[level-membership-for-cardiovascular-category]
Chapter 3 Vascular Smooth Muscle
With the evolution of an enclosed circulatory system to transport oxygenated blood, hormones, immune cells, metabolites, and waste products to and from cells in distal sites within the vertebrate body, blood vessels evolved adaptations necessary for repeated cycles of contraction and extension resulting from cardiac-driven pulsatile blood flow. These adaptations for blood vessel distensibility allow elastic conductance arteries in the macrocirculation, under the influence of the pulsatile cardiac cycle, to provide blood flow to end organs by altering the luminal diameter of the vessel. They also allow resistance arteries in the microcirculation, which experience steady flow, to regulate vasomotion at the organ level to maintain blood pressure homeostasis.1 The cells that primarily establish and orchestrate these contraction and distensible properties are vascular smooth muscle cells (VSMCs), the majority cell type within the normal vessel wall. VSMCs maintain contractile tone by a highly organized architecture of contractile/cytoskeletal proteins and associated regulatory components within the cell cytoplasm and establish distensibility by synthesis, secretion, and organization of extracellular matrix (ECM) components with elastic recoil and resilience properties.1 VSMCs within the vascular continuum have the ability to adapt expression of proteins involved in contraction and ECM synthesis according to extrinsic and intrinsic cues during different developmental stages and in disease or response to injury. This ability is due to a phenomenon known as VSMC phenotypic modulation and is a major feature that distinguishes VSMCs from terminally differentiated cells.2
Vascular smooth muscle cell phenotypic modulation is the ability to switch phenotypic characteristics from a migratory synthetic phenotype in embryonic tissue patterning to a quiescent, contractile phenotype in maintenance of vascular tone in mature vessels. Importantly, during vascular remodeling in response to injury, VSMCs can switch back to a synthetic phenotype characterized by increased VSMC proliferation and ECM synthesis. Although the ability to switch phenotypes may have evolved as an adaptive survival mechanism for VSMCs to adjust physiological responses due to changing hemodynamic demands or to repair damage after vascular injury, phenotypic modulation has important implications both during development and during vascular disease.2
This chapter will highlight how these diverse functions of VSMCs arise from both innate genetic programs and a range of diverse environmental cues that include soluble signaling factors, insoluble ECM components, physical mechanical forces, and interactions with other cell types.3 Discussion will center on the complex webs of signaling networks generated by these diverse external factors, and how these networks are regulated and integrated at multiple transcriptional and posttranslational levels to mediate the diverse functions of VSMCs in normal physiology and disease/injury pathology.
Origins of Vascular Smooth Muscle Cells During Embryonic Development
Initially in embryonic vasculature development, endothelial precursor cells form a common progenitor vessel which then gives rise to the first artery (dorsal aorta) and vein (cardinal vein) by selective sprouting and subsequent arterial-venous cell segregation4 (see also Chapter 1). The distinct molecular identities of arteries and veins are regulated by complex interactions of several signaling pathways, including sonic hedgehog (Shh), a member of the hedgehog (Hh) family of secreted morphogens; secreted growth factors in the vascular endothelial growth factor family (VEGFs)5; Notch receptors (Notch 1-4) and Notch ligands (Jagged1,2); and transmembrane proteins that can transduce cell-cell interactions into signals determining cell fates.6 Interactions of these signals induce differential expression of VEGF receptors, Ephrin ligands, and tyrosine kinase Eph receptors on the segregating arterial/venous cells, with ephrin B2 and EphB4 as markers expressed in arteries and veins, respectively.4,5 In response to VEGF signaling, endothelial cells (ECs) within these primordial vascular networks recruit mural cells, including nascent VSMCs.7
Nascent VSMCs derive from multiple and nonoverlapping embryonic origins that are reflected in different anatomical locations within the adult. Ectodermal cardiac neural crest cells give rise to the large elastic arteries (e.g., ascending and arch portions of the aorta), ductus arteriosus, and carotid arteries; proepicardium mesothelial cells produce the coronary arteries; mesodermal cells are origins for the abdominal aorta and small muscular arteries; the mesothelium forms the mesenteric vasculature; secondary heart field cells form the base of the aorta and pulmonary trunk; somite-derived cells produce the descending thoracic aorta; and satellite-like mesoangioblasts give rise to the medial layers of arteries.8 The heterogeneous mosaic of VSMCs in the vessel wall may be due in part to these diverse embryological origins of VSMCs and could be reflected in the presence of phenotypically distinct subpopulations within the media that account for VSMC plasticity.9 There is some evidence that VSMCs derived from different lineages exhibit morphologically and functionally distinct properties and respond differently to soluble factors in vitro and to morphogenetic cues in vivo,8 suggesting that the major determinants of VSMC responses to signals in vascular development are principally lineage-dependent rather than environment-dependent.8
Vascular Smooth Muscle Cell Phenotypic Modulation
Characterization of Vascular Smooth Muscle Cell Phenotypes
Given the multiple origins and distinct subpopulations of VSMCs, a compelling central question for understanding VSMC biology is how cells from these diverse embryonic origins, initially expressing lineage-specific pathways, differentiate to express the same marker genes specifically characteristic of VSMCs.8,10 Another question is how these same VSMCs, responding to both extrinsic and intrinsic cues, can alter expression of these genes (and thus molecular pathways), leading to diverse phenotypes with distinct and diverse functions. VSMC phenotypes can be loosely divided into three types: contractile/differentiated, synthetic/dedifferentiated, and inflammatory.
Contractile, differentiated vascular smooth muscle cells
Contractile or differentiated VSMCs are characterized by a repertoire of contractile proteins, contractile-regulating proteins, contractile agonist receptors, and signaling proteins responsible for contraction and maintenance of vascular tone.3,11,12 Of the VSMC “marker” proteins expressed in the contractile phenotype repertoire (Fig. 3-1), the most discriminating markers are smooth muscle myosin heavy chain (SMMHC) in conjunction with alpha-smooth muscle actin (αSMA), smoothelin, SM-22α, h1-calponin, and h-caldesmon.2 In addition to expressing these proteins associated with contractile function, contractile VSMCs exhibit differential levels of ECM components (increased collagen types 1 and IV) and matrix-modifying enzymes (decreased matrix metalloproteinases [MMPs] and increased tissue inhibitors of matrix metalloproteinases [TIMPs]). Contractile VSMCs are further characterized by an elongated spindle-shaped morphology in culture, a low proliferative rate, and expression of α1β1, α7β1 integrins and the dystrophin-glycoprotein complex (DGPC).3,13
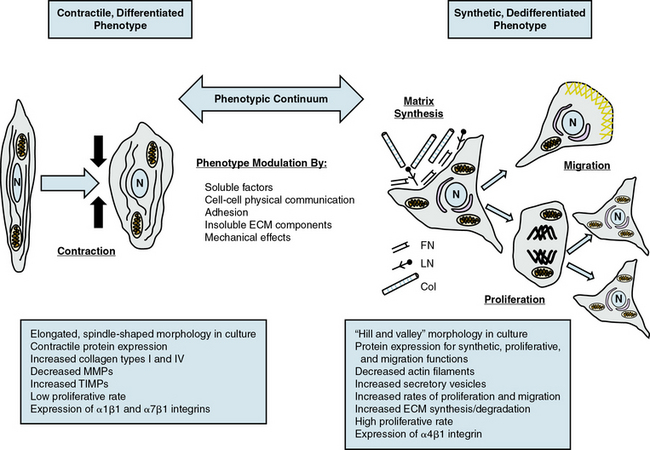
Synthetic, dedifferentiated vascular smooth muscle cells
Synthetic or dedifferentiated VSMCs have decreased expression of SMC-related genes for contractile proteins (e.g., SMMHC), with concomitant increased osteopontin, l-caldesmon, nonmuscle myosin heavy chain B, vimentin, tropomyosin 4, and cellular-retinal binding-protein-1 (CRBP-1) (see Fig. 3-1). “Positive” marker genes, such as nonmuscle myosin heavy chain (NM-B MHC) or SMMHC embryonic (SMemb) expressed specifically in embryonic or phenotypically modified VSMCs, are characteristic of dedifferentiated VSMCs in association with vascular injury.2 Other characteristics of synthetic VSMCs include decreased number of actin filaments, an increase in secretory vesicles, increased rates of proliferation and migration, extensive ECM synthesis/degradation capabilities, increased cell size and “hill-and-valley” morphology in culture, high proliferative rate, and increased expression of α4β1 integrin.
Inflammatory vascular smooth muscle cells
In addition to the phenotypic continuum between contractile and synthetic phenotypes, VSMCs can also express markers of an inflammatory phenotype in response to EC-induced recruitment of monocytes and macrophages during the progression of atherosclerosis.14 Various stimuli, including secretion of cytokines by these inflammatory cells, changes in ECM composition, oxidized low density lipoprotein (oxLDL), and VSMC interactions with monocytes/macrophages, induce expression of inflammatory cytokines, vascular cell adhesion molecule (VCAM-1) and transcription factors (NFκB) in VSMCs, leading to recruitment of inflammatory cells into the vessel wall.
Upstream Mediators of Phenotypic Modulation
Growth-inducing factors
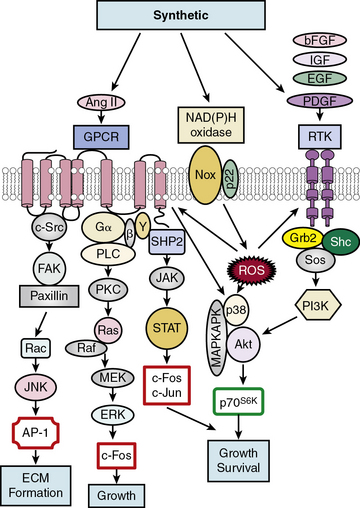
Soluble factors that include growth factors, hormones, and reactive oxygen species (ROS) serve as upstream mediators of the phenotypic switch from contractile to synthetic VSMCs, which results in large part from coordinate activation/repression of VSMC marker genes important in the contractile response2,3,15,16 (Fig. 3-2). Some of the most important growth-inducing factors include platelet-derived growth factor (PDGF), epidermal growth factor (EGF), insulin-like growth factor (IGF), and basic fibroblast growth factor (bFGF). Growth factors bind to surface membrane receptor tyrosine kinases (RTKs), triggering sequential downstream signaling pathways mediated through complex formation of activated RTKs with adaptor and signaling proteins Grb2/Shc/Sos, and activation of intracellular kinases, including phosphatidylinositol 3-kinase (PI3K), mitogen-activated protein kinases (MAPKs: extracellular signal regulated kinase, ERK1/2, p38MAPK, and c-jun NH2-terminal kinase, JNK), Akt, MAPKAPK2, and p70S6 kinase (p70S6K). These signals not only transcriptionally mediate the switch to the synthetic phenotype, but also serve to promote growth and survival. In addition, ROS such as hydrogen peroxide (H2O2) produced by activation of NADPH oxidases, multimeric enzymes containing p22phox and other subunits depending upon the specific isoform, can act as second messengers for canonical G protein–coupled receptor (GPCR) and RTK pathways.17
Differentiation-inducing factors
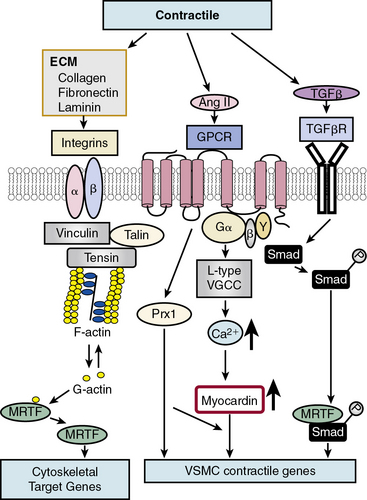
In contrast to growth factor–stimulated proliferation, the cytokine transforming growth factor β (TGF-β) and members of the bone morphogenetic protein (BMP) subgroup of this family promote the differentiated, contractile phenotype in VSMCs by inducing expression of the VSMC contractile genes αSMA and calponin (Fig. 3-3). Transforming growth factor β binds to a tetrameric complex consisting of two type I and two type II receptors, resulting in phosphorylation of Smads, transcription factors named for Caenorhabditis elegans Sma and Drosophila Mad (mothers against decapentaplegic).18 Within the TGF-β signaling pathway itself, different Smads control expression of different markers. For example, Smad3 transactivates the SM22α promoter, while Smad2 activates the αSMA gene. Other soluble factors that inhibit proliferation and increase differentiation include heparin and retinoic acid.9 Most smooth muscle differentiation markers share additional common transcriptional pathways, discussed in more detail later. For example, both TGF-β-induced phosphorylated Smads and ECM-induced activation of integrins, mediated through focal adhesion components vinculin, talin, and tensin, in concert with changes in cytoskeletal F/G actin dynamics, result in myocardin-related transcription factor (MRTF) induction of cytoskeletal/contractile genes (see Fig. 3-3).
Dual factors
One factor with a potential dual role, depending upon initial phenotype/developmental stage, is the octapeptide hormone angiotensin II (Ang II), the effector molecule of the renin–angiotensin II system.19 Angiotensin II can induce either contractile or synthetic phenotypes, with differential responses depending upon cell context and locations within the artery (see Figs. 3-2 and 3-3). Angiotensin II, binding to its GPCR AT1R, activates VSMC marker gene expression indicative of the contractile phenotype through L-type voltage-gated Ca2 + channel–induced elevations in intracellular Ca2 + concentrations, and subsequent increased myocardin transcription coactivator expression dependent upon Prx1, a homeodomain protein that promotes serum response factor (SRF) binding to conserved elements in VSMC marker gene promoters.20 In addition, Ang II binding to AT1R can induce signatures of the synthetic phenotype by activation of multiple kinase and enzyme pathways that are interconnected in signaling networks (see Fig. 3-2). These include the MAPKs; RTKs, including ROS-sensitive transactivation of epidermal growth factor receptor (EGFR); nonreceptor tyrosine kinases (c-Src/focal adhesion kinase [FAK]/paxillin/Rac/JNK/AP-1) and tyrosine phosphatases; SHP2/Janus kinase and signal transducers and activators of transcription (JAK/STAT); and GPCR classic signaling cascades (phospholipase C [PLC]/protein kinase C [PKC]/Ras/Raf/mitogen extracellular signal regulated kinase [MEK]/ERK) leading to stimulation of early growth-response genes (c-fos, c-jun), survival pathways (e.g., Akt), and ECM formation (JNK/AP-1).
Notch communication
In addition to its critical function in development, Notch signaling is also important in defining VSMC differentiation.21,22 Downstream Notch effector gene activation results in activation of “master regulators” of VSMC differentiation (myocardin, MRTFs, or SRF) or direct induction of contractile proteins SMMHC and αSMA, as well as the VSMC specific differentiation marker SM22α (also known as transgelin).6 Data regarding Notch signaling on VSMC differentiation, however, are conflicting, with some studies supporting a repressive effect, while others indicate a promoting effect on expression of VSMC marker genes SMMHC and αSMA.22 These discrepancies may be due to the antagonistic roles of Notch and the Notch effector Hairy-related transcription factor 1 (HRT1) on markers of VSMC differentiation, specifically αSMA and SMMHC.23 Hairy-related transcription factor 1 inhibits Notch/RBP-Jκ binding to the αSMA promoter in a histone deacetylase-independent manner. The context-dependent roles of Notch and HRT1 on markers of VSMC differentiation may serve to fine-tune VSMC phenotypic modulation during vascular development, injury, and disease.
There is considerable cross-talk between Notch and other signaling pathways. Notch and TGF-β cooperatively induce a functional contractile, differentiated phenotype through parallel signaling axes,24 while HRT factors block VSMC differentiation in both pathways. Other examples of cross-talk among key signaling pathways for morphogenesis (Hh, Notch) and mitogenesis (VEGF-A, PDGF) include a Shh/VEGF-A/Notch signaling axis in VSMCs in the neointima to increase growth and survival,25 and Notch-induced up-regulation of PDGFR-β to mediate growth and migration.26
Homotypic VSMC-VSMC Notch-mediated signaling pathways are also apparent in adult vascular pathologies and response to injury.22 After injury, Notch receptors are increased, along with elevated levels of HRT. Negative feedback between HRT and Notch may account for the adaptive response to injury in which initial Notch/HRT-induced suppression of the contractile phenotype is followed by arterial remodeling. As Notch/HRT signaling decreases, the contractile phenotype is reestablished.24
Transcriptional Regulation of Vascular Smooth Muscle Cell Diversity
The complex web of signaling pathways induced by these external signals—whether they are soluble, insoluble, structural, or mechanical—converge on a network of transcription factors (TFs) that coordinately regulate gene expression and act as “master switches” for growth and differentiation27 (Fig. 3-4). Transcription of VSMC-specific differentiation or proliferative genes is regulated by cooperative interaction of TFs and their coregulators, including SRF,28 myocardin and myocardin-related TFs (MRTF-A and -B),29 Ets domain transcription factors known as ternary complex factors (TCFs),30 zinc finger factors GATA630 and PRISM/PRDM6,31 and Krüppel-like factors (KLFs).32,33
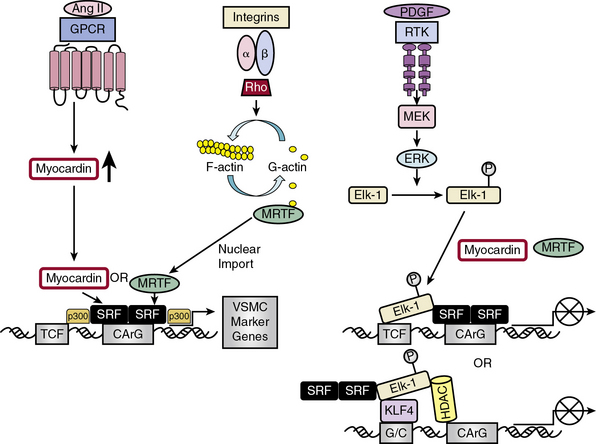
Serum response factor/myocardin axis
Serum response factor, a widely expressed member of the MADS (MCM1, agamous, deficien, SRF) box of TFs, is a nodal point linking signaling pathways to differential gene expression related either to growth or differentiation, depending upon which transcriptional partner is bound to SRF.28 Serum response factor self-dimerizes and binds with high affinity and specificity to a consensus deoxyribonucleic acid (DNA) sequence CArG box found in the promoters of cyto-contractile genes.34 More than half of the VSMC “marker” genes that define VSMC molecular signature contain CArG boxes.34 Included in these genes are three categories modulating actin filament dynamics: (1) structural (e.g., αSMA-actin, SM22α, caldesmon, SMMHC); (2) effectors of actin turnover (e.g., cofilin, gelsolin); and (3) regulators of actin dynamics (four-and-a-half LIM domains proteins [FHL1 and 2], MMP9, and myosin light chain kinase).35
Serum response factor itself is a weak activator of CArG-dependent genes.30 Potent SRF-dependent transcriptional activation is therefore dependent upon regulation at several levels: by interaction with different signal-regulated or tissue-specific regulatory SRF transcription cofactors/corepressors; by posttranslational phosphorylation, acetylation, and sumoylation, modifications that affect these interactions; and by epigenetic alterations in chromatin structure in which myocardin serves as a scaffold for recruitment of chromatin-remodeling enzymes36 that enable SRF and its cofactors to gain access to SRF target genes.8 Myocardin association with histone acetyltransferases (HATs), including p300, enhances transcription of VSMC-restricted genes, whereas association with class II histone deacetylases (HDACs) suppresses myocardin-induced transcription of VSMC marker genes36 (see Fig. 3-4).
