[level-membership-for-neurology-category]
Chapter 51A Vascular Diseases of the Nervous System
Ischemic Cerebrovascular Disease
Pathophysiology of Cerebral Ischemia
Clinical Syndromes of Cerebral Ischemia
Diagnosis and Treatment of Threatened Ischemic Stroke
Large-Artery Atherothrombotic Infarctions
Small-Vessel or Penetrating Artery Disease
Nonatherosclerotic Vasculopathies
Inherited and Miscellaneous Disorders
Infarcts of Undetermined Cause
Essential Investigations for Patients with Threatened Strokes
Preventing Stroke Recurrence: Medical Therapy
Treatment of Acute Ischemic Stroke
Epidemiology and Risk Factors
There are approximately 785,000 new or recurrent strokes annually in the United States (600,000 being first events and 185,000 being recurrent events). Some 88% of these events are ischemic strokes; 8% to 12% of ischemic strokes result in death within 30 days. On average, every 40 seconds someone in the United States has a stroke. Despite gradual declines in overall stroke death rates in many industrialized countries, stroke remains a leading cause of death and disability, particularly in the United States. Worldwide, stroke is also a leading cause of death, with stroke mortality being particularly high in Eastern Europe and Asia (World Health Organization, 2004). By 2020, 19 out of 25 million annual stroke deaths will be in developing countries (Lemogoum et al., 2005). Stroke is also the leading cause of disability in adults. Of the hundreds of thousands of stroke survivors each year, approximately 30% require assistance with activities of daily living, 20% require assistance with ambulation, and 16% require institutional care.
A number of factors that may be classified as modifiable and unmodifiable increase the risk for ischemic stroke (Table 51A.1). Nonmodifiable risk factors for stroke include older age, male gender, ethnicity, family history, and prior history of stroke. Modifiable risk factors may be subdivided into lifestyle and behavioral risk factors and non-lifestyle factors, although these two subgroups are interrelated. Presumed modifiable lifestyle risk factors include cigarette consumption and illicit drug use. Non-lifestyle risk factors include low socioeconomic status, arterial hypertension, dyslipidemia, heart disease, and asymptomatic carotid artery disease. Stroke secondary to sickle cell disease is also a modifiable non-lifestyle risk factor. Potentially modifiable risk factors (that have yet to be shown to decrease risk when modified, however) include diabetes mellitus (DM), hyperhomocysteinemia, and left ventricular hypertrophy. Less well-documented risk factors include blood markers (i.e., C-reactive protein), ankle-brachial blood pressure ratios, silent cerebral infarcts, white-matter hyperintensities on magnetic resonance imaging (MRI), and degree of carotid artery intima-media thickness. Clinicians cannot assume that these risk factors express themselves exclusively by accelerating atherosclerosis. There are also considerable data implicating hemostatic and microcirculatory disorders in stroke as well as circadian and environmental factors.
| Nonmodifiable | Modifiable |
|---|---|
| Age | Arterial hypertension |
| Gender | Transient ischemic attacks |
| Race/ethnicity | Prior stroke |
| Family history | Asymptomatic carotid bruit/stenosis |
| Genetics | Cardiac disease |
| Aortic arch atheromatosis | |
| Diabetes mellitus | |
| Dyslipidemia | |
| Cigarette smoking | |
| Alcohol consumption | |
| Increased fibrinogen | |
| Elevated homocysteine | |
| Low serum folate | |
| Elevated anticardiolipin antibodies | |
| Oral contraceptive use | |
| Obesity |
Risk Factors for Stroke
The incidence of stroke increases dramatically with advancing age, and increasing age is the most powerful risk factor for stroke. The incidence of stroke doubles each decade past 55 years of age. Half of all strokes occur in people older than 70 to 75 years. Overall, stroke incidence rate is 1.25 times greater in men than women. Men develop ischemic strokes at higher rates than women up to the age of 75 years. With an estimated 20% of the population being older than 65, greater than 10 million octogenarians, and an increasing life expectancy in the United States, it is predicted that in the near future, the incidence of stroke will reach 1 million per year. Compared to whites, African Americans have approximately a twofold increased risk of first-ever stroke. The rate of cerebral infarction is higher in African Americans and Hispanic Americans than in whites; this could be partially explained by the higher prevalence of DM and arterial hypertension experienced by these ethnic minorities. African Americans had been thought to have higher rates of intracranial atherosclerotic occlusive disease compared with whites, but this may actually reflect ascertainment bias (Sacco et al., 1995; Wityk et al., 1996). Furthermore, the stroke incidence and case fatality rates are also markedly different among the major ethnic groups in Auckland, New Zealand. Maori and Pacific Islands people have a higher rate of mortality within 28 days of stroke when compared with Europeans, especially men (Bonita et al., 1997). Chinese, Koreans, and Japanese also may have increased rates of intracranial hemorrhage and intracranial atherosclerotic cerebrovascular disease compared to whites. In comparison with the United States and Western Europe, where hemorrhagic stroke represents 20% or less of all stroke subtypes, 40% of the stroke subtypes are hemorrhagic (intracerebral or subarachnoid hemorrhage) in Japan. Conversely, in India, where ischemic stroke accounts for 80% of all strokes, 10% to 15% of strokes occur in people younger than 40 years and are mostly related to intracranial atherosclerosis.
Heredity and Risk of Stroke
Heredity seems to play a role in the pathogenesis of cerebral infarction. An increased risk is seen with a family history of stroke among first-degree relatives. Genetic factors have been linked with the pathogenesis of ischemic stroke, but specific genetic variants remain largely unknown, and some purported genetic associations have not been replicated (Chinnery et al., 2010). There are a number of genetic causes of stroke. Some inherited diseases, such as the hereditary dyslipoproteinemias, predispose to accelerated atherosclerosis. A number of inherited diseases are associated with nonatherosclerotic vasculopathies, including Ehlers-Danlos (especially type IV) syndrome, Marfan syndrome, Rendu-Osler-Weber disease, and Sturge-Weber syndrome. Familial atrial myxomas, hereditary cardiomyopathies, and hereditary cardiac conduction disorders are examples of inherited cardiac disorders that predispose to stroke. Deficiencies of protein C and S or antithrombin (AT) are examples of inherited hematological abnormalities that can cause stroke. Finally, rare inherited metabolic disorders that can cause stroke include mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), Fabry disease, and homocystinuria. The presence of the apolipoprotein epsilon-2 allele in elderly individuals and deletion of the gene for the angiotensin-converting enzyme may increase the risk for stroke, but the association with stroke subtype is unclear (Agerholm-Larsen et al., 1997; Slooter et al., 1997; Szolnoki et al., 2001). The most significant findings of the ongoing Siblings With Ischemic Stroke Study (SWISS) to date have been the relationship between age and stroke in probands and sibs, and the lack of a tight association among ischemic stroke subtypes (Adams Jr. et al., 1993) within families (Meschia et al., 2005; Meschia et al., 2006; Wiklund et al., 2007).
Common Modifiable Risk Factors
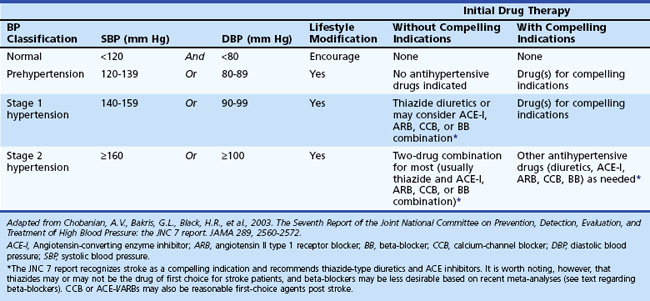
At least 25% of the adult population has arterial hypertension, defined as systolic blood pressure (SBP) greater than 140 mm Hg or diastolic blood pressure (DBP) greater than 90 mm Hg. Prehypertension is defined as SBP between 120 and 139 mm Hg or DBP between 80 and 90 mm Hg. Optimal blood pressure is defined as SBP less than 120 mm Hg and DBP less than 80 mm Hg (Chobanian et al., 2003). Similar guidelines have been developed by the European Society of Hypertension–European Society of Cardiology Guidelines Committee (2003). The JNC7 Report (Table 51A.2) emphasizes that patients at risk, including those with DM or a history of stroke, should be treated with medications. Unfortunately, arterial hypertension remains poorly treated worldwide, and in the United States many patients are either undertreated or untreated (Hajjar and Kotchen, 2003). Arterial hypertension predisposes to ischemic stroke by aggravating atherosclerosis and accelerating heart disease, increasing the relative risk for stroke an estimated three- to fourfold. The risk is greater for patients with isolated systolic hypertension and elevated pulse pressure. Arterial hypertension is also the most important modifiable risk factor for stroke and the most powerful risk factor for all forms of vascular dementia (vascular cognitive impairment). Lowering blood pressure in stroke survivors helps prevent recurrent stroke and is more important than the specific hypotensive agent used, although there is some suggestion that beta-blockers are less preferred than other agents based on recent meta-analyses (Lindholm et al., 2005). Blood pressure treatment that results in a modest reduction in SBP of 10 to 12 mm Hg and 5 to 6 mm Hg diastolic is associated with a 38% reduction in stroke incidence (MacMahon and Rodgers, 1996). Treatment of isolated systolic hypertension in the elderly is also effective for reducing stroke risk. The Systolic Hypertension in the Elderly Program showed a 36% reduction in nonfatal plus fatal stroke over 5 years in the age 60-and-older group when isolated systolic hypertension was treated. Treating systolic hypertension also slows the progression of carotid artery stenosis. The PROGRESS trial evaluated the effects of perindopril and indapamide on the risk for stroke in patients with histories of stroke or transient ischemic attack (TIA). Regardless of blood pressure at entry, patients clearly benefited from treatment (PROGRESS Collaborative Group, 2001).
Table 51A.2 JNC 7 Report: Seventh Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure
About 171 million people worldwide have type 2 DM, including 18 million Americans. It is estimated that these numbers will grow to 366 million people worldwide and 30 million Americans by 2030 (Fonseca et al., 2002). Diabetes mellitus increases the risk of ischemic cerebrovascular disease an estimated two- to fourfold as compared with the risk in people without diabetes. In addition, DM increases morbidity and mortality after stroke. Macrovascular disease is the leading cause of death among patients with DM. The mechanisms of stroke secondary to diabetes may be caused by cerebrovascular atherosclerosis, cardiac embolism, or rheological abnormalities. The excess stroke risk is independent of age or blood pressure status. Diabetes associated with arterial hypertension adds significantly to stroke risk. There is a fourfold increase in the relative risk of cardiovascular events among patients with diabetes and hypertension compared to those without the two conditions. Diabetic persons with retinopathy and autonomic neuropathy appear to be a group at particularly high risk for ischemic stroke. High insulin levels increase the risk for atherosclerosis and may represent a pathogenetic factor in cerebral small-vessel disease. Presently, no evidence exists that tighter diabetic control or normal HbA1c levels over time decrease the risk for stroke or stroke recurrence. Moreover, optimal target blood glucose levels in stroke patients remain largely unknown (Gray et al., 2004; Van den Berghe et al., 2006). The UK Glucose Insulin in Stroke Trial (GIST-UK) failed to demonstrate any clinical benefit of insulin-induced euglycemia (target glucose 72-126 mg/dL). Furthermore, mortality appeared to be higher among patients with the greatest glucose reduction (Gray et al., 2009). Moreover, in the Spectroscopic Evaluation of Lesion Evolution in Stroke: Trial of Insulin for Acute Lactic Acidosis (SELESTIAL), the infusion of glucose-potassium-insulin did not have a favorable impact on cerebral infarct growth (McCormick et al., 2010).
High total cholesterol and high low-density lipoprotein (LDL) concentration are correlated with atherosclerosis. Dyslipidemia is a recognized risk factor for ischemic stroke. Meta-analyses have suggested that ischemic stroke risk increases with increasing serum cholesterol, and the reduction in stroke risk associated with 3-hydroxy 3-methylglutaryl coenzyme A reductase inhibitor (statin) therapies is related to reduction in LDL cholesterol (Amarenco et al., 2004; Tirschwell et al., 2004). Lipid-modifying therapy with statins has definitively established that reduction of LDL cholesterol reduces cardiovascular risk. Statins benefit stroke survivors as well. Lipid-lowering agents may slow progression of atherosclerotic plaque growth and may possibly cause a regression in plaque formation.
The Scandinavian Simvastatin Survival Study (4S) (Scandinavian Simvastatin Survival Study Group, 1999) investigated cholesterol lowering in persons with coronary heart disease and hypercholesterolemia and reported a highly significant relative reduction in the total mortality rate, major coronary events, and number of cardiac revascularization procedures. Post hoc analysis also showed a 28% reduction in fatal or nonfatal stroke and TIAs (Pedersen et al., 1998). The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) study investigated cholesterol lowering with pravastatin in patients with a previous myocardial infarction (MI) or unstable angina who had cholesterol levels between 155 and 271 mg/dL and reported a remarkable reduction in MI, cardiac revascularizations, and cardiovascular deaths, as well as a 20% reduction in the risk for stroke (Long-Term Intervention with Pravastatin in Ischaemic Disease [LIPID] Study Group, 1998). Similar findings were associated with atorvastatin in the Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) trial and the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA) studies. MIRACL showed a 50% relative risk reduction (RRR) (P = .045) in stroke among high-risk coronary disease patients (Schwartz et al., 2001). The ASCOT-LLA study demonstrated a favorable trend for fatal and nonfatal stroke, with a 27% RRR in patients at low risk for coronary events (hazard ratio, 0.73; 95% confidence interval [CI], 0.56-0.96; P = .0236), though not to the prespecified endpoint of P = .01 (Sever et al., 2003).
