhibits CYP2C9 and CYP2C19 (hepatic drug-metabolizing enzymes); concomitant prescription medications, herbals, and over-the-counter products must be evaluated for detection of severe, often life-threatening interactions (Table 21-8).
TABLE 21-7
Routine Laboratory Testing in Patients Receiving Amiodarone
| TYPE OF TEST | TIME WHEN TEST IS PERFORMED |
| Liver enzyme tests | Baseline and then every 6 months |
| Thyroid function (T4 and TSH) | Baseline and then every 6 months |
| Serum creatinine and electrolytes | Baseline and then every 6 months |
| Chest radiograph | Baseline and then yearly |
| Ophthalmic evaluation | Baseline and for visual impairment or symptoms, and then every 6 months |
| Pulmonary function tests | Baseline and for unexplained dyspnea, especially in patients with underlying lung disease, and if there are suggestive abnormalities on chest radiograph |
| ECG | Baseline and then yearly |
ECG, Electrocardiogram; T4, thyroxine; TSH, thyroid-stimulating hormone.
TABLE 21-8
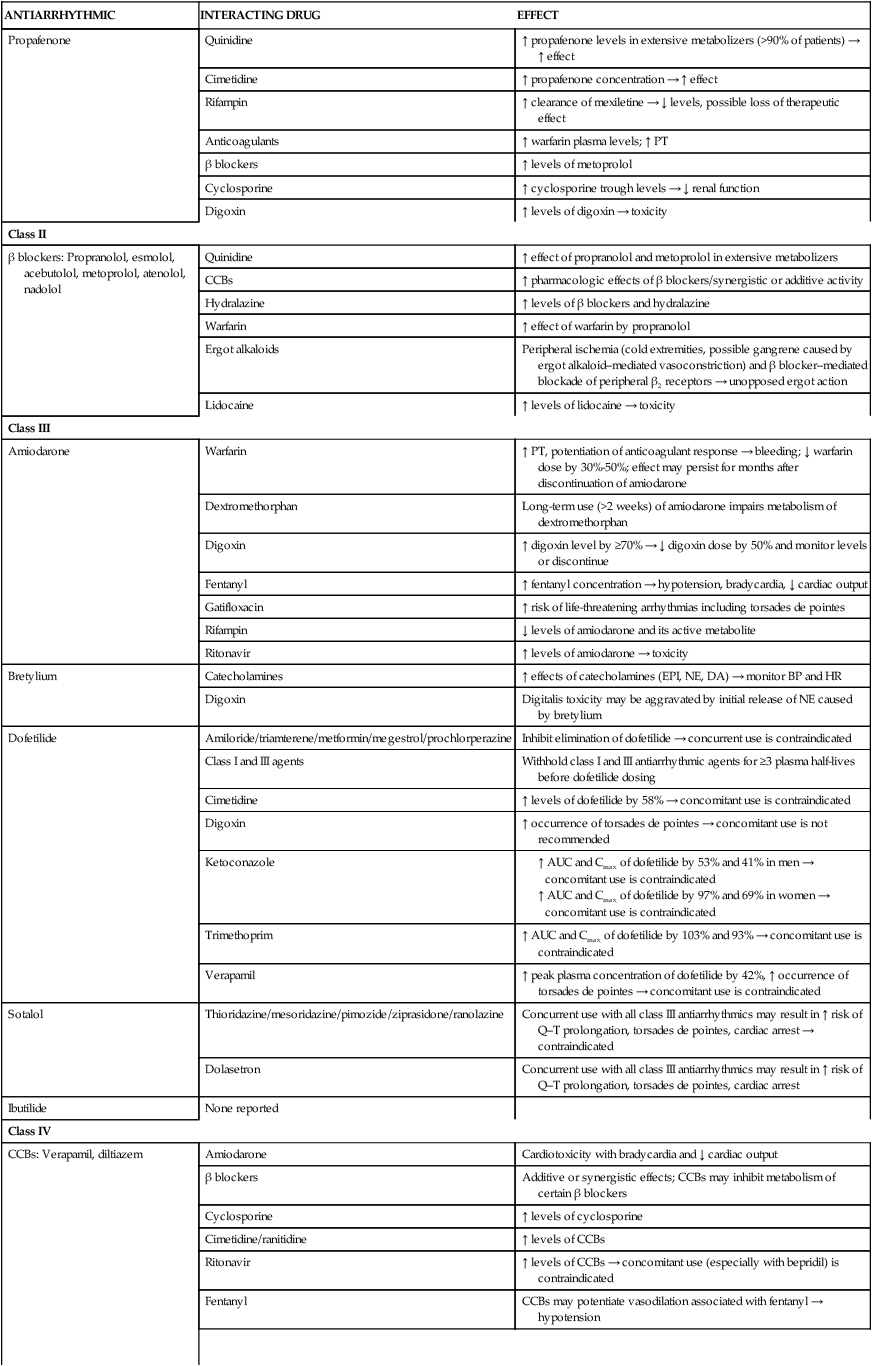
Clinically Significant Drug Interactions with Antiarrhythmics
| ANTIARRHYTHMIC | INTERACTING DRUG | EFFECT |
| Class IA | ||
| Moricizine | Cimetidine | Levels ↑ 1.4-fold, ↓ clearance by 50%—initiate moricizine at lower doses (<600 mg/day) |
| Digoxin | Additive P–R interval prolongation (not as significant as second or third AV block) | |
| Propranolol | Small additive ↑ in P–R interval; no change in overall ECG intervals | |
| Theophylline | ↑ theophylline clearance by 44%-66% and half-life by 19%-33% | |
| Diltiazem | ↑ concentration of moricizine by diltiazem; ↓ diltiazem concentration by moricizine | |
| Quinidine | Amiodarone | ↑ levels of quinidine → fatal arrhythmias |
| Antacids | ↑ levels of quinidine | |
| Barbiturates | ↓ levels and elimination half-life of quinidine | |
| Cholinergic drugs | Failure to terminate PSVT owing to inability to antagonize vagal excitation on atrium and AV node | |
| Cimetidine | ↑ levels of quinidine | |
| Rifampin | ↑ metabolism and ↓ in therapeutic effect of quinidine | |
| Sucralfate | ↓ levels of quinidine → ↓ therapeutic efficacy | |
| Succinylcholine | Prolonged effects of succinylcholine | |
| Tricyclic antidepressants | ↓ clearance of TCAs → ↑ pharmacologic effects or toxicity | |
| Procainamide | Quinidine | ↑ levels of procainamide and NAPA → ↑ pharmacologic effects |
| Amiodarone | ↑ serum concentration of procainamide or NAPA by 33% → fatal cardiac arrhythmias | |
| Anticholinergics | Additive antivagal effect on AV conduction | |
| Cimetidine/ranitidine | ↓ renal clearance → ↑ serum concentration of procainamide → monitor levels closely | |
| Quinolone antibiotics | ↑ risk of torsades de pointes | |
| Thioridazine/ziprasidone | Synergistic Q–Tc prolongation → ↑ risk of torsades de pointes | |
| Trimethoprim | ↑ procainamide and NAPA concentrations | |
| Neuromuscular blockers | Prolonged neuromuscular blockade → decrease dose of neuromuscular blocker | |
| Disopyramide | Quinidine | ↑ disopyramide levels and ↓ quinidine levels |
| Erythromycin | ↑ levels of disopyramide → ↑ Q–Tc interval (fatalities reported) | |
| Hydantoins | ↓ half-life, levels, and bioavailability of disopyramide; effects may persist for days after stopping phenytoin | |
| Class IB | ||
| Lidocaine | β blockers | ↑ levels of lidocaine → possible toxicity |
| Cimetidine | ↓ renal clearance → ↑ serum concentration of lidocaine (other H2 blockers do not seem to interact) | |
| Procainamide | Additive cardiopressant action, potential for conduction abnormalities | |
| Tocainide | Pharmacologically similar agents → ↑ incidence of ADRs | |
| Neuromuscular blockers | Prolonged neuromuscular blockade | |
| Mexiletine | Antacids/atropine/narcotics | ↓ absorption of mexiletine |
| Metoclopramide | Accelerated absorption of mexiletine | |
| Phenytoin/rifampin | ↑ clearance of mexiletine → ↓ levels | |
| Theophylline | ↑ levels of theophylline → theophylline toxicity | |
