Vascular Disorders of the Liver
Matthew M. Yeh
James M. Crawford
Introduction
Vascular diseases of the liver are less common than many other conditions, but they have assumed increasing importance in the differential diagnosis of liver disorders—in part because of improved ability to diagnose the more common chronic hepatitic and biliary liver diseases, and in part because vascular damage can play a role in the pathophysiology of virtually all liver disorders. In most liver diseases, the primary injury affects hepatocytes or duct cells, and the vascular damage is secondary. However, there are many primary disorders of the hepatic vasculature, and these are the focus of this chapter. Hepatic vascular disease is classified according to the size and type of blood vessels involved, because various etiologic types of liver disease target different portions of the hepatic vasculature.
The vasculature of the liver is unique in that it has two afferent supplies, arterial and splanchnic. The hepatic artery accounts for 30% to 40% and the portal vein accounts 60% to 70% of the hepatic blood supply. The obligate pathophysiologic outcome of disease affecting the afferent circulation is downstream ischemia. The degrees to which these two circulations are compromised account for the great variety of histologic patterns produced by vascular disease. The patterns reflect the number and size of afferent vessels involved and whether obstruction is rapid or slow. Compromise of hepatic venous outflow also creates ischemic injury, because blood flow through the liver is impeded. In this situation, however, localized or more general mechanical compression may also contribute to hepatic damage, depending on the severity of outflow obstruction.
As the predominant cell type in the liver, hepatocytes can manifest two levels of ischemic injury. Atrophy is a reduction of cell size that occurs in response to mild ischemia; it is typically seen with pure portal vein obstruction or isolated mild venous outflow obstruction. If portal vein compromise is accompanied by damage to the hepatic artery feeding the same region or to the regional hepatic vein, hepatocellular death occurs. Primary obstruction of the hepatic veins, small or large, also leads to hepatocellular death. After hepatocytes die, local collapse of the tissue leads to secondary fibrosis, a lesion referred to as parenchymal extinction. When parenchymal extinction is widespread, the accumulated lesions are recognized as cirrhosis (see Chapter 50 for greater detail). Considering that the pathology of hepatitis includes vascular damage, it may be a challenge to discern whether histologic injury to the hepatic parenchyma is a result of primary vascular disease or part of a broader pattern of hepatitis that evolves to cirrhosis.
If primary vascular injury leads to regional hepatocellular atrophy without fibrosis, other well-vascularized areas may regenerate and lead to the development of hepatocellular nodules. The combination of regional atrophy with nearby regeneration occurs in two major histologic patterns: nodular regenerative hyperplasia (NRH), in which the nodules are small, uniform, and diffuse, and large regenerative nodules, in which the nodules are large and irregularly distributed, up to and including focal nodular hyperplasia (FNH).
Hepatic arterial compromise has a direct effect on bile duct epithelial cells, because bile ducts derive their vascular supply exclusively from arteries. The most dramatic example is occlusion of the hepatic artery after liver transplantation, which leads to necrosis of the major bile ducts and loss of the organ.
Hepatic venous outflow obstruction often exhibits mixed features, because thrombi can propagate in a retrograde fashion, and the sluggish intrahepatic blood flow can lead to secondary portal vein thrombosis. This process is discussed further in the section on hepatic vein disease.
Agenesis of the portal vein and large spontaneous portosystemic shunts may be associated with other congenital anomalies.1 In patients with cirrhosis, spontaneous large-caliber shunts are usually secondary to portal hypertension (see Arterioportal and Arteriovenous Shunts).
The clinical presentation of hepatic vascular disease depends on the location of the obstruction. Obstruction of portal veins is usually clinically silent initially, but severe and prolonged obstruction may lead to varices, usually without ascites or liver failure. Obstruction of hepatic arteries is usually silent but it may result in necrosis of hepatocytes or bile ducts if combined with hypotension or with another vascular lesion, or if it occurs after organ transplantation. Obstruction of hepatic veins tends to cause increased formation of hepatic lymph, leading to ascites and, if severe, splanchnic varices and hepatic failure.
The position of the liver, between the capillary bed of the intestines and the heart, accounts for the important clinical effects of portal hypertension caused by obstruction of blood vessels within the liver. Portal hypertension is commonly classified as either cirrhotic or noncirrhotic, according to the histology of the liver parenchyma. Before 1945, approximately 40% of patients with portal hypertension were thought to have noncirrhotic portal hypertension,2 but in recent years, that percentage has decreased to less than 1%.3 The decline in noncirrhotic portal hypertension is largely the result of recognition that although the fibrosis of cirrhosis is largely reversible after successful treatment of the primary liver disease, the abnormalities in vascular pathophysiology remain.4–7 Specifically, patients with features of cirrhosis on biopsy specimens may subsequently have biopsy specimens that lack histologic criteria of cirrhosis. However, these patients often continue to have portal hypertension because more normal portal and hepatic vein anatomy may never be completely restored. Therefore, patients with “regressed” cirrhosis fall into the category of hepatic vascular disease and merit discussion in this chapter.
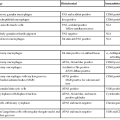
Although hepatic vessels have some unique properties, their response to stress is similar to that in other organs. Therefore, the cause of hepatic vascular disease may be considered in terms of the elements of the Virchow triad: vascular injury, obstruction, and hypercoagulable states. Tables 51.1 through 51.3 delineate the reported clinical settings in which venous thrombosis in the liver can occur; selected citations are given with the tables.
Table 51.1
Hypercoagulable States Associated with Obstruction of Large Veins in the Liver
| PV Thrombosis | HV Thrombosis | References | |
| Myeloproliferative Disease | |||
| Latent myeloproliferative disease | + | + | 207–210 |
| Polycythemia vera | + | + | 103, 203 |
| Primary myelofibrosis | + | + | 103, 203 |
| Paroxysmal nocturnal hemoglobinuria | + | + | 127, 203, 211, 212 |
| Idiopathic thrombocytosis | + | 203 | |
| Chronic myeloid leukemia | + | + | 213, 214 |
| Promyelocytic leukemia | − | + | 215, 216 |
| Multiple myeloma | − | + | 217 |
| Genetic Anomalies | |||
| Protein C deficiency | + | + | 208, 218–220 |
| Protein S deficiency | + | + | 208, 221–223 |
| Antithrombin III deficiency | + | + | 224–226 |
| Factor II G20210A | + | + | 227–229 |
| Factor V Leiden | + | + | 208, 230, 231 |
| Heparin cofactor II deficiency | + | − | 232 |
| Plasminogen deficiency | − | + | 233 |
| Dysfibrinogenemia | − | + | 234 |
| Homocystinemia | + | − | 235 |
| Other Hypercoagulable States | |||
| Pregnancy | + | + | 236, 237 |
| Oral contraceptive therapy | + | + | 208, 238–241 |
| Lupus anticoagulant or antiphospholipid antibodies | + | + | 37, 208, 242–247 |
| Idiopathic thrombocytopenic purpura | − | + | 210 |
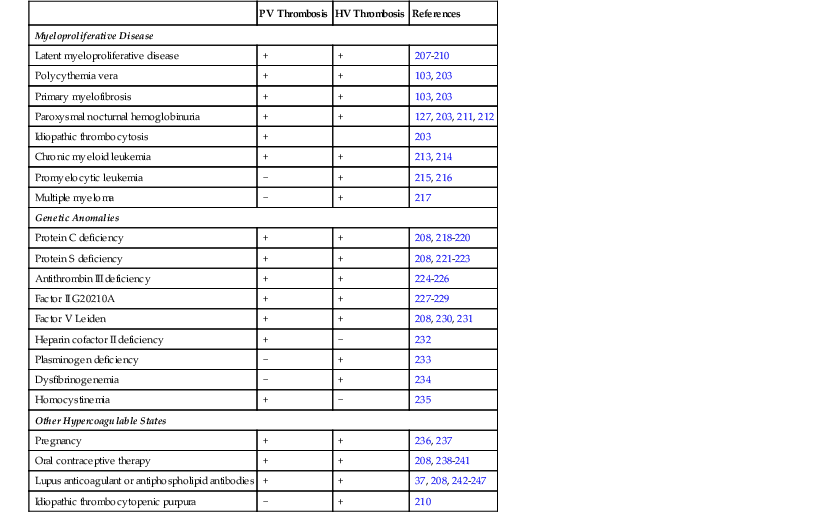
HV, Hepatic vein; PV, portal vein; +, reported cases or case series; −, no reported examples.
Table 51.2
Tumors and Other Stasis Lesions Associated with Obstruction of Large Veins in the Liver
| PV Thrombosis or Obstruction | HV Thrombosis | References | |
| Tumors | |||
| Hepatocellular carcinoma | + | + | 211, 248, 249 |
| Carcinoma of pancreas | + | − | 250, 251 |
| Renal cell carcinoma | − | + | 252 |
| Adrenal carcinoma | − | + | 253 |
| Hodgkin disease | − | + | 254 |
| Epithelioid hemangioendothelioma | − | + | 255 |
| Wilms tumor | − | + | 256 |
| Leiomyosarcoma or leiomyoma | − | + | 249, 257, 258 |
| Metastatic neoplasm | − | + | 249 |
| Other Stasis Lesions | |||
| Cirrhosis | + | + | 156, 248, 259–261 |
| Splenectomy | + | − | 262 |
| Retroperitoneal fibrosis | + | − | 263 |
| Congestive heart failure | − | + | 261 |
| Constrictive pericarditis | − | + | 264, 265 |
| Membranous obstruction of inferior vena cava | − | + | 266 |
| Superior vena cava obstruction | − | + | 267 |
| Other congenital anomalies | − | + | 268, 269 |
| Umbilical cord redundancy or placental thrombosis | − | + | 270 |
| Atrial myxoma | − | + | 271 |
| Sickle cell disease | − | + | 272 |
| Hydatid cyst | − | + | 273 |
| Hepatic abscess | − | + | 274, 275 |
| Hematoma | − | + | 276 |
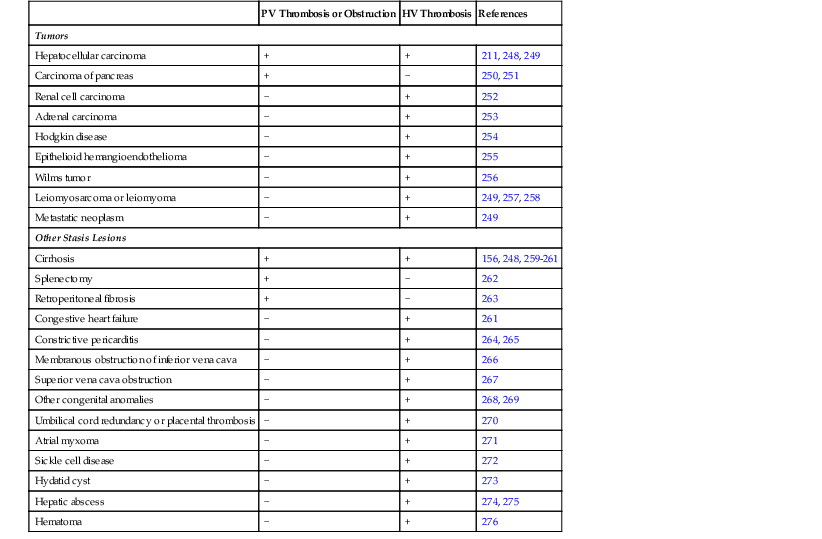
HV, Hepatic vein; PV, portal vein; +, reported cases or case series; −, no reported examples.
