[level-membership-for-critical-care-medicine-category]32
Valvular Heart Disease in Critical Care
Rheumatic heart disease accounted for 50% or more of admissions for heart disease in the first half of the twentieth century, with congenital heart disease representing the other major indication for valve surgery through the 1960s.1 With evolving technology and an increase in average life expectancy, heart valve surgery has shifted to valve repair, as well as replacement for a variety of acquired valvulopathies. The therapeutic armamentarium, once limited to minimal supportive medical therapy, then solely to valve replacement, has expanded to a variety of surgical repair techniques; a multitude of tissue and mechanical valve options for replacement; and, most recently, an expanding set of percutaneous interventions including percutaneous valve implantation and repair. In addition, various mechanical devices for temporary hemodynamic support are important adjuncts to the management of patients with valvular heart disease in the critical care setting.
Diagnosis of valvular heart disease in critical care units is challenging, with lack of a quiet environment for auscultation; comorbid conditions that affect physical findings; and stress, infection, or metabolic abnormalities that result in tachycardia and shorter intervals for evaluation of heart sounds. Patients in a low output state have softer heart sounds and murmurs as well. In addition, clinical reliance on physical examination has fallen, as has the ability to make accurate diagnosis of valve disease.2 A delay in recognizing the presence of significant valve dysfunction continues to occur frequently, and some patients who have had prior routine medical outpatient care nevertheless present with undiagnosed valvular heart disease.3 The currently accepted thresholds for mild, moderate, and severe valvular heart disease are shown in Table 32.1.
Table 32.1
Classification of Severity of Valvular Heart Disease Based on ACC/AHA Guidelines
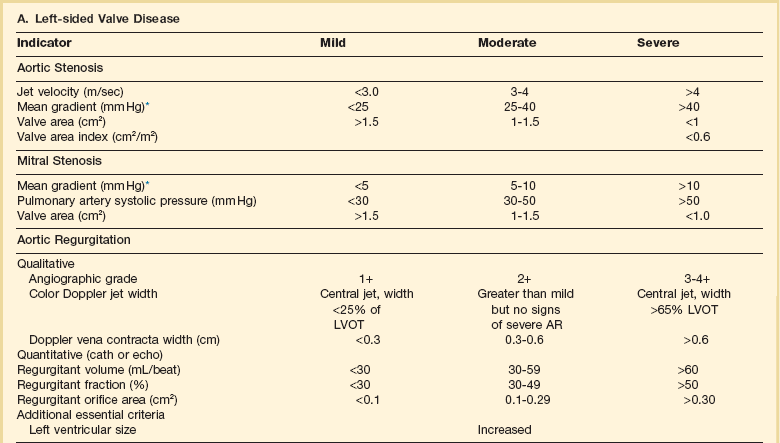
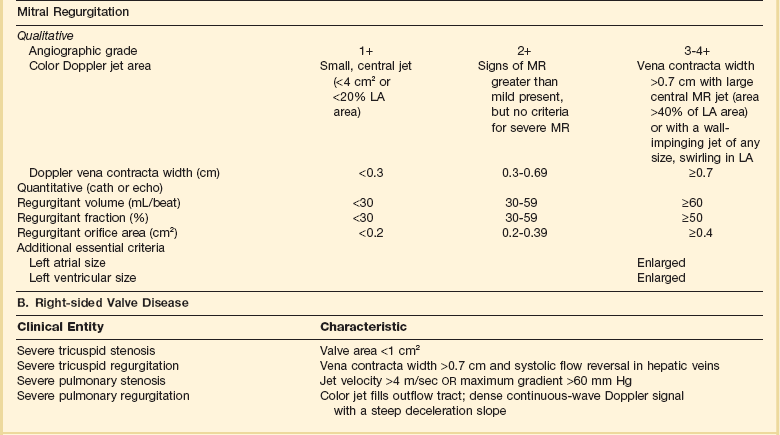
*Valve gradients are flow-dependent and when used as estimates of severity of valve stenosis should be assessed with knowledge of cardiac output or forward flow across the valve.
Modified from Bonow RO, Carabello BA, Chatterjee K, et al: ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 1998 guidelines for the management of patients with valvular heart disease) developed in collaboration with the Society of Cardiovascular Anesthesiologists and endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. J Am Coll Cardiol 2006;48:e1-148.
Aortic Stenosis
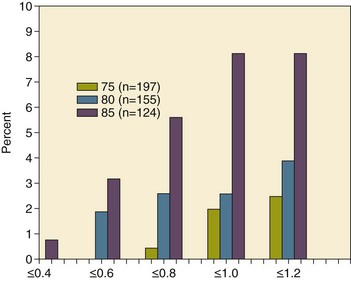
With the aging of the population, aortic stenosis (AS) has moved to the forefront in frequency of valvular heart disease encountered in older populations. The prevalence is surprisingly high; in patients 85 years of age, more than 8% of a random general population survey had Doppler-derived aortic valve area estimates of 1 cm2 or less (Fig. 32.1),4 which is consistent with severe AS by the 2006 American College of Cardiology/American Heart Association (ACC/AHA) guidelines.5 Thus AS should be considered in the differential diagnosis of each elderly critical care patient with hemodynamic instability. The possibility of AS should be dismissed only after considering physical examination and electrocardiographic and, when applicable, echocardiographic and invasive findings, all of which have potential limitations in achieving accurate diagnosis as discussed subsequently.6
Pathophysiology
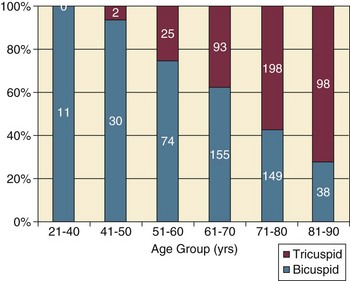
AS represents a continuum, from hemodynamically insignificant congenital and atherosclerotic disease to end-stage decompensation secondary to severe valvular obstruction. The congenital pathway is typically secondary to a bicuspid aortic valve, the most common congenital cardiac anomaly if mitral valve prolapse is excluded, occurring in approximately 1.5% of the population7 and originally described by Da Vinci in 1513.8 The most common acquired anomaly is typically referred to as degenerative disease, an atherosclerotic process that represents a continuum from aortic valve sclerosis, which is not associated with a significant gradient, to a densely calcified aortic valve with severe outflow obstruction. Although the cause of aortic valve stenosis in patients undergoing aortic valve replacement (AVR) has shifted to calcific tricuspid aortic valve disease from bicuspid and rheumatic disease, this remains an age-related phenomenon, with a predominance of bicuspid aortic valve disease in patients younger than age 70 (Fig. 32.2).9
Aortic Valve Sclerosis
Aortic valve sclerosis is an important disease entity, although usually not because of hemodynamic considerations. Defined as calcification and thickening of the aortic valve without significant outflow obstruction (gradient <20 to 25 mm Hg), it is present in nearly 30% of the population older than age 65 and nearly 50% by age 8510 and is associated with a 50% increase in 5-year cardiovascular mortality rate.11 Its incidence is as high as 15-fold greater than aortic valve stenosis; the two diagnoses can be differentiated from one another by hemodynamics and physical examination findings discussed subsequently. In keeping with the primary atherosclerotic nature of aortic sclerosis, it is associated with increasing age, male gender, hypertension, smoking, elevated low-density lipoprotein and lipoprotein(a) levels, and diabetes.10 The primary importance of aortic valve sclerosis is that it provides a window on the overall presence of vascular disease, in particular coronary artery disease.12 Thus there should be a high index of suspicion of vascular disease in any patient with aortic valve sclerosis managed in the critical care setting.
Patients with aortic sclerosis do progress to aortic valve stenosis; a study of more than 2000 patients with valve thickening showed progression to severe AS in 2.5% over an average time interval of 7 years.13 The fact that so-called degenerative disease of the aortic valve is absent in nearly half of octogenarians is also important to consider because it implies that it is not only aging but other factors that result in leaflet thickening, calcification, and stenosis. In the Helsinki Aging Study from which data are reflected in Figure 32.1, additional analysis demonstrated that not only age but also hypertension and low body mass index independently predicted calcification of the aortic valve, and age and serum ionized calcium were independently associated with valvular stenosis.14 In general the process of sclerosis and then stenosis of the aortic valve appears, like atherosclerosis in general, to be an active inflammatory process, with deposition of lipoproteins, local inflammation with T lymphocyte and macrophage infiltration, fibroblast proliferation, and eventually osteoblast and bone formation.15 Similar to other vascular disease, endothelial disruption likely leads to the initial lipid deposition in the leaflet tissues. The areas of early focal plaque formation appear at the loci of greatest stress: on the aortic side of the leaflets at the flexion points. Because bicuspid valves have greater mechanical stress, the average age at presentation of patients with bicuspid aortic valve stenosis is significantly lower than in the tricuspid aortic valve stenosis that is typically seen in an elderly population.16
An important element in understanding calcific AS is lack of commissural fusion; unlike rheumatic mitral valve stenosis, in which fusion of the commissures is the primary cause of obstructive disease, lack of mobility of the aortic valve leaflets is the primary cause of obstruction in AS. Rheumatic AS is associated with commissural fusion but is now a rare finding, even in countries where rheumatic heart disease is prevalent, and is usually associated with mitral valve involvement and typically aortic insufficiency as well. More obscure causes, such as unicuspid and quadricuspid aortic valve disease, are uncommon, although presentation can be delayed to adulthood. In the case of unicuspid aortic valves there is a strong association with AS,17 whereas with quadricuspid aortic valves there is a high incidence of significant aortic insufficiency.18
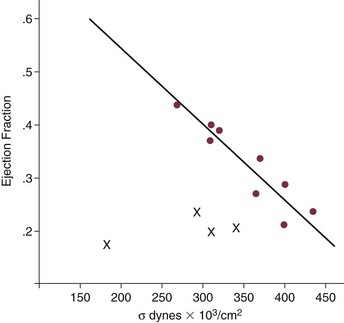
Normal aortic valve area ranges from 3 to 4 cm2. Significant resistance to outflow does not occur until the valve orifice is reduced more than 50%. Based on the simple hydraulic principle of Poiseuille’s law, a 50% reduction in valve diameter results in approximately a 16-fold increase in resistance. In practice, as the resistance to outflow rises, the left ventricle is subject to pressure overload, resulting in compensatory hypertrophy. This in turn normalizes wall stress because the latter is proportional to chamber diameter times pressure divided by wall thickness (the LaPlace principle). The degree of wall thickness is variable: In patients with inadequate hypertrophic response, wall stress is inordinately high and there is early dilation and dysfunction.19 In patients with a profound hypertrophic response disproportionate to valvular resistance, wall stress actually falls below normal and ejection fraction (EF) becomes supranormal,20 a phenomenon that appears to be more common in women with AS. The EF for any degree of wall stress is predictable,19 and patients who have disproportionately poor ejection phase indices (afterload mismatch) typically manifest left ventricular (LV) dysfunction beyond the depression that would be associated with high wall stress (Fig. 32.3),21 a phenomenon described under low-gradient, low-output AS that follows.
LV hypertrophy, although it reduces wall stress, has several features that may be deleterious. With progressive hypertrophy, diastolic pressures may rise as LV compliance decreases, resulting in increased LV filling pressures. In addition, the myocardial supply-demand relationship is deleteriously affected by hypertrophy in this setting. With increased wall stress and increased wall mass, myocardial oxygen demand is increased; at the same time coronary flow reserve is decreased in AS,22 and in later stages of the disease diastolic perfusion pressure is lowered. Because of abnormal flow reserve, conventional testing for ischemia in the setting of significant aortic valve stenosis has inadequate specificity to identify the presence or absence of hemodynamically significant concomitant coronary artery disease.23
Other features associated with AS that are important in the critical care setting include abnormal platelet function and decreased levels of von Willebrand factor,24 with associated bleeding risk that improves with AVR. Another associated cause of bleeding is gastrointestinal angiodysplasia associated with aortic valve stenosis,25 possibly exacerbated by the concomitant presence of von Willebrand syndrome.
The progression of AS is highly variable, but with the onset of moderately severe disease, the estimated mean decrease in valve area is on the order of 0.1 cm2/year.26 Certain demographics and noninvasive findings appear to predict the rate of progression, including age older than 50 years and moderate to severe valve calcification.27 The issue of progression is particularly important in patients who are asymptomatic of the disease because AVR before it is necessary is undesirable but must be weighed against the risk of sudden death. The latter has been studied in a number of longitudinal studies and is rare if patients are followed prospectively. The patients who are most likely to require early intervention are those with peak Doppler systolic velocities greater than 4 m/second or who have a rapid increase in serial transvalvular Doppler velocity measurements.26,27 This threshold has been confirmed by other series.28 Once patients become symptomatic, with a classic triad of heart failure, angina, or syncope, hemodynamic deterioration can be rapid with significant associated mortality risk.29
Diagnosis
Physical Examination
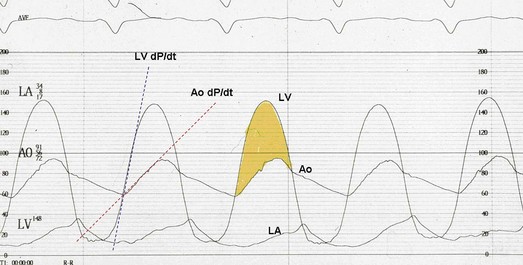
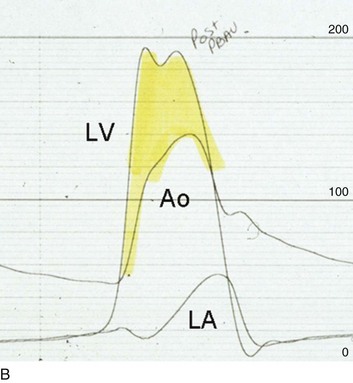
The hallmarks of the physical examination relate to the contour of the aortic pressure upstroke, with a low volume and delayed carotid upstroke (pulsus parvus et tardus), a late peaking systolic ejection murmur, and a diminished or absent aortic second sound. Figure 32.4 demonstrates the hemodynamics that manifest in an abnormal carotid pulse contour; unlike the brisk upstroke of LV pressure over time (dP/dt), the aortic pressure is slow to rise and of significantly lower volume. An ejection click may be present in young patients with congenital AS, but with increasing calcification of the valve, mobility decreases to a point at which an ejection click becomes unlikely. The aortic component of the second heart sound, reflecting deceleration and closure of the aortic valve, is diminished or absent in severe AS when the thickened aortic valve leaflets are poorly mobile, have little deceleration, and drift rather than snap shut. The murmur of AS is characteristically a crescendo-decrescendo murmur that is late peaking, consistent with the timing of the peak gradient as seen in Figure 32.4, and is typically best heard over the right upper sternum and clavicle. It reflects the high-velocity systolic jet directed into the ascending aorta. A second component, described by Gallavardin as a musical component, is best heard at the left lower sternal border. The latter confounds diagnosis because the murmur is suggestive of mitral regurgitation (MR), but in its true form it is purely secondary to aortic valve stenosis. Because handgrip raises resistance to LV ejection, the murmur should decrease if it is a true Gallavardin phenomenon, whereas when it is secondary to MR, it should increase.30
Unfortunately, none of these findings has sufficiently high sensitivity or specificity for AS or for differentiating moderate from severe disease.3 Many patients with AS, particularly the elderly with stiff noncompliant vessels, will have a wide pulse pressure even with declining cardiac output and may have systolic hypertension. Hypotension may not be seen until late stages of the disease. The loudness of the murmur, which correlates to some degree with severity, is also not specific because body habitus and a variety of other factors affect the acoustics transmitted across the chest. Most importantly, in severe AS, as the cardiac output drops, the gradient generated decreases as well. In this setting the murmur may be quite soft, although sometimes still high pitched because of the high velocity across a tight constriction. A useful means of differentiating aortic sclerosis from stenosis is that in the former the systolic ejection murmur heard over the right sternum is typically midpeaking and mild to moderate in intensity, and it features a well-preserved aortic second heart sound. Although the physical findings described are variable, presence of the aortic second heart sound tends to exclude severe calcific AS.31
Noninvasive Evaluation
In contrast to the physical examination, echocardiographic techniques have progressively improved over the past several decades and availability in the critical care setting in industrialized nations is ubiquitous. The characteristic echocardiographic features of AS are decreased valve leaflet mobility, calcification in all except congenital AS in adolescents and young adults, and an augmented Doppler velocity that generally allows accurate estimation of the gradient. In congenital bicuspid AS in young adults, when commissural fusion is dominant, a characteristic doming pattern is seen, but this disappears with progressive valve calcification. The severity of calcification correlates with extent of obstruction by middle age, and typically Doppler signal velocity with peak and mean pressure gradient and valve area by continuity equation provide an accurate overall assessment. However, the gradient is highly dependent on flow across the valve, and in low-output states the gradient may result in underestimation of severity of disease; in high-output states such as in patients with augmented cardiac output caused by inotropic stimulation, endogenous high catecholamine states, and sepsis, the gradient may be disproportionately higher than the severity of stenosis would suggest. The latest ACC/AHA guidelines describe the threshold for severe AS as antegrade jet velocity greater than 4 m/second, gradient greater than 40 mm Hg, and aortic valve area less than 1 cm2, with the caveat that the gradient and jet velocity depend on the overall transvalvular flow5 (see Table 32.1). Importantly, antegrade flow across the aortic valve does not equal cardiac output in patients with confounding conditions, including aortic insufficiency; thus, for example, a patient with mild to moderate AS and moderate aortic insufficiency may have a transvalvular gradient suggestive of severe AS. Finally, although most practitioners do not index the valve area, it is important to appreciate that in patients with large body surface areas, disease that might be considered moderate in smaller patients may be functionally severe.
Several caveats need to be considered before acceptance of noninvasive data in the critical care setting. Inadequate acoustic windows in some patients and difficulty in positioning patients on respirators and with multiple lines in place do limit echocardiographic imaging. Technical errors in recording and interpretation result in significant misdiagnosis: errors in recording angle, inadvertent imaging of MR jets instead of aortic outflow, assessing proximal velocity instead of the transvalvular signal, and selecting signals in the setting of arrhythmias that are not representative of mean heartbeats can all lead to skewed assessment of valve disease severity.32 Further, a number of other potential confounding variables can lead to overestimation or underestimation of the severity of AS,6,33 and valve area calculations by the continuity equation in a low-flow setting may be inaccurate.34 Because the continuity equation depends on measurement of the LV outflow track dimension, which is prone to inaccuracy, another measure of AS severity, the dimensionless index (the ratio of blood flow velocity across the LV outflow track to that across the aortic valve), has been widely adopted.35 A number of other techniques for determining the severity of AS supplement the most commonly used noninvasive tools, including three-dimensional echo, computed tomography,36 and magnetic resonance imaging (MRI).37 Overall, there has been a major a shift in the gold standard from catheter-derived to noninvasive-derived determination of AS severity, and cardiac catheterization is no longer indicated for hemodynamic assessment of aortic valve disease severity when noninvasive findings are unequivocal.5
The echocardiogram provides important additional information including severity of LV hypertrophy, LV function and size, and concomitant disease of other heart valves. The presence of regional wall motion abnormalities, in the absence of a conduction disturbance, suggests concomitant coronary artery disease. The electrocardiogram (ECG) in AS is typically abnormal and frequently features LV hypertrophy, with ST-segment abnormalities in the lateral leads typically described as a strain pattern.38 Although occasionally mistaken for anterior ischemia or infarction, with loss of R voltage across the precordium and occasionally with narrow QS complexes, the ECG is not specific for severity of AS. With increasing calcification of the perivalvular tissues, heart block is seen, typically late in the course of the disease.
Cardiac Catheterization
Cardiac catheterization is indicated in patients in whom the noninvasive data are equivocal, and coronary angiography is indicated in a range of settings identified in Box 32.1. The hemodynamics of AS are best recorded with a catheter or catheters placed simultaneously on either side of the aortic valve.6,39 Unfortunately, most laboratories record femoral artery pressure in lieu of central aortic pressure or do a catheter pullback in lieu of simultaneous pressure tracings; both techniques can result in significant misinterpretation of the severity of aortic valve disease.40 A variety of other errors in catheterization laboratory pressure measurements makes the catheter-derived valve area generally more variable than desirable in all except a few laboratories, including inherent errors in estimating rather than measuring oxygen consumption and in assuming that the Gorlin constant (originally established for a limited subset of patients41) and the valve area itself remain constant under varying loading conditions.34 In the catheterization laboratory as well, transvalvular flow may be underestimated if there is concomitant aortic insufficiency, resulting in overestimation of the severity of aortic valve disease.
