[level-membership-for-anesthesiology-category]
19 Valvular Heart Disease
Replacement and Repair
In analyzing the physiologic response of the left ventricle (LV) to a variety of abnormal loading conditions, it is useful to consider the concepts of afterload mismatch and preload reserve.1 These concepts frame discussions of the pathophysiology of the individual valvular lesions. Another set of physiologic concepts essential to understanding VHD and its evolution are pressure-volume loops and the linear end-systolic pressure-volume relation (ESPVR). The former provides a graphic analysis of ventricular pressure-volume relations in a single beat, whereas the latter is a method for quantifying the intrinsic contractility of the myocardium over multiple contractions and is relatively independent of changes in loading conditions (see Chapters 5, 12, 13 and 14).
Pathophysiology

Pressure-Volume Loops
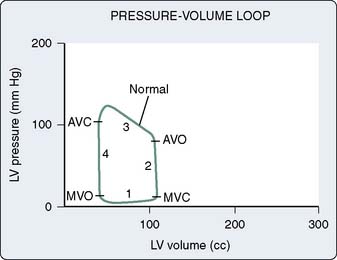
The pressure and volume changes that occur during a normal cardiac cycle are depicted in Figure 19-1. A pressure-volume loop is generated when ventricular volumes are plotted against simultaneously occurring ventricular pressures for a single contraction (Figure 19-2). Familiarity with this graphic analysis of cardiac function, the normal pressure-volume loop of the LV, provides a basis for appreciating a load-insensitive index of contractility, the ESPVR. Phase 1 in Figure 19-2 shows ventricular filling and represents the loading of the heart before contraction (i.e., preload). During early and middle diastole, filling of the ventricle is rapid, depending on the pressure gradient between the left atrium and the LV. In late diastole, the left atrium contracts (a wave), which results in the final left ventricular end-diastolic volume (LVEDV) and pressure (LVEDP). Atrial contraction in the normal heart accounts for 15% to 20% of ventricular filling. The normal ventricle accommodates large changes in ventricular volume with only a small change in ventricular diastolic pressure.
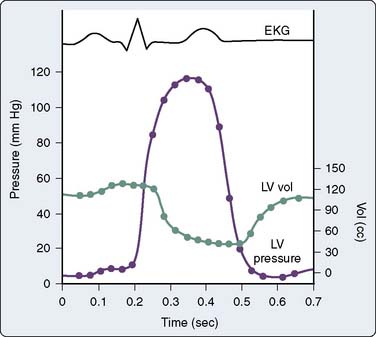
Figure 19-1 Simultaneous left ventricular (LV) volume and pressure during one cardiac cycle. EKG, electrocardiogram.
(From Barash PG, Kopriva DJ: Cardiac pump function and how to monitor it. In Thomas SJ [ed]: Manual of cardiac anesthesia. New York: Churchill Livingstone, 1984, p 1.)

Afterload Stress and Preload Reserve
Figure 19-3 shows the response of the LV to changes in afterload, with preload held constant, in the context of the pressure-volume loops from single contractions. These curves are constructed experimentally by infusing a pure α-adrenergic agonist while simultaneously measuring the corresponding end-systolic volumes (ESVs). The resultant curves describe the diastolic pressure-volume relation (ventricular compliance) and the linear ESPVR at a given level of myocardial contractility. Each counterclockwise loop represents a cardiac cycle. The stroke volume (SV) progressively decreases as the impedance to ejection increases from beats 1 to 3. This pattern continues until beat 4, when the peak ventricular systolic pressure fails to open the aortic valve and only isovolumic contraction ensues.1 This inverse relation between afterload and SV (inverse force-velocity relation) also was documented experimentally in a canine preparation in which the LVEDV was held constant (Figure 19-4).2 In the intact heart, afterload is a function of ventricular size and arterial pressure.3 Its pivotal role in cardiovascular regulation is summarized in Figure 19-5. Afterload is defined as the tension, or force per unit of cross-sectional area, in the ventricular wall during ejection.4 Laplace’s law provides a mathematical expression for the wall tension5 (in which P is the intraventricular pressure) developed in a spherical chamber of radius, R, and wall thickness, h:
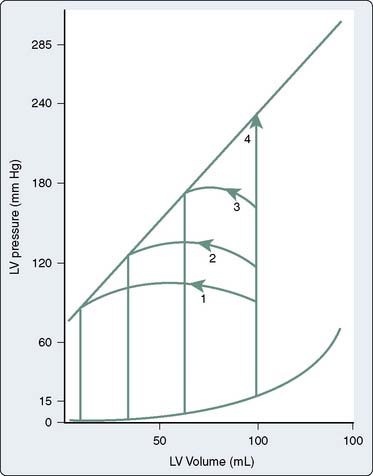
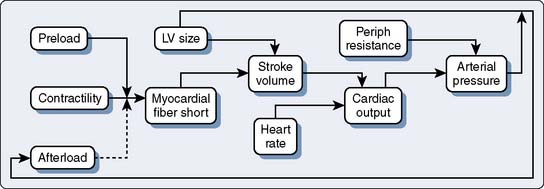
Figure 19-5 Role of afterload in cardiac regulation.
LV, left ventricular; Periph, peripheral.
(From Braunwald E: Regulation of the circulation. N Engl J Med 290:1124, 1974.)
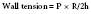
This means that ventricular afterload, a function of the constantly changing intraventricular pressure and radius, varies continuously during systole. It is extremely difficult to precisely quantify afterload in the clinical setting, and commonly used approximations such as blood pressure or systemic vascular resistance (SVR) are inherently misleading because they fail to reflect instantaneous pressure-volume variations. Left ventricular size or preload is a determinant of the SV and afterload.6 Normally, the LV compensates for increases in afterload by increases in preload. This increase in end-diastolic fiber stretch or radius further increases wall tension in accordance with Laplace’s law, resulting in a reciprocal decline in myocardial fiber shortening (i.e., inverse force-velocity relation). Despite this relative decline in fiber shortening, the SV is maintained because contractile force is augmented at the higher preload (i.e., Starling or length-active tension effect). This increased contractility at higher LVEDVs may be mediated by an increased sensitivity to the inotropic effects of extracellular calcium at longer muscle lengths.7–9
Use of this “preload reserve” allows the LV to maintain its SV in the face of an afterload stress,1 as shown in Figure 19-6.1 The increase in afterload (beat 2) elicits a compensatory increase in end-diastolic volume (EDV; beat 3), preserving SV at the higher afterload. However, when the ventricle reaches the limit of its preload reserve, it overdistends, and preload behaves as if it were fixed. SV then decreases with further increases in the systolic pressure (afterload mismatch; beat 4), a clinical corollary of the inverse force-velocity relation.

Ventricular Compliance
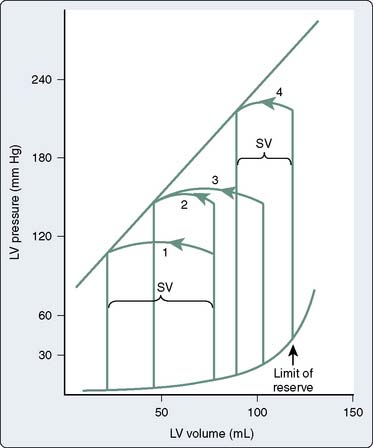
Translating this physiologic analysis to the clinical setting is complicated by a number of practical constraints. One of the most important is the inconstant relation between LVEDV and LVEDP. The diastolic pressure-volume relation for the normal mammalian LV is a curvilinear function (Figure 19-7).3,10 The slope of this curve (i.e., ratio of change in volume to change in pressure during diastole) is the ventricular compliance (dV/dP). Although the normal ventricle is extremely compliant in the physiologic range, the instantaneous compliance decreases with increments in diastolic filling. This progressively increasing slope of the pressure-volume curve becomes evident at the extremes of ventricular volume, in which succeeding increments in volume result in exponentially greater increases in end-diastolic pressure (EDP). Patients with acute aortic regurgitation (AR) experience the hemodynamic consequences of this phenomenon. The catastrophic increases in ventricular filling pressure reflect the absolute magnitude of the volume overload and are a corollary of a precipitously declining compliance relation.
Figure 19-7 The normal diastolic pressure-volume relation.
(From Spotnitz HM, Sonnenblick EH, Spiro D: Relation of ultrastructure to function in the intact heart: Sarcomere structure relative to pressure-volume curves of the intact left ventricles of dog and cat. Circ Res 18:49, 1966.)
These hemodynamic manifestations of acute shifts up and down a single compliance curve must be distinguished from chronic, pathologic alterations in ventricular compliance, which produce actual shifts of the entire curve relating diastolic pressure and volume. For example, in animal models of chronic volume overload, the entire pressure-volume curve is shifted to the right, such that substantial increases in ventricular volume are tolerated with relatively little change in EDP (Figure 19-8).11 In this example, the slope of the new pressure-volume curve (i.e., compliance) is decreased. Similarly, time-dependent, rightward shifts of the entire pressure-volume relation have been shown to occur in patients with severe ventricular volume overload resulting from chronic AR.12 In these examples, the development of a new relation between pressure and volume may reflect the physiologic process of creep, the time-dependent change in size or the dimension of tissue maintained at a constant level of stress.4
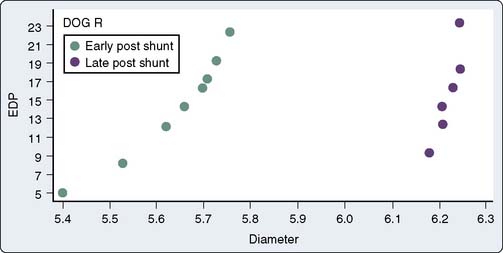
Myocardial wall thickness is an important determinant of diastolic compliance. In clinical settings of chronic pressure overload (e.g., aortic stenosis [AS], chronic hypertension), diastolic compliance and ventricular wall thickness have been shown to be linearly and inversely related (Figure 19-9).13 This may explain why the normal, thinner-walled right ventricle (RV) is more compliant than the normal LV, even though the ventricles share similar qualities of intrinsic myocardial stiffness.3,14 This association between pathologic hypertrophy of the ventricle and deterioration in its diastolic compliance is a well-documented but poorly understood phenomenon.
Figure 19-9 Relation between chamber stiffness (ΔP/ΔD, the inverse of compliance) and ventricular wall thickness.
(From Grossman W, McLaurin P, Moos SP, et al: Wall thickness and diastolic properties of the left ventricle. Circulation 49:129, 1974.)
It has become customary to characterize diastolic function in certain disease states as normal or abnormal, such as diastolic dysfunction or diastolic failure.15,16 The latter condition is a distinct pathophysiologic entity that results from an increased resistance to ventricular filling and leads to an inappropriate upward shift of the diastolic pressure-volume relation.17 For example, diastolic dysfunction is seen in ischemic cardiomyopathy, particularly when combined with pressure overload hypertrophy.18
In certain diseases, these primary derangements of diastolic function may predominate over abnormalities of diastolic function associated with ventricular hypertrophy. For example, in hypertrophic cardiomyopathy (HCM), the impaired ventricular relaxation inherent in the myopathic process appears to play the greater role in the observed abnormalities of diastolic filling because the diastolic dysfunction is often disproportionate to the degree of ventricular hypertrophy.19,20 The effects of pathologic hypertrophy on diastolic compliance and ventricular relaxation are complex and are considered in more detail later in the Aortic Stenosis and Hypertrophic Cardiomyopathy sections.
Filling of one ventricle or changes in its configuration or compliance properties can significantly alter the diastolic pressure-volume characteristics of the other ventricle.21,22 Progressive increases in right ventricular filling shift the left ventricular compliance curve up and to the left. This effect is greatest at high right ventricular filling pressures and is accentuated by the presence of the pericardium.23 With severe right ventricular distention, the interventricular septum encroaches on the LV. This encroachment reduces the size of the LV and alters its geometric configuration such that its compliance declines.21 As a result, left ventricular filling pressures may fail to reflect even directional changes in left ventricular size.24

Contractility
Myocardial contractility can be defined as the ability of the heart to generate force at a given preload.25 Although most clinicians and researchers seem to be comfortable with their intuitive notions of the cardiac inotropic state, the search for a consensus on a quantitative yardstick of ventricular inotropy has proved to be elusive. Its accurate and reproducible measurement is of more than theoretic interest because the contractile function of the heart is a key determinant of prognosis for most cardiac diseases, and it is especially important in critical decisions regarding the timing of surgical correction in patients with VHD.
Historically, methods of assessing myocardial contractility have been divided into two groups, based on analysis of the isovolumetric or the ejection phase of cardiac contraction.4 Details of their clinical determination and relative reproducibility are beyond the scope of this discussion, but more information can be found in several excellent reviews,4,26,27 as well as in Chapters 5 and 14. The isovolumetric indices include measurements such as maximal velocity of myocardial fiber shortening (Vmax), peak pressure development (dP/dt), and peak dP/dt measured at an instantaneous pressure (dP/dt/P). Although relatively insensitive to loading conditions, these tests poorly reflect basal levels of contractility and are unreliable for comparing contractility among patients or assessing directional changes in contractility in an individual patient over time.26
Ejection phase indices, such as the ejection fraction (EF), are determined, in part, by the intrinsic inotropic state and can be used to define basal levels of contractility.26 Such measurements are extremely useful in evaluating ventricular function in patients with CAD or other conditions that do not significantly alter ventricular loading conditions.28 Ejection phase indices are understandably popular because they are readily available, and they are the most widely used clinical measures of left ventricular function. However, these indices are directly proportional to preload, vary inversely with ventricular afterload, and are accordingly unreliable for assessing contractile performance in patients with most forms of VHD.
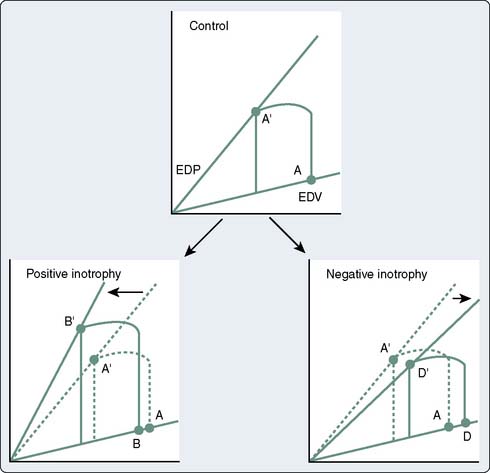
The use of the pressure-volume diagram and analysis of the ESPVR allow a more precise appreciation of left ventricular contractility, which is independent of preload.29 The extent of myocardial shortening, and therefore of end-systolic fiber length, is a direct function of afterload (i.e., inverse force-velocity relation), and myocardial contractility can be evaluated by making use of this fundamental property. In most instances, end-systolic pressure (ESP) can be substituted for afterload. Only with pathologic degrees of ventricular hypertrophy is there a major divergence between ESP and afterload. This means that for any level of contractility, the ESV to which a ventricle contracts is a linearly increasing function of ESP. A stronger ventricle contracts to a smaller ESV (i.e., empties more completely) at any given level of ventricular afterload. Changes in the inotropic state also can be viewed in the context of the idealized pressure-volume loop. Positive inotropic interventions shift the curve up and to the left, increasing the work that can be performed at any given EDV (preload). Conversely, negative inotropic interventions shift the curve down and to the right30 (Figure 19-10; see Chapters 5 and 14). These load-independent indices of contractility are largely research tools, but in the future, it may be possible to construct pressure-volume loops and quantify ESPVRs in real time with echocardiographic equipment featuring automated border detection31 (see Chapter 12).
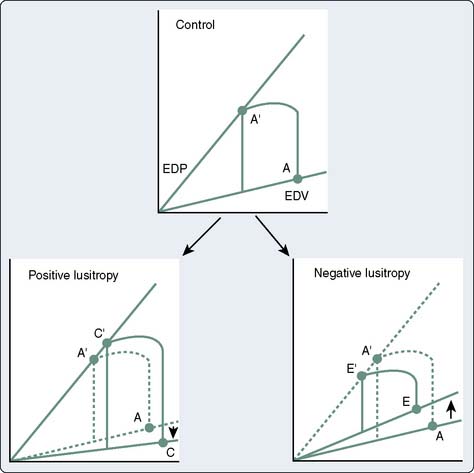
Clinical studies of patients with relatively normal loading conditions have shown that variations in the ESV reliably correlate with changes in ejection phase indices (Figure 19-11).32 The ESPVR represents an index of contractility that depends on systolic ventricular pressure (i.e., afterload) but is independent of end-diastolic length (i.e., preload).28,33,34 Pressure-volume loops also provide a framework for considering the interactions between systolic (inotropic) and diastolic (lusitropic) function. Although the preload (i.e., EDV) is an independent determinant of SV, because of the circular nature of blood flow, the SV ultimately determines venous return and the resultant preload for the next cardiac cycle.30 The “inotropically determined” ESV is the other element besides the venous return that contributes to the EDV. Just as the ESPVR uniquely describes systolic function, the end-diastolic pressure-volume relation is a manifestation of the intrinsic relaxation (lusitropic) properties of the ventricle. Positive lusitropic interventions facilitate ventricular filling and shift the end-diastolic pressure-volume relation down and to the right, and a negative lusitropic intervention shifts it up and to the left (Figure 19-12).
Aortic stenosis

