Chapter 1 Vascular Embryology and Angiogenesis
In simple terms, the cardiovascular system consists of a sophisticated pump (i.e., the heart) and a remarkable array of tubes (i.e., blood and lymphatic vessels). Arteries and arterioles (efferent blood vessels in relation to the heart) deliver oxygen, nutrients, paracrine hormones, blood and immune cells, and many other products to capillaries (small-caliber, thin-walled vascular tubes). These substances are then transported through the capillary wall into extravascular tissues where they participate in critical physiological processes. In turn, waste products are transported from the extravascular space back into blood capillaries and returned by venules and veins (afferent vessels) to the heart. Alternatively, about 10% of the fluid returned to the heart courses via the lymphatic system to the large veins.1 To develop normally, the embryo requires delivery of nutrients and removal of waste products beginning early in development; indeed, the cardiovascular system is the first organ to function during morphogenesis.
This chapter summarizes many key molecular and cellular processes and underlying signals in the morphogenesis of the different layers of the blood vessel wall and of the circulatory system in general. Specifically, for intimal development, it concentrates on early EC patterning, specification and differentiation, lumen formation, co-patterning of vessels and nonvascular tissues, and briefly discusses lymphatic vessel development. In the second section, development of the tunica media is divided into subsections examining components of the media, VSMC origins, smooth muscle cell (SMC) differentiation, and patterning of the developing VSMC layers and ECM. Finally, the chapter concludes with a succinct summary of the limited studies of morphogenesis of the blood vessel adventitia. Understanding these fundamental vascular developmental processes are important from a pathophysiological and therapeutic standpoint because many diseases almost certainly involve recapitulation of developmental programs. For instance, in many vascular disorders, mature VSMCs dedifferentiate and exhibit increased rates of proliferation, migration, and ECM synthesis through a process termed phenotypic modulation.2
Tunica Intima: Endothelium
Early Development
Development begins with fertilization of the ovum by the sperm. Chromosomes of the ovum and sperm fuse, and then a mitotic period ensues. The early 16- to 32-cell embryo, or morula, consists of a sphere of cells with an inner core termed the inner cell mass. The first segregation of the inner cell mass generates the hypoblast and epiblast. The hypoblast gives rise to the extraembryonic yolk sac and the epiblast to the amnion and the three germ layers of the embryo known as the endoderm, mesoderm, and ectoderm. The epiblast is divided into these layers in the process of gastrulation, when many of the embryonic epiblast cells invaginate through the cranial-caudal primitive streak and become the mesoderm and endoderm, while the cells that remain in the embryonic epiblast become the ectoderm. Most of the cardiovascular system derives from the mesoderm, including the initial ECs, which are first observed during gastrulation. A notable exception to mesodermal origin is SMCs of the aortic arch and cranial vessels, which instead derive from the neural crest cells of the ectoderm.3
Although ECs are thought to derive exclusively from mesodermal origins, the other germ layers may play an important role in regulating differentiation of the mesodermal cells to an EC fate. In a classic study of quail-chick intracoelomic grafts, host ECs invaded limb bud grafts, whereas in internal organ grafts, EC precursors derived from the graft itself.4 The authors hypothesized that the endoderm (i.e., from internal organ grafts) stimulates emergence of ECs from associated mesoderm, whereas the ectoderm (i.e., from limb bud grafts) may have an inhibitory influence.4 Yet the endoderm does not appear to be absolutely required for initial formation of EC precursors.5,6
The initial primitive vascular system is formed prior to the first cardiac contraction. This early vasculature develops through vasculogenesis, a two-step process in which mesodermal cells differentiate into angioblasts in situ, and these angioblasts subsequently coalesce into blood vessels.7 Early in this process, many EC progenitors apparently pass through a bipotential hemangioblast stage in which they can give rise to endothelial or hematopoietic cells. Furthermore, early EC precursors may in fact be multipotent; there is controversy whether ECs and mural cells share a common lineage.8,9
Following formation of the initial vascular plexus, more capillaries are generated through sprouting and nonsprouting angiogenesis, and the vascular system is refined through pruning and regression (reviewed in10). In the most well studied form of angiogenesis, existing blood vessels sprout new vessels, usually into areas of low perfusion, through a process involving proteolytic degradation of surrounding ECM, EC proliferation and migration, lumen formation, and EC maturation. Nonsprouting angiogenesis is often initiated by EC proliferation, which results in lumen widening.10 The lumen then splits through transcapillary ECM pillars or fusion and splitting of capillaries to generate more vessels.10 In addition, the developing vascular tree is fine-tuned by the pruning of small vessels. Although not involved in construction of the initial vascular plan, flow is an important factor in shaping vascular system maturation, determining which vessels mature and which regress. For instance, unperfused vessels will regress.
Arterial and Venous Endothelial Cell Differentiation
Classically it was thought that arterial and venous blood vessel identity was established as a result of oxygenation and hemodynamic factors such as blood pressure, shear stress, and the direction of flow. However, over the last decade, it has become increasingly evident that arterial-specific and venous-specific markers are segregated to the proper vessels quite early in the program of vascular morphogenesis. For instance, ephrinB2, a transmembrane ligand, and one of its receptors, the EphB4 tyrosine kinase, are expressed in the mouse embryo in an arterial-specific and relatively venous-specific manner, respectively, prior to the onset of angiogenesis.11–13 EphrinB2 and EphB4 are each required for normal angiogenesis of both arteries and veins.12,13 However, in mice homozygous for a tau-lacZ knock-in into the ephrinB2 or EphB4 locus (which renders the mouse null for the gene of interest), lacZ staining is restricted to arteries or veins, respectively.12,13 This result indicates that neither of these signaling partners is required for arterial and venous specification of ECs.
Furthermore, even before initial ephrinB2 and EphB4 expression and prior to the first heart beat, Notch pathway members delta C and gridlock mark presumptive ECs in the zebrafish.14–16 In this model, deltaC is a homolog of the Notch ligand gene Delta, and gridlock (grl) encodes a basic helix-loop-helix protein that is a member of the Hairy-related transcription factor family and is downstream of Notch. The lateral plate mesoderm (LPM) contains artery and vein precursors,17 and prior to vessel formation, the grl gene is expressed as two bilateral stripes in the LPM.16 Subsequently, gridlock expression is limited to the trunk artery (dorsal aorta) and excluded from the trunk vein (cardinal vein).16
In a lineage tracking experiment of the zebrafish LPM, Zhong et al. loaded one- to two-cell embryos with 4,5-dimethoxy-2-nitrobenzyl-caged fluorescent dextran.15 Between the 7- and 12-somite stage of development, a laser was used to activate a patch of 5 to 10 LPM cells with pulsations and thereby “uncage” the dye.15 The contribution of the uncaged cells and their progeny to the dorsal aorta and cardinal vein was assayed the next day.15 Among all the uncaging experiments, marked cells were found in the artery in 20% of experiments and in the vein in 32% of experiments.15 Interestingly, within a single uncaging experiment, the group of marked cells never included both arterial and venous cells, suggesting to the authors that by the 7- to 12-somite stage, an individual angioblast is destined to contribute in a mutually exclusive fashion to the arterial or venous system.15
In addition to being an early marker of arterial ECs, the Notch pathway is a key component of a signaling cascade that regulates arterial EC fate. In zebrafish, down-regulating the Notch pathway through genetic means or injection of messenger ribonucleic acid (mRNA) encoding a dominant-negative Suppressor of Hairless, a known intermediary in the Notch pathway, results in reduced ephrinB2 expression with loss of regions of the dorsal aorta.15,18 Reciprocally, contiguous regions of the cardinal vein expand and EphB4 expression increases.15 By contrast, activation of the Notch pathway results in reduced expression of flt4, a marker of venous cell identity, without an effect on arterial marker expression or dorsal aorta size.15,18 Furthermore, Lawson et al. followed up on these findings to describe a signaling cascade in which vascular endothelial growth factor (VEGF) functions upstream of Notch, and Sonic hedgehog (Shh) is upstream of VEGF.19 Taken together, these results suggest that the Shh-VEGF-Notch axis is necessary for arterial EC differentiation; however, Notch is not sufficient to induce arterial EC fate.
These studies of EC fate raise the issues of when the arterial-venous identities of ECs are specified and whether and/or when these identities become fixed. To examine these issues, Moyon et al. dissected the dorsal aorta, carotid artery, cardinal vein, or jugular vein from the embryonic day 2 to 15 (E2-15) quail and grafted the vessel into the E2 chick coelom.20 On E4, the host embryos were immunostained with arterial-specific antibodies and the quail-specific anti-EC antibody QH1 to determine whether the grafted vessels yielded ECs that colonized host arteries, veins, or neither.20 Quail vessels that were harvested until around E7 and then grafted into the chick colonized ECs in both host arteries and veins, but if harvesting was delayed after E7, plasticity of the grafted vessels decreased.20 Indeed, quail arteries or veins that were isolated after E10 and subsequently grafted almost exclusively contributed to host arteries (> 95% of QH1+ ECs) or veins (~ 90% of QH1+ ECs), respectively.20 Interestingly, when ECs were isolated by collagenase treatment from the quail E11 dorsal aorta wall and then grafted, plasticity of the ECs was restored to that of an E5 aorta (~ 60% of QH1+ EC contribution to arteries and ~ 40% contribution to veins).20 The authors reasoned that an unknown signal from the vessel wall regulates EC identity.20 A recent investigation of the origins of the coronary vascular endothelium also highlights the plasticity of ECs during early mouse development.21 This study suggests that EC sprouts from the sinus venous, the structure that returns blood to the embryonic heart, dedifferentiate as they migrate over and through the myocardium.21 Endothelial cells that invade the myocardium differentiate into the coronary arterial and capillary ECs, while those that remain on surface of the heart will redifferentiate into the coronary veins.21
Endothelial Tip and Stalk Cell Specification in Sprouting Angiogenesis
Tubular structures are essential for diverse physiological processes, and proper construction of these tubes is critical. Tube morphogenesis requires coordinated migration and growth of cells that compose the tubes; the intricate modulation of the biology of these cells invariably uses sensors that detect external stimuli.22 This information is then integrated and translated into a biological response. Important examples of such biological sensors include the growth cones of neurons and the terminal cells of the Drosophila tracheal system. Both of these sensors have long dynamic filopodia that sense and respond to external guidance cues and are critical in determining the ultimate pattern of their respective tubular structures.