Low-Gradient, Low-Output Aortic Stenosis
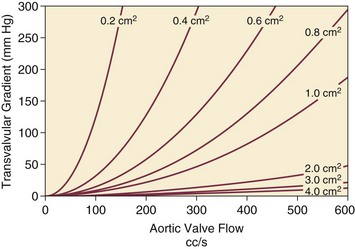
The critical care unit patient with a low to moderate gradient (typically <30 mm Hg) across the aortic valve in the setting of low cardiac output represents an important conundrum (and should be differentiated from the aortic sclerosis patient with low gradient not associated with depressed output). In general, the rules of hydraulics demonstrate that valve area is proportional to flow divided by the square root of the gradient.41 Thus, when the valve area is fixed, increased flow is associated with an exponential rise in gradient when valve areas are less than 1 cm2 (Fig. 32.5). When a patient in the critical care setting has depressed cardiac output and a low gradient across the aortic valve, there are two possible interpretations of these findings: either the patient has mild to moderate AS and poor LV function, or the patient has severe AS with a low EF appropriate to high wall stress (see Fig. 32.3) and secondary depression of cardiac output. In the case of the former, increasing flow results in better opening of the aortic valve, with only mild to moderate increase in gradient and an increase in the calculated valve area of 0.3 cm2 or greater.42 In the latter case, a fixed obstruction to outflow exists and increasing transvalvular flow results in a dramatic increase in gradient. In addition to increasing flow across the valve in these patients, typically with dobutamine infusion,43 the echocardiogram can be useful in several other ways44: valve calcification is suggestive of fixed outflow obstruction and, if severe, suggests that the underlying disease is severe AS rather than LV dysfunction. Preserved LV contractile reserve, with significant rise in stroke volume (>20%), peak velocity (>0.6 m/second), or mean transvalvular gradient (>10 mm Hg) at the time of dobutamine infusion is an additional useful marker. Lack of contractile reserve has been associated with lower operative survival rate45 (6% vs. 33% in one study), although it should not be the sole parameter for the decision on whether or not to perform AVR: patients who survive may manifest significant recovery in LV function postoperatively.46 Low contractile reserve should not preclude AVR in patients who do have severe fixed obstruction because their prognosis without surgery is abysmal.47 One other variation of low-gradient low-output AS involves patients with small hypertrophied ventricles and preserved EF. This subset, sometimes called high valvuloarterial impedance AS, has low gradient because of low stroke volume; the burgeoning evidence base does show improved results with AVR.48
Therapy
Medical Management
Despite the caveats, vasodilator therapy, including nitroprusside, has been used in the critical care setting in patients with severe LV dysfunction and severe AS.49 In a select group of patients not dependent on inotropes, cardiac output rose significantly and there was overall hemodynamic improvement. Similarly, intra-aortic balloon pumping has been used, although there are only isolated case reports of efficacy.50,51 As would be expected, both nitroprusside and intra-aortic balloon pumping result in afterload reduction and some increase in transvalvular flow along with some increase in aortic valve gradient.
Lipid-lowering therapy and converting enzyme inhibitors have been the subject of substantial investigation,52 although their role is in the chronic setting rather than during critical care. Two prospective randomized trials failed to show clear benefits of a statin53,54 in slowing disease progression. Some preliminary evidence suggests potential benefits of angiotensin-converting enzyme (ACE) inhibitors in decreasing progression of aortic valve calcification,55,56 but the available clinical data do not show slowing of AS progression.57 Both lipid-lowering and converting enzyme inhibitors need to be tested in larger populations earlier in the course of aortic valve disease.
Percutaneous Interventions
Balloon Valvuloplasty
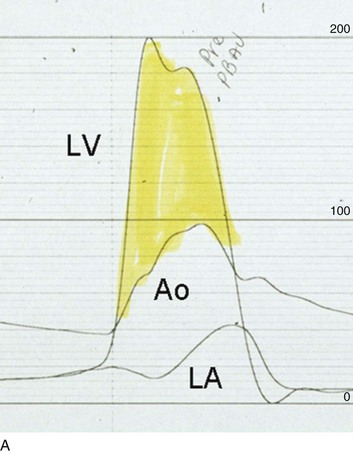
The acute hemodynamic results (Fig. 32.6) are poor compared with those of AVR, with a 50% reduction in gradient and an increase in aortic valve area of only 0.2 to 0.3 cm2.58 The in-hospital mortality rate was close to 10% in the National Heart, Lung and Blood Institute registry59 and is substantially higher in patients with hemodynamic decompensation and multiorgan failure. Most valves have restenosed within a few months, with a recent series showing an 87% mortality rate after a median period of less than 7 months,60 and no benefit for long-term outcomes has been shown. Indeed, the overall clinical course is not influenced by balloon valvotomy.61 Nevertheless, it can serve as a bridge to transcatheter AVR or surgical AVR in select patients with multiple comorbid conditions and is an alternative but low efficacy measure for patients who are not candidates for AVR. It is not an alternative to TAVR or surgery for patients who can undergo valve replacement, even for most patients at relatively high risk for the latter.
Several settings in which balloon aortic valvuloplasty was previously considered to have a role are no longer included as indications in the 2006 ACC/AHA guidelines.5 These settings include preoperative balloon dilation in patients undergoing noncardiac surgery62; in general all but decompensated patients can withstand general anesthesia as long as careful hemodynamic monitoring and anesthesia management are performed.63 Patients who require urgent noncardiac surgery in the setting of severe symptomatic AS do have a significant perioperative risk,64 and preoperative AVR should be considered if at all possible. Balloon aortic valvuloplasty has also been proposed as a tool to differentiate myocardial dysfunction from severe AS with secondary LV dysfunction in low-gradient, low-output AS.65 The latter patients typically demonstrated substantial improvement in stroke volume and EF after balloon dilation, but dobutamine stress testing is a far safer, less morbid screening tool.
Transcatheter Aortic Valve Replacement
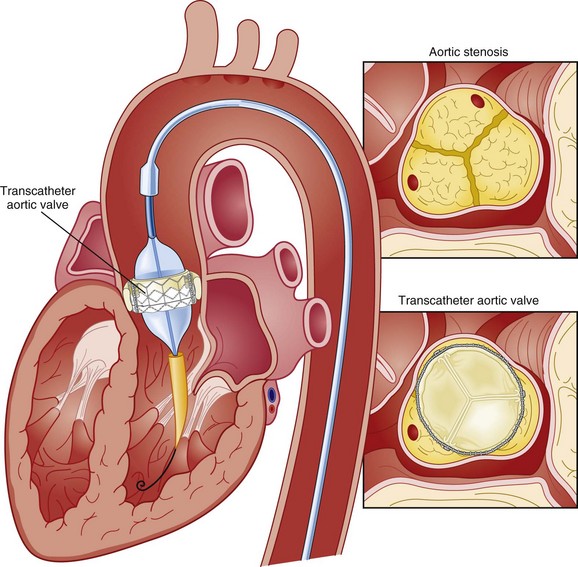
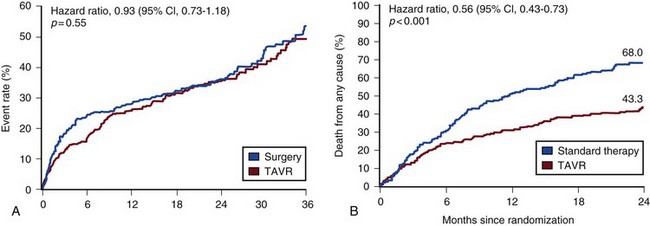
Transcatheter aortic valve replacement (TAVR), a technology developed in the past decade, has revolutionized the treatment of AS. The procedure is of increasing importance in the critical care setting. Tissue valves are sewn to either balloon expandable or self-expanding stents, which are crimped onto a catheter and advanced across the stenotic native valve. Both balloon expandable (Fig. 32.7) and self-expanding technologies are widely available outside the United States. The landmark PARTNER (Placement of AoRtic TraNscathetER valves) study had two cohorts: PARTNER A enrolled high-risk but operable patients while PARTNER B enrolled patients deemed inoperable. PARTNER A randomized patients to surgical aortic valve replacement (SAVR) versus two types of TAVRs: transfemoral if the iliac and femoral vasculature was suitable for accommodating the large catheter shafts or transapical if not.66 The results of TAVR and SAVR were similar, including mortality rate, stroke, and quality of life with published follow-up through 2 years; vascular complications were higher with TAVR and major bleeding and AF were higher with SAVR66 (Fig. 32.8A). PARTNER B randomized patients with suitable vasculature to transfemoral TAVR versus medical therapy.67 Patients who received only medical therapy (with or without balloon valvuloplasty) had a 51% 1-year mortality rate compared to those inoperable patients randomized to TAVR who had a 31% 1-year mortality rate, an approximately 40% relative reduction (Fig. 32.8B). Thus, TAVR appears to be among the most dramatically successful of all modern cardiac interventions. However, it is noteworthy that the residual mortality rate in the inoperable TAVR patients remains very high, reflecting the severe comorbid conditions in patients who met inoperability criteria. There was also an initially higher stroke risk in the TAVR patients.67 The risk of stroke in follow-up appears to be largely related to patient comorbid conditions that increase stroke risk, rather than the type of valve replacement.68 The dramatic mortality risk benefit with TAVR in inoperable patients has been shown to continue through 2 years of follow-up; because the stroke risk is higher in the surviving medically treated population, the early excess stroke risk associated with TAVR largely resolves by 24 months.69 Outside the United States, TAVR has been performed in some 70,000 patients; the Food and Drug Administration initially approved TAVR for inoperable patients based on the results of PARTNER B and subsequently approved the high-risk population treated in PARTNER A. A variety of investigational uses of the TAVR platform, such as valve-in-valve replacement for patients with bioprosthetic valve dysfunction (AS and aortic insufficiency) are being performed, primarily outside the United States70; the possibility that transcatheter valve-in-valve replacement will eventually supplant reoperation may change the algorithm for choice of tissue versus mechanical AVR.
The management of TAVR patients after valve replacement frequently takes place in the critical care unit. The higher rate of vascular complications associated with TAVR requires careful monitoring for limb ischemia, retroperitoneal hemorrhage, and a variety of other vascular complications.71 Conduction disturbances, particularly after use of a self-expanding TAVR platform, occur in a significant percentage of cases, requiring permanent pacemaker placement.72 A number of issues, such as the use of dual antiplatelet therapy and anticoagulation after TAVR remain to be fully investigated; most operators treat with dual antiplatelet therapy for periods ranging from 1 to 6 months; dual antiplatelet therapy plus anticoagulation for patients with AF is a combination with particularly high major bleeding risk, though the evidence base is incomplete, and many clinicians use only aspirin and warfarin in this setting.
Aortic Valve Replacement
AVR remains the treatment of choice for severe AS in patients who are considered to have reasonable operable risk. Class I indications are severe AS (valve area less than 1 cm2) with symptoms or, regardless of symptoms, if patients have severe AS and are undergoing coronary artery bypass, are undergoing surgery of the aorta or other heart valves, or have LV dysfunction. A long list of class II indications generally focuses on patients who do not meet class I criteria but are thought to be at increased risk of rapid disease progression or hemodynamic compromise. In addition, patients with moderate AS who undergo other cardiac surgery generally have simultaneous AVR. Survival after AVR is excellent if LV function is preserved, with operative mortality rate in ideal candidates as low as 1%. In these patients, age-matched survival is not significantly different from patients without AS.73 In the setting of LV dysfunction, however, postoperative life expectancy is relatively poor.74 Nevertheless, conservative therapy for severe AS remains an undesirable alternative: A recent review of 453 patients treated without intervention despite severe AS had 1-year, 5-year, and 10-year survival rates of only 62%, 32%, and 18%, respectively,75 with the worst survival in patients with renal failure, pulmonary hypertension, age older than 75 years, diminished EF, and congestive heart failure.
Aortic Insufficiency
The overall prevalence of moderate or severe aortic insufficiency (AI) in the United States, as shown in the Framingham Offspring Study, ranged from 0.3% in the fifth decade of life to 2.2% for patients aged 70 to 83.76 The frequency is age and male gender related. The diagnosis of AI is more difficult and the treatment issues in many ways more complex than for AS, with less of a clear dichotomy in the decision tree for valve replacement. AI needs to be considered in light of its acuity: Acute AI is usually associated with life-threatening comorbid conditions and the resultant acute regurgitation places great stress on the left ventricle and may itself be fatal. In contrast, chronic AI may evolve from clinically insignificant to requiring surgery over decades. Unlike AS, it is not primarily a disease of the aging process (although it does occur more frequently with increasing age) and is typically secondary to one of an extensive list of systemic or structural diseases that result in insufficiency of the aortic valve. Acute AI frequently requires urgent surgery, whereas chronic AI may be managed by watchful waiting and some limited options for medical therapy.
Pathophysiology
Acute AI is generally associated with leaflet involvement by endocarditis or disruption of the aortic valve’s annular structure by dissection or trauma. Chronic AI, by contrast, is caused by congenital valve abnormalities, most importantly bicuspid aortic valve disease, or by the degenerative process described earlier associated with AS. Conditions that distort the annulus and aortic root including systemic hypertension have been considered to be important causes of secondary AI, although the association between chronic AI and systemic hypertension remains to be confirmed.76,77 Rheumatic AI, almost invariably associated with mitral valve involvement as well, remains prevalent in developing countries but is an uncommon cause of AI in industrialized nations.
In general, causes of AI (Box 32.2) have been divided into those that affect the leaflets primarily and those that affect the root and annulus. The former includes bicuspid and other aortic valve abnormalities, endocarditis, rheumatic aortic valve disease, the atherosclerotic process described earlier, connective tissue disorders, antiphospholipid syndrome (Libman-Sacks endocarditis), and toxicity from anorectic drugs.78 The aortic root and annulus are affected by a variety of comorbid conditions that dilate the aortic root; Marfan and Ehlers-Danlos syndromes; osteogenesis imperfecta; chronic aortic dissection; syphilitic aortitis; connective tissue disorders; and, along with the valve leaflets, ankylosing spondylitis.
In isolated acute AI, the sudden onset of regurgitation imposes a large-volume load on the left ventricle in diastole prior to an adaptive process being in place. The abrupt rise in pressure is reflected by parallel development of left atrial and pulmonary vascular hypertension, frequently resulting in pulmonary edema. Because the compliance of a previously unaffected left ventricle is not sufficient to allow adequate dilation to absorb the regurgitant volume, the ventricle operates on the steep portion of its pressure-volume curve, with inadequate stroke volume to accommodate the high regurgitant flow. The effective (net) forward stroke volume (antegrade flow minus retrograde filling) is therefore low, compensated to some degree by tachycardia, but frequently insufficient to maintain normal cardiac output. This phenomenon is exaggerated in patients with preexistent LV hypertrophy in whom the ventricle is already operating on the steep portion of its pressure-volume curve, and particularly severe decompensation is seen when the left ventricle is small and hypertrophic. Critical care settings for the latter unfortunately include many of the common scenarios for acute AI including endocarditis in the setting of AS and aortic dissection in patients with hypertension and a dilated aortic root.5
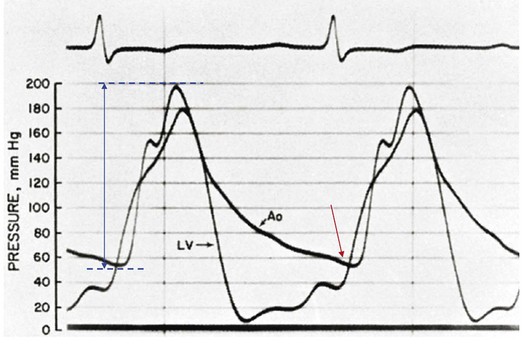
Acute severe AI results in near approximation of aortic and LV pressures at end diastole, with consequent deleterious effects on coronary perfusion of the subendocardium (Fig. 32.9). Because coronary perfusion pressure is the difference between diastolic pressure in the aortic root and subendocardial pressure in the LV cavity, the decrease in aortic diastolic pressure and rise in subendocardial pressure that are hallmarks of acute AI can result in profound subendocardial ischemia, especially in patients with preexistent hypertrophy or patients with underlying coronary artery disease. In addition, because afterload and wall stress in this setting are increased, there is a rise in myocardial oxygen demand simultaneous with a fall in supply, occasionally leading to cardiogenic shock and death. Acute AI also results in early closure of the mitral valve as LV diastolic pressure rises above left atrial pressure, a phenomenon that has potential protective benefits for the pulmonary circulation.
In contrast, chronic AI features a host of adaptive processes by the left ventricle including progressive dilation, increased compliance, and hypertrophy. As with AS, hypertrophy results in lower wall stress but AI features an increase in afterload combined with progressive LV dilation and somewhat less hypertrophy, resulting in significantly higher wall stress than seen in compensated AS.79 End-diastolic dilation of the left ventricle allows for larger stroke volumes to compensate for regurgitant fractions, which may be in the range of 50% or greater. With combined pressure and volume overload, unique among the valve disorders discussed in this chapter, LV adaptations allow for maintenance of normal overall function until ventricular remodeling and hypertrophy are no longer sufficient to maintain forward stroke volume and EF. With increasing wall stress and LV dilation, LV contractility is eventually impaired, at which point filling pressures begin to rise (or rise further) and patients become symptomatic. Coronary flow reserve in AI, as with AS, is impaired80 and, combined with increased demand and the need for additional perfusion for a hypertrophic myocardium, may result in significant ischemia. The onset of symptoms may be abrupt, especially with new onset of atrial tachyarrhythmias or sudden increase in cardiac output demand such as with exertion or infection.
Although acute AI represents a relative or absolute medical emergency, chronic AI may have an insidious course over decades. Some insight into the rate of progression of chronic AI is provided by a meta-analysis incorporated into the most recent ACC/AHA guidelines.5 In a review of nine admittedly heterogeneous studies incorporating nearly 600 primarily asymptomatic or mildly symptomatic patients with AI, average progression to symptoms with or without LV systolic dysfunction was 4.3% annually, and sudden death occurred in 0.2% annually.5 Although this rate of progression is modest, patients do not necessarily develop symptoms before developing LV dysfunction or sudden death.81 Age, end-systolic dimensions,82 and rate of deterioration of end-systolic dimension and EF83 are more sensitive tools for predicting outcome in chronic AI.
In contrast, in the setting of symptomatic AI that does not fall in the acute severe category, an annual mortality rate of 6% to 25% has been described, depending on severity of symptoms. By the 10-year follow-up, 75% had undergone AVR or died.84
Diagnosis
Physical Examination
The hallmarks of acute severe AI include features consistent with congestive heart failure and low cardiac output including tachycardia, dyspnea, and signs of impaired cardiac output such as peripheral vasoconstriction. The wide pulse pressure that is a hallmark of AI may or may not be seen, in part because it is dependent on LV compliance. The classic AI murmur may not be heard because of the limited gradient between the aorta and left ventricle during diastole when LV diastolic pressures in some cases approach systemic diastolic pressures. Tachycardia and diminished effective forward flow in acute AI may offset the augmented systolic and wide pulse pressure seen in later stages of the disease, and these patients may have normal or occasionally diminished pulses, although more moderate acute AI may feature the more classic findings. Early closure of the mitral valve may result in a soft first heart sound, and distortions of leaflet anatomy may result in lack of a distinct aortic second heart sound. In some cases absence of a second heart sound in a patient presenting with cardiogenic shock may be the most prominent physical finding.78
The dramatic physical findings in chronic AI are among the most familiar to physicians and trainees. A wide pulse pressure results primarily because of increased stroke volume, which augments systolic pressure,85 and low aortic diastolic pressures because of volume runoff into the left ventricle. This in turn results in a large variety of associated eponymous physical findings including bounding carotid and peripheral pulses (Corrigan’s and water hammer pulses, respectively); Hill’s sign (dramatically higher systolic pressure in the legs than the arms because of the exaggerated effect of harmonics on systolic pressure waveforms, variously described as having a 20 or 40 mm Hg threshold); Duroziez’s sign (to and fro murmur over the femoral arteries); and Traube’s sign (“pistol shot” sound over the femoral artery during simultaneous compression). With LV dilation, the apical impulse is displaced laterally.