Clinical Features and Natural History
Aortic stenosis (AS) is the most common cardiac valve lesion in the United States. Approximately 1% to 2% of the population is born with a bicuspid aortic valve, which is prone to stenosis with aging, and the population is aging. Clinically significant aortic valve stenosis is present in 2% of unselected individuals older than 65 years, and in 5.5% of those older than 85 years.35 Incipient aortic valve stenosis with calcification and stiffening is observed in 50% of people in the 75 to 80 age group and in up to 75% in those older than 85 years. Bicuspid aortic valve is a common congenital cardiac abnormality, occurring in approximately 1% of the population.36 The heritability coefficient is estimated at 89%, suggesting that the bicuspid aortic valve is almost entirely genetic in nature.37 It is a risk factor for premature AS and ascending aortic aneurysms. The ascending aorta in bicuspid valvular disease has the same histopathologic features as Marfan syndrome, such as medial degeneration, decreased fibrillin-1, and enhanced matrix metalloproteinase activity in the aortic wall. Recent data from the International Registry of Acute Aortic Dissection demonstrate that patients with bicuspid aortic valve have a 6.14% lifetime risk rate of aortic dissection (nine times that of the general population), compared with 40% for patients with Marfan syndrome. Yet, bicuspid aortic valvular disease is a hundred times more common than Marfan syndrome and is, therefore, responsible for a larger number of aortic dissections.36 Calcific AS has several features in common with CAD. Both conditions are more common in men, older people, and patients with hypercholesterolemia, and both result, in part, from an active inflammatory process. There is clinical evidence of an atherosclerotic hypothesis for the cellular mechanism of aortic valve stenosis. There is a clear association between clinical risk factors for atherosclerosis and the development of AS: increased lipoprotein levels, increased low-density lipoprotein (LDL) cholesterol, cigarette smoking, hypertension, diabetes mellitus, increased serum calcium and creatinine levels, and male sex.38 Homozygous familial hypercholesterolemia produces a severe form of AS in children.39 The early lesion of aortic valve sclerosis may be associated with CAD and vascular atherosclerosis. The extent of aortic valve calcification was observed as an important predictor of poor outcome in patients with AS.40 These and other studies suggest that aortic valve calcification is an inflammatory process promoted by atherosclerotic risk factors.
The early lesion of AS resembles that of the initial plaque of CAD, and there is a close correlation between calcification found in coronary arteries and the amount of calcification found in the aortic valve. Pathologic studies of aortic valves revealed the presence of LDL, suggesting a common cellular mechanism for the genesis of valvular and vascular atherosclerotic disease.38 Degenerative lesions on aortic valves contain an inflammatory infiltrate of nonfoam cells and foam cell macrophages, T lymphocytes, and other inflammatory cells. Lipid is accumulated in the fibrosa immediately below the endothelium layer on the aortic side of the valve. LDL- and apolipoprotein E–containing lipoproteins are present as well. Little is known about the synthesis of bone matrix proteins in calcific aortic valve stenosis. The calcifications are composed of hydroxyapatite on a matrix of collagen, osteopontin, and other bone matrix proteins.
A potential mechanism for this cellular inflammation process involves the combination of macrophages, LDL, and the secretion by macrophages of transforming growth factor-β and platelet-derived growth factor to stimulate the conversion of valvular fibroblast cells into osteoblasts that produce bone proteins.38 Most surprising are the findings that 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (e.g., statins) retard the progression of CAD and AS.41,42 The odds ratio of AS progression was 0.46 in 38 treated patients compared with 118 untreated patients.43 Other studies have shown similar results, and it is possible that therapy with statins or other drugs may be used to block or slow the progression of valve lesions in the future.38,44 In addition to recruitment of inflammatory cells and lipid accumulation into valves, calcification is a prominent feature that contributes to leaflet thickness and rigidity. Early development of aortic valve calcification may be related to vitamin D–receptor genotypes or mutations in the Notch gene.35 Calcification is an active process that includes osteopontin, osteonectin, osteocalcin, and other bone morphogenic proteins that regulate calcification and ossification. Active osteoblastic bone formation and osteoclastic bone resorption occur in stenotic valves. Increased fibroblasts produce collagen, leading to fibrosis. Concomitant elastin degradation also contributes to valve stiffening. Whereas normal valves are avascular, microvessels form in thickened stenotic valves, assuring the continued supply of inflammatory cells and lipids. Recent advances in genetic epidemiology have demonstrated a strong inheritability of bicuspid aortic valve with three loci on chromosomes 18q, 5q, and 13q, which likely contain genes responsible for bicuspid aortic valve.37 The mode of inheritance of other valvular diseases is unclear. No inheritability or inheritance studies have been reported for calcific aortic valve disease. In smaller genetic studies, a small number of candidate genes such as VDR, APOE, APOB, IL-10, and ESR1 have been identified as possibly playing a role in aortic valve disease. A recent breakthrough in the genetics of aortic valve disease was the identification of the Notch1 signaling pathway, involved in embryonic patterning, and that is highly expressed within the developing embryonic aortic valve. Notch1 is a repressor of Runx2, a critical regulator of osteoblast development. This supports the concept that a developmental program might be reactivated in disease processes.37
The rate of progression is, on average, a decrease in aortic valve area (AVA) of 0.1 cm2/year, and the peak instantaneous gradient increases by 10 mm Hg/year.45 The rate of progression of AS in men older than 60 is faster than in women, and it is faster in women older than 75 than in women 60 to 74 years old.46 Treatment with hemodialysis, the use of calcium supplements, and increased serum creatinine levels correlated with rapid progression of AS.47 Plasma brain natriuretic peptide, produced to a large extent by the ventricles, and the N-terminal part of the propeptide may serve as early markers of left ventricular hypertrophy (LVH), whereas atrial natriuretic peptide and the N-terminal part of the propeptide reflect atrial pressure increase.48 Repeated measurements of this marker may reflect information on the stage of AS and its hemodynamic impact.49
Angina, syncope, and congestive heart failure (CHF) are the classic symptoms of the disease, and their appearance is of serious prognostic significance because postmortem studies indicate that symptomatic AS is associated with a life expectancy of only 2 to 5 years.50–52 These early natural history studies were completed before the availability of cardiac catheterization studies, and some patients, although symptomatic, may have had objectively less severe degrees of stenosis. The “hemodynamically insignificant murmur of aortic stenosis” seen so often in cardiologic consultations does not portend such dire consequences, although it is sometimes difficult to correlate AS severity with clinical symptoms. There is evidence that patients with moderate AS (i.e., valve areas of 0.7 to 1.2 cm2) are also at increased risk for the development of complications, with the appearance of symptoms further increasing their risk.53 According to recent American College of Cardiology (ACC) and American Heart Association (AHA) guidelines, peak velocity greater than 4 m/sec, a mean gradient greater than 40 mm Hg, and a valve area less than 1.0 cm2 are considered hemodynamically severe AS.54 Aortic valve surgery should be performed promptly in symptomatic patients. In asymptomatic patients, high aortic valve calcium content and a positive exercise test are features that suggest a benefit from early aortic valve replacement (AVR).
An interesting question is whether the natural history of AS has changed significantly in recent years.55 The question is prompted by two trends. The first, at least in North America, is related to the steadily diminishing number of patients with rheumatic disease. Today, AS is essentially the bicuspid, the calcific, or what has been traditionally referred to as the senile variety. The second trend is related to the senile attribution. People are living longer, particularly those with heart disease. The typical patient with AS is older and much more likely to have other significant medical problems, including major coexisting cardiac disease, most often CAD.
Angina is a frequent and classic symptom of the disease, occurring in approximately two thirds of patients with critical AS, and about half of symptomatic patients are found to have anatomically significant CAD.56–58 However, a controversy persists about the incidence of CAD in patients with AS who do not have angina, and the controversy probably reflects this underlying sense of a change in the natural history of the disease. Some studies report that the absence of angina virtually excludes the possibility of atherosclerotic heart disease.59,60 In contrast, a 25% incidence rate of angiographically significant (> 70% obstruction) coronary occlusions was described in angina-free patients, most of whom had single-vessel disease.61 However, a much larger study found that 14% of patients with triple-vessel or left main CAD and AS presented without angina.62 Underscoring the continued lack of a consensus on this topic is the finding of a review that the reported incidence rates of significant CAD in asymptomatic patients range from 0% to 33%.63 Identification of asymptomatic patients is important because although coexistent, untreated (i.e., unbypassed) CAD has a detrimental effect on early and late survival after AVR, concomitant CABG with AVR does not increase perioperative mortality.64–67 It appears that a substantial number of patients with coexisting CAD may have symptoms other than chest pain.68 However, there do not appear to be other significant differences in the clinical or hemodynamic findings between patients with normal coronary arteries and those with coexistent coronary stenoses.
If the traditional natural history of AS is perhaps more ominous in the elderly patient with likely coexistent CAD, is there any rationale for prophylactic AVR? With steady improvement in surgical results, this question has been raised, although rigorous studies still suggest that asymptomatic patients with hemodynamically significant disease face a very low risk for sudden death before the onset of symptoms.69–71 As if by way of settling this particular question, an editorial by Braunwald72 stated that there was no role for prophylactic AVR, and that “operative treatment is the most common cause of sudden death in asymptomatic patients with aortic stenosis.” These patients warrant careful follow-up, and the case against prophylactic surgical intervention is further supported by studies showing that ventricular function is preserved and myocardial hypertrophy regresses after successful valve replacement.73–75
It is probably never too late to operate on patients with symptomatic AS.76,77 First, unlike AR, most symptomatic patients undergo valve replacement when left ventricular function is still normal. Second, even when impaired left ventricular function develops in AS, the relief of pressure overload almost always restores normal function or at least produces considerable improvement. Morbidity rates, mortality rates, and clinical results are favorable even in the oldest surgical candidates.78,79 Recent advances in operative techniques and perioperative management have contributed to excellent results after AVR in patients 80 years of age or older, with minimal incremental postoperative morbidity.80 The principal postoperative complication is respiratory failure.
The preoperative assessment of AS with Doppler echocardiography includes measurement of the AVA and the transvalvular pressure gradient.81,82 The latter is calculated from the Doppler-quantified transvalvular velocity of blood flow, which is increased in the presence of AS. This maximal velocity (v) is then inserted into the modified Bernoulli equation to determine the pressure gradient (PG) between the LV and the aorta (see Figure 19-2):
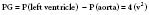
The pressure gradient is the maximal difference between the LV and aortic pressures that occurs during ventricular systole.83,84 This maximal instantaneous gradient is not the same as the peak-to-peak gradient determined by cardiac catheterization. The peak-to-peak gradient is determined by separate measurements of events that are not synchronous in real time. Of more practical interest is the fact that the best estimate of obstruction severity, as determined from pressure data alone, is the mean systolic gradient, which is calculated online by Doppler equipment85 (see Chapters 3 and 12).
The Doppler-calculated gradient is subject to the same flow limitation as invasively calculated gradients. Best understood by considering the extreme, this means that a patient with true end-stage AS would exhibit a relatively low calculated gradient because of minimal flow across a critically narrowed valve. In part because of this flow dependency, pressure gradients determined invasively or by Doppler echocardiography correctly classify AS severity in less than 50% of cases compared with estimates of AVA.86 However, the latter also can be determined by Doppler echocardiographic techniques. The preferred method requires only two Doppler-generated velocities: those proximal and those distal to the stenotic valve. These values are inserted into the continuity equation, which relates the respective velocities and cross-sectional areas proximal and distal to a stenotic area (see Figure 19-3). Specifically, the equation states:
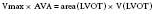
in which AVA is the aortic valve area, V is the volume, and LVOT is the left ventricular outflow tract. Several studies have demonstrated the reliability of these Doppler-determined valve areas.83–86
Although advances in Doppler technology allow completely noninvasive evaluation of a large number of patients, coronary angiography is probably indicated in all patients older than 50 years who are demonstrated to have significant AS. Angiography and, if indicated, CABG may improve the long-term outlook for patients with CAD who undergo AVR.62 Angiography also can identify a smaller number of patients whose CAD alone would warrant CABG.

Pathophysiology
The normal AVA is 2.6 to 3.5 cm2, with hemodynamically significant obstruction usually occurring at cross-sectional valve areas of 1 cm2 or less. Generally accepted criteria for critical outflow obstruction include a systolic pressure gradient greater than 50 mm Hg, with a normal cardiac output, and an AVA of less than 0.4 cm2. In view of the ominous natural history of severe AS (AVA < 0.7 cm2),55,87 symptomatic patients with this degree of AS are generally referred for immediate AVR.88 The Hakki equation is a simplification of the Gorlin equation to calculate the AVA based on the cardiac output (CO) and the peak pressure gradient (PG) across the valve.
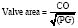
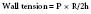
in which P is the intraventricular pressure, R is the inner radius, and h is the wall thickness.
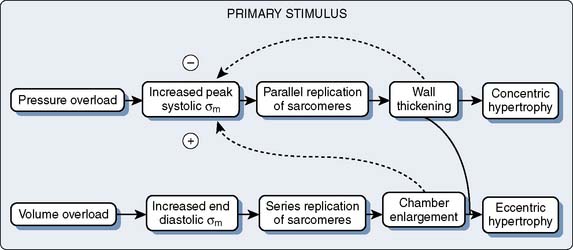
This increase of wall tension is believed to be the direct stimulus for the further parallel replication of sarcomeres, which produces the concentrically hypertrophied ventricle characteristic of chronic pressure overload.89,90 The consequences of this LVH include alterations in diastolic compliance, potential imbalances in the myocardial oxygen supply and demand relationship, and possible deterioration of the intrinsic contractile performance of the myocardium.
Figure 19-13 shows a typical pressure-volume loop for a patient with AS. Two differences from the normal curve are immediately apparent. First, the peak pressure generated during systole is much greater because of the high transvalvular pressure gradient. Second, the slope of the diastolic limb is steeper, reflecting the reduced left ventricular diastolic compliance that is associated with the increase in chamber thickness.13 Clinically, this means that small changes in diastolic volume produce relatively large increases in ventricular filling pressure.
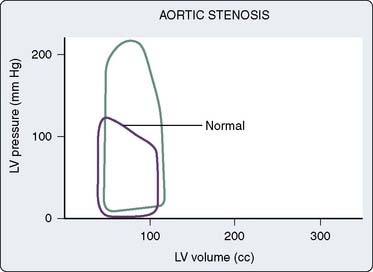
Figure 19-13 Pressure-volume loop in aortic stenosis.
(From Jackson JM, Thomas SJ, Lowenstein E: Anesthetic management of patients with valvular heart disease. Semin Anesth 1:239, 1982.)
This increased chamber stiffness places a premium on the contribution of atrial systole to ventricular filling, which in patients with AS may account for up to 40% of the LVEDV, rather than the 15% to 20% characteristic of the normal LV. Echocardiographic and radionuclide studies have documented that diastolic filling and ventricular relaxation are abnormal in patients with hypertrophy from a variety of causes, with significant prolongation of the isovolumic relaxation period being the most characteristic finding.91–94 This necessarily compromises the duration and amount of filling achieved during the early rapid diastolic filling phase and increases the relative contribution of atrial contraction to overall diastolic filling (Figure 19-14). A much greater mean left atrial (LA) pressure is necessary to distend the LV in the absence of the sinus mechanism. One treatment of junctional rhythm is volume infusion.
The systolic limb of the pressure-volume loop shows preservation of pump function, as evidenced by maintenance of the SV and EF (see Figure 19-13). It is likely that use of preload reserve and adequate LVH are the principal compensatory mechanisms that maintain forward flow. Clinical studies have confirmed that ejection performance is preserved at the expense of myocardial hypertrophy, and the adequacy of the hypertrophic response has been related to the degree to which it achieves normalization of wall stress, in accordance with the Laplace relation.95–97 LVH can be viewed as a compensatory physiologic response that completes a negative feedback loop (Figure 19-15). It is possible, however, that severe afterload stress and proportionately massive LVH could decrease subendocardial perfusion and superimpose a component of ischemic contractile dysfunction.
In patients with AS, LVH develops as a result of the increase in pressure load. The development of LVH and its regression after therapeutic interventions are accompanied by changes in the cardiac extracellular matrix guided by an increase in ECM1 gene expression during LVH and complete regression after complete correction.98
Systemic hypertension and AS represent an increase in afterload to the LV, and each contributes to left ventricular remodeling and LVH. In a large series of 193 patients with AS, 62 of whom were hypertensive, symptoms manifested with larger AVAs and lower stroke work loss.99 Patterns of left ventricular remodeling (i.e., concentric vs. eccentric remodeling and hypertrophy) were not different between hypertensive and normotensive patients. Left ventricular mass decreased by 23% 1 year after AVR, back into the normal range, in patients with normal preoperative ventricular function; diastolic function improved concomitantly. Improvements in myocardial blood flow and coronary vasodilator reserve after AVRs result from reduced extravascular compression and increased diastolic perfusion time.100–102
Ejection phase indices of contractile function are abnormal in many patients with AS.103,104 However, indices of contractile function, which are exquisitely sensitive to afterload, are inherently unreliable for quantitating the inotropic state in a disease such as AS, in which the essence of the hemodynamic insult is the severely increased ventricular afterload. This in no way excludes the possibility that a subset of patients also may experience some intrinsic depression of myocardial contractility. For example, patients with AS may be particularly at risk for superimposed, ischemic ventricular dysfunction, but this possibility can be assessed only by the application of load-insensitive measurements of contractile function.
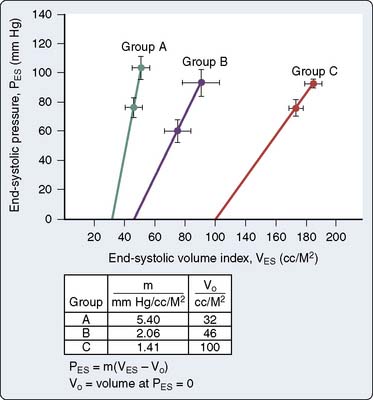
In most patients, however, load-insensitive indices of contractility (i.e., end-systolic stress-diameter determinations) are virtually identical before and after the development of hypertrophy, suggesting that the increase in chamber thickness compensates for the afterload stress, and myocardial contractility is, therefore, normal, albeit appropriate for the higher afterload. Figure 19-16 illustrates the adaptation of the LV to chronic pressure overload. Figure 19-16A shows the pressure-volume relation before (loop A) and after (loop B) the development of concentric hypertrophy. The linear ESPVR is shifted up and to the left after concentric hypertrophy occurs. However, the ESPVR may not be as accurate or load insensitive in assessing contractility at the extremes of afterload. Figure 19-16B shows normalization of this apparently supranormal contractility, when wall stress (rather than pressure) is used as a more exact measure of ventricular afterload. Concentric hypertrophy normalizes wall stress, and contractility remains unchanged. A decline in any index of myocardial contractility in patients with AS may represent relatively inadequate hypertrophy for the degree of wall stress, some intrinsic depression of contractility, or a combination of these two factors.56,105,106 The fact that most patients have normal contractility is the reason for the usually favorable response to AVR.
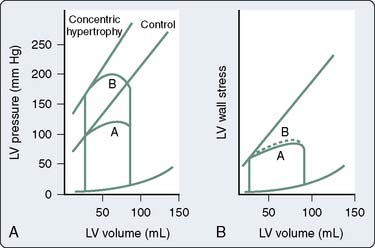
Figure 19-16 Adaptation to pressure overload.
(From Ross J Jr: Afterload mismatch in aortic and mitral valve disease: Implications for surgical therapy. J Am Coll Cardiol 5:811, 1985.)
On the supply side, the greater LVEDP of the poorly compliant ventricle inevitably narrows the diastolic coronary perfusion pressure (CPP) gradient. With severe outflow obstruction, decreases in SV and resultant systemic hypotension may critically compromise coronary perfusion. A vicious cycle may develop because ischemia-induced abnormalities of diastolic relaxation can aggravate the compliance problem and further narrow the CPP gradient.107 This sets the stage for ischemic contractile dysfunction, additional decreases in SV, and worsening hypotension.
Cardiac hypertrophy also is associated with structural abnormalities of the coronary circulation (Figure 19-17).108,109 Animal models have documented that epicardial coronary vessels do not enlarge proportionately when the LV is subjected to a chronic pressure load.110,111 LVH accompanying chronic hypertension is associated with an increased wall lumen ratio of the coronary arterioles, a change that limits vessel dilation and augments active constriction.112,113 Animal models of pressure-induced myocardial hypertrophy also have shown a decrease in the capillary density of about 20% to 30%.108,110,112–116 There is considerable evidence that in LVH caused by pressure overload, a reduction in the coronary vascular reserve may underlie episodes of myocardial ischemia117,118 (see Chapter 6).
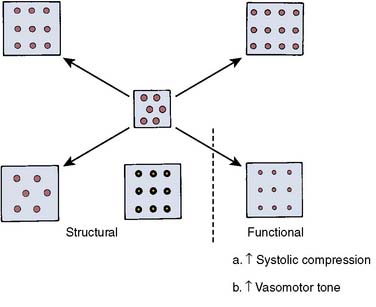
Figure 19-17 Possible coronary circulatory changes accompanying ventricular hypertrophy.
(From Marcus ML: Effects of cardiac hypertrophy on the coronary circulation. In Marcus ML [ed]: The coronary circulation in health and disease. New York: McGraw-Hill, 1983.)
Echocardiographic and hemodynamic assessments of the myocardial oxygen supply and demand ratio are not significantly different in the presence or absence of angina in patients with AS.119 However, considerable data indicate that dynamic factors relative to oxygen supply may be crucial to the pathogenesis of angina in these patients.120 Several clinical studies have documented a decrease in coronary vascular reserve in adult and pediatric patients with significant LVH or right ventricular hypertrophy (RVH).121–123 It is further postulated that repeated episodes of subendocardial ischemia may contribute to the development of subendocardial fibrosis, a component of ischemic contractile dysfunction.108
It is possible that myocardial ischemia underlies the impaired ventricular relaxation that has been documented in patients with myocardial hypertrophy because of a variety of causes.91,93,94 Prolongation of ventricular relaxation and resultant diastolic dysfunction (i.e., poor compliance) are probably universal features of myocardial ischemia in a variety of clinical settings.124–128 Experimental data suggest that ischemia-induced impairment of calcium sequestration by the sarcoplasmic reticulum may underlie the increased diastolic chamber stiffness.129 Prevention of ischemia-induced myoplasmic calcium overload may be the mechanism by which the calcium channel blockers are able to improve diastolic dysfunction in patients with CAD.130 These drugs also ameliorate ventricular relaxation and diastolic filling in patients with HCM, although the mechanism of action is controversial.91,131–133 The absence of diastolic filling abnormalities in patients with physiologic, as opposed to pathologic, hypertrophy also may reflect the relative likelihood of ischemia in the two conditions.134–136
In summary, the pathophysiologic response to chronic pressure overload is a complex one characterized by unique interaction among the divergent effects of hypertrophy on systolic and diastolic function. Mural thickening enhances systolic performance, maximizing the mechanical work performed while minimizing its metabolic cost. The price of this systolic efficiency is relatively inadequate growth of the coronary microcirculation, which contributes to relaxation abnormalities, diastolic dysfunction, and the potential for superimposition of ischemia-induced abnormalities of systolic dysfunction (Table 19-1).137 The potentially deleterious effects of LVH cannot be overemphasized. Even in the absence of AS, in which LVH can be viewed as a compensatory and potentially beneficial pathophysiologic response, LVH has been found to have an independent adverse effect on survival.138,139
| Beneficial Aspects | Detrimental Aspects |
|---|---|
| Increases ventricular work | Decreases ventricular diastolic distensibility |
| Normalizes wall stress | Impairs ventricular relaxation |
| Normalizes systolic shortening | Impairs coronary vasodilator reserve, leading to subendocardial ischemia |
From Lorell BH, Grossman W: Cardiac hypertrophy: The consequences for diastole. J Am Coll Cardiol 9:1189, 1987.

Difficulty of Low-Gradient, Low-Output Aortic Stenosis
A subset of patients with severe AS, left ventricular dysfunction, and low transvalvular gradient suffers a high operative mortality rate and poor prognosis.140 It is difficult to assess accurately the AVA in this low-flow, low-gradient AS because the calculated valve area is proportional to forward SV and because the Gorlin constant varies in low-flow states. Some patients with low-flow, low-gradient AS have a decreased AVA as a result of inadequate forward SV rather than anatomic stenosis. Surgical therapy is unlikely to benefit these patients because the underlying pathology is a weakly contractile myocardium. However, patients with severe anatomic AS may benefit from valve replacement despite the increased operative risk associated with the low-flow, low-gradient hemodynamic state.140 Guidelines from the ACC and AHA call for a dobutamine echocardiography evaluation to distinguish patients with fixed anatomic AS from those with flow-dependent AS with left ventricular dysfunction.141 Low-flow, low-gradient AS is defined as a mean gradient of less than 30 mm Hg and a calculated AVA less than 1.0 cm2.
Dobutamine echocardiography revealed three basic response patterns—fixed AS, relative AS, and absence of contractile reserve—in an initial study by deFilippi et al.142 In a series of 45 patients with low-flow, low-gradient AS, the 30-day operative mortality rate was 8% for patients with contractile reserve and 50% for those without contractile reserve.143 Dobutamine challenges during cardiac catheterization provided unique insights into low-flow, low-gradient AS, and the details are summarized in Figure 19-18.144 In Figure 19-18A, the transvalvular gradient and cardiac output increased, and the valve area did not change. Patients represented in Figure 19-18B increased their CO with little or no change in gradient, and the calculated valve area increased slightly. This group still benefited from surgery. The third group of patients had no contractile reserve because CO did not increase with dobutamine, and the transvalvular gradient decreased (see Figure 19-18C). Dobutamine infusion in the cardiac catheterization laboratory appears to be helpful in identifying patients with low-flow, low-gradient AS who have a truly fixed anatomic stenosis that may benefit from valve replacement. The findings of these studies also emphasize that contractile reserve is an important prognostic indicator in these patients and dobutamine challenge may help select patients for valve replacement. It appears that patients with contractile reserve and fixed AS have a relatively good prognosis with valve replacement. Left ventricular contractile reserve appears to be a critical variable for prognosis. Recent studies have focused on AS with low transvalvular gradients and normal ventricular EF. The pathophysiology of the low transvalvular gradient has been explained by decreased EDV caused by excessive ventricular hypertrophy accompanied by increased SVR. In a recent series of patients with severe AS (AVA < 0.8 cm2) with ventricular dysfunction (EF < 35%) and/or a low transvalvular gradient (< 30 mm Hg), mortality predictors were advanced age, low EF, renal insufficiency, and lack of AR. Regardless of ventricular function, patients who underwent AVR had a significantly better survival.36
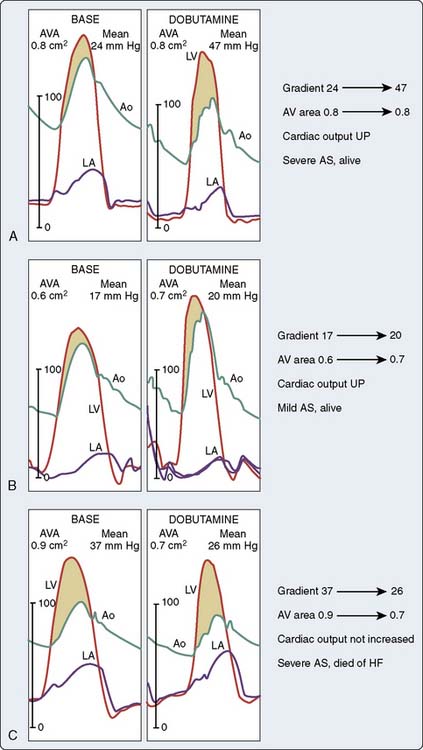
Figure 19-18 Hemodynamic tracings from three patients representing three different responses to dobutamine.
(Adapted from Nishimura RA, Grantham JA, Connolly HM, et al: Low-output, low-gradient aortic stenosis in patients with depressed left ventricular systolic function: The clinical utility of the dobutamine challenge in the catheterization laboratory. Circulation 106:809, 2002.)