Although the Heart Protection Study (HPS) of patients at high risk with DM, coronary artery disease, or other atherosclerotic vascular disease did show an overall reduction in stroke risk, a subgroup analysis of the Heart Protection Study (HPS) did not show a reduction in the risk for stroke among patients with prior cerebrovascular disease (Heart Protection Study Collaborative Group, 2004). This subgroup analysis was limited in that the mean time from event to enrollment was 4.3 years, and the LDL reduction was 38 mg/dL. Subsequently, the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) study was published (SPARCL Investigators, 2006). This was a study of 4731 patients who had suffered either a stroke or TIA, had no known coronary heart disease, had LDL cholesterol levels of 100 to 190 mg/dL, and who were randomized to placebo or 80 mg of atorvastatin. A 56-mg/dL reduction in LDL treatment was noted in patients on atorvastatin. During a median follow-up of 4.9 years, 11.2% of atorvastatin-treated patients and 13.1% of placebo patients had a fatal or nonfatal stroke for an adjusted hazard ratio of 0.84 (95% CI, 0.71-0.99). A small increase in hemorrhagic stroke was reported in the atorvastatin group. Moreover, recent studies evaluating withdrawal of statins in acute ischemic stroke showed a higher incidence of death or dependency at 90 days (Blanco et al., 2007). Current guidelines of the American Heart Association and proposed modifications of the NCEP-III guidelines would therefore suggest that all patients at risk for stroke or who have had a cerebral infarction should be treated to a goal LDL level of below 70 mg/dL (Grundy et al., 2004; Sacco et al., 2006).
Atrial fibrillation, the most common sustained cardiac arrhythmia in the general population, affects about 1% of adults, is the most common cause for cardioembolic stroke, and is a risk factor for future cardiovascular disease. An estimated 1 to 2 million Americans have chronic nonvalvular atrial fibrillation (NVAF), a condition that is associated with an overall risk for stroke of approximately five- to sixfold, and a mortality rate approximately twice that of age- and sex-matched individuals without atrial fibrillation. The prevalence of atrial fibrillation increases with advancing age and is 0.5% for patients aged 50 to 59 years and 8.8% for those aged 80 to 89 years. Approximately 70% of individuals with atrial fibrillation are between 65 and 85 years of age. NVAF is associated with a substantial risk for stroke. Heart failure, arterial hypertension, DM, prior stroke or TIA, and age older than 75 years increase the risk for embolism in patients with NVAF. High-risk patients have a 5% to 7% yearly risk for thromboembolism. The CHADS2 Score represents a validated quantification of risk, with congestive heart failure, hypertension, age older than 75 years, and a history of DM being assigned 1 point; stroke or TIA are assigned 2 points (Gage et al., 2001). Ischemic stroke rates increase from 1.9 to 18.2 events per 100 patient-years with CHADS 2 scores of 0 and 6, respectively. Warfarin therapy, with the International Normalized Ratio (INR) value adjusted to between 2.0 and 3.0, decreases the relative risk for stroke in patients with NVAF by approximately two-thirds. High-risk patients, regardless of age, sustain particular benefit from warfarin anticoagulation. Left atrial enlargement also increases the risk for stroke in men. Likewise, left ventricular hypertrophy as demonstrated by electrocardiography (ECG) in men with preexisting ischemic heart disease is a major risk factor for stroke.
Cigarette smoking is the leading cause of preventable death in the United States. Cigarette smoking is a major risk factor for coronary artery disease, stroke, and peripheral arterial disease, an independent risk factor for ischemic stroke in men and women of all ages, and a leading risk factor of carotid atherosclerosis in men. The risk for stroke in smokers is two to three times greater than in nonsmokers. The mechanisms of enhanced atherogenesis promoted by cigarette smoking are incompletely understood but include reduced capacity of the blood to deliver oxygen, cardiac arrhythmias, increased blood coagulability, and triggering of arterial thrombosis and arterial spasm. Tobacco also increases carotid artery plaque thickness. More than 5 years may be required before a reduction in stroke risk is observed after cessation of smoking. Switching to pipe or cigar smoking is of no benefit. Counseling, nicotine replacement products, varenicline (a nicotinic receptor partial agonist),and bupropion are efficacious smoking cessation treatments. Selective blockers of the cannabinoid receptor type 1, such as rimonabant, have been proposed for treatment of multiple cardiometabolic risk factors including smoking and abdominal obesity (Gelfand and Cannon, 2006).
Numerous studies have established an association between obstructive sleep apnea (OSA) and stroke; moreover the severity of OSA is much higher in stroke patients than in controls (Dyken, 2000). Habitual snoring increases the risk for stroke and adversely affects the outcome of patients admitted to the hospital with stroke. Mounting evidence also suggests that inflammation, lipoprotein(a) concentration, impaired fibrinolysis, and increased thrombotic potential are important nontraditional cardiovascular risk factors.
The aorta is the most frequent site of atherosclerosis. Protruding atheroma may be the cause of otherwise unexplained TIAs or strokes. Aortic-arch atheromatosis detected by transesophageal echocardiography is an independent risk factor for cerebral ischemia; the association is particularly strong with mobile and thick atherosclerotic plaques more than 4 mm in thickness (French Study of Aortic Plaques in Stroke Group, 1996). The prevalence of ulcerated plaques was 16.9% among patients with cerebrovascular diseases, compared to 5.1% among patients with other neurological diseases. Remarkably, ulcerated plaques were found in 61% of cryptogenic cerebral infarcts, compared to 22% of cerebral infarcts with a known cause.
Other Risk Factors for Stroke
Hemostatic factors may be important in assessing the risk for cerebrovascular disease. Elevated hematocrit, hemoglobin concentration, and increased blood viscosity may be indicators of risk for ischemic stroke. Elevation of plasma fibrinogen is an independent risk factor for the development of cerebral infarction. Epidemiological studies have shown a correlation between elevated plasma fibrinogen levels and both ischemic stroke incidents and mortality. An elevated plasma fibrinogen level may reflect progression of atherogenesis. Plasminogen activator inhibitor-1 excess and factor VII are independent risk factors for coronary heart disease. Compared with white Americans, black Americans have higher mean levels of fibrinogen, factor VIII, von Willebrand factor, and AT, and lower mean levels of protein C. Fibrinogen levels are closely correlated with other stroke risk factors such as cigarette smoking, arterial hypertension, diabetes, obesity, hematocrit levels, and spontaneous echocardiographic contrast. Antiphospholipid (aPL) antibodies are a marker for an increased risk for thrombosis, including TIAs and stroke, particularly in those younger than 50 years. In older patients, the presence of aPL (either lupus anticoagulants [LAs] or anticardiolipin [aCL]) among patients with ischemic stroke does not predict either increased risk for subsequent vascular occlusive events over 2 years or a differential response to aspirin or warfarin therapy. As such, routine screening for aPL in older patients with ischemic stroke does not appear warranted (Levine et al., 2004) The factor V Leiden mutation is associated with deep venous thrombosis in otherwise healthy individuals with additional prothrombotic risk factors. An overall association of the factor V Leiden mutation and arterial thrombosis has not been found. As opposed to homozygous mutations, heterozygous factor V Leiden and prothrombin gene mutations have no clear association with increased stroke risk (Fields and Levine, 2005). Elevated von Willebrand factor is a risk factor for MI and ischemic stroke. Elevated levels of fasting total homocysteine (normal 5-15 mM), a sulfhydryl-containing amino acid, have been associated with an increased risk for stroke and thrombotic events in case-controlled studies. Metabolism of homocysteine requires vitamin B6 (pyridoxine), vitamin B12 (cyanocobalamin), folate, and betaine. Plasma homocysteine concentrations may be reduced by the administration of folic acid alone or in combination with vitamins B6 and B12. Conversely, serum folate concentrations of 9.2 nM or less have been associated with elevated plasma levels of homocysteine, and a decreased folate concentration alone may be a risk factor for ischemic stroke, particularly among blacks (Giles et al., 1995). However, folic acid supplementation does not have a major impact on stroke reduction (Lee et al., 2010).
Stroke is uncommon among women of childbearing age, estimated at 4.4/100,000 (Petitti et al., l997). The relative risk for ischemic stroke is increased among users of high-dose estrogen oral contraceptives, particularly with coexistent arterial hypertension, cigarette smoking, and increasing age. Based on an odds ratio of 1.93, the risk is increased to 8.5/100,000, which translates into a number needed to harm of 24,000 women to cause one ischemic stroke (Gillum et al., 2000). Thus, for a healthy young woman without any other stroke risk factors, the risk of stroke associated with oral contraceptives is small and probably outweighed by their benefits. New agents containing lower doses of estrogen and progestin have reduced the frequency of oral contraceptive–related cerebral infarction. Two postmenopausal hormone replacement studies with equine estrogen (Premarin) showed no benefit in reducing the incidence of stroke in a cohort of women with coronary heart disease (Hulley et al., 1998). The Women’s Estrogen for Stroke Treatment study of estradiol replacement in postmenopausal women status post stroke also failed to show a reduction in recurrent stroke (Viscoli et al., 2001). In addition, the Women’s Health Initiative, a prospective randomized trial of estrogen therapy in healthy postmenopausal women, was halted prematurely because the risks outweighed the benefits. Absolute excess risks per 10,000 person-years attributable to estrogen plus progestin were sevenfold for coronary heart disease events, eightfold for strokes, eightfold for pulmonary thromboembolism, and eightfold for invasive cancers, whereas absolute risk reductions per 10,000 person-years were sixfold for colorectal cancers and fivefold for hip fractures (Rossouw et al., 2002). The risk for thrombosis associated with pregnancy is high in the postpartum period. The risk for cerebral infarction is increased in the 6 weeks after delivery but not during pregnancy.
A diurnal and seasonal variation of ischemic events occurs. Circadian changes in physical activity, catecholamine levels, blood pressure, blood viscosity, platelet aggregability, blood coagulability, and fibrinolytic activity may explain the circadian variations in onset of myocardial and cerebral infarction. Although an early-morning peak occurs for all subtypes of stroke, most clinical trials on the use of platelet antiaggregants or other antithrombotic agents do not take these circadian variations into account. Rhythmometric analyses support the notion that stroke is a chrono-risk disease, in which cold temperatures also represent a risk factor. A history of recent infection, particularly of bacterial origin and within 1 week of the event, is also a risk factor for ischemic stroke in patients of all ages. A number of recent reports suggest that Chlamydia pneumoniae, a causative organism of respiratory infections, may have a role in carotid and coronary atherosclerosis. Some studies have also identified an association with chronic infections with Helicobacter pylori and cytomegalovirus (Bittner, 1998; Ross, 1999). These findings have not been confirmed, however (Lindsberg and Grau, 2003).
Pathophysiology of Cerebral Ischemia
A cascade of complex biochemical events occurs seconds to minutes after cerebral ischemia. Cerebral ischemia is caused by reduced blood supply to the microcirculation. Ischemia causes impairment of brain energy metabolism, loss of aerobic glycolysis, intracellular accumulation of sodium and calcium ions, release of excitotoxic neurotransmitters, elevation of lactate levels with local acidosis, free radical production, cell swelling, overactivation of lipases and proteases, and cell death (Fisher and Ratan, 2003). Many neurons undergo apoptosis after focal brain ischemia (Choi, 1996). Ischemic brain injury is exacerbated by leukocyte infiltration and development of brain edema. Exciting new treatments for stroke target these biochemical changes.
Clinical Syndromes of Cerebral Ischemia
A number of syndromes result from ischemia involving the central nervous system (CNS) (Brazis et al., 2011).
Transient Ischemic Attacks
An estimated 400,000 individuals experience a TIA each year. A TIA is a prognostic indicator of stroke, with one-third of untreated TIA patients having a stroke within 5 years. About 1 in 10 patients with TIA experience a stroke in the next 3 months. The interval from the last TIA is an important predictor of stroke risk; of all patients who subsequently experience stroke, 21% do so within 1 month and 51% do so within 1 year of the last TIA. In one series, patients with TIA had a 3-month stroke risk of 10.5%, equal to the recurrence rate following a stroke. Furthermore, 50% of those strokes following a TIA occurred within 48 hours of TIA onset (Johnston et al., 2000). Cardiac events are the principal cause of death in patients who have had a TIA. The 5% to 6% annual mortality rate after TIA is mainly caused by MI, similar to the 4% annual cardiac mortality rate in patients with stable angina pectoris.
A TIA is a temporary and “non-marching” neurological deficit of sudden onset; attributed to focal ischemia of the brain, retina, or cochlea; and lasting less than 24 hours. Yet most TIAs last only a few minutes. Episodes that last longer than 1 hour are usually due to small infarctions. With the advent of diffusion-weighted magnetic resonance imaging (DW-MRI) sequences, the time-based definition of TIA is inadequate because infarctions are sometimes evident on DW-MRI in patients whose clinical manifestations resolved completely within a few hours. Thus, a “tissue-based” modification of the TIA definition has been proposed, suggesting that any transient episode, regardless of duration, associated with a clinically appropriate lesion by MRI be defined as a stroke, and that otherwise prolonged events (>1-6 hours in duration) be defined as stroke rather than TIA when otherwise clinically appropriate (Albers et al., 2002). Furthermore, DW-MRI is useful in predicting the risk for early stroke; patients with TIA and DW-MRI lesions are at greater risk for experiencing a subsequent stroke than patients without a lesion (Coutts et al., 2005). Johnston et al. also introduced a new unified score for risk stratification in patients with TIAs, known as the ABCD2 score. The ABCD2 score is based on age, blood pressure (≥140/90 mm Hg), clinical features, TIA duration, and diabetes (Table 51A.3). ABCD2 scores of 4 or greater indicate a moderate to high stroke risk and justify prompt hospital admission (Johnston et al., 2007).
| Age 60 or older | 1 point |
| Blood pressure ≥140/90 | 1 point |
| Clinical: | |
| Unilateral weakness | 2 points |
| Speech impairment | 1 point |
| Duration: | |
| 60 minutes or more | 2 points |
| <60 minutes | 1 point |
| Diabetes mellitus | 1 point |
Agreement between physicians to define the likelihood of a TIA, even among fellowship-trained neurologists, remains poor (Castle et al., 2010). The following symptoms are considered typical of TIAs in the carotid circulation: ipsilateral amaurosis fugax, contralateral sensory or motor dysfunction limited to one side of the body, aphasia, contralateral homonymous hemianopia, or any combination thereof. The following symptoms represent typical TIAs in the vertebrobasilar system: bilateral or shifting motor or sensory dysfunction, complete or partial loss of vision in the homonymous fields of both eyes, or any combination of these symptoms. Perioral numbness also occurs. Isolated diplopia, vertigo, dysarthria, and dysphagia should not be considered as being caused by a TIA unless they occur in combination with one another or with any of the other symptoms just listed (Box 51A.1). Older patients with isolated vertebrobasilar symptoms and a significant history of cardiovascular risk factors should, however, be evaluated for possible TIA or stroke, because they are at substantially higher risk for cerebrovascular events (Norrving et al., 1995).