| Tocainide | Cimetidine | ↓ tocainide bioavailability and peak concentration (ranitidine does not seem to interact) |
| Metoprolol | Additive effects on wedge pressure and cardiac index | |
| Rifampin | ↓ half-life, bioavailability, and clearance of tocainide | |
| Phenytoin | Amiodarone | Long-term use (>2 weeks) of amiodarone impairs metabolism of phenytoin → possible phenytoin toxicity |
| ↓ amiodarone levels may be seen | ||
| Class IC | ||
| Flecainide | Amiodarone | ↑ levels of flecainide |
| Cimetidine | ↑ bioavailability and renal excretion of flecainide | |
| Disopyramide | Disopyramide has negative inotropic actions; do not use together unless risks outweigh benefits | |
| Propranolol | Additive negative inotropic effects; levels of both agents may be increased | |
| Smoking | Smokers have greater plasma clearance than nonsmokers → use higher doses in smokers | |
| Digoxin | ↑ absorption, peak concentration, and bioavailability of digoxin | |
| Propafenone | Quinidine | ↑ propafenone levels in extensive metabolizers (>90% of patients) → ↑ effect |
| Cimetidine | ↑ propafenone concentration → ↑ effect | |
| Rifampin | ↑ clearance of mexiletine → ↓ levels, possible loss of therapeutic effect | |
| Anticoagulants | ↑ warfarin plasma levels; ↑ PT | |
| β blockers | ↑ levels of metoprolol | |
| Cyclosporine | ↑ cyclosporine trough levels → ↓ renal function | |
| Digoxin | ↑ levels of digoxin → toxicity | |
| Class II | ||
| β blockers: Propranolol, esmolol, acebutolol, metoprolol, atenolol, nadolol | Quinidine | ↑ effect of propranolol and metoprolol in extensive metabolizers |
| CCBs | ↑ pharmacologic effects of β blockers/synergistic or additive activity | |
| Hydralazine | ↑ levels of β blockers and hydralazine | |
| Warfarin | ↑ effect of warfarin by propranolol | |
| Ergot alkaloids | Peripheral ischemia (cold extremities, possible gangrene caused by ergot alkaloid–mediated vasoconstriction) and β blocker–mediated blockade of peripheral β2 receptors → unopposed ergot action | |
| Lidocaine | ↑ levels of lidocaine → toxicity | |
| Class III | ||
| Amiodarone | Warfarin | ↑ PT, potentiation of anticoagulant response → bleeding; ↓ warfarin dose by 30%-50%; effect may persist for months after discontinuation of amiodarone |
| Dextromethorphan | Long-term use (>2 weeks) of amiodarone impairs metabolism of dextromethorphan | |
| Digoxin | ↑ digoxin level by ≥70% → ↓ digoxin dose by 50% and monitor levels or discontinue | |
| Fentanyl | ↑ fentanyl concentration → hypotension, bradycardia, ↓ cardiac output | |
| Gatifloxacin | ↑ risk of life-threatening arrhythmias including torsades de pointes | |
| Rifampin | ↓ levels of amiodarone and its active metabolite | |
| Ritonavir | ↑ levels of amiodarone → toxicity | |
| Bretylium | Catecholamines | ↑ effects of catecholamines (EPI, NE, DA) → monitor BP and HR |
| Digoxin | Digitalis toxicity may be aggravated by initial release of NE caused by bretylium | |
| Dofetilide | Amiloride/triamterene/metformin/megestrol/prochlorperazine | Inhibit elimination of dofetilide → concurrent use is contraindicated |
| Class I and III agents | Withhold class I and III antiarrhythmic agents for ≥3 plasma half-lives before dofetilide dosing | |
| Cimetidine | ↑ levels of dofetilide by 58% → concomitant use is contraindicated | |
| Digoxin | ↑ occurrence of torsades de pointes → concomitant use is not recommended | |
| Ketoconazole |
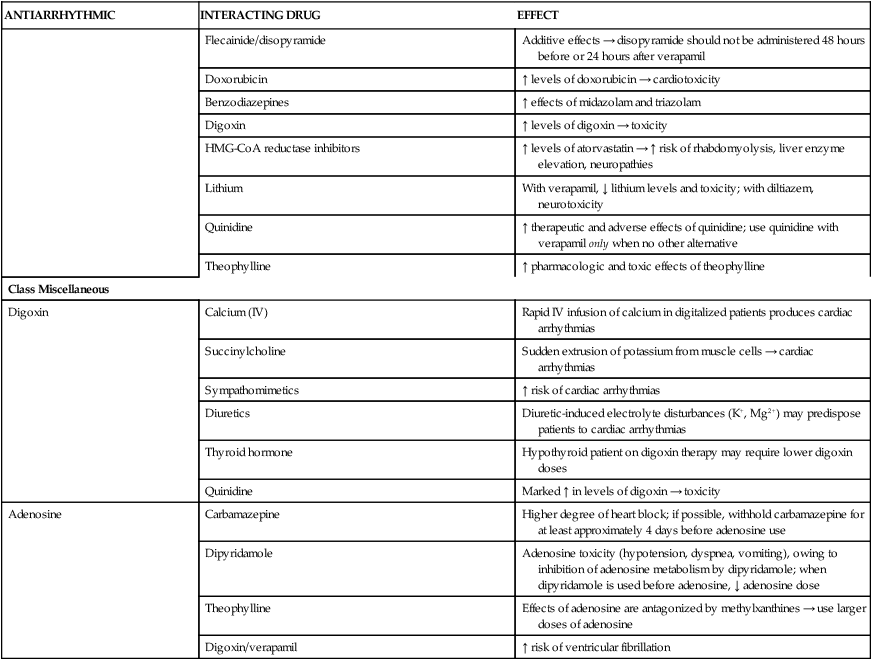
Dronedarone (multaq)
Although chemically structured similar to amiodarone, dronedarone (Multaq) differs from amiodarone by the removal of the iodine moiety and addition of a methylsulfonamide group (Figure 21-7). These structural changes result in decreased accumulation of the drug inside various tissues, leading to reduced toxicities of the thyroid gland and other organs associated with amiodarone. In addition, the modifications allow dronedarone to achieve steady-state faster than amiodarone because a shorter half-life of approximately 1 day versus more than 50 days. Similar to amiodarone, dronedarone is primarily a class III antiarrhythmic, but it shows properties of all four Vaughan Williams classes. It is indicated to reduce risk for hospitalization in patients with paroxysmal or persistent AF/AFL who are currently in sinus rhythm or pending cardioversion to sinus rhythm.14 It is available only by the oral route.