Developments in the Hemodynamic Management of Critical Aortic Stenosis Patients
The use of vasodilators is traditionally contraindicated in patients with severe AS because cardiac output is relatively fixed across a narrowed orifice. Vasodilation reduces SVR without any possibility for a compensatory increase in CO and severe hypotension would typically result. This traditional paradigm recently was re-examined in patients with fixed, severe AS (area < 1.0 cm2) and left ventricular dysfunction (EF < 0.35).145 Nitroprusside was carefully titrated to maintain a mean arterial pressure of more than 60 mm Hg with concomitant hemodynamic monitoring. The cardiac index increased over 24 hours from a mean of 1.60 to 2.52 L/min/m2 with no changes in heart rate and mean arterial pressure (Figure 19-19). The pulmonary capillary wedge pressure (PCWP) and SVR decreased, whereas SV increased.
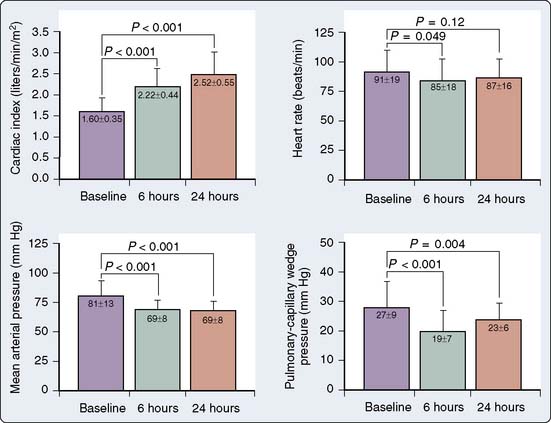
It appears that this treatment is effective to alleviate to some extent the left ventricular dysfunction component of the complete syndrome. Left ventricular dysfunction benefits derived from unloading and careful titration of the nitroprusside most likely allowed SVR to decrease without changes in mean arterial pressure. This titrated unloading may benefit patients with severe AS and left ventricular dysfunction, and it may serve as a temporary bridge to AVR or oral vasodilator therapy.145 It is unclear whether treatment with positive inotropic agents would produce similar effects with fewer risks. Adequate treatment of patients with AS, a depressed EF, and a low transvalvular gradient continues to pose a diagnostic and therapeutic challenge.146 The use of nitroprusside in AS in the absence of ventricular dysfunction may not be as effective and even be deleterious because of the prompt decrease in preload by nitroprusside.

Timing of Intervention
In asymptomatic patients with AS, it appears to be relatively safe to delay surgery until symptoms develop, but outcomes vary widely. The presence of moderate or severe valvular calcification along with a rapid increase in aortic-jet velocity identify patients with a very poor prognosis. These patients should be considered for early valve replacement rather than delaying until symptoms develop.40
Echocardiography and exercise testing may identify asymptomatic patients who are likely to benefit from surgery.147 In a study of 58 asymptomatic patients, 21 had symptoms for the first time during exercise testing.148 Guidelines for AVR in patients with AS are shown in Table 19-2.
TABLE 19–2 Recommendations for the Use of Aortic Valve Replacement in Patients with Aortic Stenosis
| Replacement Indicated | Replacement Possibly Indicated |
|---|---|
| Patients with severe aortic stenosis and any of its classic symptoms (e.g., angina, syncope, dyspnea) | Patients with moderate aortic stenosis who require coronary artery bypass surgery or surgery on the aorta or heart valves |
| Patients with severe aortic stenosis who are undergoing coronary artery bypass surgery | Asymptomatic patients with severe aortic stenosis and at least one of the following: ejection fraction of no more than 0.50, hemodynamic instability during exercise (e.g., hypotension), ventricular tachycardia; not indicated to prevent sudden death in asymptomatic patients who have none of the findings listed |
| Patients with severe aortic stenosis who are undergoing surgery on the aorta or other heart valves |
Adapted from the American Heart Association website (www.americanheart.org).
Functional outcome after AVR in patients older than 80 years is excellent, operative risk is limited, and late survival rates are good.149 In patients with severe left ventricular dysfunction and low transvalvular mean gradient, operative mortality is increased, but AVR was associated with improved functional status.150 Postoperative survival was best in younger patients and with larger prosthetic valves, whereas medium-term survival was related to improved postoperative functional class.150

Anesthetic Considerations
The described pathophysiologic principles dictate that anesthetic management be based on the avoidance of systemic hypotension, maintenance of sinus rhythm and an adequate intravascular volume, and awareness of the potential for myocardial ischemia (Box 19-1). In the absence of CHF, adequate premedication may reduce the likelihood of undue preoperative excitement, tachycardia, and the resultant potential for exacerbating myocardial ischemia and the transvalvular pressure gradient. In patients with truly critical outflow tract obstruction, however, heavy premedication with an exaggerated venodilatory response can reduce the appropriately increased LVEDV (and LVEDP) needed to overcome the systolic pressure gradient. In these patients, in particular, the additional precaution of administering supplementary oxygen may provide worthwhile insurance against the possibility of a similarly pronounced response to the sedative effects of the premedicant.
Intraoperative monitoring should include a standard five-lead electrocardiographic (ECG) system, including a V5 lead, because of the LV’s vulnerability to ischemia. A practical constraint in terms of interpretation is that these patients usually exhibit ECG changes because of preoperative LVH. The associated ST-segment abnormalities (i.e., strain pattern) may be indistinguishable from or at least very similar to those of myocardial ischemia, making the intraoperative interpretation difficult. Lead II and possibly an esophageal ECG should be readily obtainable because they may be useful for assessing the P-wave changes in the event of supraventricular arrhythmias (see Chapter 25).
Hemodynamic monitoring is controversial, and few prospective data are available on which to base an enlightened clinical decision. The central venous pressure (CVP) is a particularly poor estimate of left ventricular filling when left ventricular compliance is reduced. A normal CVP can significantly underestimate the LVEDP or PCWP. The principal risks, although minimal, of using a pulmonary artery catheter (PAC) in the patient with AS are arrhythmia-induced hypotension and ischemia. Loss of synchronous atrial contraction or a supraventricular tachyarrhythmia can compromise diastolic filling of the poorly compliant LV, resulting in hypotension and the potential for rapid hemodynamic deterioration. The threat of catheter-induced arrhythmias is significant for the patient with AS. However, accepting a low-normal CVP as evidence of good ventricular function can lead to similarly catastrophic underfilling of the LV on the basis of insufficient replenishment of surgical blood loss. To some extent, even the PCWP can underestimate the LVEDP (and LVEDV) when ventricular compliance is markedly reduced. Placement of a PAC also allows for measurement of CO, derived hemodynamic parameters, mixed venous oxygen saturation (Svo2), and possible transvenous pacing (see Chapter 14).
Patients with symptomatic AS are usually encountered only in the setting of cardiovascular surgery because of their ominous prognosis without AVR. Few studies have specifically addressed the response of these patients to the standard intravenous and inhalation induction agents; however, the responses to narcotic151,152 and nonnarcotic153,154 intravenous agents are apparently not dissimilar from those of patients with other forms of VHD. The principal benefit of a narcotic induction is the assurance of an adequate depth of anesthesia during intubation, which reliably blunts potentially deleterious reflex sympathetic responses capable of precipitating tachycardia and ischemia.
Many clinicians also prefer a pure narcotic technique for maintenance. The negative inotropy of the inhalation anesthetics is a theoretical disadvantage for a myocardium faced with the challenge of overcoming outflow tract obstruction. A more clinically relevant drawback may be the increased risk for arrhythmia-induced hypotension, particularly that associated with nodal rhythm and resultant loss of the atrium’s critical contribution to filling of the hypertrophied ventricle155,156 (see Chapter 9).
Occasionally, surgical stimulation elicits a hypertensive response despite the impedance posed by the stenotic valve and a seemingly adequate depth of narcotic anesthesia. In such patients, a judicious trial of low concentrations of an inhalation agent, used purely for control of hypertension, may prove efficacious. The ability to concurrently monitor CO is useful in this situation. The temptation to control intraoperative hypertension with vasodilators should be resisted in most cases. Given the risk for ischemia, nitroglycerin seems to be a particularly attractive drug. Its effectiveness in relieving subendocardial ischemia in patients with AS is controversial157,158; however, there is always the risk for even transient episodes of “overshoot.” The hypertrophied ventricle’s critical dependence on an adequate CPP may be unforgiving of even a momentary dip in the systemic arterial pressure.
Intraoperative hypotension, regardless of the primary cause, should be treated immediately and aggressively with a direct α-adrenergic agonist such as phenylephrine.159 The goal should be to immediately restore the CPP and then to address the underlying problem (e.g., hypovolemia, arrhythmia). After the arterial pressure responds, treatment of the precipitating event should be equally aggressive, but rapid transfusion or cardioversion should not delay the administration of a direct-acting vasoconstrictor. Patients with severe AS in whom objective signs of myocardial ischemia persist despite restoration of the blood pressure should be treated extremely aggressively. This may mean the immediate use of an inotropic agent or simply accelerating the institution of cardiopulmonary bypass (CPB).
Intraoperative myocardial ischemic injury in the patient with AS has been appreciated for some time, and there are several theories regarding its pathogenesis. The potential vulnerability of the hypertrophied myocardium to ischemic damage was initially speculated on by cardiac surgeons who described the phenomenon of irreversible ischemic contracture—what they called the stone heart—occurring after AVR in a group of patients with severe LVH.160 The anatomic and hemodynamic bases for an unfavorable myocardial oxygen supply and demand relation have already been considered, but the possibility that these patients might be experiencing prebypass ischemic injury remains a plausible, although unproven, hypothesis.
Most attention has been focused on the potential for irreversible cellular damage occurring during the period of ischemic cardiac arrest. Although there is a consensus that improved myocardial preservation has been crucial in reducing the mortality rate after CABG, there is evidence that current cardioplegic techniques may provide suboptimal protection for patients with VHD.161,162 Specifically, there is ultrastructural evidence (e.g., intracellular or mitochondrial edema) suggesting that the hypertrophied myocardium is uniquely susceptible to ischemic damage during the aortic cross-clamp interval, despite the presumed protection. Although these changes are observed in patients whose clinical course is seemingly unremarkable, such ischemic cellular damage also may underlie the frequent occurrence of postoperative low-output syndromes and the greater mortality rates associated with valvular operations.163
Although a detailed consideration of myocardial preservation is beyond the scope of this chapter (see Chapters 28 and 29), certain aspects are peculiar to the operative management of patients with AS, and it is likely that improved cardioplegic techniques will play an important role in reducing operative morbidity and mortality rates. Because the operation requires an ascending aortotomy, many surgeons routinely use direct coronary ostial cannulation for the delivery of the cardioplegic solution. Problems associated with this approach are uncommon but include the inherent hazard of injury to the coronary ostia; cannula-induced late stenosis of the left main coronary artery also has been described.164 Coronary ostial delivery requires interrupting the surgical procedure for subsequent reinfusions of the cardioplegic solution. These problems are obviated by the use of cardioplegia delivery by the coronary sinus. The retrograde technique commonly is used in conjunction with an initial dose of antegrade cardioplegia; the latter provides for a more immediate electromechanical arrest.165–167

Noncardiac Surgery in the Patient with Aortic Stenosis
Patients with AS who undergo noncardiac surgery were identified by Goldman et al148 as being at increased risk for cardiac complications, including myocardial infarction, CHF, and supraventricular tachyarrhythmias. Likewise, the ACC Task Force on perioperative evaluation of the cardiac patient identified patients with severe or symptomatic AS as being at increased risk for serious perioperative cardiac morbidity.168 It stated that symptomatic stenotic lesions are associated with the risk for perioperative severe CHF or shock and often require percutaneous valvotomy or valve replacement before noncardiac surgery to reduce cardiac risk. With the overall aging of the surgical population and given a peak incidence of significant AS in the fifth and sixth decades, it is likely that these patients will be encountered with increasing frequency in the setting of noncardiac surgery. Given the natural history of the disease and following the conservative ACC guidelines cited previously, one approach is to recommend immediate but elective AVR to all AS patients, before any noncardiac procedures. Although there is a rationale for this “shotgun” solution, a variety of ethical, practical, and economic constraints argue for a more selective approach.
A common clinical problem is the elderly, asymptomatic patient, with a harsh systolic ejection murmur, who is scheduled for noncardiac surgery. Risk assessment for the asymptomatic patient is challenging because the prognosis in the absence of AVR may be quite benign. However, symptoms correlate poorly with AS severity, which may by itself portend a more ominous natural history. In these patients, the use of a two-dimensional (2D) Doppler echocardiographic examination allows for the noninvasive assessment of the severity of stenosis57 and for some quantification of contractile function (see Chapters 1, 2, and 12). Patients with moderate AS have a greater short-term incidence of cardiovascular complications, and this risk is further increased by the presence of symptoms or objective evidence of contractile impairment.53 Armed with the echocardiographic evaluation (e.g., stenosis severity, contractile state), the clinician can make an initial assessment of relative risk. Depending on the overall clinical picture, some of these patients may warrant immediate elective AVR, whereas in others, it may be more appropriate to proceed with the noncardiac operation, with hemodynamic monitoring as dictated by the echocardiographic data and the nature of the surgery. A reality of the clinical setting is that the overall picture often seems to dictate that the anesthesiologist should proceed with the originally planned operation, albeit at increased risk for perioperative cardiac morbidity.
Hypertrophic cardiomyopathy

Hypertrophic Obstructive Cardiomyopathy
Hypertrophic obstructive cardiomyopathy is a relatively common genetic malformation of the heart with a prevalence of approximately 1 in 500 (see Chapter 22). The hypertrophy initially develops in the septum and extends to the free walls, often giving a picture of concentric hypertrophy. Asymmetric septal hypertrophy leads to a variable pressure gradient between the apical left ventricular chamber and the LVOT. The LVOT obstruction leads to increases in left ventricular pressure, which fuels a vicious cycle of further hypertrophy and increased LVOT obstruction.169 Various treatment modalities include β-adrenoceptor antagonists, calcium channel blockers, and surgical myectomy of the septum. For more than 40 years, the traditional standard treatment has been the ventricular septal myotomy-myomectomy of Morrow, in which a small amount of muscle from the subaortic septum is resected.170 Two new treatment modalities have gained popularity in recent years: dual-chamber pacing and septal reduction (ablation) therapy with ethanol.
Dual-chamber DDD pacing is based on the observation that excitation of the septum of the LV contracts it away from the apposing wall, which may reduce the LVOT gradient. The precise mechanism by which dual-chamber pacing decreases LVOT gradient or improves symptoms is uncertain. Possible mechanisms are asynchronous ventricular activation, paradoxic septal wall motion, a negative inotropic effect, an increase in ESV, decreased systolic anterior leaflet motion, altered myocardial perfusion, and regression of LVH.171 The AV interval must be sufficiently short to guarantee early right ventricular apical activation without conduction through the His-Purkinje system. Although some studies have shown a decrease in the LVOT gradient of 25%, there are still variable results with respect to improvements in exercise capacity and symptoms.171–174
Patients with severe, drug-resistant symptoms of CHF, angina, or syncope are considered for nonsurgical septal reduction therapy. In one series, patients with a resting gradient of more than 40 mm Hg or a dobutamine-induced gradient of more than 60 mm Hg were included.175 After coronary angiography to exclude significant CAD and after placement of temporary pacing wires, the LVOT gradient was measured at rest and during various interventions (e.g., Valsalva, postextrasystolic, isoproterenol, amyl nitrite). The coronary septal branches were cannulated with a small balloon-equipped catheter. The balloon was inflated to prevent overflow or spillage into the left anterior descending coronary artery. Depending on the septal branch size and area of septal hypertrophy, a dose of 2 to 5 mL ethanol was injected slowly through the balloon catheter lumen. The LVOT gradient decreased immediately in 85% to 90% of patients. Further decreases in gradient were reported after 6 months.171 The septal muscle mass was not decreased adequately through alcohol injections at the time of the intervention, but ventricular remodeling continued and led to sustained symptomatic improvement, most dramatically over the first 3 to 6 months.169 Exercise tolerance increased as well, but atrioventricular block, right bundle branch block, and left bundle branch block were frequent adverse consequences. Permanent heart blocks still occurred in 5% to 10% of cases.176

Clinical Features and Natural History
HCM is an uncommon familial disorder in which there is pronounced hypertrophy of histologically abnormal sarcomeres with characteristically disproportionate involvement of the interventricular septum.177–179 This disease has numerous pathologic and pathophysiologic features, which vary in their relative predominance in individual patients. A variety of other names have been applied to the disorder, including asymmetric septal hypertrophy, muscular subaortic stenosis, and idiopathic hypertrophic subaortic stenosis, with each emphasizing some aspect of the disease that may or may not be a prominent feature of its presentation in an individual patient. The cause and exact pattern of inheritance of the disease remain unknown, although it appears to be an autosomal dominant trait expressed with a high degree of penetrance179–181 (see Chapter 22).
Patients vary widely in their clinical presentation. The contribution of echocardiography to the diagnosis has unquestionably increased the number of asymptomatic patients who carry the diagnosis. Most patients with HCM are asymptomatic and have been seen by the echocardiographer because of relatives having clinical disease. Follow-up remains an important problem for cardiologists because sudden death or cardiac arrest may occur as the presenting symptom in slightly more than half of previously asymptomatic patients.182
Less dramatic frequently presenting complaints include dyspnea, angina, and syncope.179 The clinical picture is often similar to valvular AS. The symptoms may share a similar pathophysiologic basis (e.g., poor diastolic compliance) in the two conditions. The prognostic implications of clinical disease, however, are less certain for patients with HCM. Although cardiac arrest may be an unheralded event, other patients may have a stable pattern of angina or intermittent syncopal episodes for many years.183 Palpitations also are frequently described and may be related to a variety of underlying arrhythmias. Of patients studied with continuous ambulatory monitoring, ventricular arrhythmias occur in more than 75%, supraventricular tachycardias in 25%, and atrial fibrillation (AF) in 5% to 10%.184–186 The latter often precipitates clinical deterioration because of the dependence of the noncompliant LV on atrial systole for its filling.187
The natural history of patients with HCM also is extremely variable. These patients are all at risk for sudden death, although those with a family history of this problem form an especially high-risk group.188 Unfortunately, younger and previously asymptomatic patients with minimal subaortic gradients also may be at greater risk for sudden death because of their more frequent and vigorous physical activity.182,189 It is unknown whether vigorous physical activity in patients with HCM is an independent risk factor for sudden death, but this disease is the most frequent autopsy finding in previously healthy competitive athletes who die suddenly.190
Although patients referred to diagnostic centers for evaluation of HCM are usually young-to-middle-aged persons, a syndrome with similar clinical and echocardiographic findings has been identified in the elderly.93 Echocardiographically, these patients exhibit a similarly thickened and hypercontractile LV. Their most common symptoms are those of CHF and are thought to reflect severe reductions in the ventricular diastolic compliance. As in younger patients with classic HCM, there is marked systolic anterior motion (SAM) of the mitral apparatus. However, in HCM of the elderly, the SAM is seemingly caused or at least accentuated by a severe degree of mitral annular calcification, which results in a particularly small LVOT.191 Previously asymptomatic, these patients often experience the onset of progressively severe symptoms in the sixth decade of life, and the response to medical therapy, usually calcium channel-blocking drugs, has been poor.191 This lack of improvement with medical therapy may reflect the fact that mitral annular calcification plays a key role in producing a truly physical (i.e., less dynamic) subaortic obstruction. This also would explain the female predominance of the disease because mitral annular calcification is more common in elderly women. It is unclear whether this represents a true variant of classic HCM with late onset or it is perhaps etiologically linked to long-standing systemic hypertension, with secondary, unexplained, disproportionate involvement of the interventricular septum.93