Serum response factor interacts with cofactors in two principal families: the TCF family of Ets-domain proteins (Elk, SAP-1, and Net)37 activated by the MAPK pathway, leading to SRF binding to immediate early growth factor-inducible genes such as c-fos28; and the myocardin/MRTF-A/MRTF-B family35 to promote activation of VSMC-specific marker genes, most of which code for filamentous proteins that function in contractile activities or proteins that function in cell-matrix adhesions.10 These alternative pathways provide the “plasticity” associated with VSMC phenotypic modulation ranging from contractile functions to maintain vascular tone to synthetic or proliferative functions in response to vascular injury.29
Discovery of the cell-restricted SRF transcriptional coactivator myocardin, expressed specifically in cardiac and VSMCs, resolved the paradoxical observations that SRF can regulate mutually exclusive gene expression programs for growth or differentiation.30,34 In VSMCs, myocardin is a master regulator of SMC marker gene expression and sufficient for the smooth muscle–like contractile phenotype. Myocardin competes with Elk-1 for direct binding to SRF in VSMCs; thus, myocardin and Elk-1 can act as binary transcriptional switches that may regulate contractile vs. synthetic VSMC phenotypes30 (see Fig. 3-4). In addition, myocardin transduction leads to lower levels of the cell cycle–associated gene cyclin D1, resulting in repression of growth. Therefore, myocardin is a nodal point for two features indicative of SMC differentiation: expression of the contractile apparatus and suppression of growth.30
While myocardin functions exclusively as a transcriptional coactivator,38 additional proteins function to regulate transcriptional activity of myocardin. Hairy-related transcription factor 2 and GATA factors repress or enhance myocardin-induced transcriptional activity, depending upon cell context.30 In addition, activation of Notch receptors by Jagged1 endogenous ligand induces translocation of Notch intracellular domain (ICD) to the nucleus where it inhibits myocardin-induced SMC gene expression.29 Angiotensin II stimulation, as well as activation of L-type voltage-gated Ca2 + channels, activates SMC marker genes by inducing myocardin expression and, in the case of Ang II, increasing SRF binding to CArG elements in the promoter regions of VSMC marker genes such as αSMA.20
Serum response factor transcriptional activity is also controlled by Rho-induced actin dynamics that facilitate movement of MRTFs into or out of the nucleus29 (see Fig. 3-4). In most cell types, MRTFs form a stable complex with monomeric G-actin and remain sequestered in the cytoplasm. Myocardin-related transcription factors in VSMCs, however, are localized in the nucleus where binding to SRF in the basal state promotes contractile gene expression, and the differentiated phenotype. In response to growth factors or vascular injury, extracellular signals transduced through the Rho-actin pathway result in nuclear export of MRTF, down-regulation of SRF/MRTF-induced VSMC contractile gene expression, and promotion of mitogen-induced ERK1/2 phosphorylation of TCFs, resulting in TCF displacement of MRTFs and SRF/TCF-mediated activation of growth-responsive genes.29 These differential pathways provide a switch in which SRF target genes are differentially regulated through growth factor–induced signaling for growth (active TCF, MRTF blocked) or Rho-actin signaling for differentiation (inactive TCF, MRTF active)30 (see Fig. 3-4).
Zinc finger proteins
GATA6, a zinc finger transcription factor expressed in VSMCs, induces growth arrest by increasing expression of the general cyclin-dependent kinase inhibitor (CDKI) p21CIP1 and inhibiting S-phase entry.30 PRISM is a smooth muscle–restricted member of zinc finger proteins belonging to the PRDM (PR domain in smooth muscle) family and acts as a transcriptional repressor by interacting with class I histone deacetylases and G9a histone methyltransferases. PRISM induces the proliferative phenotype while repressing differentiation regulators myocardin and GATA6.31
One of the most intensely studied zinc finger transcriptional regulators in VSMCs is the KLF subfamily that binds to the TGF-β control element (TCE) in the regulatory sequences of target genes (reviewed in32,33,39). Vascular smooth muscle cells express four KLFs (KLF4, KLF5, KLF13, and KLF15), each with individual biological functions implicated in regulating a range of processes in both growth and differentiation.32 Individual KLFs may have opposing functions, depending upon temporal and developmental expression patterns and interactions with other factors. For example, KLF4 inhibits, whereas KLF5 and KLF13 induce, VSMC marker gene expression. Mechanisms that may account for these opposing functions of KLF factors include posttranslational modifications, interaction with specific cofactors, differential expression by growth factors, cytokines and differentiation state, or regulation by another KLF.32
KLF4 functions as both a VSMC growth repressor and a repressor for VSMC differentiation, although data on the effect of KLF4 on VSMC differentiation are conflicting33 (see Fig. 3-4). As a growth repressor, KLF4 inhibits PDGF-BB-induced mitogenic signaling and induces expression of the negative cell cycle regulator p53 and its target gene p21CIP1. As a differentiation repressor, KLF4 prevents SRF from binding to the TCE in promoters of VSMC marker genes, suppresses expression of myocardin, inhibits myocardin-induced activation of SMC marker genes, reduces SRF binding to CArG elements in SMC contractile gene promoters,33 and induces histone hypoacetylation at SMC CArG regions associated with gene silencing.40 On the other hand, there is evidence that KLF4 promotes VSMC differentiation by directly activating VSMC marker gene transcription of SM22α and αSMA.33 KLF4 thus functions as a bifunctional TF or “molecular switch” that can both activate and repress VSMC marker genes, depending upon regulation of KLF4.33
Even though the closely homologous KLF4 and KLF5 TFs share similar developmental and tissue pattern expression, they exert different, often opposing, effects on gene regulation and proliferation/differentiation.33 Whereas KLF4 is associated with growth arrest, KLF5 exerts pro-proliferative effects, particularly in vascular remodeling in response to injury. KLF5 expression, abundant in fetal VSMCs but down-regulated in the adult (reviewed in39), is induced after vascular injury by activation of immediate early response genes by Ang II and ROS.41 KLF5 in turn mediates re-expression of SMemb/NMHC-B, a marker for the dedifferentiated phenotype, and activates other critical injury response genes involved in remodeling, such as PDGF-A/B, Egr-1, plasminogen activator inhibitor 1 (PAI-1), inducible nitric oxide synthase (iNOS), and VEGFR, implicating KLF5 as a key regulator for VSMC response to injury.39 In additional injury responses, KLF5 increases cyclin D1 expression and inhibits the cyclin kinase inhibitor p21, thus leading to vascular remodeling by increased cell proliferation.42 Similar to KLF4 regulation, KLF5 expression and activity are regulated at multiple levels, including upstream Ras/MAPK, PKC, and TGF-β signaling pathways; downstream interactions with TFs, including retinoic acid receptor α (RARα), NF-κB, and peroxisome proliferator–activated receptor gamma (PPARγ); as well as posttranslational modifications that can positively or negatively regulate KLF activity.39 In addition, KLF5 activity is regulated in the nucleus by chromatin remodeling factors such as SET, a histone chaperone that inhibits the DNA binding activity of KLF5,43 p300 (a coactivator/acetylase that coactivates KLF5 transcription), and HDAC1, which inhibits KLF5 binding to DNA.32
Two additional KLFs have been identified in VSMCs: KLF13 and KLF15.32 After vascular injury, KLF13 is induced and activates the promoter for the VSMC differentiation marker SM22α, while KLF15 expression is down-regulated, implicating KLF15 as a negative regulator of VSMC proliferation and a counterbalance to the growth-promoting effects of KLF5 in vascular injury response.
Posttranscriptional Regulation of Vascular Smooth Muscle Cell Diversity: Noncoding microRNAs
Upstream signaling and downstream transcriptional pathways in VSMCs are intertwined with a multitude of micro ribonucleic acid (miRNAs) that act as “rheostats” and “switches” in regulating protein activity in development, function, and disease.44 miRNAs are small, noncoding RNAs (20-25 nucleotides in length) that associate with a miRNA-induced silencing complex (miRISC) of regulatory proteins, including Argonaute family proteins, Argonaute interacting proteins of the GW182 family, eukaryotic initiation factors (eIFs), polyA-binding complexes, decapping enzymes/ activators, and deadenylases, to induce posttranscriptional silencing of their target genes.45 These multiple components are assembled and interact in a multistep process with components of the translational machinery to inhibit translation initiation, mark mRNAs for degradation through deadenylation, and sequester targets into cytoplasmic P bodies.44 Multiple mechanistic models for miRNA-induced gene silencing have been proposed that provide insights into the molecular mechanisms of translational inhibition, deadenylation, and mRNA decay, but questions remain concerning the kinetics and ordering of these translational events and whether they are coupled or independent.45 A recent unifying model for miRNA-regulated gene repression is an attempt to reconcile the often conflicting existing data. It proposes that recruitment of Argonaute and associated GW182 proteins to miRNA induces binding to the mRNA 5′m7 Gcap, thus blocking translation initiation, potentially by mRNA deadenylation. Subsequent to miRNA-mediated deadenylation, mRNA is degraded through recruitment of decapping proteins.46 In this model, inhibition of translation initiation is linked to subsequent rapid mRNA decay in a coupled process. Because miRNAs—which in general are negative regulators of gene expression—may be almost as important as transcription factors in controlling gene expression in the pathogenesis of human diseases,47 insights into the functions of this class of noncoding RNAs are important in evaluating their potential use as therapeutic targets.45
Cardiovascular-specific, highly conserved miRNAs miR-143 and miR-145, the most abundant miRNA in the vascular wall,48 are key players in programming VSMC fate from multipotent progenitors in embryonic development and in reprogramming VSMCs during phenotypic modulation in the adult44,49 (Fig. 3-5). miR-143 and miR-145 have distinct sequences but are clustered together and transcribed as a bicistronic unit. Upstream in the genomic sequence of miR-143/145 is a conserved SRF-binding CArG box site, indicating control by SRF and myocardin.49,50 These miRNAs cooperatively feed back to modulate the actions of SRF by targeting a network of transcription factors/coactivators/corepressors. This network includes miR-145-induced repression of KLF4, a positive regulator of proliferation and myocardin repressor; miR-143-induced repression of Elk-1, a myocardin competitor and positive regulator of proliferation; and, contrary to the usual inhibitory role of miRNA, miR-145-induced stimulation of myocardin, a positive regulator of differentiation. Thus, miR-145 is necessary and sufficient for VSMC differentiation, and the miR-143/miR-145 cluster acts as an integrated signaling node to promote differentiation while concurrently repressing proliferation.49 Although mice with genetic deletions for miR-143/145 show no obvious abnormalities in early development, VSMCs in the adult exhibit both structural and phenotypic differences in injury- or stress-induced vascular remodeling. Ultrastructural analysis of arteries from miR-143/145 knockout mice shows reduced numbers of medial VSMCs with a contractile appearance, and an increase in synthetic VSMCs.51 These results suggest that miR-143 and miR-145 modulate cytoskeletal structure, actin dynamics, and modulation to a dedifferentiated phenotype50 (see Fig. 3-5). Importantly, miR-143/145 knockout mice with increased synthetic VSMCs develop spontaneous neointimal lesions in the femoral artery in the absence of hyperlipidemia and inflammation, supporting a key role for phenotypically altered VSMCs in the pathogenesis of lesion formation.51
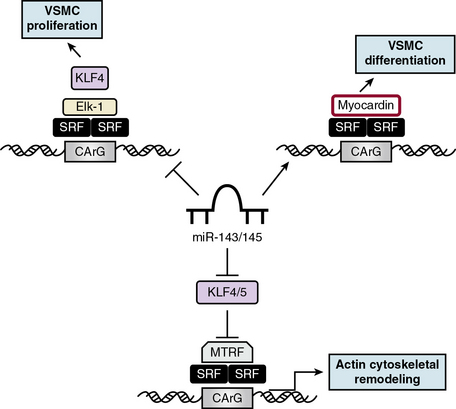
While miR-143 and miR-145 play keys roles in the contractile phenotype of VSMCs and the response to injury,52 miR-221 and miR-222 are modulators of VSMC proliferation, although largely by affecting growth-related signaling pathways rather than by controlling VSMC phenotype. miR-221 and miR-222, encoded by a gene cluster on the X chromosome, are up-regulated in VSMCs in neointimal lesions and in proliferating cultured VSMCs stimulated by PDGF-BB.53 Studies show that two CDKIs, p27KIP1 and p57KIP2, have miR-221 and miR-222 binding sites and are gene targets for miR-221 and miR-222 in the rat carotid artery in vivo.53 Thus, miR-221 and miR-222 are pro-proliferative because they repress two CDKIs, p27KIP1 and p57KIP2. Furthermore, PDGF, via miR-221 induction, inhibits VSMC differentiation via c-kit-induced inhibition of myocardin.54
Posttranslational Regulation of Vascular Smooth Muscle Cell Diversity: Epigenetics
The “epigenetic landscape” controls gene expression by chemical modifications that mark regions of chromosomes either by methylation of promoter CpG sequences in the DNA itself, or by covalent modification of histone proteins that package DNA by posttranslational addition of methyl, acetyl, phosphoryl, ubiquityl, or sumoyl groups, leading to expression/repression of transcription (reviewed in55). In VSMCs, multiple levels of epigenetic controls exist for gene expression leading to differentiation or dedifferentiation programs in healthy cells and for dysregulated gene expression in vascular disease. These epigenetic changes in VSMCs involve both DNA and histone methylation as well as histone acetylation/deacetylation. Methylation of histones, catalyzed by histone methyltransferases (HMTs), results in a tight, stable epigenetic mark between methylated histones and chromatin that can be passed to daughter cells, thus providing “epigenetic memory” that defines cell lineage and identity by controlling SRF access to VSMC-specific marker genes.55 Acetylation is controlled by HATs, which promote gene transcription by destabilizing chromatin structure to an “open,” transcriptionally active conformation, and HDACs, which promote chromatin condensation to a “closed,” transcriptionally silent conformation with restricted access to DNA. Histone acetylation/deacetylation thus serves to regulate transcription in a rapid and “on/off” manner in response to dynamic environmental changes and links the cell’s genome with new extrinsic signals.55
In VSMCs, SRF binding to CArG boxes in the promoters of SMC marker genes to promote the VSMC differentiated phenotype depends upon alterations of chromatin structure, including histone methylation and acetylation. In a model for epigenetic regulation of VSMC phenotype,56 SRF binding to CArG boxes in VSMC marker gene promoters is blocked by conditions such as PDGF-BB exposure or vascular injury. Such conditions promote KLF4-induced myocardin suppression as well as KLF4-induced recruitment of HDACs, resulting in “closed” deacetylated chromatin and transcriptional repression of VSMC marker genes. Histone methylation, in contrast, is not affected by PDGF-BB and may serve as a permanent “memory” for VSMC identity during repression of SRF-dependent transcription and can, once repressive signals are terminated, reactivate the differentiation program by recruiting myocardin/SRF complexes or HATs to VSMC marker genes for reexpression. In the absence of KLF4 activation, SRF/myocardin can bind to HAT-induced acetylated “open” chromatin at CArG boxes for transcriptional activation of VSMC marker genes, thus promoting VSMC differentiation. In addition, myocardin induces acetylation of histones in the vicinity of SRF-binding promoters in VSMC marker genes by association with p300, a ubiquitous transcriptional coactivator with its own intrinsic HAT activity, leading to synergistic activation of VSMC marker gene expression. This pro-myogenic program is antagonized and repressed by myocardin binding to class II HDACs, which strongly inhibits expression of marker genes αSMA, SM22α, SMMLCK and SMMHC. These opposing actions of HATs and HDACs on SRF/myocardin function to activate or repress, respectively, VSMC differentiation and serve to regulate transcription in a rapid and reversible manner in response to dynamic changes in the environment.55
Often, transcription mediators play roles in both classic signal transduction pathways and epigenetic programming.57 Smad proteins, for example, transmit TGF-β signals from the membrane to the nucleus to mediate gene transcription and VSMC differentiation. The balance between Smad-induced recruitment of corepressors or coactivators to TGF-β-responsive genes is associated with activation of HDAC or HAT (p300), which then alters histone acetylation. Transforming growth factor β induces histone hyperacetylation at the VSMC marker gene SM22 promoter through recruitment of HATs, Smad3, SRF, and myocardin, demonstrating a role for HATs and HDACs in TGF-β activation of VSMC differentiation.58
A proposed example of metabolic memory stored in the histone code of VSMCs is found in the dysregulation of histone H3 methylation, an epigenetic mark usually associated with transcriptional repression in type 2 diabetes.59 In VSMCs derived from type 2 diabetic db/db mice, levels of H3K9me3 (H3 lysine-9 trimethylation), as well as its HMT, are both reduced at the promoters of inflammatory genes. This loss of repressive histone marks, leading to increased inflammatory gene expression, is sustained in VSMCs from db/db mice cultured in vitro, suggesting persistence of metabolic memory. These results suggest that dysregulation in the histone code in VSMCs is a potential mechanism for increased and sustained inflammatory response in diabetic patients who continue to exhibit “metabolic memory” and vascular complications after glucose normalization.60
Influence of Cell-Cell and Cell-Matrix Interactions
Many differential VSMC functions are influenced by cell-cell and cell-matrix adhesion receptors that are altered during phenotypic modulation and during response to injury or disease. Cell-cell adhesion receptors include cadherins and gap junction connexins; cell-matrix interactions are dependent upon combinations of integrins, syndecans, and α-dystroglycan.11
Cell-Cell Adhesion Molecules: Cadherins and Gap Junction Connexins
After investment of VSMCs to the EC layer of nascent vessels, vascular stabilization, also known as maturation,61 is regulated by the sphingosine 1-phosphate (S1P) receptor S1P1, a GPCR on ECs. S1P1 activates the cell-cell adhesion molecule N-cadherin in ECs and induces formation of direct N-cadherin-based junctions between ECs and VSMCs required for vessel stabilization.61 To maintain VSMC quiescence within the vascular wall, cadherin-mediated cell-cell adherens-type junctions between VSMCs inhibit VSMC proliferation, possibly by inhibiting the transcriptional activity of β-catenin, a component of the Wnt signaling pathway, which interacts with the intracellular domain of cadherins.62 Inhibition of β-catenin or stabilization of cadherin junctions in VSMCs may be useful in treating vascular disease or injury.