Table 51.3
Vascular Injury and Inflammatory Conditions Associated with Obstruction of Large Veins in the Liver
| PV Thrombosis | HV Thrombosis | References | |
| Behçet disease | + | + | 277–279 |
| Trauma | + | + | 2, 280–283 |
| Catheterization | + | + | 285, 286 |
| Sarcoidosis | + | + | 11, 287–289 |
| Umbilical sepsis | + | − | 213 |
| Cholecystitis | + | − | 290 |
| Pylephlebitis | + | − | 291–293 |
| Congenital hepatic fibrosis | + | − | 277, 294 |
| Cytomegalovirus infection | + | − | 295 |
| Hematopoietic cell transplantation | + | − | 296 |
| Esophageal sclerotherapy | + | − | 297, 298 |
| Schistosomiasis | + | − | 299 |
| Inflammatory bowel disease | + | − | 300, 301 |
| Ventriculoatrial shunt | − | + | 302 |
| Sclerotherapy | − | + | 297 |
| Amyloidosis | − | + | 303 |
| Vasculitis or tissue inflammation | − | + | 268 |
| Tuberculosis | − | + | 304 |
| Fungal vasculitis | − | + | 305, 306 |
| Idiopathic granulomatous venulitis | − | + | 142 |
| Filariasis | − | + | 307 |
| Inflammatory bowel disease | − | + | 262, 308, 309 |
| Mixed connective tissue disease | − | + | 310 |
| Protein-losing enteropathy | − | + | 311, 312 |
| Celiac disease | − | + | 313 |
| 5q Deletion syndrome and hypereosinophilia | − | + | 141 |
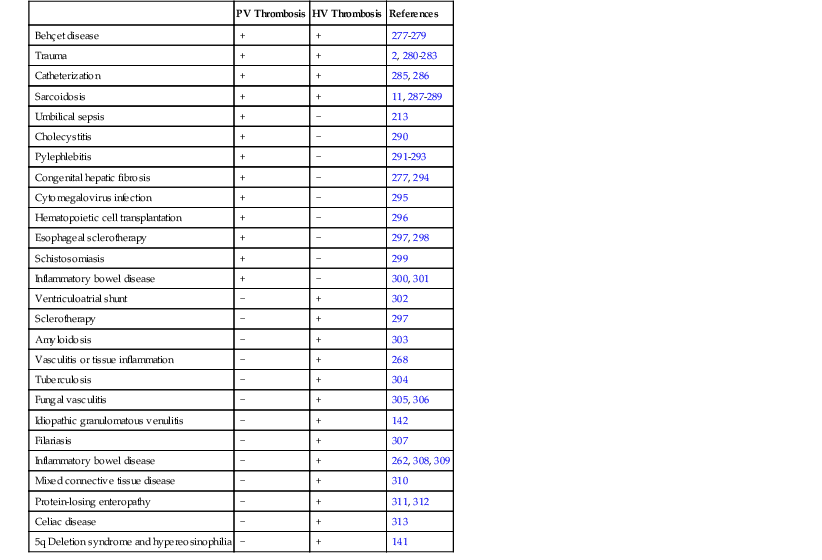
HV, Hepatic vein; PV, portal vein; +, reported cases or case series; −, no reported examples.
Identification of normal vascular structures of the liver merits a brief discussion.8 Portal veins divide dichotomously at acute angles and are accompanied by arteries and bile ducts. At the most terminal portions of the portal tree, portal veins disappear, leaving only the last traces of the hepatic artery/bile duct pairings. The smallest portal tracts, without or with portal veins, may be identified in Masson trichrome–stained sections on the basis of the delicate investment of connective tissue around the portal tract. Large hepatic veins also divide dichotomously, but the smallest hepatic veins enter larger branches at a right angle, similar to the bristles of a brush. There also is a slight preponderance of portal tracts versus hepatic veins within the hepatic parenchyma. In some disorders, portal veins are obliterated with local atrophy of the adjacent parenchyma, so that hepatic veins often “migrate” to the periportal region. When parenchymal extinction has occurred, an hepatic vein may be “tethered” to a portal tract by a short fibrous septum. For these reasons, identification of hepatic veins in some instances may be a challenge, one that can be solved by identification of elastic fibers in the wall of the vein or by tracing the vessel through multiple levels to a more recognizable centrilobular location within the parenchyma.
In the normal liver, lymphatic channels within portal tracts are difficult to identify, because they have few or no muscle fibers and are not dilated on histologic sections. Closer to the hepatic hilum, larger lymphatic channels may be identified and valves may be seen. When the hepatic vasculature is compromised, however, dilated lymphatic channels in the more distal portal tract distribution may become prominent. In cirrhosis, there may be an abundance of lymphatic channels within the fibrous septa, identified in part by the absence of erythrocytes in their lumina. Whether normal or abnormal, lymphatic endothelium stains positive with D2-40, whereas venous endothelium does not.
Portal Vein Disease (Portal Vein Obstruction)
Clinical Features
Obstruction of large portal veins most often occurs in adults with symptomatic cirrhosis, and the portal vein lesion is discovered during imaging studies. In the absence of cirrhosis, large portal vein obstruction may develop as a primary event, often termed idiopathic portal hypertension (IPH), or secondary to inflammatory conditions involving the portal vein, such as schistosomiasis or sarcoidosis.9 In these conditions, the portal vein thrombosis is typically silent until the onset of bleeding varices; ascites is usually absent. During the asymptomatic period, clinical evidence of portal hypertension, especially splenomegaly or thrombocytopenia, often leads to the diagnosis through imaging studies. The portal vein may appear to be absent, and multiple hilar collaterals may result in an appearance termed cavernous transformation. Large collaterals develop in the round ligament in 5% of patients; rarely, this manifests as a bruit or an umbilical caput medusa. Round ligament collaterals are dilated paraumbilical veins that communicate with the left portal vein. Large collaterals between the extrahepatic portal vein and the renal or adrenal veins are frequently seen. Numerous smaller collaterals are often seen during imaging or abdominal surgery. Secondary aneurysmal dilatation of the portal vein may also occur.
In contrast to this type of indolent natural history, acute portal vein obstruction accompanied by thrombosis of the mesenteric veins can be catastrophic and can lead to infarction of the intestines. This sequence often occurs in the setting of abdominal sepsis, trauma with vascular injury, cirrhosis, or growth of hepatocellular carcinoma into the main portal vein.
The portal vein branches are hypoplastic in patients with persistent ductus venosus or other congenital anomalies of the major vessels. Hepatic encephalopathy with hyperammonemia may be a presenting feature when portosystemic shunting is prominent.
Obstruction confined to the small portal veins is a rare cause of portal hypertension and tends to be milder than that seen in patients with large portal vein obstruction. Consideration is given at the end of this chapter to hepatoportal sclerosis, a condition that appears to arise from chronic damage to the large and small intrahepatic portal veins.
Pathogenesis
Large Portal Veins
Most often, portal vein obstruction occurs in the setting of another hepatic disease, particularly cirrhosis. Beyond that, thrombosis is the most frequent mechanism of large portal vein obstruction, again following the Virchow triad of thrombosis secondary to obstruction, venous inflammation, or a hypercoagulable state (see Tables 51.1 through 51.3). Hypercoagulable states causing portal vein obstruction may involve inherited abnormalities of the clotting cascade or acquired abnormalities of the platelets, as in polycythemia vera or other myeloproliferative diseases (see Table 51.1). Coincidental hypercoagulable states have also been documented to contribute to thrombosis, even when cirrhosis is present.10
Portal vein obstruction may also be related to the presence of tumor, either in the hepatic hilum and growing into the portal vein (typically hepatocellular carcinoma) or in the pancreas, or to small portal vein disease in early primary biliary cirrhosis. Injury to the portal vein vasculature may be caused by a hilar bile leak in primary sclerosing cholangitis or in biliary necrosis after transplantation, or by other events such as splanchnic sepsis, variceal sclerotherapy, or trauma. Trauma may result from blunt abdominal injury or from surgical interventions such as splenectomy, umbilical vein catheterization, portacaval shunt, insertion of a transjugular intrahepatic portosystemic shunt (TIPS), or a Kasai portoenterostomy procedure.
Small Portal Veins
Obliteration of small portal veins (obliterative portal venopathy) may develop secondary to local inflammation, thrombosis, congestive portal venopathy, or toxic injury. Local inflammation is important in any disease with portal inflammation, including chronic hepatitis, primary biliary cirrhosis, primary sclerosing cholangitis, sarcoidosis, polyarteritis nodosa, and congenital hepatic fibrosis. Thrombosis may occur as a result of retrograde propagation of thrombi from hepatic veins or in response to sluggish and reversed blood flow, usually in cirrhotic livers. Also in cirrhotic livers, high intrahepatic pressure causes congestive venous injury. A variety of vasculotoxins, including azathioprine, cyclophosphamide, methotrexate, and arsenic, can cause endothelial injury and secondary luminal obliteration. In some geographic regions, schistosomiasis is the most frequent cause of portal vein disease and portal hypertension. The vascular changes in cirrhosis are discussed in Chapter 50.
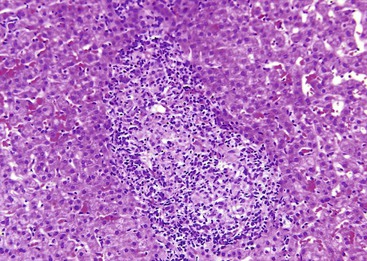
Granulomas in sarcoidosis can cause phlebitis of small portal veins.11 Medium-sized hepatic veins may also be affected. In most patients, granulomas in the portal tracts or associated portal fibrosis can cause presinusoidal resistance and portal hypertension but not cirrhosis.12,13 NRH and portal vein thrombosis may occur.14
Pathologic Features
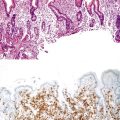
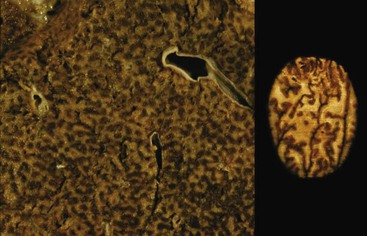
Portal vein thrombus is usually evident only in the healed state, after recanalization and fibrosis have already occurred. The healing process may be almost complete, so that no residual changes, or only slight pearly thickening of the intima, is seen grossly. In other instances, residual high-grade obstruction causes marked intraluminal fibrosis containing numerous racemose channels (Fig. 51.1). Thrombi in large and medium-sized portal veins may recanalize almost completely, leaving a layer of residual intimal fibrosis (Fig. 51.2), or they may remain largely occluded (Fig. 51.3). Multiple layers of collagen indicate recurrent thrombosis. As thrombi heal with a granulation tissue response, small arteries are often seen within the neointima. When thrombosis or inflammatory injury involves small portal veins, the vein walls usually disappear completely within a few weeks after the inciting event. The elastic trichrome stain is better than Masson trichrome for identifying residua of vein walls, which are marked by the location of muscle bundles or variation in the elastic fibers.
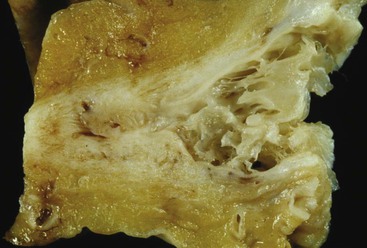
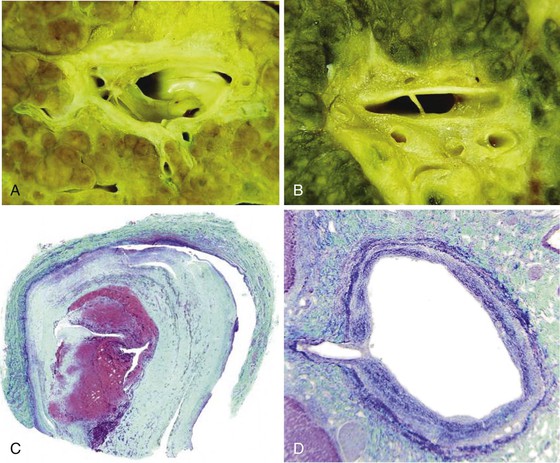
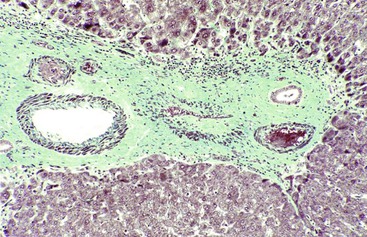
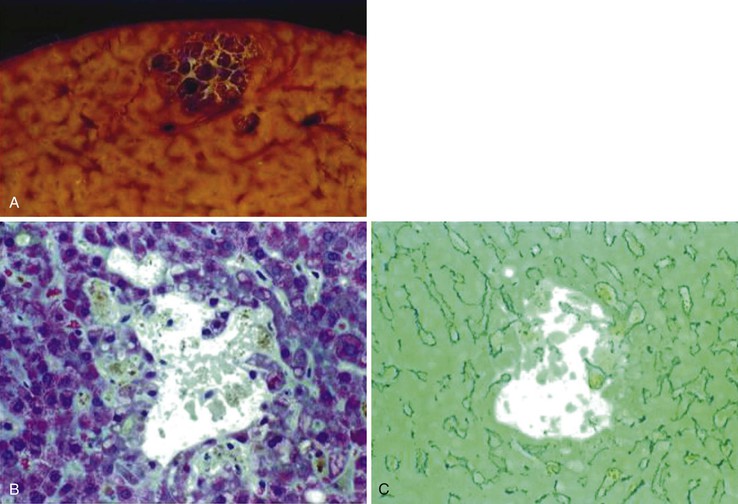
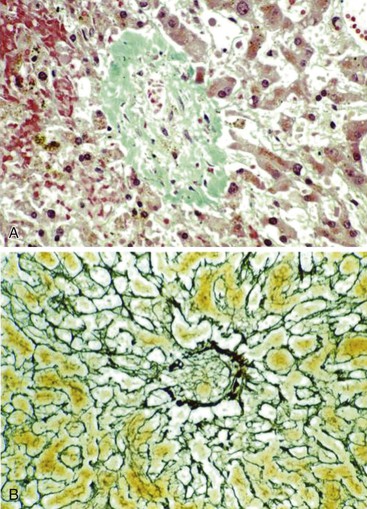
After organization of a thrombotic or inflammatory event, obliterated small portal veins have a nonspecific appearance, but several clues may indicate the cause of the original lesion. Portal granulomas may be seen in sarcoidosis and primary biliary cirrhosis; duct paucity favors the latter (Fig. 51.4). Granulomas in sarcoidosis are usually numerous, but in inactive disease, they may resorb, making diagnosis difficult. Eggs of Schistosoma species may occur either with or without granulomatous inflammation (Fig. 51.5). Eggs are few in number with Schistosoma mansoni and numerous with S. japonicum. Active or healed arteritis may suggest polyarteritis nodosa or another rheumatologic condition. Irregular and dilated ducts (Von Meyenburg complexes) are found in most portal tracts in cases of congenital hepatic fibrosis and in occasional portal tracts in polycystic disease. Thorotrast deposits in portal macrophages may be associated with obliteration of small portal veins and noncirrhotic portal hypertension (Fig. 51.6). If marked parenchymal congestion or obstruction of hepatic veins is observed, the portal vein blockage is likely secondary to stasis, with thrombosis.