Pathophysiology
In HCM, the principal pathophysiologic abnormality is myocardial hypertrophy. The hypertrophy is a primary event in these patients and occurs independently of outflow tract obstruction. Unlike in AS, the hypertrophy begets the pressure gradient, not the other way around. Histologically, the hypertrophy consists of myocardial fiber disarray, and, anatomically, there is usually disproportionate enlargement of the interventricular septum.192
Controversy exists concerning the intrinsic contractile strength of the myocardium in patients with HCM. Several studies have demonstrated normal or supranormal indices of systolic function in patients with this disease.193–195 Left ventricular ejection is rapid, with 80% of the SV being ejected very early during systole.193,196 This is true regardless of the presence, temporal location during systole, or magnitude of outflow tract obstruction. However, studies have shown that end-systolic stress is significantly reduced in relation to ESV (Figure 19-20).197,198 Reiteration of the Laplace formula for wall stress, P × R/2h, shows that, in HCM, the degree of primary hypertrophy (i.e., with increased wall thickness [h]) should minimize instantaneous left ventricular afterload. As a result, there is preservation, perhaps even elevation, of afterload-sensitive indices of systolic function, such as the EF.199 Myocardial hypertrophy, particularly to the extent that it accentuates subaortic obstruction, increases the ESPVR, widening the divergence between it and the more accurate (load-insensitive) ratio of end-systolic wall stress to ESV. High peak systolic pressures (i.e., subaortic obstruction), elevation of ejection phase indices, and low ESV (i.e., minimal afterload) may reflect uncontrolled hypertrophy of an abnormal and perhaps intrinsically depressed myocardium. Whether overall contractility is normal, supranormal, or impaired in a given patient, it is likely that there will be regional differences in a ventricle’s contractile function, which correlate with the histologic heterogeneity so characteristic of this disease.200
A consensus exists that the disease is characterized by a wide spectrum of the severity of obstruction. It is totally absent in some patients, may be variable in others, or may be critically severe. Its most distinctive qualities are its dynamic nature (depending on contractile state and loading conditions), its timing (begins early, peaks variably), and its subaortic location.193,201 Those accepting the phenomenon of physical subaortic obstruction believe that it arises from the hypertrophied septum’s encroachment on the systolic outflow tract, which is bounded anteriorly by the interventricular septum and posteriorly by the anterior leaflet of the mitral valve. In most patients with obstruction, exaggerated anterior (i.e., toward the septum) motion of the anterior mitral valve leaflet during systole accentuates the obstruction.202 The cause of this SAM is unclear. One possibility is that the mitral valve is pulled toward the septum by contraction of the papillary muscles, whose orientation is abnormal because of the hypertrophic process.203 Another theory is that vigorous contraction of the hypertrophied septum results in rapid acceleration of the blood through a simultaneously narrowed outflow tract. This could generate hydraulic forces consistent with a Venturi effect, whereby the anterior leaflet of the mitral valve would be drawn close to or within actual contact with the interventricular septum204 (Figure 19-21). This means that after the obstruction is triggered, the mitral valve leaflet is forced against the septum by the pressure difference across the orifice. However, the pressure difference further decreases orifice size and further increases the pressure difference in a time-dependent amplifying feedback loop.205 This analysis also is consistent with observations that the measured gradient is directly correlated with the duration of mitral-septal contact. Although still controversial,206–208 there appears to be good correlation between the degree of SAM and the magnitude of the pressure gradient.209,210 The SAM-septal contact also underlies the severe subaortic obstruction characteristic of HCM of the elderly, although the narrowing usually is more severe and the contribution of septal movement toward the mitral valve is usually greater.191
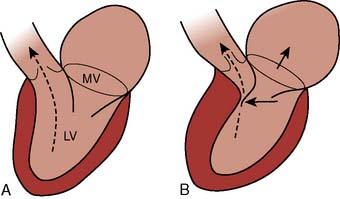
Figure 19-21 Proposed mechanism of systolic anterior motion in hypertrophic cardiomyopathy.
(From Wigle ED, Sasson Z, Henderson MA, et al: Hypertrophic cardiomyopathy: The importance of the site and the extent of hypertrophy. A review. Prog Cardiovasc Dis 28:1, 1985.)
The timing of SAM-septal contact also is important because the magnitude of the subaortic gradient is proportional to the fraction of forward flow ejected in the presence of anatomic obstruction.211 In general, large gradients occur as a result of early and prolonged SAM-septal contact, and small gradients arise from delayed and brief SAM-septal contact.204,209 In studies of patients with various degrees of subaortic obstruction, the proportion of flow ejected in the presence of a gradient has varied between 30% and 70%.209,212,213 Patients with evidence of systolic obstruction at rest have the longest intervals of systolic contact between the anterior leaflet of the mitral valve and the septum.214 However, if SAM-septal contact is restricted to late systole, a pressure gradient may occur on the basis of “cavity obliteration” in the absence of any functional obstruction to blood flow.215,216 In such patients, 95% of the SV may be ejected by the halfway point in systole, when SAM-septal contact is just beginning.193,209 Despite its seemingly critical role in producing outflow tract obstruction, SAM of the mitral valve is not a specific finding for HCM, and it may occur in patients with HCM who do not have obstruction.206,207,214
In addition to SAM, approximately two thirds of patients exhibit a constellation of structural malformations of the mitral valve.217 These malformations include increased leaflet area and elongation of the leaflets or anomalous papillary muscle insertion directly into the anterior mitral valve leaflet. HCM is not a disease process confined to cardiac muscle alone because these anatomic abnormalities of the mitral valve are unlikely to be acquired or secondary to mechanical factors.
Three basic mechanisms—increased contractility, decreased afterload, and decreased preload—exacerbate the degree of SAM-septal contact and produce the dynamic obstruction characteristic of patients with HCM. The common pathway is a reduction in ventricular volume (actively by increased contractility, directly or reflexly in response to vasodilation, or passively by reduced preload), which increases the proximity of the anterior mitral valve leaflet to the hypertrophied septum.179,183,218 Factors that usually impair contractile performance, such as myocardial depression, systemic vasoconstriction, and ventricular overdistention, characteristically improve systolic function in patients with HCM and outflow tract obstruction. Diagnostically, these paradoxes are exploited by quantifying the degree of subaortic obstruction after isoproterenol (e.g., increased inotropy, tachycardia, and decreased volume) and the Valsalva maneuver (e.g., decreased venous return and ventricular volume), both of which reliably elicit increases in the pressure gradient. In the operating room, catheter-induced ectopy or premature ventricular contractions resulting from cardiac manipulation also may transiently exacerbate the gradient by increased inotropy from postextrasystolic potentiation. Therapeutically, volume loading, myocardial depression, and vasoconstriction should minimize obstruction and augment forward flow.
Patients with HCM demonstrate critical derangements in diastolic function, which, although in some ways more subtle than the unique phenomenon of subaortic obstruction, may contribute equally to the challenge posed.219–222 These abnormalities include prolongation of the isovolumic relaxation time (i.e., aortic valve closure to mitral valve opening) and reduction in the peak velocity of left ventricular filling.91,195 Diastolic dysfunction exhibits the same heterogeneity characteristic of its systolic function, probably because its underlying cause is intrinsic to the primary myopathic process and not merely secondary to the associated chamber hypertrophy.223 Accordingly, diastolic filling abnormalities in HCM are largely independent of the magnitude of LVH and are seen even in patients with only mild, localized hypertrophy.20
Poor diastolic compliance is the most clinically apparent manifestation of the relaxation abnormalities. Left ventricular filling pressures are markedly increased despite enhanced systolic ejection and the normal or subnormal EDV. This reduced ventricular volume re-emphasizes the pivotal role played by the hypertrophied but intrinsically depressed myocardium. Reductions in afterload, mediated by hypertrophy, support the ventricle’s systolic performance, resulting in increased emptying and a small diastolic volume. However, hypertrophy also impairs relaxation, resulting in poor diastolic compliance and an increased ventricular filling pressure. The key point is that the high filling pressure does not reflect distention of a failing ventricle, even though stress-volume relations suggest that its contractility is intrinsically depressed (Figure 19-22 and Table 19-3). This disease is characterized by systolic and diastolic dysfunction.
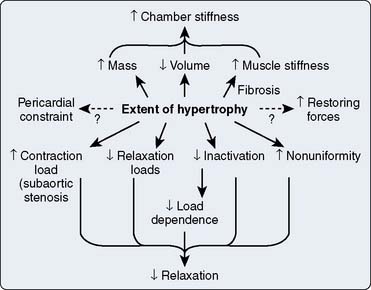
Figure 19-22 Proposed interactions between myocardial hypertrophy and other related properties of ventricular function.
(From Wigle ED, Sasson Z, Henderson MA, et al: Hypertrophic cardiomyopathy: The importance of the site and the extent of hypertrophy. A review. Prog Cardiovasc Dis 28:1, 1985.)
TABLE 19-3 Factors That Affect Left Ventricular Diastolic Filling in Hypertrophic Cardiomyopathy
| Chamber stiffness* |
| Relaxation |
| Loads |
| Contraction load |
| Subaortic stenosis |
| Relaxation loads |
| Late-systolic loading |
| End-systolic deformation (restoring forces) |
| Coronary filling |
| Ventricular filling |
| Inactivation |
| Myoplasmic calcium overload |
| Primary |
| Ischemic |
| Nonuniformity of load and inactivation in space and time† |
| Pericardial constraints and ventricular interaction |
| Effect of extent of hypertrophy on the preceding factors |
* Chamber stiffness = Myocardial mass × Myocardial stiffness/Ventricular volume.
† Nonuniformity of contraction and relaxation.
From Wigel ED, Wilansky S: Diastolic dysfunction in hypertrophic cardiomyopathy. Heart Fail 3:85, 1987.
As in patients with valvular AS, relatively high filling pressures reflect the LVEDV (i.e., degree of preload reserve) needed to overcome the outflow obstruction. Intervention with vasodilators is, therefore, inappropriate. The poor ventricular compliance also means that patients with HCM depend on a large intravascular volume and the maintenance of sinus rhythm for adequate diastolic filling. The atrial contribution to ventricular filling is even more important in HCM than in valvular AS, and it may approach 75% of total SV.132
Another similarity between HCM and valvular AS is that the combination of myocardial hypertrophy, with or without LVOT obstruction, may precipitate imbalances in the myocardial oxygen supply and demand relation. Angina-like discomfort is one of the classic symptoms of patients with HCM, and its pathogenesis has been attributed to increases in Mvo2, specifically the increased overall muscle mass and the high systolic wall tension generated by the ventricle’s ejection against the dynamic subaortic obstruction. However, as in patients with AS, there is also evidence of a compromise in myocardial oxygen supply.224,225 Patients with HCM suffer from several types of hypertrophy-related abnormalities of the coronary circulation; some are common to other conditions associated with pathologic hypertrophy (e.g., AS), and others are apparently unique to this distinctive form of hypertrophy.226 A reduced capillary density in the hypertrophied myocardium prevents flow from increasing in proportion to the increase in myocardial mass.227 It also is possible that there are primary HCM-associated abnormalities of the coronary circulation that are unrelated to myocardial hypertrophy.228 In addition to these coronary circulatory abnormalities, there is evidence of a metabolic derangement in the hypertrophied interventricular septum, whereby there is a decreased use of adequately delivered metabolic substrates.225
The reduction in coronary vascular reserve is similar to that observed in patients with valvular AS, in whom Mvo2 may be presumed to be uniformly increased.118 However, angina also occurs in patients with HCM in the absence of subaortic obstruction. Although basal Mvo2 is increased in proportion to the increased muscle mass, it is particularly interesting that clinical studies support a greater pathogenetic role for coronary circulatory abnormalities in producing ischemia in patients with nonobstructive hypertrophy.229
Hemodynamic derangements peculiar to the disease may aggravate the ventricle’s anatomic vulnerability to ischemia. The increased LVEDP for any LVEDV (i.e., poor compliance) inevitably narrows the diastolic CPP gradient. This may precipitate subendocardial ischemia in some patients with HCM, particularly those faced with the increased oxygen demand of overcoming late-systolic obstruction.219 There is evidence that hypertrophy-induced myocardial ischemia may underlie the diastolic dysfunction characteristic of HCM.230 As in patients with valvular AS, ischemia-induced abnormalities of diastolic calcium sequestration may further exacerbate relaxation abnormalities, initiating a vicious cycle (Figure 19-23).107,122,137,160,161,231
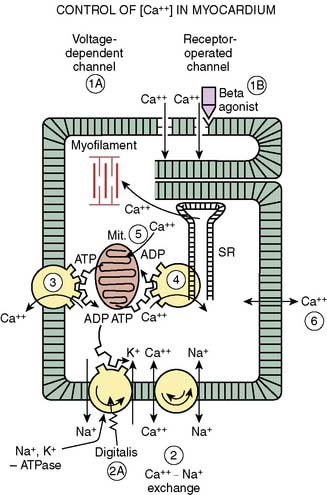
Figure 19-23 Regulation of cytoplasmic calcium.
The diastolic extrusion of calcium from the myofilaments occurs at several sites. At two of the most important sites, energy (adenosine triphosphate [ATP]) is required for the active reuptake of calcium by the sarcolemma (3) and the sarcoplasmic reticulum (SR; 4). ADP, adenosine diphosphate.
(From Braunwald E: Mechanisms of action of calcium channel blocking agents. N Engl J Med 307:1618, 1982.)
β-Blockers and calcium channel blockers form the basis of medical therapy for HCM. β-Blockade is most useful for preventing sympathetically mediated increases in the subaortic gradient and for the prevention of tachyarrhythmias, which also can exacerbate outflow obstruction. Disopyramide also has been used to reduce contractility and for its antiarrhythmic properties.232 Calcium channel blockers often prove clinically effective in patients with HCM regardless of the presence or absence of systolic obstruction.233 The mechanism of action involves improvement in diastolic relaxation, allowing an increase in LVEDV at a relatively lower LVEDP. The negative inotropy may attenuate the subaortic pressure gradient; however, in selected patients, the gradient may worsen because of pronounced and unpredictable degrees of vasodilation.234,235
Surgery—a septal myotomy or partial myomectomy by the aortic approach—is reserved for those patients who remain symptomatic despite maximal pharmacologic therapy. In a long-term retrospective study, the cumulative survival rate was significantly better in surgically than in pharmacologically treated patients.236 However, it is quite likely that pharmacologic therapy may be more appropriate for the patient with a dynamic component to their degree of subaortic obstruction.237 Further improvement in the clinical outcome of surgically treated patients may be achieved with the addition of verapamil, presumably reflecting a two-pronged attack on the systolic (myomectomy) and diastolic (verapamil) components of the disease. Enthusiasm continues for the therapeutic use of dual-chamber pacing in this disease, with some patients demonstrating reductions in their subaortic gradients.171–174,238,239 It was not an option for patients in AF.
Myomectomy usually results in significant symptomatic improvement, together with a reduction in the subaortic gradient.240 The intraoperative guidance and evaluation of the surgical result by an experienced echocardiographer are essential for the success of this procedure.241–243 In successful cases, the postmyomectomy study will show dramatic septal thinning, widening of the LVOT, and resolution of SAM and the LVOT color mosaic.
As in any disease with diverse diagnostic criteria, it is difficult to compare mortality rates among various series, but perioperative mortality rates for isolated septal myomectomy are less than 2%, although operative risks are greater when combined with other procedures.244,245 However, clinical studies of predominantly elderly patients reported mortality rates in excess of 15%. Complications other than heart block are infrequent, although more than one third of patients will experience new, clinically insignificant AR.246 It is unclear whether this new AR poses a long-term risk for these patients from the standpoint of CHF, but it probably warrants the continued use of endocarditis prophylaxis.

Anesthetic Considerations
Priorities in anesthetic management are to avoid aggravating the subaortic obstruction while remaining aware of the derangements in diastolic function that may be somewhat less amenable to direct pharmacologic manipulation (Box 19-2). It is, therefore, necessary to maintain an appropriate intravascular volume while avoiding direct or reflex increases in contractility or heart rate. The latter goals can be achieved with a deep level of general anesthesia (GA) and the associated direct myocardial depression. Regardless of the specific technique, the preservation of an adequate CPP, using vasoconstrictors rather than inotropes, is necessary to avoid myocardial ischemia. Heavy premedication is advisable with a view to avoiding anxiety-induced tachycardia or a reduction in ventricular filling. Chronic β-blockade or calcium-channel blockade, or both, should be continued up to and including the day of surgery. These medications should be restarted immediately after surgery, particularly in those patients undergoing noncardiac surgery.
Intraoperative monitoring should include an ECG system with the capability of monitoring a V5 lead and each of the six limb leads. Inspection of lead II may be helpful in the accurate diagnosis of supraventricular and junctional tachyarrhythmias, which may precipitate catastrophic hemodynamic deterioration because of the potential for inadequate ventricular filling resulting from the reduction in diastolic time or loss of the atrial contribution to ventricular filling. The latter may be crucial in patients with significantly reduced diastolic compliance.132 Abnormal Q waves have been described on the electrocardiograms in 20% to 50% of patients with HCM.179 These waves should not raise concern about a previous myocardial infarction; instead, they probably represent accentuation of normal septal depolarization or delay in depolarization of electrophysiologically abnormal cells.247 Some patients exhibit a short PR interval with initial slurring of the QRS complex, and they may be at increased risk for supraventricular tachyarrhythmias on the basis of preexcitation.248 Although the specific predisposing factors are unknown, patients with HCM are at increased risk for any type of arrhythmia in the operative setting.249
Given the pronounced abnormalities in left ventricular diastolic compliance, the CVP is likely to be an inaccurate guide to changes in left ventricular volume. However, a CVP catheter is extremely useful for the prompt administration of vasoactive drugs if they become necessary. As in valvular AS, the information provided by insertion of a PAC is worth the small arrhythmogenic risk. The potential for hypovolemia-induced exacerbation of outflow tract obstruction makes it crucial that the clinician have an accurate gauge of intravascular filling. The reduced diastolic compliance means that the PCWP will overestimate the patient’s true volume status, and a reasonable clinical objective is to maintain the PCWP in the high-normal to elevated range. A PAC with pacing capability is ideal because atrial overdrive pacing can effect immediate hemodynamic improvement in the event of episodes of junctional rhythm (see Chapters 14, 22, and 25). The absolute requirement of these patients for an adequate preload cannot be overemphasized because even abrupt positioning changes have resulted in acute hemodynamic deterioration, including acute pulmonary edema.250
Intraoperative arrhythmias require aggressive therapy. During cardiac surgery, insertion of the venous cannulae may precipitate atrial arrhythmias. Because the resultant hypotension may be severe, the surgeon should cannulate the aorta before any atrial manipulations. Supraventricular or junctional tachyarrhythmias may require immediate cardioversion if they precipitate catastrophic degrees of hypotension. Although verapamil is one drug of choice for paroxysmal atrial and junctional tachycardia, it has the potential of disastrously worsening the LVOT obstruction if it elicits excessive vasodilation or if it is used in the setting of severe hypotension.235 Cardioversion is preferable when the mean arterial pressure is already very low; the concurrent administration of phenylephrine also is advisable. This drug is almost always a low-risk, high-yield choice for the hypotensive patient with HCM. It augments perfusion, may ameliorate the pressure gradient, and often elicits a potentially beneficial vagal reflex when used to treat tachyarrhythmia-induced hypotension.
Several investigators have suggested that regional anesthesia, with its potential for accentuating peripheral vasodilation, may be relatively contraindicated in the management of patients with HCM.251,252 The theoretic constraints are similar to those for patients with valvular AS, and the same clinical caveats apply. If the vascular system is kept appropriately full and “tight” with vasopressors, it is reasonable to consider these techniques in the light of other clinical advantages they might offer the patient. Catheter techniques (e.g., continuous spinals, epidurals) may be preferable to the bolus administration of local anesthetics to achieve a finer degree of control of the anesthetic level.253 There unquestionably is the potential for a cascade of iatrogenic complications if sympatholytic hypotension is treated in a knee-jerk fashion with ephedrine, epinephrine, or a variety of other equally contraindicated β-adrenergic agonists.
Although echocardiography has undoubtedly contributed to an increased frequency of diagnosis of this disease, an occasional patient escapes detection by ultrasound examination in the course of the preoperative workup. When other objective data, including the physical examination, ECG results, and chest radiograph, show only nonspecific abnormalities, it is easy to disregard vague or atypical complaints of chest pain, presyncope, and dyspnea. This is particularly true when the symptoms might reasonably be attributed to the primary condition that brought the patient to surgery (e.g., CABG).254 Other identifiable populations are said to be at high risk for occult HCM. HCM of the elderly already has been discussed, and probably only a fraction of such patients arrive in the operating room with such a formal diagnosis. However, it is not uncommon for the anesthesiologist to encounter an elderly patient with long-standing systemic hypertension and unexplained episodes of pulmonary congestion for whom the primary physician has prescribed a digitalis preparation. Anesthesiologists occasionally may be presented with the opportunity to diagnose unsuspected HCM or one of its variants. Intraoperative events that could conceivably provoke or accentuate physiologic manifestations of the disease include hypovolemia, tachycardia, spontaneous or “manipulative” ectopy, and paradoxic responses to vasoactive drugs (e.g., inotropes, vasodilators, and vasoconstrictors) or anesthetic agents.254,255 Clues such as these allow the experienced and astute clinician to recognize the phenomenon of dynamic subaortic obstruction when it occurs in less obvious clinical settings.
Aortic regurgitation

Clinical Features and Natural History
AR may result from an abnormality of the valve itself, from bicuspid anatomy, have a rheumatic or infectious origin, or occur in association with any condition producing dilation of the aortic root and leaflet separation. Nonrheumatic valvular diseases commonly resulting in AR include infective endocarditis, trauma, and connective tissue disorders such as Marfan syndrome or cystic medionecrosis of the aortic valve.56 Aortic dissection from trauma, hypertension, or chronic degenerative processes also can result in dilatation of the root and functional incompetence (see Chapter 21).
The natural history of chronic AR is that of a long asymptomatic interval during which the valvular incompetence and secondary ventricular enlargement become progressively more severe.3,141 When symptoms do appear, they are usually those of CHF, and chest pain, if it occurs, is often nonexertional in origin. The life expectancy for patients with significant disease has historically been about 9 years, and in contrast with AS, the onset of symptoms because of AR does not portend an immediately ominous prognosis.256,257 In the absence of surgery, early recognition of AR and chronic use of vasodilators appear to be prolonging life span in this patient population.141,258
A relatively unique and problematic feature of chronic AR is that the severity of symptoms and their duration may correlate poorly with the degree of hemodynamic and contractile impairment.141,259 The issue in surgical decision making is that many patients can remain asymptomatic, during which time they are undergoing progressive deterioration in their myocardial contractility. Noninvasive diagnostic studies (e.g., radionuclide cineangiography and 2D and Doppler echocardiographic assessment of response to pharmacologic afterload stress) may facilitate the detection of early derangements in contractile function in relatively asymptomatic patients.260–262 These findings are important to the cardiologist when considering surgical referral (see Chapters 1 to 3) because patients with depressed preoperative left ventricular function have a greater perioperative mortality rate and are at increased risk for persistent postoperative heart failure (HF).96,263,264