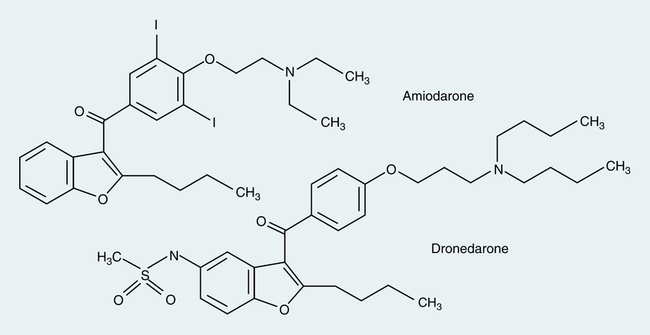
Similar to amiodarone, this medication is a CYP450 3A4 substrate and a moderator inhibitor for both CYP3A4 and CYP2D6 isoenzymes. It is contraindicated for use with potent CYP3A4 inhibitors (e.g., clarithromycin, telithromycin, cyclosporine, itraconazole, voriconazole) and inducers (e.g., carbamazepine, phenobarbital, phenytoin, rifampin). If used concurrently with nondihydropyridine calcium channel blockers (diltiazem or verapamil) or β blockers, these medications should be initiated at a lower dose to minimize risk for bradycardia or heart block. Dronedarone is also an inhibitor of P-glycoprotein, and digoxin should be avoided; however, if use of digoxin is necessary, the dose should be empirically reduced by 50% with increased monitoring for clinical response and potential adverse effects. Because of a greater than twofold increased risk in mortality found in patients with New York Heart Association (NYHA) class III and IV congestive heart failure (CHF) treated with dronedarone compared with placebo in the Antiarrhythmic Trial with Dronedarone in Moderate Severe CHF Evaluating Morbidity Decrease (ANDROMEDA) study,15 the drug is contraindicated in any patients with NYHA class IV CHF and NYHA class II-III CHF with recent decompensation requiring hospital admission or referral to a specialized CHF clinic. Other contraindications include Q–Tc greater than 500 msec, heart rate less than 50 beats/min, concomitant use of Q–T interval–prolonging medications or herbals owing to risk for torsades de pointes, and sick sinus syndrome or second-degree or third-degree AV block unless a functional pacemaker is present.
Dofetilide (tikosyn)
Dofetilide (Tikosyn) is available as an oral formulation and is indicated for the maintenance of sinus rhythm after successful conversion, but it is ineffective in paroxysmal AF. Dofetilide carries a significant risk of VAs such as torsades de pointes, associated with prolongation of the Q–T interval (duration of ventricular electrical activity). The Q–T interval can be reported as Q–Tc. This drug should be discontinued in patients with Q–Tc greater than 500 msec. The risk of torsades de pointes among patients administered dofetilide is greatest for the following patients:4
• Patients with congenital heart disease or ischemic heart disease
• Patients with diminished renal function
• Patients receiving dofetilide doses exceeding 500 mg twice daily
This medication must be adjusted to avoid renal accumulation. Drug interactions with dofetilide pose a significant problem. Agents such as cimetidine, azole antifungals, prochlorperazine, metformin, and the trimethoprim component of trimethoprim-sulfamethoxazole (Bactrim) may inhibit active tubular secretion of dofetilide and increase the plasma concentration. Therapy with dofetilide must be initiated in a facility that can provide continuous ECG monitoring and the presence of personnel trained to manage severe VAs for at least 3 days. Both the prescriber and the pharmacy must be participants in a program known as the Tikosyn in Pharmacy System (TIPS) before prescribing and dispensing dofetilide.16
Sotalol (betapace and betapace af)
Sotalol (Betapace and Betapace AF) is available only by the oral route and works by prolonging the AP duration and the relative refractory period. Sotalol can be used for supraventricular and ventricular arrhythmias. When initiating sotalol, the patient should be kept in a facility that can provide continuous ECG monitoring and the presence of personnel trained to manage severe VAs for at least 3 days.4 As with any β-blocking agent, caution must be exercised when treating patients with restrictive airway disease.