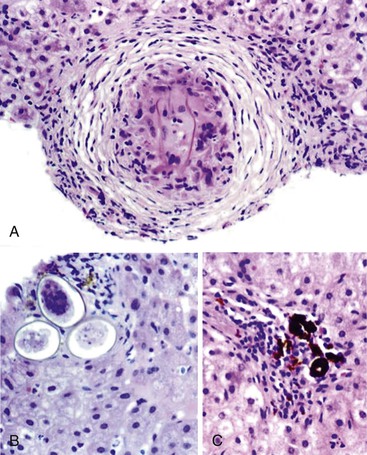
Patients with noncirrhotic portal hypertension usually have portal vein intimal fibrosis and delicate septa, suggesting regressed cirrhosis and superimposed portal vein thrombosis or congestive portal venopathy.15 After portal vein obliteration, the liver parenchyma becomes atrophic, with crowding of the portal tracts. If the region of sinusoidal dilatation is focal, it is called an infarct of Zahn (see Sinusoidal Dilatation). Atrophy may be either uniform or mixed with small regenerative nodules in a pattern referred to as NRH (discussed later).
Differential Diagnosis
Causes of portal hypertension in the absence of cirrhosis are variable. Worldwide, schistosomiasis is the most common cause. Alcohol-induced liver disease, inherited metabolic diseases, and autoimmune liver diseases can cause portal hypertension at the precirrhotic stage. However, the most important differential diagnosis is cirrhosis.
Exclusion of Cirrhosis and Regressed Cirrhosis
The diagnosis of portal vein disease does not rely on the histologic appearance alone but requires consideration of clinical and imaging information. The most useful task in a patient with portal hypertension is to confirm the presence or absence of cirrhosis, including the possibility of regressed cirrhosis (discussed in Chapter 50). This allows the investigation to be directed at the most likely cause of the cirrhosis.
Of course, biopsy specimen size is important in providing the pathologist with enough evidence to accurately exclude cirrhosis (see Chapter 50). Cirrhosis, particularly when highly regressed, is frequently missed when biopsy specimens are shorter than 2 cm in length. Regressed cirrhosis should be suspected when there is a reduction of both portal and hepatic veins, delicate remnants of fibrous septa, and an irregular arrangement of portal structures and hepatic veins, typically with hepatic veins in close approximation to portal tracts. If, in cirrhosis, small portal veins are obliterated, the acinar arrangement is normal (usually), and hepatic veins are patent, the disease likely involves only the portal veins or portal tracts. Irregularity of parenchymal atrophy and hyperplasia (i.e., NRH) suggests small vessel disease, even if this is not represented in the biopsy specimen. Similar changes may occur adjacent to mass lesions, including neoplasms and abscesses.
Patients may be diagnosed with idiopathic or noncirrhotic portal hypertension on the basis of imaging studies (no identifiable alteration in hepatic features) or biopsy evidence (no identifiable abnormality in hepatic microanatomy that would suggest regressed cirrhosis). In the latter instance, the limited amount of tissue in liver biopsy specimens may make identification of regressed cirrhosis difficult. Alternatively, the histologic manifestations of regressed cirrhosis may be termed incomplete septal cirrhosis or hepatoportal sclerosis, depending on the location of residual fibrous tissue (delicate parenchymal septa in the former, portal tracts alone in the latter).16 Incomplete septal cirrhosis is discussed in Chapter 50; hepatoportal sclerosis is discussed later in this chapter. Last, “IPH” identified clinically may represent a mixture of diseases6 or one disease with various histologic manifestations.17
Exclusion of Portal Vein Thrombosis
Liver biopsy is seldom indicated for identification of large portal vein disease, because peripheral biopsies do not sample these vessels. Recanalization of large portal vein thrombi make these lesions elusive from a clinical point of view. Prior thrombosis is suspected when there is prominent intimal fibrosis of portal veins, especially those larger than 200 µm in diameter. No specific histologic features can be used to diagnose hypercoagulable states. However, hepatomegaly, marked splenomegaly, and extramedullary hematopoiesis in the liver are often found in myeloproliferative disease, even in patients without hepatic vascular disease.
Obliteration of small portal veins is nonspecific. Lesions associated with inflammatory obliteration of small portal veins should be sought, including duct lesions of primary biliary cirrhosis, granulomas of sarcoidosis and schistosomiasis, and arteritis. Obliteration of subcapsular small portal veins is a common event among aged individuals.18 However, the finding that the majority of small portal veins are missing is likely to be significant. Congestive portal venopathy and postthrombotic scarring are often identical in appearance. Thrombosis, when present, involves many small and medium-sized portal veins, whereas congestive lesions are usually patchy in distribution and confined to the small veins. This criterion is useful only when examining large samples, such as liver explant specimens. Congestive lesions are more likely to occur in cirrhotic livers, where high-grade hepatic vein outflow obstruction is characteristically present.
Exclusion of Parasitic Disease
Schistosomes are trematode flukes. Some species may involve the liver, such as S. japonicum in Asia and S. mansoni in Africa and South America. Liver disease in schistosomiasis is attributed to the entrapment of parasitic ova in portal veins.19 The secretion of ova induces a granulomatous reaction and collagen deposition. In an individual with severe infestation of schistosomes, marked portal fibrosis ensues; this condition is known as Symmers pipestem fibrosis.20 The clinical presentation of hepatic schistosomiasis mimics IPH in that variceal bleeding and massive splenomegaly with normal liver test results and no ascites are common manifestations.9
Arterial Disease
The hepatic arterial system may be involved by systemic diseases such as amyloidosis or polyarteritis nodosa. Atherosclerosis may affect the extrahepatic arterial trunk, and the intrahepatic arteries may exhibit hyaline arteriosclerosis in older individuals. Atherosclerotic emboli may occasionally lodge in the hepatic arterial circulation. Regardless of cause, arterial disease in the liver is seldom symptomatic, mainly because of the smaller contribution of the hepatic artery to hepatic blood flow and compensation from portal veins. However, in patients with hypotension or congestive heart failure, infarcts or liver failure may occur as a result of regional arterial disease.21,22 Moreover, as noted earlier, arterial compromise leading to ischemic damage to the biliary tree may lead to failure of the entire organ.
Liver Injury Resulting from Ischemia and Shock
Forward-flow circulatory compromise to the liver may be the result of heart failure caused by acute myocardial infarction or circulatory shock resulting from hypovolemia, severe trauma, or sepsis.23 Patients with circulatory shock have both low arterial blood pressure and reduced oxygen tension in the portal veins. Typically, a sharp rise in serum aminotransferases, with or without liver failure develops in patients with shock. Enzymes typically normalize rapidly in those patients who survive. Hepatic infarction has also been associated with Budd-Chiari syndrome, hepatic trauma, hepatic transplantation,24 hepatic catheterization,25 laparoscopic cholecystectomy,26 TIPS insertion,27 and alcohol injection.28 A variety of hypercoagulable states and vascular injury syndromes may cause liver ischemia, including disseminated intravascular coagulation (DIC), sepsis, toxemia of pregnancy, the HELLP syndrome (i.e., hemolysis, elevated liver enzymes, and low platelet count), arteritis,29 sickle cell disease,30 and oral contraceptive use.31
Pathologic Features
Left-sided cardiac failure or shock may lead to the development of sharply demarcated zones of coagulative necrosis (Fig. 51.7). Zone 3 is usually most susceptible to the effects of ischemia, giving rise to the term centrilobular necrosis. Rarely, isolated zone 2 necrosis is seen.32 Single-cell calcification may occur. Apoptotic bodies are usually observed in the zone between healthy and coagulated hepatocytes. Zone 1 necrosis is typical of diseases that produce intravascular fibrin deposition, such as toxemia of pregnancy and DIC. The fibrin in these conditions may be present in arterioles, portal venules, and zone 1 sinusoids. Regardless of cause, a reticulin stain can help identify areas of architectural collapse resulting from loss of hepatocytes.
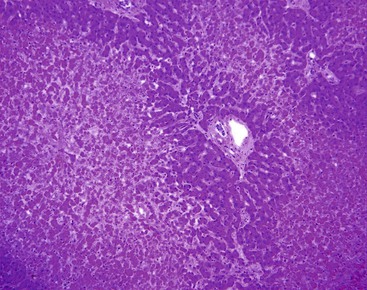
In the liver, an infarct is defined as ischemic necrosis involving two or more contiguous and complete acini; both zone 1 and zone 2 must be involved. Infarcts occur when at least two of the following vessels are involved in the same unit of liver tissue: portal vein, hepatic vein, and hepatic artery (Fig. 51.8). In the presence of hypotension, lesser degrees of vascular obstruction are necessary to produce infarction. Often, no vascular obstruction can be identified.21
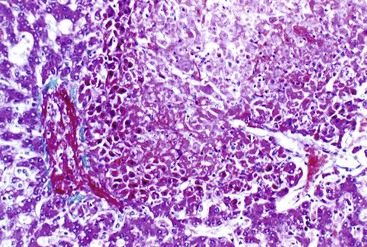
Differential Diagnosis
The differential diagnosis of zone 3 necrosis includes drug-induced injury, particularly with acetaminophen or cocaine. Amanita phalloides (mushroom) hepatotoxicity also shows zone 3 coagulative necrosis. Herpes virus infection results in necrosis that resembles infarcts except that the margins of necrosis do not follow the normal hepatic acinar landmarks and viral inclusions are usually visible. Atrophy and sinusoidal dilatation suggest underlying chronic passive congestion, as seen in patients with right-sided heart failure.33 (see Hepatic Vein Disease). Livers with marked zone 3 atrophy and/or zone 3 necrosis, combined with zone 3 hemorrhage from retrograde congestion (as from right-sided heart failure), often have a gross appearance that resembles the cut surface of a nutmeg and is termed “nutmeg liver” (Fig. 51.9). The combination on microscopy of centrilobular necrosis from forward-flow ischemia with hemorrhage from retrograde congestion is termed centrilobular hemorrhagic necrosis.
Arteritis
Large- and medium-sized hepatic arteries may be affected in polyarteritis, Wegener granulomatosis, and rheumatoid arthritis. The arteritis is usually clinically silent, although hepatic rupture has been reported.34,35 Small vessel arteritis, as in systemic lupus erythematosus (SLE) or rheumatoid arthritis, is also usually clinically silent but may result in obliteration of adjacent portal veins, leading to NRH and portal hypertension.36–38 The histology of the various types of arteritis is the same in the liver as in other organs.