Box 51A.1 Recognition of Carotid and Vertebrobasilar Transient Ischemic Attacks
Symptoms Suggestive of Vertebrobasilar Transient Ischemic Attacks
Usually bilateral weakness or clumsiness but may be unilateral or shifting
Bilateral, shifting, or crossed (ipsilateral face and contralateral body) sensory loss or paresthesias
Bilateral or contralateral homonymous visual field defects or binocular vision loss
Two or more of the following symptoms: vertigo, diplopia, dysphagia, dysarthria, and ataxia
Transient global amnesia (TGA) is characterized by a reversible antegrade and retrograde memory loss, except for a total amnesia of events that occur during the attacks and inability to learn newly acquired information. During the attacks, patients remain alert without motor or sensory impairments and often ask the same questions repeatedly. Patients are able to retain personal identity and carry on complex activities. TGA most commonly affects patients in their 50s and older. Men are affected more commonly than women. The attacks begin abruptly and without warning. A typical attack lasts several hours (mean, 3-6 hours) but seldom longer than 12 hours. Onset of TGA may follow physical exertion, sudden exposure to cold or heat, or sexual intercourse. Although a large number of conditions have been associated with transient episodes of amnesia, in most instances, TGA is of primary or unknown cause. TGA has been documented in association with epilepsy, migraine, intracranial tumors, overdose of diazepam, cardiac arrhythmias secondary to digitalis intoxication, and as a complication of cerebral and coronary angiography. Many reports have suggested a vascular causal factor for this heterogeneous syndrome. Bilateral hippocampal and parahippocampal complex ischemia, possibly of migrainous origin, in the distribution of the posterior cerebral arteries is a potential mechanism. Acute confusional migraines in children and TGA have a number of similar features. Others have suggested an epileptic causal factor for a minority of patients. Venous hypertension with transient hypoxemia in the context of incompetent internal jugular vein valves has also been suggested as a possible mechanism for TGA (Nedelmann et al., 2005). Transient amnesias have been divided into pure TGA, probable epileptic amnesia, and probable transient ischemic amnesia. In contrast to patients with TIAs, the prognosis of persons with pure TGA is benign, with no apparent increased risk for vascular endpoints. Recurrences are uncommon. Extensive evaluations are not usually required except to distinguish TGA from TIA or seizures. Treatment with platelet antiaggregants is not indicated in most patients unless there is a suspicion for transient ischemic amnesia. The use of prophylactic calcium channel blockers may be justified in patients with a potential migrainous causal factor.
Different types of microemboli (e.g., cholesterol crystals, platelet fibrin, calcium, and other forms of debris) can be seen in the retinal arterioles during or between attacks of transient monocular visual loss. Engorgement of conjunctival and episcleral vessels, corneal edema and rubeosis irides, and anterior-chamber cell flares are indicative of an underlying ischemic oculopathy. Asymmetrical hypertensive retinal changes noted on funduscopy are suggestive of a high-grade carotid artery stenosis or occlusion on the side of the less severely involved retina. Venous stasis retinopathy may occur with high-grade carotid artery stenosis or occlusion and is characterized by diminished or absent venous pulsations, dilated and tortuous retinal veins, peripheral microaneurysms, and blossom-shaped hemorrhages in the midperipheral retina. Retinal microvascular abnormalities correlate with an increased incidence of lacunar strokes (Yatsuya et al., 2010). Corneal arcus senilis may be less obvious or absent on the side of low perfusion.
Carotid Artery System Syndromes
Carotid Artery Syndromes
Amaurosis fugax may be described as a sudden onset of a fog, haze, scum, curtain, shade, blur, cloud, or mist. A curtain or shade pattern with the loss of vision moving superiorly to inferiorly is described only in 15% to 20% of patients. Less commonly, a concentric vision loss, presumed to be caused by marginal perfusion, can diminish blood flow to the retina. Most attacks are spontaneous and unrelated to positional changes. The vision loss is sudden, often brief, and painless. The duration of vision loss is usually 1 to 5 minutes and rarely lasts more than 30 minutes. After an episode of amaurosis fugax, the vision is usually fully restored, although some patients may have permanent vision loss caused by a retinal infarction (see Chapters 14 and 15).
Syndromes of the Anterior Cerebral Artery and Related Blood Vessels
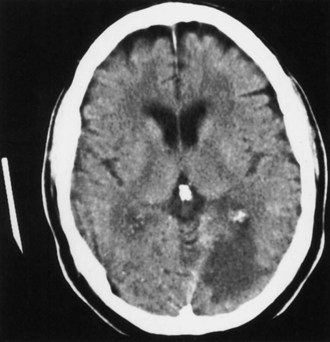
Anterior cerebral artery (ACA) territory infarctions are uncommon (Fig. 51A.1). They occur in patients with vasospasm after subarachnoid hemorrhage caused by ACA or anterior communicating artery aneurysm. Excluding these causes, the percentage of acute cerebral infarcts that are in the ACA territory is less than 3%. The characteristics of ACA infarction vary according to the site of involvement and the extent of collateral blood flow. Contralateral weakness involving primarily the lower extremity and, to a lesser extent, the arm is characteristic of infarction in the territory of the hemispheric branches of the ACA. Other characteristics include abulia, akinetic mutism (with bilateral mesiofrontal damage), impaired memory or emotional disturbances, transcortical motor aphasia (with dominant hemispheric lesions), deviation of the head and eyes toward the lesion, paratonia (gegenhalten), discriminative and proprioceptive sensory loss (primarily in the lower extremity), and sphincter incontinence. An anterior disconnection syndrome with left arm apraxia due to involvement of the anterior corpus callosum can be seen. Pericallosal branch involvement can cause apraxia, agraphia, and tactile anomia of the left hand. Infarction of the basal branches of the ACA can cause memory disorders, anxiety, and agitation. Infarction in the territory of the medial lenticulostriate artery (artery of Heubner) causes more pronounced weakness of the face and arm without sensory loss caused by this artery’s supply of portions of the anterior limb of the internal capsule.
Lacunar Syndromes
Ischemic strokes resulting from small-vessel or penetrating artery disease (lacunes) have unique clinical, radiological, and pathological features. Lacunar infarcts are small ischemic infarctions in the deep regions of the brain or brainstem that range in diameter from 0.5 to 15 mm. These infarctions result from occlusion of the penetrating arteries, chiefly the anterior choroidal, middle cerebral, posterior cerebral, and basilar arteries. Lacunar infarcts could also be the result of occlusion of penetrating arteries by atherosclerosis of the parent artery or by microembolism. Lacunes may be single or multiple, symptomatic or asymptomatic. At least 20 lacunar syndromes have been described. Lacunar syndromes are predictive of lacunar infarcts, with a positive predictive value of approximately 84% to 90% (Gan et al., 1997). The five best recognized syndromes are (1) pure motor hemiparesis, (2) pure sensory stroke, (3) sensory-motor stroke, (4) homolateral ataxia and crural paresis (ataxic hemiparesis), and (5) dysarthria–clumsy hand syndrome. Multiple lacunes may be associated with acquired cognitive decline. Headaches are uncommon in patients with lacunar infarcts.
Vertebrobasilar System Syndromes
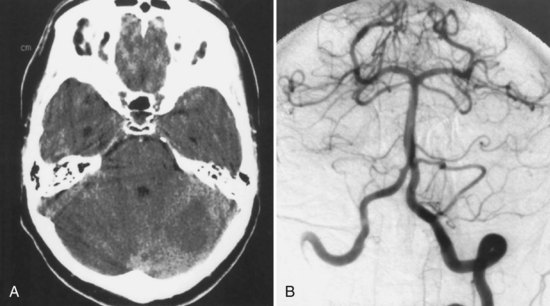
The areas of the cerebellum supplied by the posterior inferior cerebellar artery (PICA) are variable (see Chapter 19). There are several different patterns of PICA territory cerebellar infarctions. If the medial branch territory is affected, involving the vermis and vestibulocerebellum, the clinical findings include prominent vertigo, ataxia, and nystagmus. If the lateral cerebellar hemisphere is involved, patients can have vertigo, gait ataxia, limb dysmetria and ataxia, nausea, vomiting, conjugate or dysconjugate gaze palsies, miosis, and dysarthria. If the infarction is large, altered consciousness or confusion may occur as a result of cerebellar poststroke edema causing brainstem compression. Hydrocephalus or herniation may also develop with compression of the fourth ventricle. Although a PICA occlusion can be the cause of Wallenberg (lateral medullary) syndrome, this syndrome is more often due to an intracranial vertebral artery occlusion.
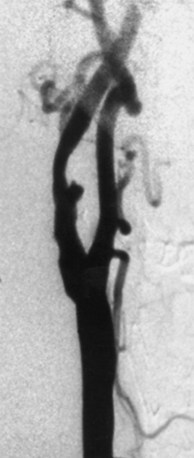
Infarction in the territory of the superior cerebellar artery (SCA) produces a dorsal cerebellar syndrome (Fig. 51A.2). Vertigo may be present, although it is less common with SCA infarcts than with the other cerebellar syndromes (Fig. 51A.3). Nystagmus is caused by involvement of the medial longitudinal fasciculus and the cerebellar pathways. An ipsilateral Horner syndrome due to involvement of the descending sympathetic tract may be present. Ipsilateral ataxia and asynergia and gait ataxia follow involvement of the superior cerebellar peduncle, brachium pontis, superior cerebellar hemisphere, and dentate nucleus. There is an intention tremor caused by involvement of the dentate nucleus and superior cerebellar peduncle. Choreiform dyskinesias may be present ipsilaterally. Contralaterally, there is hearing loss due to lateral lemniscus disruption and trunk and extremity hypalgesia and thermoanesthesia caused by spinothalamic tract involvement.
Weber syndrome is caused by infarction in the distribution of the penetrating branches of the posterior cerebral artery (PCA) affecting the cerebral peduncle, especially medially, with damage to the fascicle of cranial nerve III and the pyramidal fibers. The resultant clinical findings are contralateral hemiplegia of the face, arm, and leg secondary to corticospinal and corticobulbar tract involvement, and ipsilateral oculomotor paresis including a dilated pupil. A slight variation of this syndrome is the midbrain syndrome of Foville in which the supranuclear fibers for horizontal gaze are interrupted in the medial peduncle, causing a conjugate gaze palsy to the opposite side. Benedikt syndrome is caused by a lesion affecting the mesencephalic tegmentum in its ventral portion, with involvement of the red nucleus, brachium conjunctivum, and fascicle of cranial nerve III. This syndrome is due to infarction in the distribution of the penetrating branches of the PCA to the midbrain. The clinical manifestations are ipsilateral oculomotor paresis, usually with pupillary dilation and contralateral involuntary movements including intention tremor, hemiathetosis, or hemichorea. Claude syndrome is caused by lesions that are more dorsally placed in the midbrain tegmentum than with Benedikt syndrome. There is injury to the dorsal red nucleus, which results in more prominent cerebellar signs without the involuntary movements. Oculomotor paresis occurs. Nothnagel syndrome is characterized by an ipsilateral oculomotor palsy with contralateral cerebellar ataxia. Infarction in the distribution of the penetrating branches of the PCA to the midbrain is the cause of this syndrome. Parinaud syndrome can result from infarctions in the midbrain territory of PCA penetrating branches. This syndrome is characterized by supranuclear paralysis of eye elevation, defective convergence, convergence-retraction nystagmus, light-near dissociation, lid retraction, and skew deviation (see Chapter 19). Parinaud syndrome occurs more characteristically as a result of mass effect from midline mass lesions such as a pineal gland tumor.
Top of the basilar syndrome (see Chapter 19) is caused by infarction of the midbrain, thalamus, and portions of the temporal and occipital lobes. It is due to occlusive vascular disease, often embolic in nature, of the rostral basilar artery. Associated behavioral abnormalities include somnolence, peduncular hallucinosis, memory disturbances, or agitated delirium. Ocular findings include unilateral or bilateral paralysis of upward or downward gaze, impaired convergence, pseudoabducens palsy, convergence-retraction nystagmus, abnormalities of abduction, Collier sign (which consists of elevation and retraction of the upper eyelids), skew deviation, and oscillatory eye movements. Visual defects that may be present include hemianopia, cortical blindness, and Balint syndrome. Pupillary abnormalities are variable and may be large or small, reactive or fixed. Motor deficits may also occur.
The lateral medullary syndrome (Wallenberg syndrome) is most often due to occlusion of the intracranial segment of the vertebral artery (Fig. 51A.4). Less commonly, it is caused by occlusion of the PICA. This syndrome produces an ipsilateral Horner syndrome; loss of pain and temperature sensation in the face; weakness of the palate, pharynx, and vocal cords; and cerebellar ataxia. Contralateral to the lesion, there is hemibody loss of pain and temperature sensation. The medial medullary (Dejerine) syndrome is less common and may be caused by occlusion of the distal vertebral artery, a branch of the vertebral artery, or the lower basilar artery. Vertebral artery dissection, dolichoectasia of the vertebrobasilar system, and embolism are less common causes of the medial medullary syndrome. The findings with this syndrome include an ipsilateral lower motor neuron paralysis of the tongue and contralateral paralysis of the arm and leg. The face is often spared. In addition, there is contralateral hemibody loss of tactile, vibratory, and position sense. Occlusion of the intracranial vertebral artery can lead to a total unilateral medullary syndrome (of Babinski-Nageotte), a combination of the medial and lateral medullary syndromes.