Another type of direct intercellular junction between cells in the vasculature is the gap junction.63 Gap junctions, formed by connexin proteins between ECs and VSMCs and between VSMCs, are intercellular channels that allow movement of metabolites, small signaling molecules, and ions between cells.63–65 Of the four connexin proteins expressed in VSMCs (Cx37, Cx40, Cx43, and Cx45), Cx45 is exclusively found in VSMCs, while Cx43 is the most prominent and is essential for coordination of proliferation and migration.63 Homotypic gap junctions between VSMCs coordinate changes in membrane potential and intracellular Ca2 +, and heterotypic contacts between ECs and VSMCs at the myoendothelial junction control vascular tone by EC-mediated VSMC hyperpolarization. Notably, expression and/or activity of vascular connexins are altered in vascular diseases such as hypertension, atherosclerosis, or restenosis64 and in diabetes.63
Cell-Matrix Adhesion Molecules: Integrins and Syndecans
Integrins
Transmembrane integrin receptors are composed of combinations of α and β subunits, each combination with its own ligand-binding specificity and signaling properties. Integrins link the ECM with the actin cytoskeleton within VSMCs. The β1 subunit is the main β subunit in VSMCs in vivo and in vitro; the major α integrin subunits expressed in VSMCs in vivo are α1, α3, and α5.11 Integrin α1β1 is involved in collagen remodeling after injury, and integrin α5β1 binds to fibronectin (FN) and effects FN polymerization.
Activation of different VSMC integrins results in expression of differential phenotypic programs. Beta-1 expression contributes to maintenance of the VSMC contractile phenotype, whereas integrins α2β1, α5β1, α7β1, and αvβ3 participate in SMC migration indicative of the synthetic phenotype.11 Neointimal formation after vessel injury is reduced by blocking αvβ3, but apoptosis in the injured vessel is increased, potentially promoting plaque rupture. In addition, neointimal formation is prevented and the VSMC contractile phenotype is maintained by binding of α7β1 integrins to COMP (cartilage oligomeric matrix protein), a macromolecular ECM protein.66
Syndecan coreceptor
Syndecans are members of a family of four transmembrane heparan sulfate proteoglycans (HSPGs) consisting of a core protein covalently coupled with (GAGs).67,68 Syndecans function as coreceptors with growth factor or adhesion receptors and function to “tune” extracellular signal transfer across the cell surface to the cytoskeleton and cytoplasmic mediators to effect activation of a variety of intracellular signaling cascades. All four syndecans are expressed in the artery, and VSMC syndecans bind to ECM proteins, cell adhesion molecules, heparin-binding growth factors such as fibroblast growth factor (FGF) and EGF, lipoproteins, lipoprotein lipases, and components of the blood coagulation cascade.11 Syndecan-1 inhibits VSMC growth in response to PDGF-BB and FGF2 after vascular injury.69 Syndecan-4 has been implicated in thrombin-induced VSMC migration and proliferation by acting both as a mediator for bFGF signaling and as a cofactor for fibroblast growth factor receptor 1 (FGFR-1), suggesting that syndecan-4 is an early response gene after injury, whereas syndecan-1 is active during the proliferative and migratory phase.68
Insoluble extracellular matrix components
One of the most important functions of VSMCs is to secrete, organize, and maintain an elaborate ECM architecture, an “extended cytoskeleton” that varies according to the biomechanical stresses of the differing vascular beds. Large elastic arteries (e.g., thoracic aorta, carotid, renal arteries) are characterized by multiple concentric elastic lamellae that distribute cardiac-driven pulsatile stress evenly throughout the vessel wall. Smaller muscular arteries that experience less force (e.g., coronary, cerebral, mesenteric) contain only two elastic laminae. Elaboration of the ECM synthesized and organized by VSMCs is considered to be a major part of their “differentiated” phenotype70 because ECM components influence the same pathways regulated by growth/differentiation factors (see Fig. 3-3). Changes acquired by VSMCs during acquisition of contractile properties are in turn maintained by the ECM in “dynamic reciprocity” between the matrix and gene expression. In addition to providing a structural elastic scaffold for the extensible vessels, the ECM regulates gene expression through binding of matrix receptors on the cell surface and through acting as a reservoir for growth factors such as PDGF and FGF that regulate cell function (reviewed in71).
Extracellular matrix components are classified as fiber-forming molecules (certain collagens and elastin), non-fiber-forming or interfibrillar molecules (proteoglycans and glycoproteins), and matricellular proteins (thrombospondin-1 and -2, secreted protein acidic and rich in cysteine [SPARC/osteonectin], tenascin-C, and osteopontin) that modulate cell-matrix interactions and tissue repair.72 A list of ECM molecules and diseases resulting from ECM alterations can be found in a recent review72 (also see Chapter 4).
Basement membrane
Vascular smooth muscle cells in the intact vessel are surrounded by a basement membrane composed primarily of type IV collagen and laminin.11 Laminins are basement membrane modular glycoproteins that interact with both cells and ECM to affect proliferation, migration, and differentiation.70 Evidence from cultured VSMCs suggests that laminin induces expression of contractile proteins and moderates the proliferative response to mitogens such as PDGF through a mechanism involving the laminin receptor α7β1, which links the basement membrane to the VSMC contractile apparatus.3
Fibronectin
Fibronectin is present in developing tissues prior to collagen, and there is evidence that FN has an organizing role in ECM assembly as a “master orchestrator” for matrix assembly, organization, and stability.73,74 Fibronectin binding to α5β1 induces integrin-bound FN clustering, resulting in activation of actin polymerization, actin-myosin interactions, and signaling through kinase cascades. Thus, FN modulates VSMCs toward the synthetic phenotype.12
Collagens
Differential phenotypic modulation of VSMCs in response to different forms of collagen or to different isotypes of collagen illustrates the importance of cues from the physical and chemical ECM environment that regulate VSMC physiology in normal and disease states.75 Cells cultured on fibrillar vs. monomeric collagen type 1 exhibit very different gene expression profiles, responses to growth factors such as PDGF-BB, and migration properties.12 Fibrillar collagen type 1 promotes the contractile phenotype, whereas monomeric collagen type 1, found in the degraded matrix of vascular lesions (“atherosclerotic matrix”), activates proliferation,76 reduces contractile gene expression, and promotes a VSMC inflammatory phenotype with increased VCAM-1 expression.75 Vascular smooth muscle cells also exhibit different phenotypic profiles depending upon contact with different collagen isotypes: collagen type IV, a component of the basement membrane surrounding VSMCs (“protective” matrix), promotes expression of contractile proteins by regulating the SRF coactivator myocardin expression and mediating recruitment of SRF to CArG boxes in αSMA and SMMHC promoters.75
Elastins and elastin-associated proteins
Elastic fibers are composed of tropoelastin, fibrillin-1, and fibrillin-2 and are assembled and deposited in a tightly regulated, hierarchical manner.77,78 They provide not only unique elastomeric properties to the vessel wall but also influence phenotypes of VSMCs, directly through adhesion and indirectly through TGF-β signaling,77 to regulate migration, survival, and differentiation.78 Elastin maintains the quiescent, contractile phenotype of VSMCs by specifically regulating actin polymerization and organization via a signal transduction pathway involving Rho GTPases and their effector proteins.79 Mechanical injury or inflammation that results in focal destruction of insoluble elastin into soluble elastin-derived peptides induces VSMC dedifferentiation and migration. Elastin-derived peptides can activate cyclins/cyclin-dependent kinases, leading to cell cycle progression and proliferation found in neointimal formation.
Fibrillins
Fibrillins are large cysteine-rich glycoproteins that serve dual roles: (1) providing stability and elasticity to tissues and (2) sequestering TGF-β and BMP complexes in the ECM to limit their bioavailability, providing for spatial and temporal growth factor signaling during remodeling or repair.80,81 Mutations in the fibrillin-1 gene are found in Marfan syndrome, a heritable disease associated with disorganized elastic fibers in defective aorta and excess TGF-β signaling.78
Fibulins
Fibulins are elastic fiber–associated proteins.78 Vascular smooth muscle cells from fibulin-5 null mice exhibit enhanced proliferation and migration, indicating an inhibitory role for fibulin-5 in VSMC response to mitogenic stimuli.82 Vascular smooth muscle cell-specific deletion of the fibulin-4 gene results in large aneurysm formation exclusively in the ascending aorta and down-regulation of SMC-specific contractile proteins and transcription factors for SMC differentiation. Thus fibulin-4 may serve a dual role in both elastic fiber formation and SMC differentiation, and therefore may protect the aortic wall against aneurysm formation in vivo and may also maintain an ECM environment for VSMC differentiation. Fibulin-2 and fibulin-5 double knockout mice have vessels that exhibit disorganized internal elastic lamina and an inability to remodel after carotid artery ligation-induced injury,83 which was not observed in single knockout mice for fibulin-2 or fibulin-5. These data suggest that fibulins 2 and 5 function cooperatively to form the internal elastic laminae and protect vessel integrity.
EMILINs
EMILINs (elastin microfibril interface-located proteins) act as an extracellular negative regulator of TGF signaling.84 EMILIN null mice (Emilin1[−/−]) exhibit inhibition of cell proliferation, smaller blood vessels, altered elastic fibers,85 and increased peripheral resistance, causing hypertension.84 These data indicate a role for EMILIN in elastogenesis, maintenance of VSMC morphology, and—importantly—in blood pressure control.
Glycosaminoglycans, proteoglycans, and matricellular proteins
Glycosaminoglycans in the vascular ECM, including heparin and the related heparan sulfate, inhibit VSMC migration and proliferation. Heparin also induces expression of contractile markers for maintenance of the differentiated phenotype.3 Proteins bearing GAG chains, the proteoglycans,86 which include syndecan transmembrane HSPG and perlecan basement membrane HSPG, interact with FN in matrix assembly.74 Different proteoglycans can have opposing effects on VSMCs: the HSPG perlecan inhibits VSMC proliferation and intimal thickening by sequestering FGF-2,12,69 while versican, a chondroitin sulphate proteoglycan, promotes VSMC proliferation.87 Vasoactive agents acting through GPCRs such as endothelin-1 and Ang II stimulate elongation of GAG chains on the proteoglycan core proteins.88 These elongated GAG chains exhibit enhanced binding to low-density lipoprotein (LDL), providing a mechanism for atherogenic lipid retention in the vessel wall. Finally, matricellular proteins (e.g., thrombospondins, tenascins, SPARC), are thought to be “antiadhesive proteins” with effects on VSMC migration and adhesion.70 CCN (cysteine-rich protein, Cyr 61/CCN1) is a family of secreted matricellular proteins that mediate cellular responses to environmental stimuli through interaction with a variety of cell surface proteins and adhesion receptors including Notch receptors and integrins.89 CCN1, which is up-regulated in the VSMCs of injured arteries, stimulates VSMC proliferation through CCN1/α6β1 integrin interactions.90 Knockdown of CCN1 in injury models suppresses neointimal hyperplasia. In contrast, CCN3 protein inhibits VSMC proliferation in a TGF-β-independent manner by increasing the CDKI p21, partly through Notch signaling, thus suppressing neointimal thickening.91 These contrasting roles for pro-proliferative CCN1/α6β1 integrin signaling and antiproliferative CCN3/Notch signaling in VSMCs offer therapeutic strategies for reducing neointimal hyperplasia.91
Matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases
Matrix metalloproteinases are zinc-containing enzymes that, along with extracellular proteases in the plasminogen activation system, induce remodeling of VSMC cell-matrix and cell-cell interactions (reviewed in92–94) and release ECM-bound growth factors, cytokines, and proteolyzed ECM fragments, or “matrikines,” with cytokine-like properties into the ECM. Members of the MMP family found in vascular tissues (listed in Ref. 95) include interstitial collagenases, basement membrane gelatinases, stromelysins, matrilysins, and membrane type (MT)-MMPs and metalloelastase (see Chapter 4). In the vascular wall, production of pro-MMP-2, MMP-14, and TIMP-1 and -2 is constitutive,96 while other MMPs can be induced by inflammatory cytokines (interleukin [IL]-1 and -4 and tumor necrosis factor α [TNF-α]), hemodynamics, vessel injury, and ROS.93 In addition, MMPs can act synergistically with growth factors such as PDGF and FGF-2.
Matrix metalloproteinase induced remodeling of basement membrane components laminin, polymerized type IV collagen, and HSPGs promotes a VSMC migratory phenotype. In addition, MMP cleavage and shedding of non-matrix substrates—in particular, adherens junction cadherins—act to remove physical constraints on cell movement.93 Furthermore, ECM remodeling enables integrin signaling from the cell surface to focal adhesions, modulating cell cycle components cyclin D1 and p21/p27 CDKIs.96
In vascular remodeling, MMP activities are tightly regulated at several levels: transcriptional level, activation of pro-forms, interaction with specific ECM components, and inhibition by TIMPs. Modulation of MMP activity is evident in VSMC migration and neointima formation after injury, plaque destabilization in atherosclerosis, aneurysm formation, hypertension, and coronary restenosis.95 In atherosclerosis, MMPs have potential either to promote plaque instability, as in advanced plaques of hypercholesterolemia models, or to stabilize plaques by increasing VSMC migration/proliferation. Up-regulation of MMPs in VSMCs may contribute to aneurysm formation.3
Mechanical effects
Data on VSMC phenotypic modulation by the mechanical environment indicate that continuous cyclic mechanical strain acting directly on VSMCs increases collagen and fibronectin synthesis, possibly by paracrine release of TGF-β1, resulting in increased ECM remodeling indicative of a VSMC synthetic phenotype.12 In contrast, some studies have shown that mechanical strain can also stimulate expression of contractile genes.3 Although MAPK signaling pathways are induced following initiation of cyclic strain, mechanisms for this induction are unclear. Activation of ion channels and tyrosine kinases, and paracrine release of soluble mediators such as Ang II, PDGF, and IGF, are postulated to play a role.3
Mechanical signals play a role in stimulating cell cycle progression. Actin filament polymerization and organization induced by integrin ligation generate intracellular mechanical tensional forces that promote cell cycle progression.97 In addition, “stiffness,” or compliance of the ECM, can direct cellular functions through integrin-dependent signaling pathways involving FAK, the canonical mediator of integrin signaling, Rho family GTPase Rac and cyclin D1.98
Phenotype-Specific Vascular Smooth Muscle Cell Functions
Contraction
In nearly all cases, stimulation of VSMC with contractile agents results in activation of a specific GPCR (Fig. 3-6). The immediate response is activation of PLC, which cleaves the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) to release inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3, in turn, binds to its receptor (a channel) on the sarcoplasmic reticulum (SR), creating an open conformation and translocating Ca2 + to the cytoplasm. Simultaneously, receptor activation depolarizes the plasma membrane by altering the activity of pumps such as the sodium/potassium–adenosine triphosphate (Na+/K+-ATPase), and channels that include Ca2 +-sensitive K+ channels and TRP channels.99 Membrane depolarization leads to activation of voltage-dependent L-type Ca2 + channels, calcium influx, and a more sustained but less robust elevation of cytosolic calcium. Moreover, Ca2 + entry through these channels activates ryanodine receptors on the SR, further increasing Ca2 + release into the cytosol.
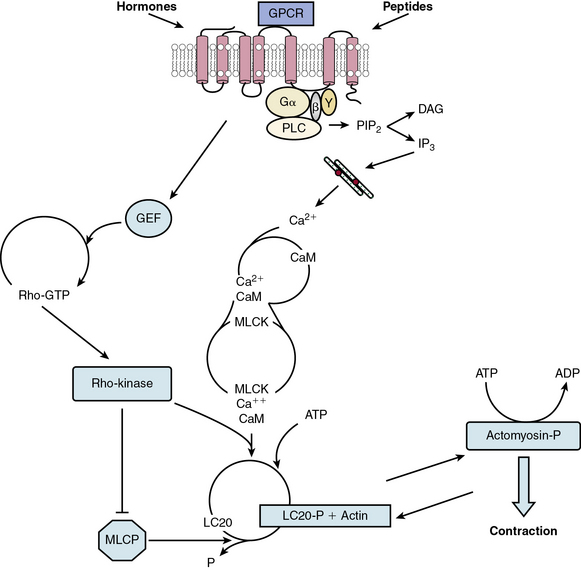
Figure 3-6 Model for contraction cascade in vascular smooth muscle cell (VSMC).
(Adapted from Griendling K, Harrison D, Alexander R: Biology of the vessel wall. In Fuster V, Walsh R, O’Rourke R, Poole-Wilson P, editors. Hurst’s the heart. 12th ed. New York, 2008, McGraw-Hill, pp 135–154.)
The increased cytoplasmic calcium binds to calmodulin (CaM) at a ratio of four calcium ions to one CaM molecule. Calmodulin then undergoes a conformational change, and binds to and activates myosin light chain kinase (MLCK), the enzyme responsible for phosphorylation of the 20-kD regulatory myosin light chain (LC20) on serine 19. Activated LC20 facilitates actin-mediated myosin adenosine triphosphate (ATPase) activity and cyclic interaction of myosin and actin,100 leading to contraction. Contraction is maintained even when calcium drops, suggesting that LC20 becomes sensitized to calcium, likely by inhibition of myosin phosphatase (see later discussion).101
Because the increase in intracellular calcium caused by vasoconstrictors is largely responsible for activation of the contractile apparatus, essential mechanisms exist to limit Ca2 + entry and clear Ca2 + from the cytosol. Ryanodine receptors cluster to release calcium sparks, which in turn stimulate Ca2 +-activated large conductance K channels (BK channels) to cause hyperpolarization and limit L-type calcium channel activity.102 Additionally, the sarcoplasmic reticulum Ca2 +-ATPase (SERCA) mediates Ca2 + reuptake into the SR and serves to maximize Ca2 + extrusion from the cell because the newly taken-up SR Ca2 + is released in a directed manner towards the plasma membrane, where a plasma membrane Ca2 +-ATPase extrudes Ca2 + from the cell. Importantly, SERCA is inhibited by CaM kinase II–mediated phosphorylation.103
Recently, ROS and reactive nitrogen species (RNS) have emerged as effective modulators of contractile signaling.104 Specifically, high levels of ROS oxidize SERCA, thereby inhibiting its activity. Hydrogen peroxide applied externally increases IP3 receptor-mediated release of Ca2 + into the cytosol, and activation of NADPH oxidases by contractile agonists sensitizes the IP3 receptor to IP3. Ryanodine receptors are also redox-sensitive. S-nitrosylation activates them, and exposure to endogenous levels of ROS and RNS can protect these receptors from inhibition by CaM at high concentrations of calcium. Both hydrogen peroxide and superoxide can stimulate Ca2 + entry via L-type or T-type calcium channels (including TRP channels), but S-nitrosylation by nitric oxide (NO) is inhibitory. Thus, in general, ROS and RNS inhibit Ca2 + pumps and activate Ca2 + entry and release, resulting in an increase in intracellular Ca2 + concentration.