Hepatic Artery Obstruction
Hepatic artery obstruction, caused by thrombosis, arteritis, or surgical ligation, is usually well-tolerated unless hypotension or DIC is also present, in which case infarction may occur. After liver transplantation, however, the ducts of the implanted liver do not have the rich, anastomosing arterial bed that was present in the native liver and therefore are dependent on blood flow from an intact hepatic artery. Thrombosis of this vessel may lead to ischemia of the bile ducts, leakage of bile, and necrosis of the perihilar parenchyma39 (Fig. 51.10). The resulting necrotic debris often harbors Candida or other microorganisms. Partial biliary obstruction (stricture or bile cast syndrome) often leads to liver failure, necessitating replacement of the organ.40 Hepatic artery cannulation and infusion with floxuridine or other agents may also lead to biliary ischemia and eventually to large duct stenosis.41,42
Arterioportal and Arteriovenous Shunts
Large shunts between the splanchnic artery and the portal or the hepatic vein most commonly develop many months or years after penetrating trauma (e.g., gunshot wound, liver biopsy).43,44 Large shunts may also occur as the result of a developmental anomaly. In some patients, they are found early in life; in others, especially with hereditary hemorrhagic telangiectasia, the shunts develop progressively during several decades. Increased arterial flow to the liver may be inapparent but can manifest with a bruit, high-output congestive heart failure, ascites, diarrhea, weight loss, protein-losing enteropathy, or hemobilia.45 When the shunt involves the portal vein, portal hypertension is the most important effect. The diagnosis depends on detecting a dilated high-flow channel by Doppler ultrasonography or other imaging studies. On arteriography, one may see an apparent doubling of the vascular tree, possibly because of early retrograde filling of the portal veins that run parallel to the arteries. The portal vein branches may be obliterated. Histologically, numerous congested capillaries and arterioles, either within or adjacent to portal tracts, is characteristic.46 Hereditary hemorrhagic telangiectasia is discussed further in Chapter 54.
Sinusoidal Disease
The sinusoids play a critical role as conduits of blood and nutrients to hepatocytes. The normal sinusoidal wall is composed of highly fenestrated endothelial cells and a delicate fibrillar matrix without a well-defined basement membrane or occlusive pericytes.47 Stellate cells reside in the subendothelial space of Disse. These cells contain droplets of retinoyl esters and produce collagen in response to inflammation. They also have contractile properties that are activated by endothelin 1 and inhibited by nitric oxide.48,49 The sinusoids must adapt to physiologic and pathologic alterations in arterial and venous blood flow. In chronic liver diseases, the stellate cells are activated and transformed into myofibroblasts, which lay down extracellular matrix causing hepatic fibrosis and, through their contractile activity, increase resistance to blood flow through the sinuosoids. Many types of disorders involve histologic changes of the sinusoids and small veins.
Sinusoidal Dilatation
Sinusoidal diameter and hepatocyte size are fairly uniform within the normal liver, although there is a slight widening of sinusoidal diameter in zone 3. This uniformity is lost when there is local obstruction of portal or hepatic veins, which leads to hepatocellular atrophy and sinusoidal dilatation and is often accompanied by a local compensatory increase in arterial blood flow. The resulting localized increase in the blood space is seen macroscopically as a darkened region of liver parenchyma, termed infarct of Zahn.50 When many adjacent obstructive portal vein lesions occur, hepatocyte atrophy causes the portal tracts to become crowded together. Typically, these lesions are seen adjacent to neoplasms, which compromise regional blood flow because of their compressive mass effect, or with focal portal vein thrombosis.
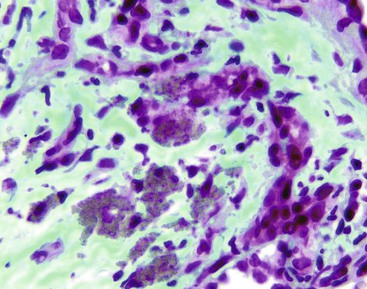
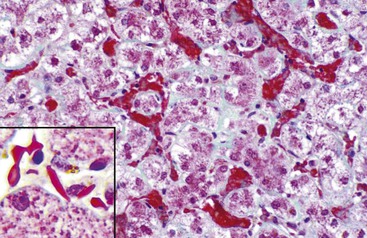
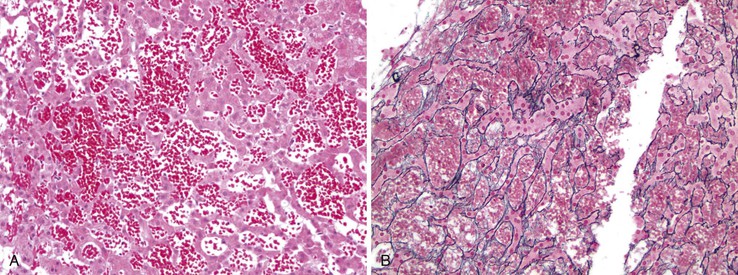
In chronic congestive heart failure and constrictive pericarditis with retrograde impediment to hepatic venous outflow, a diffuse increase in sinusoidal pressure leads to zone 3 hepatocellular atrophy and sinusoidal dilatation (Fig. 51.11). With time, pericellular fibrosis develops, obliterating small hepatic veins; rarely, cirrhosis that features a pericentral pattern of fibrous septation develops. Sinusoidal dilatation is also seen in patients with chronic wasting illnesses51 such as tuberculosis or acquired immunodeficiency syndrome (AIDS)51,52; with malignancies, notably Hodgkin disease53 or renal cell carcinoma54,55; and within nodules of severe cirrhosis. Dilatation of zone 1 and 2 sinusoids occurs during pregnancy and in women taking oral contraceptives.56,57 The mechanism of this effect may be related to mild diffuse angiogenesis and increased arterial blood flow. Sickle cell disease characteristically shows small clumps of sickled red blood cells within sinusoids (Fig. 51.12). In this disease, sinusoidal fibrosis is often seen. Cirrhosis is rare; when present, it is caused by coincidental viral or other liver disease.
A sharp delineation between well-preserved zone 1 hepatocyte plates and atrophic zone 2 and zone 3 plates, with sinusoidal dilatation, may result not from retrograde obstruction to blood flow but from forward-flow underperfusion. This underperfusion may be caused by vascular disease affecting the portal veins and hepatic arteries (discussed earlier) or may occur in the setting of modest compromise of the vascular anastomoses in a hepatic transplantation graft.58 Finally, parenchymal lesions that create local scarring and “traction” on adjacent regions of the liver may create secondary sinusoidal dilatation unrelated to the physiology of blood flow. The most dramatic features of “traction dilatation” are seen in tertiary syphilis, in which gummatous lesions create a characteristic hepar lobatum macroscopically, with interspersed fibrotic scars and dilated sinusoidal spaces in the hepatic parenchyma surrounding the gummata. More limited examples may be seen adjacent to other mass lesions of the liver if there is scarring of the surrounding parenchyma.
Peliosis Hepatis
Peliosis hepatis is defined as the presence of blood-filled spaces in the liver resulting from focal rupture of sinusoidal walls.59 The term was initially used to describe grossly visible lesions, but it is now also applied to microscopic lesions. Severe sinusoidal dilatation may resemble peliosis. The difference, by definition, is that peliosis is caused by rupture of the sinusoidal walls, whereas these walls are intact in sinusoidal dilatation.
Clinical Features
Peliosis may be minimal, asymptomatic, and grossly inapparent or severe in cases of cholestasis, liver failure, portal hypertension, development of a mass lesion, or spontaneous rupture. Calcifications may develop and can be seen radiologically.60 Peliosis has been associated with exposure to a variety of drugs, including anabolic steroids, tamoxifen, corticosteroids, azathioprine, methotrexate, 6-thioguanine, 6-mercaptopurine, vinyl chloride, arsenic, and Thorotrast, and it may be seen in hairy cell leukemia.61–63 Bartonella infection can cause bacillary peliosis, which occurs mainly in immunosuppressed patients.
Pathologic Features
The endothelial lining may be lost during lesion development, but it is usually regained in chronic lesions.64 Severe peliosis is characterized by separation of the sinusoidal parenchyma from the portal tracts. The portal tracts appear similar to exfoliated branches of a tree in winter (Fig. 51.13). In Bartonella infection (discussed later), the organisms may be seen as a vague haze on hematoxylin and eosin (H&E)-stained sections but are well visualized with a Warthin-Starry stain.65 Peliotic change to the sinusoidal vasculature may also be observed in various tumors, particularly hepatocellular adenoma, hepatocellular carcinoma, and angiosarcoma. Therefore, it is important to examine the surrounding liver and the endothelial lining for these lesions.
Peliosis may be mistaken for hemangioma. However, in the latter condition, the blood-filled spaces are lined by a robust vascular wall, and the portal tracts do not extend into the blood-filled cavities, as they do in peliosis.
Bacillary Angiomatosis
Infection with Bartonella (formerly known as Rochalimaea) may manifest as vascular proliferative lesions in patients with HIV infection and other immunocompromised hosts.66–68 Bacillary angiomatosis may affect the liver (called bacillary peliosis hepatis), spleen (bacillary splenitis), and skin. Necrotizing granulomatous disease is the other form of Bartonella infection. In patients with malignancy who are receiving chemotherapy or are immunocompromised caused by HIV infection or organ transplantation, Bartonella infection should be considered in the differential diagnosis of a febrile illness.
Bacillary peliosis of the liver generates peliotic foci of necrosis within the parenchyma, characterized by multiple cystic blood-filled spaces or lakes.69 Although peliosis has been described in patients receiving azathioprine and cyclosporine after organ transplantation and in those receiving anabolic androgenic and estrogenic steroids,70–73 the finding of aggregates of Bartonella bacilli highlighted by a Warthin-Starry silver stain with a mixture of inflammatory cells in the background of fibromyxoid stroma helps support a diagnosis of infection caused by Bartonella.65 Serologic analysis, culture, and, ultimately, polymerase chain reaction (PCR) testing of peripheral blood or liver tissue may be confirmatory.74
Sinusoidal Injury, Fibrosis, and Arterialization
Because of their close proximity to hepatocytes, sinusoids are injured in all forms of acute and chronic hepatitis. Injury is most often appreciated when contiguous hepatocytes are lost, a phenomenon referred to as parenchymal extinction. Hepatocyte loss that spans across all acinar zones is termed bridging necrosis. When necrotic regions collapse, the portal tracts and veins become closely approximated to each other. This is readily appreciated with a reticulin stain, because collapse of the intervening parenchymal plates with complete loss of hepatocytes is evident. If dropout involves only zone 3 hepatocytes, the liver cell plates may be “empty,” but there may be little or no collapse of the reticulin framework, resulting in a lesion termed “evacuation of the liver cell plates.”75 This lesion is most commonly seen in patients with allograft rejection, acetaminophen toxicity, or chronic hepatitis.
Sinusoids are normally lined by CD34-negative endothelial cells. In chronic liver disease, the endothelium becomes CD34 positive, first in endothelium near the portal tracts and later throughout the lobules (e.g., in cirrhosis). This is termed arterialization (or capillarization) of the sinusoids. Other features of arterialization include decreased fenestration of the endothelial cells, increased collagen and other matrix proteins in the space of Disse, and loss of microvilli on the surface of hepatocytes.76,77 Sinusoidal fibrosis is detected in early and active disease, usually in association with activated hepatic stellate cells, which stain positively for α-smooth muscle actin.
Sinusoidal fibrosis may be found in any type of chronic liver disease, although it is typically most prominent in patients with alcoholic disease, nonalcoholic steatohepatitis,78–80 hepatic vein thrombosis, vitamin A toxicity,81,82 congenital syphilis, sickle cell disease, or Gaucher disease. CD34-positive sinusoidal endothelial cells are present to a variable degree in cirrhosis, hepatocellular carcinoma, adenomas, FNH, and NRH.