Posterior Cerebral Artery Syndromes
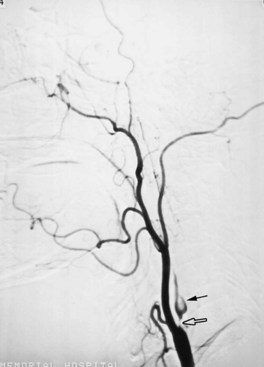
More complex visual changes may occur, including formed or unformed visual hallucinations, visual and color agnosias, or prosopagnosia. Finally, some alteration of sensation with PCA hemispheral infarctions occurs, including paresthesias or altered position, pain, and temperature sensations. Infarction in the distribution of the callosal branches of the PCA involving the left occipital region and the splenium of the corpus callosum produces alexia without agraphia (Fig. 51A.5). In this syndrome, patients can write, speak, and spell normally but are unable to read words and sentences. The ability to name letters and numbers may be intact, but there can be inability to name colors, objects, and photographs. Right-hemispheric PCA territory infarctions may cause contralateral visual-field neglect. Amnesia may be present with PCA infarctions that involve the left medial temporal lobe or when there are bilateral mesiotemporal infarctions. In addition, an agitated delirium may occur with unilateral or bilateral penetrating mesiotemporal infarctions. Large infarctions of the left posterior temporal artery territory may produce an anomic or transcortical sensory aphasia.
Diagnosis and Treatment of Threatened Ischemic Stroke
An ischemic stroke develops when there is interrupted cerebral blood flow to an area of the brain. Ischemic strokes account for approximately 80% to 88% of all strokes. Ischemic strokes may result from (1) large-artery atherosclerotic disease resulting in stenosis or occlusion, (2) small-vessel or penetrating artery disease (lacunes), (3) cardiogenic or artery-to-artery embolism, (4) nonatherosclerotic vasculopathies, (5) hypercoagulable disorders, and (6) infarcts of undetermined causes. The most recognized mechanistic classification is the TOAST (Trial of Org 10172 in Acute Stroke Treatment) classification (Madden et al., 1995). However, a rigid classification of ischemic stroke subtypes is difficult to establish because of the frequent occurrence of mixed syndromes.
Large-Artery Atherothrombotic Infarctions
Large-artery atherothrombotic infarctions almost always occur in patients who already have significant risk factors for cerebrovascular atherosclerosis (see Table 51A.1). Atherothrombosis is multifactorial, comorbidities frequently overlap, and risk factors are often additive. For example, arterial hypertension is often associated with hyperlipidemia, hyperglycemia, elevated fibrinogen levels, excessive weight, and left ventricular hypertrophy on ECG. A resting ankle-brachial index of less than 0.90 is indicative of generalized atherosclerosis (Zheng et al., 1997). Persons with a stroke are at high risk for development of other vascular complications. After a stroke, there is a 25% chance of a fatal thrombotic event in 3 years. Many of these deaths are due to MI. The mechanisms of large-artery atherothrombotic infarction often reflect plaque complication, ulceration with artery-to-artery embolization (see Fig. 51A.5), or thrombosis in the setting of preexisting arterial stenosis. Arteriogenic embolism is a common mechanism of cerebral ischemia. Embolism from ulcerated carotid artery atherosclerotic plaques is a common cause of cerebral infarction. In situ thrombosis occurs in the proximal carotid artery, distal vertebral artery, and lower or middle basilar artery (Fig. 51A.6). Atherosclerotic involvement of the intracranial portion of the vertebrobasilar system frequently occurs in tandem and is the common pathological mechanism associated with the syndrome of vertebrobasilar territory infarction; this may also arise in association with hypercoagulable states. Hypoperfusion secondary to hemodynamic alterations also may trigger these events. Whether the pathogenesis of stroke due to intracranial arterial stenosis is different from stroke due to extracranial arterial disease is unsettled.
In patients with risk factors for atherosclerosis, the cholesterol-rich minimally raised fatty streak may progress to a fibrous plaque that can evolve into a complicated plaque with intraplaque hemorrhage, extensive necrosis, calcification, and subsequent thrombosis (Fig. 51A.7). The infiltration of the fibrous cap by foam cells may contribute to the rupture of the atherosclerotic carotid artery plaque. Atherosclerosis is often segmental and asymmetrical, and earlier lesions tend to occur in areas of low shear stress, such as the outer aspect of the carotid artery bulb. Atherosclerosis primarily affects the larger extracranial and intracranial vessels, particularly the bifurcation of the common carotid artery and proximal internal carotid artery, the carotid siphon, MCA stem, origin of the vertebral arteries (V1), intracranial segment of the vertebral arteries (V4), and basilar artery. The distribution of cerebral atherosclerosis may be different in certain ethnic groups. Stenosis of major intracranial arteries is more prevalent among blacks and Asians than in whites, but this may also reflect ascertainment biases.
Small-Vessel or Penetrating Artery Disease
Lacunes usually occur in patients with long-standing arterial hypertension, current cigarette smoking, and DM. The most frequent sites of involvement are the putamen, basis pontis, thalamus, posterior limb of the internal capsule, and caudate nucleus. Multiple lacunae are strongly associated with arterial hypertension and DM. Available evidence suggests that structural changes of the cerebral vasculature caused by arterial hypertension are characterized by fibrinoid angiopathy, lipohyalinosis, and microaneurysm formation. Accelerated hypertensive arteriolar damage of the small penetrating arteries is operative in a large number of patients with lacunar infarction. However, microatheroma of the ostium of a penetrating artery, arterial or cardiac embolism, or changes in hemorheology can be pathophysiologically operative in the remainder of cases. The mere association of a lacunar syndrome in a patient with arterial hypertension and diabetes is insufficient for a diagnosis of lacunar infarct, and other causes of ischemic stroke must be excluded. In particular, the presence of sensorimotor stroke, limb weakness and sudden onset in a patient with atrial fibrillation, and large striatocapsular infarctions should be distinguished from lacunar infarcts, because they frequently have potential cardioembolic sources or coexistent severe carotid or MCA stenosis, and they often present with signs and symptoms of cortical dysfunction (Arboix et al., 2010; Nicolai et al., 1996). Control of hypertension, prevention of microangiopathy, a better understanding of the ideal hemodynamic profile, and judicious use of platelet antiaggregants are essential in the management of patients with lacunar infarcts.
Cardiogenic Embolism
Congenital heart disease is probably the most common cardiac disorder causing ischemic stroke in children. With the increased survival of many children with congenital heart disease, strokes are being seen with increased frequency. Children with congenital heart disease and a low hemoglobin concentration are at special risk for arterial strokes; those with a high hematocrit are more likely to experience cerebral venous thrombosis (Perloff, 1998). Emboli from cardiac sources may be silent or cause severe neurological deficit or death. Although most types of heart disease may produce cerebral embolism, certain cardiac disorders are more likely to be associated with emboli (Box 51A.2).
Box 51A.2 Sources of Cardioembolism
Rheumatic mitral valve disease
Nonbacterial thrombotic endocarditis
Filamentous strands of the mitral valve
Aneurysms of the sinus of Valsalva
Intracardiac tumors (atrial myxoma, rhabdomyoma, papillary fibroelastoma)
Intracardiac defects with paradoxical embolism
Cyanotic congenital heart disease
Fontan procedure or its modifications (cavopulmonary anastomosis)
Mitochondrial encephalomyopathies (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes; myoclonic epilepsy and ragged-red fibers; Kearns-Sayre syndrome)
Coronary artery bypass grafting
An embolic stroke occurs in approximately 1% of hospitalized patients with acute MI. Left ventricular thrombi are commonly associated with recent anterior wall transmural MI. Echocardiographic studies have demonstrated that approximately one-third to one-half of acute anterior MIs but less than 4% of acute inferior MIs develop left ventricular thrombi. Almost all episodes of embolism occur within 3 months following acute MI, with 85% of emboli developing in the first 4 weeks. A decreased ejection fraction is an independent predictor of an increased risk for stroke following MI (Loh et al., 1997). Patients with acute MI who receive thrombolytic therapy have a small risk for ischemic stroke. A direct comparison of tissue plasminogen activator (tPA) and streptokinase for MI shows an excess of strokes with tPA. Although the prevalence of left ventricular thrombi in individuals with left ventricular aneurysms is high, the frequency of systemic embolism is low. Dilated or congestive cardiomyopathy may result from arterial hypertension or a variety of inflammatory, infectious, immune, metabolic, toxic, and neuromuscular disorders. The global impairment of ventricular performance predisposes to stasis and thrombus formation. Patients commonly have signs of impaired left ventricular systolic function, and less than half have diastolic heart failure. Occasionally, patients have atrial fibrillation. A cerebral embolism may be the presenting manifestation of heart failure. Embolism occurs in approximately 18% of patients with dilated cardiomyopathy not receiving anticoagulants. Patients with idiopathic hypertrophic subaortic stenosis may also present with stroke. Thromboembolism is not uncommon in patients with congestive heart failure. Mitochondrial disorders are seldom associated with dilated cardiomyopathies, but cerebral infarction is a complication of mitochondrial encephalomyopathies (MELAS, myoclonic epilepsy and ragged-red fibers, and Kearns-Sayre syndrome). Stroke in Kearns-Sayre syndrome is likely secondary to embolism. Apical aneurysms also complicate Chagas cardiomyopathy, with resultant cerebral embolism.
Most cases of mitral stenosis are due to rheumatic heart disease. Systemic emboli occur in 9% to 14% of patients with mitral stenosis, with 60% to 75% having cardioembolic cerebral ischemia. Systemic embolism may be the first symptom of mitral stenosis, particularly if it is associated with atrial fibrillation. Aortic valve calcification with or without stenosis is not a major risk factor for stroke, but cases have been described (Boon et al., 1996; Oliveira-Filho et al., 2000). Cerebral embolism is a rare but described occurrence in patients with bicuspid aortic valves. Individuals with mitral annular calcification (MAC) have a twofold risk for stroke compared with those without MAC, but stroke rates are low. Mitral valve prolapse affects 3% to 4% of adults and when uncomplicated does not seem to increase their risk for stroke. Neurological ischemic events appear to occur more commonly among men older than 50 years who have auscultatory findings of a systolic murmur and thick mitral valve leaflets on echocardiography. Thromboembolic phenomena complicating infective endocarditis may be systemic (left-sided endocarditis) or pulmonary (right-sided endocarditis). Vegetations are detected by transthoracic echocardiography in 54% to 87% of patients with infective endocarditis and are associated with an increased risk for embolism (Eishi et al., 1995). Transesophageal echocardiography is the gold standard, however, with detection rates of better than 90% (Reynolds et al., 2003). Systemic emboli may occur in nearly half of patients with nonbacterial thrombotic endocarditis, a condition characterized by the presence of multiple small, sterile thrombotic vegetations most frequently involving the mitral and aortic valves. The risk for thromboembolism is higher with mechanical prosthetic heart valves than with biological prosthetic heart valves. Thromboemboli are more common with prosthetic heart valves in the mitral position than with prosthetic heart valves in the aortic position. The rate of systemic embolism in patients with mechanical heart valves receiving anticoagulant therapy is 4% per year in the mitral position and 2% per year in the aortic position. Filamentous strands attached to the mitral valve appear to represent a risk for cerebral embolism, particularly in young patients, but the risk for recurrent cerebral ischemia is incompletely understood. The association between cerebral embolism and giant Lambl excrescences or aneurysms of the sinus of Valsalva is low.
Cerebral and systemic embolism also may occur in the setting of the sick sinus syndrome. Patients at greatest risk for embolization have bradytachyarrhythmias; left atrial spontaneous echocardiographic contrast and decreased atrial ejection force increase stroke risk (Mattioli et al., 1997). Patients with sick sinus syndrome may experience cerebral ischemia or systemic embolism even after pacemaker insertion. Ventricular-inhibited (VVI) pacing is associated with a higher risk for embolic complications than atrial or dual-chamber pacing. The risk for thromboembolism is also higher among patients in chronic atrial flutter (Seidl et al., 1998; Wood et al., 1997).
A paradoxical embolism caused by a right-to-left shunt through a PFO or ASA can be responsible for stroke and other ischemic cerebral events. A PFO provides opportunity for right-to-left shunting during transient increases in the right atrial pressure. A PFO is present in 35% of subjects without stroke between the ages of 1 and 29 years, in 25% of people between the ages of 30 and 79 years, and in 20% between the ages of 80 and 99 years. A PFO is more common in patients with stroke than in matched controls. Patients with no identifiable cause for ischemic stroke (cryptogenic stroke) and PFO may have a larger PFO with more extensive right-to-left shunting than patients with stroke of determined cause. Platelet antiaggregants, oral anticoagulants, transcatheter closure of the PFO, or surgical closure of the PFO have been recommended. Preliminary results of the CLOSURE I trial show that PFO closure using the Starflex device is not superior to best medical therapy for preventing recurrent stroke or TIA (NMT Medical, 2010). Cerebrovascular complications of percutaneous PFO closure can include air embolism or thromboembolism, atrial fibrillation, infective endocarditis, or delayed cardioembolic stroke due to thrombus formation around the device. A study found no difference in the time to primary endpoints between aspirin and warfarin (mean INR = 2.04) (Homma et al., 2002). ASA also might be a source of cerebral emboli. Only one study has reported that the coexistence of PFO and ASA increases the risk for embolic stroke (Messe et al., 2004). PFO and ASA also have been associated with mitral valve prolapse. Strokes are sometimes severe, but recurrences are uncommon in the context of PFO-related stroke. Pulmonary arteriovenous malformations can also be a source of paradoxical embolism causing cerebral ischemia and occur in 15% to 20% of patients with Rendu-Osler-Weber syndrome.
The preponderance of posterior circulation ischemia following cardiac catheterization is unexplained. Stroke occurs after coronary artery bypass grafting with a frequency ranging between 1% and 5% (Fig. 51A.8). Two-thirds of strokes occur by the second postoperative day and predominantly involve the cerebral hemispheres; brainstem/cerebellar infarcts and lacunar strokes are less common. The cause of postoperative stroke is multifactorial; hypoperfusion, ventricular thrombus, and emboli are probable causal factors, although embolic causes are the most likely mechanisms. Clamp manipulation during coronary artery bypass surgery also may favor the release of aortic atheromatous debris. Epiaortic ultrasound studies demonstrate an increased stroke rate associated with an increased severity of aortic atherosclerosis. Strokes following coronary artery bypass grafting rarely relate to carotid artery stenosis. Carotid artery occlusion, but not carotid artery stenosis, increases the risk for stroke following coronary artery bypass grafting (Mickleborough et al., 1996). Thromboembolic phenomena can complicate cardiac surgery using cardiopulmonary bypass with deep hypothermia and cardiac arrest. Stroke is a potential complication of cardioversion for atrial fibrillation. Cerebral embolism may also complicate valvuloplasty; the risk is greater for aortic rather than mitral valvuloplasty. Strokes may follow heart transplantation, the use of ventricular support systems and artificial hearts, and the use of the extracorporeal membrane oxygenator. Stroke following inadvertent placement of left-sided heart pacemaker leads is an unusual complication. Ischemic myelopathy is a rare complication of the intraaortic balloon pump. Aortic dissection or hematoma may lead to an occlusion of a major radicular branch or local occlusion of the artery of Adamkiewicz (see Chapter 51D).