The intensity of the classic diastolic decrescendo murmur heard over the left midsternal border generally correlates well with the severity of AI,86 although in later stages of decompensation (or in acute AI), when the gradient between the aorta and the left ventricle disappears in later stages of diastole, the murmur typically shortens. When the murmur is better heard over the right (rather than left) sternal border, the cause of the AI may be secondary to a dilated aortic root rather than a primary leaflet abnormality.87 Two other murmurs are frequently heard with moderate to severe AI: a systolic ejection murmur consistent with increased transvalvular flow and on occasion an Austin-Flint murmur. The latter is the apical diastolic rumble occasionally confused with mitral stenosis and thought to originate from vibration of the anterior mitral leaflet caused by the diastolic regurgitant jet originating at the aortic valve; it has been described only in the setting of severe AI.87 In rheumatic AI, if a diastolic rumble is heard, mitral stenosis should be excluded. An S3 gallop is most likely to be present when severe AI occurs in a setting of depressed LV function88 but may merely reflect substantial volume loading; the finding is not specific.
Noninvasive Evaluation
As with AS, noninvasive assessment has replaced cardiac catheterization for determining severity of AI. It provides both structural and physiologic information including visualization of the leaflets, annulus, and aortic root; semiquantitative assessment of the severity of AI; and characterization of LV size, hypertrophy, and function, as well as other structural heart disease. Transesophageal echocardiography has superior diagnostic sensitivity to transthoracic echo for certain parameters, in particular for detection of valvular vegetations.89 A wide range of techniques for measurement of AI severity including width of the color Doppler jet compared with size of the LV outflow tract, width of the vena contracta, regurgitant volume, regurgitant fraction, and regurgitant orifice area are described in Chapter 8. The correlation of these parameters with severity of AI is listed in Table 32.1. Additional findings of importance are rate of jet velocity deceleration as diastasis is achieved between aortic and LV diastolic pressures and duration of diastolic flow reversal in the aorta.90 In addition to echocardiography, cine MRI can assess severity of AI, size of chamber volumes, LV mass, wall thickness, and systolic function.91 LV size and function have also been followed serially by radionuclide ventriculography.92
The size and function of the LV have been used to set thresholds at which AVR should be considered. The ACC/AHA guidelines5 and the European Society of Cardiology guidelines93 have set slightly different values for class I and II recommendations. The echocardiogram-based recommendations, which vary with clinical settings, use thresholds that include LV end-diastolic dimension greater than 70 to 75 mm and end-systolic dimension greater than 50 to 55 mm, aortic root dimension greater than 50 to 55 mm (the lower threshold is for patients with bicuspid valves or Marfan syndrome),93 and EF less than 50%. As the observational data have grown, these numbers have shifted slightly from the familiar “rule of 55s,” which recommended surgery for asymptomatic patients with EF less than 55% or LV end-systolic dimension greater than 55 mm. An important consideration is that the end-systolic and end-diastolic dimensions should be adjusted downward for patients with small body surface area. Keeping in mind that these threshold values can be confounded by comorbid conditions that affect chamber size and function including ischemic and other forms of cardiomyopathy, as well as multivalvular disease, is important.
The ECG was once an important tool in the serial assessment of patients with AI. Similar to AS, features of LV hypertrophy may be present. Progression of LV dilation and dysfunction were thought to correlate with development of an LV strain pattern, a phenomenon that has been confirmed.94 As a result, the use of digitalis glycosides, which can cause a similar strain pattern, was discouraged in order to maintain relative specificity of the finding. Conduction disturbances remain an important feature, particularly heart block in the setting of endocarditis, discussed subsequently.
Cardiac Catheterization
As with the other valvular diseases discussed in this chapter, cardiac catheterization is indicated only for patients with inconclusive noninvasive findings or when noninvasive findings are discordant with the rest of the clinical picture. Typical findings include a wide aortic pulse pressure and an exaggerated rise in LV diastolic pressure, reflecting continuous retrograde filling during diastole (see Fig. 32.9). Cardiac catheterization in this setting normally includes aortic root angiography; although the latter requires a significant dye load (typically 30 mL/second for 2 seconds) to be diagnostically accurate. The hallmark of severe AI is greater opacification of the left ventricle than the aorta. The indications for coronary angiography in this setting are listed in Box 32.1.
Therapy
Medical Management
In acute AI, medical therapy is targeted to stabilization pending AVR. Nitroprusside has been the standard for reducing peripheral vascular resistance and improving net forward flow across the aortic valve since the 1970s.95 The use of arterial vasodilators is more complex when the patient is already hypotensive, and a combination of inotropes and afterload reduction should be considered. Although afterload reduction in this setting is highly beneficial, the otherwise optimal combination of afterload reduction and augmented pressure normally provided by intra-aortic balloon pumping in cardiogenic shock (see Chapters 7 and 30) is not a viable choice in AI. Counterpulsation during diastole increases the severity of AI with potentially catastrophic consequences; it represents an absolute contraindication to the intra-aortic balloon pump.
In chronic AI, the evidence base is incomplete and somewhat controversial. The general consensus is that asymptomatic patients with mild to at most moderate AI with normal LV function who are not hypertensive do not require or benefit from vasodilator therapy.81,83 Only when patients are not surgical candidates is vasodilator use in chronic AI a class I indication.5 Class II indications are short-term treatment to optimize the patient’s hemodynamic status prior to AVR and, more controversially, long-term therapy for asymptomatic patients with severe AI but preserved LV function. Two important studies with divergent results have been performed: One, using nifedipine, appeared to demonstrate some slowing of progression to need for AVR compared with patients on digoxin.96 The second, a placebo-controlled study, failed to demonstrate clear benefits of vasodilator therapy with either nifedipine or an ACE inhibitor,97 but there are concerns regarding the size of the study, dose of drug used, and modest severity of disease at baseline. Oral vasodilators that have been described as having potential therapeutic benefit include several dihydropyridine calcium channel blockers (nifedipine and felodipine), as well as hydralazine. The appropriateness of vasodilator therapy regardless of severity of disease is clearer in patients with systemic hypertension, in whom reduction of afterload has dual benefits. The dose of drug should be sufficient to show at least some reduction in systolic pressure. There is no evidence base to recommend other forms of medical therapy, such as nitrates or drugs commonly used to treat congestive heart failure to prevent progression of disease in chronic asymptomatic AI. In the critical care setting, however, inotropes and vasodilators are appropriate if patients have congestive heart failure or a low output state, or both.
Aortic Valve Replacement
Two factors primarily determine when a patient should be referred for valve replacement: the presence of symptoms and findings of LV systolic dysfunction.5 Surgery results in superior long-term survival than medical therapy alone for symptomatic patients.84,98 Similarly, patients with LV dysfunction have an inverse correlation between EF and mortality risk.99 Three ACC/AHA class I indications for surgery, all for patients with severe AI, exist: (1) patients who have symptoms regardless of LV function; (2) patients with LV systolic dysfunction (EF less than or equal to 50%) regardless of symptoms; and (3) patients undergoing coronary bypass surgery or surgery on other heart valves or the aorta.
The question of whether it is ever too late to perform AVR in patients with AI has been raised.85 Because the disease involves high afterload and wall stress, there is postoperative improvement in EF100 even in patients with severe baseline LV dysfunction. In general there is substantial reversal of the physiologic adaptations required by chronic severe AI in the postoperative state.101 Nevertheless, delay in referring patients for surgery is deleterious, with significant negative impact on outcomes102; the prognosis is adversely affected both by the severity and duration of LV dysfunction.103 This includes a fourfold increase in operative mortality rate for patients with EF less than 35% (14%) compared with those with EF greater than 50% (3.7%). Thus the trend has been for earlier referral of symptomatic patients and more aggressive noninvasive monitoring for early detection of deterioration in LV function or dimensions.
Special consideration should be given to patients with acute aortic valve endocarditis. Deteriorating hemodynamics, as well as extension of infection as manifest by progressive heart block and ring abscess formation on echo (Fig. 32.10), are among several factors that mandate early surgery104; despite the semielective nature of the operation (with an operative mortality rate close to 10%) and the theoretical risk of an infected prosthesis, the long-term outcomes have been satisfactory, with 75% 10-year survival rate after hospital discharge.105
Mitral Regurgitation
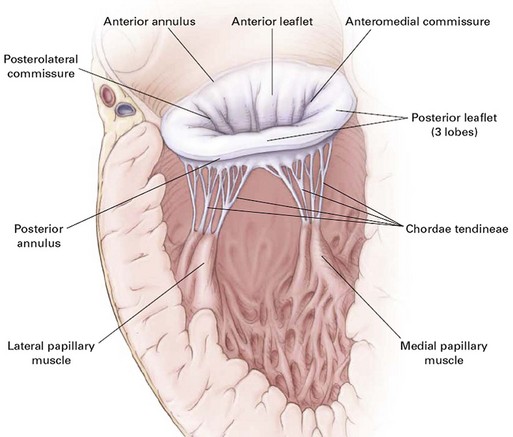
Anatomically and physiologically, the mitral valve is the most complex of the heart valves, dependent for function on a complex interaction between leaflets, annulus, commissures, and the supporting apparatus consisting of chordae tendineae tethered to the papillary muscles (Fig. 32.11). Distortion or dysfunction of any of the elements and changes in function and geometry of the left ventricle and left atrium can all have a significant impact on flow across the mitral valve. In addition, unlike other valvulopathies, the mitral valve is highly dependent on the myocardial circulation. In contrast with the relatively passive nature of opening and closing of the aortic valve, mitral valve motion is an active process reflecting not just cyclic pressure variations but also tethering and dynamic interaction with the supporting apparatus. As with AI, mitral insufficiency (MR) has acute and chronic manifestations and chronic (rather than acute) insufficiency is more common. However, acute MR has a much larger variety of causes than acute AI and the overall incidence is higher. Data from the Framingham Offspring Study revealed a prevalence of moderate or greater MR from 0.9% of women in the fifth decade of life, to 11.2% of men between ages 70 and 83, and correlated with age, low body mass, and systemic hypertension.76 As with AI, ventricular function predicts overall outcomes. Chronic MR can both cause and be the result of LV remodeling and heart failure and, when secondary to LV dysfunction, is associated with a significantly poorer overall prognosis than LV dysfunction without secondary MR.106
Pathophysiology
Fundamental understanding of the pathophysiology of MR has expanded substantially in the past 2 decades. In particular, awareness of the role of the subvalvular apparatus in preserving LV function and data demonstrating the need for earlier referral to surgery have resulted in significant changes in clinical and surgical practice, leading to substantial improvement in outcomes.107
As with AI, MR results in volume overloading of the LV. Unlike AI, the afterload is not increased and overall LV ejection is enhanced rather than impaired by loading conditions, although in significant MR half or more of the ejection is retrograde into the left atrium. The incompetent mitral valve acts as a second pathway for blood ejection, which reduces afterload in a setting in which preload is increased, thus potentiating ejection via the Frank-Starling mechanism. The combination ultimately may raise EF to supranormal early in the course of the disease. As a result, patients with “normal” EF may already have significant impairment in LV function.108 Adaptations to the volume loading include lengthening of the myocytes, which increases LV volume. Progressive MR eventually results in contractile dysfunction. Relatively early in the course of the disease, the sympathetic nervous system is activated as a compensatory mechanism to volume loading and decreased forward ejection.109 Chronic high sympathetic tone results in secondary myocardial damage110 and has been postulated as the rationale for potential benefit from beta blockade in chronic MR.111 Over time, although some hypertrophy develops, the predominant response to volume overload without pressure overload is enlargement of chamber size. Extrapolating the LaPlace relationship, which is manifestly protective of wall stress in AS, the progressive expansion of LV dimensions in MR without similar compensatory hypertrophy results in increasing wall stress, which in turn results in increasing MR. In a similar manner, a second vicious circle is caused by the effect of MR on LV chamber size, which, as it enlarges, results in stretching of the annulus, further exacerbating MR.112 Thus chronic MR can both cause and be the result of LV remodeling and heart failure.
The causes of MR vary by geography, with mitral valve prolapse being the most common in industrialized nations. Rheumatic MR remains common in developing countries. The causes of acute MR include ischemic MR, endocarditis, chordal rupture, papillary muscle dysfunction, and a variety of diseases affecting prosthetic valves (Box 32.3). The most likely causes of acute MR in the critical care setting are ischemia, including acute myocardial infarction and infectious endocarditis, and should be considered in any patient who develops sudden hemodynamic decompensation in the peri-infarction period or who has known endocarditis. Chronic MR is secondary to a variety of structural, degenerative, connective tissue, inflammatory, and ischemic disorders, as well as congenital abnormalities outlined in Box 32.3.
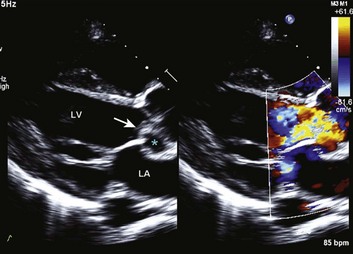
The classic teaching has been that acute MR secondary to ischemia was caused by papillary muscle dysfunction, most likely secondary to ischemia of the posteromedial papillary muscle, which is dependent on a single blood supply from the right coronary artery. The anterolateral papillary muscle typically has dual circulation from the left anterior descending and circumflex coronary arteries, specifically from a diagonal branch and a circumflex marginal branch. The source of papillary muscle circulation is in fact variable, and on occasion the anterior papillary muscle also has a single blood supply.113 The role of impaired circulation to the papillary muscle as the cause of ischemic MR has been revisited,114 and evidence to date suggests that ischemic MR is secondary to alterations in ventricular geometry and leaflet tethering rather than dysfunction caused solely by impaired circulation to the papillary muscle during ischemia,115 although alterations in papillary muscle geometry can be seen in chronic ischemic MR.116 The hemodynamics of dynamic, ischemic MR are shown in Figure 32.12.
Analysis of thrombolysis in myocardial infarction data showed a 27% incidence of MR most commonly in the setting of anterior rather than inferior myocardial infarction, although these values vary widely by criteria and techniques used for screening, with the incidence ranging from 11% to 59%.117 A Mayo Clinic review of more than 1300 acute myocardial infarction patients found moderate to severe MR in 12% and found it to be an independent predictor of both heart failure and survival.118 It is associated with multivessel coronary artery disease, as well as prior infarction.119
Papillary muscle rupture is a rare but potentially catastrophic cause of flail leaflets and represents a surgical emergency.120 In addition to the typical acute myocardial infarction setting, it may be secondary to trauma and isolated other causes such as postpartum Ehlers-Danlos syndrome.121 Other causes of acute MR to consider (see Box 32.3) include spontaneous chordal rupture, infectious endocarditis with distortion of the valve leaflets or chordal rupture, and prosthetic valve dysfunction including paravalvular prosthetic valve leak. In patients with sudden onset of acute MR, retrograde flow is typically into a normal-sized and relatively noncompliant left atrium unless preexisting atrial hypertension has been present. The sudden volume loading immediately places the left atrium on the steep portion of its pressure-volume curve, which commonly causes acute pulmonary edema. As is the case with acute AI, the left ventricle is unable to adapt acutely and effective forward stroke volume is decreased. In the setting of myocardial infarction in particular, patients with acute MR may develop cardiogenic shock.
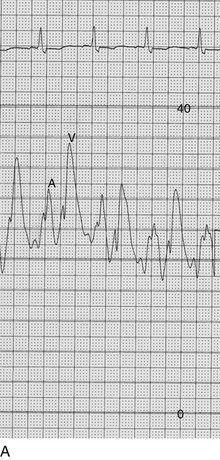
The classic hemodynamic response, a giant V wave, is shown in Figure 32.13. Differentiating a giant V wave from a prominent pulmonary artery pressure waveform can be difficult in the critical care setting, and repeated attempts to obtain a wedge pressure when the catheter is already in the wedge position may result in iatrogenic pulmonary artery hemorrhage.
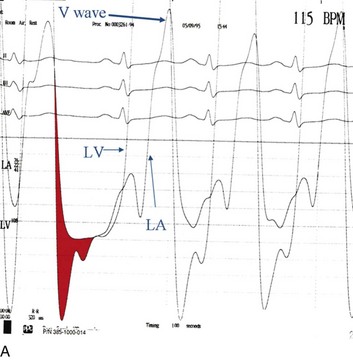
Diagnosis
Physical Examination
In acute MR the abrupt rise of left atrial pressure results from torrential retrograde flow, although the gradient between the left atrium and left ventricle may narrow substantially near the end of systole because of the substantial rise in left atrial pressure as shown in Figure 32.13. Thus the systolic murmur may decrease in intensity prior to the onset of the aortic second heart sound. A systolic thrill may be noted. The sudden and severe left atrial hypertension typically results in pulmonary edema with accompanying physical findings, as well as an increase in the pulmonic second sound and paradoxical splitting of the second heart sound. Because the adaptive mechanisms are not yet in place, features of LV volume overload are not seen, although a prominent precordial impulse, reflecting increased (albeit retrograde) ejection, may be noted. Unlike AI, the pulse pressure is not increased and may be reduced to reflect the lower forward stroke volume.
In chronic MR, precordial features of volume overload include a displaced point of maximal impulse and prominent precordial pulsation. An S3 gallop is heard, especially with marked dilation of the left ventricle,88 and a diastolic flow murmur representing increased transvalvular flow (rather than mitral stenosis) may be present. The first heart sound may be soft, reflecting lack of brisk apposition of the mitral valve leaflets. The murmur of MR commences immediately following the first heart sound, reflecting the observation that a significant amount of regurgitation takes place during what would otherwise be isovolumic systole (which normally commences immediately following mitral valve closure). The murmur of chronic MR is characteristically blowing; may be medium or high pitched; is best heard at the apex; radiates to the axilla and left scapula (when severe); and extends throughout systole, reflecting the presence of a relatively high gradient throughout LV ejection.
The intensity of the MR murmur reflects a number of phenomena relating to the size of the gradient, compliance of the left ventricle and left atrium, and contractility of the left ventricle but does not correlate well with extent of MR; thus, the murmur may be difficult to hear or inaudible, especially in the critical care setting.122
Noninvasive Evaluation
Echocardiography provides visualization of the multiple structures responsible for causing MR including the valve leaflets, annulus, chordae tendineae papillary muscles, and left ventricle (see Fig. 32.11). Transesophageal echo provides superior imaging of the valve including assessment for vegetations in case endocarditis is suspected and better visualization of flail leaflets; it is particularly useful when the transthoracic views are incomplete or inadequate. In general, transesophageal echo is superior for fully assessing characteristics of the mitral regurgitant jet123 and provides additional information in evaluating mitral valve morphology and motion. As with AI, Doppler methods allow assessment of the width of the regurgitant jet and size of the vena contracta, and semiquantitative measures of the regurgitant volume, regurgitant jet, and effective orifice area (see Table 32.1) are discussed in detail in Chapter 8. In patients presenting with acute heart failure and a hyperdynamic left ventricle, acute MR should always be considered.
Cardiac Catheterization
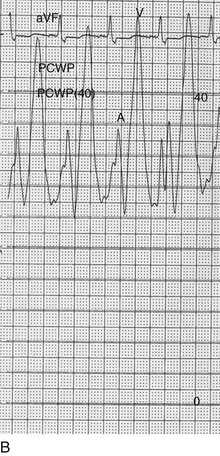
Although the indications for cardiac catheterization (see Box 32.1) are similar to those for AS and AI, an additional consideration is included as a class I indication: to assess pulmonary artery pressures at rest and if necessary with exercise to evaluate appropriateness for surgery (see valve replacement indications later). The size of the V wave is not an accurate indicator of MR severity and is not used as a decision-making criterion for surgery because it may be unremarkable even in severe MR when there is high left atrial compliance. In addition, the size of the V wave is typically recorded by wedge rather than direct left atrial pressure, which can result in significant underestimation of the severity of MR (Fig. 32.14). Right heart catheterization and oxygen saturation sampling allow differentiation between the two most common causes of abrupt mechanical deterioration in acute MI: severe MR and ventricular septal defect. Both conditions can feature a large V wave because of sudden volume loading of a noncompliant left atrium: secondary to retrograde flow across the mitral valve in the case of acute MR and antegrade flow of shunted blood from the LV across the pulmonary circulation in the case of a ventricular septal defect.