Pathophysiology
Left ventricular volume overload is the pathognomonic feature of chronic AR. The degree of volume overload is determined by the magnitude of the regurgitant flow, which is related to the size of the regurgitant orifice, the aorta-ventricular pressure gradient, and the diastolic time. It has been suggested that the size of the regurgitant aortic orifice is constant and independent of changes in loading conditions.265 However, in other valvular lesions (e.g., AS, mitral regurgitation), the orifice size is not constant and is dependent on the hemodynamic state. There is some evidence, in an experimental model of acute AR, that the regurgitant orifice area can change with increases or decreases in the transvalvular pressure gradient.266 When AR occurs in the absence of valvular fibrosis or calcification (valve elasticity is preserved), reduction of regurgitant area may be one of the mechanisms of the beneficial effect of afterload reduction.
The hemodynamic effects of changes in heart rate are also less straightforward than might be assumed.267 Theoretically, tachycardia should maximize net forward flow by shortening the regurgitant diastolic time interval. This hypothesis, first offered by Corrigan in 1832, may be the pathophysiologic basis for the seemingly paradoxic observation that patients with AR may tolerate exercise despite having symptoms of pulmonary congestion at rest. Sympathetically mediated vasodilation, increased inotropy, and tachycardia may contribute to the increased CO achieved during isometric exercise in patients with AR.268 Chronically, similar, reflex-induced hemodynamic changes may contribute to the beneficial effects of arteriolar dilators.269 The peripheral vasculature directly affects regurgitant volume through reflex changes in heart rate; it also can alter volume loading by effects on the diastolic time. It plays a key role in the overall pathophysiology of AR270,271 (Figure 19-24).
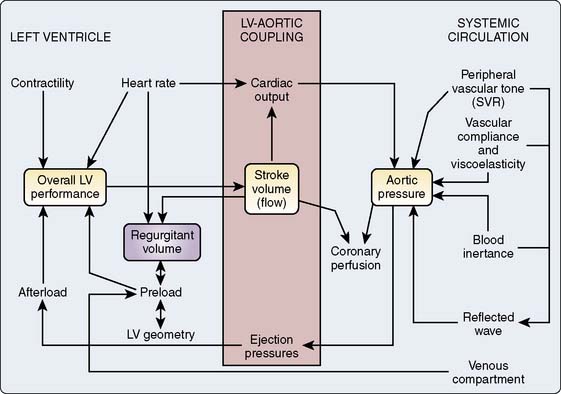
Despite these observations, the beneficial effects of shortened diastolic time alone may be less clear-cut. Angiographic study of subjects with chronic AR has shown that pacing-induced tachycardia can decrease the SV, LVEDV, and LVEDP and can increase cardiac output,272 but that the ratio of the regurgitant volume to the total SV may remain unchanged. This occurs because tachycardia shortens the diastolic period per stroke, but increases the number of strokes per minute, leaving the net diastolic time per minute relatively unchanged. Similar observation has been made with radionuclide studies that demonstrated that tachycardia can increase CO and decrease LVEDV; however, pulmonary arterial pressure (PAP) and PCWP may not change with increasing heart rate.273 This divergence between the effects of tachycardia on left ventricular volume (decreased) and filling pressures (unchanged) may reflect the pronounced rightward shift of the left ventricular pressure-volume loop that is characteristic of long-standing AR.
Chronically, AR results in a state of left ventricular volume and pressure overload. Progressive volume overloading from AR increases end-diastolic wall tension (i.e., ventricular afterload) and stimulates the serial replication of sarcomeres, producing a pattern of eccentric ventricular hypertrophy.97,264 This dilation of the ventricle, in accordance with Laplace’s law, also increases the systolic wall tension, stimulating some concentric hypertrophy. The result is normalization of the ratio of ventricular wall thickness to cavitary radius.97 This process of eccentric hypertrophy results in the greatest absolute degrees of cardiomegaly seen in valve disease. EDV may be three to four times normal, and very high cardiac outputs can be sustained.274
Figure 19-25 shows the pressure-volume loops for acute and chronic AR. In the chronic form, the diastolic pressure-volume curve is shifted far to the right. This permits a tremendous increase in LVEDV with minimal change in filling pressure, a property frequently described as high diastolic compliance.274 However, animal models of chronic left ventricular volume overloading instead suggest that the entire diastolic pressure-volume curve is shifted to the right (see Figure 19-8).11 This accounts for the apparent paradox of high ventricular volumes at relatively low filling pressures.
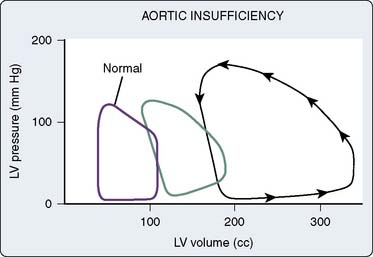
Figure 19-25 Pressure-volume loop in aortic regurgitation (AR).
Acute AR (middle loop); chronic AR (right loop). LV, left ventricular.
(Adapted from Jackson JM, Thomas SJ, Lowenstein E: Anesthetic management of patients with valvular heart disease. Semin Anesth 1:239, 1982.)
The parallel shifts of the entire curve relating diastolic pressure and volume are a manifestation of the physiologic process known as “creep.”4 This refers to the time-dependent increase in dimension that occurs as a result of an applied stress—in this case, volume overload. Because of this chronic adaptation, left ventricular filling pressures are in the low-to-normal range and are relatively insensitive to changes in intravascular volume. This is not true of greater filling pressures, for which increases are a reliable guide to volume overload and ventricular distention.
Because the increase in preload is compensated for by ventricular hypertrophy, CO is maintained by the Frank-Starling mechanism, and cardiac failure is not seen. This is true despite probable decreases in contractility.258 There is virtually no isovolumic diastolic phase because the ventricle is filling throughout diastole. The isovolumic phase of systole also is brief because of the low aortic diastolic pressure. This minimal impedance to the forward ejection of a large SV allows for the performance of maximal myocardial work at a minimum of oxygen consumption. Eventually, however, progressive volume overload increases ventricular EDV to the point that compensatory hypertrophy is no longer sufficient to compensate, and a decline in systolic function occurs. As systolic function declines, end-systolic dimension increases further, left ventricular wall stress increases, and left ventricular function is further compromised by the excessive ventricular afterload. At this point, the decline of ventricular function is progressive and can be quite rapid. As is shown in Figure 19-26A, in the patient with compensated AR, the eccentrically dilated ventricle maintains its SV and EF by using preload reserve; wall stress is only slightly increased. In Figure 19-26B, the limit of preload reserve has been reached and ventricular dysfunction has shifted the end-systolic wall stress relation to the right. As a result of the higher wall stress, the SV and EF inevitably decline. In patients in whom the EF has become less than 25% or the end-systolic diameter larger than 60 mm, irreversible myocardial changes are likely to have occurred.141
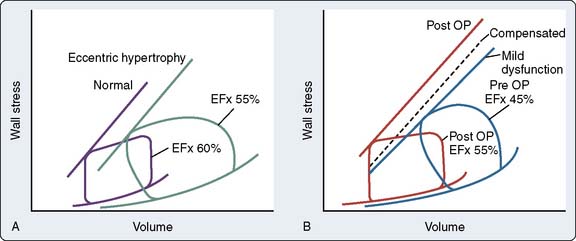
Figure 19-26 Adaptation to volume overload.
(From Ross J Jr: Afterload mismatch in aortic and mitral valve disease: Implications for surgical therapy. J Am Coll Cardiol 5:811, 1985.)
The primary determinants of Mvo2—contractility and wall tension—usually are not significantly increased with chronic AR, although calculated myocardial work may be twice normal. Actual Mvo2 may be only minimally increased because the greater proportion of myocardial work is spent in the energy-efficient process of fiber shortening, with little increase occurring in oxygen-consuming tension development275 (see Figure 19-26).
Despite the relatively normal Mvo2, angina can occur in one third of patients with severe AR, even in the absence of CAD.276 Patients with chronic AR may be at risk for myocardial ischemia caused by hypertrophy-induced abnormalities of the coronary circulation. The increase in total myocardial mass can increase baseline Mvo2, and there is evidence that total coronary blood flow, although increased, fails to keep pace with the increase in myocardial mass. Evidence suggests that the insidious development of contractile dysfunction may, in part, have an ischemic basis.277
Despite these rather favorable considerations in terms of myocardial energetics, an increase in Mvo2 may pose a threat to the patient with chronic AR because of decreased coronary vascular reserve.278 Although this phenomenon has been more thoroughly documented in patients with chronic pressure-induced hypertrophy, some studies indicate that the coronary vascular reserve is similarly compromised in patients with left ventricular volume overload.279,280 The process of hypertrophy may be a double-edged sword from the standpoint of the myocardial oxygen balance. Hypertrophy minimizes oxygen consumption as measured by wall stress, but increases basal Mvo2 and may elicit derangements in the quality or quantity of the coronary vasculature.
It also is important to remember that intraoperatively, patients with chronic AR may be at risk for acute ischemia with episodes of significant bradycardia. As bradycardia prolongs diastolic time, it increases regurgitant flow, and left ventricular diastolic pressure and wall tension increase rapidly. Simultaneously, the CPP is decreased as aortic runoff occurs during diastole and diastolic ventricular pressure is increased.281 Under these conditions, myocardial perfusion pressure may be insufficient. Clinically, very rapid decompensation can occur. The ischemic ventricle can dilate rapidly such that progressively increased end-systolic dimensions are seen, and ischemia and ventricular failure become a positive feedback loop.

Surgical Decision Making
Although ejection phase indices (e.g., EF) are most familiar to anesthesiologists, they are inherently unreliable for quantifying ventricular function in the setting of chronic volume overloading, because the Starling mechanism continues to support increases in the SV long after the onset of intrinsic myocardial depression. Left ventricular dilation, compensatory hypertrophy, and minimal afterload may normalize the EF, even though contractility is significantly depressed.258 Preload can be exquisitely sensitive to changes in heart rate and systemic vascular tone. Ventricular performance, therefore, may not be adequately reflected by the EF.282,283 The often-reported regurgitant volume or regurgitant fraction are similarly rate and load dependent, and they may correlate poorly with the underlying inotropic state.284
A variety of end-systolic indices have been examined in chronic AR, and these provide a more load-independent assessment of the inotropic state. Although the left ventricular end-systolic volume (LVESV) is preload independent, it does vary with myocardial contractility and afterload. An increased ESV may reflect a depressed inotropic state, or it may increase because of an increased left ventricular afterload resulting from increased chamber size or pressure.282 There also is evidence that the relative load independence of the LVESV is limited when ventricular hypertrophy is primarily eccentric.25 However, with long-standing volume overload, virtually all studies have found that an increased end-systolic dimension correlates with a poor prognosis and a significantly depressed contractile state.261,285
When valve surgery is performed after ventricular decompensation has occurred, the immediate and long-term results are marginal. Much of the increased mortality is a function of postoperative deaths from HF.264,286 When valve surgery occurs before significant ventricular decompensation, ventricular recovery is remarkable, with remodeling and a reduction in left ventricular size beginning as early as 2 weeks after surgery.141,264 Because delays in surgical intervention can be catastrophic and because the response to surgery is so good, there is evidence of a shift toward earlier valve replacement, and surgery in relatively asymptomatic patients has been advocated.264 Based on review of the published literature, the ACC and the AHA have provided practice guidelines for surgical intervention in chronic AR.141 Valve surgery is recommended for asymptomatic patients with left ventricular systolic dysfunction. Surgery also should be considered if ventricular dilation has occurred in the asymptomatic patient, even if the EF is normal. In patients who are symptomatic but have normal ventricular function, the ACC and AHA recommend further evaluation for an unrelated cause and observation. In these cases, serial echocardiographic assessment is appropriate. Symptomatic patients with left ventricular dysfunction should undergo surgery.13

Acute Aortic Regurgitation
In acute AR, sudden diastolic volume overload of a nonadapted LV results in a precipitous increase in the EDP because the ventricle is operating on the steepest portion of the diastolic pressure-volume curve (see Figure 19-25).287 In severe acute AR, the LVEDP can equilibrate with aortic diastolic pressure and exceed the LA pressure in late diastole. This may be sufficient to cause closure of the mitral valve before atrial systole.288 This is an important echocardiographic finding indicative of severe AR.289 Although this phenomenon initially shields the pulmonary capillaries from the full force of the dramatically increased LVEDP, the protection may be short-lived.290 Severe left ventricular distention often follows and produces mitral annular enlargement and functional mitral regurgitation.
The inevitable decline in SV in acute decompensating AR elicits a reflex sympathetic response so that tachycardia and a high SVR are common. Moderate tachycardia beneficially shortens the regurgitant time without reducing the transmitral filling volume.291 Vasoconstriction, however, preserves CPP at the expense of increasing the aorta-ventricular gradient and regurgitation.
As may be expected, patients with acute AR may be at greater risk for myocardial ischemia. As with chronic AR and bradycardia, coronary perfusion may be compromised by the combination of a low diastolic arterial pressure and the precipitously increased LVEDP. This narrowing of CPP may be so severe that the phasic epicardial blood flow may change to a predominantly systolic pattern with severe acute AR.292 Dissection of the coronary ostia is rare but frequently causes fatality in patients with acute AR. In addition to the structural impediment to myocardial oxygen delivery, catastrophic hypotension and high LVEDP combine to cause accentuated ischemia and ventricular dilation. Immediate surgical correction is the only hope for salvaging these patients, who often prove refractory to inotropes and vasodilators. Attempts at stabilizing the ischemic component of their injury with the intra-aortic balloon are usually contraindicated because augmenting the diastolic pressure worsens regurgitation.
Acute AR most commonly is due to infective endocarditis or aortic dissection, and intraoperative transesophageal echocardiography (TEE) has assumed increasing importance both in diagnosis of acute AR and in decisions regarding its surgical management.293 Transesophageal echocardiographic studies are highly sensitive and specific for the diagnosis of infective endocarditis, and are significantly more sensitive than transthoracic echocardiography.294 TEE has been shown to be particularly useful in the diagnosis of abscesses associated with endocarditis,295 and may detect previously unsuspected abnormalities (Roger Click, MD, Mayo Clinic, personal communication). Although an area of active investigation, there is currently no completely satisfactory noninvasive method for quantifying the severity of AR. Premature closure of the mitral valve, determination of the pressure half-time of AR, and color-flow estimation of both the regurgitant jet’s width and its size in relation to the LVOT are commonly described techniques.296–298 (Refer to Chapters 12 and 13 for further discussion.)

Anesthetic Considerations
Intraoperative monitoring should include an ECG system with the capability of monitoring a lateral precordial lead because ischemia is a potential hazard (Box 19-3). For most valvular procedures, a PAC provides useful information. A PAC allows determination of basal filling pressures and cardiac output, which is particularly useful in chronic AR given the potential unreliability of the clinical history and EF. Equally important is the ability to accurately monitor ventricular preload and CO response to pharmacologic interventions. The aggressive use of vasodilators often is appropriate therapy perioperatively for the failing ventricle, but their use can compromise the preload to which the ventricle has chronically adjusted. Concurrent preload augmentation, guided by the pulmonary artery diastolic pressure or PCWP, may be crucial to optimize CO when afterload is pharmacologically manipulated299–301 (see Chapters 10, 12 to 14, 32, and 34). The other requirement for a PAC is to allow for pacing when it is anticipated. The deleterious effects of significant bradycardia in AR have been described. In patients who arrive in the operating room with heart rates less than 70 beats/min or in patients for whom rapid epicardial pacing may be difficult to establish (e.g., redo operations), placement of a pacing wire probably is indicated. Typically, only a ventricular wire would be appropriate; it is more reliable than atrial pacing, and in AR, the atrial contribution to ventricular diastolic volume usually is not essential. Capturing the ventricle with a PAC-based, transvenous wire can be difficult because of the very large ventricular cavity size in patients with chronic AR.
Mitral regurgitation

Clinical Features and Natural History
Unlike mitral stenosis (MS), which is almost always the result of rheumatic valve disease, mitral regurgitation may result from a variety of disease processes that affect the valve leaflets, the chordae tendineae, the papillary muscles, the valve annulus, or the LV. Mitral regurgitation can be classified as organic or functional. Organic mitral regurgitation describes diseases that result in distortion, disruption, or destruction of the mitral leaflets or chordal structures. In Western countries, degenerative processes that lead to leaflet prolapse with or without chordal rupture represent the most common cause of mitral regurgitation.300 Other causes of organic mitral regurgitation include infective endocarditis, mitral annular calcification, rheumatic valve disease, and connective tissue disorders such as Marfan or Ehlers-Danlos syndrome. Much less common causes of organic mitral regurgitation include congenital mitral valve clefts, diet-drug or ergotamine toxicity, and carcinoid valve disease with metabolically active pulmonary tumors or right-to-left intracardiac shunting.302 Mitral valve prolapse (MVP) is a common (2.4%) disorder with a strong hereditary component. Several genes play a pivotal role in heart valve formation, including calcineurin, Wnt/beta-catenin signaling, fibroblast growth factor 4 (FGF-4), the homeobox gene Sox4, and the downstream modulator of transforming growth factor-β superfamily signaling Smad6. Genotypic linkage was found to chromosomes 11, 13, and 16. The recent finding of a mutation in familial mitral valve prolapse not related to connective tissue syndromes, an X-linked filament A mutation, suggests that mitral valve prolapse may be the final common outcome of multiple genetic defects.
Because it can be caused by a wide variety of disease processes, the natural history of mitral regurgitation is quite variable.302 Even among patients with acute-onset disease, the clinical course depends on the mechanism of regurgitation and the response to treatment. For instance, patients with acute, severe mitral regurgitation caused by a ruptured papillary muscle have a dismal outcome without surgery.302 However, the clinical course of acute mitral regurgitation caused by endocarditis could be favorable if the patient responds well to antibiotic therapy.302 Although those with chronic mitral regurgitation usually enter an initial, often asymptomatic, compensated phase, the time course for progression to left ventricular dysfunction and symptomatic HF is unpredictable.302 The literature reflects the wide variability in the natural history of mitral regurgitation, with published 5-year survival rates for patients with mitral regurgitation of 27% to 97%.303 Selection bias, small study populations, and poorly defined degrees of mitral regurgitation likely explain these discrepancies.304 Later studies better define the clinical course of certain subgroups of patients with mitral regurgitation. For instance, Ling et al305 examined the natural history of patients with mitral regurgitation because of flail leaflets. Among this select group, the investigators observed an excess annual mortality rate of 6.3% with a combined rate of death or surgery of 90% within a 10-year period.305 Patients with flail mitral valve leaflets also experience an increased risk for sudden death.306 Grigioni et al306 reported a 1.8% per year rate of sudden death among patients with flail mitral leaflets who were being medically managed. The rate of sudden death diminished after surgical intervention in this population.306
The application of quantitative Doppler echocardiography to the prospective study of mitral regurgitation has allowed researchers to document the progressive nature of this condition. Enriquez-Sarano et al307 found that, on average, the regurgitant volume increases 7.5 mL and the effective regurgitant orifice increases 5.9 mm2 each year. However, as with the natural history of mitral regurgitation in general, there was significant variability in the rate of progression among patients. Rapid progression of its severity occurred among patients who developed flail leaflets, but mitral regurgitation regressed in 11% of those studied.307

Pathophysiology
The presentation of patients with mitral regurgitation varies depending on the pathophysiology of the specific condition, which is affected by the mechanism, severity, and acuity of the mitral regurgitation. In cases of acute, severe mitral regurgitation, such as in patients with a ruptured papillary muscle after AMI, the sudden increase in preload enhances left ventricular contractility by the Frank-Starling mechanism. Despite the increased preload, left ventricular size initially is normal. Normal left ventricular size combined with the ability to eject into a low-pressure circuit (i.e., the left atrium) results in decreased afterload in the acute setting. The measured LVEF in cases of sudden, severe mitral regurgitation may approach 75%, although forward SV is reduced.302 However, because the left atrium has not yet dilated in response to the large regurgitant volume, LA pressure increases acutely and may lead to pulmonary vascular congestion, pulmonary edema, and dyspnea.302
Many patients with mitral regurgitation, particularly those whose valvular incompetence develops more slowly, may enter a chronic, compensated phase. In this phase, chronic volume overload triggers left ventricular cavity enlargement by promoting eccentric hypertrophy (see Figure 19-15). Increased preload continues to augment left ventricular systolic performance. At the same time, the left atrium dilates in response to the ongoing regurgitant volume. Although LA dilatation maintains a low-pressure circuit that facilitates left ventricular systolic ejection, the increased radius of the left ventricular cavity leads to increased wall tension according to the law of Laplace, which was given earlier.
Unlike the case of sudden, acute mitral regurgitation, which is characterized by normal left ventricular size and reduced afterload, afterload remains in the normal range in the chronic, compensated phase of mitral regurgitation.308 The changes seen in chronic, compensated mitral regurgitation are represented graphically in Figure 19-27. The isovolumic contraction phase is shortened as the blood is ejected into the low-pressure left atrium early in the cycle of ventricular systole. Because the dilated, compliant LV accepts increased diastolic volumes at low or normal filling pressures, some physicians believe that left ventricular diastolic function is enhanced in patients with chronic mitral regurgitation.308 Whereas dilatation of the left heart chambers permits left ventricular filling at low or normal pressures, chamber enlargement progressively dilates the mitral annulus, potentially increasing the effective regurgitant orifice and regurgitant volume over time. Patients may tolerate the phase of chronic, compensated mitral regurgitation for many years because increased preload maintains left ventricular ejection performance. If symptoms develop during this time, they often are related to diminished forward CO because the regurgitant fraction may exceed 50%. Rather than dyspnea or signs of pulmonary congestion, fatigue and weakness predominate.
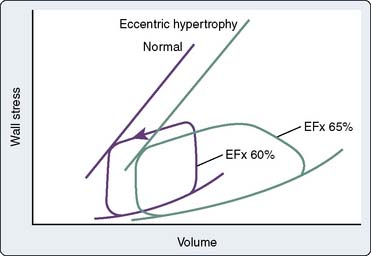
Figure 19-27 Volume overload with mitral regurgitation (MR).
(From Ross J Jr: Afterload mismatch in aortic and mitral valve disease: Implications for surgical therapy. J Am Coll Cardiol 5:811, 1985.)
With the eventual decline in left ventricular systolic function, patients enter a decompensated phase. Progressive left ventricular dilatation increases wall stress and afterload, causing further deterioration in left ventricular performance,302 mitral annular dilatation, and worsening of the mitral regurgitation. Left ventricular end-systolic pressure increases. The increased left ventricular filling pressures result in increase of LA pressures and, given time, pulmonary vascular congestion, pulmonary hypertension, and right ventricular dysfunction. In addition to fatigue and weakness, patients with decompensated, chronic mitral regurgitation also may report dyspnea and orthopnea. It is difficult to predict when a patient with mitral regurgitation is likely to decompensate clinically. The progression of disease in any given patient depends on the underlying cause of mitral regurgitation, its severity, the response of the LV to volume overload, and possibly the effect of medical management.302,309
Assessment of left ventricular function in patients with mitral regurgitation is controversial but critically important for clinical decision making.303 As in AR, the altered loading conditions that characterize mitral regurgitation confound traditional, load-dependent measures of left ventricular systolic function such as EF. Although numerous methods of assessing left ventricular function have been proposed, no consensus exists.303 Despite its limitations, EF remains the most studied variable of left ventricular function in patients with mitral regurgitation and the most predictive of clinical outcome. For instance, preoperative EF is the most important prognostic factor for long-term survival after surgery for mitral regurgitation.310 Preoperative EF also predicts postoperative EF and the occurrence of CHF after the surgical correction of mitral regurgitation.311,312 Because of the combination of increased preload and the ability to eject into the low-pressure left atrium, a normally functioning LV should display an increased EF in the presence of significant mitral regurgitation. Conversely, an EF considered normal in a patient with competent valves may represent diminished left ventricular function in the setting of mitral regurgitation. In patients with severe mitral regurgitation, an EF in the range of 50% to 60% likely represents significant left ventricular dysfunction and is itself an indication for surgery.141 Enriquez-Sarano et al310 reported that the postoperative survival after mitral repair or replacement declines after the preoperative EF decreases to less than 60%.
A number of studies provide insight into the origin of left ventricular dysfunction in the setting of mitral regurgitation. In animal models of the disease, myocardial mass accumulates mainly through a decrease in the natural degradation of cellular elements, rather than by an increased rate of protein synthesis.313–315 In contrast, myocardial protein synthesis increases within hours of the creation of simulated AS and left ventricular pressure overload.316 Histologic preparations from animals with experimental mitral regurgitation and left ventricular dysfunction also show a 35% reduction in contractile elements within individual myocytes.317 Examination of pressure-volume loops from patients with AS and mitral regurgitation is also insightful (see Figures 19-13 and 19-27). If the area contained within each loop is examined, it can be seen that this area, or stroke work, is similar for the two disease states. However, patients with AS generally display a greater degree of myocardial hypertrophy when compared with patients suffering from mitral regurgitation. Stroke work per gram of myocardium is greater in patients with mitral regurgitation, a finding that also may explain left ventricular dysfunction in this patient population.308