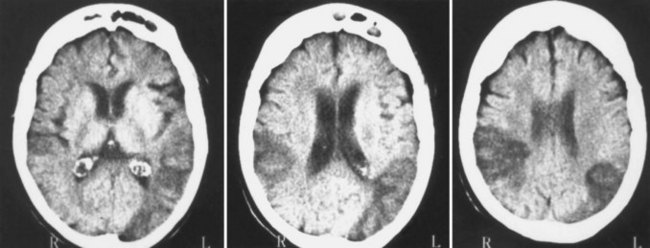
Nonatherosclerotic Vasculopathies
Although the majority of arterial disorders leading to stroke are caused by atherosclerosis, several nonatherosclerotic vasculopathies can be responsible for a minority of ischemic strokes. These vasculopathies include cervicocephalic arterial dissections, traumatic cerebrovascular disease, radiation vasculopathy, moyamoya, fibromuscular dysplasia (FMD), and cerebral vasculitis (Boxes 51A.3 and 51A.4). Together, these uncommon conditions represent 5% of all ischemic strokes. They are relatively more common in children and young adults.
Box 51A.4
Classification of Cerebral Vasculitides
Vasculitis associated with collagen vascular disease:
Vasculitis associated with other systemic diseases:
From Biller, J., Sparks, L.H., 1993. Diagnosis and management of cerebral vasculitis. In: Adams Jr., H.P. (Ed.), Handbook of Cerebrovascular Diseases. Marcel Dekker, New York, p. 150.
Dissections
Cervicocephalic arterial dissections have been reported after blunt or penetrating trauma and also are associated with FMD, Marfan syndrome, Ehlers-Danlos syndrome type IV, pseudoxanthoma elasticum, coarctation of the aorta, Menkes disease, α1-antitrypsin deficiency, cystic medial degeneration, reticular fiber deficiency, accumulation of mucopolysaccharides, osteogenesis imperfecta, adult polycystic kidney disease, elevated arterial elastase content, lentiginosis, atherosclerosis, extreme vessel tortuosity, moyamoya syndrome, homocystinuria, the 677TT genotype of the 5,10-methylenetetrahydrofolate reductase gene (MTHFR 677TT), pharyngeal infections, sympathomimetic drug abuse, and luetic arteritis. Most typically, cervicocephalic arterial dissections occur either spontaneously or in relation to minor trauma (Debette and Leys, 2009).
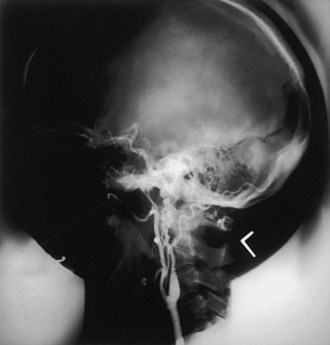
Cervicocephalic arterial dissections should be considered in the differential diagnosis of ischemic stroke in any young adult, particularly when traditional risk factors are absent. Diagnosis is based on arteriographic findings, although high-resolution MRI with T1 fat suppression sequences and magnetic resonance angiography (MRA), computed tomographic angiography (CTA), and extracranial and transcranial Doppler ultrasound are rapidly replacing contrast angiography for the diagnosis of cervicocephalic arterial dissections, particularly in cases of carotid artery involvement (Fig. 51A.9).
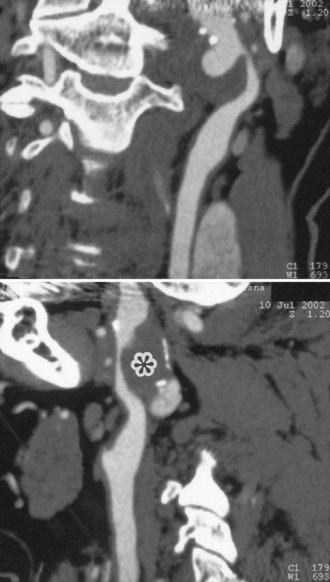
Features on arteriography include the presence of a “pearl-and-string” sign; double-lumen sign; short, smooth, tapered occlusion; or pseudoaneurysm formation. MRI demonstrates the intramural hematoma and the false lumen of the dissected artery (Fig. 51A.10). MRA, CTA, or ultrasound studies can be helpful in monitoring the course of dissection.
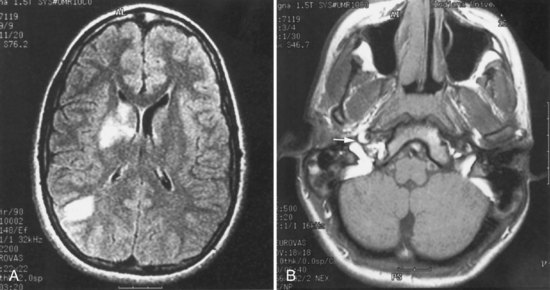
Therapeutic interventions have included immediate anticoagulation with heparin, followed by a 3- to 6-month course of warfarin, then platelet antiaggregants instead of or following warfarin. Angioplasty and stenting or surgical correction are reserved for selected individuals with pseudoaneurysms or those who have multiple recurrent events following adequate medical therapy. Although anticoagulants are often empirically recommended, their value in patients with extracranial cervicocephalic arterial dissections has not been firmly established. Anticoagulation should be withheld in patients with intracranial dissections (particularly involving the vertebrobasilar circulation) because of the risk for subarachnoid hemorrhage. The Cervical Artery Dissection in Stroke Study (CADISS) is an ongoing randomized multicenter open-treatment trial comparing anticoagulant and antiplatelet therapies for acute (within 7 days) symptomatic extracranial carotid or vertebral artery dissection (Kasner and Dreier, 2009).
Trauma
Trauma is a leading cause of cerebrovascular mortality in the United States (see Chapter 50B). Blunt or penetrating traumatic cerebrovascular disease may result in cervicocephalic arterial dissection, arterial thrombosis, arterial rupture, pseudoaneurysm formation, or development of an arteriovenous fistula. Internal carotid artery thrombosis also may follow maxillary and mandibular angle fractures. Carotid artery trauma may cause hematoma formation of the lateral neck, retinal or hemispheric ischemia, and Horner syndrome. Neurological deficits may be mild or devastating. Comatose patients with carotid arterial injuries with a Glasgow Coma Scale score of 8 or less do poorly regardless of treatment (see Chapter 5). Missing the diagnosis may lead to devastating results. A thorough evaluation of the airway, oropharynx, and esophagus is needed. Arteriography is indicated in most instances, although CTA has become a preferred screening modality for diagnosis; thereafter, surgical repair or angioplasty may be needed (Nuñez et al., 2004).
Moyamoya
Moyamoya is a chronic progressive nonatherosclerotic, noninflammatory, nonamyloid occlusive intracranial vasculopathy of unknown cause. Pathologically, there is fibrocellular intimal thickening, smooth muscle cell proliferation, and increased elastin accumulation, resulting in stenosis of the suprasellar intracranial internal carotid arteries. There is also thinning of the media and a tortuous and often multilayered internal elastic lamina. Thrombotic lesions may be seen in major cerebral arteries. There are also numerous perforating and anastomotic branches around the circle of Willis. Intracranial aneurysms may be seen at the circle of Willis or in the peripheral vessels (Yamamoto et al., 1997). Moyamoya disease is sometimes distinguished from moyamoya syndrome associated with a number of different putative causes. Cases have been associated with neonatal anoxia, trauma, basilar meningitis, tuberculous meningitis, leptospirosis, cranial radiation therapy for optic pathway gliomas, neurofibromatosis type 1, tuberous sclerosis, encephalotrigeminal angiomatosis (Sturge-Weber syndrome), phakomatosis pigmentovascularis type IIIb, brain tumors, FMD, polyarteritis nodosa, Marfan syndrome, Turner syndrome, pseudoxanthoma elasticum, hypomelanosis of Ito, Williams syndrome, cerebral dissecting and saccular aneurysms, sickle cell disease, β-thalassemia, Fanconi anemia, hereditary spherocytosis, lupus anticoagulant, Sneddon syndrome, homocystinuria, oral contraceptives, factor XII deficiency, type I glycogenosis, reduced form of nicotinamide adenine dinucleotide phosphate (NADP)–coenzyme Q reductase deficiency, renal artery stenosis, Down syndrome, Apert syndrome, Graves disease, coarctation of the aorta, Alagille syndrome, hyperphosphatasia, Schimke immuno-osseous dysplasia, primary oxalosis, pulmonary sarcoidosis, and Hirschsprung disease.
Moyamoya disease has a bimodal age distribution, with peaks in the first and fourth decades of life. Childhood moyamoya is characterized by ischemic manifestations, whereas adult moyamoya disease presents with hemorrhagic manifestations. Moyamoya affects children, adolescents, and young adults most frequently. Diagnosis is based on a distinct arteriographic appearance as described by Suzuki’s 6 angiographic stages: (1) stenosis of the carotid fork, (2) initial appearance of moyamoya vessels at the base of the brain, (3) intensification of moyamoya vessels, (4) minimization of moyamoya vessels, (5) reduction of moyamoya vessels, and (6) disappearance of moyamoya vessels (collaterals only from external carotid arteries) (Fig. 51A.11). Moyamoya is characterized by progressive bilateral stenosis of the distal internal carotid arteries, extending to the proximal ACA and MCA, often with involvement of the circle of Willis and development of an extensive collateral (parenchymal, leptomeningeal, and transdural) network at the base of the brain like a cloud or puff of smoke (moyamoya). Intracranial aneurysms, particularly located in the posterior circulation, may be present.
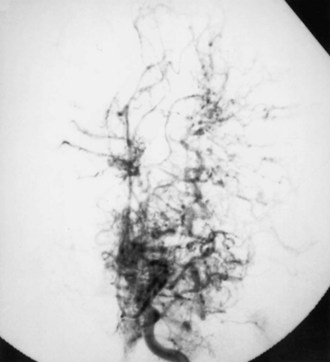
Fibromuscular Dysplasia
The diagnosis of cervicocephalic FMD may be made on the basis of MRA, CTA, or conventional catheter cerebral angiography. Cervicocephalic FMD occurs most often in the extracranial carotid artery and is bilateral in approximately two-thirds of cases. The lesions of medial fibroplasia account for the characteristic “string of beads” angiographic appearance seen in approximately 90% of cases (Fig. 51A.12).
Inflammatory Vasculitides
Inflammatory vasculitides can involve any size of vessel, including the precapillary arterioles and postcapillary venules. Many infectious and multisystem noninfectious inflammatory diseases cause cerebral vasculitis (see Table 51A.6). Cerebral vasculitis is a consideration in young patients with ischemic or hemorrhagic stroke; patients with recurrent stroke; patients with stroke associated with encephalopathic features; and patients with stroke accompanied by fever, multifocal neurological events, mononeuritis multiplex, palpable purpura, or abnormal urinary sediment. Other manifestations of cerebral vasculitis include headaches, seizures, and cognitive deterioration. Laboratory studies typically show anemia of chronic disease, leukocytosis, and an elevated erythrocyte sedimentation rate. The diagnosis of vasculitis usually requires confirmation by arteriography or biopsy. Overall, these disorders have a poor prognosis, but corticosteroids and other immunosuppressive agents have improved the survival rate (Biller and Grau, 2004).
Infections and Stroke
Fungal arteritis may result in aneurysms, pseudoaneurysms, thrombus formation, and cerebral infarction. Complications of acute purulent meningitis include intracranial arteritis and thrombophlebitis of the major venous sinuses and cortical veins. Intracranial arterial stenoses have been associated with a complicated clinical course. Varicella-zoster may cause a virus-induced necrotizing arteritis similar to granulomatous angiitis. Cerebral infarction is a complication of acquired immunodeficiency syndrome (AIDS) and may result from vasculitis, meningovascular syphilis, varicella-zoster virus vasculitis, opportunistic infections, infective endocarditis, aneurysmal dilation of major cerebral arteries, nonbacterial thrombotic endocarditis, aPL antibodies, or other hypercoagulable states, or from hyperlipidemia resulting from protease inhibitors and other factors such as HIV-1-related malignancy, cancer chemotherapy, and thrombotic thrombocytopenic purpura (TTP). Large-artery cerebrovascular occlusions have been found in association with meningoencephalitis caused by free living amebae. Other infectious agents known to produce cerebral infarcts include Mycoplasma pneumoniae, coxsackie 9 virus, California encephalitis virus, mumps paramyxovirus, hepatitis C virus, Borrelia burgdorferi, Rickettsia typhi group, cat-scratch disease, Trichinella infection, and the larval stage (cysticercus) of Taenia solium. Cerebrovascular involvement in neurocysticercosis is usually ischemic and is caused by chronic meningitis, arteritis, or endarteritis of small vessels. Unilateral or bilateral carotid artery occlusion can complicate necrotizing fasciitis of the parapharyngeal space. Infection with Chlamydia pneumoniae accelerates the process of atherosclerosis in animal studies; treatment with azithromycin has been shown to reduce the degree of atherosclerotic lesions in a rabbit model (Moazed et al., 1999; Muhlestein et al., 1998).
Drug Abuse and Stroke
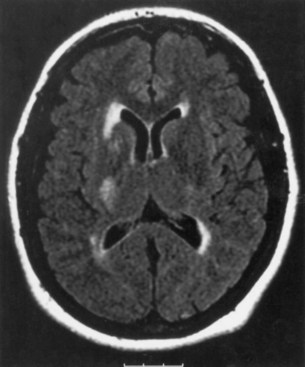
Ischemic stroke is a complication of illicit drug use and use of over-the-counter sympathomimetic drugs. Stroke mechanisms associated with the use of illicit drugs are multifactorial, including foreign-body embolization, vasculitis, vasospasm, acute onset of arterial hypertension or arterial hypotension, endothelial damage, accelerated atherosclerosis, hyper- or hypocoagulability, cardiac arrhythmias, embolism from an MI, or AIDS. The substances implicated most commonly are the amphetamines, cocaine (freebase or “crack”), phenylpropanolamine, pentazocine (Talwin) in combination with Pyribenzamine (“T’s and blues”), phencyclidine, heroin, anabolic steroids, and glue sniffing. Ischemic or hemorrhagic strokes may follow within hours of cocaine use, whether the drug is smoked, snorted, or injected (Fig. 51A.13) (see Chapters 51E and 58). The risk for intracerebral hemorrhage, especially among young women, has led to removal from the American market of phenylpropanolamine (Kernan et al., 2000). Ephedra, also called ma-huang, widely used in weight-loss products, has been associated with high blood pressure, heart attacks, and strokes. Stroke in young athletes may also be the result of anabolic-androgen steroid abuse and recombinant erythropoietin (“blood doping”) administration.