Similarly, endothelial tip cells are located at the ends of angiogenic sprouts and are polarized with long filopodia that play both a sensory and motor role22 (Fig. 1-1). In a classic study published over 30 years ago, Ausprunk and Folkman reported that on the day after V2 carcinoma implantation into the rabbit cornea, ECs of the host limbal vessels displayed surface projections that resembled “regenerating ECs,”23 consistent with what is now classified as tip cell filopodia. Tip cells are post-mitotic and express high levels of actin, platelet derived growth factor-β (PDGF-β), and vascular endothelial growth factor receptor-2 (VEGFR-2).22 Proximal to the tip cells are stalk cells that also express VEGFR-2 but, unlike tip cells, are proliferative22 (see Fig. 1-1). During initiation of sprouting angiogenesis, endothelial tip cells develop initial projections prior to stalk cell proliferation.23
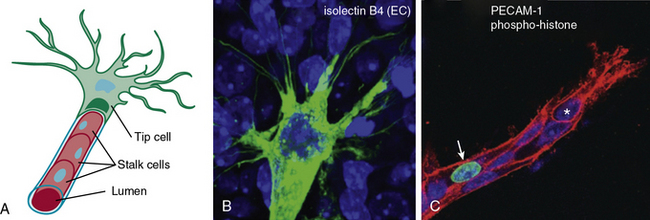
Figure 1-1 Endothelial tip and stalk cells.
(Redrawn with permission from Gerhardt H, Golding M, Fruttiger M, et al: VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161:1163–1177, 2003.)
The mouse retina model has been widely utilized in studies of angiogenesis and is an excellent model for studying different aspects of blood vessel development: retinal vasculature is visible externally and develops postnatally through a stereotyped sequence of well-described steps. In addition, at most time points, the retina simultaneously includes sprouting at the vascular front and remodeling at the core. The VEGF pathway is critical for guiding angiogenic sprouts, and in the retina, expression of the ligand VEGF-A is limited to astrocytes, with the highest levels at the leading edge of the front of the extending EC plexus,22 suggesting that the astrocytes lay down a road map for the ECs to follow.24 Vascular endothelial growth factor-A signals through VEGFR-2 on tip and stalk cells. Interestingly, proper distribution of VEGF-A is required for tip cell filopodia extension and tip cell migration, while the absolute concentration, but not the gradient, of VEGF-A appears to be critical for stalk cell proliferation.22
Similar to sprouting angiogenesis, budding Drosophila trachea airways encompass tip cells that lead branch outgrowth and lagging cells that form the branch tube. Ghabrial and Krasnow used this system to address a fundamental question that commonly arises in a variety of disciplines ranging from politics to sports, and in this case to biology: “What does it take to become a leader?”25 An elegant genetic mosaic analysis showed that tracheal epithelial cells are assigned to the role of tip (i.e., leader) or stalk (i.e., follower) cell in the dorsal branch as a result of a competition for FGF activity.25 Those cells with the highest FGF activity become tip cells, and those with lower activity are relegated to the stalk position.25 Furthermore, Notch pathway–mediated lateral inhibition plays an important role in limiting the number of leading cells.25
Similarly, the Notch pathway is also critical in assigning ECs in sprouting angiogenesis to tip and stalk positions (Fig. 1-2; reviewed in26). The Notch ligand Dll4 is specifically expressed in arterial and capillary ECs, and in the developing mouse retina, Dll4 is enriched in tip cells, while Notch activity is greatest in stalk cells.26–28 Attenuation of Notch activity through genetic (i.e., dll4[+/−]) or pharmacological (i.e., γ-secretase inhibitors) approaches results in increased capillary sprouting and branching, filopodia formation, and tip cell marker expression.26,29 Importantly, VEGF appears to induce dll4 expression in vivo; injection of soluble VEGFR1, which functions as a VEGF sink, into the eyes of mice reduces Dll4 transcript levels.29
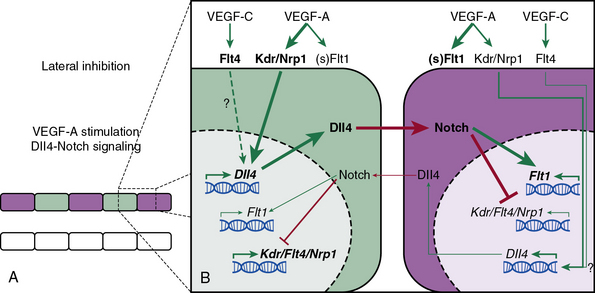
Figure 1-2 Notch-mediated lateral inhibition of neighboring endothelial cells (ECs).
(Redrawn with permission from Phng LK, Gerhardt H: Angiogenesis: a team effort coordinated by notch. Dev Cell 16:196–208, 2009.)
Furthermore, as with the investigations of tip and stalk cells in the Drosophila dorsal airway branches,25 mosaic analyses indicate that competition between cells (in this case for Notch activity) is critical in determining the division of labor in sprouting angiogenesis. Genetic mosaic analysis involves mixing at least two populations of genetically distinct cells in the early embryo, and subsequently comparing the contribution of each cell population to a specific structure or process. Notably, mosaic analysis is usually complementary to experiments with total knockouts and in fact can often be more informative because complete removal of a gene may impair interpretation by grossly distorting the tissue architecture or eliminating competition between cells that harbor differing levels of a gene product.
Experiments using mosaic analysis of Notch pathway mutants in a wildtype background indicate that the Notch pathway acts in a cell autonomous fashion to limit the number of tip cells. In comparison to wildtype ECs in the mouse retina, ECs that are genetically engineered to have reduced or no notch1 receptor expression are enriched in the tip cell population.27
Mosaic studies of Notch signaling components in the developing zebrafish intersegmental vessels (ISVs) are also informative. ISVs traverse between the somites from the dorsal aorta to the dorsal longitudinal anastomotic vessel (DLAV) and are widely used in investigation of blood vessel development. The ISV has been classified as consisting of three (or four) cells in distinct positions: a base cell that contributes to the dorsal aortic cell, a connector cell that courses through the somites, and the most dorsal cell that contributes to the DLAV.30,31 Lateral plate mesoderm angioblasts contribute to the ECs of all the trunk vasculature, including the dorsal aorta, posterior cardinal vein, ISVs, DLAV, and the subintestinal venous vessels. Precursors destined for the ISVs and DLAV initially migrate to the midline dorsal aorta and then between somites to their ultimate positions.30,31 Siekmann and Lawson generated mosaic zebrafish by transplanting into early wildtype embryos marked cells from embryos either lacking the key Notch signaling component recombining protein suppressor of hairless (Rbpsuh) or expressing an activated form of Notch.31 Interestingly, rbpsuh-deficient cells were excluded from the dorsal aorta and enriched in the DLAV position.31 In turn, transplanted cells harboring activated Notch mutations were excluded from the DLAV in mosaics and instead preferentially localized to the base cell and dorsal aorta positions.31
Taken together, the findings indicate that in sprouting angiogenesis, ECs compete for the tip position through Notch-mediated lateral inhibition of neighboring cells26 (see Fig. 1-2). Tip cells express high levels of Dll4, which engages Notch receptors on neighboring cells and thereby inhibits these neighboring cells from developing tip cell characteristics. Furthermore, in the developing retina, the expression of Dll4 is regulated by VEGF-A, which is secreted by astrocytes in response to hypoxia.