Ischemic Mitral Regurgitation
Clinicians increasingly are recognizing the importance of IMR because it is quite common. The incidence rate of mitral regurgitation after AMI as detected by echocardiography ranges from 39% to 73%.309 Echocardiographic studies also demonstrate that in 13% to 29% of cases, the IMR is at least moderate in severity.309 The presence of IMR also portends a worse clinical outcome. For instance, patients with IMR after AMI experience a twofold to sixfold increase in cardiovascular mortality in the first year after the ischemic event.318 Patients with severe IMR have a 1-year mortality rate that approaches 50%.319 However, in evaluating patients with mitral regurgitation presumed to be of ischemic origin, clinicians should attempt to identify the severity of the regurgitation, the time of onset, and the likely mechanism of valvular incompetence, because these factors influence prognosis and treatment. For instance, patients with mild IMR in the early phase of an AMI rarely experience hemodynamic sequelae from this mitral leakage, whereas those with ischemic rupture of a papillary muscle often present with acute CHF and have a poor prognosis without surgical intervention.
Investigators have identified certain patient characteristics associated with an increased incidence of IMR. For instance, patients with advanced age, inferior or posterior AMI, spherical left ventricular geometry, and multiple-vessel CAD tend to display greater rates of IMR.309,320 Other factors associated with increased rates of IMR include LA enlargement, mitral annular dilatation, and decreased EF.309 Frank rupture of a papillary muscle, which occurs in 1% of AMI cases, usually involves the posteromedial papillary muscle because its blood supply is derived from a single coronary artery.
The understanding of the pathophysiologic basis of IMR continues to evolve. Early investigators proposed ischemic papillary muscle dysfunction as a primary cause of IMR.321–323 Later studies indicated that isolated ischemia of one or both papillary muscles does not cause significant mitral regurgitation.324–327 Instead, ischemia of the papillary muscle and the adjacent myocardium is necessary to produce IMR.309,324,326,327 Myocardial ischemia may result in focal or global left ventricular bulging and, with time, ventricular remodeling to a more spherical shape. Such geometric changes cause outward migration of the papillary muscles. The finding most strongly correlated with chronic IMR is outward papillary muscle displacement.309,328 When the papillary muscles are displaced outward, the point of mitral leaflet coaptation moves apically and away from the mitral annulus, resulting in the appearance of valve tenting. Besides outward bulging of the LV, scarring and retraction of the papillary muscles also may produce mitral leaflet tethering, with the net effect of incomplete leaflet coaptation and valvular incompetence. Some investigators believe that papillary muscle dysfunction may reduce the degree of IMR in certain patients.329,330 Komeda et al330 suggested that reduced contractility of the papillary muscles may counteract the tethering effect of ischemic myocardium, thereby allowing leaflet coaptation to occur closer to the mitral annulus. An additional potential mechanism of IMR is decreased contractility of the posterior mitral annulus. During systole, annular contraction reduces the mitral orifice area by 25%.331–333 Because the anterior portion of the mitral annulus is more fibrous, posterior annular contraction plays a greater role in reducing the size of the mitral orifice. Loss of posterior annular contraction may contribute to mitral regurgitation in the setting of myocardial ischemia.
The clinical approach to IMR depends on its underlying mechanism. Timely surgical intervention often is warranted in cases of papillary muscle rupture. For patients with an intact mitral apparatus who present with IMR in the setting of AMI, early reperfusion therapy improves regional and global left ventricular function, reduces ventricular dilatation, and decreases the likelihood of adverse remodeling and associated papillary muscle displacement.329,334–337 The resultant improvements in ventricular function and geometry combine to reduce the incidence of IMR. Clinicians often prescribe angiotensin-converting enzyme inhibitors to patients with ischemic heart disease. Chronic angiotensin-converting enzyme inhibitor therapy may decrease the incidence and severity of IMR by preventing left ventricular remodeling, although data to support this theory are lacking.329

Assessment of Mitral Regurgitation
Clinicians may suspect mitral regurgitation on the basis of current symptoms, medical history, or findings on physical examination. Echocardiography with the capability of 2D and Doppler (including color-flow) imaging represents the diagnostic modality of choice for the assessment of mitral regurgitation. Transthoracic echocardiogram (TTE) is readily available in most areas, is noninvasive, and provides detailed information about the mechanism of mitral regurgitation, its severity, and its impact on cardiac chamber size and function. Additional information available from TTE includes calculation of right ventricular systolic pressure based on the peak velocity of the tricuspid regurgitant signal and an estimate of right atrial pressure. Whenever possible, echocardiographers should attempt to provide a quantitative assessment of mitral regurgitation severity. Techniques that allow the determination of effective regurgitant orifice, regurgitant volume, or regurgitant fraction include the proximal isovelocity surface area (PISA) method and the continuity equation. Other echocardiographic findings such as pulmonary vein systolic flow reversals suggest severe mitral regurgitation, and they should be reported if present. Assessment of the size of the regurgitant jet in the left atrium may provide a gross estimation of mitral regurgitation severity but is limited by factors such as LA size and pressure, machine settings such as color-flow gain and aliasing velocity, and the orientation of the regurgitant jet itself. Eccentric regurgitant jets propagating along the wall of the left atrium often appear smaller than a centrally directed jet of equivalent severity. When transthoracic echocardiography is suboptimal, patients may be referred for TEE. Because of the proximity of the transducer to the mitral valve, TEE often produces superior images of the mitral valve. When performing intraoperative TEE examinations, anesthesiologists should recall that altered loading conditions, such as anesthetic-induced decreases in SVR, might favor forward cardiac output and diminish the observed amount of mitral regurgitation. More detailed discussions of the echocardiographic evaluation of the mitral valve are presented in Chapters 1, 2, 12 and 13.
Cardiac catheterization with left ventriculography may also be used to evaluate mitral regurgitation. This invasive procedure often is reserved for cases in which the echocardiographic data are suboptimal, conflicting, or discordant with the clinical findings.302 Assessment of severity by ventriculography requires analysis of the amount of contrast material that enters the left atrium after injection into the LV. However, the amount of contrast material appearing in the left atrium depends on the severity of mitral regurgitation and on the volume of the left atrium, the catheter position, and the rate of contrast injection.302 Right-heart catheterization may or may not demonstrate the presence of v waves in patients with significant mitral regurgitation. A high degree of LA compliance makes the appearance of prominent v waves less likely (see Chapter 3).
Other diagnostic tests commonly obtained in patients with mitral regurgitation include an electrocardiogram and chest radiograph. ECG findings such as AF, LA enlargement, and ST-segment abnormalities may be observed in patients with mitral regurgitation, but they are not specific.301 Similarly, chest radiographs may identify enlargement of the left heart chambers and pulmonary vascular congestion, but such findings are not specific for mitral regurgitation.

Surgical Decision Making
Just as progress in the understanding of the pathophysiology of mitral regurgitation has evolved, so too has the surgical approach to this disease process. A high operative mortality associated with the surgical correction of mitral regurgitation in the 1980s led many clinicians to manage patients conservatively.303,304,338 Because favorable loading conditions and high LA compliance allow even patients with significant mitral regurgitation to remain asymptomatic for long periods, it is likely that many patients did not undergo surgery until the onset of disabling symptoms. Studies show that more severe preoperative symptoms are associated with a lower EF and a greater incidence of postoperative CHF.304,311,312 Historically, poor outcomes after surgery for mitral regurgitation might have occurred because clinicians did not appreciate the true degree of left ventricular dysfunction at the time of surgery in symptomatic patients. An EF of less than 60% in the setting of severe mitral regurgitation represents significant left ventricular dysfunction and predicts a worse outcome with surgery or medical management.305,310 Surgical techniques common in the 1980s probably also contributed to unfavorable postoperative outcomes. For instance, although the mechanisms are incompletely understood, resection of the subvalvular apparatus contributes to decreased left ventricular systolic performance after mitral replacement.339
In part because of improved surgical techniques, the operative mortality rate for patients with organic mitral regurgitation who are younger than 75 years is about 1% in some centers.310 Besides preservation of the subvalvular apparatus, valve repair represents another surgical technique associated with improved postoperative outcome.304,340 Although not applicable to all patients, such as those with advanced rheumatic disease, the popularity of valve repairs continues to grow. Studies indicate numerous benefits associated with mitral repair. For instance, after accounting for baseline characteristics, patients who undergo mitral repair instead of replacement experience lower operative mortality and better long-term survival, largely because of improved postoperative left ventricular function.304,340 The survival benefit that accompanies valve repair also is observed among patients undergoing combined valve and CABG surgery.340 Valve repair does not increase the likelihood of reoperation when compared with replacement.340 Although originally used most often for posterior leaflet disease, surgeons now routinely repair anterior mitral leaflets with good success.341 When repairing anterior leaflet prolapse, surgeons may insert artificial chordae.342,343 The approach to flail or prolapsing posterior mitral leaflet segments often involves resection of a portion of the leaflet.343 In addition to resecting a portion of the leaflet and plicating the redundant tissue, an annuloplasty ring often is placed to reduce mitral orifice size and return the annulus to a more anatomic shape.343 Some surgeons favor a flexible, partial, posterior annuloplasty band, which may allow improved systolic contraction of the posterior annulus and better postoperative left ventricular function.332,343,344
Timely and appropriate surgical referral helps improve the perioperative outcome of patients with mitral regurgitation. To appropriately refer patients for surgery, clinicians should have an understanding of the factors that influence surgical risk in this population.303 The factors that correlated best with increased operative risk in patients with significant mitral regurgitation included age older than 75, severe preoperative symptoms of CHF, and concomitant CAD.310,345,346 Even though they represent a high-risk group, patients with severe mitral regurgitation and symptoms of HF should still be referred for surgery, because valve repair or replacement offers a survival advantage compared with medical management.303,305 Similarly, although perioperative risk remains increased, patients with evidence of left ventricular dysfunction, such as an EF less than 60% or an end-systolic left ventricular diameter greater than 45 mm, should be referred for surgery to prevent further, possibly irreversible deterioration in ventricular performance.141,303 Asymptomatic patients without evidence of left ventricular dysfunction also should be considered for surgery if conditions such as AF, ventricular tachycardia, or pulmonary hypertension are identified or the effective regurgitant orifice is greater than 40 mm.2,141,303 Institutional experience, particularly with techniques such as valve repair, is an important consideration for clinicians contemplating early surgical referral.303

Minimally Invasive Mitral Valve Surgery
Beginning in the mid-1990s, several groups adopted a minimally invasive approach to mitral valve repair.347,348 Typically performed via lower ministernotomies or right parasternal incisions, aortic cannulation for CPB was accomplished via the chest. The venous cannula was placed either via the femoral vein347 or the right atrium.348 Standard surgical repair techniques were utilized. In reporting a series of 707 minimally invasive mitral valve repairs, McClure et al347 noted an operative mortality rate of 0.4% and an incidence rate of stroke of 2%. Failed repair necessitating reoperation occurred in 4.8% of cases, with long-term follow-up demonstrating 83% survival beyond 11 years. When compared with conventional sternotomy for mitral repair, these authors also noted reductions in hospital length of stay, aortic cross-clamp time, and total CPB time in their minimally invasive cases.347,349 Additional benefits of these minimally invasive approaches were reported by Svensson et al.348 From a cohort of patients who underwent minimally invasive mitral repair between 1995 and 2004, the authors selected 590 cases that were matched using propensity scores with 590 patients who received mitral repair via conventional sternotomy. This study demonstrated improved pain scores and FEV1 (forced expiratory volume in 1 second), together with reductions in perioperative bleeding and transfusion requirements in the minimally invasive group. Procedural success was not reduced by the adoption of a minimally invasive approach, and in fact, there was a trend toward fewer patients with 3+ or 4+ residual mitral regurgitation at 1- and 5-year follow-up in this cohort.
The trend toward even less invasive mitral repair continued in the late 1990s with the advent of thoracoscopic and robotic-assisted procedures. In 1996, Carpentier350 performed the first video-assisted mitral valve repair via a minithoracotomy with fibrillatory arrest. The era of robotic-assisted mitral repair began the following year when Mohr351 used a voice-controlled robotic arm or AESOP (automated endoscopic system for optimal positioning) to provide thoracoscopic visualization of the mitral valve during a repair that was accomplished through a 4-cm right thoracotomy. Then, in 1998, Carpentier used a prototype robotic system (da Vinci; Intuitive Surgical, Mountain View, CA) to perform a mitral repair.352 Although approved 2 years earlier for general laparoscopic surgery, the da Vinci system was given FDA approval for mitral valve surgery in the United States in 2002.353 Since that time, there has been a rapid increase in literature reports related to robotic-assisted cardiac surgery. In fact, within a 6-year period, more than 200 articles were published related to robotic-assisted cardiac surgery, including 60 from a single author.354 Several centers have published series of more than 100 robotic cases.355–357
Currently, the concept of minimally invasive mitral surgery generally refers to valve repairs accomplished via a 3- or 4-cm right inframammary incision in the fourth or fifth intercostal space (Figure 19-28). Several additional, 1-cm incisions surround the primary incision and facilitate placement of robotic arms or other thoracoscopic instruments. The arterial cannula for CPB is inserted in the femoral artery (Figure 19-29). Venous cannulation is also accomplished via the femoral route using TEE guidance (Figure 19-30). Supplementary venous drainage is used in some centers by inserting either a 15 to 17 French right internal jugular vein cannula or specialized PAC with multiple end holes that drains to the venous reservoir during CPB. Cardioplegia may be given either antegrade into the aortic root or retrograde via the coronary sinus. Surgeons typically administer antegrade cardioplegia by one of two methods. The first involves the placement of a catheter tip into the ascending aorta through a right parasternal stab incision under thoracoscopic vision. This method is similar to standard antegrade cardioplegia administration in median sternotomy cases. A long-shafted aortic cross-clamp placed through a stab incision in the right lateral chest wall is used to occlude the aorta distal to the cardioplegia cannula. The second method of antegrade cardioplegia administration utilizes a specialized endoaortic cannula inserted via the femoral artery. A balloon near the distal end of this cannula is positioned in the ascending aorta using TEE guidance. Inflation of the balloon occludes the ascending aorta while antegrade cardioplegia delivery commences at the distal tip of the device. Although the choice of antegrade cardioplegia system varies between surgeons and institutions, use of the endoaortic clamp and cardioplegia delivery system have been associated with increased morbidity, cost, and cross-clamp times.358 Retrograde cardioplegia may be given, if desired, by means of a coronary sinus catheter that has been percutaneously placed via the right internal jugular vein. When using a right minithoracotomy approach, the actual repair may be performed using long-handled, thoracoscopic instruments (Figure 19-31) or robotic assistance. Though referred to as “robotic,” systems such as the da Vinci are probably more appropriately described as telemanipulators. As such, these devices receive direct input from the hands and feet of the surgeon who is seated at a remote console and translate these motions to end-effectors within the chest of the patient (Figure 19-32). Proponents of robotic-assisted mitral repair cite a number of advantages of this approach compared with minimally invasive thoracoscopic surgery.353 When seated at the remote console of a robotic device, the surgeon has near-stereoscopic vision compared with viewing a 2D image on a television screen. In addition, robotic devices provide motion scaling and tremor filtration to smooth movements. Because the robotic arms have articulating “wrists” at their distal ends, the surgeon can achieve 7 degrees of freedom of movement within the chest, similar to open surgery. By comparison, long-handled thoracoscopic instruments, which are often oriented nearly parallel to one another, afford only 4 degrees of freedom. Both thoracoscopic and robotic-assisted approaches utilize the same operative techniques as standard open repairs. Thus, techniques such as leaflet resection, chordal insertion or transfer, sliding plasties, edge-to-edge repair, and annuloplasty band insertion may be used by experienced surgeons.
The procedural success and operative mortality of robotic-assisted mitral repair in larger series appear relatively comparable with traditional surgical approaches. In reporting their first 300 robotic-assisted mitral repairs, Chitwood et al355 noted a 30-day mortality of 0.7% with a stroke rate of 0.7%. Mean CPB time was 159 minutes with a mean aortic cross-clamp time of 122 minutes. Eighty-nine concomitant procedures were performed, such as the Maze procedure and closure of atrial level shunts. At a mean follow-up of 815 days, 93% of patients were found to have residual mitral regurgitation that ranged from none to mild. Technical failure, such as annuloplasty band dehiscence, led to reoperation in 3.3% of cases. Hospital length of stay averaged 5.2 days. Cheng et al356 recently described the outcomes of their first 120 robotic-assisted mitral repair cases. Similar to Chitwood et al,355 Cheng et al356 reported a 30-day mortality rate of less than 1%. Mean CPB time was 157 minutes with a mean aortic cross-clamp time of 117 minutes. Fifty-three concomitant procedures were performed in these patients. Failed repair necessitating reoperation was encountered in 5% of cases. Citing a procedural learning curve, the authors noted that all failed repairs occurred within the first 74 cases. Average hospital length of stay was 6.3 days.
Just as catheter-based techniques have been developed to treat valvular AS, efforts also are under way to develop catheter-based interventions for mitral regurgitation. Currently, the two primary percutaneous techniques under investigation involve edge-to-edge repair and annuloplasty band insertion.359 The edge-to-edge repair, popularized in open mitral operations by Alfieri, uses a percutaneously delivered clip to secure the anterior leaflet to the posterior leaflet, thereby creating a double-orifice mitral valve. Although several proprietary devices are under development by different manufacturers, the MitraClip (Evalve, Menlo Park, CA) is currently the subject of investigation in the phase II multicenter EVEREST II (Endovascular Valve Edge-to-edge Repair Study) trial.360
Although transventricular devices are under investigation, the most popular approach to percutaneous mitral annuloplasty relies on the anatomic proximity of the coronary sinus to the posterior mitral annulus. Tension applied to a device anchored in the coronary sinus has effectively limited mitral annular size both in animal models and early human experience.360