Sinusoidal Obstruction Syndrome (Veno-occlusive Disease)
Sinusoidal obstruction syndrome (SOS) occurs most commonly as a complication of myeloablative regimens that are used to prepare patients for hematopoietic stem cell transplantation (i.e., bone marrow transplantation).83 Its frequency and severity have decreased during recent years, largely as a result of changing transplantation regimens.84 SOS is defined by the presence of prominent obstructive, nonthrombotic lesions of the small hepatic veins in individuals exposed to either radiation or a hepatotoxin. The definition has been modified, and the term SOS has replaced the former term, veno-occlusive disease (VOD). Specifically, the initial lesion is caused by injury to the endothelial cells of sinusoids and small veins,85 leading to hemorrhage into the liver parenchyma and into the walls of hepatic veins. Because of this acute phenomenon, the disorder is termed sinusoidal obstruction syndrome.86 Among survivors of SOS, the sinusoidal lesions become less apparent with time, and the major residual lesion is fibrous obliteration of small hepatic veins. This residual pattern of disease is the one that was characterized in the original description of VOD87; the acute sinusoidal lesions were not appreciated histologically or experimentally until many years later.86,88
Clinical Features
Patients with early disease exhibit tender hepatomegaly, ascites, rapid weight gain, and hepatic failure with elevated serum bilirubin.85,89 The onset of this toxic syndrome can be diagnosed on clinical grounds, and percutaneous liver biopsy may be too risky a procedure given the potential of an evolving coagulopathy. If histologic diagnosis is needed to exclude other conditions, transjugular biopsy can provide a tissue sample and enable measurement of the hepatic venous pressure gradient.90,91 A gradient greater than 10 mm Hg is considered highly specific for SOS in patients undergoing hematopoietic cell transplantation.
SOS occurs most commonly in patients being prepared for bone marrow transplantation with myeloablative doses of radiomimetic drugs and irradiation.86,92 The drugs most often implicated are cyclophosphamide, busulfan, and gemtuzumab ozogamicin (Mylotarg93). Occasionally, SOS develops in patients given low doses of other drugs and toxins, such as azathioprine, cysteamine, dacarbazine, dactinomycin, carmustine (BCNU), 6-mercaptopurine, 6-thioguanine, dimethylbusulfan, cytosine arabinoside, indicine-N-oxide, mustine (methchlorethamine hydrochloride), doxorubicin, urethane, vincristine, mitomycin-C, oxaliplatin,94 etoposide, arsenic, Thorotrast, and intraarterial flurodeoxyuridine.95–97 Patients who survive acute SOS usually recover completely, but the residual lesions of VOD may remain in the liver for a long period. Portal hypertension or cirrhosis is rare. If cirrhosis becomes clinically manifest, it is usually associated with the presence of other disease (e.g., chronic hepatitis C).98
The initial description of “VOD” was in subjects exposed to pyrrolizidine alkaloids. These compounds are found in plants of the genera Senecio, Heliotropium, Crotalaria, and many others. Epidemics of pyrollizidine alkaloid toxicity occur mainly in arid climates, where toxin-containing plants may overgrow crops during periods of drought. Livestock may be affected when grazing, and humans may be affected by eating bread derived from these crops. Herbal medicines created from toxic plants, commonly called “bush tea,” can cause severe disease, especially in young children, and death may occur. Cirrhosis can occur in survivors of pyrrolizidine alkaloid–induced VOD.99
SOS has also been observed in the use of oxaliplatin to treat colorectal cancer metastatic to liver. The underlying mechanism is thought to be toxic injury to the sinusoidal endothelial cells by oxaliplatin.94
Pathologic Features
During the first 2 weeks of SOS, the liver exhibits marked diffuse hemorrhage.100 In the early phase, cirrhosis that develops in survivors of pyrrolizidine toxicity is indistinguishable histologically from other causes of cirrhosis not related to SOS.99
The early histologic changes, occurring as early as 1 week after cytoreductive or myeloablative therapy, include a widened and edematous subendothelial zone of the terminal hepatic venule that contains entrapped red blood cells, resulting in concentric narrowing of the venular lumina.100 Clusters of debris from necrotic hepatocytes, dilated and destroyed sinusoids, and hemorrhage into the space of Disse can be seen surrounding the injured terminal hepatic venules (Fig. 51.14). The necrotic debris may also fill the venular lumina. In this early phase, the hepatic veins are often difficult to locate because of the degree of perivenous hemorrhage. Use of special stains, such as Masson trichrome or reticulin, may help demonstrate the presence and location of the hepatic veins. Examination of tissue under polarized light may also help identify collagen bundles that surround the hepatic veins.
As SOS evolves, hepatic stellate cells are activated, and collagen fibers are deposited in the sinusoidal spaces and hepatic venules. After several weeks, sinusoidal congestion and dropout (loss) of zone 3 hepatocytes develop, with sinusoidal fibrosis and intimal fibrosis of hepatic veins that are typically less than 200 µm in diameter (Fig. 51.15); this constitutes the “VOD” lesion. Bridging fibrosis connecting central veins can be seen in advanced stages.91 The degree of sinusoidal sclerosis and venous luminal narrowing is associated with the severity of the clinical signs and symptoms.100,101
Differential Diagnosis
Although acute or chronic graft-versus-host disease (GVHD) needs to be considered in patients who have undergone hematopoietic stem cell (bone marrow) transplantation, the histologic patterns of injury are different from SOS. The injuries of GVHD center in the portal tracts, namely, bile duct damage and endotheliitis in acute GVHD and paucity of bile ducts in chronic GVHD.101,102
Sinusoidal Cellular Infiltration
Extramedullary hematopoiesis, characterized by the presence of megakaryocytes, normoblasts, and other hematopoietic cells, may be found in the liver of normal infants, in patients who have undergone cardiac bypass surgery, and in patients with myeloproliferative disorders.103 Mast cells may be numerous in mastocytosis and, most likely because of obliterative hepatic venopathy, may be associated with portal hypertension.104 Malignant neoplasms, including lymphoma and hairy cell leukemia, breast cancer, and malignant melanoma, may infiltrate the sinusoids and cause ischemia and hepatic failure.62,105–107 Inherited storage disorders, including especially Gaucher disease and Neimann-Pick disease (see Chapter 54), may also cause marked enlargement of Kupffer cells. However, these storage conditions do not readily cause portal hypertension, despite the hepatomegaly that develops. The splenomegaly and hypersplenism in these storage conditions is primary rather than secondary to portal hypertension.
Amyloidosis and Light Chain Deposition Disease
Hepatic amyloidosis is a rare disease that manifests as an infiltrative disease involving the liver. More than 90% of patients with amyloidosis have amyloid in the liver.108,109 Amyloidosis is usually seen in a systemic form, but 10% to 20% of cases are localized.
Clinical Features
Patients with amyloidosis may have hepatomegaly, cholestasis, hepatic failure, ascites, or portal hypertension.110,111 Other features include renal failure, nephrotic syndrome, and cardiomyopathy. Light chain deposition disease may also present with hepatomegaly, cholestasis, or hepatic failure.112,113 Among patients with amyloidosis, those with hepatic involvement have significantly higher levels of alkaline phosphatase and C-reactive protein than those without hepatic amyloidosis.114
Pathologic Features
Amyloid is deposited in the matrix space or connective tissue of the portal tracts, or within the sinusoidal space of Disse, and within thickened vascular walls of arteries or veins. There is typically great variation in both the severity and the distribution of hepatic amyloid deposits (Fig. 51.16). Although arterial and sinusoidal deposits are most frequent, small globular deposits may occur in the portal tracts or parenchymal regions and may represent the initial stage of the sinusoidal pattern of involvement.115–117 Bile ducts and peribiliary glands may show subepithelial deposits.118,119 The pattern of distribution does not distinguish between AA and AL amyloidosis.108 Apolipoprotein A1 amyloidosis usually shows interstitial deposits in the portal tracts.120 In transthyretin amyloidosis, hepatic deposition is largely confined to nerves, so that clinical evidence of liver disease is usually absent.
In advanced cases with massive infiltration, the liver is enlarged and has a rubbery elastic consistency. Severe sinusoidal involvement impedes hepatocellular nutrition, leading to atrophy and dropout of hepatocytes, especially in zone 1.108 Venous involvement may lead to accentuation of hepatocyte loss and condensation of amyloid into a confluent mass (amyloidoma). NRH may occur in patients with amyloidosis.121
Amyloid deposits are usually easily recognized on routine staining. Amyloid deposits stain weakly with eosin and with periodic acid–Schiff (PAS) stain with diastase digestion (d-PAS); they are usually positive with Congo red stain. Minimal involvement may be visualized with Congo red staining followed by examination under polarized light to detect apple-green birefringence. Thioflavin-T stain and an immunostain for the P component are more sensitive than Congo red at detecting small amounts of amyloid. Congophilia of AA amyloid is abolished by permanganate treatment. With regard to the differential diagnosis, light chain deposition disease may involve the sinusoidal spaces in a manner that resembles amyloid on H&E and d-PAS stains.112 However, these deposits have a granular ultrastructure, whereas amyloid deposits are fibrillar.
The diagnosis of amyloidosis is often established in biopsy specimens of the rectum, abdominal fat pad, labial salivary glands, or liver. Injection of radiolabeled serum amyloid P component, followed by scintigraphy, is a sensitive and specific method for establishing a diagnosis of both AA and AL amyloidosis. This method avoids the need for biopsy and demonstrates the entire distribution of amyloid deposits within the human body.119
Hepatic Vein Disease
Hepatic venous outflow obstruction is the physiologic event of impaired efflux of blood from the liver. Congestive hepatopathy is defined as histologic lesions caused by hepatic venous outflow obstruction at sites including the sinusoids and portal and hepatic veins of all sizes. The causes of hepatic venous outflow obstruction are many (see Tables 51.1 through 51.3). The term congestive hepatopathy has been used to describe the hepatic effects of congestive heart failure122 but also in patients with primary intrahepatic obstruction. To avoid confusion, the latter condition may be designated intrahepatic congestive hepatopathy. However, the lesions are qualitatively identical regardless of whether the obstruction is primarily in the heart, the large hepatic veins (e.g., in Budd-Chiari syndrome), or in small hepatic veins (most forms of chronic liver disease).
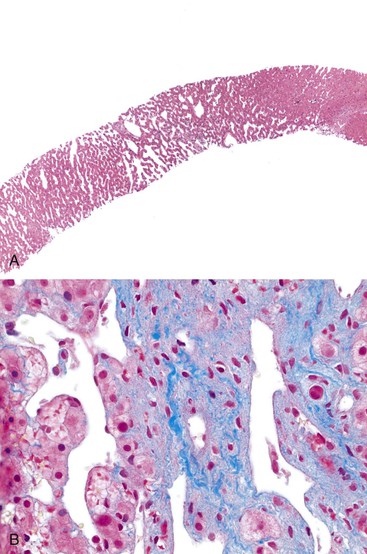
The concept of congestive hepatopathy is useful because it distinguishes primary vascular pathophysiology from the diagnostically nonspecific vascular lesions observed in chronic liver disease. Another important feature of congestive hepatopathy is that congestive injury to the vascular bed leads to further outflow obstruction in a positive feedback loop. In the case of vascular pathophysiology secondary to chronic liver disease, the obstructive complications of vascular damage contribute to the irreversibility of late-stage cirrhosis (see Chapter 50).
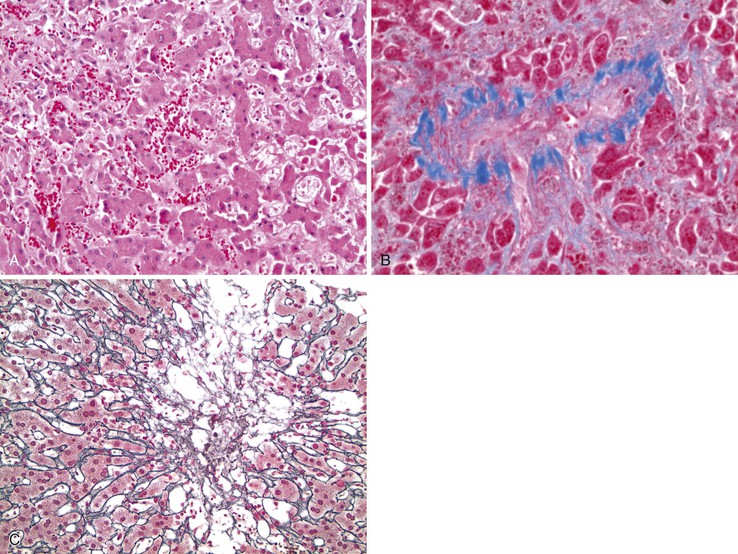
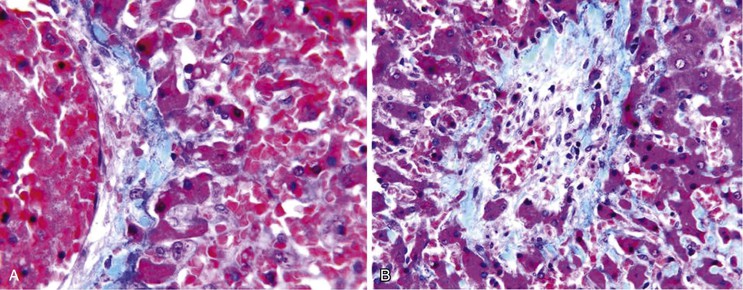
The histologic features of congestive hepatopathy include congestive hepatic venopathy, congestive portal venopathy, congestive sinusoidal injury, and interstitial edema. The histologic appearance of these lesions varies in severity and chronicity. For congestive sinusoidal injury, the mildest form is sinusoidal dilatation, with or without hepatocellular atrophy. More severe disease is marked by hemorrhage into the liver cell plates, apoptosis or frank necrosis of sinusoidal endothelial cells and hepatocytes, and collapse of the tissue (parenchymal extinction) (Fig. 51.17). In chronic lesions, sinusoidal fibrosis (pericellular fibrosis) develops. Venous lesions also show a spectrum of changes, from intimal edema and hemorrhage to intimal fibrosis with luminal obstruction. Figure 51.18 shows an example of chronic congestive hepatopathy in a patient with schistosomiasis.