Ibutilide (corvert)
Ibutilide (Corvert) is available as an IV formulation and is an alternative to electrical cardioversion. Ibutilide is the first antiarrhythmic agent indicated for rapid conversion of AF/AFL of recent onset by the FDA. In clinical trials, ibutilide was more effective for the treatment of AFL than AF (more than 50% versus less than 40%). Class I antiarrhythmics and other class III antiarrhythmics should not be given with this medication or within 4 hours of an ibutilide infusion because of the potential for prolonged refractoriness. Because AF has the potential to form clots within the atrium of the heart, patients must be adequately anticoagulated before chemical cardioversion to reduce the risk of stroke. Patients who fail electrical cardioversion require lifelong anticoagulation.4 There is also evidence (TIME study) to suggest that prophylaxis of magnesium can enhance the efficacy of ibutilide and decrease the incidence of torsades de pointes by more than 30%. Before initiation, all electrolytes must be maintained within normal limits, and continuous ECG monitoring is required because of the high incidence of ventricular fibrillation (2.7% to 4.9%).1,17,18
Miscellaneous
Digoxin (lanoxin)
Digoxin (Lanoxin) has direct AV-blocking effects and vagotonic properties, which aid in reducing the heart rate. Although digoxin prolongs the relative refractory period of the AV node and reduces the number of impulses through the AV node, it is not regarded as a first-line agent for AF.4,11 Digoxin does not have a rapid onset of effect, especially for the management of an acute condition such as AF; it requires approximately 2 hours to achieve maximal effect. Additionally, digoxin has the potential to shorten the refractory period of atrial muscles, allowing electrical impulses to be conducted throughout the myocardium and ultimately potentiating episodes of AF. It is less effective than β blockers and calcium channel blockers during states of increased sympathetic tone, such as in exercise and stress. Digoxin is not regarded as a first-line agent for the control of ventricular rate in AF except in patients with impaired left ventricular function or heart failure.11
Adenosine (adenocard)
Rapid administration of adenosine (Adenocard) is implemented to terminate SVTs only. Adenosine has a half-life of approximately 12 seconds, and because of its ultrashort half-life, adenosine is best administered through a central line for rapid arrival at the site of action, or, if given through a brachial line, the arm should be held in the upright position followed almost instantly by a saline flush. Bronchospasms, dyspnea, hyperpnea, and cough have been reported after administration of IV adenosine in patients with asthma and chronic obstructive pulmonary disease; these symptoms are generally benign and short-lasting.19
Management and pharmacotherapy of advanced cardiac life support
Sudden cardiac death
Sudden cardiac death (SCD) is a leading cause of death in the United States and Canada and is defined as an episode of VF, pulseless ventricular tachycardia, pulseless electrical activity (PEA), or asystole, all of which are life-threatening arrhythmias.20 Although the fatalities associated with episodes of SCD are unacceptably high, an individual may be resuscitated, and it is common to encounter patients having a “history” of SCD. The goal in treating SCD is to restore sinus rhythm, to prevent further episodes of SCD, and to prevent impairment of neurologic function. Several studies have shown benefits in mortality reduction by minimizing time to defibrillation and by delivery of cardiopulmonary resuscitation (CPR).16
In a patient with VF, survival decreases by 7% to 10% for every minute that passes from the time of symptom onset to defibrillation.16 When CPR is initiated, the decline in survival occurs at a more gradual rate of approximately 3% to 4% for every minute between onset of symptoms and time to defibrillation.16 Needless to say, efficient and timely delivery of defibrillation and CPR is imperative for successful management of SCD.
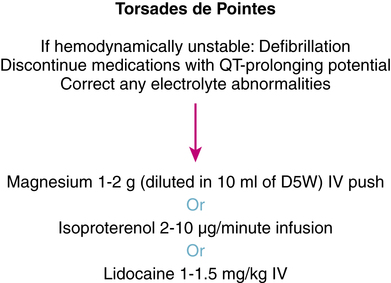
After beginning CPR and attempting defibrillation, health care workers may begin establishing other therapeutic modalities such as IV access; medication therapy and the insertion of an advanced airway should be considered. VF and pulseless ventricular tachycardia are managed primarily by defibrillation and CPR and secondarily by pharmacotherapy; conversely, asystole and PEA are not managed by defibrillation and are managed first by CPR only and second by pharmacotherapy as depicted in the algorithms in Figures 21-8, 21-9, and 21-10. It may be prudent to review the national consensus guidelines for further details of advanced cardiac life support algorithms.