Molecular Determinants of Branching
The pattern of many branched structures, such as the vasculature, is critical for function; diverse branched structures use similar signaling pathways to generate their specific patterns. A number of well-studied systems such as the Drosophila trachea, mammalian lung, ureteric bud (UB), and the vasculature consist of hierarchical tubes, progressing from larger to smaller diameter, that transport important gas and/or fluid constituents. The molecular strategies underlying morphogenesis of these patterns often include receptor tyrosine kinase–mediated signaling as well as fine-tuning with inhibitors of these signaling pathways.32,33
In the Drosophila embryo, trachealess selects the trachea primordia and induces conversion of planar epithelium into tracheal sacs that express breathless (btl), the fibroblast growth factor receptor (FGFR) homolog.33,34 The FGF ligand branchless (bnl) is expressed dynamically at positions surrounding the tracheal system, in a pattern which determines where and in which direction a new branch will form.35 Furthermore, loss of bnl prevents branching, and misexpression of bnl induces mislocalized branching.35 Signaling through this FGF receptor pathway is critical for the migration of cells and change in cell shape inherent in formation of primary or secondary airway branches.33,34 Furthermore, tertiary airways consist of a single highly ramified cell whose pattern is not inherently fixed, but instead adapts to tissue oxygen needs in an FGF-dependent manner.36 Finally, as a means of fine-tuning Drosophila airway patterning, branchless induces sprouty, an inhibitor of FGFR signaling, which blocks branching.37,38
Evolutionary conservation of these signaling pathways is striking because the FGF pathway is also essential for determining branch patterning in the mammalian airway system (e.g., the lung). In the mouse, trachea and lung bronchi bud from gut wall epithelium at about E9.5,39 Subsequently, three distinct branching subroutines are repeated in various combinations to generate a highly stereotyped, complex, tree-like structure40 that facilitates gas exchange. In early embryogenesis, the visceral mesenchyme adjacent to the heart expresses FGF10, and FGF10 binds endodermal FGFR2b, the mouse ortholog of Drosophila breathless.32 FGF10 null mice lack lungs and have a blind trachea.41 Similarly, FGFR2b(−/−) mice form underdeveloped lungs that undergo apoptosis.42 Akin to the Drosophila tracheal system, sprouty is a key component of an FGF-induced negative-feedback loop in the lung.38 In response to FGF10, FGFR2b induces Sprouty2 tyrosine phosphorylation and activation, and active Sprouty2 inhibits signaling downstream of FGFR2b.32 In addition, carefully regulated levels of the morphogens sonic hedgehog and bone morphogenic protein (BMP) 4 modulate the branching of lung airways.32
As with the Drosophila and mammalian airway systems, generation of the metanephric kidney requires signals conveyed through epithelial receptor tyrosine kinase. The metanephric mesenchyme secretes glial-derived neurotrophic factor (GDNF), which activates the receptor tyrosine kinase Ret and its membrane-anchored co-receptor Gdnf family receptor alpha 1 (Gfra1), thereby inducing the UB to evaginate from the nephric duct.43,44 These components are required for UB branching because UB outgrowth fails in mice null for Gdnf, Gfra1, or Ret.43 Furthermore, RET is frequently mutated in humans with renal agenesis.45 In addition, FGFR2b is also highly expressed on UB epithelium, and FGFR2b-mediated signaling regulates UB branching.32 FGF7 and FGF10 are expressed in mesenchymal tissue surrounding the UB, and FGFR2b binds with comparable affinity to these ligands.32 As with lung development, BMP4-mediated signaling modulates the branching of the renal system.32
The most well-studied molecular determinants of vascular branching are the VEGF family of ligands (VEGF-A, -B, -C, and -D) and endothelial receptor tyrosine kinases (VEGFR1, 2, and 3).46 VEGF has been shown to be a potent EC mitogen and motogen and vascular permeability factor, and the level of VEGF is strictly regulated in development; VEGF heterozygous mice die around E11.5 with impaired angiogenesis and blood island formation.47,48 During embryogenesis, VEGFRs are expressed in proliferating ECs and the ligands in adjacent tissues. For instance, secretion of VEGF by the ventricular neuroectoderm is thought to induce capillary ingrowth from the perineural vascular plexus.49 Mice null for VEGFR2 or VEGFR1 die around E9.0, with VEGFR2(−/−) mice lacking yolk-sac blood islands and vasculogenesis50 and VEGFR1(−/−) mice displaying disorganized vascular channels and blood islands.51 Although VEGFR3 expression eventually restricts to lymphatic ECs, its broad vascular endothelial expression early in development is critical for embryonic morphogenesis. Indeed, VEGF3 null mice undergo vasculogenesis and angiogenesis; however, the lumens of large vessels are defective, resulting in pericardial effusion and cardiovascular failure by E9.5.52 As with hypoxia-induced FGF-dependent tertiary branching in the Drosophila airway,36 low oxygen levels induce vascular EC branching through hypoxia-inducible factor-1 alpha (HIF-1α)-mediated expression of VEGFR2.53 VEGFR1 is thought to largely function as a negative regulator of VEGF signaling by sequestering VEGF-A. The affinity of VEGFR1 for VEGF-A is higher than that of VEGFR2, and VEGFR1 kinase domain mutants are viable.46
Although generally not as well studied as the role of the VEGF pathway in vessel branching, other signaling pathways, such as those mediated by FGF, Notch, and other guidance factors, are also likely to play important roles. For instance, transgenic FGF expression in myocardium augmented coronary artery branching and blood flow, whereas expression of a dominant-negative FGFR1 in retinal pigmented epithelium reduced the density and branching of retinal vessels.32 Furthermore, a murine homolog of sprouty was shown to inhibit small blood vessel branching and sprouting in mouse embryo cultures.54 The role of the Notch pathway was discussed earlier in the section on endothelial tip and stalk cells. The role of guidance cues initially described in the nervous system is discussed later in the section on neurons and vessels. Finally, the maturation of branches to a more stable state that is resistant to pruning is thought to largely be regulated by signaling pathways that modulate EC branch coverage by mural cells. Interestingly, two of the most important such pathways involve receptor tyrosine kinases such as the angiopoietin-Tie and PDGF ligand receptor pathways.
Vascular Lumenization
Endothelial cells at the tips of newly formed branches lack lumens, but as the vasculature matures, formation of a lumen is an essential step in generating tubes that can transport products. Angioblasts initially migrate and coalesce to form a solid cord that is subsequently hollowed out to generate a lumen through a mechanism that has recently become controversial. Around 100 years ago, researchers first suggested that vascular lumenization in the embryo occurs through an intracellular process involving vacuole formation.55 Seventy years later, Folkman and Haudenschild developed the first method for long-term culture of ECs, and bovine or human ECs cultured in the presence of tumor-conditioned medium were shown to form lumenized tubes (56and references therein). In this and similar in vitro approaches, an individual cell forms Cdc42+ pinocytic vacuoles that coalesce, extend longitudinally, and then join the vacuole of neighboring ECs to progressively generate an extended lumen.56–58 Subsequently, a study using two-photon high-resolution time-lapse microscopy suggested that the lumens of zebrafish ISVs are generated through a similar mechanism of endothelial intracellular vacuole coalescence, followed by intercellular vacuole fusion.59
Recently, however, a number of studies have called this intracellular vacuole coalescence model into question, and instead support an alternate model in which the lumen is generated extracellularly (reviewed in60). One such investigation61 suggests that in contrast to what had been thought previously,30,31 ECs are not arranged serially along the longitudinal axis of the zebrafish ISV, but instead overlap with one another substantially; the circumference of an ISV at a given longitudinal position usually traverses multiple cells. If the lumen of a vessel were derived intracellularly in a unicellular tube, the tube would be “seamless” (as in the terminal cells of Drosophila airways62) and only have intercellular junctions at the proximal and distal ends of the cells. However, in the 30 hours post fertilization zebrafish, junctional proteins zona occludens 1 (ZO-1) and VE-cadherin are co-expressed, often in two medial “stripes” along the longitudinal axis of the ISV, suggesting that ECs align and overlap along extended regions of the ISV.61 Thus, the lumen is extracellular—that is, in between adjacent cells, not within the cytoplasm of a single cell.
In addition, recent investigations show that EC polarization is a prerequisite for lumen formation, and both the Par3 complex and VE-cadherin play a critical role in establishing polarity.63 Endothelial-specific knockdown of β1-integrin reduces levels of Par3 and leads to a multilayered endothelium with cuboidal-shaped ECs and frequent occlusion of midsized vascular lumens.63 VE-cadherin is a transmembrane EC-specific cell adhesion molecule that fosters homotypic interactions between neighboring ECs, and in vascular cords, VE-cadherin is distributed broadly in the apical membrane (reviewed in60). VE-cadherin deletion is embryonic lethal in the mouse; development of VE-cadherin(−/−) embryonic vessels arrests at the cord stage and does not proceed to lumenization.60,64,65 Under normal conditions, during polarization, junctions form at the lateral regions of the apical membrane as VE-cadherin translocates to these regions, which also harbor ZO-1.60 VE-cadherin is required for the apical accumulation of de-adhesive molecules, such as the highly glycosylated podocalyxin/gp135, which likely contributes to lumen formation through cell-cell repulsion. In addition to anchoring neighboring ECs, VE-cadherin also is linked through β-catenin, plakoglobin, and α-catenin to the F-actin cytoskeleton.60
Although establishing polarity of the ECs is a critical step, it is insufficient to induce lumen formation. Indeed, in VEGF-A(+/−) mice, ECs of the dorsal aorta polarize, but this vessel does not lumenize.65 VEGF-A activates Rho-associated protein kinases (ROCKs) that induce nonmuscle myosin II light chain phosphorylation, thereby enhancing recruitment of nonmuscle myosin to the apical membrane.65 Actomyosin complexes at the apical surface are thought to play an important role in pulling the apical membranes of neighboring cells apart, thus generating an extracellular lumen.63
Another important component of the process of EC cord lumenization is the dynamic dissolution and formation of inter-EC junctions. Egfl7 is an EC-derived secreted protein that promotes EC motility and is required for tube formation.66 The knockdown of Egfl7 in zebrafish impairs angioblasts from dissolving their junctions, preventing them from separating, which is required for tube formation.66 Interestingly, a recent study suggests that excessive cell-cell junctions in migratory angioblasts may explain the delayed migration of these cells in endodermless zebrafish.5
Neurons and Vessels
The similarities between the vasculature and neurons extend well beyond the cell biology of their respective sensors (i.e., tip cells, growth cones). Interestingly, in many organs, vascular and neural networks are closely aligned67 (Fig. 1-3). In a landmark paper, Mukouyama et al. investigated vascular and neural development of the mouse limb, in which skin arteries but not veins are specifically aligned with peripheral nerves.68 As in many developing vascular beds, the vasculature of the mouse limb initially consists of an EC plexus that, in the case of the limb bud, is present prior to peripheral nerve invasion. Subsequently, nerve invasion and vascular plexus remodeling ensue, resulting in formation of larger vessels, and most nerve-associated vessels express arterial markers such as ephrinB2, Neuropilin1 (Nrp1), and/or Connexin40 (CX40).68 The semaphorins are a family of important axon guidance factors, and mice null for Semaphorin3A (Sema3A) display disorganized peripheral nerve growth. Interestingly, in Sema3A(−/−) mice, small-diameter blood vessels align with this disorganized array of peripheral nerves and express Nrp1 and CX40.68 In contrast, Neurogenin1/Neurogenin2 compound homozygous nulls have essentially no peripheral nerves or associated Schwann cells in the limb skin and have markedly reduced arterial marker expression in small-diameter vessels.68 Finally, to specifically examine the role of Schwann cells, the authors investigated mice with homozygous null mutations in erbB3, a co-receptor for the axon-derived signal Neuregulin-1.68 These erbB3(−/−) mice lack peripheral Schwann cells and, similar to Sema3A null mice, have a disordered pattern of axon growth. However, in contrast to Sema3A null mice, there is a marked reduction in both arterial marker expression and association of blood vessels with the disordered peripheral nerves in erbB3(−/−) limb skin. Furthermore, Schwann cells isolated from wildtype limb skin express VEGF, and in co-culture, Schwann cells induce undifferentiated ECs to express ephrinB2 in a VEGFR2-dependent manner.68 Taken together, the mouse limb skin provides an example of how neurons and/or neural-associated tissues such as Schwann cells can modulate the patterning and differentiation of arterial networks.