Stroke and Systemic Vasculitides
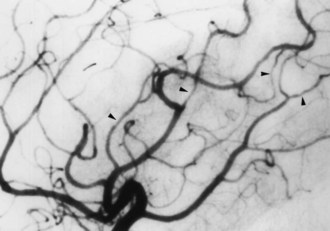
Ischemic stroke is also a complication of a variety of multisystem vasculitides. Stroke in patients with systemic lupus erythematosus may be attributable to cardiogenic embolism (nonbacterial verrucous or Libman-Sacks endocarditis, which occurs in the ventricular surface of the mitral valve), aPL antibodies, or underlying vasculopathy, or nephrotic syndrome associated with lupus nephritis, or less often to an immune-mediated vasculitis (Fig. 51A.14) (see Chapter 49A).
Behçet syndrome may involve vessels of any size. Venous thrombosis is more frequent than occlusive arterial compromise. Affected patients are mainly of Mediterranean or East Asian origin and may have a history of iritis, uveitis, and oral, genital, and mucocutaneous ulcerations. Cerebrovascular complications include strokes, carotid aneurysm formation, and cerebral venous thrombosis. Cogan syndrome is a rare condition characterized by nonsyphilitic interstitial keratitis, vestibular dysfunction, and deafness. Complications include aortic insufficiency and mesenteric ischemia. The angiitic form of sarcoidosis primarily affects the eyes, meninges, and cerebral arteries and veins. Kohlmeier-Degos disease or malignant atrophic papulosis is a multisystem occlusive vasculopathy characterized by cutaneous, gastrointestinal, and neurological manifestations; it may be complicated by ischemic or hemorrhagic strokes. Cerebral vasculitis may also complicate the course of children with acute poststreptococcal glomerulonephritis. The multisystem vasculitides are described in more detail in Chapter 49A.
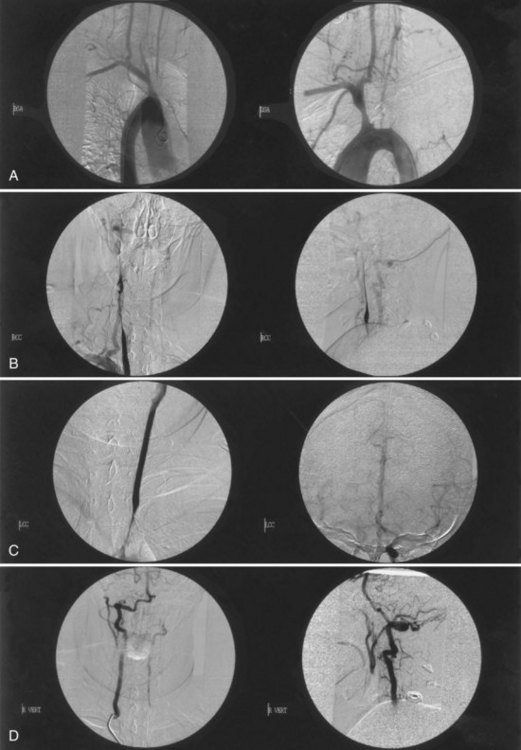
Takayasu arteritis is a chronic inflammatory arteriopathy of the aorta and its major branches, as well as the pulmonary artery. The cause is unknown, but an immune mechanism is suspected. The disease, prevalent in young women of Asian, Mexican, or Native American ancestry, develops insidiously, causing stenosis, occlusion, aneurysmal dilatation, or coarctation of the involved vessels. The disease has two phases. In the acute or “prepulseless” phase, nonspecific systemic manifestations are present. Patients have rashes, erythema nodosum, fever, myalgias, arthritis, pleuritis, carotidynia, and elevated erythrocyte sedimentation rate. Months or years later, the second or occlusive phase develops and is characterized by multiple arterial occlusions. Patients may have cervical bruits, absent carotid or radial pulses, asymmetrical blood pressure recordings, and arterial hypertension. Neurological symptoms result from CNS or retinal ischemia associated with stenosis or occlusion of the aortic arch and arch vessels, or arterial hypertension caused by aortic coarctation or renal artery stenosis. Visual disturbances are most often bilateral. The diagnosis can be confirmed by MRA or CTA, but the most accurate assessment still requires aortography (Fig. 51A.15).
Patients with active disease are treated with oral glucocorticoids; cyclophosphamide, azathioprine, or methotrexate may be needed in special circumstances. Surgical treatment (angioplasty or bypass) of severely stenotic vessels may be required (Kerr et al., 1999).
Cranial (giant cell or temporal) arteritis is a polysymptomatic systemic large-vessel arteritis with a predilection to involve carotid artery branches (see Chapter 69). Thromboangiitis obliterans, also known as Buerger disease, is a rare segmental, inflammatory, obliterative angiopathy of unknown cause. The condition involves small and medium arteries and veins. It is suspected in young men who smoke and have a history of superficial migratory thrombophlebitis presenting with distal limb ischemia accompanied by digital gangrene. The disorder is characterized by remissions and exacerbations. Cerebral involvement is uncommon. Strokes can result from isolated angiitis of the CNS. Symptoms of large-vessel involvement include stroke-like presentations. Small-vessel involvement may be manifested as a mass lesion in the brain or multifocal encephalopathy (Fig. 51A.16). The erythrocyte sedimentation rate is usually normal or minimally elevated (see Chapter 51E).
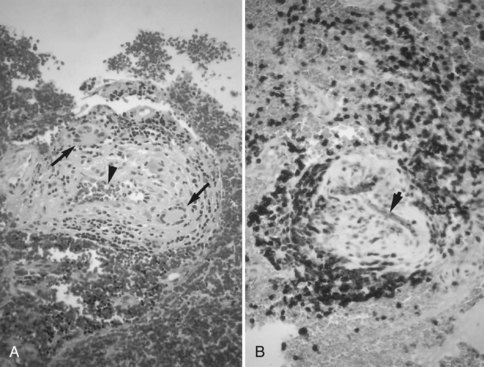
Migraine and Stroke
Migraine (see Chapters 18 and 69) affects women more often than men and may start during childhood or adolescence. Epidemiological studies suggest a nonrandom association of both headache and migraine with stroke, particularly among young women. This rare association was limited to women younger than age 35 in a large Italian case-controlled study (Carolei et al., 1996). The possible association between migraine headache and stroke was also evaluated by the Physician’s Health Study; physicians reporting migraine had increased risks of subsequent total stroke and ischemic stroke compared with those not reporting migraines (Buring et al., 1995). In the Women’s Health Study, migraine with aura raised the risk of stroke by 108% (95% CI, 30%-231%). Migraine without aura raised the stroke risk by approximately 25%. The risk of MI was also increased (Kurth et al., 2005; Woodward, 2009). The risk of white-matter abnormalities on MRI is also increased, particularly among subjects with migraine with aura (Kruit et al., 2004). The risk of intracerebral hemorrhage is not increased in persons who have migraines.
The International Headache Society Classification and Diagnostic Criteria require that to establish a diagnosis of migrainous infarction, one or more migrainous aura symptoms must be present and not fully reversed within 7 days from onset, and must be associated with neuroimaging confirmation of ischemic infarction (see Chapter 73). This definition implies that a firm diagnosis of migraine with aura has been made in the past. Also, the clinical manifestations judged to be the result of a migrainous infarction must be those typical of previous attacks for that individual, and finally, other causes of infarction, including those related to migraine therapy, need to be excluded by appropriate investigations.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a familial nonarteriosclerotic, nonamyloid microangiopathy characterized by migraine with aura, recurrent subcortical ischemic strokes starting in mid-adulthood, leading to pseudobulbar palsy, cognitive decline, subcortical dementia, and early white matter hyperintensities on MRI. CADASIL is caused by simple missense mutations or small deletions in the Notch 3 gene on chromosome 19q12 encoding a transmembrane receptor Notch 3. The Notch 3 mutation has been thought to be one of the most common human mutations. Pathologically, there is a characteristic granular osmiophilic material in arterial walls, including dermal arteries (Kalimo et al., 2002). A subtype of migraine known as familial hemiplegic migraine and characterized by transient weakness or frank paralysis during the aura has also been mapped close to the CADASIL locus (Hutchinson et al., 1995). The newer acronym, CADASILM (cerebral autosomal dominant arteriopathy with subcortical infarcts, leukoencephalopathy, and migraine), refers to a subvariety of CADASIL characterized by the high frequency of migraine (Verin et al., 1995). A clinical trial of donepezil in CADASIL patients with subcortical vascular cognitive impairment showed no improvement in general cognition, but improvement in some executive functions such as processing speed and attention (Schneider, 2008).
Inherited and Miscellaneous Disorders
Homocystinuria, an inborn error of amino acid metabolism, is an unusual cause of stroke (Box 51A.5). Three specific enzyme deficiencies responsible for homocystinuria have been identified: cystathionine-β-synthetase, homocysteine methyltransferase, and methylene tetrahydrofolate reductase. The accumulation of homocysteine in the blood leads to endothelial injury and premature atherosclerosis. Patients with homocystinuria may display a marfanoid habitus, malar flush, livedo reticularis, ectopia lentis, myopia, glaucoma, optic atrophy, psychiatric abnormalities, mental retardation, spasticity, seizures, osteoporosis, and a propensity for intracranial arterial or venous thrombosis. Death may result from pulmonary embolism, MI, or stroke. Raised levels of plasma homocysteine may be an independent risk factor for cerebrovascular disease, coronary artery disease, and peripheral arterial occlusive disease. Elevated levels of homocysteine can be effectively reduced with the administration of folic acid, occasionally requiring the addition of pyridoxine (vitamin B6) and vitamin B12. However, the treatment of high-normal homocysteine levels with folic acid and B-complex vitamins has not been associated with decreased stroke or coronary risk (Lonn et al., 2006; Toole et al., 2004). Other agents that may reduce homocysteine include choline, betaine, estrogen, and acetylcysteine.
Box 51A.5 Inherited and Miscellaneous Disorders Causing Cerebral Infarction
Sneddon syndrome consists of widespread livedo reticularis and ischemic cerebrovascular manifestations. A number of reports have documented a hereditary transmission and a link between Sneddon syndrome and aPL antibodies. An association with systemic lupus erythematosus (SLE) is also described. However, the etiopathogenesis remains unknown, although an immune mechanism is suspected. Endothelial cells could be the primary target tissue. Antiendothelial cell antibodies may be present (Frances et al., 1995).
Microangiopathy of brain, retina, and inner ear (Susac syndrome), also known as retinocochleocerebral vasculopathy, is a very rare microcirculatory syndrome that affects mainly adult women (Susac, 2004). The syndrome is unrelated to arterial hypertension or diabetes and is characterized by arteriolar branch occlusions of the brain, retina, and inner ear, with resultant encephalopathy, vision loss, vestibular dysfunction, tinnitus, vertigo, and asymmetrical sensorineural hearing loss. CSF examination may be normal or show mild inflammatory response. MRI findings include multifocal white-matter hyperintensities with preferential involvement for the central fibers of the corpus callosum. Brain biopsy may show multifocal brain microinfarcts in both gray and white matter. The cause of Susac syndrome is unknown, but signs and symptoms have been attributed to a disturbance of coagulation, microembolism, or both. Treatment with corticosteroids, cyclophosphamide, azathioprine, plasmapheresis, or anticoagulant therapy is empirical, but branch retinal artery occlusions and CNS infarctions may recur despite the treatment. Hyperbaric oxygen treatment may be an option for refractory visual symptoms.
Cerebral amyloid angiopathy occurs both sporadically or in rare instances as a hereditary disorder. Cerebral amyloid angiopathy is characterized by the localized deposition of amyloid in the media and adventitia of small arteries and arterioles of the cerebral cortex and meninges in the elderly. Cerebral amyloid angiopathy is more commonly associated with lobar hemorrhage than with ischemic stroke, but it has been associated with an increased frequency of cerebral infarction in patients with Alzheimer disease (Olichney et al., 1995). Biopsy of the involved cortex and leptomeninges is the only definitive way to diagnose cerebral amyloid angiopathy.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 51A Vascular Diseases of the Nervous System
Ischemic Cerebrovascular Disease
Pathophysiology of Cerebral Ischemia
Clinical Syndromes of Cerebral Ischemia
Diagnosis and Treatment of Threatened Ischemic Stroke
Large-Artery Atherothrombotic Infarctions
Small-Vessel or Penetrating Artery Disease
Nonatherosclerotic Vasculopathies
Inherited and Miscellaneous Disorders
Infarcts of Undetermined Cause
Essential Investigations for Patients with Threatened Strokes
Preventing Stroke Recurrence: Medical Therapy
Treatment of Acute Ischemic Stroke
Epidemiology and Risk Factors
There are approximately 785,000 new or recurrent strokes annually in the United States (600,000 being first events and 185,000 being recurrent events). Some 88% of these events are ischemic strokes; 8% to 12% of ischemic strokes result in death within 30 days. On average, every 40 seconds someone in the United States has a stroke. Despite gradual declines in overall stroke death rates in many industrialized countries, stroke remains a leading cause of death and disability, particularly in the United States. Worldwide, stroke is also a leading cause of death, with stroke mortality being particularly high in Eastern Europe and Asia (World Health Organization, 2004). By 2020, 19 out of 25 million annual stroke deaths will be in developing countries (Lemogoum et al., 2005). Stroke is also the leading cause of disability in adults. Of the hundreds of thousands of stroke survivors each year, approximately 30% require assistance with activities of daily living, 20% require assistance with ambulation, and 16% require institutional care.