Myosin light chain phosphatase (MLCP) is also a vital regulator of vascular contraction. It is a multimeric enzyme composed of a regulatory myosin-binding subunit (MYPT1), a catalytic subunit (PP1c), and a 20-kD protein (M20). The activity of MLCP is largely regulated by Rho kinase-mediated phosphorylation of MYPT1 on Thr695, either directly or via Rho-kinase activation of ZIP kinase.101 Myosin light chain phosphatase activity can also be inhibited by CPI-17 (PKC-potentiated PP1 inhibitory protein of 17 kD), which when phosphorylated by PKC, acts as a pseudosubstrate, binds to PP1c, and competes with LC20 for phosphorylation. Inhibition of MLCP activity enhances contraction, as mentioned, by inducing Ca2 + sensitization of the contractile apparatus.105
Rho kinase has thus emerged as an important part of the contraction cascade.106 In addition to its role in enhancing contraction, such as in response to Ang II, it is a major regulator of relaxation. Its activator, the small-molecular-weight GTPase RhoA, is a target of NO, which by activating protein kinase G (PKG), inactivates Rho, thus indirectly inhibiting Rho kinase, increasing MLCP activity, and inhibiting contraction.
It is noteworthy that paracrine factors such as NO secreted by neighboring ECs represent the major mechanism of vasorelaxation. Shear stress forces and hormones such as acetylcholine or bradykinin stimulate ECs to secrete NO, which in turn initiates VSMC relaxation.3,107,108 Nitric oxide induces relaxation of smooth muscle potentially via a number of pathways, the most important of which depend on its ability to release cyclic guanosine monophosphate (cGMP). It can directly (via S-nitrosylation of cysteine residues) or indirectly (through PKG) activate BK channels,109 thus causing membrane hyperpolarization and reducing influx through L-type Ca2 + channels. In addition, PKG phosphorylates IP3 receptor-associated PKG-I substrate (IRAG), which inhibits Ca2 + release from IP3 receptors. Nitric oxide also increases Ca2 + uptake via S-glutathionylation of SERCA and decreases the Ca2 + sensitivity of contractile proteins. This pathway is perturbed in diabetic animal models, in which high levels of ROS derived from NADPH oxidase 4 irreversibly oxidize SERCA, rendering it insensitive to NO.110 In addition to regulating Ca2 + levels, NO-mediated activation of PKG can phosphorylate PP1c and/or MYPT1 to block vasoconstrictor-mediated inhibition of MLCP.
Other relaxing factors secreted by endothelial cells include hydrogen peroxide, prostaglandins, and epoxyeicosatrienoic acids (EETs). In addition, perivascular adventitial adipocytes (PVAs) have also been shown to secrete factors that influence contractility (reviewed in111). These cytokines, collectively known as adipokines, are both vasoactive and pro- and antiinflammatory, and include cytokines TNF-α, IL-6, chemokines (IL-8 and monocyte chemoattractant protein [MCP-1]) and hormones (leptin, resistin, and adiponectin).111,112
Proliferation
Vascular smooth muscle cell proliferation is important in early vascular development and in repair mechanisms in response to injury. However, excessive VSMC proliferation contributes to pathology, not only in vascular proliferative diseases such as atherosclerosis but also, ironically, as a consequence of the intervention procedures used to treat these occlusive atherosclerotic diseases and their complications, including postangioplasty restenosis, vein bypass graft failure, and transplant failure.113
Vascular smooth muscle cell proliferation can be regulated by myriad soluble and insoluble factors that activate a variety of intracellular signaling pathways such as MAPK or Janus kinase/signal transducers, tyrosine phosphorylation, and mitogen-activated proteins.114,115 Regardless of the initial proliferative stimulus, these signaling pathways ultimately converge onto the cell cycle116 (Fig. 3-7). The four distinct phases of the cell cycle are: (1) Gap 1 (G1) in which factors necessary for DNA replication are assembled; (2) DNA replication or S phase; (3) Gap 2 (G2) in preparation for mitosis; and (4) mitosis or M phase. Restriction points in the cell cycle exist at transitions between G1/S and G2/M. Progression through the cell cycle phases is regulated by cyclin-dependent kinases (CDKs) and their regulatory cyclin subunits. Cyclins D/E and CDK2, 4, and 5 control G1, cyclin A and CDK2 control the S phase along with the DNA polymerase cofactor PCNA, and cyclins A/B and CDK1 control the M phase. Cyclin-dependent kinases such as p27KIP1 and p21CIP1 bind to and inhibit the activation of cyclin-CDK complexes (see Fig. 3-7). Activities of these enzymes depend upon phosphorylation status of CDKs, levels of expression of cyclins, and nuclear translocation of cyclin-CDK complexes. One regulatory protein is survivin, which competitively interacts with the CDK4/p16INK4a complex to form a CDK4/survivin complex, thus inducing CDK2/cyclin E activation and S-phase entry and cell cycle progression.117 Transcription factors that transactivate CDKs and CDKIs also mediate cell cycle progression. It is known that p53, GAX, and GATA-6 induce p21CIP1 expression, leading to G1 phase arrest, and E2F transcription factors control the G1/S transition regulated by the retinoblastoma protein Rb, the product of the rb tumor suppressor gene. Rb exerts its negative regulation on the cell cycle by binding to E2F transcription factors, rendering them ineffective as transcription factors. When the Rb/E2F complex is phosphorylated by CDKs in early G1, the complex is dissociated, leaving E2F available to activate genes required for S-phase DNA synthesis.116 It is worth noting that the HDAC inhibitor trichostatin A blocks proliferation by induction of the cell cycle inhibitor p21CIP1 and suppression of Rb protein phosphorylation, leading to subsequent cell cycle arrest at the G1/S phase.117
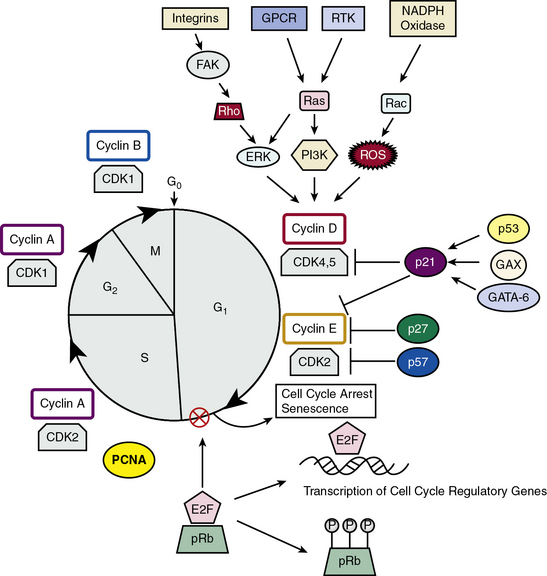
Figure 3-7 Model for cell cycle regulation in Vascular smooth muscle cells (VSMCs).
(Adapted from Fuster JJ, Fernandez P, Gonzalez-Navarro H, et al: Control of cell proliferation in atherosclerosis: insights from animal models and human studies. Cardiovasc Res 86:254–264, 2010; and Dzau VJ, Braun-Dullaeus RC, Sedding DG: Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med 8:1249–1256, 2002.)
In addition to cell cycle regulatory proteins, telomerase activity is required for VSMC proliferation. Telomeres are noncoding DNA TTAGGG repeat sequences at the ends of chromosomes that cap and stabilize chromosomes against degradation, recombination, or fusion.118 Associated with telomeric DNA are protein complexes, including telomerase, that synthesize new telomeric DNA in cells with high proliferative potential. Telomerase consists of an RNA component and two protein components, one of which is telomerase reverse transcriptase (TERT), the catalytic component and limiting factor for telomerase activation. When telomerase expression is low, telomere attrition with each mitotic cycle results in chromosome shortening and instability, replicative senescence, and growth arrest. In VSMCs, posttranslational phosphorylation of TERT is linked to telomerase activation, and levels of telomerase expression and activity correlate with proliferation.118 Importantly, telomerase activation and telomere maintenance have been associated with excessive VSMC proliferation in both animal and human vascular injury and disease;118 disruption of telomerase activity reduces this proliferative response.
Growth of VSMC is initiated by exposure of the cells to pro-proliferative signals. Classical growth factors activate RTKs, either directly or via GPCR-mediated transactivation.116,117 Growth factors in VSMCs binding to RTKs include PDGF, bFGF, IGF-1, TGF-β, EGF, and hypoxia-inducible factor (HIF), and mitogens that activate GPCRs include hormones such as Ang II,15 endothelin, or oxidized LDL. Activation of these receptors stimulates sequential signaling cascades mediated by Ras, p70S6K, Rac/NADPH/ROS, PI3K/Akt, MEK/ERK, or MAPKK/p38MAPK, which induce cyclin D1 expression.115 Src homology 2–containing protein tyrosine phosphatase 2 (SHP2), a member of the non-receptor protein tyrosine phosphatase family, dephosphorylates tyrosine residues on target proteins in response to growth factors, hormones, and cytokines.119 In VSMCs, SHP2 is a positive mediator of IGF-1- and LPA-induced MAPK signaling pathways; SHP2 has negative effects on EGF- and Ang II-induced Akt signaling, implicating SHP2 in modulating cell cycle progression, growth, and migration.
An important integration point in growth factor signaling is mTOR (mammalian target of rapamycin), which regulates protein synthesis, cell cycle progression, and proliferation.117 Mammalian target of rapamycin is a protein kinase that regulates translation initiation through effectors p70S6K and eIF4E, leading to protein synthesis necessary for cell division. Rapamycin, an immunosuppressive macrolide antibiotic, inhibits mTOR downstream signaling cascades, with reductions in protein synthesis leading to cell cycle arrest.116 In VSMCs, rapamycin inhibits the mTOR/p70S6K signaling axis, promotes a VSMC differentiated, contractile phenotype by regulating transcription of contractile proteins, and induces expression of the antiproliferative CDKIs p21CIP and p27KIP to inhibit cell cycle progression.117 Use of rapamycin (sirolimus)-coated coronary stents is highly effective in reducing the postangioplasty restenosis rate in interventional cardiology.120
Ion channels for Ca2 +, Mg2 +, and K+ are also activated by growth factors and mediate proliferation. Transient increases in Ca2 + concentration, together with subsequent Ca2 + binding to its intracellular receptor CaM, are universally required for proliferation.121 The mechanism for the Ca2 + sensitivity of this G1-to-S transition involves the Ca2 +-dependent binding of CaM to cyclin E and activation of CDK2 to promote G1/S transition and VSMC proliferation (reviewed in122,123). Elevated levels of Mg2 + increase expression of cyclin D1 and CDK4 and decrease activation of p21CIP1 and p27KIP1 through an ERK1/2-dependent, p38 MAPK-independent pathway.124 Changes in VSMC K+ channel expression profiles and activity are linked to cell cycle progression, implicating these ion channels as “internal timers” of VSMC cell division.125 Growth factor–induced release of Ca2 + from intracellular Ca2 + storage organelle activates and up-regulates intermediate-conductance Ca2 +-activated K+ (IKCa)-type K+ channels, the predominant Ca2 +-sensitive K+ channel in proliferating VSMCs.126 In addition, voltage-gated K+ channels KV1.3127 and KV3.4128 are up-regulated in proliferating VSMCs. Blockade of these Ca2 +-activated and voltage-gated K+ channels inhibits proliferation and attenuates vascular disease/injury–induced remodeling in rodents.129
Signals from insoluble ECM-activated integrins and from soluble growth factor mitogens converge and jointly regulate upstream cytoplasmic signaling networks to mediate expression of cyclin D1 and cyclin E and associated CDK4/6 and CDK2 in the G1 phase, the part of the cell cycle most affected by extracellular stimuli.130 In addition, joint RTK/integrin complex signaling networks impact G1 phase regulation by inhibiting p21CIP1 and p27KIP1, resulting in Rb phosphorylation and induction of E2F-dependent genes, with progression to autonomous stages of the cell cycle (S, G2, and M) that are independent of external stimuli.
As noted previously, Notch proteins are also important regulators of VSMC proliferation (reviewed in22). Notch4/HRT-induced repression of p27KIP1 and Notch3/HRT1-induced repression of p21CIP1, as well as up-regulation of Akt signaling, an anti-apoptosis pathway, result in promotion of VSMC proliferation. Furthermore, Notch1 is critical in mediating neointimal formation and remodeling after vascular injury.
Peroxisome proliferator-activated receptors (PPARs), nuclear hormone receptors with regulatory roles in lipid and glucose metabolism, are beneficial in VSMCs by targeting genes for cell cycle progression, cellular senescence, and apoptosis to inhibit proliferation and neointimal formation in atherosclerosis and postangioplasty restenosis (reviewed in131). Activation of PPARα suppresses G1-to-S progression by inducing expression of p16INK4a (a CDKI), thereby inhibiting phosphorylation of Rb.132 This antiproliferative effect is mediated by repression of telomerase activity by inhibiting E2F binding sites in the TERT promoter.133 Another PPAR isotype, PPARγ, also blocks G1-to-S cell cycle transition by preventing degradation of p27KIP1, resulting in inhibition of pRb phosphorylation and suppression of E2F-regulated genes responsible for DNA replication.131 Similar to PPARα, PPARγ also inhibits telomerase activity in VSMCs by inhibition of early response gene Ets-1-dependent transactivation of the TERT promoter.131 Thiazolidinediones (TZD), PPARγ agonists used clinically in the treatment of type 2 diabetes mellitus, decrease VSMC proliferation and prevent atherosclerosis in murine models of the disease.131
Cyclic adenosine 3′,5′-monophosphate (cAMP) and cGMP are second messengers in myriad signal transduction pathways.134 In VSMCs, cAMP serves as an antagonist both to mitogenic signaling pathways (by inhibiting MAPK, PI3 kinase, and mTOR signaling axes) and to cell cycle progression (by down-regulating cyclins or up-regulating CDKI p27KIP1). An additional antiproliferative effect is due to down-regulation of S-phase kinase-associated protein-2 (Skp2) mediated by inhibition of FAK phosphorylation and adhesion-dependent signaling. Skp2 is a ubiquitin ligase subunit that targets p27KIP1 for proteasomal degradation, thus promoting VSMC proliferation.135
A more recently appreciated pathway that controls VSMC growth involves miRNAs. The potential involvement of these molecules was first noted in balloon-injured rat carotid arteries, where several miRNAs, including miR-21, are up-regulated compared with control arteries (reviewed in136). Cell culture models show that miR-21 is a pro-proliferative and anti-apoptotic regulator of VSMCs, with target genes phosphatase and tensin homology deleted from chromosome 10 (PTEN), programmed cell death 4 (PDCD4), and Bcl-2. miR-21 has opposite effects on PTEN and Bcl-2: overexpression down-regulates PTEN and up-regulates Bcl-2. PTEN modulates VSMCs through PI3K and Akt signaling pathways, while Bcl-2 mediates its downstream signaling through AP-1.
Finally, cell-cell junctions, as described above for cadherins and gap junction connexins, and cell-matrix contacts can greatly influence VSMC proliferation (reviewed in115). Normally, resident VSMCs, surrounded by and binding to polymerized collagen type 1 fibrils through α2β1 integrins, exhibit low proliferation indices, are arrested in the G1 phase of the cell cycle, and are refractory to mitogenic stimuli. In this quiescent state, levels of cell cycle regulatory proteins are modulated to inhibit the G1/S transition: cyclin E and CDK2 phosphorylation is inhibited, while CDKIs are up-regulated and suppress cyclin E/CDK2 activity. Additionally, p70S6K, a potent stimulator of mitogenesis and a regulator of p27KIP1, is suppressed. In contrast, VSMCs on monomeric collagen matrices are responsive to growth factor signals which result in increased cyclin E–associated kinase activity and cell proliferation. These differential responses of VSMCs to structurally distinct forms of collagen type 1 are reflected in the differential regulation of cell cycle proteins and the differential response to mitogenic stimuli. Therefore, perturbations or degradation of the collagen matrix, as found in sites of monomeric collagen in vascular lesions, result in altered VSMC proliferation, response to mitogens, and neointimal formation.76
Migration
Smooth muscle migration is an essential element of wound repair, but unchecked migration and proliferation can contribute to neointimal thickening and development of atherosclerotic plaques. A number of promigratory and antimigratory molecules regulate VSMC migration, including peptide growth factors, ECM components, and cytokines.137 The extent of migration is also influenced by physical factors such as shear stress, stretch, and matrix stiffness. PDGF-BB, bFGF, and S1P are among the most potent pro-migratory stimuli in the vascular system. Intracellular signaling cascades initiated by these growth factors act in concert with those activated by integrin receptor interaction with matrix to mediate the migratory response. Matrix surrounding the migrating cell must be degraded by MMPs to allow a pathway into which the cell can protrude. Important promigratory matrix components include collagen I and IV, osteopontin, and laminin. Matrix interactions can also be antimigratory, as with the formation of stable focal adhesions, activation of TIMPs, and heparin.
When a cell begins to migrate, a number of coordinated events must take place in a cyclic fashion138 (Fig. 3-8). Signaling mechanisms that regulate migration have mostly been studied in fibroblasts, but recently many have been confirmed in VSMCs. Migration requires specialized signaling domains at the front and rear of the cell. When confronted with a migratory stimulus, the cell senses the gradient and establishes polarity. Plasma membrane in the form of lamellipodia is then extended in the direction of movement. This process is controlled by reorganization of the actin cytoskeleton just under the protruding membrane. New focal complexes are formed in the lamellipodia via cytoskeletal remodeling and integrin interaction with the matrix. The cell body begins to contract, powered by engagement and phosphorylation of myosin II, and focal adhesions in the rear of the cell become detached, leading to retraction of the “tail” of the cell. Finally, adhesion receptors are recycled by endocytosis and vesicular transport. Successful migration is thus dependent on proper temporal and spatial activation of many molecules, most of which are related to cytoskeletal elements.