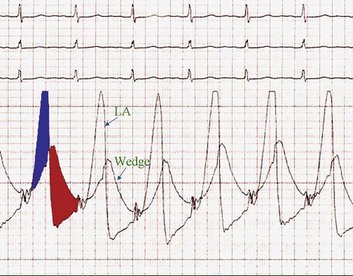
Therapy
Medical Management
For acute, severe, symptomatic MR, the primary medical therapy consists of aggressive vasodilation with nitroprusside. It provides improved aortic flow, diminishes MR, and decreases left-sided filling pressures. In the setting of acute MR with shock, nitroprusside alone is usually considered undesirable because of poor end-organ perfusion if further hypotension is caused by the drug. Intra-aortic balloon pumping is highly effective in this setting124 (see Chapter 7) because it lowers aortic impedance, improves cardiac output, and decreases regurgitant fraction at the same time as it restores blood pressure. A combination of an inotrope (typically dobutamine; a vasoconstrictor should be avoided) and nitroprusside can have similar salutary effects. When the acute MR is dynamic (i.e., occurs and resolves abruptly), the cause is frequently acute ischemia, and consideration should be given to evaluating the coronary circulation. In a small number of cases, acute coronary intervention has been documented to alleviate MR in the setting of acute ischemia.125
Therapy for chronic MR is more controversial. Because afterload is typically not increased, the benefits of afterload reduction are without clear physiologic foundation. As such, ACE inhibitors have consistently been shown not to be effective for chronic MR.126–128 When concomitant systemic hypertension exists, physiologic benefits appear more convincing.129 If patients have LV dysfunction superimposed on chronic MR, medical therapy with ACE inhibitors may be beneficial, and several studies have observed functional improvement with resychronization in this setting.130 As with other valvular disorders, new-onset AF can result in abrupt hemodynamic deterioration and cardioversion should be considered with adjunctive pharmacotherapy to maintain sinus rhythm and rate control. Because of the sympathetic activation previously discussed, beta blockers appear to be beneficial131,132 and may have additive benefit when used in combination with ACE inhibitors.133 Both classes of drugs appear to be therapeutic when used after mitral valve repair.134
Mitral Valve Surgery
Three types of surgeries are performed on the mitral valve: repair, replacement with preservation of the subvalvular apparatus and attachments, and replacement with transection of the chordae. Mitral valve repair has been consistently shown to improve outcomes compared with mitral valve replacement, both acutely and long term, with a fourfold lower operative mortality rate and a 30% improvement in 10-year survival rate.135 With mitral valve replacement, patients who nevertheless have the chordal apparatus preserved have superior long-term outcomes compared with those who have the chords transected.136 An additional benefit of repair is the avoidance of anticoagulation if patients are in sinus rhythm compared with replacement with a mechanical valve when anticoagulation is required.
In general, the timing of surgery for chronic MR should be prior to the onset of LV dysfunction. The most commonly used markers are symptoms and echocardiographic parameters including LVEF less than 60%.5 Patients who develop class III or IV symptoms, even transiently, have a nearly 10-fold increase in annual mortality rate,137 with evidence that early surgery in the setting of severe MR improves overall survival. The 60% threshold for surgery has been shown to correlate with postoperative LV function138 as well as survival.139 More recently the effective regurgitant orifice has been shown to be strongly predictive of 5-year mortality risk, with patients having an orifice of 40 mm2 or greater having an odds ratio of death greater than 5:1 compared with patients with orifice size of 20 mm2 or less.140 Preoperative end-systolic diameter of 40 mm or less has also been associated with superior outcomes. Patients with AF in general have a worse prognosis,141 although in large part because of embolic events rather than LV dysfunction, as do patients with pulmonary hypertension or right-sided heart failure, or both.142 Mitral valve surgery, even in the setting of depressed EF, may result in postoperative return to normal function.143 Comparison of long-term outcomes between operated and medically treated patients with asymptomatic MR has invariably favored the former,140 although much of the data depend on nonrandomized registry data. In general, the key to improved function and outcomes is preserving the integrity of the submitral apparatus,144 and comparison of successful repair versus replacement has indicated that high-volume institutions (>140 mitral operations/year) have the best results with nearly twice the rate of successful repair as low-volume institutions.145
A common teaching has been that repair or replacement of the mitral valve eliminates the “pop-off valve” represented by incompetent leaflets, resulting in dramatically increased afterload postoperatively. In turn, if significant preoperative LV dysfunction were present, the left ventricle would fail, with hemodynamic deterioration and subsequent death. In fact, careful investigation has demonstrated that postoperative deterioration of LV function was secondary to disruption of chordal integrity146 and elimination of the favorable effects of the submitral apparatus on valvular-ventricular interaction rather than the elimination of a low-pressure decompression pathway. Afterload following mitral valve surgery with chordal transection does in fact increase, whereas patients subjected to mitral valve surgery with preservation of the chordal apparatus had decreased LV chamber volume and, in keeping with the LaPlace equation, reduced afterload.147 Thus the recommendations regarding LVEF for surgery have changed dramatically, and successful repair of mitral valves in series of patients with EF less than 25% has been reported with operative risk of less than or equal to 5%.148 The operative risk is in the 1% to 2% range for symptomatic patients with LV dysfunction but higher EF.149 Similarly, the Society of Thoracic Surgeons database has reported mortality rates of approximately 5% with EF less than or equal to 30% but 3% with EF greater than 30%.150 Propensity analysis of patients eligible for mitral valve repair who did or did not undergo the procedure has not shown a survival advantage in patients with mean EF less than 25%,134 although a randomized trial has not been reported. Similarly, concomitant mitral valve surgery in patients undergoing coronary artery bypass graft (CABG) with moderate or greater MR did not clearly improve short-term survival.151 Combining mitral valve surgery with a mesh placed around the heart to help prevent dilation and thus lower wall stress may have additional benefit for patients with MR and LV dysfunction.149 Only in the setting of an EF of less than 30% or end-systolic dimension greater than 55 mm, as well as anatomy or surgical experience that suggests chordal preservation is unlikely, do the current guidelines recommend medical therapy alone.
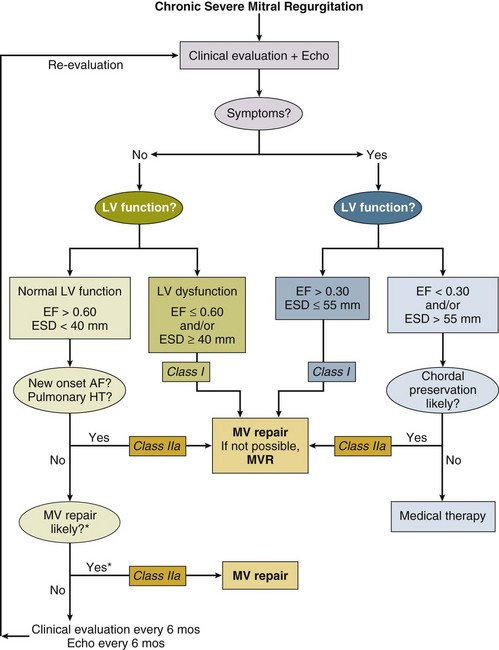
In symptomatic acute severe MR, mitral valve surgery is a class I indication regardless of level of LV function. Both with infective endocarditis,104 as well as with papillary muscle rupture, surgery is mandated to be performed with as little delay as possible.152 However, the overall risk of mortality when MR accompanies acute myocardial infarction is substantially increased,153 a phenomenon noted in patients undergoing emergent percutaneous intervention as well.154 Chronic severe MR is much more complex, and the management algorithm is outlined in Figure 32.15.
Future Directions
A number of innovative technologies are being developed for percutaneous treatment of MR. The most extensively studied is based on a catheter-based edge-to-edge repair of the mitral leaflets designed to mimic a surgical procedure developed by Alfieri and colleagues.155 The EVEREST (Endovascular Valve Edge-to-edge REpair STudy) II trial156 compared 184 patients randomized to percutaneous versus 95 patients randomized to surgical mitral valve repair and had promising though somewhat nuanced results: although there was good safety profile and similar quality of life improvement, grade 2 or greater mitral insufficiency was present in nearly half the percutaneous repair patients at 1 year, compared with only 17% of the surgical repair patients. The surgical patients did have more adverse events, primarily blood transfusions. The surgical approach appeared clearly superior for patients with degenerative MR, with relative parity in outcomes in functional MR patients (those with structurally normal mitral valves, but typically dilatation of the mitral annulus). The Alfieri procedure is an uncommonly performed type of mitral valve repair, and as with most repair procedures typically involves placement of a prosthetic ring, an option not available with the percutaneous approach, and potentially a limitation of its long-term applicability as an isolated intervention.157 In this context, it is worth noting that several percutaneous technologies are under development to constrict the coronary sinus that at least partially surrounds the mitral valve158; they thereby attempt to mimic the mechanical effects of a surgically placed mitral ring. For now, percutaneous edge-to-edge repair has had fair acceptance outside the United States and is being performed primarily in the setting of functional MR. A number of other technologies are under development.
Hypertrophic Obstructive Cardiomyopathy
Although a disease of the heart muscle rather than a primary valvular disorder, hypertrophic obstructive cardiomyopathy (HOCM) has important features of LV outflow obstruction and concomitant MR. The clinical presentation, physical examination, noninvasive and invasive findings, and overall management differ in general from AS or MR, and HOCM represents an important consideration for patients in critical care units,159 where patients with a systolic murmur and hypotension, syncope, or exercise-induced cardiac arrest in particular should have HOCM included in the differential diagnosis.
Pathophysiology
The hallmark of HOCM is dynamic obstruction of the LV outflow tract with eccentric hypertrophy; many variations exist, and obstruction is not always at the subvalvular level. Only approximately one fourth to one third of patients with hypertrophic cardiomyopathy (HCM) have a resting gradient of 30 mm Hg or greater.160 HCM is found in approximately 0.2% of the population161 and is defined as hypertrophy of the left ventricle that is not secondary to hypertrophic stimuli such as systemic hypertension or AS. Contrary to conventional wisdom, the majority of patients with HCM were found to have provokable gradients with exercise in a single large-volume center162 (likely with some skewing from the nature of its referral population). The genetics of HCM have been studied extensively, and although complex, there is a familial association in a majority of patients. Although the clinical course may be generally benign in patients without risk factors for adverse events,163 it is the most common cardiac cause of sudden death in young adults,164 is associated with cardiac arrest in athletes,165 and leads to heart failure, rhythm disturbances, and occasional sudden death in older patients. The patients at high risk for sudden death, composing only about 10% to 20% of those within the HCM population, include those with prior cardiac arrest or sustained ventricular tachycardia, those with a family history of HCM and sudden death, hypotensive response to exercise, and severe LVH with wall thickness of 30 mm or greater.166 A resting gradient across the outflow tract of more than 30 mm Hg alone predicts an increased risk of sudden death or development of moderate to severe congestive heart failure,160 and there may be higher risk with very high gradients.167 Because of impaired coronary flow reserve168 and severe hypertrophy, myocardial ischemia is a potentially important factor in acute patient management; there is also an association between HOCM and myocardial coronary bridging.169
Diagnosis
Noninvasive Evaluation
Abnormalities of mitral valve function are common. Systolic anterior motion of the mitral valve is associated with further obstruction170; in addition, MR is a typical secondary finding in HOCM. The ECG features LV hypertrophy, strain, and prominent Q waves in the inferior and precordial leads, with narrow QRS complexes being common. The echocardiogram is diagnostic for HOCM, providing both an anatomic basis and physiologic evidence of the degree of obstruction (Fig. 32.16).
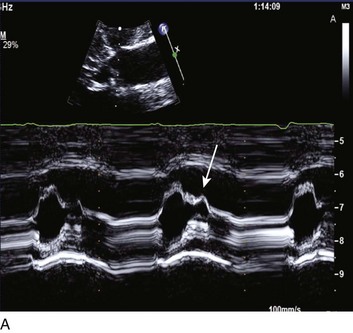
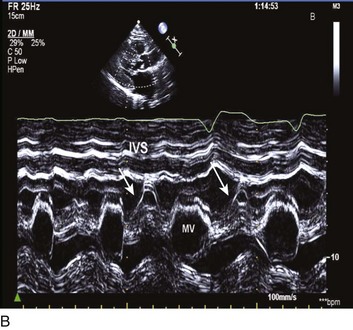
Cardiac Catheterization
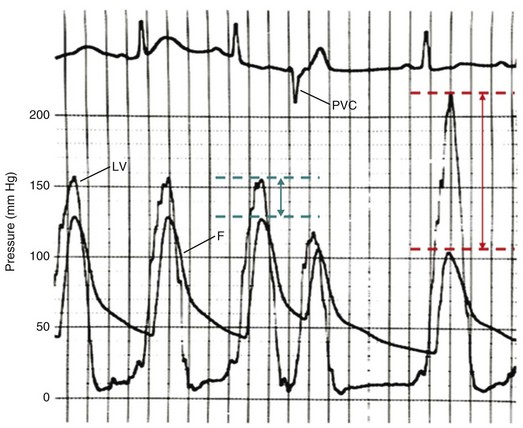
Cardiac catheterization in HOCM may reveal the standard triad of the Brockenbrough-Braunwald-Morrow sign in the postextrasystolic heartbeat: increase in the gradient between the left ventricle and the aorta, decrease in systemic blood pressure, and narrowing of the pulse pressure (Fig. 32.17). Other stimuli that increase outflow obstruction and increase gradient are Valsalva maneuver and amyl nitrite inhalation, in addition to postextrasystolic potentiation.
Therapy
Medical Management
The management of critically ill patients with HOCM focuses on several core principles: optimizing LV volumes, specifically increasing preload while not decreasing afterload, decreasing contractility, and maintaining the patient in sinus rhythm. Thus the typical regimen of diuretics, vasodilators, and inotropes used in most patients presenting with congestive heart failure who do not have HOCM can severely exacerbate the outflow gradient and lead to further decompensation or death171 (as can intra-aortic balloon pumping); somewhat counterintuitively in this setting, negative inotropes, in particular beta blockers, combined with volume loading can stabilize patients.172,173 Other negative inotropes including verapamil and disopyramide appear to have favorable hemodynamic effects and may represent an alternative to beta blockade.174 In the patient in shock, if vasopressors are required, agents such as phenylephrine, which are primarily peripheral vasoconstrictors, should be used rather than inotropic agents. Verapamil has negative inotropic benefits and some utility as an antiarrhythmic agent, as well as diastolic relaxation properties, and has been used successfully in both chronic therapy and acutely decompensated patients with HOCM.175 Verapamil does have some potentially undesirable vasodilator properties and can cause synergistic depression of conduction and contractility in combination with beta blockade; the effects of combined therapy, if used, should be monitored closely.176
Patients with hypertrophic heart disease are highly dependent on the atrial contribution to cardiac output,177 and patients with obstruction in particular depend on the additional LV filling from atrial contraction. AF is a cause of acute decompensation in HOCM patients, and cardioversion should be considered acutely. AF occurred in approximately one fourth of patients with HCM followed for 9 years and is predictive of heart failure–related death and stroke.178 In terms of the latter, patients with HCM and AF appear particularly predisposed to thromboembolic events179; anticoagulation has been shown to reduce the incidence significantly.180 Left atrial size and function predict the onset of AF.181 The Maze procedure and catheter ablation of left atrial pathways have been used successfully in small series to prevent recurrent AF.182,183 Amiodarone has been particularly effective for the maintenance of sinus rhythm in the population with AF and HCM184 and may also have additional efficacy in preventing sudden death in patients with episodes of nonsustained ventricular tachycardia.185,186 Ventricular fibrillation appears to be the primary cause of sudden death and can be prevented with implantable defibrillators for either primary or secondary prevention, with activation of the devices in 5% to 11% of patients annually depending on indications for placement187; episodes of sustained ventricular tachycardia and a history of prior sudden death place patients in the highest risk category.
Percutaneous Intervention or Surgery
Once patients develop refractory symptoms despite pharmacologic management and have significant obstruction to LV outflow, surgical myomectomy or alcohol septal ablation must be considered. The choice is controversial and depends on LV and coronary anatomy, comorbid conditions, severity of coexistent mitral valve or other potentially surgical heart disease, and a variety of other clinical issues; equivalence of outcomes with the less invasive percutaneous approach has not been demonstrated.188 Typical thresholds for intervention are a resting peak instantaneous gradient of 50 mm Hg or more, although depending on degree of symptoms and provokability, a variety of other thresholds have been applied.166 Mitral valve replacement can frequently be avoided, especially in patients without intrinsic mitral disease, in whom MR is largely abolished by myomectomy and relief of the outflow gradient.189 In patients with intrinsic mitral valve disease or for a variety of other anatomic considerations, mitral valve surgery including use of low-profile prostheses is used.166 Dual-chamber pacing, once considered therapeutically effective,190 with theoretical benefit from asynchronous septal contraction and secondary reduction in outflow tract gradient, has not been shown to have clear therapeutic benefit in randomized clinical trials, with the most likely functional benefits occurring in a limited subset of elderly patients.191
Mitral Valve Stenosis
Mitral valve stenosis (MS) is primarily a rheumatic disorder and, as such, has become increasingly uncommon in industrialized nations. Nevertheless, the disease remains prevalent in developing countries and a small but significant number of patients continue to present in the critical care setting throughout the world. Rheumatic MS involves disease of both the valve leaflets and submitral apparatus and may involve the valve ring as well (Fig. 32.18). Although MS is primarily caused by rheumatic deformity and scarring of the valve leaflets and subvalve with fusion of the commissures, a variety of disorders result in leaflet and annular calcification that also create obstruction between the left atrium and left ventricle. Most notably, mitral annular calcification, present in 6% of patients undergoing routine echocardiography,192 is more prevalent in the elderly, women, smokers, and patients on dialysis (who have high parathyroid hormone levels). It may present with severe nonrheumatic mitral stenosis, although MR is a more common association.192,193 A variety of uncommon other causes account for less than 1% of cases of MS.194
Pathophysiology
The physiologic hallmark of MS is obstruction to left atrial outflow resulting in rising left atrial and pulmonary pressures (the extent of the latter depends to a significant degree on left atrial compliance). The normal mitral valve is in the range of 4 to 6 cm2, and patients typically do not become symptomatic until valve areas decrease significantly below 2 cm2 (see Table 32.1). As valve area decreases, an additional pressure gradient is required to maintain transvalvular flow (see Fig. 32.5). Because antegrade flow is entirely during diastole, and the diastolic filling period shortens or lengthens disproportionately with increase or decrease in heart rate. respectively, the gradient is highly flow dependent. In MS, as with virtually all valvulopathies, cardiac output is potentially highly dependent on maintenance of sinus rhythm; because the influence on cardiac output is exaggerated by the need for active left atrial pumping across a stenotic orifice, atrial contraction is particularly important in MS. A frequent mode of presentation is acute congestive heart failure in a patient with MS who suddenly develops AF; the concomitant loss of atrial contraction and high ventricular rate with short diastolic filling period frequently results in pulmonary edema.
Diagnosis
Physical Examination
The diagnosis of MS on physical examination is frequently missed, in part because it is uncommonly seen by most practitioners outside developing countries. Besides findings suggestive of congestive heart failure with a low output in the critical care setting, several findings correlate strongly with MS. A right ventricular lift may occur in patients with advanced and chronic pulmonary hypertension, and occasionally a palpable pulmonary second sound is felt. Both S1 and P2 are increased early in the course of the disease; with progressive deformity of the subvalvular apparatus, S1 may become quite soft as mitral valve closure velocity decreases. The opening snap is preserved until the end stages of MS, and the severity of the disease correlates inversely with the length of time between S2 and the opening snap. The diastolic rumble may not be heard if the patient is not placed in the lateral decubitus position and is difficult to hear in the critical care setting. Presystolic accentuation is the result of increased flow in late diastole caused by atrial contraction in patients in sinus rhythm. The diagnosis is often missed in both the outpatient and critical care settings,195 and patients are sometimes treated for years for bronchitis, pneumonia, or asthma, when in fact the presentation is congestive heart failure with the underlying disease process being MS.
