[level-membership-for-pathology-category]
14 URINARY SYSTEM
The urinary system has three critical functions: (1) to clear the blood of nitrogenous and other waste metabolic products by filtration and excretion; (2) to balance the concentration of body fluids and electrolytes, also by filtration and excretion; and (3) to recover by reabsorption small molecules (amino acids, glucose, and peptides), ions (Na+, C1−, Ca2+, PO3–), and water, in order to maintain blood homeostasis (Greek homoios, similar; stasis, standing).
The kidneys is also an endocrine organ. It produces erythropoietin, a stimulant of red blood cell production in bone marrow (for the role of erythropoietin, see Chapter 6, Blood and Hematopoiesis). It also activates 1,25-hydroxycholecalciferol, a vitamin D derivative involved in the control of calcium metabolism (see vitamin D metabolism in Chapter 19, Endocrine System).
Organization of the renal vascular system
We start our discussion by focusing on the vascularization of the kidneys (Figure 14-1).
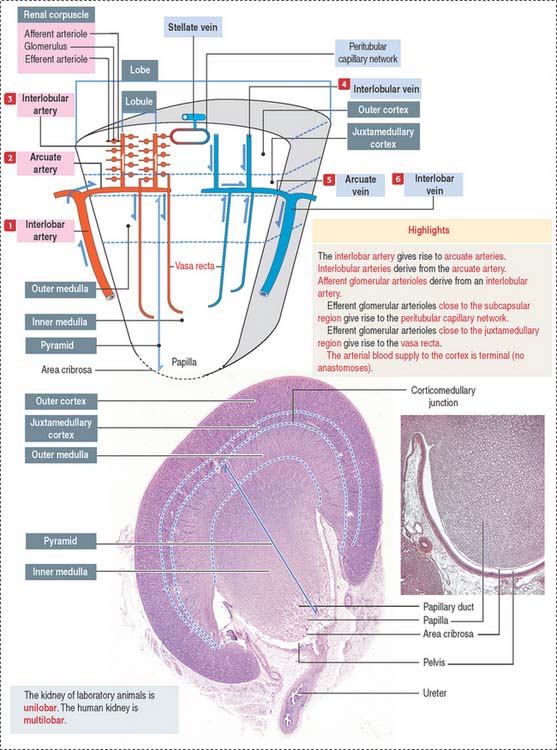
At the corticomedullary junction, interlobar arteries give off several branches at right angles, changing their vertical path to a horizontal direction to form the arcuate arteries, running along the corticomedullary boundary. The renal arterial architecture is terminal. There are no anastomoses between interlobular arteries. This is an important concept in renal pathology for understanding focal necrosis as a consequence of an arterial obstruction. For example, renal infarct can be caused by atherosclerotic plaques in the renal artery or embolization of atherosclerotic plaques in the aorta.
Vertical branches emerging from the arcuate arteries, the interlobular arteries, penetrate the cortex. As interlobular arteries ascend toward the outer cortex, they branch several times to form the afferent glomerular arterioles (see Figure 14-1).
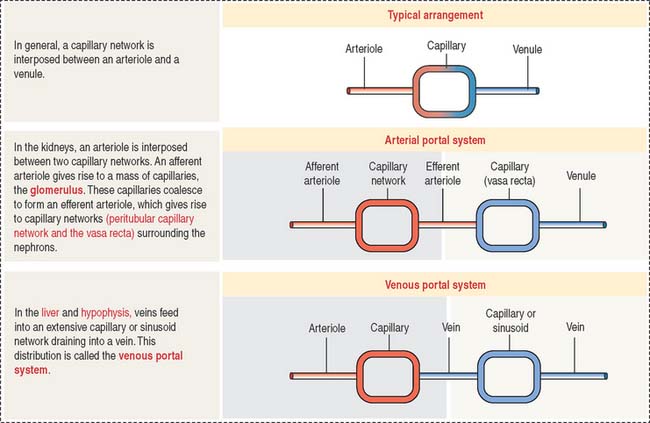
The afferent glomerular arteriole, in turn, forms the glomerular capillary network, enveloped by the two-layered capsule of Bowman, and continues as the efferent glomerular arteriole. This particular arrangement, a capillary network flanked by two arterioles (instead of an arteriole and a venule) is called the glomerulus or arterial portal system. As discussed in Chapter 12, Cardiovascular System, the glomerular arterial portal system (Figure 14-2) is structurally and functionally distinct from the venous portal system of the liver.