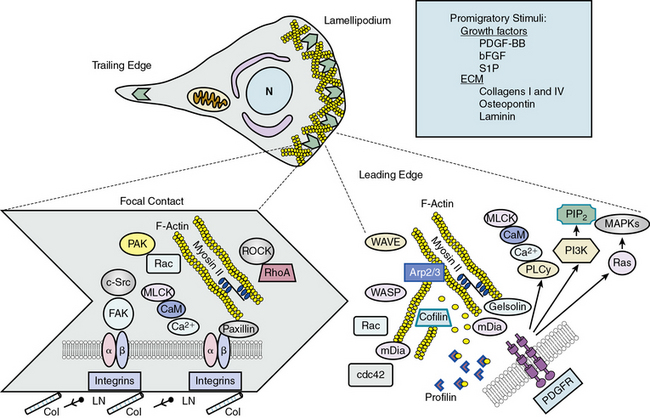
Much is known or inferred about the signaling mechanisms activated by PDGF in migrating cells.137 When PDGF-BB binds to PDGFRs, receptor autophosphorylation creates binding sites for phospholipase Cγ, which mobilizes calcium; PI3K, which forms the membrane-targeting lipid PIP2; and Ras, which activates MAPKs. Nucleation of new actin filaments at the leading edge is initiated by binding of nucleation promoting factors verprolin-homologous protein (WAVE) and Wiskott-Aldrich’s syndrome protein (WASP) to actin-related protein ARP2/3; phosphorylation of the actin binding coronin; and dissociation of actin capping proteins, many of which are regulated by PIP2. Extension of new actin filaments is promoted by formins (mDia1 and mDia2), which act on the plus end of actin filaments in coordination with profilin. Regulation of mDia proteins is largely via conformational changes induced by the small G-proteins RhoA and cdc42. Profilin increases nucleotide exchange on G-actin monomers, thus enhancing actin polymerization. Severing of existing actin filaments is a consequence of activation of gelsolin and cofilin, which limit filament length and initiate turnover of existing filaments. Rac also regulates actin reorganization in the lamellipodium, perhaps by activation of p21-activated kinase (PAK)-mediated phosphorylation of actin binding proteins. The result of these complicated, coordinated events is protrusion of lamellipodia in the direction of the detected migratory stimulus (see Fig. 3-8).
Once lamellipodial protrusion has occurred, it is necessary for the cell to create new contacts with the matrix and dissolve ones no longer needed. These nascent focal contacts provide traction for eventual contraction of the cell body and propulsion of the cell forward.137 Very little is known about focal adhesion composition in VSMCs, but signaling at focal adhesions is coordinated by integrin interaction with the matrix, integrin clustering, activation of a series of protein tyrosine kinases including integrin-linked kinase (ILK), FAK and Src, and interaction with the cortical F-actin cytoskeleton. Phosphorylation of focal adhesion components including FAK and paxillin occurs during VSMC migration, as does turnover of focal adhesion proteins by membrane-type metalloproteinases. Regulation of focal adhesion turnover is also intimately related to the microtubular network.
The final major event in cell migration is contraction of the cell body. Similar to contraction in differentiated cells, cell body contraction is initiated through calcium-mediated activation of MLCK and MLC phosphorylation following matrix interaction. RhoA and Rho kinase may also play a role because pharmacological inhibition of Rho kinase blocks migration of VSMCs.139 Current theory suggests that myosin II generates traction forces on the matrix, and the matrix in turn regulates myosin II activation.137
Inflammation
As noted earlier, VSMCs can assume an inflammatory phenotype that is found primarily in atherosclerotic lesions. These cells are found in the media of the vessel wall and express both markers of differentiation and inflammatory genes such as VCAM-1 and exhibit activated NF-κB signaling.140 One of the primary stimuli for development of this inflammatory phenotype is oxidized LDL, but ECs activated by disturbed flow also contribute to inflammatory changes in VSMC by secreting proinflammatory cytokines.14
Proinflammatory gene expression in VSMC, as in other cell types, is largely a consequence of posttranscriptional regulation of inflammatory gene expression by the stress-activated protein kinase p38MAPK and transcriptional regulation by proinflammatory transcription factors such as NF-κB and STAT1/3. Both of these pathways are activated by ROS, which have been shown to be increased in inflammatory regions of plaques as a result of macrophage infiltration as well as direct stimulation of VSMCs by cytokines. Stimulation of cytokine receptors activates p38MAPK, which controls proinflammatory protein levels by MAPKAPK-2 mediated phosphorylation of adenylate uridylate–rich elements (AREs) binding proteins such as tristetraprolin (TTP), thus promoting mRNA stability of TNF-α.141 Many other inflammatory gene mRNAs, including MCP-1, IL-1β, IL-8, intercellular adhesion molecule 1 (ICAM-1), and VCAM-1, also contain AREs. It should be noted that ARE binding proteins can both stabilize and destabilize mRNA: HuR protects ARE-containing transcripts from degradation, but AUF1 destabilizes its targets. p38MAPK can also regulate inflammatory protein expression by translational regulation via activation of MAPK signal-integrating kinase-1 (Mnk-1), which phosphorylates the translation initiation factor eIF-4E and enhances its affinity for the mRNA cap.142 Transcriptional regulation of proinflammatory gene expression is largely a consequence of activation of the NF-κB pathway. Commonly, the p65-p50 heterodimer is the transactivating factor that binds to NF-κB-containing elements to increase proinflammatory gene transcription. Regulation of gene expression by STATs is a consequence of the canonical tyrosine kinase receptor activation of JAK, and subsequent phosphorylation of STAT followed by translocation to the nucleus.
Another major environmental factor that contributes to maintenance of the VSMC proinflammatory phenotype is the matrix milieu in which cells exist. In atherosclerotic plaques, VSMCs begin to secrete collagen I and collagen III, but also, as a result of NF-κB activation, express MMP-1, MMP-3, and MMP-9, which degrade collagen fibrils to the monomeric form, thus promoting an inflammatory phenotype, as evidenced by an increase in VCAM-1 expression.75 A similar response is seen to osteopontin, which is also increased in atherosclerosis.143 The effects of these matrix proteins on VSMCs are mediated by binding to specific integrins, most likely α5β1 or αvβ3.14 The nonintegrin matrix receptor CD44, which binds to hyaluronic acid in the matrix, has also been implicated in the transition to the proinflammatory phenotype, as shown by its ability to stimulate VCAM-1 expression.144
Senescence, Apoptosis, and Autophagy
In response to aging and oxidative stress, cells that have accumulated damaged organelles/proteins/DNA due to limitations in DNA repair or antioxidant mechanisms rely on two processes to avoid replication and passing the damage to daughter cells: permanently arresting the cell cycle (senescence), or programmed cell death, including apoptosis (self-killing) or autophagy (self-eating).145
Senescent cells are permanently arrested in the G1 phase of the cell cycle and exhibit specific senescence-associated markers such as β-galactosidase, heterochromatin foci, and accumulation of lipofuscin granules. Unlike quiescent cells, senescent cells are not responsive to growth factors.146 Multiple stresses, including DNA-damaging radiation or chemicals, mitochondrial dysfunction, and oxidant stress, can invoke two types of senescence programs: stress-induced premature senescence (SIPS) and replicative senescence associated with accelerated telomere uncapping or shortening.147 These diverse stimulatory pathways converge onto two effector pathways: the tumor suppressor protein p53 and the Rb pathways; p53 is normally targeted to proteasome-mediated degradation by mouse double minute 2 MDM2). Mitogenic stress or DNA damage suppresses MDM2 activity, resulting in p53-mediated activation of the CDKI p21 and cell cycle arrest.145 In the second pathway, stress or damage activates Rb, which then binds to and inhibits E2F, a transcription factor required for the G1 phase/S phase transition to cell cycle progression (see Fig. 3-7). These two senescence pathways exhibit cross-talk at the level of p53 and can overlap death pathways. Senescent cells release degradative proteases, growth factors, and inflammatory cytokines, which impact on neighboring cells.
In VSMCs, DNA damage caused by ROS (e.g., superoxide, hydrogen peroxide, hydroxyl radicals) incites rapid (within days) SIPS. There are increased levels of ROS in all diseased layers of an atherosclerotic lesion, particularly in the plaque itself,147 and senescent VSMCs have been identified in injured arteries and in the intima of atherosclerotic plaques.148
Many of the changes in senescent VSMCs are reminiscent of changes indicative in age-related vascular disease, implicating cellular senescence in vascular pathologies.148 Therefore, a model for how senescence contributes to vascular disease emerges. Atherogenic stimuli such as Ang II initially stimulate proliferation, followed by mitogen-induced SIPS or replicative senescence via telomere uncapping. Inflammatory cytokine/chemokine release by senescent VSMCs results in ECM degradation. The decreased cellularity and increased inflammation contribute to plaque instability.148
Senescent VSMCs are also implicated in vascular calcification. They exhibit enhanced expression of osteoblastic genes such as alkaline phosphatase (ALP), type 1 collagen, and RUNX-2, while expression of matrix Gla protein (MGP), an anticalcification factor, is down-regulated.149
Apoptosis, the controlled activation of proteases and hydrolases within an intact cell’s plasma membrane boundary so that neighboring cells are not affected and an immune response is not triggered,150 is an important mechanism for blood vessel remodeling during proliferative vascular disease and after therapeutic interventions (e.g., angioplasty/stenting of arteries, vein bypass graft surgery).151 Mitogens such as thrombin or PDGF can induce proliferative episodes in VSMCs within atherosclerotic lesions (reviewed in152). Proliferation is counterbalanced by death-inducing VSMC apoptosis triggered by a variety of proinflammatory mediators, cytokines, oxidized lipids, and free radicals produced by immune cells within the plaque. These proinflammatory mediators activate caspases, components of the extrinsic death receptor pathway (e.g., Fas/CD95 TRAIL [TNF-related apoptosis-inducing ligand]), and/or cause intrinsic mitochondrial dysfunction in VSMCs under the control of Bcl family members (reviewed in153).
Interactions among mitogenic, apoptotic, and survival signals produce a variety of lesion characteristics and determine whether there is a fragile fibrous cap poised for rupture, a lipid-rich necrotic core, or a fibrotic and calcified core (reviewed in152). High percentages of apoptotic VSMCs within atherosclerotic plaques are one of the major causes of plaque rupture due to decreased cellularity in the media and thinning of the fibrous cap. In addition, reduced phagocytotic clearance of apoptotic VSMCs, resulting in necrotic VSMCs, and low levels of VSMC apoptosis over extended periods of hyperlipidemia induce viable VSMC release of IL-6 and MCP-1 to produce chronic inflammation.154 Apoptotic VSMCs also generate thrombin, promoting coagulation.152
Vascular smooth muscle cell apoptosis has also been associated with other lesion characteristics including inflammation, calcification, thrombosis, and aneurysms (reviewed in156). In vivo, VSMC apoptosis causes release of cytokines and MCP-1, recruiting macrophages. Vascular calcification has been associated with inorganic phosphate–induced VSMC apoptosis and subsequent generation of VSMC-derived matrix vesicles that serve as the nidus for calcification (reviewed in156). Statins restore the Gas6-mediated survival pathway and inhibit VSMC calcification by preventing apoptosis.
In addition to apoptosis, autophagy, a survival process by which the cell degrades its own components, such as damaged organelles or long-lived aberrant or aggregated proteins,145 contributes to pathology in atherosclerotic plaques. Ultrastructural analysis of VSMCs in the fibrous cap of advanced plaques reveals characteristics of cells undergoing autophagic degradation.157 Because autophagy is a survival mechanism and not a death pathway, VSMC autophagy in the fibrous cap may function in plaque stability and protection from oxidative stress.157 If oxidative stress damages lysosomal membranes, lysosome/autophagic vacuole fusion is impaired and apoptosis ensues.
Stem/Progenitor Cells
The ability of stem cells to differentiate into a variety of cell types has led to research on the potential efficacy of using pluripotent embryonic stem cells as a source of VSMCs for regenerative cell-based therapies and tissue engineering in injury/disease repair. Research on the role of putative resident adult stem cells in bone marrow and/or unipotent lineage committed VSMC progenitor cells within the circulating blood, vascular wall, or other peripheral tissues in the development of the neointima in atherosclerotic lesions is also ongoing.158–161
Pluripotent embryonic stem cells (ESCs) form embryoid bodies in vitro that contain isolated areas of contractile SMCs induced by endogenous TGF-β.162 Because undifferentiated ESCs have the potential to form teratocarcinomas, the ability to isolate pure populations of differentiated cells is essential for use of ESCs in tissue-engineering applications. An alternative method for tissue regeneration is to reprogram somatic cells to resemble ESCs. Somatic cells can be induced to form pluripotent stem cells (iPS) by addition of defined factors such as Sox2, Oct4, KLF4, and c-myc (reviewed in163).
Multipotent adipose-derived mesenchymal stem cells (MSCs) are candidates for a VSMC source for tissue-engineered blood vessels because these MSCs can be easily obtained from human lipoaspirate, readily expanded in culture, and differentiated into contractile VSMC-like cells in culture media containing both TGF-β and BMP-4.164
Initial hypotheses for the origin of neointimal VSMCs proposed that injury-induced growth factors and ECM proteolysis caused a VSMC phenotypic switch from a quiescent, contractile phenotype to a synthetic type, resulting in proliferation and migration of a small number of clonal or oligoclonal VSMCs from the underlying media into the intima where remodeling led to plaque formation and lumen occlusion. Subsequent evidence suggested that circulating bone marrow–derived SMC progenitor cells may contribute to normal vascular injury repair and formation of the neointima in vascular lesions.2,158 However, the origin of intimal VSMCs from bone marrow–derived progenitor cells in the blood in response to injury or disease has been disputed.165,166 In long-term studies of transplanted bone marrow cells into lethally irradiated mice with wire injury, the bone marrow–derived cells, initially found in high numbers in the neointima, were not stable residents, and the few remaining after 16 weeks did not exhibit definitive VSMC marker proteins calponin and SM MHC. Additionally, the adventitial layer of the wall serves as a niche for wall-derived MSCs and VSMC progenitor cells,161 including resident stem cell antigen-1 (Sca-1)-positive cells, maintained in the adventitia by Shh signaling and myocardin transcriptional corepressors, which are capable of differentiating into VSMCs.167 This population of Sca1 + progenitor cells in the arterial adventitia could contribute to vessel wall remodeling in injury/disease. The prevailing hypothesis is that neointimal VSMCs originate from the injured media and also from local resident progenitors in the adventitia.166
The nature of VSMC phenotype plasticity, exemplified in distinct genetic expression patterns of marker genes and thus in differential functions, complicates the definition and identification of VSMCs derived from bone marrow resident and circulating stem/progenitor cells.168 The safe and effective use of regenerative VSMCs in translational clinical therapy for cardiovascular disease awaits further methodologies for identifying, producing, and isolating cells that will differentiate into VSMCs.
1 Wagenseil J.E., Mecham R.P. Vascular extracellular matrix and arterial mechanics. Physiol Rev. 2009;89(3):957–989.
2 Owens G.K., Kumar M.S., Wamhoff B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801.
3 Beamish J.A., He P., Kottke-Marchant K., et al. Molecular regulation of contractile smooth muscle cell phenotype: implications for vascular tissue engineering. Tissue Eng Part B Rev. 2010;16(5):467–491.
4 Herbert S.P., Huisken J., Kim T.N., et al. Arterial-venous segregation by selective cell sprouting: an alternative mode of blood vessel formation. Science. 2009;326(5950):294–298.
5 Swift M.R., Weinstein B.M. Arterial-venous specification during development. Circ Res. 2009;104(5):576–588.
6 Anderson L.M., Gibbons G.H. Notch: a mastermind of vascular morphogenesis. J Clin Invest. 2007;117(2):299–302.
7 Jain R.K. Molecular regulation of vessel maturation. Nat Med. 2003;9(6):685–693.
8 Majesky M.W. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27(6):1248–1258.
9 Hao H., Gabbiani G., Bochaton-Piallat M.-L. Arterial smooth muscle cell heterogeneity: implications for atherosclerosis and restenosis development. Arterioscler Thromb Vasc Biol. 2003;23(9):1510–1520.
10 Larsson E., McLean S.E., Mecham R.P., et al. Do two mutually exclusive gene modules define the phenotypic diversity of mammalian smooth muscle? Mol Genet Genomics. 2008;280(2):127–137.
11 Moiseeva E.P. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res. 2001;52(3):372–386.
12 Rensen S.S., Doevendans P.A., van Eys G.J. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007;15(3):100–108.
13 Rzucidlo E.M. Signaling pathways regulating vascular smooth muscle cell differentiation. Vascular. 2009;17(Suppl 1):S15–S20.
14 Orr A.W., Hastings N.E., Blackman B.R., et al. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res. 2010;47(2):168–180.
15 Berk B.C. Vascular smooth muscle growth: autocrine growth mechanisms. Physiol Rev. 2001;81(3):999–1030.
16 Griendling K., Harrison D., Alexander R. Biology of the vessel wall. In: Fuster V., Walsh R., O’Rourke R., Poole-Wilson P. Hurst’s the heart. ed 12. New York: McGraw Hill; 2008:135–154.
17 Garrido A.M., Griendling K.K. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol. 2009;302(2):148–158.
18 Pardali E., Goumans M.-J., ten Dijke P. Signaling by members of the TGF-β family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20(9):556–567.
19 Mehta P.K., Griendling K.K. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292(1):C82–C97.
20 Yoshida T., Owens G.K. Molecular determinants of vascular smooth muscle cell diversity. Circ Res. 2005;96(3):280–291.
21 High F.A., Zhang M., Proweller A., et al. An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Invest. 2007;117(2):353–363.
22 Gridley T. Notch signaling in the vasculature. Curr Top Dev Biol. 2010;92:277–309.
23 Tang Y., Urs S., Liaw L. Hairy-related transcription factors inhibit Notch-induced smooth muscle α-actin expression by interfering with Notch intracellular domain/CBF-1 complex interaction with the CBF-1-binding site. Circ Res. 2008;102(6):661–668.
24 Tang Y., Urs S., Boucher J., et al. Notch and transforming growth factor-beta (TGFβ) signaling pathways cooperatively regulate vascular smooth muscle cell differentiation. J Biol Chem. 2010;285(23):17556–17563.
25 Morrow D., Cullen J.P., Liu W., et al. Sonic hedgehog induces Notch target gene expression in vascular smooth muscle cells via VEGF-A. Arterioscler Thromb Vasc Biol. 2009;29(7):1112–1118.
26 Jin S., Hansson E.M., Tikka S., et al. Notch signaling regulates platelet-derived growth factor receptor-β expression in vascular smooth muscle cells. Circ Res. 2008;102(12):1483–1491.
27 Wang D.-Z., Olson E.N. Control of smooth muscle development by the myocardin family of transcriptional coactivators. Curr Opin Genet Dev. 2004;14(5):558–566.
28 Miano J.M., Long X., Fujiwara K. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol. 2007;292(1):C70–C81.
29 Parmacek M.S. Myocardin-related transcription factors: critical coactivators regulating cardiovascular development and adaptation. Circ Res. 2007;100(5):633–644.
30 Pipes G.C., Creemers E.E., Olson E.N. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20(12):1545–1556.
31 Davis C.A., Haberland M., Arnold M.A., et al. PRISM/PRDM6, a transcriptional repressor that promotes the proliferative gene program in smooth muscle cells. Mol Cell Biol. 2006;26(7):2626–2636.
32 Haldar S.M., Ibrahim O.A., Jain M.K. Kruppel-like factors (KLFs) in muscle biology. J Mol Cell Cardiol. 2007;43(1):1–10.
33 Zheng B., Han M., Wen J.K. Role of Krüppel-like factor 4 in phenotypic switching and proliferation of vascular smooth muscle cells. IUBMB Life. 2010;62(2):132–139.