Noninvasive Evaluation
The echocardiogram is the primary tool for diagnosis of MS. The typical hockey stick appearance of the rheumatic anterior mitral leaflet is caused by partial restriction of mobility (Fig. 32.19). Other important characteristics of the mitral leaflets are degree of thickening, mobility, and calcification, all of which, along with extent of subvalvular disease, help to address suitability for balloon dilation or commissurotomy rather than valve replacement.196 It is important to differentiate rheumatic MS from severe nonrheumatic MS, as shown in Figure 32.19. Transesophageal echo provides the requisite sensitivity to detect left atrial thrombus prior to balloon commissurotomy or cardioversion. Severity of mitral stenosis is graded by physiologic and anatomic criteria: The former depend on Doppler interrogation of transvalvular flow, with peak velocity used to estimate the gradient; rate of decompression of the left atrial pressure, or pressure half time, correlates inversely with valve area. The valve area is frequently directly measured using planimetry of a cross-sectional view. Because these methods have potential for error, a number of alternative methodologies have been proposed,197–199 although there is no agreement on a gold standard.200 As with AS, a dramatic increase in gradient accompanies increased flow related to exercise or dobutamine infusion201 (Figs. 32.20 and 32.21). Cardiac catheterization may be required if the noninvasive data are inconclusive; there is a tendency for the pulmonary wedge pressure/LV gradient to overestimate the severity of MS compared with left atrial/LV gradient measurement202 (Fig. 32.22). Cardiac catheterization can also be used to assess pulmonary artery pressures and response to exercise if the clinical and noninvasive pictures are discordant.5
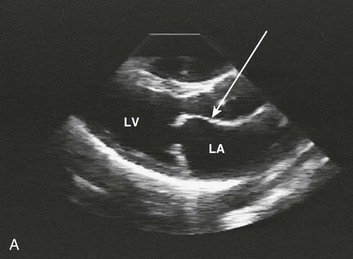
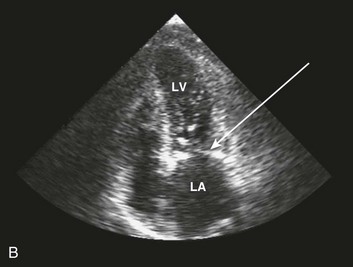
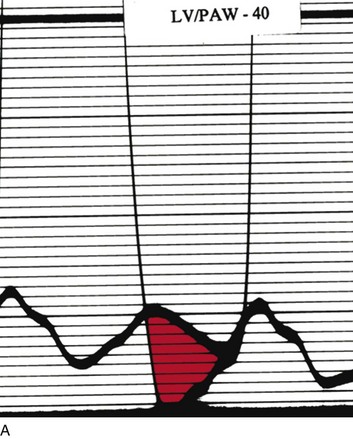
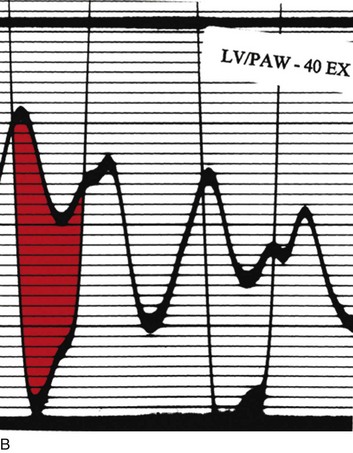
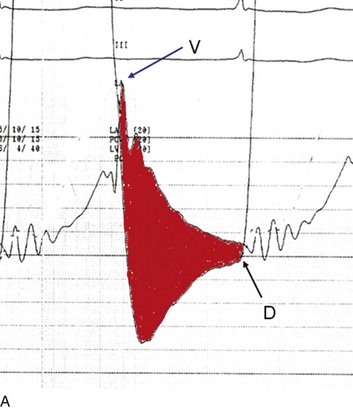
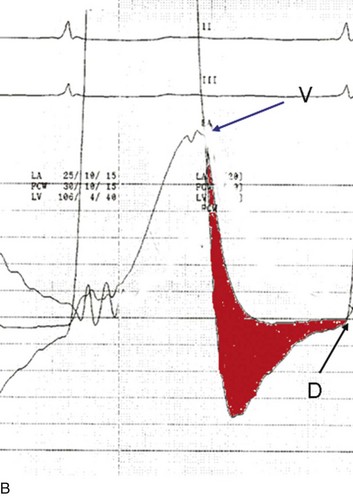
Figure 32.22 Effect of using pulmonary wedge pressure (A) or direct left atrial pressure (B) to assess gradient (in red) between the left atrium and left ventricle in mitral stenosis (40 mm Hg scale). Using the wedge pressure, the gradient appears to be approximately twice as great as when left atrial pressure is used. Rapid decompression of left atrial pressure is shown in the tracing on the right. The findings are consistent with mixed mitral stenosis and regurgitation, with the dominant physiology being secondary to mitral insufficiency. In both tracings, diastasis (D) is noted by end diastole, a feature not consistent with severe mitral stenosis except in marked bradycardia. The V wave height is similar in both tracings, unlike in Figure 32.12. The two tracings shown here were recorded a few seconds apart.
Therapy
Medical Therapy
Approximately 30% of patients with MS are thought to develop AF, with the risk increased by progressive increase in left atrial size. The risk of embolic events, especially stroke, is substantially higher than in AF patients without MS203 (up to 15% in unanticoagulated patients), possibly in part because of a hypercoagulopathy and increased platelet activation associated with MS,204 making the use of anticoagulation essential. It is not uncommon for the presenting finding to be an embolic event in a previously undiagnosed MS patient. Even in the setting of sinus rhythm,5 if spontaneous echo contrast is seen in the MS patient or left atrial size is greater than 55 mm, anticoagulation may decrease stroke risk204 and it should also be used in the sinus rhythm patient with prior embolic event or with left atrial thrombus detected by transesophageal echocardiography.
In the setting of new-onset AF and acute decompensation, cardioversion should be considered. Regardless of chronicity, heart rate slowing is essential; digoxin, beta blockers, diltiazem, or verapamil alone or in combination may be beneficial. Rate control only at rest is inadequate; many patients with low resting heart rates become tachycardic with minimal exercise. This is a vicious circle because the tachycardia may be a response to inadequate stroke volume; the tachycardia then lowers the diastolic filling period and further exacerbates the gradient. Control of tachycardia is important beyond its hemodynamic benefit; it also helps avoid tachycardia-induced cardiomyopathy.205
Percutaneous Intervention or Surgery
Percutaneous balloon mitral valvuloplasty is equivalent or superior to surgery in patients with favorable valve anatomy,206,207 characterized by mild to moderate impairment of leaflet mobility, valve thickness, valve calcification, and subvalvular disease, the components of the Wilkins-Weyman echo score.208 The scoring system has been shown to correlate with long-term outcomes.209 Surgery should be reserved for patients with unfavorable anatomy, more than mild MR, or persistent thrombus in the left atrium despite anticoagulation.5 The threshold for percutaneous or surgical intervention is a mitral valve area of 1.5 cm2 or less. If patients are symptomatic and have favorable anatomy or if they are asymptomatic but have pulmonary hypertension (>50 mm Hg systolic at rest or >60 mm Hg with exercise), they have class I indications. Patients who have unfavorable anatomy and are functional class III or IV are considered to have a class II indication, especially if they are poor candidates for surgery. Asymptomatic patients with new-onset AF and favorable anatomy and symptomatic patients with mild MS (valve area >1.5 cm2) who have pulmonary artery pressure greater than 60 mm Hg, wedge pressure 25 mm Hg or greater, or gradient 15 mm Hg or greater during exercise are also considered class II candidates.5
Acknowledgment
The author wishes to acknowledge Priscilla Peters for kindly providing Figures 32.10, 32.16, and 32.19.
References
1. Passik, CS, Ackermann, DM, Pluth, JR, Edwards, WD. Temporal changes in the causes of aortic stenosis: A surgical pathologic study of 646 cases. Mayo Clin Proc. 1987; 62:119–123.
2. Mangione, S, Nieman, LZ, Gracely, E, Kaye, D. The teaching and practice of cardiac auscultation during internal medicine and cardiology training. A nationwide survey. Ann Intern Med. 1993; 119(1):47–54.
3. Das, P, Pocock, C, Chambers, J. The patient with a systolic murmur: Severe aortic stenosis may be missed during cardiovascular examination. Q J Med. 2000; 93(10):685–688.
4. Lindroos, M, Kupari, M, Heikkila, J, Tilvis, R. Prevalence of aortic valve abnormalities in the elderly: An echocardiographic study of a random population sample. J Am Coll Cardiol. 1993; 21(5):1220–1225.
5. Bonow, RO, Carabello, BA, Chatterjee, K, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing Committee to Revise the 1998 guidelines for the management of patients with valvular heart disease) developed in collaboration with the Society of Cardiovascular Anesthesiologists endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. J Am Coll Cardiol. 2006; 48(3):e1–148.
6. Turi, ZG. Whom do you trust? Misguided faith in the catheter- or Doppler-derived aortic valve gradient. Cathet Cardiovasc Interv. 2005; 65(2):180–182.
7. Braverman, AC, Guven, H, Beardslee, MA, et al. The bicuspid aortic valve. Curr Probl Cardiol. 2005; 30(9):470–522.
8. Da Vinci L: Notes on the valves of the heart and flow of blood within it, with illustrative drawings, 1513.
9. Roberts, WC, Ko, JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation. 2005; 111(7):920–925.
10. Stewart, BF, Siscovick, D, Lind, BK, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J Am Coll Cardiol. 1997; 29(3):630–634.
11. Otto, CM, Lind, BK, Kitzman, DW, et al. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med. 1999; 341(3):142–147.
12. Carabello, BA. Aortic sclerosis—A window to the coronary arteries? N Engl J Med. 1999; 341(3):193–195.
13. Cosmi, JE, Kort, S, Tunick, PA, et al. The risk of the development of aortic stenosis in patients with “benign” aortic valve thickening. Arch Intern Med. 2002; 162(20):2345–2347.
14. Lindroos, M, Kupari, M, Valvanne, J, et al. Factors associated with calcific aortic valve degeneration in the elderly. Eur Heart J. 1994; 15(7):865–870.
15. Freeman, RV, Otto, CM. Spectrum of calcific aortic valve disease: Pathogenesis, disease progression, and treatment strategies. Circulation. 2005; 111(24):3316–3326.
16. Beppu, S, Suzuki, S, Matsuda, H, et al. Rapidity of progression of aortic stenosis in patients with congenital bicuspid aortic valves. Am J Cardiol. 1993; 71(4):322–327.
17. Novaro, GM, Mishra, M, Griffin, BP. Incidence and echocardiographic features of congenital unicuspid aortic valve in an adult population. J Heart Valve Dis. 2003; 12(6):674–678.
18. Tutarel, O. The quadricuspid aortic valve: A comprehensive review. J Heart Valve Dis. 2004; 13(4):534–537.
19. Gunther, S, Grossman, W. Determinants of ventricular function in pressure-overload hypertrophy in man. Circulation. 1979; 59(4):679–688.
20. Carroll, JD, Carroll, EP, Feldman, T, et al. Sex-associated differences in left ventricular function in aortic stenosis of the elderly. Circulation. 1992; 86:1099–1107.
21. Carabello, BA, Green, LH, Grossman, W, et al. Hemodynamic determinants of prognosis of aortic valve replacement in critical aortic stenosis and advanced congestive heart failure. Circulation. 1980; 62(1):42–48.
22. Nemes, A, Forster, T, Varga, A, et al. How can coronary flow reserve be altered by severe aortic stenosis? Echocardiography. 2002; 19(8):655–659.
23. Nemes, A, Forster, T, Thury, A, et al. The comparative value of the aortic atherosclerosis and the coronary flow velocity reserve evaluated by stress transesophageal echocardiography in the prediction of patients with aortic stenosis with coronary artery disease. Int J Cardiovasc Imaging. 2003; 19(5):371–376.
24. Vincentelli, A, Susen, S, Le Tourneau, T, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med. 2003; 349(4):343–349.
25. Pate, GE, Mulligan, A. An epidemiological study of Heyde’s syndrome: An association between aortic stenosis and gastrointestinal bleeding. J Heart Valve Dis. 2004; 13(5):713–716.
26. Otto, CM, Burwash, IG, Legget, ME, et al. Prospective study of asymptomatic valvular aortic stenosis. Clinical, echocardiographic, and exercise predictors of outcome. Circulation. 1997; 95(9):2262–2270.
27. Rosenhek, R, Binder, T, Porenta, G, et al. Predictors of outcome in severe, asymptomatic aortic stenosis. N Engl J Med. 2000; 343(9):611–617.
28. Pellikka, PA, Sarano, ME, Nishimura, RA, et al. Outcome of 622 adults with asymptomatic, hemodynamically significant aortic stenosis during prolonged follow-up. Circulation. 2005; 111(24):3290–3295.
29. Iivanainen, AM, Lindroos, M, Tilvis, R, et al. Natural history of aortic valve stenosis of varying severity in the elderly. Am J Cardiol. 1996; 78(1):97–101.
30. Giles, TD, Martinez, EC, Burch, GE. Gallavardin phenomenon in aortic stenosis. A possible mechanism. Arch Intern Med. 1974; 134(4):747–749.
31. Munt, B, Legget, ME, Kraft, CD, et al. Physical examination in valvular aortic stenosis: Correlation with stenosis severity and prediction of clinical outcome. Am Heart J. 1999; 137(2):298–306.
32. Baumgartner, H. Hemodynamic assessment of aortic stenosis: Are there still lessons to learn? J Am Coll Cardiol. 2006; 47(1):138–140.
33. Baumgartner, H, Stefenelli, T, Niederberger, J, et al. “Overestimation” of catheter gradients by Doppler ultrasound in patients with aortic stenosis: A predictable manifestation of pressure recovery. J Am Coll Cardiol. 1999; 33(6):1655–1661.
34. Burwash, IG, Thomas, DD, Sadahiro, M, et al. Dependence of Gorlin formula and continuity equation valve areas on transvalvular volume flow rate in valvular aortic stenosis. Circulation. 1994; 89(2):827–835.
35. Adegunsoye, A, Mundkur, M, Nanda, NC, Hage, FG. Echocardiographic evaluation of calcific aortic stenosis in the older adult. Echocardiography. 2011; 28(1):117–129.
36. Bamgartner, H. Is there a role for multislice computed tomography in aortic stenosis? Eur Heart J. 2006; 27(24):2923–2924.
37. John, AS, Dill, T, Brandt, RR, et al. Magnetic resonance to assess the aortic valve area in aortic stenosis: How does it compare to current diagnostic standards? J Am Coll Cardiol. 2003; 42(3):519–526.
38. Yankopoulos, NA, Haisty, WK, Pipberger, HV. Computer analysis of the orthogonal electrocardiogram and vectorcardiogram in 257 patients with aortic valve disease. Am J Cardiol. 1977; 40(5):707–715.
39. Bae, JH, Lerman, A, Yang, E, Rihal, C. Feasibility of a pressure wire and single arterial puncture for assessing aortic valve area in patients with aortic stenosis. J Invasive Cardiol. 2006; 18(8):359–362.
40. Assey, ME, Zile, MR, Usher, BW, et al. Effect of catheter positioning on the variability of measured gradient in aortic stenosis. Cathet Cardiovasc Diagn. 1993; 30(4):287–292.
41. Gorlin, R, Gorlin, SG. Hydraulic formula for calculation of the area of the stenotic mitral valve, other cardiac valves, and central circulatory shunts. Am Heart J. 1951; 41:1–29.
42. deFilippi, CR, Willett, DL, Brickner, ME, et al. Usefulness of dobutamine echocardiography in distinguishing severe from nonsevere valvular aortic stenosis in patients with depressed left ventricular function and low transvalvular gradients. Am J Cardiol. 1995; 75(2):191–194.
43. Burwash, IG. Low-flow, low-gradient aortic stenosis: From evaluation to treatment. Curr Opin Cardiol. 2007; 22(2):84–91.
44. Grayburn, PA. Assessment of low-gradient aortic stenosis with dobutamine. Circulation. 2006; 113(5):604–606.
45. Monin, JL, Quere, JP, Monchi, M, et al. Low-gradient aortic stenosis: Operative risk stratification and predictors for long-term outcome: A multicenter study using dobutamine stress hemodynamics. Circulation. 2003; 108(3):319–324.
46. Quere, JP, Monin, JL, Levy, F, et al. Influence of preoperative left ventricular contractile reserve on postoperative ejection fraction in low-gradient aortic stenosis. Circulation. 2006; 113(14):1738–1744.
47. Lange, RA, Hillis, LD. Dobutamine stress echocardiography in patients with low-gradient aortic stenosis. Circulation. 2006; 113(14):1718–1720.
48. Jander, N, Minners, J, Holme, I, et al. Outcome of patients with low-gradient “severe” aortic stenosis and preserved ejection fraction. Circulation. 2011; 123(8):887–895.
49. Khot, UN, Novaro, GM, Popovic, ZB, et al. Nitroprusside in critically ill patients with left ventricular dysfunction and aortic stenosis. N Engl J Med. 2003; 348(18):1756–1763.
50. Folland, ED, Kemper, AJ, Khuri, SF, et al. Intraaortic balloon counterpulsation as a temporary support measure in decompensated critical aortic stenosis. J Am Coll Cardiol. 1985; 5(3):711–716.
51. Theleman, KP, Grayburn, PA, Roberts, WC. Mitral “annular” calcium forming a complete circle “O” causing mitral stenosis in association with a stenotic congenitally bicuspid aortic valve and severe coronary artery disease. Am J Geriatr Cardiol. 2006; 15(1):58–61.
52. Baumgartner, H. Aortic stenosis: Medical and surgical management. Heart. 2005; 91(11):1483–1488.
53. Cowell, SJ, Newby, DE, Prescott, RJ, et al. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005; 352(23):2389–2397.
54. Rossebo, AB, Pedersen, TR, Boman, K, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008; 359(13):1343–1356.
55. O’Brien, KD, Probstfield, JL, Caulfield, MT, et al. Angiotensin-converting enzyme inhibitors and change in aortic valve calcium. Arch Intern Med. 2005; 165(8):858–862.
56. Ngo, DT, Sverdlov, AL, Horowitz, JD. Prevention of aortic valve stenosis: A realistic therapeutic target? Pharmacol Ther. 2012; 135(1):78–93.
57. Rosenhek, R, Rader, F, Loho, N, et al. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation. 2004; 110(10):1291–1295.
58. Kuntz, RE, Tosteson, AN, Berman, AD, et al. Predictors of event-free survival after balloon aortic valvuloplasty. N Engl J Med. 1991; 325:17–23.
59. NHLBI Balloon Valvuloplasty Registry. Percutaneous balloon aortic valvuloplasty. Acute and 30-day follow-up results in 674 patients from the NHLBI Balloon Valvuloplasty Registry. Circulation. 1991; 84:2383–2397.
60. Klein, A, Lee, K, Gera, A, et al. Long-term mortality, cause of death, and temporal trends in complications after percutaneous aortic balloon valvuloplasty for calcific aortic stenosis. J Intervent Cardiol. 2006; 19(3):269–275.
61. Lieberman, EB, Bashore, TM, Hermiller, JB, et al. Balloon aortic valvuloplasty in adults: Failure of procedure to improve long-term survival. J Am Coll Cardiol. 1995; 26(6):1522–1528.
62. Roth, RB, Palacios, IF, Block, PC. Percutaneous aortic balloon valvuloplasty: Its role in the management of patients with aortic stenosis requiring major noncardiac surgery. J Am Coll Cardiol. 1989; 13:1039–1041.
63. Torsher, LC, Shub, C, Rettke, SR, Brown, DL. Risk of patients with severe aortic stenosis undergoing noncardiac surgery. Am J Cardiol. 1998; 81(4):448–452.
64. Christ, M, Sharkova, Y, Geldner, G, Maisch, B. Preoperative and perioperative care for patients with suspected or established aortic stenosis facing noncardiac surgery. Chest. 2005; 128(4):2944–2953.
65. Safian, RD, Warren, SE, Berman, AD, et al. Improvement in symptoms and left ventricular performance after balloon aortic valvuloplasty in patients with aortic stenosis and depressed left ventricular ejection fraction. Circulation. 1988; 78:1181–1191.
66. Smith, CR, Leon, MB, Mack, MJ, et al. Transcatheter versus surgical aortic-valve replacement in high-risk patients. N Engl J Med. 2011; 364(23):2187–2198.
67. Leon, MB, Smith, CR, Mack, M, et al. Transcatheter aortic-valve implantation for aortic stenosis in patients who cannot undergo surgery. N Engl J Med. 2010; 363(17):1597–1607.
68. Miller, DC, Blackstone, EH, Mack, MJ, et al. Transcatheter (TAVR) versus surgical (AVR) aortic valve replacement: Occurrence, hazard, risk factors, and consequences of neurologic events in the PARTNER trial. J Thorac Cardiovasc Surg. 2012; 143(4):832–843.
69. Makkar, RR, Fontana, GP, Jilaihawi, H, et al. Transcatheter aortic-valve replacement for inoperable severe aortic stenosis. N Engl J Med. 2012; 366(18):1696–1704.
70. Azadani, AN, Tseng, EE. Transcatheter heart valves for failing bioprostheses: State-of-the-art review of valve-in-valve implantation. Circ Cardiovasc Intervent. 2011; 4(6):621–628.
71. Généreux, P, Webb, JG, Svensson, LG, et al. Vascular complications after transcatheter aortic valve replacement: Insights from the PARTNER (Placement of AoRTic TraNscathetER Valve) Trial. J Am Coll Cardiol. 2012; 60(12):1043–1052.
72. Gilard, M, Eltchaninoff, H, Iung, B, et al. Registry of transcatheter aortic-valve implantation in high-risk patients. N Engl J Med. 2012; 366(18):1705–1715.