When venous outflow obstruction is severe, the portal vein serves as an alternative outflow tract, and outflow obstruction may worsen when there is pathology in the portal vein (e.g., thrombosis). Dilatation of the hepatic arteries and angiogenesis are other responses to venous outflow obstruction. In severe and advanced venous outflow obstruction, cirrhosis may develop as an end point.
Thrombosis of Large Hepatic Veins (Budd-Chiari Syndrome)
Only in the last 10 years has agreement been reached on a unified terminology whereby Budd-Chiari syndrome refers to hepatic venous outflow tract obstruction irrespective of the level or the etiology of the obstruction.124–126 Obstruction of the hepatic venous outflow tract can occur in the small hepatic veins, in large hepatic veins, in the inferior vena cava, or in a combination of large hepatic veins and inferior vena cava.127 The predominant form of Budd-Chiari syndrome in certain regions of Asia is pure obstruction of the inferior vena cava or a combination of obstruction of the inferior vena cava and the hepatic vein.128 Pure obstruction of the hepatic vein is the prevalent form of thrombosis in Western countries.129
Budd-Chiari syndrome occurs in the setting of prothrombotic conditions or risk factors underlying venous thrombosis. Myeloproliferative disorders account for as many as half of the cases130–132 and include such conditions as polycythemia vera, essential thrombocythemia, and idiopathic myelofibrosis.133 There is a significant association between myeloproliferative disorders and a somatic point mutation of the JAK2 tyrosine kinase (i.e., JAK2 V617F).134–137 The prevalence of JAK2 mutation is approximately 9% in Budd-Chiari syndrome.138 Factor V Leiden mutation, antiphospholipid syndrome, and the G20210A prothrombin gene mutation are the next most common prothrombotic factors.139 Other risk factors associated with Budd-Chiari syndrome include Behçet disease,140 hypereosinophilic syndrome,141 granulomatous venulitis,142 and pregnancy.143
Clinical Features
Symptomatic obstruction of all three hepatic veins (Budd-Chiari syndrome) typically manifests with painful hepatomegaly, ascites, fever, and liver failure. Gastrointestinal bleeding, jaundice, and hepatic encephalopathy are uncommon. Portal venous obstruction is a common manifestation when the disease is severe.144–146 Increasingly, patients are discovered with minimal symptoms and with imaging studies that reveal involvement of only one or two of the main hepatic veins.147 Thrombi in large hepatic veins often recanalize early in the course of the Budd-Chiari syndrome, so that some patients have cryptogenic cirrhosis and patent large hepatic veins on imaging studies at presentation. Obstruction of the vena cava is associated with dilated veins in the abdominal wall and chest and edema of the legs.148 Serum transaminases, alkaline phosphatase, bilirubin, albumin, and prothrombin can be normal or increased. The protein level in ascitic fluid varies, but if it is higher than 3.0 g/dL, Budd-Chiari syndrome should be considered. However, the protein level can also be elevated in cardiac or pericardiac disease. Imaging studies such as ultrasonography with Doppler flow provide initial evidence of Budd-Chiari syndrome in most cases. Other imaging modalities include hepatic scintigraphy, computed tomography, and magnetic resonance imaging.139 These modalities are most optimally used when the obstruction occurs in large hepatic veins.
The disease course is typically chronic, with less severe hepatic dysfunction compared with isolated hepatic vein disease.149 Thrombosis typically recurs, leading to episodic worsening of disease. As a result of either sluggish blood flow or reversal of blood flow in the portal veins, thrombosis of the large portal veins occurs in 10% to 20% of patients with hepatic vein obstruction. Blood from the caudate lobe drains directly into the inferior vena cava via the inferior right hepatic vein. If this vein is spared from the thrombotic process, the caudate lobe may undergo segmental hyperplasia.150 Caudate lobe hyperplasia or large regenerative nodules elsewhere in the liver may resemble a neoplasm clinically.144 Long-standing Budd-Chiari syndrome has been shown to be a cause of hepatocellular carcinoma, especially in patients who have thrombotic involvement of the inferior vena cava.149
Pathogenesis
Thrombosis is the most common cause of large hepatic vein obstruction. This condition may occur in an otherwise normal liver or in a liver with an obstructing lesion such as a neoplasm or cirrhosis. Amoebic abscess is an important cause of hepatic vein disease in developing countries. Sarcoidosis may involve medium- and large-sized hepatic veins. As indicated in Table 51.1, hypercoagulable states are often associated with hepatic vein thrombosis. Oral contraceptives and pregnancy are important stimuli of the coagulation cascade. Other predisposing factors include paroxysmal nocturnal hemoglobinuria, factor V Leiden, prothrombin G20210A, and protein C deficiency. Frequently, patients may have more than one risk factor, typically an underlying hypercoagulable state with an acute initiating event such as commencement of oral contraceptive use, pregnancy, trauma, or infection.151
Membranous Obstruction of the Vena Cava
The term membranous obstruction of the vena cava refers to a short or long segmental narrowing that is believed to represent organized thrombus that has undergone partial resorption.152 It is not to be confused with the eustachian valve, which is a normal vein valve that in some individuals is encountered in the inferior vena cava, just proximal to the effluence of the hepatic veins and below the diaphragm. Although this condition is rare in Western countries, the majority of patients with Budd-Chiari syndrome in developing countries have membranous obstruction, known as obliterative hepatocavopathy.153 These patients do not usually have a recognizable hypercoagulable state. Recurrent infections or other environmental factors probably initiate caval injury; hepatic outflow obstruction occurs only after thrombi extend to the ostia of the hepatic veins.153 In this subgroup of patients, hepatocellular carcinoma is a frequent complication and results from their otherwise chronic liver disease.
Pathologic Features
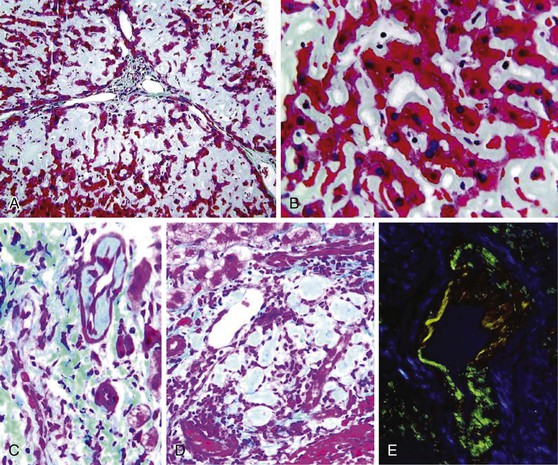
The gross and microscopic features of Budd-Chiari syndrome are shown in Figure 51.19. At the macroscopic level, acute obstruction of all three hepatic veins causes massive hepatomegaly with prominent engorgement of the liver parenchyma. With time, the affected lobes become smaller, whereas unaffected regions undergo compensatory hypertrophy, a phenomenon that most frequently affects the caudate lobe.
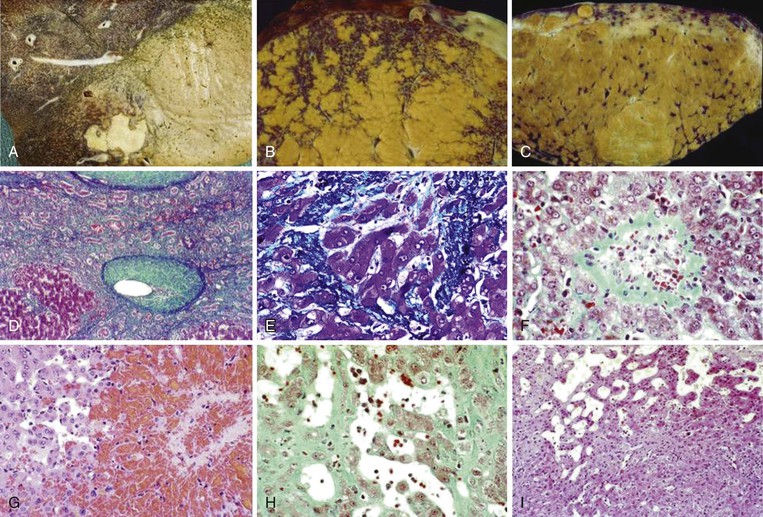
At the microscopic level, centrizonal sinusoidal congestion and dilatation, atrophy and dropout of hepatocytes, and perivenular fibrosis are seen. Large- and medium-sized hepatic veins show intimal fibrosis, often in the form of multiple layers, suggesting recurrent thrombosis. Most veins achieve some degree of recanalization with the formation of multiple luminal channels or delicate webs. Secondary thrombosis of small and large portal veins may be observed as well. The type of parenchymal injury that occurs in cases of pure hepatic vein obstruction typically leads to a characteristic venocentric pattern of septation, termed reverse nodularity. The reason is that hepatocyte necrosis occurs mainly near the obstructed hepatic veins, which results in a central vein–to–central vein pattern of necrosis and, subsequently, fibrosis. The portal tracts are seldom incorporated into the fibrous septa.
When secondary portal vein thrombosis occurs, venoportal septa may develop, leading ultimately to a “venoportal” pattern of cirrhosis. By way of context, venoportal cirrhosis is the type seen in the cirrhosis that develops in the setting of chronic hepatitis.
Regions of liver only mildly affected by venous outflow obstruction may develop a pattern of atrophy and compensatory hyperplasia that can be mistaken for NRH. Indeed, large regenerative nodules in better-vascularized areas of the liver are detected in more than 50% of cases of hepatic vein thrombosis. Although the nodules are usually few in number and typically 1 to 2 cm in diameter, in some livers they may be numerous and can reach several centimeters in maximum diameter. Clinically and radiologically, these nodules may be misinterpreted as neoplasms or as FNH.
Liver biopsies are helpful for diagnosing hepatic vein outflow obstruction. A wedged hepatic venous pressure measurement may also be obtained.91 A biopsy is sometimes used to determine the severity of the disease, including the presence of necrosis and fibrosis.127 Biopsies obtained via the transvenous route may detect thrombotic material sampled from the lumen of the hepatic vein. Regional variation in pathologic features may lead to spurious conclusions. Therefore, biopsies taken for this purpose should be obtained from two sites.
Differential Diagnosis
Budd-Chiari syndrome should be suspected when one sees severe congestion or hemorrhage within the liver cell plates, recent thrombi within hepatic veins of any size, or intimal fibrous thickening of hepatic veins larger than 100 µm in diameter. Integration of the clinical history and imaging studies with the histologic findings is important to establish a correct diagnosis.
In settings of acute or chronic obstruction to hepatic venous outflow, the differential diagnosis for Budd-Chiari syndrome includes SOS (after bone marrow transplantation or after exposure to pyrrolizidine alkaloids), pericarditis, and congestive heart disease. The pathologic features of congestive heart failure, constrictive pericarditis, and chronic SOS may be quite similar to those caused by large hepatic vein thrombosis. Oxaliplatin-based systemic neoadjuvant chemotherapy in patients with metastatic colorectal cancer can cause injury to the hepatic microvasculature that leads to sinusoidal obstruction; this injury is manifested by perisinusoidal fibrosis that may mimic Budd-Chiari syndrome.94,154 Some other forms of drug-induced injury manifest with thrombi within small-sized hepatic veins.155
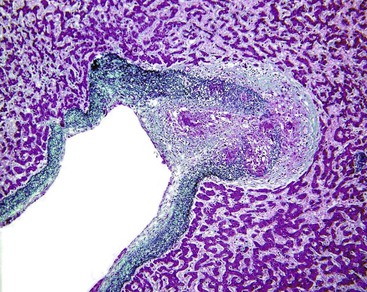
Care must be taken to examine whether any of the following are present: vasculitis, neoplasm, granulomas (Fig. 51.20), sickled red cells, intraparenchymal abscess, or infectious agents, such as amebae or fungi. The presence of extramedullary hematopoiesis may suggest a myeloproliferative disease. Most hypercoagulable states require laboratory tests for diagnosis. Bone marrow culture may reveal the presence of a subclinical myeloproliferative disease if red blood cell colonies form, in vitro, without exogenous erythropoietin stimulation. Protein S and C deficiencies are difficult to diagnose because hepatic failure may result in a decrease of the serum levels of these proteins. Genetic tests are available for some types of anomalies of the coagulation cascade.