A number of factors that may be classified as modifiable and unmodifiable increase the risk for ischemic stroke (Table 51A.1). Nonmodifiable risk factors for stroke include older age, male gender, ethnicity, family history, and prior history of stroke. Modifiable risk factors may be subdivided into lifestyle and behavioral risk factors and non-lifestyle factors, although these two subgroups are interrelated. Presumed modifiable lifestyle risk factors include cigarette consumption and illicit drug use. Non-lifestyle risk factors include low socioeconomic status, arterial hypertension, dyslipidemia, heart disease, and asymptomatic carotid artery disease. Stroke secondary to sickle cell disease is also a modifiable non-lifestyle risk factor. Potentially modifiable risk factors (that have yet to be shown to decrease risk when modified, however) include diabetes mellitus (DM), hyperhomocysteinemia, and left ventricular hypertrophy. Less well-documented risk factors include blood markers (i.e., C-reactive protein), ankle-brachial blood pressure ratios, silent cerebral infarcts, white-matter hyperintensities on magnetic resonance imaging (MRI), and degree of carotid artery intima-media thickness. Clinicians cannot assume that these risk factors express themselves exclusively by accelerating atherosclerosis. There are also considerable data implicating hemostatic and microcirculatory disorders in stroke as well as circadian and environmental factors.
| Nonmodifiable | Modifiable |
|---|---|
| Age | Arterial hypertension |
| Gender | Transient ischemic attacks |
| Race/ethnicity | Prior stroke |
| Family history | Asymptomatic carotid bruit/stenosis |
| Genetics | Cardiac disease |
| Aortic arch atheromatosis | |
| Diabetes mellitus | |
| Dyslipidemia | |
| Cigarette smoking | |
| Alcohol consumption | |
| Increased fibrinogen | |
| Elevated homocysteine | |
| Low serum folate | |
| Elevated anticardiolipin antibodies | |
| Oral contraceptive use | |
| Obesity |
Risk Factors for Stroke
The incidence of stroke increases dramatically with advancing age, and increasing age is the most powerful risk factor for stroke. The incidence of stroke doubles each decade past 55 years of age. Half of all strokes occur in people older than 70 to 75 years. Overall, stroke incidence rate is 1.25 times greater in men than women. Men develop ischemic strokes at higher rates than women up to the age of 75 years. With an estimated 20% of the population being older than 65, greater than 10 million octogenarians, and an increasing life expectancy in the United States, it is predicted that in the near future, the incidence of stroke will reach 1 million per year. Compared to whites, African Americans have approximately a twofold increased risk of first-ever stroke. The rate of cerebral infarction is higher in African Americans and Hispanic Americans than in whites; this could be partially explained by the higher prevalence of DM and arterial hypertension experienced by these ethnic minorities. African Americans had been thought to have higher rates of intracranial atherosclerotic occlusive disease compared with whites, but this may actually reflect ascertainment bias (Sacco et al., 1995; Wityk et al., 1996). Furthermore, the stroke incidence and case fatality rates are also markedly different among the major ethnic groups in Auckland, New Zealand. Maori and Pacific Islands people have a higher rate of mortality within 28 days of stroke when compared with Europeans, especially men (Bonita et al., 1997). Chinese, Koreans, and Japanese also may have increased rates of intracranial hemorrhage and intracranial atherosclerotic cerebrovascular disease compared to whites. In comparison with the United States and Western Europe, where hemorrhagic stroke represents 20% or less of all stroke subtypes, 40% of the stroke subtypes are hemorrhagic (intracerebral or subarachnoid hemorrhage) in Japan. Conversely, in India, where ischemic stroke accounts for 80% of all strokes, 10% to 15% of strokes occur in people younger than 40 years and are mostly related to intracranial atherosclerosis.
Heredity and Risk of Stroke
Heredity seems to play a role in the pathogenesis of cerebral infarction. An increased risk is seen with a family history of stroke among first-degree relatives. Genetic factors have been linked with the pathogenesis of ischemic stroke, but specific genetic variants remain largely unknown, and some purported genetic associations have not been replicated (Chinnery et al., 2010). There are a number of genetic causes of stroke. Some inherited diseases, such as the hereditary dyslipoproteinemias, predispose to accelerated atherosclerosis. A number of inherited diseases are associated with nonatherosclerotic vasculopathies, including Ehlers-Danlos (especially type IV) syndrome, Marfan syndrome, Rendu-Osler-Weber disease, and Sturge-Weber syndrome. Familial atrial myxomas, hereditary cardiomyopathies, and hereditary cardiac conduction disorders are examples of inherited cardiac disorders that predispose to stroke. Deficiencies of protein C and S or antithrombin (AT) are examples of inherited hematological abnormalities that can cause stroke. Finally, rare inherited metabolic disorders that can cause stroke include mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), Fabry disease, and homocystinuria. The presence of the apolipoprotein epsilon-2 allele in elderly individuals and deletion of the gene for the angiotensin-converting enzyme may increase the risk for stroke, but the association with stroke subtype is unclear (Agerholm-Larsen et al., 1997; Slooter et al., 1997; Szolnoki et al., 2001). The most significant findings of the ongoing Siblings With Ischemic Stroke Study (SWISS) to date have been the relationship between age and stroke in probands and sibs, and the lack of a tight association among ischemic stroke subtypes (Adams Jr. et al., 1993) within families (Meschia et al., 2005; Meschia et al., 2006; Wiklund et al., 2007).
Common Modifiable Risk Factors
At least 25% of the adult population has arterial hypertension, defined as systolic blood pressure (SBP) greater than 140 mm Hg or diastolic blood pressure (DBP) greater than 90 mm Hg. Prehypertension is defined as SBP between 120 and 139 mm Hg or DBP between 80 and 90 mm Hg. Optimal blood pressure is defined as SBP less than 120 mm Hg and DBP less than 80 mm Hg (Chobanian et al., 2003). Similar guidelines have been developed by the European Society of Hypertension–European Society of Cardiology Guidelines Committee (2003). The JNC7 Report (Table 51A.2) emphasizes that patients at risk, including those with DM or a history of stroke, should be treated with medications. Unfortunately, arterial hypertension remains poorly treated worldwide, and in the United States many patients are either undertreated or untreated (Hajjar and Kotchen, 2003). Arterial hypertension predisposes to ischemic stroke by aggravating atherosclerosis and accelerating heart disease, increasing the relative risk for stroke an estimated three- to fourfold. The risk is greater for patients with isolated systolic hypertension and elevated pulse pressure. Arterial hypertension is also the most important modifiable risk factor for stroke and the most powerful risk factor for all forms of vascular dementia (vascular cognitive impairment). Lowering blood pressure in stroke survivors helps prevent recurrent stroke and is more important than the specific hypotensive agent used, although there is some suggestion that beta-blockers are less preferred than other agents based on recent meta-analyses (Lindholm et al., 2005). Blood pressure treatment that results in a modest reduction in SBP of 10 to 12 mm Hg and 5 to 6 mm Hg diastolic is associated with a 38% reduction in stroke incidence (MacMahon and Rodgers, 1996). Treatment of isolated systolic hypertension in the elderly is also effective for reducing stroke risk. The Systolic Hypertension in the Elderly Program showed a 36% reduction in nonfatal plus fatal stroke over 5 years in the age 60-and-older group when isolated systolic hypertension was treated. Treating systolic hypertension also slows the progression of carotid artery stenosis. The PROGRESS trial evaluated the effects of perindopril and indapamide on the risk for stroke in patients with histories of stroke or transient ischemic attack (TIA). Regardless of blood pressure at entry, patients clearly benefited from treatment (PROGRESS Collaborative Group, 2001).
Table 51A.2 JNC 7 Report: Seventh Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure
About 171 million people worldwide have type 2 DM, including 18 million Americans. It is estimated that these numbers will grow to 366 million people worldwide and 30 million Americans by 2030 (Fonseca et al., 2002). Diabetes mellitus increases the risk of ischemic cerebrovascular disease an estimated two- to fourfold as compared with the risk in people without diabetes. In addition, DM increases morbidity and mortality after stroke. Macrovascular disease is the leading cause of death among patients with DM. The mechanisms of stroke secondary to diabetes may be caused by cerebrovascular atherosclerosis, cardiac embolism, or rheological abnormalities. The excess stroke risk is independent of age or blood pressure status. Diabetes associated with arterial hypertension adds significantly to stroke risk. There is a fourfold increase in the relative risk of cardiovascular events among patients with diabetes and hypertension compared to those without the two conditions. Diabetic persons with retinopathy and autonomic neuropathy appear to be a group at particularly high risk for ischemic stroke. High insulin levels increase the risk for atherosclerosis and may represent a pathogenetic factor in cerebral small-vessel disease. Presently, no evidence exists that tighter diabetic control or normal HbA1c levels over time decrease the risk for stroke or stroke recurrence. Moreover, optimal target blood glucose levels in stroke patients remain largely unknown (Gray et al., 2004; Van den Berghe et al., 2006). The UK Glucose Insulin in Stroke Trial (GIST-UK) failed to demonstrate any clinical benefit of insulin-induced euglycemia (target glucose 72-126 mg/dL). Furthermore, mortality appeared to be higher among patients with the greatest glucose reduction (Gray et al., 2009). Moreover, in the Spectroscopic Evaluation of Lesion Evolution in Stroke: Trial of Insulin for Acute Lactic Acidosis (SELESTIAL), the infusion of glucose-potassium-insulin did not have a favorable impact on cerebral infarct growth (McCormick et al., 2010).
High total cholesterol and high low-density lipoprotein (LDL) concentration are correlated with atherosclerosis. Dyslipidemia is a recognized risk factor for ischemic stroke. Meta-analyses have suggested that ischemic stroke risk increases with increasing serum cholesterol, and the reduction in stroke risk associated with 3-hydroxy 3-methylglutaryl coenzyme A reductase inhibitor (statin) therapies is related to reduction in LDL cholesterol (Amarenco et al., 2004; Tirschwell et al., 2004). Lipid-modifying therapy with statins has definitively established that reduction of LDL cholesterol reduces cardiovascular risk. Statins benefit stroke survivors as well. Lipid-lowering agents may slow progression of atherosclerotic plaque growth and may possibly cause a regression in plaque formation.
The Scandinavian Simvastatin Survival Study (4S) (Scandinavian Simvastatin Survival Study Group, 1999) investigated cholesterol lowering in persons with coronary heart disease and hypercholesterolemia and reported a highly significant relative reduction in the total mortality rate, major coronary events, and number of cardiac revascularization procedures. Post hoc analysis also showed a 28% reduction in fatal or nonfatal stroke and TIAs (Pedersen et al., 1998). The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) study investigated cholesterol lowering with pravastatin in patients with a previous myocardial infarction (MI) or unstable angina who had cholesterol levels between 155 and 271 mg/dL and reported a remarkable reduction in MI, cardiac revascularizations, and cardiovascular deaths, as well as a 20% reduction in the risk for stroke (Long-Term Intervention with Pravastatin in Ischaemic Disease [LIPID] Study Group, 1998). Similar findings were associated with atorvastatin in the Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) trial and the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA) studies. MIRACL showed a 50% relative risk reduction (RRR) (P = .045) in stroke among high-risk coronary disease patients (Schwartz et al., 2001). The ASCOT-LLA study demonstrated a favorable trend for fatal and nonfatal stroke, with a 27% RRR in patients at low risk for coronary events (hazard ratio, 0.73; 95% confidence interval [CI], 0.56-0.96; P = .0236), though not to the prespecified endpoint of P = .01 (Sever et al., 2003).
Although the Heart Protection Study (HPS) of patients at high risk with DM, coronary artery disease, or other atherosclerotic vascular disease did show an overall reduction in stroke risk, a subgroup analysis of the Heart Protection Study (HPS) did not show a reduction in the risk for stroke among patients with prior cerebrovascular disease (Heart Protection Study Collaborative Group, 2004). This subgroup analysis was limited in that the mean time from event to enrollment was 4.3 years, and the LDL reduction was 38 mg/dL. Subsequently, the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) study was published (SPARCL Investigators, 2006). This was a study of 4731 patients who had suffered either a stroke or TIA, had no known coronary heart disease, had LDL cholesterol levels of 100 to 190 mg/dL, and who were randomized to placebo or 80 mg of atorvastatin. A 56-mg/dL reduction in LDL treatment was noted in patients on atorvastatin. During a median follow-up of 4.9 years, 11.2% of atorvastatin-treated patients and 13.1% of placebo patients had a fatal or nonfatal stroke for an adjusted hazard ratio of 0.84 (95% CI, 0.71-0.99). A small increase in hemorrhagic stroke was reported in the atorvastatin group. Moreover, recent studies evaluating withdrawal of statins in acute ischemic stroke showed a higher incidence of death or dependency at 90 days (Blanco et al., 2007). Current guidelines of the American Heart Association and proposed modifications of the NCEP-III guidelines would therefore suggest that all patients at risk for stroke or who have had a cerebral infarction should be treated to a goal LDL level of below 70 mg/dL (Grundy et al., 2004; Sacco et al., 2006).
Atrial fibrillation, the most common sustained cardiac arrhythmia in the general population, affects about 1% of adults, is the most common cause for cardioembolic stroke, and is a risk factor for future cardiovascular disease. An estimated 1 to 2 million Americans have chronic nonvalvular atrial fibrillation (NVAF), a condition that is associated with an overall risk for stroke of approximately five- to sixfold, and a mortality rate approximately twice that of age- and sex-matched individuals without atrial fibrillation. The prevalence of atrial fibrillation increases with advancing age and is 0.5% for patients aged 50 to 59 years and 8.8% for those aged 80 to 89 years. Approximately 70% of individuals with atrial fibrillation are between 65 and 85 years of age. NVAF is associated with a substantial risk for stroke. Heart failure, arterial hypertension, DM, prior stroke or TIA, and age older than 75 years increase the risk for embolism in patients with NVAF. High-risk patients have a 5% to 7% yearly risk for thromboembolism. The CHADS2 Score represents a validated quantification of risk, with congestive heart failure, hypertension, age older than 75 years, and a history of DM being assigned 1 point; stroke or TIA are assigned 2 points (Gage et al., 2001). Ischemic stroke rates increase from 1.9 to 18.2 events per 100 patient-years with CHADS 2 scores of 0 and 6, respectively. Warfarin therapy, with the International Normalized Ratio (INR) value adjusted to between 2.0 and 3.0, decreases the relative risk for stroke in patients with NVAF by approximately two-thirds. High-risk patients, regardless of age, sustain particular benefit from warfarin anticoagulation. Left atrial enlargement also increases the risk for stroke in men. Likewise, left ventricular hypertrophy as demonstrated by electrocardiography (ECG) in men with preexisting ischemic heart disease is a major risk factor for stroke.