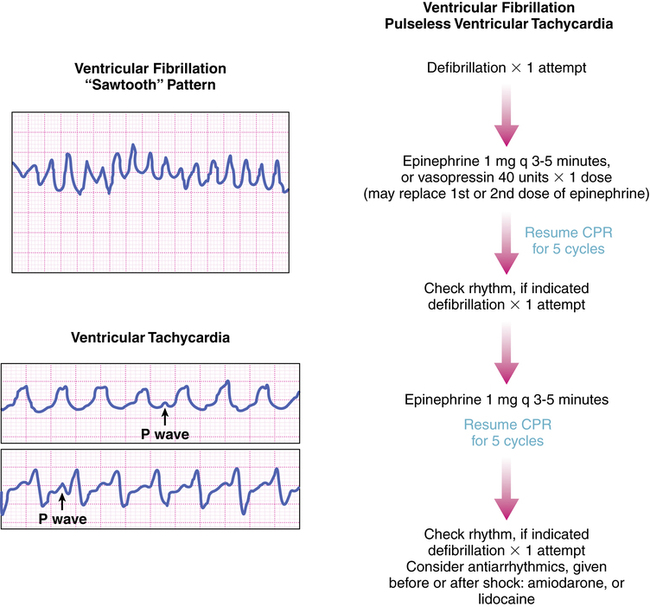
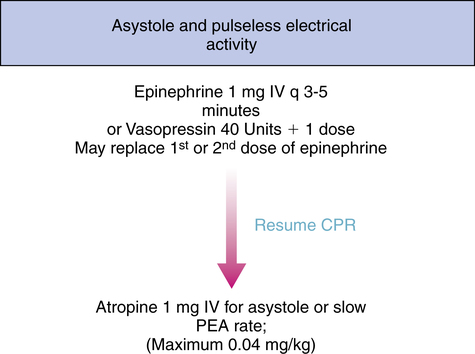
Sodium bicarbonate
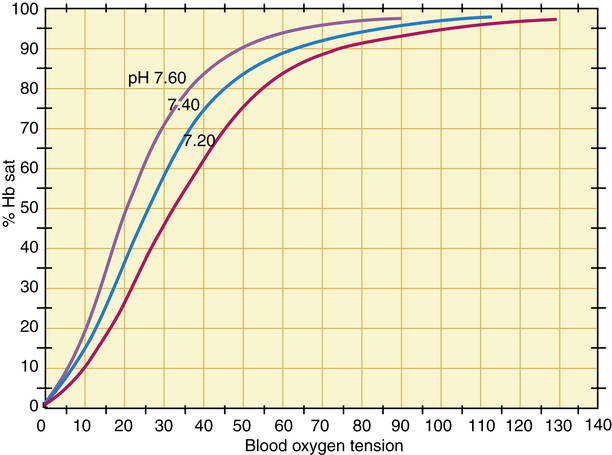
The presence of carbon dioxide helps the release and delivery of oxygen from hemoglobin, also known as the Bohr effect. When comparing the oxygen dissociation curves of a serum sample with carbon dioxide and another with no carbon dioxide, oxygen is able to dissociate more readily in the former state, as depicted in Figure 21-11.
Alternative routes of medication administration
Intraosseous route
In the face of life-threatening medical emergencies, in which there is a dire need for medication and fluid delivery, it is incumbent on the health care worker to provide vascular access in the most efficient and safest way possible. Often, IV access is difficult if not impossible in infants and young children, elderly patients with circulatory collapse, and IV drug abusers. In such situations, an intraosseous (IO) needle may be inserted with relative ease, even in the most poorly perfused patients. The 2010 American Heart Association guidelines17 for cardiopulmonary resuscitation and emergency cardiovascular care recommend IO therapy as an alternative to direct IV therapy.
Endotracheal route
The following should be done when administering the above-listed agents by the endotracheal route:
• The patient should be positioned horizontally, as opposed to being in the Trendelenburg position, and chest compressions should cease.
• A catheter should be inserted into the endotracheal tube and allowed to pass the tip of the tube. The medication solution should be sprayed down the tube, followed by 5 to 10 rapid ventilations with a respirator bag.
• Medications should be diluted with approximately 10 mL of distilled water or normal saline. Endotracheal absorption is greater with distilled water, but distilled water has a negative impact on the partial pressure of oxygen. Generally, the systemic absorption of these medications is reduced via the endotracheal route, and the dose administered should always be 2 to 2.5 times the usual IV dose except for vasopressin; the vasopressin IV dose of 40 U may be given via the endotracheal route.
 SELF-ASSESSMENT QUESTIONS
SELF-ASSESSMENT QUESTIONS
1. In which phase of the cardiac cycle does ventricular contraction occur?
2. Identify three functions that regulate mean arterial pressure.
3. Which measurements, taken by a pulmonary artery catheter, are estimates of intravascular volume?
4. Hypotension is first managed by what mode of therapy?
5. What vasopressor acts only on the α receptors within the vasculature?
6. Which agents exert an inotropic effect on the heart?
7. What electrolyte abnormality may potentiate the adverse effects of digoxin?
8. What Vaughan Williams class of antiarrhythmics acts on the fast sodium channels in the myocardium?
9. What antiarrhythmic agent is structurally similar to amiodarone but has an improved side-effect profile?
10. In patients taking dofetilide, at what Q–Tc interval should the drug be discontinued because the risk for torsades de pointes becomes too great?
11. Which antiarrhythmic agent is highly associated with the development of lupus erythematosus?
12. Identify the four categories of sudden cardiac death.
13. What medication is indicated for treatment of asystole and PEA but not VF or pulseless VT during cardiac arrest?
14. What are the two alternative routes of medication administration during cardiac arrest?
15. When medications are administered via the endotracheal route during cardiac arrest, the dose should be increased by how many times the usual intravenous dose?
 CLINICAL SCENARIO #1
CLINICAL SCENARIO #1 CLINICAL SCENARIO #2
CLINICAL SCENARIO #2[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]CHAPTER 21
Vasopressors, inotropes, and antiarrhythmic agents
After reading this chapter, the reader will be able to:
1. Define terms that pertain to vasopressors, inotropes, and antiarrhythmic drugs.
2. List the various components that make up blood pressure
3. Compare and contrast the mechanism of action of inotropes and vasopressors
4. Describe the various drug interactions that may occur with the use of vasopressors and inotropes
5. Design an algorithm for the management of hypotension
6. Describe the normal conduction of the heart
7. Define nonpharmacologic methods of treating dysrhythmias
8. Compare and contrast the categories of the Vaughan Williams classification system
9. Define the mechanism of action of digoxin
10. List all the dysrhythmias associated with cardiac arrest
11. Design an algorithm that may be used in the management of ventricular fibrillation and pulseless ventricular tachycardia
12. Design an algorithm that may be used in the management of torsades de pointes
13. Describe the proper dosing technique of intravenous magnesium therapy in the management of torsades de pointes
14. List the routes of administering medications during cardiac arrest
Group of cardiac medications that are classified according to mechanism of action; in some instances, they may have multiple mechanisms of action. The most common classification system of antiarrhythmics is the Vaughan Williams classification system, which is divided into four distinct categories and a miscellaneous section.
Irregular (faster or slower) heartbeat; the term arrhythmia is used more frequently than dysrhythmia.
Link between atrial depolarization and ventricular depolarization.
Presence of carbon dioxide aids in the release and delivery of oxygen from hemoglobin.
Amount of blood that is pumped out of the heart per unit of time.
Endogenous products that are secreted into the bloodstream and travel to nerve endings to stimulate an excitatory response.