73. Lindblom, D, Lindblom, U, Qvist, J, Lundstrom, H. Long-term relative survival rates after heart valve replacement. J Am Coll Cardiol. 1990; 15(3):566–573.
74. Connolly, HM, Oh, JK, Schaff, HV, et al. Severe aortic stenosis with low transvalvular gradient and severe left ventricular dysfunction: Result of aortic valve replacement in 52 patients. Circulation. 2000; 101(16):1940–1946.
75. Varadarajan, P, Kapoor, N, Bansal, RC, Pai, RG. Clinical profile and natural history of 453 nonsurgically managed patients with severe aortic stenosis. Ann Thorac Surg. 2006; 82(6):2111–2115.
76. Singh, JP, Evans, JC, Levy, D, et al. Prevalence and clinical determinants of mitral, tricuspid, and aortic regurgitation (the Framingham Heart Study). Am J Cardiol. 1999; 83(6):897–902.
77. Kim, M, Roman, MJ, Cavallini, MC, et al. Effect of hypertension on aortic root size and prevalence of aortic regurgitation. Hypertension. 1996; 28(1):47–52.
78. Bekeredjian, R, Grayburn, PA. Valvular heart disease: Aortic regurgitation. Circulation. 2005; 112(1):125–134.
79. Grossman, W, Jones, D, McLaurin, LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. 1975; 56(1):56–64.
80. Ardehali, A, Segal, J, Cheitlin, MD. Coronary blood flow reserve in acute aortic regurgitation. J Am Coll Cardiol. 1995; 25(6):1387–1392.
81. Borer, JS, Hochreiter, C, Herrold, EM, et al. Prediction of indications for valve replacement among asymptomatic or minimally symptomatic patients with chronic aortic regurgitation and normal left ventricular performance. Circulation. 1998; 97(6):525–534.
82. Tarasoutchi, F, Grinberg, M, Spina, GS, et al. Ten-year clinical laboratory follow-up after application of a symptom-based therapeutic strategy to patients with severe chronic aortic regurgitation of predominant rheumatic etiology. J Am Coll Cardiol. 2003; 41(8):1316–1324.
83. Bonow, RO, Lakatos, E, Maron, BJ, Epstein, SE. Serial long-term assessment of the natural history of asymptomatic patients with chronic aortic regurgitation and normal left ventricular systolic function. Circulation. 1991; 84(4):1625–1635.
84. Dujardin, KS, Enriquez-Sarano, M, Schaff, HV, et al. Mortality and morbidity of aortic regurgitation in clinical practice. A long-term follow-up study. Circulation. 1999; 99(14):1851–1857.
85. Carabello, BA. Is it ever too late to operate on the patient with valvular heart disease? J Am Coll Cardiol. 2004; 44(2):376–383.
86. Desjardins, VA, Enriquez-Sarano, M, Tajik, AJ, et al. Intensity of murmurs correlates with severity of valvular regurgitation. Am J Med. 1996; 100(2):149–156.
87. Harvey, WP. Cardiac pearls. Dis Mon. 1994; 40(2):41–113.
88. Tribouilloy, CM, Enriquez-Sarano, M, Mohty, D, et al. Pathophysiologic determinants of third heart sounds: A prospective clinical and Doppler echocardiographic study. Am J Med. 2001; 111(2):96–102.
89. Baddour, LM, Wilson, WR, Bayer, AS, et al. Infective endocarditis: Diagnosis, antimicrobial therapy, and management of complications: A statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: Endorsed by the Infectious Diseases Society of America. Circulation. 2005; 111(23):e394–e434.
90. Zoghbi, WA, Enriquez-Sarano, M, Foster, E, et al. Recommendations for evaluation of the severity of native valvular regurgitation with two-dimensional and Doppler echocardiography. J Am Soc Echocardiogr. 2003; 16(7):777–802.
91. Kozerke, S, Schwitter, J, Pedersen, EM, Boesiger, P. Aortic and mitral regurgitation: Quantification using moving slice velocity mapping. J Magn Reson Imaging. 2001; 14(2):106–112.
92. Bonow, RO. Radionuclide angiography in the management of asymptomatic aortic regurgitation. Circulation. 1991; 84(3 Suppl):1296–1302.
93. Vahanian, A, Baumgartner, H, Bax, J, et al. Guidelines on the management of valvular heart disease: The Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology. Eur Heart J. 2007; 28(2):230–268.
94. Badano, L, Rubartelli, P, Giunta, L, et al. Relation between ECG strain pattern and left ventricular morphology, left ventricular function, and DPTI/SPTI ratio in patients with aortic regurgitation. J Electrocardiol. 1994; 27(3):189–197.
95. Miller, RR, Vismara, LA, DeMaria, AN, et al. Afterload reduction therapy with nitroprusside in severe aortic regurgitation: Improved cardiac performance and reduced regurgitant volume. Am J Cardiol. 1976; 38(5):564–567.
96. Scognamiglio, R, Rahimtoola, SH, Fasoli, G, et al. Nifedipine in asymptomatic patients with severe aortic regurgitation and normal left ventricular function. N Engl J Med. 1994; 331(11):689–694.
97. Evangelista, A, Tornos, P, Sambola, A, et al. Long-term vasodilator therapy in patients with severe aortic regurgitation. N Engl J Med. 2005; 353(13):1342–1349.
98. Klodas, E, Enriquez-Sarano, M, Tajik, AJ, et al. Optimizing timing of surgical correction in patients with severe aortic regurgitation: Role of symptoms. J Am Coll Cardiol. 1997; 30(3):746–752.
99. Chaliki, HP, Mohty, D, Avierinos, JF, et al. Outcomes after aortic valve replacement in patients with severe aortic regurgitation and markedly reduced left ventricular function. Circulation. 2002; 106(21):2687–2693.
100. Taniguchi, K, Nakano, S, Kawashima, Y, et al. Left ventricular ejection performance, wall stress, and contractile state in aortic regurgitation before and after aortic valve replacement. Circulation. 1990; 82(3):798–807.
101. Roman, MJ, Klein, L, Devereux, RB, et al. Reversal of left ventricular dilatation, hypertrophy, and dysfunction by valve replacement in aortic regurgitation. Am Heart J. 1989; 118(3):553–563.
102. Tornos, P, Sambola, A, Permanyer-Miralda, G, et al. Long-term outcome of surgically treated aortic regurgitation: Influence of guideline adherence toward early surgery. J Am Coll Cardiol. 2006; 47(5):1012–1017.
103. Bonow, RO, Picone, AL, McIntosh, CL, et al. Survival and functional results after valve replacement for aortic regurgitation from 1976 to 1983: Impact of preoperative left ventricular function. Circulation. 1985; 72(6):1244–1256.
104. Middlemost, S, Wisenbaugh, T, Meyerowitz, C, et al. A case for early surgery in native left-sided endocarditis complicated by heart failure: Results in 203 patients. J Am Coll Cardiol. 1991; 18(3):663–667.
105. Pompilio, G, Brockmann, C, Bruneau, M, et al. Long-term survival after aortic valve replacement for native active infective endocarditis. Cardiovasc Surg. 1998; 6(2):126–132.
106. Trichon, BH, Felker, GM, Shaw, LK, et al. Relation of frequency and severity of mitral regurgitation to survival among patients with left ventricular systolic dysfunction and heart failure. Am J Cardiol. 2003; 91(5):538–543.
107. Carabello, BA. The pathophysiology of mitral regurgitation. J Heart Valve Dis. 2000; 9(5):600–608.
108. Starling, MR, Kirsh, MM, Montgomery, DG, Gross, MD. Impaired left ventricular contractile function in patients with long-term mitral regurgitation and normal ejection fraction. J Am Coll Cardiol. 1993; 22(1):239–250.
109. Mehta, RH, Supiano, MA, Oral, H, et al. Compared with control subjects, the systemic sympathetic nervous system is activated in patients with mitral regurgitation. Am Heart J. 2003; 145(6):1078–1085.
110. Nagatsu, M, Zile, MR, Tsutsui, H, et al. Native beta-adrenergic support for left ventricular dysfunction in experimental mitral regurgitation normalizes indexes of pump and contractile function. Circulation. 1994; 89(2):818–826.
111. Tsutsui, H, Spinale, FG, Nagatsu, M, et al. Effects of chronic beta-adrenergic blockade on the left ventricular and cardiocyte abnormalities of chronic canine mitral regurgitation. J Clin Invest. 1994; 93(6):2639–2648.
112. Carabello, BA. Ischemic mitral regurgitation and ventricular remodeling. J Am Coll Cardiol. 2004; 43(3):384–385.
113. Kim, TH, Seung, KB, Kim, PJ, et al. Images in cardiovascular medicine. Anterolateral papillary muscle rupture complicated by the obstruction of a single diagonal branch. Circulation. 2005; 112(16):e269–e270.
114. Kaul, S, Spotnitz, WD, Glasheen, WP, Touchstone, DA. Mechanism of ischemic mitral regurgitation. An experimental evaluation. Circulation. 1991; 84(5):2167–2180.
115. Levine, RA, Schwammenthal, E. Ischemic mitral regurgitation on the threshold of a solution: From paradoxes to unifying concepts. Circulation. 2005; 112(5):745–758.
116. Jouan, J, Tapia, M, Cook, C, et al. Ischemic mitral valve prolapse: Mechanisms and implications for valve repair. Eur J Cardiothorac Surg. 2004; 26(6):1112–1117.
117. Bursi, F, Enriquez-Sarano, M, Jacobsen, SJ, Roger, VL. Mitral regurgitation after myocardial infarction: A review. Am J Med. 2006; 119(2):103–112.
118. Bursi, F, Enriquez-Sarano, M, Nkomo, VT, et al. Heart failure and death after myocardial infarction in the community: The emerging role of mitral regurgitation. Circulation. 2005; 111(3):295–301.
119. Birnbaum, Y, Chamoun, AJ, Conti, VR, Uretsky, BF. Mitral regurgitation following acute myocardial infarction. Coron Artery Dis. 2002; 13(6):337–344.
120. Nishimura, RA, Gersh, BJ, Schaff, HV. The case for an aggressive surgical approach to papillary muscle rupture following myocardial infarction: “From paradise lost to paradise regained. ”. Heart. 2000; 83(6):611–613.
121. Seve, P, Dubreuil, O, Farhat, F, et al. Acute mitral regurgitation caused by papillary muscle rupture in the immediate postpartum period revealing Ehlers-Danlos syndrome type IV. J Thorac Cardiovasc Surg. 2005; 129(3):680–681.
122. Schreiber, TL, Fisher, J, Mangla, A, Miller, D. Severe “silent” mitral regurgitation. A potentially reversible cause of refractory heart failure. Chest. 1989; 96(2):242–246.
123. Castello, R, Fagan, L, Jr., Lenzen, P, et al. Comparison of transthoracic and transesophageal echocardiography for assessment of left-sided valvular regurgitation. Am J Cardiol. 1991; 68(17):1677–1680.
124. Dekker, AL, Reesink, KD, van der Veen, FH, et al. Intra-aortic balloon pumping in acute mitral regurgitation reduces aortic impedance and regurgitant fraction. Shock. 2003; 19(4):334–338.
125. Le Feuvre, C, Metzger, JP, Lachurie, ML, et al. Treatment of severe mitral regurgitation caused by ischemic papillary muscle dysfunction: Indications for coronary angioplasty. Am Heart J. 1992; 123(4 Pt 1):860–865.
126. Host, U, Kelbaek, H, Hildebrandt, P, et al. Effect of ramipril on mitral regurgitation secondary to mitral valve prolapse. Am J Cardiol. 1997; 80(5):655–658.
127. Levine, HJ, Gaasch, WH. Vasoactive drugs in chronic regurgitant lesions of the mitral and aortic valves. J Am Coll Cardiol. 1996; 28(5):1083–1091.
128. Wisenbaugh, T, Sinovich, V, Dullabh, A, Sareli, P. Six month pilot study of captopril for mildly symptomatic, severe isolated mitral and isolated aortic regurgitation. J Heart Valve Dis. 1994; 3(2):197–204.
129. Tischler, MD, Rowan, M, LeWinter, MM. Effect of enalapril therapy on left ventricular mass and volumes in asymptomatic chronic, severe mitral regurgitation secondary to mitral valve prolapse. Am J Cardiol. 1998; 82(2):242–245.
130. Breithardt, OA, Sinha, AM, Schwammenthal, E, et al. Acute effects of cardiac resynchronization therapy on functional mitral regurgitation in advanced systolic heart failure. J Am Coll Cardiol. 2003; 41(5):765–770.
131. Capomolla, S, Febo, O, Gnemmi, M, et al. Beta-blockade therapy in chronic heart failure: Diastolic function and mitral regurgitation improvement by carvedilol. Am Heart J. 2000; 139(4):596–608.
132. Starling, MR. Is prophylactic beta-adrenergic blockade appropriate in mitral regurgitation: Impact of cellular pathophysiology. Adv Cardiol. 2004; 41:25–35.
133. Nemoto, S, Hamawaki, M, De Freitas, G, Carabello, BA. Differential effects of the angiotensin-converting enzyme inhibitor lisinopril versus the beta-adrenergic receptor blocker atenolol on hemodynamics and left ventricular contractile function in experimental mitral regurgitation. J Am Coll Cardiol. 2002; 40(1):149–154.
134. Wu, AH, Aaronson, KD, Bolling, SF, et al. Impact of mitral valve annuloplasty on mortality risk in patients with mitral regurgitation and left ventricular systolic dysfunction. J Am Coll Cardiol. 2005; 45(3):381–387.
135. Enriquez-Sarano, M, Schaff, HV, Orszulak, TA, et al. Valve repair improves the outcome of surgery for mitral regurgitation. A multivariate analysis. Circulation. 1995; 91(4):1022–1028.
136. Horskotte, D, Schulte, HD, Bircks, W, Strauer, BE. The effect of chordal preservation on late outcome after mitral valve replacement: A randomized study. J Heart Valve Dis. 1993; 2(2):150–158.
137. Ling, LH, Enriquez-Sarano, M, Seward, JB, et al. Clinical outcome of mitral regurgitation due to flail leaflet. N Engl J Med. 1996; 335(19):1417–1423.
138. Matsumura, T, Ohtaki, E, Tanaka, K, et al. Echocardiographic prediction of left ventricular dysfunction after mitral valve repair for mitral regurgitation as an indicator to decide the optimal timing of repair. J Am Coll Cardiol. 2003; 42(3):458–463.
139. Enriquez-Sarano, M, Tajik, AJ, Schaff, HV, et al. Echocardiographic prediction of survival after surgical correction of organic mitral regurgitation. Circulation. 1994; 90(2):830–837.
140. Enriquez-Sarano, M, Avierinos, JF, Messika-Zeitoun, D, et al. Quantitative determinants of the outcome of asymptomatic mitral regurgitation. N Engl J Med. 2005; 352(9):875–883.
141. Eguchi, K, Ohtaki, E, Matsumura, T, et al. Pre-operative atrial fibrillation as the key determinant of outcome of mitral valve repair for degenerative mitral regurgitation. Eur Heart J. 2005; 26(18):1866–1872.
142. Borer, JS, Hochreiter, CA, Supino, PG, et al. Importance of right ventricular performance measurement in selecting asymptomatic patients with mitral regurgitation for valve surgery. Adv Cardiol. 2002; 39:144–152.
143. Starling, MR. Effects of valve surgery on left ventricular contractile function in patients with long-term mitral regurgitation. Circulation. 1995; 92(4):811–818.
144. Wisenbaugh, T. Unexpected, dismal left ventricular function after surgery for mitral regurgitation: There is just no excuse for it anymore. J Am Coll Cardiol. 2003; 42(3):464–465.
145. Gammie, JS, O’Brien, SM, Griffith, BP, et al. Influence of hospital procedural volume on care process and mortality for patients undergoing elective surgery for mitral regurgitation. Circulation. 2007; 115(7):881–887.
146. Sarris, GE, Miller, DC. Valvular-ventricular interaction: the importance of the mitral chordae tendineae in terms of global left ventricular systolic function. J Card Surg. 1988; 3(3):215–234.
147. Rozich, JD, Carabello, BA, Usher, BW, et al. Mitral valve replacement with and without chordal preservation in patients with chronic mitral regurgitation. Mechanisms for differences in postoperative ejection performance. Circulation. 1992; 86(6):1718–1726.
148. Bolling, SF. Mitral reconstruction in cardiomyopathy. J Heart Valve Dis. 2002; 11(Suppl 1):S26–S31.
149. Acker, MA, Bolling, S, Shemin, R, et al. Mitral valve surgery in heart failure: Insights from the Acorn Clinical Trial. J Thorac Cardiovasc Surg. 2006; 132(3):568–577.
150. Haan, CK, Cabral, CI, Conetta, DA, et al. Selecting patients with mitral regurgitation and left ventricular dysfunction for isolated mitral valve surgery. Ann Thorac Surg. 2004; 78(3):820–825.
151. Trichon, BH, Glower, DD, Shaw, LK, et al. Survival after coronary revascularization, with and without mitral valve surgery, in patients with ischemic mitral regurgitation. Circulation. 2003; 108(Suppl 1):II103–II110.
152. Minami, H, Mukohara, N, Obo, H, et al. Papillary muscle rupture following acute myocardial infarction. Jpn J Thorac Cardiovasc Surg. 2004; 52(8):367–371.
153. Lamas, GA, Mitchell, GF, Flaker, GC, et al. Clinical significance of mitral regurgitation after acute myocardial infarction. Survival and Ventricular Enlargement Investigators. Circulation. 1997; 96(3):827–833.
154. Pellizzon, GG, Grines, CL, Cox, DA, et al. Importance of mitral regurgitation in patients undergoing percutaneous coronary intervention for acute myocardial infarction: The Controlled Abciximab and Device Investigation to Lower Late Angioplasty Complications (CADILLAC) trial. J Am Coll Cardiol. 2004; 43(8):1368–1374.
155. Alfieri, O, Maisano, F, De Bonis, M, et al. The double-orifice technique in mitral valve repair: A simple solution for complex problems. J Thorac Cardiovasc Surg. 2001; 122(4):674–681.
156. Feldman, T, Foster, E, Glower, DD, et al. Percutaneous repair or surgery for mitral regurgitation. N Engl J Med. 2011; 364(15):1395–1406.
157. Maisano, F, Vigano, G, Blasio, A, et al. Surgical isolated edge-to-edge mitral valve repair without annuloplasty: Clinical proof of the principle for an endovascular approach. EuroIntervention. 2006; 2(2):181–186.
158. Perlowski, A, St., Goar, F, Glower, DG, Feldman, T. Percutaneous therapies for mitral regurgitation. Curr Probl Cardiol. 2012; 37(2):42–68.
159. Chockalingam, A, Dorairajan, S, Bhalla, M, Dellsperger, KC. Unexplained hypotension: The spectrum of dynamic left ventricular outflow tract obstruction in critical care settings. Crit Care Med. 2009; 37(2):729–734.
160. Maron, MS, Olivotto, I, Betocchi, S, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003; 348(4):295–303.
161. Maron, BJ, Gardin, JM, Flack, JM, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995; 92(4):785–789.
162. Maron, MS, Olivotto, I, Zenovich, AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006; 114(21):2232–2239.
163. Spirito, P, Chiarella, F, Carratino, L, et al. Clinical course and prognosis of hypertrophic cardiomyopathy in an outpatient population. N Engl J Med. 1989; 320(12):749–755.
164. Epstein, SE, Maron, BJ. Sudden death and the competitive athlete: Perspectives on preparticipation screening studies. J Am Coll Cardiol. 1986; 7:220–230.
165. Kim, JH, Malhotra, R, Chiampas, G, et al. Cardiac arrest during long-distance running races. N Engl J Med. 2012; 366(2):130–140.
166. Maron, BJ. Hypertrophic cardiomyopathy: A systematic review. JAMA. 2002; 287(10):1308–1320.
167. Maki, S, Ikeda, H, Muro, A, et al. Predictors of sudden cardiac death in hypertrophic cardiomyopathy. Am J Cardiol. 1998; 82(6):774–778.
168. Krams, R, Ten Cate, FJ, Carlier, SG, et al. Diastolic coronary vascular reserve: A new index to detect changes in the coronary microcirculation in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004; 43(4):670–677.
169. Sorajja, P, Ommen, SR, Nishimura, RA, et al. Myocardial bridging in adult patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2003; 42(5):889–894.
170. Sherrid, MV, Gunsburg, DZ, Moldenhauer, S, Pearle, G. Systolic anterior motion begins at low left ventricular outflow tract velocity in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2000; 36(4):1344–1354.