34 Miano J.M. Deck of CArGs. Circ Res. 2008;103(1):13–15.
35 Olson E.N., Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11(5):353–365.
36 Liu N., Olson E.N. Coactivator control of cardiovascular growth and remodeling. Curr Opin Cell Biol. 2006;18(6):715–722.
37 Posern G., Treisman R. Actin’ together: serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol. 2006;16(11):588–596.
38 Parmacek M.S. Myocardin: dominant driver of the smooth muscle cell contractile phenotype. Arterioscler Thromb Vasc Biol. 2008;28(8):1416–1417.
39 Dong J.-T., Chen C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell Mol Life Sci. 2009;66(16):2691–2706.
40 Kawai-Kowase K., Owens G.K. Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells. Am J Physiol Cell Physiol. 2007;292(1):C59–C69.
41 Liu Y. Wen J-K, Dong L-H, et al: Kruppel-like factor (KLF) 5 mediates cyclin D1 expression and cell proliferation via interaction with c-Jun in Ang II-induced VSMCs. Acta Pharmacol Sin. 2009;31(1):10–18.
42 Suzuki T., Sawaki D., Aizawa K., et al. Kruppel-like factor 5 shows proliferation-specific roles in vascular remodeling, direct stimulation of cell growth, and inhibition of apoptosis. J Biol Chem. 2009;284(14):9549–9557.
43 Nagai R., Suzuki T., Aizawa K., et al. Significance of the transcription factor KLF5 in cardiovascular remodeling. J Thromb Haemost. 2005;3(8):1569–1576.
44 Liu N., Olson E.N. MicroRNA regulatory networks in cardiovascular development. Dev Cell. 2010;18(4):510–525.
45 Eulalio A., Huntzinger E., Izaurralde E. Getting to the root of miRNA-mediated gene silencing. Cell. 2008;132(1):9–14.
46 Djuranovic S., Nahvi A., Green R. A parsimonious model for gene regulation by miRNAs. Science. 2011;331(6017):550–553.
47 Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233.
48 Cheng Y., Liu X., Yang J., et al. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105(2):158–166.
49 Cordes K.R., Sheehy N.T., White M.P., et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460(7256):705–710.
50 Xin M., Small E.M., Sutherland L.B., et al. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23(18):2166–2178.
51 Boettger T., Beetz N., Kostin S., et al. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest. 2009;119(9):2634–2647.
52 Song Z., Li G. Role of specific microRNAs in regulation of vascular smooth muscle cell differentiation and the response to injury. J Cardiovasc Transl Res. 2010;3(3):246–250.
53 Liu X., Cheng Y., Zhang S., et al. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009;104(4):476–487.
54 Davis B.N., Hilyard A.C., Nguyen P.H., et al. Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J Biol Chem. 2009;284(6):3728–3738.
55 McDonald O.G., Owens G.K. Programming smooth muscle plasticity with chromatin dynamics. Circ Res. 2007;100(10):1428–1441.
56 McDonald O.G., Wamhoff B.R., Hoofnagle M.H., et al. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest. 2006;116(1):36–48.
57 Mohammad H.P., Baylin S.B. Linking cell signaling and the epigenetic machinery. Nat Biotech. 2010;28(10):1033–1038.
58 Qiu P., Ritchie R.P., Gong X.Q., et al. Dynamic changes in chromatin acetylation and the expression of histone acetyltransferases and histone deacetylases regulate the SM22α transcription in response to Smad3-mediated TGFβ1 signaling. Biochem Biophys Res Commun. 2006;348(2):351–358.
59 Villeneuve L.M., Reddy M.A., Lanting L.L., et al. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc Natl Acad Sci U S A. 2008;105(26):9047–9052.
60 Ceriello A., Ihnat M.A., Thorpe J.E. The “metabolic memory”: is more than just tight glucose control necessary to prevent diabetic complications? J Clin Endocrinol Metab. 2009;94(2):410–415.
61 Paik J.H., Skoura A., Chae S.S., et al. Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev. 2004;18(19):2392–2403.
62 George S.J., Dwivedi A. MMPs, cadherins, and cell proliferation. Trends Cardiovasc Med. 2004;14(3):100–105.
63 Figueroa X.F., Duling B.R. Gap junctions in the control of vascular function. Antioxid Redox Signal. 2009;11(2):251–266.
64 Brisset A.C., Isakson B.E., Kwak B.R. Connexins in vascular physiology and pathology. Antioxid Redox Signal. 2009;11(2):267–282.
65 Johnstone S., Isakson B., Locke D. Biological and biophysical properties of vascular connexin channels. Int Rev Cell Mol Biol. 2009;278:69–118.
66 Wang L., Zheng J., Du Y., et al. Cartilage oligomeric matrix protein maintains the contractile phenotype of vascular smooth muscle cells by interacting with α7β1 integrin. Circ Res. 2010;106(3):514–525.
67 Tkachenko E., Rhodes J.M., Simons M. Syndecans: new kids on the signaling block. Circ Res. 2005;96(5):488–500.
68 Alexopoulou A.N., Multhaupt H.A.B., Couchman J.R. Syndecans in wound healing, inflammation and vascular biology. Int J Biochem Cell Biol. 2007;39(3):505–528.
69 Fukai N., Kenagy R.D., Chen L., et al. Syndecan-1: an inhibitor of arterial smooth muscle cell growth and intimal hyperplasia. Arterioscler Thromb Vasc Biol. 2009;29(9):1356–1362.
70 Kelleher C.M., McLean S.E., Mecham R.P. Vascular extracellular matrix and aortic development. Curr Top Dev Biol. 2004;62:153–188.
71 Hynes R.O. The extracellular matrix: not just pretty fibrils. Science. 2009;326(5957):1216–1219.
72 Järveläinen H., Sainio A., Koulu M., et al. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol Rev. 2009;61(2):198–223.
73 Mao Y., Schwarzbauer J.E. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005;24(6):389–399.
74 Singh P., Carraher C., Schwarzbauer J.E. Assembly of fibronectin extracellular matrix. Annu Rev Cell Dev Biol. 2010;26:397–419.
75 Orr A.W., Lee M.Y., Lemmon J.A., et al. Molecular mechanisms of collagen isotype-specific modulation of smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2009;29(2):225–231.
76 Koyama H., Raines E.W., Bornfeldt K.E., et al. Fibrillar collagen inhibits arterial smooth muscle proliferation through regulation of Cdk2 inhibitors. Cell. 1996;87(6):1069–1078.
77 Kielty C.M., Sherratt M.J., Shuttleworth C.A. Elastic fibres. J Cell Sci. 2002;115(14):2817–2828.
78 Kielty C.M. Elastic fibres in health and disease. Expert Rev Mol Med. 2006;8(19):1–23.
79 Karnik S.K., Brooke B.S., Bayes-Genis A., et al. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130(2):411–423.
80 Ramirez F., Rifkin D.B. Extracellular microfibrils: contextual platforms for TGFβ and BMP signaling. Curr Opin Cell Biol. 2009;21(5):616–622.
81 Ramirez F., Sakai L. Biogenesis and function of fibrillin assemblies. Cell Tissue Res. 2010;339(1):71–82.
82 Yanagisawa H., Davis E.C. Unraveling the mechanism of elastic fiber assembly: the roles of short fibulins. Int J Biochem Cell Biol. 2010;42(7):1084–1093.
83 Chapman S.L., Sicot F.-X., Davis E.C., et al. Fibulin-2 and fibulin-5 cooperatively function to form the internal elastic lamina and protect from vascular injury. Arterioscler Thromb Vasc Biol. 2010;30(1):68–74.
84 Zacchigna L., Vecchione C., Notte A., et al. Emilin1 links TGF-β maturation to blood pressure homeostasis. Cell. 2006;124(5):929–942.
85 Zanetti M., Braghetta P., Sabatelli P., et al. EMILIN-1 deficiency induces elastogenesis and vascular cell defects. Mol Cell Biol. 2004;24(2):638–650.
86 Couchman J.R. Transmembrane signaling proteoglycans. Annu Rev Cell Dev Biol. 2010;26:89–114.
87 Wight T.N. Arterial remodeling in vascular disease: a key role for hyaluronan and versican. Front Biosci. 2008;13:4933–4937.
88 Ballinger M.L., Ivey M.E., Osman N., et al. Endothelin-1 activates ETA receptors on human vascular smooth muscle cells to yield proteoglycans with increased binding to LDL. Atherosclerosis. 2009;205(2):451–457.
89 Chen C.-C., Lau L.F. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol. 2009;41(4):771–783.
90 Matsumae H., Yoshida Y., Ono K., et al. CCN1 knockdown suppresses neointimal hyperplasia in a rat artery balloon injury model. Arterioscler Thromb Vasc Biol. 2008;28(6):1077–1083.
91 Shimoyama T., Hiraoka S., Takemoto M., et al. CCN3 inhibits neointimal hyperplasia through modulation of smooth muscle cell growth and migration. Arterioscler Thromb Vasc Biol. 2010;30(4):675–682.
92 Newby A.C. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev. 2005;85(1):1–31.
93 Newby A.C. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc Res. 2006;69(3):614–624.
94 Nagase H., Visse R., Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69(3):562–573.
95 Raffetto J.D., Khalil R.A. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008;75(2):346–359.
96 Newby A.C. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc Med. 2007;17(8):253–258.
97 Assoian R.K., Klein E.A. Growth control by intracellular tension and extracellular stiffness. Trends Cell Biol. 2008;18(7):347–352.
98 Klein E.A., Yin L., Kothapalli D., et al. Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr Biol. 2009;19(18):1511–1518.
99 Watanabe H., Murakami M., Ohba T., et al. TRP channel and cardiovascular disease. Pharmacol Ther. 2008;118(3):337–351.
100 Akata T. Cellular and molecular mechanisms regulating vascular tone. Part 1: basic mechanisms controlling cytosolic Ca2 + concentration and the Ca2 +-dependent regulation of vascular tone. J Anesth. 2007;21(2):220–231.
101 Kim H.R., Appel S., Vetterkind S., et al. Smooth muscle signalling pathways in health and disease. J Cell Mol Med. 2008;12(6A):2165–2180.
102 Essin K., Gollasch M. Role of ryanodine receptor subtypes in initiation and formation of calcium sparks in arterial smooth muscle: comparison with striated muscle. 2009. J Biomed Biotechnol
103 Sathish V., Thompson M.A., Bailey J.P., et al. Effect of proinflammatory cytokines on regulation of sarcoplasmic reticulum Ca2 + reuptake in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L26–L34.
104 Trebak M., Ginnan R., Singer H.A., et al. Interplay between calcium and reactive oxygen/nitrogen species: an essential paradigm for vascular smooth muscle signaling. Antioxid Redox Signal. 2010;12(5):657–674.
105 Berk B. Vascular smooth muscle. In: Creager M., Dzau V., Loscalzo J. Vascular medicine: a companion to Braunwald’s heart disease. ed 1. Philadelphia: Saunders-Elsevier; 2006:17–30.
106 Hilgers R.H.P., Webb R.C. Molecular aspects of arterial smooth muscle contraction: focus on Rho. Exp Biol Med. 2005;230(11):829–835.
107 Masaki T. Historical review: endothelin. Trends Pharmacol Sci. 2004;25(4):219–224.
108 Tsutsui M., Shimokawa H., Otsuji Y., et al. Pathophysiological relevance of NO signaling in the cardiovascular system: novel insight from mice lacking all NO synthases. Pharmacol Ther. 2010;128(3):499–508.
109 Gao Y., Yang Y., Guan Q., et al. IL-1beta modulate the Ca(2 +)-activated big-conductance K channels (BK) via reactive oxygen species in cultured rat aorta smooth muscle cells. Mol Cell Biochem. 2010;338(1–2):59–68.
110 Tong X., Hou X., Jourd’heuil D., et al. Upregulation of Nox4 by TGFβ1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ Res. 2010;107(8):975–983.
111 Rajsheker S., Manka D., Blomkalns A.L., et al. Crosstalk between perivascular adipose tissue and blood vessels. Curr Opin Pharmacol. 2010;10(2):191–196.
112 Zhang H., Cui J., Zhang C. Emerging role of adipokines as mediators in atherosclerosis. World J Cardiol. 2010;2(11):370–376.
113 Fuster J.J., Fernandez P., Gonzalez-Navarro H., et al. Control of cell proliferation in atherosclerosis: insights from animal models and human studies. Cardiovasc Res. 2010;86(2):254–264.
114 Griendling K.K., Ushio-Fukai M., Lassegue B., et al. Angiotensin II signaling in vascular smooth muscle: new concepts. Hypertension. 1997;29(1):366–370.
115 Schwartz M.A., Assoian R.K. Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J Cell Sci. 2001;114(14):2553–2560.
116 Dzau V.J., Braun-Dullaeus R.C., Sedding D.G. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002;8(11):1249–1256.
117 Marsboom G., Archer S.L. Pathways of proliferation: new targets to inhibit the growth of vascular smooth muscle cells. Circ Res. 2008;103(10):1047–1049.
118 Fuster J.J., Andres V. Telomere biology and cardiovascular disease. Circ Res. 2006;99(11):1167–1180.
119 Kandadi M.R., Stratton M.S., Ren J. The role of Src homology 2 containing protein tyrosine phosphatase 2 in vascular smooth muscle cell migration and proliferation. Acta Pharmacol Sin. 2010;31(10):1277–1283.
120 Abizaid A. Sirolimus-eluting coronary stents: a review. Vasc Health Risk Manag. 2007;3(2):191–201.
121 Kahl C.R., Means A.R. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr Rev. 2003;24(6):719–736.
122 Koledova V.V., Khalil R.A. Ca2 +, calmodulin, and cyclins in vascular smooth muscle cell cycle. Circ Res. 2006;98(10):1240–1243.
123 Choi J., Husain M. Calmodulin-mediated cell cycle regulation: new mechanisms for old observations. Cell Cycle. 2006;5(19):2183–2186.
124 Touyz R.M., Yao G. Modulation of vascular smooth muscle cell growth by magnesium—role of mitogen—activated protein kinases. J Cell Physiol. 2003;197(3):326–335.
125 Burg E.D., Remillard C.V., Yuan J.X.J. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: pharmacotherapeutic implications. Br J Pharmacol. 2008;153(S1):S99–S111.
126 Neylon C.B. Potassium channels and vascular proliferation. Vasc Pharmacol. 2002;38(1):35–41.
127 Cidad P., Moreno-Dominguez A., Novensa L., et al. Characterization of ion channels involved in the proliferative response of femoral artery smooth muscle cells. Arterioscler Thromb Vasc Biol. 2010;30(6):1203–1211.
128 Miguel-Velado E., Perez-Carretero F.D., Colinas O., et al. Cell cycle-dependent expression of Kv3.4 channels modulates proliferation of human uterine artery smooth muscle cells. Cardiovasc Res. 2010;86(3):383–391.
129 Jackson W.F. KV1.3: a new therapeutic target to control vascular smooth muscle cell proliferation. Arterioscler Thromb Vasc Biol. 2010;30(6):1073–1074.
130 Assoian R.K., Schwartz M.A. Coordinate signaling by integrins and receptor tyrosine kinases in the regulation of G1 phase cell-cycle progression. Curr Opin Genet Dev. 2001;11(1):48–53.
131 Gizard F., Bruemmer D. Transcriptional control of vascular smooth muscle cell proliferation by peroxisome proliferator-activated receptor-gamma: therapeutic implications for cardiovascular diseases. 2008. PPAR Res
132 Gizard F., Amant C., Barbier O., et al. PPARα inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. J Clin Invest. 2005;115(11):3228–3238.
133 Gizard F., Nomiyama T., Zhao Y., et al. The PPARα/p16INK4a pathway inhibits vascular smooth muscle cell proliferation by repressing cell cycle-dependent telomerase activation. Circ Res. 2008;103(10):1155–1163.
134 Koyama H., Bornfeldt K.E., Fukumoto S., et al. Molecular pathways of cyclic nucleotide-induced inhibition of arterial smooth muscle cell proliferation. J Cell Physiol. 2001;186(1):1–10.
135 Wu Y.-J., Sala-Newby G.B., Shu K.-T., et al. S-phase kinase-associated protein-2 (Skp2) promotes vascular smooth muscle cell proliferation and neointima formation in vivo. J Vasc Surg. 2009;50(5):1135–1142.
136 Cheng Y., Zhang C. MicroRNA-21 in cardiovascular disease. J Cardiovasc Trans l Res. 2010;3(3):251–255.
137 Gerthoffer W.T. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007;100(5):607–621.
138 San Martín A., Griendling K.K. Redox control of vascular smooth muscle migration. Antioxid Redox Signal. 2010;12(5):625–640.
139 Seasholtz T.M., Majumdar M., Kaplan D.D., et al. Rho and Rho kinase mediate thrombin-stimulated vascular smooth muscle cell DNA synthesis and migration. Circ Res. 1999;84(10):1186–1193.
140 Landry D.B., Couper L.L., Bryant S.R., et al. Activation of the NF-kappa B and I kappa B system in smooth muscle cells after rat arterial injury. Induction of vascular cell adhesion molecule-1 and monocyte chemoattractant protein-1. Am J Pathol. 1997;151(4):1085–1095.
141 Sun L., Stoecklin G., Van Way S., et al. Tristetraprolin (TTP)-14–3–3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-alpha mRNA. J Biol Chem. 2007;282(6):3766–3777.
142 Pyronnet S. Phosphorylation of the cap-binding protein eIF4E by the MAPK-activated protein kinase Mnk1. Biochem Pharmacol. 2000;60(8):1237–1243.
143 Yin B.L., Hao H., Wang Y.Y., et al. Downregulating osteopontin reduces angiotensin II-induced inflammatory activation in vascular smooth muscle cells. Inflamm Res. 2009;58(2):67–73.
144 Cuff C.A., Kothapalli D., Azonobi I., et al. The adhesion receptor CD44 promotes atherosclerosis by mediating inflammatory cell recruitment and vascular cell activation. J Clin Invest. 2001;108(7):1031–1040.
145 Vicencio J.M., Galluzzi L., Tajeddine N., et al. Senescence, apoptosis or autophagy? When a damaged cell must decide its path–a mini-review. Gerontology. 2008;54(2):92–99.
146 Campisi J., d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740.
147 Gorenne I., Kavurma M., Scott S., et al. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc Res. 2006;72(1):9–17.
148 Minamino T., Miyauchi H., Yoshida T., et al. Vascular cell senescence and vascular aging. J Mol Cell Cardiol. 2004;36(2):175–183.
149 Burton D.G., Matsubara H., Ikeda K. Pathophysiology of vascular calcification: pivotal role of cellular senescence in vascular smooth muscle cells. Exp Gerontol. 2010;45(11):819–824.
150 Taylor R.C., Cullen S.P., Martin S.J. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–241.
151 Muto A., Fitzgerald T.N., Pimiento J.M., et al. Smooth muscle cell signal transduction: implications of vascular biology for vascular surgeons. J Vasc Surg. 2007;45(6 Suppl 1):A15–A24.