Obliteration of small hepatic veins and congestion of the liver parenchyma are frequent findings in cirrhosis of any cause. Ironically, the long-standing venocentric cirrhosis caused by hepatic vein thrombosis may show little or no congestion, possibly because of the perivenous scarring or the parenchymal regeneration that occurs. As a result, accurate histologic differentiation of hepatic vein thrombosis from other types of cirrhosis often requires large enough tissue samples to allow for an evaluation of the large hepatic veins. In any case, histologic evaluation can be difficult, because a liver with severe cirrhosis of any etiology often shows intimal thickening of medium- and large-sized hepatic veins due to congestive venopathy or thrombosis.156
Congestive Heart Failure
Long-standing obstruction to hepatic venous outflow, as in right-sided heart failure or constrictive pericarditis, can cause all of the features of congestive hepatopathy described earlier in patients with hepatic vein thrombosis. In congestive heart failure, the degree of thrombosis is usually less severe than in patients with hepatic vein thrombosis without cardiac disease.157 Typical microscopic changes include mild hepatocyte atrophy and sinusoidal dilatation. In specimens with evidence of obliteration of hepatic veins, fibrous septation is often present as well. However, nodules completely surrounded only by fibrous septa rarely occur.158 Therefore, only cardiac sclerosis may develop, as opposed to cardiac cirrhosis when portal vein thrombosis is superimposed (see Chapter 50).
Small Hepatic Vein Disease (Obliterative Hepatic Venopathy)
Injury to small hepatic veins may be caused by phlebitis, thrombosis, or luminal obstruction, and it may result in complete disappearance of the vessels. Obliterative hepatic venopathy may occur in patients with many types of disorders, such as chronic viral hepatitis, steatohepatitis,159 hepatic vein thrombosis,144 congestive heart failure,157 toxin- or radiation-induced injury,160 mast cell disease,104 sarcoidosis,11 granulomatous phlebitis,161 chronic granulomatous disease,162 and inflammatory pseudotumor.162 Identical lesions may also occur adjacent to hepatic neoplasms and abscesses. Recurring fibro-obliterative venopathy is a rare form of diffuse small hepatic vein occlusion that was reported in liver transplants in two published cases.163,164
The pathogenesis of obliterative hepatic venopathy varies according to the specific etiology and includes the effects of local inflammation, thrombosis, and congestive hepatic venopathy. Local inflammation is the cause in most cases of chronic hepatitis, steatohepatitis, and hepatic abscesses (Fig. 51.21). In congestive heart failure, in late cirrhosis, and in tissue adjacent to neoplasms, the pathogenesis of venopathy is related to hepatic congestion venopathy. In patients with hepatic vein thrombosis, the mechanism of small vessel injury is usually related to either congestive venopathy or propagation of thrombi from large-sized vessels. The vessel lesions that occur in patients with chronic hepatitis and cirrhosis are discussed in Chapter 50. The specific etiology may be indicated by the histologic milieu in which the venopathy occurs, such as steatohepatitis, chronic viral or autoimmune hepatitis, sarcoidosis-related granulomas, or vascular thrombosis.
Nodular Hyperplasia and Other Tumor-Like Conditions
Nodular hyperplasia is a family of lesions in which benign, apparently regenerative, hepatocytes form nodules as a response to parenchymal injury or vascular compromise in the liver. The lesions are classified according to their size, distribution, and histologic appearance.165,166 Clinically and pathologically, they should be distinguished from neoplastic liver nodules, which are discussed more fully in Chapter 55.
Large Regenerative Nodules
Large regenerative nodules measure from 1 cm to many centimeters in diameter.165 They may develop in patients with massive hepatic necrosis, cirrhosis, Budd-Chiari syndrome (see Fig. 51.19), hereditary hemorrhagic telangiectasia, portal vein absence, or portal vein thrombosis. Partial nodular transformation is an obsolete term that was originally applied to noncirrhotic livers with a large perihilar nodule in patients with portal hypertension. It is now believed that this condition represents the presence of large regenerative nodules in response to portal vein thrombosis.
Pathologic Features
Large regenerative nodules are composed of hepatocytes that range from completely normal to markedly reactive in appearance. The nodules are supplied by portal tracts that contain arteries and ducts, either with or without portal veins. Lobar hyperplasia in Budd-Chiari syndrome, primary sclerosing cholangitis, and biliary atresia are physiologically similar to large regenerative nodules but are delimited, in part, by the hepatic capsule. Marked regenerative activity, particularly in the presence of cholestasis, may be difficult to distinguish from dysplasia. However, regenerative nodules show cells with a low nucleus-to-cytoplasm ratio, hepatocyte plates that are usually less than two cells thick, absence of atypical mitoses, and retention of reticulin that are usually associated with portal structures. Regenerative nodules that occur in the absence of a portal vein may be confused with FNH.
Focal Nodular Hyperplasia
FNH is, essentially, a large regenerative nodule with characteristic clinical and histologic features. This lesion is discussed more fully in Chapter 55.
Clinical Features
FNH is the most frequent type of large regenerative nodule, and it is the most common benign solid liver lesion, occurring in approximately 3% of the adult population. Ninety percent of patients are women of child-bearing age, two thirds of whom have a history of oral contraceptive use. FNH also occurs in children and in the elderly. Although they usually are an incidental finding, the lesions can cause pain and in rare instances can hemorrhage. Hepatic hemangioma coexists in 20% of patients. A variety of systemic vascular anomalies have been found in association with FNH, including absence of portal vein, dystrophic systemic arteries (often with spontaneous rupture), and vascular anomalies in the brain.167 Astrocytoma and meningioma have also been reported in patients with FNH. Hepatic imaging studies characteristically show increased arterial flow involving a large artery, centripetal flow, and prolonged opacification.
Pathogenesis
Early observers reported that FNH resembles cirrhosis and therefore may represent a reaction to tissue injury.168,169 The nodularity of FNH is thought to be a manifestation of hepatocellular hyperplasia in response to increased arterial blood flow.170,171 The cells in FNH have been shown to be polyclonal.172 The consistent presence of a single large feeding artery led to the suggestion that FNH may represent a hyperplastic response to the effects of altered blood flow from a large anomalous artery.173 However, the presence of arteriovenous shunts in FNH indicates that the artery may be a secondary phenomenon and that the loss of portal veins and ducts characteristic of this lesion may also occur secondary to shunts.174
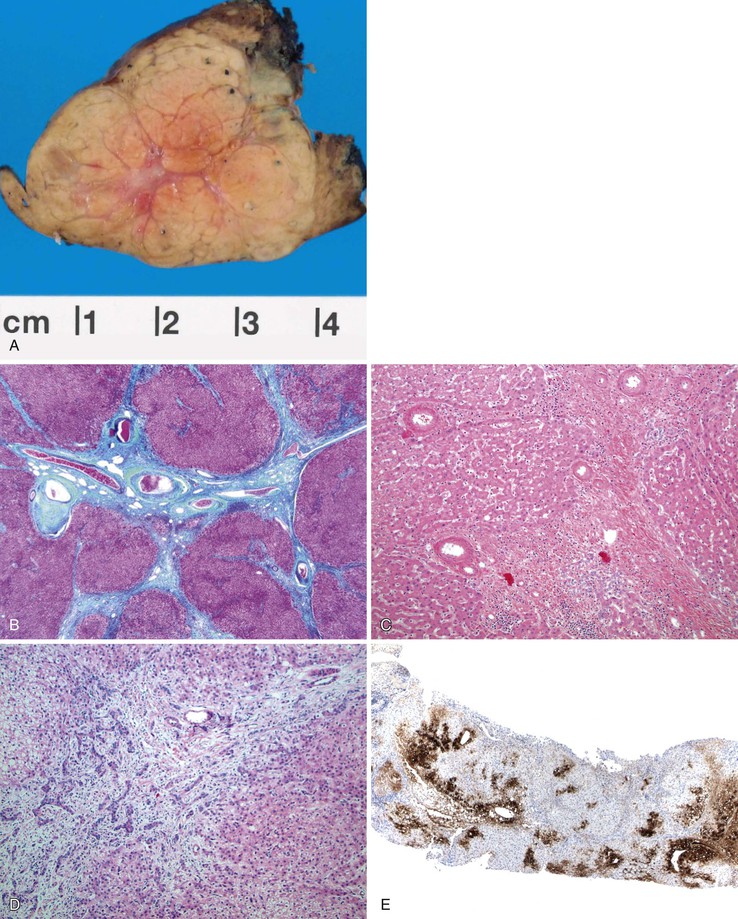
Pathologic Features
FNH nodules range from a few millimeters to larger than 5 cm, occasionally occupying an entire lobe of the liver (Fig. 51.22). Although in most cases of FNH they are solitary, approximately 20% occur as multiple lesions. Grossly, the lesion is well demarcated and not encapsulated. The cut surface shows a central, star-shaped scar with radiating septa. Microscopically, thick fibrous septa are invariably present within a nodule of hyperplasic hepatocytes arranged in cords as much as two cells thick; they are revealed by a reticulin stain. A bile ductular reaction is present on the border between the fibrous septa and parenchyma; this is demonstrated by a cytokeratin 7 (CK7) stain. Many thick-walled arteries are also present within the fibrous septa. Inflammatory infiltrates of mononuclear cells are also present in the fibrous septa. Changes of chronic cholestasis with cholate stasis and accumulation of copper (demonstrated by the rhodanine stain) or copper-binding protein (demonstrated by the Victoria blue stain) are also very common.175 In equivocal cases when the typical features such as fibrous septa, ductular reaction, and thick-walled arteries are lacking, glutamine synthetase stain showing characteristic broad and anastomosing regions mimicking a “maplike” pattern is diagnostic.176 FNH-like nodules that contain portal veins or bile ducts may represent large regenerative nodules as a precursor to FNH.177
Differential Diagnosis
Clinically and pathologically, FNH should be distinguished from hepatic adenoma (see Chapter 55). This is not usually difficult, unless one is evaluating only biopsy specimens. Adenomas are more uniform in appearance, without alternating areas of hepatocyte hyperplasia and atrophy. Adenomas also have less periarterial collagen than FNH. Most adenomas do not contain bile ducts or ductules, although a telangiectatic variant may contain these structures.168 FNH exhibits neither hemorrhage nor malignant transformation, unlike hepatocellular adenoma.
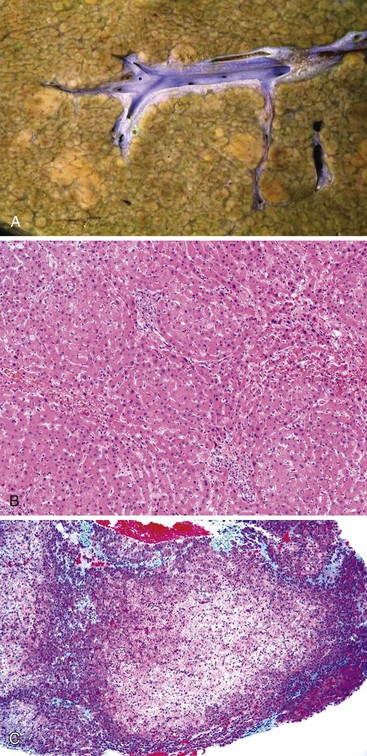
Nodular Regenerative Hyperplasia
NRH is defined by the presence of multiple 1- to 2-mm nodules separated by regions of hepatocyte atrophy with little or no fibrous septation (Fig. 51.23). NRH develops in circumstances in which the intrahepatic circulation is heterogeneous, particularly when small portal veins are obliterated and the hepatic circulation is replaced by arterial flow. Mild outflow obstruction, as in congestive heart failure or small hepatic vein obstruction, may contribute to the development of NRH. However, major outflow obstruction causes more severe degrees of hepatocyte loss, which leads to cirrhosis rather than NRH. The prevalence of NRH ranges from 0.72% to 2.6% in autopsy series.178–180 The risk for NRH increases with age.178
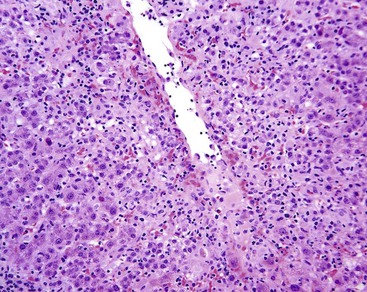
Clinically, NRH may be asymptomatic, or it may be associated with portal hypertension, usually without ascites. Many diseases have been associated with NRH. They are commonly associated with prominent portal tract inflammation, suggesting a causal relationship of this feature with NRH. These diseases include primary biliary cirrhosis, polyarteritis nodosa, systemic sclerosis, SLE, rheumatoid arteritis, chronic granulomatous disease, cystinosis, and mastocytosis.178,181 NRH may develop adjacent to mass lesions as well.