171. Ghani, MF, Parker, BM. Hypotension, heart block and reversed pulsus alternans in a patient with hypertrophic subaortic stenosis following digitalis and diuretic therapy. Chest. 1974; 65(6):695–698.
172. Hamid, MS, Gimeno, JR, Valdes, M, Elliott, PM. Reversal of acute pulmonary oedema with beta-blockers in hypertrophic cardiomyopathy. Eur J Echocardiogr. 2003; 4(1):71–72.
173. Sherrid, MV, Pearle, G, Gunsburg, DZ. Mechanism of benefit of negative inotropes in obstructive hypertrophic cardiomyopathy. Circulation. 1998; 97(1):41–47.
174. Sherrid, MV, Barac, I, McKenna, WJ, et al. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005; 45(8):1251–1258.
175. Cohen, IL, Fein, IA, Nabi, A. Reversal of cardiogenic shock and asystole in a septic patient with hypertrophic cardiomyopathy on verapamil. Crit Care Med. 1990; 18(7):775–776.
176. Borja, J, Izquierdo, I, Guindo, J. Hypertrophic cardiomyopathy. Combination of beta blockers and verapamil may be risky. BMJ. 2006; 333(7558):97.
177. Yamaji, K, Fujimoto, S, Yutani, C, et al. Does the progression of myocardial fibrosis lead to atrial fibrillation in patients with hypertrophic cardiomyopathy? Cardiovasc Pathol. 2001; 10(6):297–303.
178. Olivotto, I, Cecchi, F, Casey, SA, et al. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation. 2001; 104(21):2517–2524.
179. Shigematsu, Y, Hamada, M, Mukai, M, et al. Mechanism of atrial fibrillation and increased incidence of thromboembolism in patients with hypertrophic cardiomyopathy. Jpn Circ J. 1995; 59(6):329–336.
180. Maron, BJ, Olivotto, I, Bellone, P, et al. Clinical profile of stroke in 900 patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002; 39(2):301–307.
181. Losi, MA, Betocchi, S, Aversa, M, et al. Determinants of atrial fibrillation development in patients with hypertrophic cardiomyopathy. Am J Cardiol. 2004; 94(7):895–900.
182. Chen, MS, McCarthy, PM, Lever, HM, et al. Effectiveness of atrial fibrillation surgery in patients with hypertrophic cardiomyopathy. Am J Cardiol. 2004; 93(3):373–375.
183. Duytschaever, M, Vijgen, J, Tavernier, R. Atrial fibrillation in hypertrophic cardiomyopathy: Another indication for circumferential pulmonary vein ablation? Acta Cardiol. 2006; 61(2):193–196.
184. Robinson, K, Frenneaux, MP, Stockins, B, et al. Atrial fibrillation in hypertrophic cardiomyopathy: A longitudinal study. J Am Coll Cardiol. 1990; 15(6):1279–1285.
185. Cecchi, F, Olivotto, I, Montereggi, A, et al. Prognostic value of non-sustained ventricular tachycardia and the potential role of amiodarone treatment in hypertrophic cardiomyopathy: Assessment in an unselected non-referral based patient population. Heart. 1998; 79(4):331–336.
186. Spirito, P, Seidman, CE, McKenna, WJ, Maron, BJ. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997; 336(11):775–785.
187. Maron, BJ, Shen, WK, Link, MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000; 342(6):365–373.
188. Ralph-Edwards, A, Woo, A, McCrindle, BW, et al. Hypertrophic obstructive cardiomyopathy: Comparison of outcomes after myectomy or alcohol ablation adjusted by propensity score. J Thorac Cardiovasc Surg. 2005; 129(2):351–358.
189. Yu, EH, Omran, AS, Wigle, ED, et al. Mitral regurgitation in hypertrophic obstructive cardiomyopathy: Relationship to obstruction and relief with myectomy. J Am Coll Cardiol. 2000; 36(7):2219–2225.
190. Fananapazir, L, Epstein, ND, Curiel, RV, et al. Long-term results of dual-chamber (DDD) pacing in obstructive hypertrophic cardiomyopathy. Evidence for progressive symptomatic and hemodynamic improvement and reduction of left ventricular hypertrophy. Circulation. 1994; 90(6):2731–2742.
191. Maron, BJ, Nishimura, RA, McKenna, WJ, et al. Assessment of permanent dual-chamber pacing as a treatment for drug-refractory symptomatic patients with obstructive hypertrophic cardiomyopathy. A randomized, double-blind, crossover study (M-PATHY). Circulation. 1999; 99(22):2927–2933.
192. Movahed, MR, Saito, Y, Ahmadi-Kashani, M, Ebrahimi, R. Mitral annulus calcification is associated with valvular and cardiac structural abnormalities. Cardiovasc Ultrasound. 2007; 5:14.
193. Willens, HJ, Chirinos, JA, Hennekens, CH. Prevalence and clinical correlates of mitral annulus calcification in Hispanics and non-Hispanic whites. J Am Soc Echocardiogr. 2007; 20(2):191–196.
194. Bonow, RO, Braunwald, E. Valvular heart disease. In: Zipes DP, Libby P, Bonow RO, Braunwald E, eds. Braunwald’s Heart Disease; A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier; 2005:1553–1632.
195. Thibault, GE. Clinical problem-solving. Studying the classics. N Engl J Med. 1995; 333(10):648–652.
196. Wilkins, GT, Weyman, AE, Abascal, VM, et al. Percutaneous balloon dilatation of the mitral valve: An analysis of echocardiographic variables related to outcome and the mechanism of dilatation. Br Heart J. 1988; 60:299–308.
197. Abaci, A, Oguzhan, A, Unal, S, et al. Application of the vena contracta method for the calculation of the mitral valve area in mitral stenosis. Cardiology. 2002; 98(1-2):50–59.
198. Messika-Zeitoun, D, Brochet, E, Holmin, C, et al. Three-dimensional evaluation of the mitral valve area and commissural opening before and after percutaneous mitral commissurotomy in patients with mitral stenosis. Eur Heart J. 2007; 28(1):72–79.
199. Uzun, M, Baysan, O, Erinc, K, et al. A simple different method to use proximal isovelocity surface area (PISA) for measuring mitral valve area. Int J Cardiovasc Imaging. 2005; 21(6):633–640.
200. Palacios, IF. What is the gold standard to measure mitral valve area postmitral balloon valvuloplasty? Cathet Cardiovasc Diagn. 1994; 33(4):315–316.
201. Mohan, JC, Patel, AR, Passey, R, et al. Is the mitral valve area flow-dependent in mitral stenosis? A dobutamine stress echocardiographic study. J Am Coll Cardiol. 2002; 40(10):1809–1815.
202. Schoenfeld, MH, Palacios, IF, Hutter, AM, Jr., et al. Underestimation of prosthetic mitral valve areas: Role of transseptal catheterization in avoiding unnecessary repeat mitral valve surgery. J Am Coll Cardiol. 1985; 5:1387–1392.
203. Carabello, BA. Modern management of mitral stenosis. Circulation. 2005; 112(3):432–437.
204. Topaloglu, S, Boyaci, A, Ayaz, S, et al. Coagulation, fibrinolytic system activation and endothelial dysfunction in patients with mitral stenosis and sinus rhythm. Angiology. 2007; 58(1):85–91.
205. Schumacher, B, Luderitz, B. Rate issues in atrial fibrillation: Consequences of tachycardia and therapy for rate control. Am J Cardiol. 1998; 82(8A):29N–36N.
206. Reyes, VP, Raju, BS, Wynne, J, et al. Percutaneous balloon valvuloplasty compared with open surgical commissurotomy for mitral stenosis. N Engl J Med. 1994; 331:961–967.
207. Turi, ZG, Reyes, VP, Raju, BS, et al. Percutaneous balloon versus surgical closed commissurotomy for mitral stenosis. A prospective, randomized trial. Circulation. 1991; 83:1179–1185.
208. Heger, JJ, Wann, LS, Weyman, AE, et al. Long-term changes in mitral valve area after successful mitral commissurotomy. Circulation. 1979; 59:443–448.
209. Palacios, IF, Sanchez, PL, Harrell, LC, et al. Which patients benefit from percutaneous mitral balloon valvuloplasty? Prevalvuloplasty and postvalvuloplasty variables that predict long-term outcome. Circulation. 2002; 105(12):1465–1471.
210. Wilson, W, Taubert, KA, Gewitz, M, et al. Prevention of infective endocarditis: Guidelines from the American Heart Association: A guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007; 116(15):1736–1754.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]32
Valvular Heart Disease in Critical Care
Rheumatic heart disease accounted for 50% or more of admissions for heart disease in the first half of the twentieth century, with congenital heart disease representing the other major indication for valve surgery through the 1960s.1 With evolving technology and an increase in average life expectancy, heart valve surgery has shifted to valve repair, as well as replacement for a variety of acquired valvulopathies. The therapeutic armamentarium, once limited to minimal supportive medical therapy, then solely to valve replacement, has expanded to a variety of surgical repair techniques; a multitude of tissue and mechanical valve options for replacement; and, most recently, an expanding set of percutaneous interventions including percutaneous valve implantation and repair. In addition, various mechanical devices for temporary hemodynamic support are important adjuncts to the management of patients with valvular heart disease in the critical care setting.
Diagnosis of valvular heart disease in critical care units is challenging, with lack of a quiet environment for auscultation; comorbid conditions that affect physical findings; and stress, infection, or metabolic abnormalities that result in tachycardia and shorter intervals for evaluation of heart sounds. Patients in a low output state have softer heart sounds and murmurs as well. In addition, clinical reliance on physical examination has fallen, as has the ability to make accurate diagnosis of valve disease.2 A delay in recognizing the presence of significant valve dysfunction continues to occur frequently, and some patients who have had prior routine medical outpatient care nevertheless present with undiagnosed valvular heart disease.3 The currently accepted thresholds for mild, moderate, and severe valvular heart disease are shown in Table 32.1.
Table 32.1
Classification of Severity of Valvular Heart Disease Based on ACC/AHA Guidelines
*Valve gradients are flow-dependent and when used as estimates of severity of valve stenosis should be assessed with knowledge of cardiac output or forward flow across the valve.
Modified from Bonow RO, Carabello BA, Chatterjee K, et al: ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 1998 guidelines for the management of patients with valvular heart disease) developed in collaboration with the Society of Cardiovascular Anesthesiologists and endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. J Am Coll Cardiol 2006;48:e1-148.
Aortic Stenosis
With the aging of the population, aortic stenosis (AS) has moved to the forefront in frequency of valvular heart disease encountered in older populations. The prevalence is surprisingly high; in patients 85 years of age, more than 8% of a random general population survey had Doppler-derived aortic valve area estimates of 1 cm2 or less (Fig. 32.1),4 which is consistent with severe AS by the 2006 American College of Cardiology/American Heart Association (ACC/AHA) guidelines.5 Thus AS should be considered in the differential diagnosis of each elderly critical care patient with hemodynamic instability. The possibility of AS should be dismissed only after considering physical examination and electrocardiographic and, when applicable, echocardiographic and invasive findings, all of which have potential limitations in achieving accurate diagnosis as discussed subsequently.6
Pathophysiology
AS represents a continuum, from hemodynamically insignificant congenital and atherosclerotic disease to end-stage decompensation secondary to severe valvular obstruction. The congenital pathway is typically secondary to a bicuspid aortic valve, the most common congenital cardiac anomaly if mitral valve prolapse is excluded, occurring in approximately 1.5% of the population7 and originally described by Da Vinci in 1513.8 The most common acquired anomaly is typically referred to as degenerative disease, an atherosclerotic process that represents a continuum from aortic valve sclerosis, which is not associated with a significant gradient, to a densely calcified aortic valve with severe outflow obstruction. Although the cause of aortic valve stenosis in patients undergoing aortic valve replacement (AVR) has shifted to calcific tricuspid aortic valve disease from bicuspid and rheumatic disease, this remains an age-related phenomenon, with a predominance of bicuspid aortic valve disease in patients younger than age 70 (Fig. 32.2).9
Aortic Valve Sclerosis
Aortic valve sclerosis is an important disease entity, although usually not because of hemodynamic considerations. Defined as calcification and thickening of the aortic valve without significant outflow obstruction (gradient <20 to 25 mm Hg), it is present in nearly 30% of the population older than age 65 and nearly 50% by age 8510 and is associated with a 50% increase in 5-year cardiovascular mortality rate.11 Its incidence is as high as 15-fold greater than aortic valve stenosis; the two diagnoses can be differentiated from one another by hemodynamics and physical examination findings discussed subsequently. In keeping with the primary atherosclerotic nature of aortic sclerosis, it is associated with increasing age, male gender, hypertension, smoking, elevated low-density lipoprotein and lipoprotein(a) levels, and diabetes.10 The primary importance of aortic valve sclerosis is that it provides a window on the overall presence of vascular disease, in particular coronary artery disease.12 Thus there should be a high index of suspicion of vascular disease in any patient with aortic valve sclerosis managed in the critical care setting.
Patients with aortic sclerosis do progress to aortic valve stenosis; a study of more than 2000 patients with valve thickening showed progression to severe AS in 2.5% over an average time interval of 7 years.13 The fact that so-called degenerative disease of the aortic valve is absent in nearly half of octogenarians is also important to consider because it implies that it is not only aging but other factors that result in leaflet thickening, calcification, and stenosis. In the Helsinki Aging Study from which data are reflected in Figure 32.1, additional analysis demonstrated that not only age but also hypertension and low body mass index independently predicted calcification of the aortic valve, and age and serum ionized calcium were independently associated with valvular stenosis.14 In general the process of sclerosis and then stenosis of the aortic valve appears, like atherosclerosis in general, to be an active inflammatory process, with deposition of lipoproteins, local inflammation with T lymphocyte and macrophage infiltration, fibroblast proliferation, and eventually osteoblast and bone formation.15 Similar to other vascular disease, endothelial disruption likely leads to the initial lipid deposition in the leaflet tissues. The areas of early focal plaque formation appear at the loci of greatest stress: on the aortic side of the leaflets at the flexion points. Because bicuspid valves have greater mechanical stress, the average age at presentation of patients with bicuspid aortic valve stenosis is significantly lower than in the tricuspid aortic valve stenosis that is typically seen in an elderly population.16
An important element in understanding calcific AS is lack of commissural fusion; unlike rheumatic mitral valve stenosis, in which fusion of the commissures is the primary cause of obstructive disease, lack of mobility of the aortic valve leaflets is the primary cause of obstruction in AS. Rheumatic AS is associated with commissural fusion but is now a rare finding, even in countries where rheumatic heart disease is prevalent, and is usually associated with mitral valve involvement and typically aortic insufficiency as well. More obscure causes, such as unicuspid and quadricuspid aortic valve disease, are uncommon, although presentation can be delayed to adulthood. In the case of unicuspid aortic valves there is a strong association with AS,17 whereas with quadricuspid aortic valves there is a high incidence of significant aortic insufficiency.18
Normal aortic valve area ranges from 3 to 4 cm2. Significant resistance to outflow does not occur until the valve orifice is reduced more than 50%. Based on the simple hydraulic principle of Poiseuille’s law, a 50% reduction in valve diameter results in approximately a 16-fold increase in resistance. In practice, as the resistance to outflow rises, the left ventricle is subject to pressure overload, resulting in compensatory hypertrophy. This in turn normalizes wall stress because the latter is proportional to chamber diameter times pressure divided by wall thickness (the LaPlace principle). The degree of wall thickness is variable: In patients with inadequate hypertrophic response, wall stress is inordinately high and there is early dilation and dysfunction.19 In patients with a profound hypertrophic response disproportionate to valvular resistance, wall stress actually falls below normal and ejection fraction (EF) becomes supranormal,20 a phenomenon that appears to be more common in women with AS. The EF for any degree of wall stress is predictable,19 and patients who have disproportionately poor ejection phase indices (afterload mismatch) typically manifest left ventricular (LV) dysfunction beyond the depression that would be associated with high wall stress (Fig. 32.3),21 a phenomenon described under low-gradient, low-output AS that follows.
LV hypertrophy, although it reduces wall stress, has several features that may be deleterious. With progressive hypertrophy, diastolic pressures may rise as LV compliance decreases, resulting in increased LV filling pressures. In addition, the myocardial supply-demand relationship is deleteriously affected by hypertrophy in this setting. With increased wall stress and increased wall mass, myocardial oxygen demand is increased; at the same time coronary flow reserve is decreased in AS,22 and in later stages of the disease diastolic perfusion pressure is lowered. Because of abnormal flow reserve, conventional testing for ischemia in the setting of significant aortic valve stenosis has inadequate specificity to identify the presence or absence of hemodynamically significant concomitant coronary artery disease.23
Other features associated with AS that are important in the critical care setting include abnormal platelet function and decreased levels of von Willebrand factor,24 with associated bleeding risk that improves with AVR. Another associated cause of bleeding is gastrointestinal angiodysplasia associated with aortic valve stenosis,25 possibly exacerbated by the concomitant presence of von Willebrand syndrome.
The progression of AS is highly variable, but with the onset of moderately severe disease, the estimated mean decrease in valve area is on the order of 0.1 cm2/year.26 Certain demographics and noninvasive findings appear to predict the rate of progression, including age older than 50 years and moderate to severe valve calcification.27 The issue of progression is particularly important in patients who are asymptomatic of the disease because AVR before it is necessary is undesirable but must be weighed against the risk of sudden death. The latter has been studied in a number of longitudinal studies and is rare if patients are followed prospectively. The patients who are most likely to require early intervention are those with peak Doppler systolic velocities greater than 4 m/second or who have a rapid increase in serial transvalvular Doppler velocity measurements.26,27 This threshold has been confirmed by other series.28 Once patients become symptomatic, with a classic triad of heart failure, angina, or syncope, hemodynamic deterioration can be rapid with significant associated mortality risk.29
Diagnosis
Physical Examination
The hallmarks of the physical examination relate to the contour of the aortic pressure upstroke, with a low volume and delayed carotid upstroke (pulsus parvus et tardus), a late peaking systolic ejection murmur, and a diminished or absent aortic second sound. Figure 32.4 demonstrates the hemodynamics that manifest in an abnormal carotid pulse contour; unlike the brisk upstroke of LV pressure over time (dP/dt), the aortic pressure is slow to rise and of significantly lower volume. An ejection click may be present in young patients with congenital AS, but with increasing calcification of the valve, mobility decreases to a point at which an ejection click becomes unlikely. The aortic component of the second heart sound, reflecting deceleration and closure of the aortic valve, is diminished or absent in severe AS when the thickened aortic valve leaflets are poorly mobile, have little deceleration, and drift rather than snap shut. The murmur of AS is characteristically a crescendo-decrescendo murmur that is late peaking, consistent with the timing of the peak gradient as seen in Figure 32.4, and is typically best heard over the right upper sternum and clavicle. It reflects the high-velocity systolic jet directed into the ascending aorta. A second component, described by Gallavardin as a musical component, is best heard at the left lower sternal border. The latter confounds diagnosis because the murmur is suggestive of mitral regurgitation (MR), but in its true form it is purely secondary to aortic valve stenosis. Because handgrip raises resistance to LV ejection, the murmur should decrease if it is a true Gallavardin phenomenon, whereas when it is secondary to MR, it should increase.30
Unfortunately, none of these findings has sufficiently high sensitivity or specificity for AS or for differentiating moderate from severe disease.3 Many patients with AS, particularly the elderly with stiff noncompliant vessels, will have a wide pulse pressure even with declining cardiac output and may have systolic hypertension. Hypotension may not be seen until late stages of the disease. The loudness of the murmur, which correlates to some degree with severity, is also not specific because body habitus and a variety of other factors affect the acoustics transmitted across the chest. Most importantly, in severe AS, as the cardiac output drops, the gradient generated decreases as well. In this setting the murmur may be quite soft, although sometimes still high pitched because of the high velocity across a tight constriction. A useful means of differentiating aortic sclerosis from stenosis is that in the former the systolic ejection murmur heard over the right sternum is typically midpeaking and mild to moderate in intensity, and it features a well-preserved aortic second heart sound. Although the physical findings described are variable, presence of the aortic second heart sound tends to exclude severe calcific AS.31
Noninvasive Evaluation
In contrast to the physical examination, echocardiographic techniques have progressively improved over the past several decades and availability in the critical care setting in industrialized nations is ubiquitous. The characteristic echocardiographic features of AS are decreased valve leaflet mobility, calcification in all except congenital AS in adolescents and young adults, and an augmented Doppler velocity that generally allows accurate estimation of the gradient. In congenital bicuspid AS in young adults, when commissural fusion is dominant, a characteristic doming pattern is seen, but this disappears with progressive valve calcification. The severity of calcification correlates with extent of obstruction by middle age, and typically Doppler signal velocity with peak and mean pressure gradient and valve area by continuity equation provide an accurate overall assessment. However, the gradient is highly dependent on flow across the valve, and in low-output states the gradient may result in underestimation of severity of disease; in high-output states such as in patients with augmented cardiac output caused by inotropic stimulation, endogenous high catecholamine states, and sepsis, the gradient may be disproportionately higher than the severity of stenosis would suggest. The latest ACC/AHA guidelines describe the threshold for severe AS as antegrade jet velocity greater than 4 m/second, gradient greater than 40 mm Hg, and aortic valve area less than 1 cm2, with the caveat that the gradient and jet velocity depend on the overall transvalvular flow5 (see Table 32.1). Importantly, antegrade flow across the aortic valve does not equal cardiac output in patients with confounding conditions, including aortic insufficiency; thus, for example, a patient with mild to moderate AS and moderate aortic insufficiency may have a transvalvular gradient suggestive of severe AS. Finally, although most practitioners do not index the valve area, it is important to appreciate that in patients with large body surface areas, disease that might be considered moderate in smaller patients may be functionally severe.