152 Geng Y.-J., Libby P. Progression of atheroma: a struggle between death and procreation. Arterioscler Thromb Vasc Biol. 2002;22(9):1370–1380.
153 Gupta S., Kass G.E., Szegezdi E., et al. The mitochondrial death pathway: a promising therapeutic target in diseases. J Cell Mol Med. 2009;13(6):1004–1033.
154 Clarke M.C.H., Talib S., Figg N.L., et al. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ Res. 2010;106(2):363–372.
155 Clarke M., Bennett M. Defining the role of vascular smooth muscle cell apoptosis in atherosclerosis. Cell Cycle. 2006;5(20):2329–2331.
156 Son B.K., Akishita M., Iijima K., et al. Mechanism of pi-induced vascular calcification. J Atheroscler Thromb. 2008;15(2):63–68.
157 Martinet W., De Meyer G.R. Autophagy in atherosclerosis. Curr Atheroscler Rep. 2008;10(3):216–223.
158 Hirschi K.K., Majesky M.W. Smooth muscle stem cells. Anat Rec Part A: Disc Mol Cell Evo Biol. 2004;276A(1):22–33.
159 Margariti A., Zeng L., Xu Q. Stem cells, vascular smooth muscle cells and atherosclerosis. Histol Histopathol. 2006;21(9):979–985.
160 Orlandi A., Bennett M. Progenitor cell-derived smooth muscle cells in vascular disease. Biochem Pharmacol. 2010;79(12):1706–1713.
161 Ergün S., Tilki D., Klein D. Vascular wall as a reservoir for different types of stem and progenitor cells. Antioxid Redox Signal. 2011. Jan 7
162 Sinha S., Wamhoff B.R., Hoofnagle M.H., et al. Assessment of contractility of purified smooth muscle cells derived from embryonic stem cells. Stem Cells. 2006;24(7):1678–1688.
163 Nandan M.O., Yang V.W. The role of Kruppel-like factors in the reprogramming of somatic cells to induced pluripotent stem cells. Histol Histopathol. 2009;24(10):1343–1355.
164 Wang C., Yin S., Cen L., et al. Differentiation of adipose-derived stem cells into contractile smooth muscle cells induced by transforming growth factor-beta1 and bone morphogenetic protein-4. Tissue Eng Part A. 2010;16(4):1201–1213.
165 Daniel J.-M., Bielenberg W., Stieger P., et al. Time-course analysis on the differentiation of bone marrow-derived progenitor cells into smooth muscle cells during neointima formation. Arterioscler Thromb Vasc Biol. 2010;30(10):1890–1896.
166 Hoglund V.J., Dong X.R., Majesky M.W. Neointima formation: a local affair. Arterioscler Thromb Vasc Biol. 2010;30(10):1877–1879.
167 Passman J.N., Dong X.R., Wu S.P., et al. A sonic hedgehog signaling domain in the arterial adventitia supports resident Sca1 + smooth muscle progenitor cells. Proc Natl Acad Sci U S A. 2008;105(27):9349–9354.
168 Dotsenko O. Stem/Progenitor cells, atherosclerosis and cardiovascular regeneration. Open Cardiovasc Med J. 2010;4:97–104.
169 Raines E.W., Bornfeldt K.E. Integrin α7β1 COMPels smooth muscle cells to maintain their quiescence. Circ Res. 2010;106(3):427–429.
170 Hilenski L., Griendling K., Alexander R. Angiotensin AT1 receptors. In: Re R., DiPette D., Schiffrin E., Sowers J. Molecular mechanisms in hypertension. London: Taylor and Francis; 2006:25–40.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 3 Vascular Smooth Muscle
With the evolution of an enclosed circulatory system to transport oxygenated blood, hormones, immune cells, metabolites, and waste products to and from cells in distal sites within the vertebrate body, blood vessels evolved adaptations necessary for repeated cycles of contraction and extension resulting from cardiac-driven pulsatile blood flow. These adaptations for blood vessel distensibility allow elastic conductance arteries in the macrocirculation, under the influence of the pulsatile cardiac cycle, to provide blood flow to end organs by altering the luminal diameter of the vessel. They also allow resistance arteries in the microcirculation, which experience steady flow, to regulate vasomotion at the organ level to maintain blood pressure homeostasis.1 The cells that primarily establish and orchestrate these contraction and distensible properties are vascular smooth muscle cells (VSMCs), the majority cell type within the normal vessel wall. VSMCs maintain contractile tone by a highly organized architecture of contractile/cytoskeletal proteins and associated regulatory components within the cell cytoplasm and establish distensibility by synthesis, secretion, and organization of extracellular matrix (ECM) components with elastic recoil and resilience properties.1 VSMCs within the vascular continuum have the ability to adapt expression of proteins involved in contraction and ECM synthesis according to extrinsic and intrinsic cues during different developmental stages and in disease or response to injury. This ability is due to a phenomenon known as VSMC phenotypic modulation and is a major feature that distinguishes VSMCs from terminally differentiated cells.2
Vascular smooth muscle cell phenotypic modulation is the ability to switch phenotypic characteristics from a migratory synthetic phenotype in embryonic tissue patterning to a quiescent, contractile phenotype in maintenance of vascular tone in mature vessels. Importantly, during vascular remodeling in response to injury, VSMCs can switch back to a synthetic phenotype characterized by increased VSMC proliferation and ECM synthesis. Although the ability to switch phenotypes may have evolved as an adaptive survival mechanism for VSMCs to adjust physiological responses due to changing hemodynamic demands or to repair damage after vascular injury, phenotypic modulation has important implications both during development and during vascular disease.2
This chapter will highlight how these diverse functions of VSMCs arise from both innate genetic programs and a range of diverse environmental cues that include soluble signaling factors, insoluble ECM components, physical mechanical forces, and interactions with other cell types.3 Discussion will center on the complex webs of signaling networks generated by these diverse external factors, and how these networks are regulated and integrated at multiple transcriptional and posttranslational levels to mediate the diverse functions of VSMCs in normal physiology and disease/injury pathology.
Origins of Vascular Smooth Muscle Cells During Embryonic Development
Initially in embryonic vasculature development, endothelial precursor cells form a common progenitor vessel which then gives rise to the first artery (dorsal aorta) and vein (cardinal vein) by selective sprouting and subsequent arterial-venous cell segregation4 (see also Chapter 1). The distinct molecular identities of arteries and veins are regulated by complex interactions of several signaling pathways, including sonic hedgehog (Shh), a member of the hedgehog (Hh) family of secreted morphogens; secreted growth factors in the vascular endothelial growth factor family (VEGFs)5; Notch receptors (Notch 1-4) and Notch ligands (Jagged1,2); and transmembrane proteins that can transduce cell-cell interactions into signals determining cell fates.6 Interactions of these signals induce differential expression of VEGF receptors, Ephrin ligands, and tyrosine kinase Eph receptors on the segregating arterial/venous cells, with ephrin B2 and EphB4 as markers expressed in arteries and veins, respectively.4,5 In response to VEGF signaling, endothelial cells (ECs) within these primordial vascular networks recruit mural cells, including nascent VSMCs.7
Nascent VSMCs derive from multiple and nonoverlapping embryonic origins that are reflected in different anatomical locations within the adult. Ectodermal cardiac neural crest cells give rise to the large elastic arteries (e.g., ascending and arch portions of the aorta), ductus arteriosus, and carotid arteries; proepicardium mesothelial cells produce the coronary arteries; mesodermal cells are origins for the abdominal aorta and small muscular arteries; the mesothelium forms the mesenteric vasculature; secondary heart field cells form the base of the aorta and pulmonary trunk; somite-derived cells produce the descending thoracic aorta; and satellite-like mesoangioblasts give rise to the medial layers of arteries.8 The heterogeneous mosaic of VSMCs in the vessel wall may be due in part to these diverse embryological origins of VSMCs and could be reflected in the presence of phenotypically distinct subpopulations within the media that account for VSMC plasticity.9 There is some evidence that VSMCs derived from different lineages exhibit morphologically and functionally distinct properties and respond differently to soluble factors in vitro and to morphogenetic cues in vivo,8 suggesting that the major determinants of VSMC responses to signals in vascular development are principally lineage-dependent rather than environment-dependent.8
Vascular Smooth Muscle Cell Phenotypic Modulation
Characterization of Vascular Smooth Muscle Cell Phenotypes
Given the multiple origins and distinct subpopulations of VSMCs, a compelling central question for understanding VSMC biology is how cells from these diverse embryonic origins, initially expressing lineage-specific pathways, differentiate to express the same marker genes specifically characteristic of VSMCs.8,10 Another question is how these same VSMCs, responding to both extrinsic and intrinsic cues, can alter expression of these genes (and thus molecular pathways), leading to diverse phenotypes with distinct and diverse functions. VSMC phenotypes can be loosely divided into three types: contractile/differentiated, synthetic/dedifferentiated, and inflammatory.
Contractile, differentiated vascular smooth muscle cells
Contractile or differentiated VSMCs are characterized by a repertoire of contractile proteins, contractile-regulating proteins, contractile agonist receptors, and signaling proteins responsible for contraction and maintenance of vascular tone.3,11,12 Of the VSMC “marker” proteins expressed in the contractile phenotype repertoire (Fig. 3-1), the most discriminating markers are smooth muscle myosin heavy chain (SMMHC) in conjunction with alpha-smooth muscle actin (αSMA), smoothelin, SM-22α, h1-calponin, and h-caldesmon.2 In addition to expressing these proteins associated with contractile function, contractile VSMCs exhibit differential levels of ECM components (increased collagen types 1 and IV) and matrix-modifying enzymes (decreased matrix metalloproteinases [MMPs] and increased tissue inhibitors of matrix metalloproteinases [TIMPs]). Contractile VSMCs are further characterized by an elongated spindle-shaped morphology in culture, a low proliferative rate, and expression of α1β1, α7β1 integrins and the dystrophin-glycoprotein complex (DGPC).3,13
Synthetic, dedifferentiated vascular smooth muscle cells
Synthetic or dedifferentiated VSMCs have decreased expression of SMC-related genes for contractile proteins (e.g., SMMHC), with concomitant increased osteopontin, l-caldesmon, nonmuscle myosin heavy chain B, vimentin, tropomyosin 4, and cellular-retinal binding-protein-1 (CRBP-1) (see Fig. 3-1). “Positive” marker genes, such as nonmuscle myosin heavy chain (NM-B MHC) or SMMHC embryonic (SMemb) expressed specifically in embryonic or phenotypically modified VSMCs, are characteristic of dedifferentiated VSMCs in association with vascular injury.2 Other characteristics of synthetic VSMCs include decreased number of actin filaments, an increase in secretory vesicles, increased rates of proliferation and migration, extensive ECM synthesis/degradation capabilities, increased cell size and “hill-and-valley” morphology in culture, high proliferative rate, and increased expression of α4β1 integrin.
Inflammatory vascular smooth muscle cells
In addition to the phenotypic continuum between contractile and synthetic phenotypes, VSMCs can also express markers of an inflammatory phenotype in response to EC-induced recruitment of monocytes and macrophages during the progression of atherosclerosis.14 Various stimuli, including secretion of cytokines by these inflammatory cells, changes in ECM composition, oxidized low density lipoprotein (oxLDL), and VSMC interactions with monocytes/macrophages, induce expression of inflammatory cytokines, vascular cell adhesion molecule (VCAM-1) and transcription factors (NFκB) in VSMCs, leading to recruitment of inflammatory cells into the vessel wall.
Upstream Mediators of Phenotypic Modulation
Growth-inducing factors
Soluble factors that include growth factors, hormones, and reactive oxygen species (ROS) serve as upstream mediators of the phenotypic switch from contractile to synthetic VSMCs, which results in large part from coordinate activation/repression of VSMC marker genes important in the contractile response2,3,15,16 (Fig. 3-2). Some of the most important growth-inducing factors include platelet-derived growth factor (PDGF), epidermal growth factor (EGF), insulin-like growth factor (IGF), and basic fibroblast growth factor (bFGF). Growth factors bind to surface membrane receptor tyrosine kinases (RTKs), triggering sequential downstream signaling pathways mediated through complex formation of activated RTKs with adaptor and signaling proteins Grb2/Shc/Sos, and activation of intracellular kinases, including phosphatidylinositol 3-kinase (PI3K), mitogen-activated protein kinases (MAPKs: extracellular signal regulated kinase, ERK1/2, p38MAPK, and c-jun NH2-terminal kinase, JNK), Akt, MAPKAPK2, and p70S6 kinase (p70S6K). These signals not only transcriptionally mediate the switch to the synthetic phenotype, but also serve to promote growth and survival. In addition, ROS such as hydrogen peroxide (H2O2) produced by activation of NADPH oxidases, multimeric enzymes containing p22phox and other subunits depending upon the specific isoform, can act as second messengers for canonical G protein–coupled receptor (GPCR) and RTK pathways.17
Differentiation-inducing factors
In contrast to growth factor–stimulated proliferation, the cytokine transforming growth factor β (TGF-β) and members of the bone morphogenetic protein (BMP) subgroup of this family promote the differentiated, contractile phenotype in VSMCs by inducing expression of the VSMC contractile genes αSMA and calponin (Fig. 3-3). Transforming growth factor β binds to a tetrameric complex consisting of two type I and two type II receptors, resulting in phosphorylation of Smads, transcription factors named for Caenorhabditis elegans Sma and Drosophila Mad (mothers against decapentaplegic).18 Within the TGF-β signaling pathway itself, different Smads control expression of different markers. For example, Smad3 transactivates the SM22α promoter, while Smad2 activates the αSMA gene. Other soluble factors that inhibit proliferation and increase differentiation include heparin and retinoic acid.9 Most smooth muscle differentiation markers share additional common transcriptional pathways, discussed in more detail later. For example, both TGF-β-induced phosphorylated Smads and ECM-induced activation of integrins, mediated through focal adhesion components vinculin, talin, and tensin, in concert with changes in cytoskeletal F/G actin dynamics, result in myocardin-related transcription factor (MRTF) induction of cytoskeletal/contractile genes (see Fig. 3-3).
Dual factors
One factor with a potential dual role, depending upon initial phenotype/developmental stage, is the octapeptide hormone angiotensin II (Ang II), the effector molecule of the renin–angiotensin II system.19 Angiotensin II can induce either contractile or synthetic phenotypes, with differential responses depending upon cell context and locations within the artery (see Figs. 3-2 and 3-3). Angiotensin II, binding to its GPCR AT1R, activates VSMC marker gene expression indicative of the contractile phenotype through L-type voltage-gated Ca2 + channel–induced elevations in intracellular Ca2 + concentrations, and subsequent increased myocardin transcription coactivator expression dependent upon Prx1, a homeodomain protein that promotes serum response factor (SRF) binding to conserved elements in VSMC marker gene promoters.20 In addition, Ang II binding to AT1R can induce signatures of the synthetic phenotype by activation of multiple kinase and enzyme pathways that are interconnected in signaling networks (see Fig. 3-2). These include the MAPKs; RTKs, including ROS-sensitive transactivation of epidermal growth factor receptor (EGFR); nonreceptor tyrosine kinases (c-Src/focal adhesion kinase [FAK]/paxillin/Rac/JNK/AP-1) and tyrosine phosphatases; SHP2/Janus kinase and signal transducers and activators of transcription (JAK/STAT); and GPCR classic signaling cascades (phospholipase C [PLC]/protein kinase C [PKC]/Ras/Raf/mitogen extracellular signal regulated kinase [MEK]/ERK) leading to stimulation of early growth-response genes (c-fos, c-jun), survival pathways (e.g., Akt), and ECM formation (JNK/AP-1).
Notch communication
In addition to its critical function in development, Notch signaling is also important in defining VSMC differentiation.21,22 Downstream Notch effector gene activation results in activation of “master regulators” of VSMC differentiation (myocardin, MRTFs, or SRF) or direct induction of contractile proteins SMMHC and αSMA, as well as the VSMC specific differentiation marker SM22α (also known as transgelin).6 Data regarding Notch signaling on VSMC differentiation, however, are conflicting, with some studies supporting a repressive effect, while others indicate a promoting effect on expression of VSMC marker genes SMMHC and αSMA.22 These discrepancies may be due to the antagonistic roles of Notch and the Notch effector Hairy-related transcription factor 1 (HRT1) on markers of VSMC differentiation, specifically αSMA and SMMHC.23 Hairy-related transcription factor 1 inhibits Notch/RBP-Jκ binding to the αSMA promoter in a histone deacetylase-independent manner. The context-dependent roles of Notch and HRT1 on markers of VSMC differentiation may serve to fine-tune VSMC phenotypic modulation during vascular development, injury, and disease.
There is considerable cross-talk between Notch and other signaling pathways. Notch and TGF-β cooperatively induce a functional contractile, differentiated phenotype through parallel signaling axes,24 while HRT factors block VSMC differentiation in both pathways. Other examples of cross-talk among key signaling pathways for morphogenesis (Hh, Notch) and mitogenesis (VEGF-A, PDGF) include a Shh/VEGF-A/Notch signaling axis in VSMCs in the neointima to increase growth and survival,25 and Notch-induced up-regulation of PDGFR-β to mediate growth and migration.26
Homotypic VSMC-VSMC Notch-mediated signaling pathways are also apparent in adult vascular pathologies and response to injury.22 After injury, Notch receptors are increased, along with elevated levels of HRT. Negative feedback between HRT and Notch may account for the adaptive response to injury in which initial Notch/HRT-induced suppression of the contractile phenotype is followed by arterial remodeling. As Notch/HRT signaling decreases, the contractile phenotype is reestablished.24
Transcriptional Regulation of Vascular Smooth Muscle Cell Diversity
The complex web of signaling pathways induced by these external signals—whether they are soluble, insoluble, structural, or mechanical—converge on a network of transcription factors (TFs) that coordinately regulate gene expression and act as “master switches” for growth and differentiation27 (Fig. 3-4). Transcription of VSMC-specific differentiation or proliferative genes is regulated by cooperative interaction of TFs and their coregulators, including SRF,28 myocardin and myocardin-related TFs (MRTF-A and -B),29 Ets domain transcription factors known as ternary complex factors (TCFs),30 zinc finger factors GATA630 and PRISM/PRDM6,31 and Krüppel-like factors (KLFs).32,33
Serum response factor/myocardin axis
Serum response factor, a widely expressed member of the MADS (MCM1, agamous, deficien, SRF) box of TFs, is a nodal point linking signaling pathways to differential gene expression related either to growth or differentiation, depending upon which transcriptional partner is bound to SRF.28 Serum response factor self-dimerizes and binds with high affinity and specificity to a consensus deoxyribonucleic acid (DNA) sequence CArG box found in the promoters of cyto-contractile genes.34 More than half of the VSMC “marker” genes that define VSMC molecular signature contain CArG boxes.34 Included in these genes are three categories modulating actin filament dynamics: (1) structural (e.g., αSMA-actin, SM22α, caldesmon, SMMHC); (2) effectors of actin turnover (e.g., cofilin, gelsolin); and (3) regulators of actin dynamics (four-and-a-half LIM domains proteins [FHL1 and 2], MMP9, and myosin light chain kinase).35
Serum response factor itself is a weak activator of CArG-dependent genes.30 Potent SRF-dependent transcriptional activation is therefore dependent upon regulation at several levels: by interaction with different signal-regulated or tissue-specific regulatory SRF transcription cofactors/corepressors; by posttranslational phosphorylation, acetylation, and sumoylation, modifications that affect these interactions; and by epigenetic alterations in chromatin structure in which myocardin serves as a scaffold for recruitment of chromatin-remodeling enzymes36 that enable SRF and its cofactors to gain access to SRF target genes.8 Myocardin association with histone acetyltransferases (HATs), including p300, enhances transcription of VSMC-restricted genes, whereas association with class II histone deacetylases (HDACs) suppresses myocardin-induced transcription of VSMC marker genes36 (see Fig. 3-4).