Agent affecting the rate of contraction of the heart.
Diastolic blood pressure (DBP)
Lowest pressure reached prior to ventricular ejection.
An agent that influences the conduction of electrical impulses. A positive dromotropic agent enhances the conduction of electrical impulses to the heart.
Agent affecting the strength of muscular contraction.
Pressure that drives blood into the tissues averaged over the entire cardiac cycle.
Enzyme responsible for the breakdown of cyclic adenosine 3′,5′-monophosphate (cAMP).
Episode of ventricular fibrillation, pulseless ventricular tachycardia, pulseless electrical activity, or asystole leading to loss of life.
Peak pressure reached during ventricular ejection.
Overly rapid heartbeat, usually defined as greater than 100 beats/min in adults.
Agent causing dilation of blood vessels.
Agent causing contraction of capillaries and arteries.
Cardiac condition in which normal ventricular contractions are replaced by coarse or fine, rapid movements of the ventricular muscle.
Overview of cardiovascular system
The cardiovascular system regulates blood flow to the various regions of the body. Blood flow generally travels via a pressure gradient, shifting from areas of higher pressure to lower pressure. The central nervous system (CNS) relays electrical impulses through sensory receptors found systemically within the vasculature, affecting vascular tone and causing shunting of blood to and from various organ systems within the body. Vascular tone is regulated via the sympathetic nervous system and the circulation of neurotransmitters and hormones, such as epinephrine, vasopressin, and angiotensin. Several factors exert an effect on vascular tone as a response to tissue perfusion and circulatory volume. Hypotension is commonly present in patients with autonomic dysfunction and shock. Shock is a life-threatening medical emergency characterized by organ hypoperfusion leading to decreased delivery of oxygen and nutrients to tissues throughout the body. There are six types of shock and their effect on hemodynamic parameters can be seen in Table 21-1.
TABLE 21-1
Hemodynamic Changes in Various Shock States
| HEMODYNAMIC PARAMETER | HYPOVOLEMIC/HEMORRHAGIC | NEUROGENIC | CARDIOGENIC | SEPTIC/DISTRIBUTIVE |
| HR | ↑ | ↔ | ↔/↑ | ↑ |
| MAP | ↓ | ↑/↓ | ↑ | ↓ |
| CVP (5-12 mm Hg) | ↓ | ↓ | ↑ | ↓ |
| PCWP (10-12 mm Hg) | ↓ | ↓ | ↑ | ↓ |
| CO (5-7 L/min) | ↓ | ↔/↓ | ↓ | ↑ |
| SVR (80-1440 dyn•sec•cm−5) | ↑ | ↓ | ↑ | ↓ |
Factors affecting blood pressure
Typical measurement of blood pressure is relative to a recurring cardiac cycle of atrial and ventricular contractions and relaxations (Figure 21-1). The cycle is divided into the systolic phase and the diastolic phase. The systolic phase is the portion in which ventricular contraction occurs, resulting in ejection of blood through the aorta. Conversely, diastole is the period of ventricular relaxation and blood filling. Systolic blood pressure (SBP) is the peak pressure reached during ventricular ejection, and diastolic blood pressure (DBP) is the lowest pressure reached right before ventricular ejection. Arterial pressure is typically recorded as SBP/DBP, for example, 120/80 mm Hg. Mean arterial pressure (MAP) refers to the pressure that drives blood into the tissues averaged over the entire cardiac cycle. Because the cardiac cycle is pulsatile rather than continuous and because two-thirds of the normal cardiac cycle is spent in diastole, MAP is not the arithmetic mean of the SBP and DBP. MAP is defined as the product of cardiac output (CO) and systemic vascular resistance (SVR), as follows:
< ?xml:namespace prefix = "mml" />
The use of therapies such as fluids, vasopressors, and inotropes to maintain cardiovascular stability is directed toward altering each of these components, as seen in Table 21-2. The various vasopressors currently on the market have different affinities for the various receptors located within the body and exert different effects on the hemodynamic parameters, as seen in Table 21-3. Vasopressors and inotropes are not always first-line therapy; on the contrary, fluids are the mainstays for improving hypotensive episodes. Vasopressors and inotropes have considerable side effects, and certain medications interact with various vasopressors and inotropes, leading to alterations in hemodynamic parameters as seen in Table 21-4.
TABLE 21-2
Cardiac Drugs: Dosing, Pharmacokinetics, and Hemodynamic Effects
| PHARMACOKINETICS | HEMODYNAMIC EFFECTS | ||||||||
| AGENT | DOSE RANGE | ONSET (min) | DURATION | HALF-LIFE | HR | MAP | PCWP | SVR | CO |
| Amrinone | 0.75 mg/kg bolus, then 2.5-15 μg/kg/min | 5-10 | 0.5-2 hr | 4.8-8.3 hr | ↓ | 0-↓ | 0-↓ | 0-↓ | ↓-0-↑ |
| Dobutamine (Dobutrex) | 2-20 μg/kg/min | 1-2 | 10-15 min | 2 min | 0* | 0-↓ | ↓ | ↓ | ↑ |
| Dopamine (Inotropin) | 1-5 μg/kg/min | 5 | <10 min | 2 min | 0 | 0 | 0 | 0 | 0 |
| 5-15 μg/kg/min | 5 | <10 min | 2 min | ↑ | 0-↓ | 0-↑ | ↑ | ↑ | |
| >15 μg/kg/min | 5 | <10 min | 2 min | ↑ | 0-↓ | 0-↑ | ↑ | ↑ | |
| Epinephrine (Adrenalin) | 0.01-0.1 μg/kg/min | 1 | 3-5 min | 3-5 min | ↑ | ↑ | 0-↓ | ↓†-↑* | ↑ |
| 0.1 μg/kg/min | ↑↑ | ↑↑ | ↑ | ↑↑ | ↑↑ | ||||
| Norepinephrine (Levophed) | 0.5-30 μg/min | 1-3 | 5-10 min | 1-2 min | 0-↑ | ↑↑↑ | ↑↑ | ↑↑↑ | 0-↓ |
| Phenylephrine (Neo-Synephrine) | 0.5-5 μg/kg/min | 10-15 | 1-3 hr | 2-3 hr | ↓ | ↑ | ↑ | ↑ | ↓ |
| Milrinone (Primacor) | 50 μg/kg bolus, then 0.375-0.75 μg/kg/min | 90 | 3-5 hr | 2.3 hr | 0-↑ | ↓ | ↓ | ↓ | ↑ |
| Vasopressin (Pitressin) | 0.04 U/min | 30-60 | 30-60 min | 10-20 min | 0 | ↑ | ↑ | ↑ | ↓? |
TABLE 21-3
Inotropes and Vasopressors: Receptor Affinity
| DRUG | α | β1 | β2 | DA |
| Dopamine (Inotropin) | + to +++* | +++* | + | 0/+ |
| Dobutamine (Dobutrex) | 0 to +* | 0 to +* | + | 0 |
| Epinephrine (Adrenalin) | +++* | +++ | ++* | 0 |
| Isoproterenol (Isuprel) | 0 | +++ | +++ | 0 |
| Norepinephrine (Levophed) | +++ | ++ | ++ | 0 |
| Phenylephrine (Neo-Synephrine) | +++ | 0 | 0 | 0 |
DA, Dopamine; 0, no effect; +, slight effect; ++, moderate effect; +++, pronounced effect.