Several caveats need to be considered before acceptance of noninvasive data in the critical care setting. Inadequate acoustic windows in some patients and difficulty in positioning patients on respirators and with multiple lines in place do limit echocardiographic imaging. Technical errors in recording and interpretation result in significant misdiagnosis: errors in recording angle, inadvertent imaging of MR jets instead of aortic outflow, assessing proximal velocity instead of the transvalvular signal, and selecting signals in the setting of arrhythmias that are not representative of mean heartbeats can all lead to skewed assessment of valve disease severity.32 Further, a number of other potential confounding variables can lead to overestimation or underestimation of the severity of AS,6,33 and valve area calculations by the continuity equation in a low-flow setting may be inaccurate.34 Because the continuity equation depends on measurement of the LV outflow track dimension, which is prone to inaccuracy, another measure of AS severity, the dimensionless index (the ratio of blood flow velocity across the LV outflow track to that across the aortic valve), has been widely adopted.35 A number of other techniques for determining the severity of AS supplement the most commonly used noninvasive tools, including three-dimensional echo, computed tomography,36 and magnetic resonance imaging (MRI).37 Overall, there has been a major a shift in the gold standard from catheter-derived to noninvasive-derived determination of AS severity, and cardiac catheterization is no longer indicated for hemodynamic assessment of aortic valve disease severity when noninvasive findings are unequivocal.5
The echocardiogram provides important additional information including severity of LV hypertrophy, LV function and size, and concomitant disease of other heart valves. The presence of regional wall motion abnormalities, in the absence of a conduction disturbance, suggests concomitant coronary artery disease. The electrocardiogram (ECG) in AS is typically abnormal and frequently features LV hypertrophy, with ST-segment abnormalities in the lateral leads typically described as a strain pattern.38 Although occasionally mistaken for anterior ischemia or infarction, with loss of R voltage across the precordium and occasionally with narrow QS complexes, the ECG is not specific for severity of AS. With increasing calcification of the perivalvular tissues, heart block is seen, typically late in the course of the disease.
Cardiac Catheterization
Cardiac catheterization is indicated in patients in whom the noninvasive data are equivocal, and coronary angiography is indicated in a range of settings identified in Box 32.1. The hemodynamics of AS are best recorded with a catheter or catheters placed simultaneously on either side of the aortic valve.6,39 Unfortunately, most laboratories record femoral artery pressure in lieu of central aortic pressure or do a catheter pullback in lieu of simultaneous pressure tracings; both techniques can result in significant misinterpretation of the severity of aortic valve disease.40 A variety of other errors in catheterization laboratory pressure measurements makes the catheter-derived valve area generally more variable than desirable in all except a few laboratories, including inherent errors in estimating rather than measuring oxygen consumption and in assuming that the Gorlin constant (originally established for a limited subset of patients41) and the valve area itself remain constant under varying loading conditions.34 In the catheterization laboratory as well, transvalvular flow may be underestimated if there is concomitant aortic insufficiency, resulting in overestimation of the severity of aortic valve disease.
Low-Gradient, Low-Output Aortic Stenosis
The critical care unit patient with a low to moderate gradient (typically <30 mm Hg) across the aortic valve in the setting of low cardiac output represents an important conundrum (and should be differentiated from the aortic sclerosis patient with low gradient not associated with depressed output). In general, the rules of hydraulics demonstrate that valve area is proportional to flow divided by the square root of the gradient.41 Thus, when the valve area is fixed, increased flow is associated with an exponential rise in gradient when valve areas are less than 1 cm2 (Fig. 32.5). When a patient in the critical care setting has depressed cardiac output and a low gradient across the aortic valve, there are two possible interpretations of these findings: either the patient has mild to moderate AS and poor LV function, or the patient has severe AS with a low EF appropriate to high wall stress (see Fig. 32.3) and secondary depression of cardiac output. In the case of the former, increasing flow results in better opening of the aortic valve, with only mild to moderate increase in gradient and an increase in the calculated valve area of 0.3 cm2 or greater.42 In the latter case, a fixed obstruction to outflow exists and increasing transvalvular flow results in a dramatic increase in gradient. In addition to increasing flow across the valve in these patients, typically with dobutamine infusion,43 the echocardiogram can be useful in several other ways44: valve calcification is suggestive of fixed outflow obstruction and, if severe, suggests that the underlying disease is severe AS rather than LV dysfunction. Preserved LV contractile reserve, with significant rise in stroke volume (>20%), peak velocity (>0.6 m/second), or mean transvalvular gradient (>10 mm Hg) at the time of dobutamine infusion is an additional useful marker. Lack of contractile reserve has been associated with lower operative survival rate45 (6% vs. 33% in one study), although it should not be the sole parameter for the decision on whether or not to perform AVR: patients who survive may manifest significant recovery in LV function postoperatively.46 Low contractile reserve should not preclude AVR in patients who do have severe fixed obstruction because their prognosis without surgery is abysmal.47 One other variation of low-gradient low-output AS involves patients with small hypertrophied ventricles and preserved EF. This subset, sometimes called high valvuloarterial impedance AS, has low gradient because of low stroke volume; the burgeoning evidence base does show improved results with AVR.48
Therapy
Medical Management
Despite the caveats, vasodilator therapy, including nitroprusside, has been used in the critical care setting in patients with severe LV dysfunction and severe AS.49 In a select group of patients not dependent on inotropes, cardiac output rose significantly and there was overall hemodynamic improvement. Similarly, intra-aortic balloon pumping has been used, although there are only isolated case reports of efficacy.50,51 As would be expected, both nitroprusside and intra-aortic balloon pumping result in afterload reduction and some increase in transvalvular flow along with some increase in aortic valve gradient.
Lipid-lowering therapy and converting enzyme inhibitors have been the subject of substantial investigation,52 although their role is in the chronic setting rather than during critical care. Two prospective randomized trials failed to show clear benefits of a statin53,54 in slowing disease progression. Some preliminary evidence suggests potential benefits of angiotensin-converting enzyme (ACE) inhibitors in decreasing progression of aortic valve calcification,55,56 but the available clinical data do not show slowing of AS progression.57 Both lipid-lowering and converting enzyme inhibitors need to be tested in larger populations earlier in the course of aortic valve disease.
Percutaneous Interventions
Balloon Valvuloplasty
The acute hemodynamic results (Fig. 32.6) are poor compared with those of AVR, with a 50% reduction in gradient and an increase in aortic valve area of only 0.2 to 0.3 cm2.58 The in-hospital mortality rate was close to 10% in the National Heart, Lung and Blood Institute registry59 and is substantially higher in patients with hemodynamic decompensation and multiorgan failure. Most valves have restenosed within a few months, with a recent series showing an 87% mortality rate after a median period of less than 7 months,60 and no benefit for long-term outcomes has been shown. Indeed, the overall clinical course is not influenced by balloon valvotomy.61 Nevertheless, it can serve as a bridge to transcatheter AVR or surgical AVR in select patients with multiple comorbid conditions and is an alternative but low efficacy measure for patients who are not candidates for AVR. It is not an alternative to TAVR or surgery for patients who can undergo valve replacement, even for most patients at relatively high risk for the latter.
Several settings in which balloon aortic valvuloplasty was previously considered to have a role are no longer included as indications in the 2006 ACC/AHA guidelines.5 These settings include preoperative balloon dilation in patients undergoing noncardiac surgery62; in general all but decompensated patients can withstand general anesthesia as long as careful hemodynamic monitoring and anesthesia management are performed.63 Patients who require urgent noncardiac surgery in the setting of severe symptomatic AS do have a significant perioperative risk,64 and preoperative AVR should be considered if at all possible. Balloon aortic valvuloplasty has also been proposed as a tool to differentiate myocardial dysfunction from severe AS with secondary LV dysfunction in low-gradient, low-output AS.65 The latter patients typically demonstrated substantial improvement in stroke volume and EF after balloon dilation, but dobutamine stress testing is a far safer, less morbid screening tool.
Transcatheter Aortic Valve Replacement
Transcatheter aortic valve replacement (TAVR), a technology developed in the past decade, has revolutionized the treatment of AS. The procedure is of increasing importance in the critical care setting. Tissue valves are sewn to either balloon expandable or self-expanding stents, which are crimped onto a catheter and advanced across the stenotic native valve. Both balloon expandable (Fig. 32.7) and self-expanding technologies are widely available outside the United States. The landmark PARTNER (Placement of AoRtic TraNscathetER valves) study had two cohorts: PARTNER A enrolled high-risk but operable patients while PARTNER B enrolled patients deemed inoperable. PARTNER A randomized patients to surgical aortic valve replacement (SAVR) versus two types of TAVRs: transfemoral if the iliac and femoral vasculature was suitable for accommodating the large catheter shafts or transapical if not.66 The results of TAVR and SAVR were similar, including mortality rate, stroke, and quality of life with published follow-up through 2 years; vascular complications were higher with TAVR and major bleeding and AF were higher with SAVR66 (Fig. 32.8A). PARTNER B randomized patients with suitable vasculature to transfemoral TAVR versus medical therapy.67 Patients who received only medical therapy (with or without balloon valvuloplasty) had a 51% 1-year mortality rate compared to those inoperable patients randomized to TAVR who had a 31% 1-year mortality rate, an approximately 40% relative reduction (Fig. 32.8B). Thus, TAVR appears to be among the most dramatically successful of all modern cardiac interventions. However, it is noteworthy that the residual mortality rate in the inoperable TAVR patients remains very high, reflecting the severe comorbid conditions in patients who met inoperability criteria. There was also an initially higher stroke risk in the TAVR patients.67 The risk of stroke in follow-up appears to be largely related to patient comorbid conditions that increase stroke risk, rather than the type of valve replacement.68 The dramatic mortality risk benefit with TAVR in inoperable patients has been shown to continue through 2 years of follow-up; because the stroke risk is higher in the surviving medically treated population, the early excess stroke risk associated with TAVR largely resolves by 24 months.69 Outside the United States, TAVR has been performed in some 70,000 patients; the Food and Drug Administration initially approved TAVR for inoperable patients based on the results of PARTNER B and subsequently approved the high-risk population treated in PARTNER A. A variety of investigational uses of the TAVR platform, such as valve-in-valve replacement for patients with bioprosthetic valve dysfunction (AS and aortic insufficiency) are being performed, primarily outside the United States70; the possibility that transcatheter valve-in-valve replacement will eventually supplant reoperation may change the algorithm for choice of tissue versus mechanical AVR.
The management of TAVR patients after valve replacement frequently takes place in the critical care unit. The higher rate of vascular complications associated with TAVR requires careful monitoring for limb ischemia, retroperitoneal hemorrhage, and a variety of other vascular complications.71 Conduction disturbances, particularly after use of a self-expanding TAVR platform, occur in a significant percentage of cases, requiring permanent pacemaker placement.72 A number of issues, such as the use of dual antiplatelet therapy and anticoagulation after TAVR remain to be fully investigated; most operators treat with dual antiplatelet therapy for periods ranging from 1 to 6 months; dual antiplatelet therapy plus anticoagulation for patients with AF is a combination with particularly high major bleeding risk, though the evidence base is incomplete, and many clinicians use only aspirin and warfarin in this setting.
Aortic Valve Replacement
AVR remains the treatment of choice for severe AS in patients who are considered to have reasonable operable risk. Class I indications are severe AS (valve area less than 1 cm2) with symptoms or, regardless of symptoms, if patients have severe AS and are undergoing coronary artery bypass, are undergoing surgery of the aorta or other heart valves, or have LV dysfunction. A long list of class II indications generally focuses on patients who do not meet class I criteria but are thought to be at increased risk of rapid disease progression or hemodynamic compromise. In addition, patients with moderate AS who undergo other cardiac surgery generally have simultaneous AVR. Survival after AVR is excellent if LV function is preserved, with operative mortality rate in ideal candidates as low as 1%. In these patients, age-matched survival is not significantly different from patients without AS.73 In the setting of LV dysfunction, however, postoperative life expectancy is relatively poor.74 Nevertheless, conservative therapy for severe AS remains an undesirable alternative: A recent review of 453 patients treated without intervention despite severe AS had 1-year, 5-year, and 10-year survival rates of only 62%, 32%, and 18%, respectively,75 with the worst survival in patients with renal failure, pulmonary hypertension, age older than 75 years, diminished EF, and congestive heart failure.
Aortic Insufficiency
The overall prevalence of moderate or severe aortic insufficiency (AI) in the United States, as shown in the Framingham Offspring Study, ranged from 0.3% in the fifth decade of life to 2.2% for patients aged 70 to 83.76 The frequency is age and male gender related. The diagnosis of AI is more difficult and the treatment issues in many ways more complex than for AS, with less of a clear dichotomy in the decision tree for valve replacement. AI needs to be considered in light of its acuity: Acute AI is usually associated with life-threatening comorbid conditions and the resultant acute regurgitation places great stress on the left ventricle and may itself be fatal. In contrast, chronic AI may evolve from clinically insignificant to requiring surgery over decades. Unlike AS, it is not primarily a disease of the aging process (although it does occur more frequently with increasing age) and is typically secondary to one of an extensive list of systemic or structural diseases that result in insufficiency of the aortic valve. Acute AI frequently requires urgent surgery, whereas chronic AI may be managed by watchful waiting and some limited options for medical therapy.
Pathophysiology
Acute AI is generally associated with leaflet involvement by endocarditis or disruption of the aortic valve’s annular structure by dissection or trauma. Chronic AI, by contrast, is caused by congenital valve abnormalities, most importantly bicuspid aortic valve disease, or by the degenerative process described earlier associated with AS. Conditions that distort the annulus and aortic root including systemic hypertension have been considered to be important causes of secondary AI, although the association between chronic AI and systemic hypertension remains to be confirmed.76,77 Rheumatic AI, almost invariably associated with mitral valve involvement as well, remains prevalent in developing countries but is an uncommon cause of AI in industrialized nations.
In general, causes of AI (Box 32.2) have been divided into those that affect the leaflets primarily and those that affect the root and annulus. The former includes bicuspid and other aortic valve abnormalities, endocarditis, rheumatic aortic valve disease, the atherosclerotic process described earlier, connective tissue disorders, antiphospholipid syndrome (Libman-Sacks endocarditis), and toxicity from anorectic drugs.78 The aortic root and annulus are affected by a variety of comorbid conditions that dilate the aortic root; Marfan and Ehlers-Danlos syndromes; osteogenesis imperfecta; chronic aortic dissection; syphilitic aortitis; connective tissue disorders; and, along with the valve leaflets, ankylosing spondylitis.
In isolated acute AI, the sudden onset of regurgitation imposes a large-volume load on the left ventricle in diastole prior to an adaptive process being in place. The abrupt rise in pressure is reflected by parallel development of left atrial and pulmonary vascular hypertension, frequently resulting in pulmonary edema. Because the compliance of a previously unaffected left ventricle is not sufficient to allow adequate dilation to absorb the regurgitant volume, the ventricle operates on the steep portion of its pressure-volume curve, with inadequate stroke volume to accommodate the high regurgitant flow. The effective (net) forward stroke volume (antegrade flow minus retrograde filling) is therefore low, compensated to some degree by tachycardia, but frequently insufficient to maintain normal cardiac output. This phenomenon is exaggerated in patients with preexistent LV hypertrophy in whom the ventricle is already operating on the steep portion of its pressure-volume curve, and particularly severe decompensation is seen when the left ventricle is small and hypertrophic. Critical care settings for the latter unfortunately include many of the common scenarios for acute AI including endocarditis in the setting of AS and aortic dissection in patients with hypertension and a dilated aortic root.5
Acute severe AI results in near approximation of aortic and LV pressures at end diastole, with consequent deleterious effects on coronary perfusion of the subendocardium (Fig. 32.9). Because coronary perfusion pressure is the difference between diastolic pressure in the aortic root and subendocardial pressure in the LV cavity, the decrease in aortic diastolic pressure and rise in subendocardial pressure that are hallmarks of acute AI can result in profound subendocardial ischemia, especially in patients with preexistent hypertrophy or patients with underlying coronary artery disease. In addition, because afterload and wall stress in this setting are increased, there is a rise in myocardial oxygen demand simultaneous with a fall in supply, occasionally leading to cardiogenic shock and death. Acute AI also results in early closure of the mitral valve as LV diastolic pressure rises above left atrial pressure, a phenomenon that has potential protective benefits for the pulmonary circulation.
In contrast, chronic AI features a host of adaptive processes by the left ventricle including progressive dilation, increased compliance, and hypertrophy. As with AS, hypertrophy results in lower wall stress but AI features an increase in afterload combined with progressive LV dilation and somewhat less hypertrophy, resulting in significantly higher wall stress than seen in compensated AS.79 End-diastolic dilation of the left ventricle allows for larger stroke volumes to compensate for regurgitant fractions, which may be in the range of 50% or greater. With combined pressure and volume overload, unique among the valve disorders discussed in this chapter, LV adaptations allow for maintenance of normal overall function until ventricular remodeling and hypertrophy are no longer sufficient to maintain forward stroke volume and EF. With increasing wall stress and LV dilation, LV contractility is eventually impaired, at which point filling pressures begin to rise (or rise further) and patients become symptomatic. Coronary flow reserve in AI, as with AS, is impaired80 and, combined with increased demand and the need for additional perfusion for a hypertrophic myocardium, may result in significant ischemia. The onset of symptoms may be abrupt, especially with new onset of atrial tachyarrhythmias or sudden increase in cardiac output demand such as with exertion or infection.
Although acute AI represents a relative or absolute medical emergency, chronic AI may have an insidious course over decades. Some insight into the rate of progression of chronic AI is provided by a meta-analysis incorporated into the most recent ACC/AHA guidelines.5 In a review of nine admittedly heterogeneous studies incorporating nearly 600 primarily asymptomatic or mildly symptomatic patients with AI, average progression to symptoms with or without LV systolic dysfunction was 4.3% annually, and sudden death occurred in 0.2% annually.5 Although this rate of progression is modest, patients do not necessarily develop symptoms before developing LV dysfunction or sudden death.81 Age, end-systolic dimensions,82 and rate of deterioration of end-systolic dimension and EF83 are more sensitive tools for predicting outcome in chronic AI.
In contrast, in the setting of symptomatic AI that does not fall in the acute severe category, an annual mortality rate of 6% to 25% has been described, depending on severity of symptoms. By the 10-year follow-up, 75% had undergone AVR or died.84
Diagnosis
Physical Examination
The hallmarks of acute severe AI include features consistent with congestive heart failure and low cardiac output including tachycardia, dyspnea, and signs of impaired cardiac output such as peripheral vasoconstriction. The wide pulse pressure that is a hallmark of AI may or may not be seen, in part because it is dependent on LV compliance. The classic AI murmur may not be heard because of the limited gradient between the aorta and left ventricle during diastole when LV diastolic pressures in some cases approach systemic diastolic pressures. Tachycardia and diminished effective forward flow in acute AI may offset the augmented systolic and wide pulse pressure seen in later stages of the disease, and these patients may have normal or occasionally diminished pulses, although more moderate acute AI may feature the more classic findings. Early closure of the mitral valve may result in a soft first heart sound, and distortions of leaflet anatomy may result in lack of a distinct aortic second heart sound. In some cases absence of a second heart sound in a patient presenting with cardiogenic shock may be the most prominent physical finding.78
The dramatic physical findings in chronic AI are among the most familiar to physicians and trainees. A wide pulse pressure results primarily because of increased stroke volume, which augments systolic pressure,85
[/not-level-membership-for-critical-care-medicine-category]