Serum response factor interacts with cofactors in two principal families: the TCF family of Ets-domain proteins (Elk, SAP-1, and Net)37 activated by the MAPK pathway, leading to SRF binding to immediate early growth factor-inducible genes such as c-fos28; and the myocardin/MRTF-A/MRTF-B family35 to promote activation of VSMC-specific marker genes, most of which code for filamentous proteins that function in contractile activities or proteins that function in cell-matrix adhesions.10 These alternative pathways provide the “plasticity” associated with VSMC phenotypic modulation ranging from contractile functions to maintain vascular tone to synthetic or proliferative functions in response to vascular injury.29
Discovery of the cell-restricted SRF transcriptional coactivator myocardin, expressed specifically in cardiac and VSMCs, resolved the paradoxical observations that SRF can regulate mutually exclusive gene expression programs for growth or differentiation.30,34 In VSMCs, myocardin is a master regulator of SMC marker gene expression and sufficient for the smooth muscle–like contractile phenotype. Myocardin competes with Elk-1 for direct binding to SRF in VSMCs; thus, myocardin and Elk-1 can act as binary transcriptional switches that may regulate contractile vs. synthetic VSMC phenotypes30 (see Fig. 3-4). In addition, myocardin transduction leads to lower levels of the cell cycle–associated gene cyclin D1, resulting in repression of growth. Therefore, myocardin is a nodal point for two features indicative of SMC differentiation: expression of the contractile apparatus and suppression of growth.30
While myocardin functions exclusively as a transcriptional coactivator,38 additional proteins function to regulate transcriptional activity of myocardin. Hairy-related transcription factor 2 and GATA factors repress or enhance myocardin-induced transcriptional activity, depending upon cell context.30 In addition, activation of Notch receptors by Jagged1 endogenous ligand induces translocation of Notch intracellular domain (ICD) to the nucleus where it inhibits myocardin-induced SMC gene expression.29 Angiotensin II stimulation, as well as activation of L-type voltage-gated Ca2 + channels, activates SMC marker genes by inducing myocardin expression and, in the case of Ang II, increasing SRF binding to CArG elements in the promoter regions of VSMC marker genes such as αSMA.20
Serum response factor transcriptional activity is also controlled by Rho-induced actin dynamics that facilitate movement of MRTFs into or out of the nucleus29 (see Fig. 3-4). In most cell types, MRTFs form a stable complex with monomeric G-actin and remain sequestered in the cytoplasm. Myocardin-related transcription factors in VSMCs, however, are localized in the nucleus where binding to SRF in the basal state promotes contractile gene expression, and the differentiated phenotype. In response to growth factors or vascular injury, extracellular signals transduced through the Rho-actin pathway result in nuclear export of MRTF, down-regulation of SRF/MRTF-induced VSMC contractile gene expression, and promotion of mitogen-induced ERK1/2 phosphorylation of TCFs, resulting in TCF displacement of MRTFs and SRF/TCF-mediated activation of growth-responsive genes.29 These differential pathways provide a switch in which SRF target genes are differentially regulated through growth factor–induced signaling for growth (active TCF, MRTF blocked) or Rho-actin signaling for differentiation (inactive TCF, MRTF active)30 (see Fig. 3-4).
Zinc finger proteins
GATA6, a zinc finger transcription factor expressed in VSMCs, induces growth arrest by increasing expression of the general cyclin-dependent kinase inhibitor (CDKI) p21CIP1 and inhibiting S-phase entry.30 PRISM is a smooth muscle–restricted member of zinc finger proteins belonging to the PRDM (PR domain in smooth muscle) family and acts as a transcriptional repressor by interacting with class I histone deacetylases and G9a histone methyltransferases. PRISM induces the proliferative phenotype while repressing differentiation regulators myocardin and GATA6.31
One of the most intensely studied zinc finger transcriptional regulators in VSMCs is the KLF subfamily that binds to the TGF-β control element (TCE) in the regulatory sequences of target genes (reviewed in32,33,39). Vascular smooth muscle cells express four KLFs (KLF4, KLF5, KLF13, and KLF15), each with individual biological functions implicated in regulating a range of processes in both growth and differentiation.32 Individual KLFs may have opposing functions, depending upon temporal and developmental expression patterns and interactions with other factors. For example, KLF4 inhibits, whereas KLF5 and KLF13 induce, VSMC marker gene expression. Mechanisms that may account for these opposing functions of KLF factors include posttranslational modifications, interaction with specific cofactors, differential expression by growth factors, cytokines and differentiation state, or regulation by another KLF.32
KLF4 functions as both a VSMC growth repressor and a repressor for VSMC differentiation, although data on the effect of KLF4 on VSMC differentiation are conflicting33 (see Fig. 3-4). As a growth repressor, KLF4 inhibits PDGF-BB-induced mitogenic signaling and induces expression of the negative cell cycle regulator p53 and its target gene p21CIP1. As a differentiation repressor, KLF4 prevents SRF from binding to the TCE in promoters of VSMC marker genes, suppresses expression of myocardin, inhibits myocardin-induced activation of SMC marker genes, reduces SRF binding to CArG elements in SMC contractile gene promoters,33 and induces histone hypoacetylation at SMC CArG regions associated with gene silencing.40 On the other hand, there is evidence that KLF4 promotes VSMC differentiation by directly activating VSMC marker gene transcription of SM22α and αSMA.33 KLF4 thus functions as a bifunctional TF or “molecular switch” that can both activate and repress VSMC marker genes, depending upon regulation of KLF4.33
Even though the closely homologous KLF4 and KLF5 TFs share similar developmental and tissue pattern expression, they exert different, often opposing, effects on gene regulation and proliferation/differentiation.33 Whereas KLF4 is associated with growth arrest, KLF5 exerts pro-proliferative effects, particularly in vascular remodeling in response to injury. KLF5 expression, abundant in fetal VSMCs but down-regulated in the adult (reviewed in39), is induced after vascular injury by activation of immediate early response genes by Ang II and ROS.41 KLF5 in turn mediates re-expression of SMemb/NMHC-B, a marker for the dedifferentiated phenotype, and activates other critical injury response genes involved in remodeling, such as PDGF-A/B, Egr-1, plasminogen activator inhibitor 1 (PAI-1), inducible nitric oxide synthase (iNOS), and VEGFR, implicating KLF5 as a key regulator for VSMC response to injury.39 In additional injury responses, KLF5 increases cyclin D1 expression and inhibits the cyclin kinase inhibitor p21, thus leading to vascular remodeling by increased cell proliferation.42 Similar to KLF4 regulation, KLF5 expression and activity are regulated at multiple levels, including upstream Ras/MAPK, PKC, and TGF-β signaling pathways; downstream interactions with TFs, including retinoic acid receptor α (RARα), NF-κB, and peroxisome proliferator–activated receptor gamma (PPARγ); as well as posttranslational modifications that can positively or negatively regulate KLF activity.39 In addition, KLF5 activity is regulated in the nucleus by chromatin remodeling factors such as SET, a histone chaperone that inhibits the DNA binding activity of KLF5,43 p300 (a coactivator/acetylase that coactivates KLF5 transcription), and HDAC1, which inhibits KLF5 binding to DNA.32
Two additional KLFs have been identified in VSMCs: KLF13 and KLF15.32 After vascular injury, KLF13 is induced and activates the promoter for the VSMC differentiation marker SM22α, while KLF15 expression is down-regulated, implicating KLF15 as a negative regulator of VSMC proliferation and a counterbalance to the growth-promoting effects of KLF5 in vascular injury response.
Posttranscriptional Regulation of Vascular Smooth Muscle Cell Diversity: Noncoding microRNAs
Upstream signaling and downstream transcriptional pathways in VSMCs are intertwined with a multitude of micro ribonucleic acid (miRNAs) that act as “rheostats” and “switches” in regulating protein activity in development, function, and disease.44 miRNAs are small, noncoding RNAs (20-25 nucleotides in length) that associate with a miRNA-induced silencing complex (miRISC) of regulatory proteins, including Argonaute family proteins, Argonaute interacting proteins of the GW182 family, eukaryotic initiation factors (eIFs), polyA-binding complexes, decapping enzymes/ activators, and deadenylases, to induce posttranscriptional silencing of their target genes.45 These multiple components are assembled and interact in a multistep process with components of the translational machinery to inhibit translation initiation, mark mRNAs for degradation through deadenylation, and sequester targets into cytoplasmic P bodies.44 Multiple mechanistic models for miRNA-induced gene silencing have been proposed that provide insights into the molecular mechanisms of translational inhibition, deadenylation, and mRNA decay, but questions remain concerning the kinetics and ordering of these translational events and whether they are coupled or independent.45 A recent unifying model for miRNA-regulated gene repression is an attempt to reconcile the often conflicting existing data. It proposes that recruitment of Argonaute and associated GW182 proteins to miRNA induces binding to the mRNA 5′m7 Gcap, thus blocking translation initiation, potentially by mRNA deadenylation. Subsequent to miRNA-mediated deadenylation, mRNA is degraded through recruitment of decapping proteins.46 In this model, inhibition of translation initiation is linked to subsequent rapid mRNA decay in a coupled process. Because miRNAs—which in general are negative regulators of gene expression—may be almost as important as transcription factors in controlling gene expression in the pathogenesis of human diseases,47 insights into the functions of this class of noncoding RNAs are important in evaluating their potential use as therapeutic targets.45
Cardiovascular-specific, highly conserved miRNAs miR-143 and miR-145, the most abundant miRNA in the vascular wall,48 are key players in programming VSMC fate from multipotent progenitors in embryonic development and in reprogramming VSMCs during phenotypic modulation in the adult44,49 (Fig. 3-5). miR-143 and miR-145 have distinct sequences but are clustered together and transcribed as a bicistronic unit. Upstream in the genomic sequence of miR-143/145 is a conserved SRF-binding CArG box site, indicating control by SRF and myocardin.49,50 These miRNAs cooperatively feed back to modulate the actions of SRF by targeting a network of transcription factors/coactivators/corepressors. This network includes miR-145-induced repression of KLF4, a positive regulator of proliferation and myocardin repressor; miR-143-induced repression of Elk-1, a myocardin competitor and positive regulator of proliferation; and, contrary to the usual inhibitory role of miRNA, miR-145-induced stimulation of myocardin, a positive regulator of differentiation. Thus, miR-145 is necessary and sufficient for VSMC differentiation, and the miR-143/miR-145 cluster acts as an integrated signaling node to promote differentiation while concurrently repressing proliferation.49 Although mice with genetic deletions for miR-143/145 show no obvious abnormalities in early development, VSMCs in the adult exhibit both structural and phenotypic differences in injury- or stress-induced vascular remodeling. Ultrastructural analysis of arteries from miR-143/145 knockout mice shows reduced numbers of medial VSMCs with a contractile appearance, and an increase in synthetic VSMCs.51 These results suggest that miR-143 and miR-145 modulate cytoskeletal structure, actin dynamics, and modulation to a dedifferentiated phenotype50 (see Fig. 3-5). Importantly, miR-143/145 knockout mice with increased synthetic VSMCs develop spontaneous neointimal lesions in the femoral artery in the absence of hyperlipidemia and inflammation, supporting a key role for phenotypically altered VSMCs in the pathogenesis of lesion formation.51
While miR-143 and miR-145 play keys roles in the contractile phenotype of VSMCs and the response to injury,52 miR-221 and miR-222 are modulators of VSMC proliferation, although largely by affecting growth-related signaling pathways rather than by controlling VSMC phenotype. miR-221 and miR-222, encoded by a gene cluster on the X chromosome, are up-regulated in VSMCs in neointimal lesions and in proliferating cultured VSMCs stimulated by PDGF-BB.53 Studies show that two CDKIs, p27KIP1 and p57KIP2, have miR-221 and miR-222 binding sites and are gene targets for miR-221 and miR-222 in the rat carotid artery in vivo.53 Thus, miR-221 and miR-222 are pro-proliferative because they repress two CDKIs, p27KIP1 and p57KIP2. Furthermore, PDGF, via miR-221 induction, inhibits VSMC differentiation via c-kit-induced inhibition of myocardin.54
Posttranslational Regulation of Vascular Smooth Muscle Cell Diversity: Epigenetics
The “epigenetic landscape” controls gene expression by chemical modifications that mark regions of chromosomes either by methylation of promoter CpG sequences in the DNA itself, or by covalent modification of histone proteins that package DNA by posttranslational addition of methyl, acetyl, phosphoryl, ubiquityl, or sumoyl groups, leading to expression/repression of transcription (reviewed in55). In VSMCs, multiple levels of epigenetic controls exist for gene expression leading to differentiation or dedifferentiation programs in healthy cells and for dysregulated gene expression in vascular disease. These epigenetic changes in VSMCs involve both DNA and histone methylation as well as histone acetylation/deacetylation. Methylation of histones, catalyzed by histone methyltransferases (HMTs), results in a tight, stable epigenetic mark between methylated histones and chromatin that can be passed to daughter cells, thus providing “epigenetic memory” that defines cell lineage and identity by controlling SRF access to VSMC-specific marker genes.55 Acetylation is controlled by HATs, which promote gene transcription by destabilizing chromatin structure to an “open,” transcriptionally active conformation, and HDACs, which promote chromatin condensation to a “closed,” transcriptionally silent conformation with restricted access to DNA. Histone acetylation/deacetylation thus serves to regulate transcription in a rapid and “on/off” manner in response to dynamic environmental changes and links the cell’s genome with new extrinsic signals.55
In VSMCs, SRF binding to CArG boxes in the promoters of SMC marker genes to promote the VSMC differentiated phenotype depends upon alterations of chromatin structure, including histone methylation and acetylation. In a model for epigenetic regulation of VSMC phenotype,56 SRF binding to CArG boxes in VSMC marker gene promoters is blocked by conditions such as PDGF-BB exposure or vascular injury. Such conditions promote KLF4-induced myocardin suppression as well as KLF4-induced recruitment of HDACs, resulting in “closed” deacetylated chromatin and transcriptional repression of VSMC marker genes. Histone methylation, in contrast, is not affected by PDGF-BB and may serve as a permanent “memory” for VSMC identity during repression of SRF-dependent transcription and can, once repressive signals are terminated, reactivate the differentiation program by recruiting myocardin/SRF complexes or HATs to VSMC marker genes for reexpression. In the absence of KLF4 activation, SRF/myocardin can bind to HAT-induced acetylated “open” chromatin at CArG boxes for transcriptional activation of VSMC marker genes, thus promoting VSMC differentiation. In addition, myocardin induces acetylation of histones in the vicinity of SRF-binding promoters in VSMC marker genes by association with p300, a ubiquitous transcriptional coactivator with its own intrinsic HAT activity, leading to synergistic activation of VSMC marker gene expression. This pro-myogenic program is antagonized and repressed by myocardin binding to class II HDACs, which strongly inhibits expression of marker genes αSMA, SM22α, SMMLCK and SMMHC. These opposing actions of HATs and HDACs on SRF/myocardin function to activate or repress, respectively, VSMC differentiation and serve to regulate transcription in a rapid and reversible manner in response to dynamic changes in the environment.55
Often, transcription mediators play roles in both classic signal transduction pathways and epigenetic programming.57 Smad proteins, for example, transmit TGF-β signals from the membrane to the nucleus to mediate gene transcription and VSMC differentiation. The balance between Smad-induced recruitment of corepressors or coactivators to TGF-β-responsive genes is associated with activation of HDAC or HAT (p300), which then alters histone acetylation. Transforming growth factor β induces histone hyperacetylation at the VSMC marker gene SM22 promoter through recruitment of HATs, Smad3, SRF, and myocardin, demonstrating a role for HATs and HDACs in TGF-β activation of VSMC differentiation.58
A proposed example of metabolic memory stored in the histone code of VSMCs is found in the dysregulation of histone H3 methylation, an epigenetic mark usually associated with transcriptional repression in type 2 diabetes.59 In VSMCs derived from type 2 diabetic db/db mice, levels of H3K9me3 (H3 lysine-9 trimethylation), as well as its HMT, are both reduced at the promoters of inflammatory genes. This loss of repressive histone marks, leading to increased inflammatory gene expression, is sustained in VSMCs from db/db mice cultured in vitro, suggesting persistence of metabolic memory. These results suggest that dysregulation in the histone code in VSMCs is a potential mechanism for increased and sustained inflammatory response in diabetic patients who continue to exhibit “metabolic memory” and vascular complications after glucose normalization.60
Influence of Cell-Cell and Cell-Matrix Interactions
Many differential VSMC functions are influenced by cell-cell and cell-matrix adhesion receptors that are altered during phenotypic modulation and during response to injury or disease. Cell-cell adhesion receptors include cadherins and gap junction connexins; cell-matrix interactions are dependent upon combinations of integrins, syndecans, and α-dystroglycan.11
Cell-Cell Adhesion Molecules: Cadherins and Gap Junction Connexins
After investment of VSMCs to the EC layer of nascent vessels, vascular stabilization, also known as maturation,61 is regulated by the sphingosine 1-phosphate (S1P) receptor S1P1, a GPCR on ECs. S1P1 activates the cell-cell adhesion molecule N-cadherin in ECs and induces formation of direct N-cadherin-based junctions between ECs and VSMCs required for vessel stabilization.61 To maintain VSMC quiescence within the vascular wall, cadherin-mediated cell-cell adherens-type junctions between VSMCs inhibit VSMC proliferation, possibly by inhibiting the transcriptional activity of β-catenin, a component of the Wnt signaling pathway, which interacts with the intracellular domain of cadherins.62 Inhibition of β-catenin or stabilization of cadherin junctions in VSMCs may be useful in treating vascular disease or injury.
Another type of direct intercellular junction between cells in the vasculature is the gap junction.63 Gap junctions, formed by connexin proteins between ECs and VSMCs and between VSMCs, are intercellular channels that allow movement of metabolites, small signaling molecules, and ions between cells.63–65
[/not-level-membership-for-cardiovascular-category]