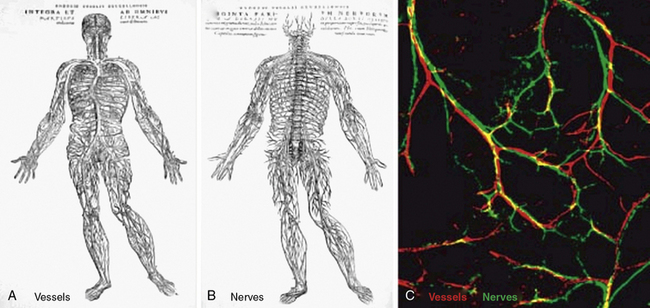
Figure 1-3 Parallels in vessel and nerve patterning.
(Redrawn with permission from Carmeliet P, Tessier-Lavigne M: Common mechanisms of nerve and blood vessel wiring. Nature 436:193–200, 2005; and Mukouyama YS, Shin D, Britsch S, et al: Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the skin. Cell 109:693–705, 2002.)
An alternative compelling potential mechanism underlying the alignment of vascular and neural networks is mutual guidance, in which the patterning of these tissues is regulated by a third structure. For instance, in the developing lung, some airways are accompanied by a closely juxtaposed artery and neuron (unpublished results). Guidance cues are integral in regulating neural patterning through their actions as attractants or repellants in short (cell- or matrix-bound) or long (diffusible) range,67 and a wealth of recent investigations have demonstrated that members of the four families of axon guidance cues (i.e., netrins, semaphorins, ephrins, slits) and their receptors play critical roles in vascular patterning.67,69 Both the nervous and vascular systems express Nrps and Eph receptors, whereas Robo4, UNC5B, and PlexinD1 expression is mostly confined to the vasculature69 (Fig. 1-4). Thus, in some locations, neurons and vessels may be co-patterned by similar guidance cues emitted by adjacent non-neuronal, nonvascular structures.
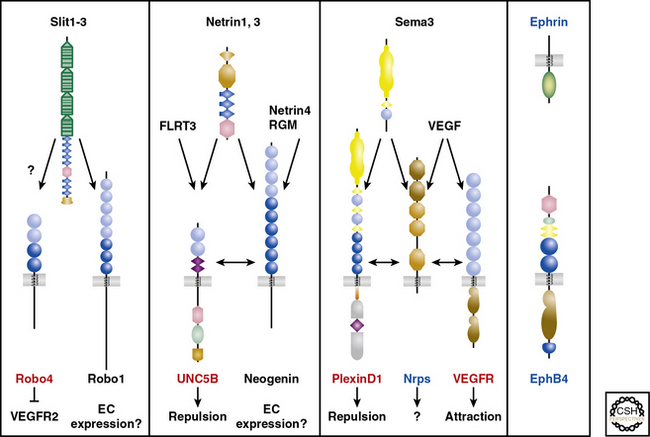
Vascular Induction of Nonvascular Tissue Development
In addition to neural patterning of the vasculature and mutual guidance of neurons and vessels, signals emitted from cells of the developing vasculature may modulate development of neurons and other nonvascular tissues. Studies with artemin (ARTN), a member of the GDNF family of ligands, and GDNF family receptor (GFR) α3/ret receptor complexes implicate vessels as playing a critical role in patterning sympathetic neurons.70,71 In mice with the tau-lacZ gene “knocked in” to the ARTN or GFRα3 locus, X-gal and anti-lacZ immunohistochemical stains indicate that ARTN is expressed in VSMCs, and GFRα3 is expressed throughout the sympathetic nervous system.71 ARTN null mice have disrupted sympathetic neuroblast migration and impaired target tissue innervation, resulting in ptosis.71 Because blood vessels may indirectly influence development of adjacent nonvascular tissues through delivery of growth factors or inhibitors, it is imperative to evaluate the role of vascular tissues and/or vessel-derived signals in the absence of blood flow. Cultured rat VSMCs and sympathetic ganglia express ARTN and GFRα3, respectively, and co-culturing femoral arteries with sympathetic ganglia promotes neurite growth in a largely ARTN-dependent manner.70 Furthermore, ARTN-coated beads placed adjacent to sympathetic chains in whole embryo mouse cultures induce robust neurite outgrowth towards the ectopic source of ARTN.71
In addition to induction of neural networks, the vasculature plays an important role in shaping morphogenesis of other tissues, including endodermal-derived organs. For instance, shortly after the initial specification and proliferation of hepatic cells in the endodermal epithelium, the early nascent liver bud invades the adjacent septum transversum mesenchyme. Prior to invasion, discontinuous angioblasts that have not yet formed tubes comprise a loose network located between the early epithelial and mesenchymal layers.72 Matsumoto et al. argue that this primitive vasculature interacts with nascent liver cells “prior to blood vessel formation and function.”72 VEGFR-2(−/−) embryos lack ECs,50 and their early hepatic endodermal cells fail to both proliferate adequately and invade the septum transversum mesenchyme.72 Furthermore, experiments with liver bud explants isolated from VEGFR-2 null mice or cultured in the presence of EC inhibitors show that ECs specifically induce hepatic cell proliferation.72 Similarly, the dorsal aorta has been implicated as playing an important role in development of the dorsal pancreatic bud, which gives rise to the body and tail of the pancreas.73 In co-culture experiments, dorsal aortic tissue induces dorsal endodermal expression of pancreatic transcription factors as well as hormones such as insulin.73–75 Removal of aortic precursors in Xenopus embryos or deletion of VEGFR-2 in mice results in failure to form the dorsal pancreatic bud or to express insulin, respectively.74,75 In addition to directly influencing pancreatic morphogenesis, aortic ECs have an indirect effect by promoting survival of nearby mesenchymal cells, which in turn signal to the dorsal pancreatic bud.76 Furthermore, a case study of a patient with coarctation of the aorta and dorsal pancreas agenesis demonstrates the clinical relevance of these developmental studies.77
Lymphatic Vessel Development
Based on her experiments over 100 years ago, Florence Sabin proposed the “centrifugal model” in which lymphatic sacs derive from veins, and vessels sprouting from these sacs give rise to the lymphatic vasculature.78,79 Recently, histological, marker, and lineage studies have yielded findings supportive of Sabin’s model (reviewed in1). The homeobox transcription factor Sox18 (sex-determining region Y box 18) is a molecular switch that turns on the differentiation of venous ECs to a lymphatic EC fate,80 and mutations in SOX18 underlie lymphatic abnormalities in the human disorder hypotrichosis-lymphedema-telangectasia.81 Sox18 induces expression of a number of lymphatic markers, including the homeobox gene Prox1,80 which is absolutely required to initiate lymphatic vessel morphogenesis.1 Lymphatic development begins in the lateral parts of the cardinal veins with EC expression of Sox18, followed by Prox1 expression, and subsequently these Sox18+/Prox1+ ECs sprout laterally and form lymph sacs.1,80 The peripheral lymphatic vasculature then results from centrifugal sprouting from the lymph sacs and remodeling of the LEC capillary plexus. The Tie2-GFP transgene is expressed specifically in blood ECs and not in LECs or undifferentiated mesenchyme, whereas lineage tracing with the transgenic Tie2-Cre strain and the R26R lacZ cre reporter marks LECs, further supporting a venous origin for lymphatics.82 Interestingly, the venous identity of lymphatic precursors is critical; deletion of COUP-TFII in ECs results in arterialization of veins and inhibition of LEC specification of cardinal vein ECs.82,83
Tunica Media: Smooth Muscle and Extracellular Matrix
Cellular and Extracellular Matrix Components
Although differences exist between VSMCs and pericytes (Table 1-1), in general these mural cell types are considered to exist along a continuum and lack rigid distinctions (reviewed in84). Pericytes are imbedded in the basement membrane of capillary ECs, and thus may be characterized as having an intimal location, whereas VSMCs are separated from the basement membrane in the media. Vascular smooth muscle cells are oriented circumferentially around the vessel, whereas pericytes have an irregular orientation. Pericytes contact multiple ECs and are thought to play important roles in intercellular communication, microvessel structure, and phagocytosis; VSMCs are important in regulating vascular tone. Molecular markers of these cell types are overlapping, but the commonly used markers of pericytes include platelet-derived growth factor receptor beta (PDGFR-β), neuron glial 2 (NG2), and regulator of G-protein signaling 5 (RGS5). The markers of VSMCs include alpha–smooth muscle actin (αSMA) and smooth muscle myosin heavy chain (SMMHC).
Table 1-1 Vascular Mural Cells: Pericytes and Vascular Smooth Muscle Cells*
| Characteristic | Pericyte | VSMC |
|---|---|---|
| Vessel size | Smaller | Larger |
| Vascular wall location | Within endothelial BM | Media |
| Orientation in vessel wall | Irregular | Circumferential |
| “Function” | Intercellular communication Microvessel structure Phagocytosis (in CNS) |
Vascular tone |
| “Canonical” markers | PDGFR-β, NG2, RGS5 | αSMA, SMMHC |
BM, basement membrane; CNS, central nervous system; NG2, neuron glial 2; PDGFR-β, platelet derived growth factor receptor beta; RGS5, regulator of G-protein signaling; αSMA, alpha-smooth muscle actin; SMMHC, smooth muscle myosin heavy chain; VSMC, vascular smooth muscle cell.