Cigarette smoking is the leading cause of preventable death in the United States. Cigarette smoking is a major risk factor for coronary artery disease, stroke, and peripheral arterial disease, an independent risk factor for ischemic stroke in men and women of all ages, and a leading risk factor of carotid atherosclerosis in men. The risk for stroke in smokers is two to three times greater than in nonsmokers. The mechanisms of enhanced atherogenesis promoted by cigarette smoking are incompletely understood but include reduced capacity of the blood to deliver oxygen, cardiac arrhythmias, increased blood coagulability, and triggering of arterial thrombosis and arterial spasm. Tobacco also increases carotid artery plaque thickness. More than 5 years may be required before a reduction in stroke risk is observed after cessation of smoking. Switching to pipe or cigar smoking is of no benefit. Counseling, nicotine replacement products, varenicline (a nicotinic receptor partial agonist),and bupropion are efficacious smoking cessation treatments. Selective blockers of the cannabinoid receptor type 1, such as rimonabant, have been proposed for treatment of multiple cardiometabolic risk factors including smoking and abdominal obesity (Gelfand and Cannon, 2006).
Numerous studies have established an association between obstructive sleep apnea (OSA) and stroke; moreover the severity of OSA is much higher in stroke patients than in controls (Dyken, 2000). Habitual snoring increases the risk for stroke and adversely affects the outcome of patients admitted to the hospital with stroke. Mounting evidence also suggests that inflammation, lipoprotein(a) concentration, impaired fibrinolysis, and increased thrombotic potential are important nontraditional cardiovascular risk factors.
The aorta is the most frequent site of atherosclerosis. Protruding atheroma may be the cause of otherwise unexplained TIAs or strokes. Aortic-arch atheromatosis detected by transesophageal echocardiography is an independent risk factor for cerebral ischemia; the association is particularly strong with mobile and thick atherosclerotic plaques more than 4 mm in thickness (French Study of Aortic Plaques in Stroke Group, 1996). The prevalence of ulcerated plaques was 16.9% among patients with cerebrovascular diseases, compared to 5.1% among patients with other neurological diseases. Remarkably, ulcerated plaques were found in 61% of cryptogenic cerebral infarcts, compared to 22% of cerebral infarcts with a known cause.
Other Risk Factors for Stroke
Hemostatic factors may be important in assessing the risk for cerebrovascular disease. Elevated hematocrit, hemoglobin concentration, and increased blood viscosity may be indicators of risk for ischemic stroke. Elevation of plasma fibrinogen is an independent risk factor for the development of cerebral infarction. Epidemiological studies have shown a correlation between elevated plasma fibrinogen levels and both ischemic stroke incidents and mortality. An elevated plasma fibrinogen level may reflect progression of atherogenesis. Plasminogen activator inhibitor-1 excess and factor VII are independent risk factors for coronary heart disease. Compared with white Americans, black Americans have higher mean levels of fibrinogen, factor VIII, von Willebrand factor, and AT, and lower mean levels of protein C. Fibrinogen levels are closely correlated with other stroke risk factors such as cigarette smoking, arterial hypertension, diabetes, obesity, hematocrit levels, and spontaneous echocardiographic contrast. Antiphospholipid (aPL) antibodies are a marker for an increased risk for thrombosis, including TIAs and stroke, particularly in those younger than 50 years. In older patients, the presence of aPL (either lupus anticoagulants [LAs] or anticardiolipin [aCL]) among patients with ischemic stroke does not predict either increased risk for subsequent vascular occlusive events over 2 years or a differential response to aspirin or warfarin therapy. As such, routine screening for aPL in older patients with ischemic stroke does not appear warranted (Levine et al., 2004) The factor V Leiden mutation is associated with deep venous thrombosis in otherwise healthy individuals with additional prothrombotic risk factors. An overall association of the factor V Leiden mutation and arterial thrombosis has not been found. As opposed to homozygous mutations, heterozygous factor V Leiden and prothrombin gene mutations have no clear association with increased stroke risk (Fields and Levine, 2005). Elevated von Willebrand factor is a risk factor for MI and ischemic stroke. Elevated levels of fasting total homocysteine (normal 5-15 mM), a sulfhydryl-containing amino acid, have been associated with an increased risk for stroke and thrombotic events in case-controlled studies. Metabolism of homocysteine requires vitamin B6 (pyridoxine), vitamin B12 (cyanocobalamin), folate, and betaine. Plasma homocysteine concentrations may be reduced by the administration of folic acid alone or in combination with vitamins B6 and B12. Conversely, serum folate concentrations of 9.2 nM or less have been associated with elevated plasma levels of homocysteine, and a decreased folate concentration alone may be a risk factor for ischemic stroke, particularly among blacks (Giles et al., 1995). However, folic acid supplementation does not have a major impact on stroke reduction (Lee et al., 2010).
Stroke is uncommon among women of childbearing age, estimated at 4.4/100,000 (Petitti et al., l997). The relative risk for ischemic stroke is increased among users of high-dose estrogen oral contraceptives, particularly with coexistent arterial hypertension, cigarette smoking, and increasing age. Based on an odds ratio of 1.93, the risk is increased to 8.5/100,000, which translates into a number needed to harm of 24,000 women to cause one ischemic stroke (Gillum et al., 2000). Thus, for a healthy young woman without any other stroke risk factors, the risk of stroke associated with oral contraceptives is small and probably outweighed by their benefits. New agents containing lower doses of estrogen and progestin have reduced the frequency of oral contraceptive–related cerebral infarction. Two postmenopausal hormone replacement studies with equine estrogen (Premarin) showed no benefit in reducing the incidence of stroke in a cohort of women with coronary heart disease (Hulley et al., 1998). The Women’s Estrogen for Stroke Treatment study of estradiol replacement in postmenopausal women status post stroke also failed to show a reduction in recurrent stroke (Viscoli et al., 2001). In addition, the Women’s Health Initiative, a prospective randomized trial of estrogen therapy in healthy postmenopausal women, was halted prematurely because the risks outweighed the benefits. Absolute excess risks per 10,000 person-years attributable to estrogen plus progestin were sevenfold for coronary heart disease events, eightfold for strokes, eightfold for pulmonary thromboembolism, and eightfold for invasive cancers, whereas absolute risk reductions per 10,000 person-years were sixfold for colorectal cancers and fivefold for hip fractures (Rossouw et al., 2002). The risk for thrombosis associated with pregnancy is high in the postpartum period. The risk for cerebral infarction is increased in the 6 weeks after delivery but not during pregnancy.
A diurnal and seasonal variation of ischemic events occurs. Circadian changes in physical activity, catecholamine levels, blood pressure, blood viscosity, platelet aggregability, blood coagulability, and fibrinolytic activity may explain the circadian variations in onset of myocardial and cerebral infarction. Although an early-morning peak occurs for all subtypes of stroke, most clinical trials on the use of platelet antiaggregants or other antithrombotic agents do not take these circadian variations into account. Rhythmometric analyses support the notion that stroke is a chrono-risk disease, in which cold temperatures also represent a risk factor. A history of recent infection, particularly of bacterial origin and within 1 week of the event, is also a risk factor for ischemic stroke in patients of all ages. A number of recent reports suggest that Chlamydia pneumoniae, a causative organism of respiratory infections, may have a role in carotid and coronary atherosclerosis. Some studies have also identified an association with chronic infections with Helicobacter pylori and cytomegalovirus (Bittner, 1998; Ross, 1999). These findings have not been confirmed, however (Lindsberg and Grau, 2003).
Pathophysiology of Cerebral Ischemia
A cascade of complex biochemical events occurs seconds to minutes after cerebral ischemia. Cerebral ischemia is caused by reduced blood supply to the microcirculation. Ischemia causes impairment of brain energy metabolism, loss of aerobic glycolysis, intracellular accumulation of sodium and calcium ions, release of excitotoxic neurotransmitters, elevation of lactate levels with local acidosis, free radical production, cell swelling, overactivation of lipases and proteases, and cell death (Fisher and Ratan, 2003). Many neurons undergo apoptosis after focal brain ischemia (Choi, 1996). Ischemic brain injury is exacerbated by leukocyte infiltration and development of brain edema. Exciting new treatments for stroke target these biochemical changes.
Clinical Syndromes of Cerebral Ischemia
A number of syndromes result from ischemia involving the central nervous system (CNS) (Brazis et al., 2011).
Transient Ischemic Attacks
An estimated 400,000 individuals experience a TIA each year. A TIA is a prognostic indicator of stroke, with one-third of untreated TIA patients having a stroke within 5 years. About 1 in 10 patients with TIA experience a stroke in the next 3 months. The interval from the last TIA is an important predictor of stroke risk; of all patients who subsequently experience stroke, 21% do so within 1 month and 51% do so within 1 year of the last TIA. In one series, patients with TIA had a 3-month stroke risk of 10.5%, equal to the recurrence rate following a stroke. Furthermore, 50% of those strokes following a TIA occurred within 48 hours of TIA onset (Johnston et al., 2000). Cardiac events are the principal cause of death in patients who have had a TIA. The 5% to 6% annual mortality rate after TIA is mainly caused by MI, similar to the 4% annual cardiac mortality rate in patients with stable angina pectoris.
A TIA is a temporary and “non-marching” neurological deficit of sudden onset; attributed to focal ischemia of the brain, retina, or cochlea; and lasting less than 24 hours. Yet most TIAs last only a few minutes. Episodes that last longer than 1 hour are usually due to small infarctions. With the advent of diffusion-weighted magnetic resonance imaging (DW-MRI) sequences, the time-based definition of TIA is inadequate because infarctions are sometimes evident on DW-MRI in patients whose clinical manifestations resolved completely within a few hours. Thus, a “tissue-based” modification of the TIA definition has been proposed, suggesting that any transient episode, regardless of duration, associated with a clinically appropriate lesion by MRI be defined as a stroke, and that otherwise prolonged events (>1-6 hours in duration) be defined as stroke rather than TIA when otherwise clinically appropriate (Albers et al., 2002). Furthermore, DW-MRI is useful in predicting the risk for early stroke; patients with TIA and DW-MRI lesions are at greater risk for experiencing a subsequent stroke than patients without a lesion (Coutts et al., 2005). Johnston et al. also introduced a new unified score for risk stratification in patients with TIAs, known as the ABCD2 score. The ABCD2 score is based on age, blood pressure (≥140/90 mm Hg), clinical features, TIA duration, and diabetes (Table 51A.3). ABCD2 scores of 4 or greater indicate a moderate to high stroke risk and justify prompt hospital admission (Johnston et al., 2007).
| Age 60 or older | 1 point |
| Blood pressure ≥140/90 | 1 point |
| Clinical: | |
| Unilateral weakness | 2 points |
| Speech impairment | 1 point |
| Duration: | |
| 60 minutes or more | 2 points |
| <60 minutes | 1 point |
| Diabetes mellitus | 1 point |
Agreement between physicians to define the likelihood of a TIA, even among fellowship-trained neurologists, remains poor (Castle et al., 2010). The following symptoms are considered typical of TIAs in the carotid circulation: ipsilateral amaurosis fugax, contralateral sensory or motor dysfunction limited to one side of the body, aphasia, contralateral homonymous hemianopia, or any combination thereof. The following symptoms represent typical TIAs in the vertebrobasilar system: bilateral or shifting motor or sensory dysfunction, complete or partial loss of vision in the homonymous fields of both eyes, or any combination of these symptoms. Perioral numbness also occurs. Isolated diplopia, vertigo, dysarthria, and dysphagia should not be considered as being caused by a TIA unless they occur in combination with one another or with any of the other symptoms just listed (Box 51A.1). Older patients with isolated vertebrobasilar symptoms and a significant history of cardiovascular risk factors should, however, be evaluated for possible TIA or stroke, because they are at substantially higher risk for cerebrovascular events (Norrving et al., 1995).
Box 51A.1 Recognition of Carotid and Vertebrobasilar Transient Ischemic Attacks
Symptoms Suggestive of Vertebrobasilar Transient Ischemic Attacks
Usually bilateral weakness or clumsiness but may be unilateral or shifting
Bilateral, shifting, or crossed (ipsilateral face and contralateral body) sensory loss or paresthesias
Bilateral or contralateral homonymous visual field defects or binocular vision loss
Two or more of the following symptoms: vertigo, diplopia, dysphagia, dysarthria, and ataxia
Transient global amnesia (TGA) is characterized by a reversible antegrade and retrograde memory loss, except for a total amnesia of events that occur during the attacks and inability to learn newly acquired information. During the attacks, patients remain alert without motor or sensory impairments and often ask the same questions repeatedly. Patients are able to retain personal identity and carry on complex activities. TGA most commonly affects patients in their 50s and older. Men are affected more commonly than women. The attacks begin abruptly and without warning. A typical attack lasts several hours (mean, 3-6 hours) but seldom longer than 12 hours. Onset of TGA may follow physical exertion, sudden exposure to cold or heat, or sexual intercourse. Although a large number of conditions have been associated with transient episodes of amnesia, in most instances, TGA is of primary or unknown cause. TGA has been documented in association with epilepsy, migraine, intracranial tumors, overdose of diazepam, cardiac arrhythmias secondary to digitalis intoxication, and as a complication of cerebral and coronary angiography. Many reports have suggested a vascular causal factor for this heterogeneous syndrome. Bilateral hippocampal and parahippocampal complex ischemia, possibly of migrainous origin, in the distribution of the posterior cerebral arteries is a potential mechanism. Acute confusional migraines in children and TGA have a number of similar features. Others have suggested an epileptic causal factor for a minority of patients. Venous hypertension with transient hypoxemia in the context of incompetent internal jugular vein valves has also been suggested as a possible mechanism for TGA (Nedelmann et al., 2005
[/not-level-membership-for-neurology-category]