TABLE 21-4
| PRECIPITANT DRUG* | EFFECT | OBJECT DRUG* | COMMENTS |
| Dobutamine, Isoproterenol, Norepinephrine | |||
| Bretylium | ↑ | Dobutamine, isoproterenol, norepinephrine | Concomitant use may potentiate effects of vasopressors, causing arrhythmias |
| Halogenated hydrocarbon anesthetics | |||
| Guanethidine | May increase pressor response, causing severe hypertension | ||
| Oxytocic drugs | |||
| Tricyclic antidepressants | |||
| Phenylephrine | |||
| Bretylium | ↑ | Phenylephrine | Concomitant use may potentiate effects of vasopressors |
| Guanethidine | |||
| Halogenated hydrocarbon anesthetics | |||
| Oxytocic drugs | |||
| Tricyclic antidepressants | ↔ | Tricyclic antidepressants may increase effects of phenylephrine | |
| Dopamine | |||
| Dopamine | ↓ | Guanethidine | Antihypertensive effects of guanethidine may be reversed |
| Phenytoin | Concomitant use may lead to seizures, severe hypotension, and bradycardia | ||
| Tricyclic antidepressants | Dopamine | Tricyclic antidepressants may increase effects of dopamine | |
| Halogenated hydrocarbon anesthetics | ↑ | Dopamine | May sensitize myocardium to actions of vasopressors, causing arrhythmia |
| MAOIs | Dopamine is metabolized by MAOIs. MAOIs increase pressor response to dopamine by 6-fold to 20-fold | ||
| Oxytocic drugs | Concomitant use may cause severe hypertension | ||
| Epinephrine | |||
| Cardiac glycosides | ↑ | Epinephrine | May sensitize myocardium to actions of vasopressors, causing arrhythmia |
| Halogenated hydrocarbon anesthetics | |||
| Levothyroxine antihistamines (chlorpheniramine, diphenhydramine) | |||
| MAOIs | Concomitant use may cause severe hypertension | ||
| Methyldopa | |||
| Oxytocic drugs | |||
| Reserpine | |||
| Sympathomimetics | |||
| Tricyclic antidepressants | |||
| β blockers | |||
| α blockers | ↓ | Epinephrine | Vasoconstricting and hypertensive effects of pressor may be reversed |
| Chlorpromazine | |||
| Diuretics | |||
| Epinephrine | Guanethidine | Epinephrine may antagonize effects of guanethidine, resulting in decreased antihypertensive effects | |
| Digoxin | |||
| Amiodarone | ↑ | Digoxin | Amiodarone may increase digoxin blood level; reduce digoxin dose by 50% |
| β blockers | Combination may cause advanced or complete heart block | ||
| Calcium channel blockers | |||
| Calcium | Rapid administration of intravenous calcium may result in fatal arrhythmias | ||
| Succinylcholine | Succinylcholine may cause sudden extrusion of K+ from muscle cells, leading to arrhythmias | ||
| Sympathomimetics | Combination may cause increased risk of cardiac arrhythmias | ||
| Thiazide and loop diuretics | Diuretic-induced electrolyte disturbances may predispose to digitalis toxicity | ||
| Thyroid hormones | ↓ | Digoxin | Thyroid hormones may reduce digoxin blood levels; hypothyroid patients may require higher dose of digoxin |
MAOIs, Monoamine oxidase inhibitors.
*Precipitant drug refers to the drug that causes the interaction; object drug refers to the drug affected by the interaction. ↑, Object drug increased; ↓, object drug decreased; ↔, object drug unaffected.
Agents used in the management of shock
Catecholamines
Norepinephrine (levophed) and epinephrine (adrenalin chloride)
Norepinephrine (Levophed) and epinephrine (Adrenalin Chloride) are endogenous catecholamines that are secreted by the adrenal medulla. Epinephrine is ultimately synthesized by the catalytic actions of tyrosine hydroxylase, which converts the amino acid tyrosine to levodopa and subsequently to dopamine, norepinephrine, and epinephrine.1,2 These neurotransmitters travel to sympathetic nerve endings, where they are released to stimulate other nerve fibers and to stimulate an excitatory response. Norepinephrine and epinephrine stimulate α receptors on the vasculature and β receptors, also located within the vasculature as well as in the myocardium. α receptors within the vasculature cause vasoconstriction, whereas β receptors cause vasodilation. Most vascular beds within the body contain β receptors; however, they are outnumbered by α receptors, so any epinephrine and norepinephrine stimulation of β receptors is negligible or of no effect, yielding a net response of vasoconstriction. In addition, β receptors more densely populate the myocardium compared with α receptors, leading to a net effect of tachycardia.3,4
Isoproterenol (isuprel)
Isoproterenol (Isuprel) is a synthetic catecholamine used for the treatment of symptomatic bradycardia or torsades de pointes. Isoproterenol works solely as an agonist of β receptors. By stimulating β1-adrenergic receptors, it exerts pronounced inotropic and chronotropic effects. By stimulating β2-adrenergic receptors, it leads to smooth muscle relaxation of the bronchi, skeletal muscle, vasculature, and gastrointestinal tract. Venous return to the heart is also increased by vasodilation of the venous bed. The use of isoproterenol is limited because of its pronounced stimulatory effect on the heart rate.4
Vasopressin
The 2008 practice guidelines5 published by the Society of Critical Care Medicine do not recommend vasopressin as the initial vasopressor of choice or as a lone agent. Precaution stems from the fact that vasopressin infusion may decrease splanchnic blood flow. Other settings in which vasopressin may be used include diabetes insipidus at doses of 5 to 10 U given intramuscularly or subcutaneously and repeated two or three times per day. Vasopressin has also been used to treat esophageal bleeding at doses up to 2 U/min. Caution is also needed when treating conditions other than shock; myocardial ischemia may ensue as a result of the potent vasoconstrictive properties at higher doses.