* Differences between pericytes and VSMCs are noted, but in general, these mural cell types lack rigid distinctions and are considered to exist along a continuum.62 See text for details.
Vascular Smooth Muscle Cell Origins
The origins of VSMCs are diverse and differ among blood vessels and even within specific regions of individual blood vessels (Fig. 1-5; reviewed in3). Interestingly, the borders between SMCs of different lineages are sharp, with little mixing among cells of different origins. Smooth muscle cells of the aorticopulmonary septum, aortic arch, and cranial vessels derive from neural crest cells of the ectoderm, and descending aorta SMCs originate from the mesoderm.3 Using hoxB6-cre to mark cells derived from the LPM, Wasteson et al. suggest that these cells are the source of descending aortic ECs and that the ventral wall of the descending aorta is temporarily inhabited at around E9.5 for about 1 day with early SMCs that derive from the LPM.85 Subsequently, Meox1-cre, which labels cells derived from both the presomitic paraxial mesoderm and the somites, marks SMCs that replace the LPM-derived aortic wall cells.85 Thus, in the adult descending aorta, ECs and SMCs derive from distinct mesodermal populations, the LPM and the presomitic/somitic mesoderm, respectively. Importantly, another investigation using a powerful and distinct approach, clonal analysis, previously showed that aortic SMCs share a lineage with paraxial mesoderm-derived skeletal muscle cells.86 Here, a nlaacZ reporter containing a duplication of the lacZ coding sequence that yields a truncated inactive β-galactosidase enzyme was targeted to the α-cardiac actin locus.86 The nlaacZ reporter requires a very rare intragenic recombination event that is heritable and random in order to generate a functional lacZ gene.86 X-gal staining showed that only 2% of nlaacZ embryos analyzed had labeled cells in the dorsal aorta; of these, two thirds had concomitant labeling in the somitic-derived myotome.86 Finally, Topouzis and Majesky suggest that the lineage of SMC populations has important functional implications.87 In response to transforming growth factor (TGF)-β stimulation, ectodermally-derived E14 chick embryo aortic arch SMCs increase deoxyribonucleic acid (DNA) synthesis, while the growth of mesodermally derived abdominal aortic SMCs was inhibited.87
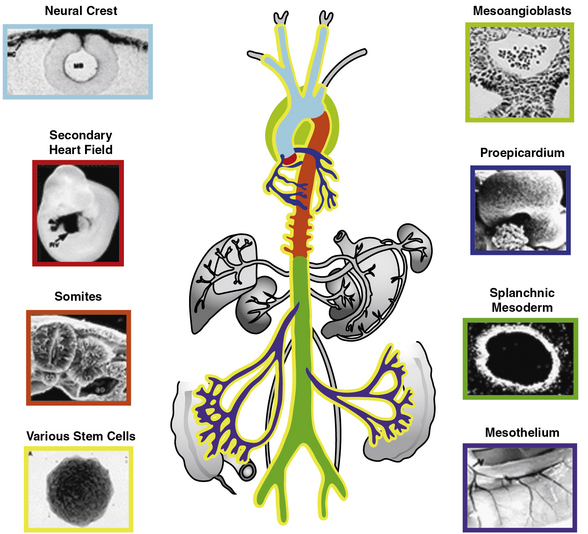
Figure 1-5 Developmental origins of vascular smooth muscles.
(Redrawn with permission from Majesky MW: Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol 27:1248–1258, 2007.)
Coronary artery SMCs are critical players in atherosclerotic heart disease, and there has been significant investigation into their origin from the proepicardium (reviewed in88). The proepicardium is a transient tissue that forms on the pericardial surface of the septum transversum in the E9.5 mouse and, through a fascinating process, gives rise to epicardial cells that migrate as a mesothelial sheet over the myocardium. Signals emanating from the myocardial cells induce an epithelial-to-mesenchymal transition (EMT) in which some epicardial cells lose their cell-cell adhesion and invade the myocardium. Furthermore, lineage labeling with dyes and viral vectors and more recently with genetic approaches using the Wilms tumor1 (Wt1)-cre has illustrated that the proepicardium and epicardium contribute to the coronary artery SMC lineage.88,89
Similar to these studies of the coronary artery, investigations of other organs suggest the mesothelium could more generally be an important source of VSMCs. For instance, Wilm et al. showed that expression of the Wt1 protein in the developing gut is limited to the serosal mesothelium, and a Wt1-cre yeast artificial chromosome (YAC) transgene marked a lineage of cells that includes the SMCs of gut and mesenteric major blood vessels.90 Using the Wt1-cre YAC transgene and a panel of cre reporters, lung mesothelium was implicated as the source of about a third of all pulmonary vascular cells expressing αSMA.91
More recently, the etiology of pulmonary artery SMCs has become controversial. Morimoto et al. reported that embryos with the same Wt1-cre YAC transgene and a R26R-YFP cre reporter have only rare YFP+ lung VSMCs.92 Furthermore, using the Tie1-cre, these authors suggest that most SMCs of the proximal pulmonary arteries arise from ECs.92 Transdifferentiation of ECs into VSMCs has been raised previously in developmental and disease contexts.8,9,93 For instance, embryonic stem cell–derived Flk1+ cells have the potential to differentiate into ECs or mural cells.9 However, our recent results with the VE-cadherin-cre94 and mTomato/mGFP cre reporter95 indicate that ECs are not a significant source of the E18.5 pulmonary arterial SMCs. Additional experiments indicate that instead, these cells largely derive from local mesenchyme.96
Smooth Muscle Cell Differentiation
A critical component of characterizing the morphogenesis of any tissue (e.g., vascular smooth muscle) is defining morphological and molecular criteria that constitute the differentiated phenotype of specific cell types (e.g., VSMCs) that make up the tissue. Early undifferentiated cells that are presumed to be destined to the VSMC fate have prominent endoplasmic reticulum and Golgi, a euchromatic nucleus, and lack a distinctly filamentous cytoplasm.97 In contrast, mature VMSCs have a heterochromatic nucleus, myofilaments, and decreased synthetic organelles.97 In addition to these morphological changes, differentiation of SMCs is marked by expression of a number of contractile and cytoskeletal proteins. αSMA is the most abundant protein of SMCs, comprising 40% of the total protein in a differentiated SMC.2 αSMA is an early marker of SMCs but is nonspecific; it is expressed in skeletal muscle and a variety of other cell types, and is temporarily expressed in cardiac muscle during development.2,98 The actin and tropomyosin binding protein transgelin (also known as SM22α) is another early marker of SMCs and a more specific marker of adult SMCs; however, it also is expressed in the other muscle types during development.98 The two isoforms of SMMHC are expressed slightly later during development than αSMA and SM22α, and in contrast to these other markers, SMMHC expression is limited to the SMC lineage.99 Smoothelin is another cytoskeletal protein that is also specific for SMCs but is not expressed until very late in the differentiation process when the cells are part of a contractile tissue.100
Studies of VSMC development or even SMCs in the mature blood vessel are challenging because these cells can assume a variety of phenotypes, depending on their environment.2 During the early stages of blood vessel development, many VSMCs rapidly proliferate, migrate substantial distances, and synthesize large amounts of ECM components. In contrast, more mature VSMCs are predominantly sedentary and nonproliferative and express contractile proteins but do not generate significant ECM. However, the distinctions between these synthetic and contractile states are not always firm. Even adult VSMCs are not terminally differentiated, so in many vascular diseases, extracellular cues are implicated in inducing VSMCs to assume a dedifferentiated state through a process termed phenotypic modulation.2
Underlying these phenotypes, the gene expression program of SMCs toggles between a differentiated contractile set of genes and a distinct, undifferentiated, synthetic and proliferative set of genes.101 Expression of almost all smooth muscle contractile and cytoskeletal genes is modulated by the ubiquitous transcription factor serum response factor (SRF). Serum response factor binds the 10-base-pair DNA consensus sequence CC(A/T)6GG known as the CArG box (i.e., C, AT rich, G box), which is found in the regulatory regions of virtually all smooth muscle genes. In fact, for most SMC genes, there are at least two CArG boxes. However, the CArG box sequence is also found within the 23-base-pair serum response enhancer element of early growth response genes such as the c-fos proto-oncogene.101 Because SRF is ubiquitous and the cis-regulatory CArG element is present in both growth and differentiation genes, a higher order of control is required to determine which of these disparate gene sets are expressed in a specific cell at a given time period.
Control of expression of contractile and cytoskeletal SMC genes is regulated through a competition for SRF between the transcriptional coactivator myocardin and ternary complex factors.102 Myocardin is a master regulator of SMC differentiation in that ectopic expression of this factor in nonmuscle cells is sufficient to induce activation of the SMC differentiation gene program.103 In addition, murine embryos null for myocardin lack VSMC differentiation and die at mid-gestation.104 Counterbalancing this effect of myocardin is the ternary complex factor Elk-1, which acts as a myogenic repressor by competing with myocardin for a common docking site on SRF, thereby preventing induction of SMC differentiation gene expression.102
Patterning of Developing Vascular Smooth Muscle Cell Layers
Although a number of recent investigations describe the molecular mechanisms regulating SMC differentiation, there are relatively few studies of the patterning of morphogenesis of SMC layers of a developing blood vessel (reviewed in97). Consequently, little is known about recruitment of SMCs and/or their precursors to the vascular wall, investment of these cells around the nascent EC tube, and the pattern of differentiation of VSMC precursors within or in proximity to the vascular wall. Limited relevant studies have mostly focused on histology and αSMA expression in the developing aortic wall. Early in development, the dorsal aortae exist as parallel tubes that subsequently fuse to generate the single descending aorta. The early EC tube is surrounded by loose undifferentiated mesenchymal cells, and as the aorta matures, expression of αSMA proceeds in a cranial-to-caudal direction.105,106 Within a cross-section of the descending aorta, the location of initial mesenchymal cell consolidation and αSMA expression depends on the cranial-caudal position: proximally these processes initially occur on the dorsal aspect of the aorta, whereas more distally they are first noted on the ventral side.105,106 Studies published 40 years ago indicate that within the chick aortic media, outer layers mature initially with condensation and elongation of early presumptive SMCs and accumulation of elastic tissue.107,108 In contrast, in rodent or quail aortae, cells immediately adjacent to the EC layer are the first to consolidate and express SMC markers; subsequently, additional layers of SMCs are added.105,106,109,110
Recently we have undertaken a meticulous investigation of murine pulmonary artery morphogenesis and found that the medial and adventitial wall of this vessel is constructed radially from inside out by sequential induction and recruitment of successive layers.96 The inner layer undergoes a series of morphological and molecular transitions that lasts about 3 days in order to build a relatively mature SMC layer. After this process commences in the first layer, the next layer initiates and completes a similar process. Finally, this developmental program arrests midway through construction of the outer layer to generate a relatively “undifferentiated” adventitial cell layer.