Pathologically, the capsular surface of the liver often reveals minimal shallow irregularities that may be mistaken for cirrhosis. Microscopically, NRH is composed of regenerating nodules of hepatocytes of normal or slightly regenerative appearance that abut each other without intervening fibrosis. The area between nodules usually shows atrophy of the hepatocytes and condensation of reticulin, which can impart a false impression of fibrous tissue. A reticulin stain may help to delineate the nodular contour as well as the compressed and atrophic hepatocytic cords; Masson trichrome stain demonstrates absence of fibrous septa. Portal structures are usually present, but small portal veins may be obliterated. Sinusoids may be arterialized, and therefore may show positivity with the CD34 stain.
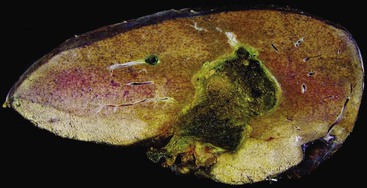
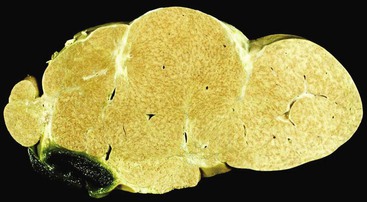
Hepar Lobatum
Hepar lobatum is a condition in which the presence of deep furrows in the liver capsule causes the lobes to be segmented. This gross anatomic finding is usually accompanied by almost normal-appearing liver parenchyma. Hepar lobatum may result from any disease that causes focal obstruction of adjacent large portal and hepatic veins, most often Hodgkin disease, metastatic breast carcinoma, or syphilitic gummata (Fig. 51.24). Hepar lobatum develops when the primary lesion causes obstruction of vessels while tumor growth remains slow and minimal.
Fibrotic Conditions with Vascular Pathophysiology
Cirrhosis is the most important fibrotic condition of the liver associated with impaired blood flow. However, two noncirrhotic conditions are also associated with impaired blood flow in which fibrosis is prominent and portal hypertension occurs.
Congenital Hepatic Fibrosis
The occurrence of splenomegaly and portal hypertension in an otherwise healthy child or adolescent may prompt evaluation for the presence of clinically undetected cirrhosis or inherited conditions. Congenital hepatic fibrosis is a condition in which dense portal tract–based bridging fibrous septa develop during the course of childhood and adolescence (see Chapter 54). Sclerosis of portal tracts impairs the flow of portal vein blood into the liver. However, this condition is not considered true cirrhosis. The parenchyma of the liver is not damaged, and regenerative nodules do not develop.
Hepatoportal Sclerosis
In the period 1884 to 1910, Banti proposed the term morbus Banti for a condition characterized by the presence of primary cryptogenic splenomegaly and anemia not associated with hematologic disease.182 In the first half of the 20th century, the association of portal hypertension and splenomegaly with “Banti disease” was expanded to include patients with cirrhosis or portal vein thrombosis.183,184 In the 1960s, investigators in India established a disease entity termed noncirrhotic portal hypertension,185 which is characterized histologically by “obliterative portovenopathy.” A similar type of noncirrhotic sclerosis of intrahepatic portal veins was described in 1965 by Mikkelsen and associates16 and given the term hepatoportal sclerosis. Working in Calcutta, Basu and colleagues established that patients with noncirrhotic intrahepatic portal vein sclerosis had a more favorable prognosis than patients with cirrhosis,186 and they termed this condition idiopathic portal hypertension. Ultimately, the diagnostic names given to this condition included Banti disease, hepatoportal sclerosis, noncirrhotic portal fibrosis, obliterative portal venopathy, noncirrhotic intrahepatic portal hypertension, benign intrahepatic portal hypertension, and idiopathic presinusoidal portal hypertension.184,187–189
The geographic distribution of IPH is not homogeneous throughout the world. It is rare in the West, and most series have been reported from Asian countries including India and Japan. However, the prevalence appears to have significantly decreased in Japan after 1970.184
Definition
Hepatoportal sclerosis denotes a condition consisting of portal hypertension, splenomegaly, and anemia (caused by hypersplenism) in a patient without cirrhosis but with dense portal tract fibrosis and obliteration of portal veins.184 By definition, parasites, myeloproliferative disease, and radiographic evidence of occlusion of the major hepatic and portal veins are excluded. However, some studies also describe IPH as a condition that results from thrombosis of the intrahepatic portal veins190 and hepatoportal sclerosis as an “abnormality of intrahepatic portal veins with portal fibrosis and nodular regeneration.”191 Thus, confusion persists regarding the definition of this condition. To create further confusion, dense portal tract fibrosis also may be the residuum of regressed cirrhosis (discussed in Chapter 50) or a remnant lesion of intrahepatic portal vein thrombosis (discussed earlier in this chapter).
The underlying etiology of hepatoportal sclerosis remains unclear. Factors such as infection, autoimmunity, and hypercoagulopathy have been suggested based on epidemiologic or experimental studies.192–198 The key issue remains whether portal tract sclerosis can arise de novo and generate “true” hepatoportal sclerosis.
Chronic exposure to arsenic, which may occur after years of arsenic ingestion at concentrations of 0.01 mg/kg/day, does, in fact, result in a “true” hepatoportal sclerosis.199 For example Nevens and co-workers200 described eight patients who had been treated for psoriasis and had received arsenic-containing Fowler solution. The total arsenic intake varied from 4 to 16 g, and the interval between treatment and the onset of portal hypertension ranged from 2 to 16 years. Datta and colleagues201 reported the development of noncirrhotic portal hypertension in nine patients from northern India, all of whom had consumed high levels of arsenic from contaminated drinking water, adulterated opium, or indigenous medicines. The hypothesis has been advanced that arsenic can cause hepatoportal sclerosis by inducing chronic damage to the intrahepatic portal veins; whether this involves bone fide thrombosis of these veins is not known.
Other conditions associated with noncirrhotic portal hypertension in which the reported histologic lesion is “hepatoportal sclerosis” include latent or overt myeloproliferative syndromes,202 a variety of prothrombotic states,201 human immunodeficiency virus (HIV) infection,203 and the rare Adams-Oliver syndrome.191 In Japan, a role for immunologic abnormalities has also been suggested.184
Clinical Features
Clinically, hepatoportal sclerosis is manifested by portal hypertension, splenomegaly, and pancytopenia resulting from hypersplenism. Aside from splenomegaly, anemia, and noncirrhotic portal hypertension,184 patients usually have normal or near-normal serum transaminases, bilirubin, and alkaline phosphatase levels. Hepatic function is intact. Esophageal varices are often demonstrable by endoscopy or radiography, and the hepatic veins are patent. The extrahepatic portal vein is patent, usually with the formation of collateral vessels. There may be normal or slightly elevated wedge hepatic venous pressure. Not all of these radiographic studies need to be performed to invoke the diagnosis, nor is a liver biopsy (percutaneous or transjugular) always necessary. Nevertheless, by definition, associated clinical evidence of portal hypertension should be unequivocal.
Assuming that other conditions are excluded, the prognosis for patients with hepatoportal sclerosis is generally good, particularly if bleeding from esophageal varices can be managed conservatively. Massive bleeding caused by varices is the most common cause of death. Therefore, in severe cases, mitigation of variceal bleeding is critical, either endoscopically or surgically.
Pathologic Features
The pathologic changes of hepatoportal sclerosis are attributable to persistent circulatory insufficiency of the portal venous system.204 Macroscopically, the liver may be normal in size or mildly atrophic, with an irregular undulating and finely wrinkled surface contour.184 Surface features of atrophy are not uniformly distributed, and there may be distinct disproportionality in the sizes of the liver lobes. Definite nodule formation is uncommon. On cut surface of a resected or autopsied liver, the trunk of the portal vein and major intrahepatic portal vein branches may be dilated and may show thickening of the muscular wall. Fibrosis of portal tracts may extend throughout the corpus of the liver, commonly forming spikes or wedges. Conspicuous phlebosclerosis and perivascular fibrosis in the portal tracts may be present, along with newly formed thrombi. The distribution of hepatic veins may be irregular, and they may be sclerosed or obliterated in atrophic regions.
The cardinal pathologic features of IPH are attributed to compromise of the intrahepatic terminal portal radicles with significant fibrosis of the portal tracts and atrophy of the hepatic parenchyma resulting from insufficient perfusion from the portal system.184 Microscopically, the primary features are marked portal fibrosis, obliteration of intrahepatic terminal portal vein radicles, and a variable degree of parenchymal atrophy (Fig. 51.25). The lobular architecture is maintained, but occasional portal-to-portal or portal-to-central bridging septa may be present.135,203 Some cases show extensive ischemic changes in the parenchyma. Portal veins, when identified, exhibit thickening and sclerosis of the wall, with a reduction in luminal diameter and muscular hypertrophy or fibrosis. In many portal tracts, portal vein radicles are completely obliterated by fibroelastosis. These changes are best demonstrated with an elastin stain. In other portal tracts, organized thrombi with recanalization may be present. The bile ducts may show concentric periductal fibrosis. Marked dilatation of the sinusoids may also be present, resulting in “megasinusoids,” which are believed to result from portal hypertension. When one is examining the portal tracts of livers with this condition, hepatic arteries and bile ducts serve as valuable anatomic landmarks. Portal veins usually exhibit much larger diameter than the hepatic arteries. However, in hepatoportal sclerosis, the calibers of affected portal veins are similar to those of the hepatic arteries or even smaller. Although thrombosis of the portal veins is a known phenomenon, it is very rare, accounting for only as much as 3% in reported series.205
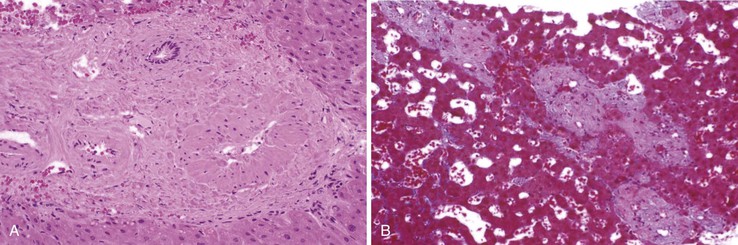
The hepatic parenchyma between affected portal tracts is atrophic, with shrunken hepatocytes and dilated sinusoids. Abnormally dilated blood vessels may be seen connecting residual portal veins to sinusoids. It is thought that increased portal pressure may aggravate the formation of these abnormal vessels,205 which appear to serve as collaterals formed in response to the blockage of the portal blood flow.206 In affected regions, phlebosclerosis and occlusion of the terminal hepatic venules or major branches of the hepatic veins may also be present; this condition is considered to be a result of the persistent insufficiency of the portal blood supply. The hepatic parenchyma commonly shows vague nodularity with hyperplasia but without surrounding fibrous septa. Collagen deposition within the sinusoidal spaces that surround the hepatocytes in a patchy fashion may occasionally be seen. However, hepatoportal sclerosis does not evolve to cirrhosis.
Differential Diagnosis
To establish a diagnosis of hepatoportal sclerosis, cirrhosis, especially highly regressed cirrhosis, should be excluded, along with overt extrahepatic portal vein thrombosis, hepatic vein thrombosis, and the presence of intrabiliary parasites and schistosomiasis. This disorder remains a diagnosis of exclusion. Only then can reassurance of a benign clinical course be conveyed to the patient.
Acknowledgment
We are indebted to Ian R. Wanless, MD, FRCPC, for his extensive work on this chapter in prior editions of this book.