Anesthetic Considerations
Patients who present to the operating room with mitral regurgitation may differ significantly with respect to duration of disease, symptoms, hemodynamic stability, ventricular function, and involvement of the right heart and pulmonary circulation (Box 19-4). For instance, a patient with severe mitral regurgitation caused by acute papillary muscle rupture may enter the operating room in cardiogenic shock with pulmonary congestion requiring intra-aortic balloon pump augmentation. Another patient with a newly diagnosed flail posterior mitral leaflet may enter the surgical suite with relatively preserved left ventricular function and no symptoms whatsoever. In the latter patient, the compliance of the left atrium may have prevented pulmonary vascular congestion, pulmonary hypertension, and right ventricular dysfunction. Despite the differences in presentation, the general management goals remain similar and include maintenance of forward cardiac output and reduction in the mitral regurgitant fraction. The anesthesiologist also must seek to optimize right ventricular function, in part by avoiding increases in pulmonary vascular congestion and pulmonary hypertension. Various degrees of intervention are needed to achieve these hemodynamic management goals, depending on the patient’s presentation.
Information obtained during the preoperative interview and examination provides the anesthesiologist with important insight into the patient’s degree of hemodynamic compromise. For example, a patient who reports dyspnea at rest or with minimal activity may have significant pulmonary vascular congestion and possibly compromised right ventricular function. This information combined with the estimated pulmonary artery systolic pressure derived from the preoperative TTE report helps the anesthesiologist prepare for potential right-heart dysfunction intraoperatively. In such a patient, the anesthesiologist avoids heavy premedication with its attendant risk for obtundation, hypoventilation, and further increase of PAPs. Auscultation of the heart may reveal rhythm disturbances such as AF and the systolic murmur of mitral regurgitation. Clinicians should recall, however, that in cases of acute, severe mitral regurgitation, a significant increase in LA pressure decreases the systolic pressure gradient between the left heart chambers, and the murmur of mitral regurgitation may be diminished or even absent.302
Invasive hemodynamic monitoring provides the anesthesiologist with a wealth of important information. Arterial catheters are essential for monitoring beat-to-beat changes in blood pressure that occur in response to a variety of surgical and anesthetic manipulations. PACs facilitate many aspects of intraoperative patient management. Intraoperative use of a PAC allows the anesthesiologist to more carefully optimize left-sided filling pressures. Although the PCWP and pulmonary artery diastolic pressure depend on LA and left ventricular compliance and filling, examination of intraoperative trends in these variables enhances the ability of the anesthesiologist to provide appropriate levels of preload while avoiding volume overload. Periodic determination of cardiac output allows a more objective assessment of the patient’s response to interventions such as fluid administration or inotropic infusion. The presence or size of a v wave on a PCWP tracing does not reliably correlate with the severity of mitral regurgitation, because this finding depends on LA compliance. Just as in the management of patients with aortic valvular regurgitation, another benefit of PAC insertion is the ability to introduce a ventricular pacing wire to rapidly counteract hemodynamically significant bradycardia. In patients with right ventricular compromise, monitoring trends in the CVP recording also may be helpful. Tricuspid regurgitation (TR) detected through analysis of the CVP tracing may suggest right ventricular dilatation, which may be caused by pulmonary hypertension.361
Intraoperative TEE provides invaluable information during the surgical correction of mitral regurgitation. It reliably identifies the mechanism of mitral regurgitation, thereby guiding the surgical approach,362 and it objectively demonstrates the size and function of the cardiac chambers. TEE can readily identify the cause of hemodynamic derangements, facilitating proper intervention. For instance, the appearance of SAM of the mitral apparatus immediately after valve repair allows the anesthesiologist to intervene with volume infusion and medications such as esmolol or phenylephrine as appropriate. In rare circumstances when hemodynamically significant SAM persists despite these interventions, the surgeon may elect to further repair or even replace the mitral valve. TEE also identifies concomitant pathology that may warrant surgical attention, such as atrial level shunts and additional valve disease (see Chapters 12 and 13).
During minimally invasive and robotic-assisted mitral valve surgery, intraoperative TEE is essential. The use of a right minithoracotomy for these procedures precludes bypass cannulation via the chest. Instead, femoral arterial and venous cannulation are selected, with or without supplementary venous drainage from the superior vena cava or pulmonary artery. Real-time TEE imaging typically guides cannulation for CPB. If an endoaortic balloon clamp is used, the echocardiographer ensures that the balloon is correctly positioned in the ascending aorta. If a transthoracic aortic cross-clamp is chosen, the aortic cannula inserted into the femoral artery generally is not visualized with TEE. However, confirmation of guidewire placement in the descending aorta may be requested to exclude accidental passage into the contralateral iliac artery. The desired position of the tip of the femoral venous cannula is variable; some prefer the tip within the superior vena cava, whereas others select the right atrium or inferior vena cava-right atrial junction. Whatever the chosen position for the venous cannula, TEE imaging can identify a malpositioned cannula or guidewire (Figure 19-33). Tamponade after perforation of the LA appendage by a guidewire that was placed across a patent foramen ovale has been reported.363 TEE also is invaluable during the percutaneous placement of coronary sinus catheters for retrograde cardioplegia delivery (Figure 19-34A and B). Patients who may benefit from retrograde cardioplegia, such as those with significant AR, may be identified by the intraoperative echocardiographer.
In addition to specific TEE considerations related to cannulation procedures, the selection of a minimally invasive or robotic-assisted approach to mitral repair necessitates other changes in anesthetic management. Though not universally used, one-lung ventilation is preferred in many centers. This may be achieved by the usual methods, such as a double-lumen endotracheal tube or bronchial blocker. Impaired oxygenation is not uncommon when one-lung ventilation is used during the termination of CPB during these procedures.364 The delivery of cardioplegia requires special attention from both the surgical and anesthesia teams. If an endoaortic balloon clamp system is used, one or more methods should be used to verify its position. In addition to TEE, some centers place arterial catheters in both the right and left radial arteries; dampening of the right radial arterial waveform could signify balloon migration toward the innominate artery. If retrograde cardioplegia is administered via a percutaneously placed catheter, its position should be well documented by TEE, with or without fluoroscopy. Coronary sinus pressure should be monitored at baseline, during balloon inflation, and during cardioplegia administration. External patches for defibrillation are generally applied before patient positioning. A multimodal approach to analgesia may facilitate early extubation in these patients. Some centers include a regional technique such as intrathecal opioids or paravertebral blocks as part of the anesthetic plan.
The intraoperative management of patients with mitral regurgitation before the institution of CPB focuses on optimizing forward cardiac output, minimizing the mitral regurgitant volume, and preventing deleterious increases in PAPs. Maintaining adequate left ventricular preload is essential. An enlarged LV that operates on a higher portion of the Frank-Starling curve requires adequate filling. At the same time, excessive volume administration is to be avoided because it may cause unwanted dilatation of the mitral annulus and worsening of the mitral regurgitation. Excessive fluid administration may precipitate right ventricular failure in patients with pulmonary vascular congestion and pulmonary hypertension. Optimization of preload is aided by analysis of data obtained from PAC measurements and TEE images. Because significant left ventricular dysfunction is present in many patients with mitral regurgitation, anesthesiologists often select specific induction and maintenance regimens to avoid further depressing left ventricular function. For this reason, large doses of narcotics have been popular in the past.151,152 Other researchers have shown that smaller doses of narcotics combined with vasodilating inhalation anesthetics also produce acceptable intraoperative hemodynamics.365,366 By reducing the amount of narcotics administered, the addition of a vasodilating inhalation agent to the anesthetic regimen may allow for faster extubation of the trachea after surgery. With the current trend toward early referral of asymptomatic patients for mitral repair, anesthetic regimens that reduce the duration of postoperative mechanical ventilation may be advantageous.
In patients with severe left ventricular dysfunction, infusions of inotropic medications such as dopamine, dobutamine, or even epinephrine may be required to maintain an adequate cardiac output. Phosphodiesterase inhibitors such as milrinone also may augment systolic ventricular performance and reduce pulmonary and peripheral vascular resistances. By reducing pulmonary and peripheral vascular resistance, forward cardiac output is facilitated. Nitroglycerin and sodium nitroprusside represent two additional options for reducing the impedance to ventricular ejection. If patients prove refractory to inotropic and vasodilator therapy, insertion of an intra-aortic balloon pump should be strongly considered (see Chapters 10, 13, 27, 32, and 34).
Because severe mitral regurgitation may result in pulmonary hypertension and right ventricular dysfunction, anesthesiologists should tailor their intraoperative management strategies accordingly. Hypercapnia, hypoxia, and acidosis increase PAPs and should be avoided. Mild hyperventilation may be beneficial in some patients. The effect of nitrous oxide on pulmonary vascular resistance (PVR) and pulmonary hypertension is controversial. Some studies show no change in PVR when administered to anesthetized patients with CAD or VHD.367–369 Other studies in patients with MS demonstrate an increase in PVR after nitrous oxide administration, and in vitro evidence suggests that nitrous oxide increases norepinephrine release from the pulmonary artery.370–372
Patients with severe right ventricular dysfunction after CPB can prove exceptionally difficult to manage. Besides avoiding the factors known to increase PVR, only a few options exist for these patients. Inotropic agents with vasodilating properties such as dobutamine, isoproterenol, and milrinone augment right ventricular systolic performance and decrease PVR, but their use often is confounded by systemic hypotension. Prostaglandin E1 (PGE1) reliably reduces PVR and undergoes extensive first-pass metabolism in the pulmonary circulation.373,374 Although PGE1 reduces PAPs after CPB, systemic hypotension requiring infusions of vasoconstrictors through an LA catheter also has occurred.375–378 Inhaled nitric oxide represents another alternative available for the treatment of right ventricular failure in the setting of pulmonary hypertension. Nitric oxide reliably relaxes the pulmonary vasculature and is then immediately bound to hemoglobin and inactivated. Studies indicate that systemic hypotension during nitric oxide therapy is unlikely.379,380 (See Chapter 24.)
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
19 Valvular Heart Disease
Replacement and Repair
In analyzing the physiologic response of the left ventricle (LV) to a variety of abnormal loading conditions, it is useful to consider the concepts of afterload mismatch and preload reserve.1 These concepts frame discussions of the pathophysiology of the individual valvular lesions. Another set of physiologic concepts essential to understanding VHD and its evolution are pressure-volume loops and the linear end-systolic pressure-volume relation (ESPVR). The former provides a graphic analysis of ventricular pressure-volume relations in a single beat, whereas the latter is a method for quantifying the intrinsic contractility of the myocardium over multiple contractions and is relatively independent of changes in loading conditions (see Chapters 5, 12, 13 and 14).
Pathophysiology
Pressure-Volume Loops
The pressure and volume changes that occur during a normal cardiac cycle are depicted in Figure 19-1. A pressure-volume loop is generated when ventricular volumes are plotted against simultaneously occurring ventricular pressures for a single contraction (Figure 19-2). Familiarity with this graphic analysis of cardiac function, the normal pressure-volume loop of the LV, provides a basis for appreciating a load-insensitive index of contractility, the ESPVR. Phase 1 in Figure 19-2 shows ventricular filling and represents the loading of the heart before contraction (i.e., preload). During early and middle diastole, filling of the ventricle is rapid, depending on the pressure gradient between the left atrium and the LV. In late diastole, the left atrium contracts (a wave), which results in the final left ventricular end-diastolic volume (LVEDV) and pressure (LVEDP). Atrial contraction in the normal heart accounts for 15% to 20% of ventricular filling. The normal ventricle accommodates large changes in ventricular volume with only a small change in ventricular diastolic pressure.
Figure 19-1 Simultaneous left ventricular (LV) volume and pressure during one cardiac cycle. EKG, electrocardiogram.
(From Barash PG, Kopriva DJ: Cardiac pump function and how to monitor it. In Thomas SJ [ed]: Manual of cardiac anesthesia. New York: Churchill Livingstone, 1984, p 1.)
Afterload Stress and Preload Reserve
Figure 19-3 shows the response of the LV to changes in afterload, with preload held constant, in the context of the pressure-volume loops from single contractions. These curves are constructed experimentally by infusing a pure α-adrenergic agonist while simultaneously measuring the corresponding end-systolic volumes (ESVs). The resultant curves describe the diastolic pressure-volume relation (ventricular compliance) and the linear ESPVR at a given level of myocardial contractility. Each counterclockwise loop represents a cardiac cycle. The stroke volume (SV) progressively decreases as the impedance to ejection increases from beats 1 to 3. This pattern continues until beat 4, when the peak ventricular systolic pressure fails to open the aortic valve and only isovolumic contraction ensues.1 This inverse relation between afterload and SV (inverse force-velocity relation) also was documented experimentally in a canine preparation in which the LVEDV was held constant (Figure 19-4).2 In the intact heart, afterload is a function of ventricular size and arterial pressure.3 Its pivotal role in cardiovascular regulation is summarized in Figure 19-5. Afterload is defined as the tension, or force per unit of cross-sectional area, in the ventricular wall during ejection.4 Laplace’s law provides a mathematical expression for the wall tension5 (in which P is the intraventricular pressure) developed in a spherical chamber of radius, R, and wall thickness, h:
Figure 19-5 Role of afterload in cardiac regulation.
LV, left ventricular; Periph, peripheral.
(From Braunwald E: Regulation of the circulation. N Engl J Med 290:1124, 1974.)
This means that ventricular afterload, a function of the constantly changing intraventricular pressure and radius, varies continuously during systole. It is extremely difficult to precisely quantify afterload in the clinical setting, and commonly used approximations such as blood pressure or systemic vascular resistance (SVR) are inherently misleading because they fail to reflect instantaneous pressure-volume variations. Left ventricular size or preload is a determinant of the SV and afterload.6 Normally, the LV compensates for increases in afterload by increases in preload. This increase in end-diastolic fiber stretch or radius further increases wall tension in accordance with Laplace’s law, resulting in a reciprocal decline in myocardial fiber shortening (i.e., inverse force-velocity relation). Despite this relative decline in fiber shortening, the SV is maintained because contractile force is augmented at the higher preload (i.e., Starling or length-active tension effect). This increased contractility at higher LVEDVs may be mediated by an increased sensitivity to the inotropic effects of extracellular calcium at longer muscle lengths.7–9
Use of this “preload reserve” allows the LV to maintain its SV in the face of an afterload stress,1 as shown in Figure 19-6.1 The increase in afterload (beat 2) elicits a compensatory increase in end-diastolic volume (EDV; beat 3), preserving SV at the higher afterload. However, when the ventricle reaches the limit of its preload reserve, it overdistends, and preload behaves as if it were fixed. SV then decreases with further increases in the systolic pressure (afterload mismatch; beat 4), a clinical corollary of the inverse force-velocity relation.
Ventricular Compliance
Translating this physiologic analysis to the clinical setting is complicated by a number of practical constraints. One of the most important is the inconstant relation between LVEDV and LVEDP. The diastolic pressure-volume relation for the normal mammalian LV is a curvilinear function (Figure 19-7).3,10 The slope of this curve (i.e., ratio of change in volume to change in pressure during diastole) is the ventricular compliance (dV/dP). Although the normal ventricle is extremely compliant in the physiologic range, the instantaneous compliance decreases with increments in diastolic filling. This progressively increasing slope of the pressure-volume curve becomes evident at the extremes of ventricular volume, in which succeeding increments in volume result in exponentially greater increases in end-diastolic pressure (EDP). Patients with acute aortic regurgitation (AR) experience the hemodynamic consequences of this phenomenon. The catastrophic increases in ventricular filling pressure reflect the absolute magnitude of the volume overload and are a corollary of a precipitously declining compliance relation.
Figure 19-7 The normal diastolic pressure-volume relation.
(From Spotnitz HM, Sonnenblick EH, Spiro D: Relation of ultrastructure to function in the intact heart: Sarcomere structure relative to pressure-volume curves of the intact left ventricles of dog and cat. Circ Res 18:49, 1966.)
These hemodynamic manifestations of acute shifts up and down a single compliance curve must be distinguished from chronic, pathologic alterations in ventricular compliance, which produce actual shifts of the entire curve relating diastolic pressure and volume. For example, in animal models of chronic volume overload, the entire pressure-volume curve is shifted to the right, such that substantial increases in ventricular volume are tolerated with relatively little change in EDP (Figure 19-8).11 In this example, the slope of the new pressure-volume curve (i.e., compliance) is decreased. Similarly, time-dependent, rightward shifts of the entire pressure-volume relation have been shown to occur in patients with severe ventricular volume overload resulting from chronic AR.12 In these examples, the development of a new relation between pressure and volume may reflect the physiologic process of creep, the time-dependent change in size or the dimension of tissue maintained at a constant level of stress.4
Myocardial wall thickness is an important determinant of diastolic compliance. In clinical settings of chronic pressure overload (e.g., aortic stenosis [AS], chronic hypertension), diastolic compliance and ventricular wall thickness have been shown to be linearly and inversely related (Figure 19-9).13 This may explain why the normal, thinner-walled right ventricle (RV) is more compliant than the normal LV, even though the ventricles share similar qualities of intrinsic myocardial stiffness.3,14 This association between pathologic hypertrophy of the ventricle and deterioration in its diastolic compliance is a well-documented but poorly understood phenomenon.
Figure 19-9 Relation between chamber stiffness (ΔP/ΔD, the inverse of compliance) and ventricular wall thickness.
(From Grossman W, McLaurin P, Moos SP, et al: Wall thickness and diastolic properties of the left ventricle. Circulation 49:129, 1974.)
It has become customary to characterize diastolic function in certain disease states as normal or abnormal, such as diastolic dysfunction or diastolic failure.15,16 The latter condition is a distinct pathophysiologic entity that results from an increased resistance to ventricular filling and leads to an inappropriate upward shift of the diastolic pressure-volume relation.17 For example, diastolic dysfunction is seen in ischemic cardiomyopathy, particularly when combined with pressure overload hypertrophy.18
In certain diseases, these primary derangements of diastolic function may predominate over abnormalities of diastolic function associated with ventricular hypertrophy. For example, in hypertrophic cardiomyopathy (HCM), the impaired ventricular relaxation inherent in the myopathic process appears to play the greater role in the observed abnormalities of diastolic filling because the diastolic dysfunction is often disproportionate to the degree of ventricular hypertrophy.19,20 The effects of pathologic hypertrophy on diastolic compliance and ventricular relaxation are complex and are considered in more detail later in the Aortic Stenosis and Hypertrophic Cardiomyopathy sections.
Filling of one ventricle or changes in its configuration or compliance properties can significantly alter the diastolic pressure-volume characteristics of the other ventricle.21,22 Progressive increases in right ventricular filling shift the left ventricular compliance curve up and to the left. This effect is greatest at high right ventricular filling pressures and is accentuated by the presence of the pericardium.23 With severe right ventricular distention, the interventricular septum encroaches on the LV. This encroachment reduces the size of the LV and alters its geometric configuration such that its compliance declines.21 As a result, left ventricular filling pressures may fail to reflect even directional changes in left ventricular size.24
Contractility
Myocardial contractility can be defined as the ability of the heart to generate force at a given preload.25 Although most clinicians and researchers seem to be comfortable with their intuitive notions of the cardiac inotropic state, the search for a consensus on a quantitative yardstick of ventricular inotropy has proved to be elusive. Its accurate and reproducible measurement is of more than theoretic interest because the contractile function of the heart is a key determinant of prognosis for most cardiac diseases, and it is especially important in critical decisions regarding the timing of surgical correction in patients with VHD.
Historically, methods of assessing myocardial contractility have been divided into two groups, based on analysis of the isovolumetric or the ejection phase of cardiac contraction.4 Details of their clinical determination and relative reproducibility are beyond the scope of this discussion, but more information can be found in several excellent reviews,4,26,27 as well as in Chapters 5 and 14. The isovolumetric indices include measurements such as maximal velocity of myocardial fiber shortening (Vmax), peak pressure development (dP/dt), and peak dP/dt measured at an instantaneous pressure (dP/dt/P). Although relatively insensitive to loading conditions, these tests poorly reflect basal levels of contractility and are unreliable for comparing contractility among patients or assessing directional changes in contractility in an individual patient over time.26
Ejection phase indices, such as the ejection fraction (EF), are determined, in part, by the intrinsic inotropic state and can be used to define basal levels of contractility.26 Such measurements are extremely useful in evaluating ventricular function in patients with CAD or other conditions that do not significantly alter ventricular loading conditions.28 Ejection phase indices are understandably popular because they are readily available, and they are the most widely used clinical measures of left ventricular function. However, these indices are directly proportional to preload, vary inversely with ventricular afterload, and are accordingly unreliable for assessing contractile performance in patients with most forms of VHD.
The use of the pressure-volume diagram and analysis of the ESPVR allow a more precise appreciation of left ventricular contractility, which is independent of preload.29 The extent of myocardial shortening, and therefore of end-systolic fiber length, is a direct function of afterload (i.e., inverse force-velocity relation), and myocardial contractility can be evaluated by making use of this fundamental property. In most instances, end-systolic pressure (ESP) can be substituted for afterload. Only with pathologic degrees of ventricular hypertrophy is there a major divergence between ESP and afterload. This means that for any level of contractility, the ESV to which a ventricle contracts is a linearly increasing function of ESP. A stronger ventricle contracts to a smaller ESV (i.e., empties more completely) at any given level of ventricular afterload. Changes in the inotropic state also can be viewed in the context of the idealized pressure-volume loop. Positive inotropic interventions shift the curve up and to the left, increasing the work that can be performed at any given EDV (preload). Conversely, negative inotropic interventions shift the curve down and to the right30 (Figure 19-10; see Chapters 5 and 14). These load-independent indices of contractility are largely research tools, but in the future, it may be possible to construct pressure-volume loops and quantify ESPVRs in real time with echocardiographic equipment featuring automated border detection31 (see Chapter 12).
Clinical studies of patients with relatively normal loading conditions have shown that variations in the ESV reliably correlate with changes in ejection phase indices (Figure 19-11).32 The ESPVR represents an index of contractility that depends on systolic ventricular pressure (i.e., afterload) but is independent of end-diastolic length (i.e., preload).28,33,34 Pressure-volume loops also provide a framework for considering the interactions between systolic (inotropic) and diastolic (lusitropic) function. Although the preload (i.e., EDV) is an independent determinant of SV, because of the circular nature of blood flow, the SV ultimately determines venous return and the resultant preload for the next cardiac cycle.30 The “inotropically determined” ESV is the other element besides the venous return that contributes to the EDV. Just as the ESPVR uniquely describes systolic function, the end-diastolic pressure-volume relation is a manifestation of the intrinsic relaxation (lusitropic) properties of the ventricle. Positive lusitropic interventions facilitate ventricular filling and shift the end-diastolic pressure-volume relation down and to the right, and a negative lusitropic intervention shifts it up and to the left (Figure 19-12).
Aortic stenosis
Clinical Features and Natural History
Aortic stenosis (AS) is the most common cardiac valve lesion in the United States. Approximately 1% to 2% of the population is born with a bicuspid aortic valve, which is prone to stenosis with aging, and the population is aging. Clinically significant aortic valve stenosis is present in 2% of unselected individuals older than 65 years, and in 5.5% of those older than 85 years.35 Incipient aortic valve stenosis with calcification and stiffening is observed in 50% of people in the 75 to 80 age group and in up to 75% in those older than 85 years. Bicuspid aortic valve is a common congenital cardiac abnormality, occurring in approximately 1% of the population.36 The heritability coefficient is estimated at 89%, suggesting that the bicuspid aortic valve is almost entirely genetic in nature.37 It is a risk factor for premature AS and ascending aortic aneurysms. The ascending aorta in bicuspid valvular disease has the same histopathologic features as Marfan syndrome, such as medial degeneration, decreased fibrillin-1, and enhanced matrix metalloproteinase activity in the aortic wall. Recent data from the International Registry of Acute Aortic Dissection demonstrate that patients with bicuspid aortic valve have a 6.14% lifetime risk rate of aortic dissection (nine times that of the general population), compared with 40% for patients with Marfan syndrome. Yet, bicuspid aortic valvular disease is a hundred times more common than Marfan syndrome and is, therefore, responsible for a larger number of aortic dissections.36 Calcific AS has several features in common with CAD. Both conditions are more common in men, older people, and patients with hypercholesterolemia, and both result, in part, from an active inflammatory process. There is clinical evidence of an atherosclerotic hypothesis for the cellular mechanism of aortic valve stenosis. There is a clear association between clinical risk factors for atherosclerosis and the development of AS: increased lipoprotein levels, increased low-density lipoprotein (LDL) cholesterol, cigarette smoking, hypertension, diabetes mellitus, increased serum calcium and creatinine levels, and male sex.38 Homozygous familial hypercholesterolemia produces a severe form of AS in children.39 The early lesion of aortic valve sclerosis may be associated with CAD and vascular atherosclerosis. The extent of aortic valve calcification was observed as an important predictor of poor outcome in patients with AS.40 These and other studies suggest that aortic valve calcification is an inflammatory process promoted by atherosclerotic risk factors.