This inside-outside radial patterning is likely to involve an EC-derived signal and result from one or more potential mechanisms. For instance, in the morphogen gradient model,111 an EC-derived signal diffuses through the media and adventitia and, depending on discrete concentration thresholds, induces responses in the cells of these compartments, such as changes in morphology, gene expression, and/or proliferation. Alternatively, in the relay mechanism,112 a short-range or plasma membrane-bound EC signal induces adjacent cells, which in turn propagate the signal through either secreting a morphogen or inducing their neighbors, and so on (i.e., “the bucket brigade model”). Such a bucket brigade mediated by the Notch ligand Jagged1 on SMCs is implicated in regulating ductus arteriosus closure in a recently published report.113 Finally, our recent results suggest a third mechanism in which some of the progeny of inner-layer SMCs migrate radially outward to contribute to the next layer(s) of SMCs.96
A number of signaling pathways involving an EC-derived signal and mesenchymal receptors have been implicated in vascular wall morphogenesis (reviewed in114). The PDGF pathway is perhaps the most well-studied pathway in vascular mural cell development, with a ligand expressed in ECs (PDGF-β) and receptors expressed in undifferentiated mesenchyme (PDGFR-α and -β) and pericytes (PDGFR-β). Mice null for PDGF-β or PDGFR-β have reduced SMC coverage of medium-sized arteries and lack pericytes, which results in microvascular hemorrhages and perinatal lethality.115–118 In addition, when co-cultured with ECs, undifferentiated embryonic mesenchymal 10 T1/2 cells are induced to express SMC markers and elongate in a TGF-β-dependent manner.119 Similar changes are also induced by directly treating 10 T1/2 cells with TGF-β1.119 Furthermore, the Notch pathway has been shown to play important roles in arterial SMC differentiation in vivo (reviewed in120), and EC-derived Jagged1 is required for normal aortic and yolk sac vessel SMC differentiation.121 In human adults, the receptor Notch3 is specifically expressed in arterial SMCs, and at birth, blood vessels of Notch3 null mice and wildtype mice are indistinguishable.122,123 However, Notch3 is required for postnatal maturation of the tunica media of small vessels in mice.122 Furthermore, NOTCH3 mutations in humans cause the CADASIL (cerebral autosomal dominant arteriopathy with stroke and dementia) syndrome, characterized clinically by adult-onset recurrent subcortical ischemic strokes and vascular dementia, and pathologically by degeneration and eventual loss of VSMCs.123,124 Finally, it is important to note that other signaling pathways, such as those mediated by angiopoietin-Tie and S1P ligand-receptor pairs, do not involve an EC-derived ligand and/or mesenchymal receptors but play important roles in SMC development.114
Extracellular Matrix: Collagen and Elastic Fibers
In addition to maturation of cellular constituents of the blood vessel wall, proper formation of the ECM is also critical for vascular function. Gene expression profiling of the developing mouse aorta revealed dynamic expression of most structural matrix proteins: an initial major increase of expression at E14 is often followed by a brief decrease at postnatal day 0 (P0), and then a steady rise for about 2 weeks, and finally a decline to low levels at 2 to 3 months that persist into adulthood.125,126 Within the tunica media, circumferential collagen fibers have high tensile strength and bear most of the stressing forces at or above physiological blood pressures.126 Seventeen collagens are expressed in the developing murine aortic wall, and deletions in a number of them result in vascular phenotypes.126 Furthermore, COLLAGEN3A1 mutations in humans are responsible for Ehlers-Danlos syndrome type IV, with vascular manifestations that include vessel fragility and large-vessel aneurysm and rupture.126
In contrast to collagen, elastin has low tensile strength, is distensible, and distributes stress throughout the wall, including onto collagen fibers.126 Elastin is the major protein of the arterial wall, comprising up to 50% of the dry weight of the aorta.127 Vascular smooth muscle cells secrete tropoelastin monomers that undergo posttranslational modifications, cross-linking, and are organized into circumferential elastic lamellae in the tunica media. These elastic lamellae alternate with rings of VSMCs to form lamellar units. Eln(+/−) mice have a normal lifespan despite being hypertensive and having a 50% reduction in elastin mRNA.128,129 In comparison to wildtype, the Eln(+/−) aorta has thinner elastic lamellae but a 35% increase in the number of lamellar units, which results in a similar tension per lamellar unit.129,130 More dramatically, humans hemizygous for the ELN-null mutant have a 2.5-fold increase in lamellar units and suffer an obstructive arterial disease, supravalvular aortic stenosis.129 Similarly, at the end of gestation in the mouse, subendothelial cells of Eln(−/−) arteries are hyperproliferative, resulting in increased numbers of αSMA+ cells and reduced luminal diameter, with lethality by P4.5.131 Furthermore, it is conceivable that localized disruption of elastin in the mature artery results in focal SMC phenotypic modulation and consequent neointima formation132 (Fig. 1-6).
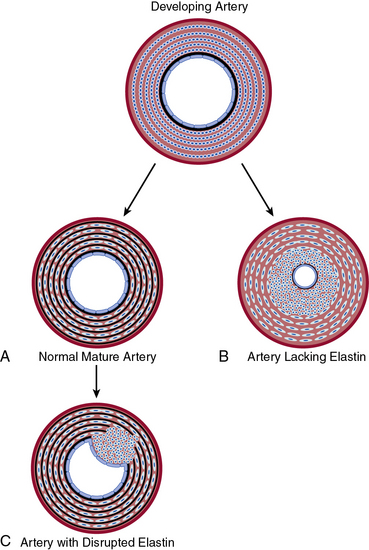
Figure 1-6 Elastin–vascular smooth muscle cell (VSMC) interactions in development and disease.
A, During normal development, concentric rings of elastic lamellae form around arterial lumen. Elastin signals VSMCs to localize around elastic lamellae and remain in a quiescent, contractile state. B, In the absence of elastin, this morphogenic signal is lost, resulting in pervasive subendothelial migration and proliferation of VSMCs that occlude vascular lumen. C, Karnik et al. propose that vascular injury of the mature artery may focally disrupt elastin, releasing smooth muscle cells (SMCs) to dedifferentiate, migrate, and proliferate and thereby contribute to neointimal formation.132
(Redrawn with permission from Karnik SK, Brooke BS, Bayes-Genis A, et al: a critical role for elastin signaling in vascular morphogenesis and disease. Development 130:411–423, 2003.)
Finally, microfibrils are fibrous structures intimately associated with elastic fibers surrounding the elastin core. Fibrillin1 is the major structural component of microfibrils, and its temporal pattern of expression during aortic development is similar to that of most structural proteins (e.g., elastin), except the peak expression of fibrillin1 occurs at P0.125 Mutations in the human FBN1 gene result in Marfan syndrome, with vascular manifestations that include aortic root aneurysm and dissection.133
Tunica Adventitia: Fibroblasts and Loose Connective Tissue
Owing to a striking paucity of studies, very little is known about development of the outer layer of blood vessels, which is referred to as the tunica adventitia or tunica externa. The tunica externa is composed of loose connective tissue (mostly collagen), and the predominant cell type is the fibroblast. Diffusion of nutrients from the lumen to the adventitia and outer media is inadequate in larger vessels, so the adventitia of these vessels also includes small arteries known as the vaso vasorum that supply a capillary network extending through the adventitia and into the media. The adventitia of coronary vessels is thought to arise from the epicardium, based on experiments with quail-chick transplants.134 Quail epicardial cells grafted into the pericardial space of the E2 chick undergo EMT and contribute to both coronary vascular SMCs (consistent with findings discussed earlier regarding VSMC origins in the tunica media) and coronary perivascular fibroblasts.134
Recently a number of studies have investigated a population of adventitial cells expressing stem cell markers. These investigations are largely a result of a paradigm shift: classically, the adventitia was considered a passive supportive tissue, but more recently, adventitial fibroblast and progenitor cells have been implicated as playing an important role in neointimal formation during vascular disease.135,136 A niche for cells expressing the stem cell marker CD34 (but not the EC marker CD31) has been identified in the interface between the media and adventitia of human internal thoracic arteries.137 The intensively studied growth factor Shh is expressed in this vascular “stem cell” niche of medium and large-sized arteries of the perinatal mouse.138 Patched-1 (Ptc1) and patched-2 (Ptc2) are Shh target genes, and their gene products are Shh receptors. β-Galactosidase staining in Shh reporter mice, Ptc1lacZ or Ptc2lacZ, suggests that Shh signaling is active in the adventitia during the late embryonic period and early postnatal period.138 Cells expressing the stem cell marker Sca1 are located in the adventitia of the mouse between the aortic and pulmonary trunks, initially in the late embryonic stages and persisting into adulthood, and Shh signaling appears to be critical for this population of cells because the number of adventitial Sca1+ cells is greatly diminished in Shh null mice.138 In sum, the adventitia is likely to be an important tissue in vascular development and disease; however, its role in these processes is critically understudied.