Inotropic agents
Dobutamine
Dobutamine is indicated for the short-term treatment of decompensated heart failure secondary to depressed contractility. Dobutamine is a synthetic catecholamine that is chemically related to dopamine; however, in contrast to dopamine, it is not metabolized to norepinephrine, and it does not stimulate dopamine receptors.4 Its pharmacologic actions are due to the effects of its racemic components. The (R)-isomer is responsible for its activity on the β1 and β2 receptors, causing predominant positive inotropic and chronotropic effects and vasodilatory effects, respectively. This combination of effects enhances cardiac output and stroke volume. The (S)-isomer is responsible for its activity on the α1 receptors, causing vasoconstriction.1,6 The vasodilatory β2-adrenergic effects counterbalance the vasoconstrictive α1 effects, leading to minor changes in systemic vascular resistance usually seen at lower doses. With increasing doses, the β2-vasodilatory actions predominate over the α1-vasoconstrictive effect, causing a decrease in systemic and pulmonary vascular resistance. The decline in systemic and pulmonary vascular resistance may also be secondary to enhanced cardiac output.
As an inotropic agent, dobutamine has adverse cardiac effects, which include arrhythmias, increase in myocardial oxygen consumption and demand, tachycardia, and hypotension. A limiting factor when dobutamine is used for more than 72 hours is tachyphylaxis; this may be due to a downregulation of β1 receptors and may be overcome by increasing the dose. In patients with sulfite sensitivity, allergic reactions such as anaphylaxis or life-threatening asthmatic episodes may occur because dobutamine formulations contain sulfites.4
Phosphodiesterase inhibitors: inamrinone and milrinone
Milrinone is the phosphodiesterase inhibitor most commonly used in practice today because it has a shorter half-life than inamrinone and is less likely to cause thrombocytopenia. It undergoes renal elimination with an elimination half-life of 1 to 3 hours in patients with normal renal function; steady-state concentrations are reached in 4 to 6 hours if initiated without a loading dose. The risk of hypotension occurring is higher when a loading dose is given. Milrinone may be given as an initial intravenous (IV) bolus dose of 50 μg/kg administered slowly over 10 minutes followed by continuous infusion at a rate of 0.375 to 0.75 μg/kg/min and titrated to effect. Dosage adjustment should be made in patients with severe cardiac failure or renal impairment because of the considerable reduction in clearance.1,4,6
Cardiac glycosides: digoxin (lanoxin)
In contrast, digitalis toxicity may cause hyperkalemia by its inhibitory actions on the Na+,K+-ATPase pump. Digoxin toxicity may manifest as serious life-threatening ventricular arrhythmias, including premature ventricular contractions, AV junctional rhythm, bigeminal rhythm, and second-degree AV blockade. Bradycardia may also occur early on in the setting of digoxin toxicity. The initial symptoms of digitalis toxicity are nausea, vomiting, anorexia, and abdominal pain. These symptoms may be due to a direct effect on the gastrointestinal tract or result from CNS stimulation of the chemoreceptor trigger zone. Other rare but possible neuropsychiatric effects may manifest as disorientation and hallucination, especially in elderly patients, and visual disturbances such as yellow-green halos. Digoxin immune Fab is the antidote used to facilitate the speedy elimination of digoxin from the body. Digoxin immune Fab is indicated in the setting of life-threatening toxicity such as ventricular arrhythmias, bradyrhythmias, ingestion of greater than 10 mg in adults or 4 mg in children, a steady state level greater than 10 ng/mL, progressive elevation of potassium, or a potassium level greater than 5 mEq/L.7
Electrophysiology of myocardium
Electrical activity is initiated by an innate pacemaker located at the sinoatrial (SA) node. Electrical potential exists across the cell membrane, and it changes in response to transmembrane movement of Na+, K+, Ca2+, and Cl− ions. These ions mediate the process of myocardial contraction and relaxation. When an electrical stimulus is evoked from the SA node, it generates an action potential (AP). Once generated, the AP produces a local current, which evokes further APs along the myocardium. An AP elicits myocardial depolarization or contraction. The link between atrial depolarization and ventricular depolarization is a portion of the conduction system called the atrioventricular (AV) node. The AV node slows down the electrical impulse to ensure that atrial excitation is completed before ventricular excitation. After leaving the AV node, the impulse travels to the wall between the two ventricles via the conducting system fibers known as the bundle of His. From the bundle of His, the cardiac conduction system bifurcates into three main bundle branches: the right bundle and two left bundles. These bundle branches form a conduction network, referred to as Purkinje fibers (Figure 21-2). The conduction system innervates the myocardium and causes changes in membrane polarization of the muscle fiber.8
An AP (Figure 21-3) can be divided into the following five different phases:
Phase 0: Initial rapid depolarization of myocardial tissues secondary to an abrupt transmembrane influx of sodium through “fast” sodium channels
Phase 1: Fast sodium channels are inactivated; this, coupled with the movement of K+ and Cl− ions, leads to a transient net outward current and the beginning of repolarization
Phase 2: “Plateau” phase, maintained by a balance between calcium influx and potassium efflux
Phase 3: Calcium channels close, but membrane remains permeable to potassium, resulting in cellular repolarization
Phase 4: Cell returns to its “resting” state; the resting membrane potential is reached through gradual depolarization related to a constant sodium influx balanced by a decreasing efflux of potassium