The early lesion of AS resembles that of the initial plaque of CAD, and there is a close correlation between calcification found in coronary arteries and the amount of calcification found in the aortic valve. Pathologic studies of aortic valves revealed the presence of LDL, suggesting a common cellular mechanism for the genesis of valvular and vascular atherosclerotic disease.38 Degenerative lesions on aortic valves contain an inflammatory infiltrate of nonfoam cells and foam cell macrophages, T lymphocytes, and other inflammatory cells. Lipid is accumulated in the fibrosa immediately below the endothelium layer on the aortic side of the valve. LDL- and apolipoprotein E–containing lipoproteins are present as well. Little is known about the synthesis of bone matrix proteins in calcific aortic valve stenosis. The calcifications are composed of hydroxyapatite on a matrix of collagen, osteopontin, and other bone matrix proteins.
A potential mechanism for this cellular inflammation process involves the combination of macrophages, LDL, and the secretion by macrophages of transforming growth factor-β and platelet-derived growth factor to stimulate the conversion of valvular fibroblast cells into osteoblasts that produce bone proteins.38 Most surprising are the findings that 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (e.g., statins) retard the progression of CAD and AS.41,42 The odds ratio of AS progression was 0.46 in 38 treated patients compared with 118 untreated patients.43 Other studies have shown similar results, and it is possible that therapy with statins or other drugs may be used to block or slow the progression of valve lesions in the future.38,44 In addition to recruitment of inflammatory cells and lipid accumulation into valves, calcification is a prominent feature that contributes to leaflet thickness and rigidity. Early development of aortic valve calcification may be related to vitamin D–receptor genotypes or mutations in the Notch gene.35 Calcification is an active process that includes osteopontin, osteonectin, osteocalcin, and other bone morphogenic proteins that regulate calcification and ossification. Active osteoblastic bone formation and osteoclastic bone resorption occur in stenotic valves. Increased fibroblasts produce collagen, leading to fibrosis. Concomitant elastin degradation also contributes to valve stiffening. Whereas normal valves are avascular, microvessels form in thickened stenotic valves, assuring the continued supply of inflammatory cells and lipids. Recent advances in genetic epidemiology have demonstrated a strong inheritability of bicuspid aortic valve with three loci on chromosomes 18q, 5q, and 13q, which likely contain genes responsible for bicuspid aortic valve.37 The mode of inheritance of other valvular diseases is unclear. No inheritability or inheritance studies have been reported for calcific aortic valve disease. In smaller genetic studies, a small number of candidate genes such as VDR, APOE, APOB, IL-10, and ESR1 have been identified as possibly playing a role in aortic valve disease. A recent breakthrough in the genetics of aortic valve disease was the identification of the Notch1 signaling pathway, involved in embryonic patterning, and that is highly expressed within the developing embryonic aortic valve. Notch1 is a repressor of Runx2, a critical regulator of osteoblast development. This supports the concept that a developmental program might be reactivated in disease processes.37
The rate of progression is, on average, a decrease in aortic valve area (AVA) of 0.1 cm2/year, and the peak instantaneous gradient increases by 10 mm Hg/year.45 The rate of progression of AS in men older than 60 is faster than in women, and it is faster in women older than 75 than in women 60 to 74 years old.46 Treatment with hemodialysis, the use of calcium supplements, and increased serum creatinine levels correlated with rapid progression of AS.47 Plasma brain natriuretic peptide, produced to a large extent by the ventricles, and the N-terminal part of the propeptide may serve as early markers of left ventricular hypertrophy (LVH), whereas atrial natriuretic peptide and the N-terminal part of the propeptide reflect atrial pressure increase.48 Repeated measurements of this marker may reflect information on the stage of AS and its hemodynamic impact.49
Angina, syncope, and congestive heart failure (CHF) are the classic symptoms of the disease, and their appearance is of serious prognostic significance because postmortem studies indicate that symptomatic AS is associated with a life expectancy of only 2 to 5 years.50–52 These early natural history studies were completed before the availability of cardiac catheterization studies, and some patients, although symptomatic, may have had objectively less severe degrees of stenosis. The “hemodynamically insignificant murmur of aortic stenosis” seen so often in cardiologic consultations does not portend such dire consequences, although it is sometimes difficult to correlate AS severity with clinical symptoms. There is evidence that patients with moderate AS (i.e., valve areas of 0.7 to 1.2 cm2) are also at increased risk for the development of complications, with the appearance of symptoms further increasing their risk.53 According to recent American College of Cardiology (ACC) and American Heart Association (AHA) guidelines, peak velocity greater than 4 m/sec, a mean gradient greater than 40 mm Hg, and a valve area less than 1.0 cm2 are considered hemodynamically severe AS.54 Aortic valve surgery should be performed promptly in symptomatic patients. In asymptomatic patients, high aortic valve calcium content and a positive exercise test are features that suggest a benefit from early aortic valve replacement (AVR).
An interesting question is whether the natural history of AS has changed significantly in recent years.55 The question is prompted by two trends. The first, at least in North America, is related to the steadily diminishing number of patients with rheumatic disease. Today, AS is essentially the bicuspid, the calcific, or what has been traditionally referred to as the senile variety. The second trend is related to the senile attribution. People are living longer, particularly those with heart disease. The typical patient with AS is older and much more likely to have other significant medical problems, including major coexisting cardiac disease, most often CAD.
Angina is a frequent and classic symptom of the disease, occurring in approximately two thirds of patients with critical AS, and about half of symptomatic patients are found to have anatomically significant CAD.56–58 However, a controversy persists about the incidence of CAD in patients with AS who do not have angina, and the controversy probably reflects this underlying sense of a change in the natural history of the disease. Some studies report that the absence of angina virtually excludes the possibility of atherosclerotic heart disease.59,60 In contrast, a 25% incidence rate of angiographically significant (> 70% obstruction) coronary occlusions was described in angina-free patients, most of whom had single-vessel disease.61 However, a much larger study found that 14% of patients with triple-vessel or left main CAD and AS presented without angina.62 Underscoring the continued lack of a consensus on this topic is the finding of a review that the reported incidence rates of significant CAD in asymptomatic patients range from 0% to 33%.63 Identification of asymptomatic patients is important because although coexistent, untreated (i.e., unbypassed) CAD has a detrimental effect on early and late survival after AVR, concomitant CABG with AVR does not increase perioperative mortality.64–67 It appears that a substantial number of patients with coexisting CAD may have symptoms other than chest pain.68 However, there do not appear to be other significant differences in the clinical or hemodynamic findings between patients with normal coronary arteries and those with coexistent coronary stenoses.
If the traditional natural history of AS is perhaps more ominous in the elderly patient with likely coexistent CAD, is there any rationale for prophylactic AVR? With steady improvement in surgical results, this question has been raised, although rigorous studies still suggest that asymptomatic patients with hemodynamically significant disease face a very low risk for sudden death before the onset of symptoms.69–71 As if by way of settling this particular question, an editorial by Braunwald72 stated that there was no role for prophylactic AVR, and that “operative treatment is the most common cause of sudden death in asymptomatic patients with aortic stenosis.” These patients warrant careful follow-up, and the case against prophylactic surgical intervention is further supported by studies showing that ventricular function is preserved and myocardial hypertrophy regresses after successful valve replacement.73–75
It is probably never too late to operate on patients with symptomatic AS.76,77 First, unlike AR, most symptomatic patients undergo valve replacement when left ventricular function is still normal. Second, even when impaired left ventricular function develops in AS, the relief of pressure overload almost always restores normal function or at least produces considerable improvement. Morbidity rates, mortality rates, and clinical results are favorable even in the oldest surgical candidates.78,79 Recent advances in operative techniques and perioperative management have contributed to excellent results after AVR in patients 80 years of age or older, with minimal incremental postoperative morbidity.80 The principal postoperative complication is respiratory failure.
The preoperative assessment of AS with Doppler echocardiography includes measurement of the AVA and the transvalvular pressure gradient.81,82 The latter is calculated from the Doppler-quantified transvalvular velocity of blood flow, which is increased in the presence of AS. This maximal velocity (v) is then inserted into the modified Bernoulli equation to determine the pressure gradient (PG) between the LV and the aorta (see Figure 19-2):
The pressure gradient is the maximal difference between the LV and aortic pressures that occurs during ventricular systole.83,84 This maximal instantaneous gradient is not the same as the peak-to-peak gradient determined by cardiac catheterization. The peak-to-peak gradient is determined by separate measurements of events that are not synchronous in real time. Of more practical interest is the fact that the best estimate of obstruction severity, as determined from pressure data alone, is the mean systolic gradient, which is calculated online by Doppler equipment85 (see Chapters 3 and 12).
The Doppler-calculated gradient is subject to the same flow limitation as invasively calculated gradients. Best understood by considering the extreme, this means that a patient with true end-stage AS would exhibit a relatively low calculated gradient because of minimal flow across a critically narrowed valve. In part because of this flow dependency, pressure gradients determined invasively or by Doppler echocardiography correctly classify AS severity in less than 50% of cases compared with estimates of AVA.86 However, the latter also can be determined by Doppler echocardiographic techniques. The preferred method requires only two Doppler-generated velocities: those proximal and those distal to the stenotic valve. These values are inserted into the continuity equation, which relates the respective velocities and cross-sectional areas proximal and distal to a stenotic area (see Figure 19-3). Specifically, the equation states:
in which AVA is the aortic valve area, V is the volume, and LVOT is the left ventricular outflow tract. Several studies have demonstrated the reliability of these Doppler-determined valve areas.83–86
Although advances in Doppler technology allow completely noninvasive evaluation of a large number of patients, coronary angiography is probably indicated in all patients older than 50 years who are demonstrated to have significant AS. Angiography and, if indicated, CABG may improve the long-term outlook for patients with CAD who undergo AVR.62 Angiography also can identify a smaller number of patients whose CAD alone would warrant CABG.
Pathophysiology
The normal AVA is 2.6 to 3.5 cm2, with hemodynamically significant obstruction usually occurring at cross-sectional valve areas of 1 cm2 or less. Generally accepted criteria for critical outflow obstruction include a systolic pressure gradient greater than 50 mm Hg, with a normal cardiac output, and an AVA of less than 0.4 cm2. In view of the ominous natural history of severe AS (AVA < 0.7 cm2),55,87 symptomatic patients with this degree of AS are generally referred for immediate AVR.88 The Hakki equation is a simplification of the Gorlin equation to calculate the AVA based on the cardiac output (CO) and the peak pressure gradient (PG) across the valve.
in which P is the intraventricular pressure, R is the inner radius, and h is the wall thickness.
This increase of wall tension is believed to be the direct stimulus for the further parallel replication of sarcomeres, which produces the concentrically hypertrophied ventricle characteristic of chronic pressure overload.89,90 The consequences of this LVH include alterations in diastolic compliance, potential imbalances in the myocardial oxygen supply and demand relationship, and possible deterioration of the intrinsic contractile performance of the myocardium.
Figure 19-13 shows a typical pressure-volume loop for a patient with AS. Two differences from the normal curve are immediately apparent. First, the peak pressure generated during systole is much greater because of the high transvalvular pressure gradient. Second, the slope of the diastolic limb is steeper, reflecting the reduced left ventricular diastolic compliance that is associated with the increase in chamber thickness.13 Clinically, this means that small changes in diastolic volume produce relatively large increases in ventricular filling pressure.
Figure 19-13 Pressure-volume loop in aortic stenosis.
(From Jackson JM, Thomas SJ, Lowenstein E: Anesthetic management of patients with valvular heart disease. Semin Anesth 1:239, 1982.)
This increased chamber stiffness places a premium on the contribution of atrial systole to ventricular filling, which in patients with AS may account for up to 40% of the LVEDV, rather than the 15% to 20% characteristic of the normal LV. Echocardiographic and radionuclide studies have documented that diastolic filling and ventricular relaxation are abnormal in patients with hypertrophy from a variety of causes, with significant prolongation of the isovolumic relaxation period being the most characteristic finding.91–94 This necessarily compromises the duration and amount of filling achieved during the early rapid diastolic filling phase and increases the relative contribution of atrial contraction to overall diastolic filling (Figure 19-14). A much greater mean left atrial (LA) pressure is necessary to distend the LV in the absence of the sinus mechanism. One treatment of junctional rhythm is volume infusion.
The systolic limb of the pressure-volume loop shows preservation of pump function, as evidenced by maintenance of the SV and EF (see Figure 19-13). It is likely that use of preload reserve and adequate LVH are the principal compensatory mechanisms that maintain forward flow. Clinical studies have confirmed that ejection performance is preserved at the expense of myocardial hypertrophy, and the adequacy of the hypertrophic response has been related to the degree to which it achieves normalization of wall stress, in accordance with the Laplace relation.95–97 LVH can be viewed as a compensatory physiologic response that completes a negative feedback loop (Figure 19-15). It is possible, however, that severe afterload stress and proportionately massive LVH could decrease subendocardial perfusion and superimpose a component of ischemic contractile dysfunction.
In patients with AS, LVH develops as a result of the increase in pressure load. The development of LVH and its regression after therapeutic interventions are accompanied by changes in the cardiac extracellular matrix guided by an increase in ECM1 gene expression during LVH and complete regression after complete correction.98
Systemic hypertension and AS represent an increase in afterload to the LV, and each contributes to left ventricular remodeling and LVH. In a large series of 193 patients with AS, 62 of whom were hypertensive, symptoms manifested with larger AVAs and lower stroke work loss.99 Patterns of left ventricular remodeling (i.e., concentric vs. eccentric remodeling and hypertrophy) were not different between hypertensive and normotensive patients. Left ventricular mass decreased by 23% 1 year after AVR, back into the normal range, in patients with normal preoperative ventricular function; diastolic function improved concomitantly. Improvements in myocardial blood flow and coronary vasodilator reserve after AVRs result from reduced extravascular compression and increased diastolic perfusion time.100–102
Ejection phase indices of contractile function are abnormal in many patients with AS.103,104 However, indices of contractile function, which are exquisitely sensitive to afterload, are inherently unreliable for quantitating the inotropic state in a disease such as AS, in which the essence of the hemodynamic insult is the severely increased ventricular afterload. This in no way excludes the possibility that a subset of patients also may experience some intrinsic depression of myocardial contractility. For example, patients with AS may be particularly at risk for superimposed, ischemic ventricular dysfunction, but this possibility can be assessed only by the application of load-insensitive measurements of contractile function.
In most patients, however, load-insensitive indices of contractility (i.e., end-systolic stress-diameter determinations) are virtually identical before and after the development of hypertrophy, suggesting that the increase in chamber thickness compensates for the afterload stress, and myocardial contractility is, therefore, normal, albeit appropriate for the higher afterload. Figure 19-16 illustrates the adaptation of the LV to chronic pressure overload. Figure 19-16A shows the pressure-volume relation before (loop A) and after (loop B) the development of concentric hypertrophy. The linear ESPVR is shifted up and to the left after concentric hypertrophy occurs. However, the ESPVR may not be as accurate or load insensitive in assessing contractility at the extremes of afterload. Figure 19-16B shows normalization of this apparently supranormal contractility, when wall stress (rather than pressure) is used as a more exact measure of ventricular afterload. Concentric hypertrophy normalizes wall stress, and contractility remains unchanged. A decline in any index of myocardial contractility in patients with AS may represent relatively inadequate hypertrophy for the degree of wall stress, some intrinsic depression of contractility, or a combination of these two factors.56,105,106 The fact that most patients have normal contractility is the reason for the usually favorable response to AVR.
Figure 19-16 Adaptation to pressure overload.
(From Ross J Jr: Afterload mismatch in aortic and mitral valve disease: Implications for surgical therapy. J Am Coll Cardiol 5:811, 1985.)
On the supply side, the greater LVEDP of the poorly compliant ventricle inevitably narrows the diastolic coronary perfusion pressure (CPP) gradient. With severe outflow obstruction, decreases in SV and resultant systemic hypotension may critically compromise coronary perfusion. A vicious cycle may develop because ischemia-induced abnormalities of diastolic relaxation can aggravate the compliance problem and further narrow the CPP gradient.107 This sets the stage for ischemic contractile dysfunction, additional decreases in SV, and worsening hypotension.
Cardiac hypertrophy also is associated with structural abnormalities of the coronary circulation (Figure 19-17).108,109 Animal models have documented that epicardial coronary vessels do not enlarge proportionately when the LV is subjected to a chronic pressure load.110,111 LVH accompanying chronic hypertension is associated with an increased wall lumen ratio of the coronary arterioles, a change that limits vessel dilation and augments active constriction.112,113 Animal models of pressure-induced myocardial hypertrophy also have shown a decrease in the capillary density of about 20% to 30%.108,110,112–116 There is considerable evidence that in LVH caused by pressure overload, a reduction in the coronary vascular reserve may underlie episodes of myocardial ischemia117,118 (see Chapter 6).
Figure 19-17 Possible coronary circulatory changes accompanying ventricular hypertrophy.
(From Marcus ML: Effects of cardiac hypertrophy on the coronary circulation. In Marcus ML [ed]: The coronary circulation in health and disease. New York: McGraw-Hill, 1983.)
Echocardiographic and hemodynamic assessments of the myocardial oxygen supply and demand ratio are not significantly different in the presence or absence of angina in patients with AS.119 However, considerable data indicate that dynamic factors relative to oxygen supply may be crucial to the pathogenesis of angina in these patients.120 Several clinical studies have documented a decrease in coronary vascular reserve in adult and pediatric patients with significant LVH or right ventricular hypertrophy (RVH).121–123 It is further postulated that repeated episodes of subendocardial ischemia may contribute to the development of subendocardial fibrosis, a component of ischemic contractile dysfunction.108
It is possible that myocardial ischemia underlies the impaired ventricular relaxation that has been documented in patients with myocardial hypertrophy because of a variety of causes.91,93,94 Prolongation of ventricular relaxation and resultant diastolic dysfunction (i.e., poor compliance) are probably universal features of myocardial ischemia in a variety of clinical settings.124–128 Experimental data suggest that ischemia-induced impairment of calcium sequestration by the sarcoplasmic reticulum may underlie the increased diastolic chamber stiffness.129 Prevention of ischemia-induced myoplasmic calcium overload may be the mechanism by which the calcium channel blockers are able to improve diastolic dysfunction in patients with CAD.130 These drugs also ameliorate ventricular relaxation and diastolic filling in patients with HCM, although the mechanism of action is controversial.91,131–133 The absence of diastolic filling abnormalities in patients with physiologic, as opposed to pathologic, hypertrophy also may reflect the relative likelihood of ischemia in the two conditions.134–136
In summary, the pathophysiologic response to chronic pressure overload is a complex one characterized by unique interaction among the divergent effects of hypertrophy on systolic and diastolic function. Mural thickening enhances systolic performance, maximizing the mechanical work performed while minimizing its metabolic cost. The price of this systolic efficiency is relatively inadequate growth of the coronary microcirculation, which contributes to relaxation abnormalities, diastolic dysfunction, and the potential for superimposition of ischemia-induced abnormalities of systolic dysfunction (Table 19-1).137 The potentially deleterious effects of LVH cannot be overemphasized. Even in the absence of AS, in which LVH can be viewed as a compensatory and potentially beneficial pathophysiologic response, LVH has been found to have an independent adverse effect on survival.138,139
| Beneficial Aspects | Detrimental Aspects |
|---|---|
| Increases ventricular work | Decreases ventricular diastolic distensibility |
| Normalizes wall stress | Impairs ventricular relaxation |
| Normalizes systolic shortening | Impairs coronary vasodilator reserve, leading to subendocardial ischemia |
From Lorell BH, Grossman W: Cardiac hypertrophy: The consequences for diastole. J Am Coll Cardiol 9:1189, 1987.
Difficulty of Low-Gradient, Low-Output Aortic Stenosis
A subset of patients with severe AS, left ventricular dysfunction, and low transvalvular gradient suffers a high operative mortality rate and poor prognosis.140 It is difficult to assess accurately the AVA in this low-flow, low-gradient AS because the calculated valve area is proportional to forward SV and because the Gorlin constant varies in low-flow states. Some patients with low-flow, low-gradient AS have a decreased AVA as a result of inadequate forward SV rather than anatomic stenosis. Surgical therapy is unlikely to benefit these patients because the underlying pathology is a weakly contractile myocardium. However, patients with severe anatomic AS may benefit from valve replacement despite the increased operative risk associated with the low-flow, low-gradient hemodynamic state.140 Guidelines from the ACC and AHA call for a dobutamine echocardiography evaluation to distinguish patients with fixed anatomic AS from those with flow-dependent AS with left ventricular dysfunction.141 Low-flow, low-gradient AS is defined as a mean gradient of less than 30 mm Hg and a calculated AVA less than 1.0 cm2.
Dobutamine echocardiography revealed three basic response patterns—fixed AS, relative AS, and absence of contractile reserve—in an initial study by deFilippi et al.142 In a series of 45 patients with low-flow, low-gradient AS, the 30-day operative mortality rate was 8% for patients with contractile reserve and 50% for those without contractile reserve.143 Dobutamine challenges during cardiac catheterization provided unique insights into low-flow, low-gradient AS, and the details are summarized in Figure 19-18.144 In Figure 19-18A, the transvalvular gradient and cardiac output increased, and the valve area did not change. Patients represented in Figure 19-18B increased their CO with little or no change in gradient, and the calculated valve area increased slightly. This group still benefited from surgery. The third group of patients had no contractile reserve because CO did not increase with dobutamine, and the transvalvular gradient decreased (see Figure 19-18C). Dobutamine infusion in the cardiac catheterization laboratory appears to be helpful in identifying patients with low-flow, low-gradient AS who have a truly fixed anatomic stenosis that may benefit from valve replacement. The findings of these studies also emphasize that contractile reserve is an important prognostic indicator in these patients and dobutamine challenge may help select patients for valve replacement. It appears that patients with contractile reserve and fixed AS have a relatively good prognosis with valve replacement. Left ventricular contractile reserve appears to be a critical variable for prognosis. Recent studies have focused on AS with low transvalvular gradients and normal ventricular EF. The pathophysiology of the low transvalvular gradient has been explained by decreased EDV caused by excessive ventricular hypertrophy accompanied by increased SVR. In a recent series of patients with severe AS (AVA < 0.8 cm2) with ventricular dysfunction (EF < 35%) and/or a low transvalvular gradient (< 30 mm Hg), mortality predictors were advanced age, low EF, renal insufficiency, and lack of AR. Regardless of ventricular function, patients who underwent AVR had a significantly better survival.36
Figure 19-18 Hemodynamic tracings from three patients representing three different responses to dobutamine.
(Adapted from Nishimura RA, Grantham JA, Connolly HM, et al: Low-output, low-gradient aortic stenosis in patients with depressed left ventricular systolic function: The clinical utility of the dobutamine challenge in the catheterization laboratory. Circulation 106:809, 2002.)
Developments in the Hemodynamic Management of Critical Aortic Stenosis Patients
The use of vasodilators is traditionally contraindicated in patients with severe AS because cardiac output is relatively fixed across a narrowed orifice. Vasodilation reduces SVR without any possibility for a compensatory increase in CO and severe hypotension would typically result. This traditional paradigm recently was re-examined in patients with fixed, severe AS (area < 1.0 cm2) and left ventricular dysfunction (EF < 0.35).145
[/not-level-membership-for-anesthesiology-category]