1 Tammela T., Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140(4):460–476.
2 Owens G.K., Kumar M.S., Wamhoff B.R. Molecular regulation of vascular differentiation in development and disease. Physiol Rev. 2004;84(3):767–801.
3 Majesky M.W. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27(6):1248–1258.
4 Pardanaud L., Yassine F., Dieterlen-Lievre F. Relationship between vasculogenesis, angiogenesis and haemopoiesis during avian ontogeny. Development. 1989;105(3):473–485.
5 Jin S.W., Beis D., Mitchell T., et al. Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development. 2005;132(23):5199–5209.
6 Vokes S.A., Krieg P.A. Endoderm is required for vascular endothelial tube formation, but not for angioblast specification. Development. 2002;129(3):775–785.
7 Risau W., Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73–91.
8 DeRuiter M.C., Poelmann R.E., VanMunsteren J.C., et al. Embryonic endothelial cells transdifferentiate into mesenchymal cells expressing smooth muscle actins in vivo and in vitro. Circ Res. 1997;80(4):444–451.
9 Yamashita J., Itoh H., Hirashima M., et al. Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature. 2000;408(6808):92–96.
10 Risau W. Mechanisms of angiogenesis. Nature. 1997;386(6626):671–674.
11 Adams R.H., Wilkinson G.A., Weiss C., et al. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999;13(3):295–306.
12 Gerety S.S., Wang H.U., Chen Z.F., et al. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol Cell. 1999;4(3):403–414.
13 Wang H.U., Chen Z.F., Anderson D.J. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93(5):741–753.
14 Smithers L., Haddon C., Jiang Y.J., et al. Sequence and embryonic expression of deltaC in the zebrafish. Mech Dev. 2000;90(1):119–123.
15 Zhong T.P., Childs S., Leu J.P., et al. Gridlock signalling pathway fashions the first embryonic artery. Nature. 2001;414(6860):216–220.
16 Zhong T.P., Rosenberg M., Mohideen M.A., et al. Gridlock, an HLH gene required for assembly of the aorta in zebrafish. Science. 2000;287(5459):1820–1824.
17 Noden D.M. Embryonic origins and assembly of blood vessels. Am Rev Respir Dis. 1989;140(4):1097–1103.
18 Lawson N.D., Scheer N., Pham V.N., et al. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development. 2001;128(19):3675–3683.
19 Lawson N.D., Vogel A.M., Weinstein B.M. Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell. 2002;3(1):127–136.
20 Moyon D., Pardanaud L., Yuan L., et al. Plasticity of endothelial cells during arterial-venous differentiation in the avian embryo. Development. 2001;128(17):3359–3370.
21 Red-Horse K., Ueno H., Weissman I.L., et al. Coronary arteries form by developmental reprogramming of venous cells. Nature. 2010;464(7288):549–553.
22 Gerhardt H., Golding M., Fruttiger M., et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161(6):1163–1177.
23 Ausprunk D.H., Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc Res. 1977;14(1):53–65.
24 Stone J., Itin A., Alon T., et al. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci. 1995;15(7 Pt 1):4738–4747.
25 Ghabrial A.S., Krasnow M.A. Social interactions among epithelial cells during tracheal branching morphogenesis. Nature. 2006;441(7094):746–749.
26 Phng L.K., Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell. 2009;16(2):196–208.
27 Hellstrom M., Phng L.K., Hofmann J.J., et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445(7129):776–780.
28 Shutter J.R., Scully S., Fan W., et al. Dll4, a novel Notch ligand expressed in arterial endothelium. Genes Dev. 2000;14(11):1313–1318.
29 Suchting S., Freitas C., le Noble F., et al. The Notch ligand delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci U S A. 2007;104(9):3225–3230.
30 Childs S., Chen J.N., Garrity D.M., et al. Patterning of angiogenesis in the zebrafish embryo. Development. 2002;129(4):973–982.
31 Siekmann A.F., Lawson N.D. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature. 2007;445(7129):781–784.
32 Horowitz A., Simons M. Branching morphogenesis. Circ Res. 2008;103(8):784–795.
33 Affolter M., Caussinus E. Tracheal branching morphogenesis in Drosophila: new insights into cell behaviour and organ architecture. Development. 2008;135(12):2055–2064.
34 Metzger R.J., Krasnow M.A. Genetic control of branching morphogenesis. Science. 1999;284(5420):1635–1639.
35 Sutherland D., Samakovlis C., Krasnow M.A. Branchless encodes a Drosophila FGF homolog that controls tracheal cell migration and the pattern of branching. Cell. 1996;87(6):1091–1101.
36 Jarecki J., Johnson E., Krasnow M.A. Oxygen regulation of airway branching in Drosophila is mediated by branchless FGF. Cell. 1999;99(2):211–220.
37 Hacohen N., Kramer S., Sutherland D., et al. Sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell. 1998;92(2):253–263.
38 Mason J.M., Morrison D.J., Basson M.A., et al. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006;16(1):45–54.
39 Cardoso W.V., Lu J. Regulation of early lung morphogenesis: questions, facts and controversies. Development. 2006;133(9):1611–1624.
40 Metzger R.J., Klein O.D., Martin G.R., et al. The branching programme of mouse lung development. Nature. 2008;453(7196):745–750.
41 Min H., Danilenko D.M., Scully S.A., et al. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev. 1998;12(20):3156–3161.
42 De Moerlooze L., Spencer-Dene B., Revest J.M., et al. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127(3):483–492.
43 Costantini F., Kopan R. Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev Cell. 2010;18(5):698–712.
44 Dressler G.R. Advances in early kidney specification, development and patterning. Development. 2009;136(23):3863–3874.
45 Skinner M.A., Safford S.D., Reeves J.G., et al. Renal aplasia in humans is associated with RET mutations. Am J Hum Genet. 2008;82(2):344–351.
46 Olsson A.K., Dimberg A., Kreuger J., et al. VEGF receptor signalling in control of vascular function. Nat Rev Mol Cell Biol. 2006;7(5):359–371.
47 Carmeliet P., Ferreira V., Breier G., et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380(6573):435–439.
48 Ferrara N., Carver-Moore K., Chen H., et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380(6573):439–442.
49 Breier G., Albrecht U., Sterrer S., et al. Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development. 1992;114(2):521–532.
50 Shalaby F., Rossant J., Yamaguchi T.P., et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376(6535):62–66.
51 Fong G.H., Rossant J., Gertsenstein M., et al. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376(6535):66–70.
52 Dumont D.J., Jussila L., Taipale J., et al. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science. 1998;282(5390):946–949.
53 Coulon C., Georgiadou M., Roncal C., et al. From vessel sprouting to normalization: role of the prolyl hydroxylase domain protein/hypoxia-inducible factor oxygen-sensing machinery. Arterioscler Thromb Vasc Biol. 2010;30(12):2331–2336.
54 Lee S.H., Schloss D.J., Jarvis L., et al. Inhibition of angiogenesis by a mouse sprouty protein. J Biol Chem. 2001;276(6):4128–4133.
55 Downs K.M. Florence Sabin and the mechanism of blood vessel lumenization during vasculogenesis. Microcirculation. 2003;10(1):5–25.
56 Folkman J., Haudenschild C. Angiogenesis by capillary endothelial cells in culture. Trans Ophthalmol Soc U K. 1980;100(3):346–353.
57 Davis G.E., Bayless K.J. An integrin and Rho GTPase-dependent pinocytic vacuole mechanism controls capillary lumen formation in collagen and fibrin matrices. Microcirculation. 2003;10(1):27–44.
58 Bayless K.J., Davis G.E. The Cdc42 and Rac1 GTPases are required for capillary lumen formation in three-dimensional extracellular matrices. J Cell Sci. 2002;115(Pt 6):1123–1136.
59 Kamei M., Saunders W.B., Bayless K.J., et al. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature. 2006;442(7101):453–456.
60 Zeeb M., Strilic B., Lammert E. Resolving cell-cell junctions: lumen formation in blood vessels. Curr Opin Cell Biol. 2010;22(5):626–632.
61 Blum Y., Belting H.G., Ellertsdottir E., et al. Complex cell rearrangements during intersegmental vessel sprouting and vessel fusion in the zebrafish embryo. Dev Biol. 2008;316(2):312–322.
62 Lubarsky B., Krasnow M.A. Tube morphogenesis: making and shaping biological tubes. Cell. 2003;112(1):19–28.
63 Zovein A.C., Luque A., Turlo K.A., et al. Beta1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev Cell. 2010;18(1):39–51.
64 Carmeliet P., Lampugnani M.G., Moons L., et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98(2):147–157.
65 Strilic B., Kucera T., Eglinger J., et al. The molecular basis of vascular lumen formation in the developing mouse aorta. Dev Cell. 2009;17(4):505–515.
66 Parker L.H., Schmidt M., Jin S.W., et al. The endothelial-cell-derived secreted factor Egfl7 regulates vascular tube formation. Nature. 2004;428(6984):754–758.
67 Carmeliet P., Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature. 2005;436(7048):193–200.
68 Mukouyama Y.S., Shin D., Britsch S., et al. Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the skin. Cell. 2002;109(6):693–705.
69 Adams R.H., Eichmann A. Axon guidance molecules in vascular patterning. Cold Spring Harb Perspect Biol. 2010;2(5):a001875.
70 Damon D.H., Teriele J.A., Marko S.B. Vascular-derived artemin: a determinant of vascular sympathetic innervation? Am J Physiol Heart Circ Physiol. 2007;293(1):H266–H273.
71 Honma Y., Araki T., Gianino S., et al. Artemin is a vascular-derived neurotropic factor for developing sympathetic neurons. Neuron. 2002;35(2):267–282.
72 Matsumoto K., Yoshitomi H., Rossant J., et al. Liver organogenesis promoted by endothelial cells prior to vascular function. Science. 2001;294(5542):559–563.
73 Eberhard D., Kragl M., Lammert E. “Giving and taking”: endothelial and beta-cells in the islets of Langerhans. Trends Endocrinol Metab. 2010;21(8):457–463.
74 Lammert E., Cleaver O., Melton D. Induction of pancreatic differentiation by signals from blood vessels. Science. 2001;294(5542):564–567.
75 Yoshitomi H., Zaret K.S. Endothelial cell interactions initiate dorsal pancreas development by selectively inducing the transcription factor Ptf1a. Development. 2004;131(4):807–817.
76 Jacquemin P., Yoshitomi H., Kashima Y., et al. An endothelial-mesenchymal relay pathway regulates early phases of pancreas development. Dev Biol. 2006;290(1):189–199.
77 Kapa S., Gleeson F.C., Vege S.S. Dorsal pancreas agenesis and polysplenia/heterotaxy syndrome: a novel association with aortic coarctation and a review of the literature. JOP. 2007;8(4):433–437.
78 Sabin F.R. On the origin of the lymphatic system from the veins and the development of the lymph hearts and thoracic duct in the pig. Am J Anat. 1902;1:367–391.
79 Sabin F.R. The lymphatic system in human embryos, with a consideration of the morphology of the system as a whole. Am J Anat. 1909;9:43–91.
80 Francois M., Caprini A., Hosking B., et al. Sox18 induces development of the lymphatic vasculature in mice. Nature. 2008;456(7222):643–647.
81 Irrthum A., Devriendt K., Chitayat D., et al. Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. Am J Hum Genet. 2003;72(6):1470–1478.
82 Srinivasan R.S., Dillard M.E., Lagutin O.V., et al. Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes Dev. 2007;21(19):2422–2432.
83 You L.R., Lin F.J., Lee C.T., et al. Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature. 2005;435(7038):98–104.
84 Armulik A., Abramsson A., Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97(6):512–523.
85 Wasteson P., Johansson B.R., Jukkola T., et al. Developmental origin of smooth muscle cells in the descending aorta in mice. Development. 2008;135(10):1823–1832.
86 Esner M., Meilhac S.M., Relaix F., et al. Smooth muscle of the dorsal aorta shares a common clonal origin with skeletal muscle of the myotome. Development. 2006;133(4):737–749.
87 Topouzis S., Majesky M.W. Smooth muscle lineage diversity in the chick embryo. Two types of aortic smooth muscle cell differ in growth and receptor-mediated transcriptional responses to transforming growth factor-beta. Dev Biol. 1996;178(2):430–445.
88 Majesky M.W. Development of coronary vessels. Curr Top Dev Biol. 2004;62:225–259.
89 Zhou B., Ma Q., Rajagopal S., et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature. 2008;454(7200):109–113.
90 Wilm B., Ipenberg A., Hastie N.D., et al. The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development. 2005;132(23):5317–5328.
91 Que J., Wilm B., Hasegawa H., et al. Mesothelium contributes to vascular smooth muscle and mesenchyme during lung development. Proc Natl Acad Sci U S A. 2008;105(43):16626–16630.
92 Morimoto M., Liu Z., Cheng H.T., et al. Canonical Notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. J Cell Sci. 2010;123(Pt 2):213–224.
93 Arciniegas E., Frid M.G., Douglas I.S., et al. Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293(1):L1–L8.
94 Alva J.A., Zovein A.C., Monvoisin A., et al. VE-cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn. 2006;235(3):759–767.
95 Muzumdar M.D., Tasic B., Miyamichi K., et al. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45(9):593–605.
96 Greif DM, Hum JN, An AC, et al: Radial construction of the arterial wall. Manuscript in preparation.
97 Hungerford J.E., Little C.D. Developmental biology of the vascular smooth muscle cell: building a multilayered vessel wall. J Vasc Res. 1999;36(1):2–27.
98 Li L., Miano J.M., Cserjesi P., et al. SM22 alpha, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ Res. 1996;78(2):188–195.
99 Miano J.M., Cserjesi P., Ligon K.L., et al. Smooth muscle myosin heavy chain exclusively marks the smooth muscle lineage during mouse embryogenesis. Circ Res. 1994;75(5):803–812.
100 van der Loop F.T., Schaart G., Timmer E.D., et al. Smoothelin, a novel cytoskeletal protein specific for smooth muscle cells. J Cell Biol. 1996;134(2):401–411.
101 Miano J.M. Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol. 2003;35(6):577–593.
102 Wang Z., Wang D.Z., Hockemeyer D., et al. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature. 2004;428(6979):185–189.
103 Wang Z., Wang D.Z., Pipes G.C., et al. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci U S A. 2003;100(12):7129–7134.
104 Li S., Wang D.Z., Wang Z., et al. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc Natl Acad Sci U S A. 2003;100(16):9366–9370.
105 de Ruiter M.C., Poelmann R.E., van Iperen L., et al. The early development of the tunica media in the vascular system of rat embryos. Anat Embryol (Berl). 1990;181(4):341–349.
106 Hungerford J.E., Owens G.K., Argraves W.S., et al. Development of the aortic vessel wall as defined by vascular smooth muscle and extracellular matrix markers. Dev Biol. 1996;178(2):375–392.
107 el-Maghraby A.A., Gardner D.L. Development of connective-tissue components of small arteries in the chick embryo. J Pathol. 1972;108(4):281–291.
108 Kadar A., Gardner D.L., Bush V. The relation between the fine structure of smooth-muscle cells and elastogenesis in the chick-embryo aorta. J Pathol. 1971;104(4):253–260.
109 Nakamura H. Electron microscopic study of the prenatal development of the thoracic aorta in the rat. Am J Anat. 1988;181(4):406–418.
110 Takahashi Y., Imanaka T., Takano T. Spatial and temporal pattern of smooth muscle cell differentiation during development of the vascular system in the mouse embryo. Anat Embryol (Berl). 1996;194(5):515–526.
111 Turing A.M. The chemical basis of morphogenesis. Philos Trans R Soc Lond B Biol Sci. 1952;237(641):37–72.
112 Reilly K.M., Melton D.A. Short-range signaling by candidate morphogens of the TGF beta family and evidence for a relay mechanism of induction. Cell. 1996;86(5):743–754.
113 Feng X., Krebs L.T., Gridley T. Patent ductus arteriosus in mice with smooth muscle-specific Jag1 deletion. Development. 2010;137:4191–4199.
114 Gaengel K., Genove G., Armulik A., et al. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29(5):630–638.
115 Hellstrom M., Kalen M., Lindahl P., et al. Role of PDGF-β and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126(14):3047–3055.
116 Leveen P., Pekny M., Gebre-Medhin S., et al. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8(16):1875–1887.
117 Lindahl P., Johansson B.R., Leveen P., et al. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277(5323):242–245.
118 Soriano P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994;8(16):1888–1896.
119 Hirschi K.K., Rohovsky S.A., D’Amore P.A. PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol. 1998;141(3):805–814.
120 Gridley T. Notch signaling in vascular development and physiology. Development. 2007;134(15):2709–2718.
121 High F.A., Lu M.M., Pear W.S., et al. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci U S A. 2008;105(6):1955–1959.
122 Domenga V., Fardoux P., Lacombe P., et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004;18(22):2730–2735.
123 Wang T., Baron M., Trump D. An overview of Notch3 function in vascular smooth muscle cells. Prog Biophys Mol Biol. 2008;96(1–3):499–509.
124 Joutel A., Corpechot C., Ducros A., et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383(6602):707–710.
125 McLean S.E., Mecham B.H., Kelleher C.M., et al. Extracellular matrix gene expression in the developing mouse aorta. Adv Dev Biol. 2005;15:81–128.
126 Kelleher C.M., McLean S.E., Mecham R.P. Vascular extracellular matrix and aortic development. Curr Top Dev Biol. 2004;62:153–188.
127 Parks W.C., Pierce R.A., Lee K.A., et al. Elastin. Adv Mol Cell Biol. 1993;6:133–181.
128 Faury G., Pezet M., Knutsen R.H., et al. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest. 2003;112(9):1419–1428.
129 Li D.Y., Faury G., Taylor D.G., et al. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest. 1998;102(10):1783–1787.
130 Wagenseil J.E., Nerurkar N.L., Knutsen R.H., et al. Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteries. Am J Physiol Heart Circ Physiol. 2005;289(3):H1209–H1217.
131 Li D.Y., Brooke B., Davis E.C., et al. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393(6682):276–280.
132 Karnik S.K., Brooke B.S., Bayes-Genis A., et al. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130(2):411–423.
133 Ramirez F., Dietz H.C. Marfan syndrome: from molecular pathogenesis to clinical treatment. Curr Opin Genet Dev. 2007;17(3):252–258.
134 Dettman R.W., Denetclaw W., Ordahl C.P., et al. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev Biol. 1998;193(2):169–181.
135 Hu Y., Zhang Z., Torsney E., et al. Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE-deficient mice. J Clin Invest. 2004;113(9):1258–1265.
136 Sartore S., Chiavegato A., Faggin E., et al. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circ Res. 2001;89(12):1111–1121.
137 Zengin E., Chalajour F., Gehling U.M., et al. Vascular wall resident progenitor cells: a source for postnatal vasculogenesis. Development. 2006;133(8):1543–1551.
138 Passman J.N., Dong X.R., Wu S.P., et al. A sonic hedgehog signaling domain in the arterial adventitia supports resident Sca1+ smooth muscle progenitor cells. Proc Natl Acad Sci U S A. 2008;105(27):9349–9354.