Vasa recta
Difference between lobe and lobule
A renal lobule is a cortical structure that can be defined in two different ways (see Figure 14-1): (1) The renal lobule is a portion of the cortex flanked by two adjacent ascending interlobular arteries. Each interlobular artery gives rise to a series of glomeruli, each consisting of an afferent glomerular arteriole, a capillary network, and the efferent glomerular arteriole. (2) The renal lobule consists of a single collecting duct (of Bellini) and the surrounding nephrons that drain into it. The straight portions of the nephrons, together with the single collecting duct, is called a medullary ray (of Ferrein). A medullary ray is the axis of the lobule (Figure 14-3).
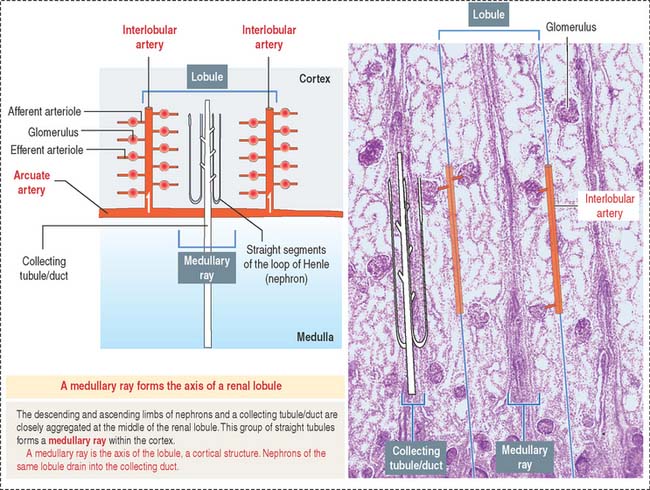
Note that the cortex has many lobules and that each lobule has a single medullary ray.
The uriniferous tubule consists of a nephron and a collecting duct
Each kidneys has about 1.3 million uriniferous tubules surrounded by a stroma containing loose connective tissue, blood vessels, lymphatics, and nerves. Each uriniferous tubule consists of two embryologically distinct segments (Figure 14-4): (1) the nephron and (2) the collecting duct.
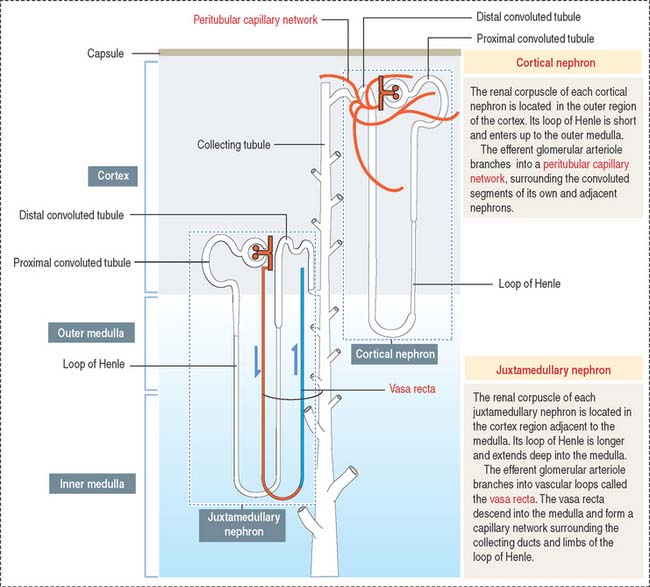
Depending on the distribution of renal corpuscles, nephrons can be either cortical or juxtamedullary. Renal tubules derived from cortical nephrons have a short loop of Henle that penetrates just up to the outer medulla. Renal tubules from juxtamedullary nephrons have a long loop of Henle projecting into the inner medulla (Figure 14-5).
The renal corpuscle
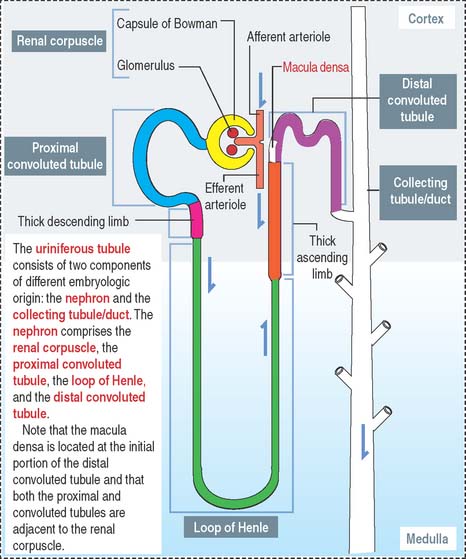
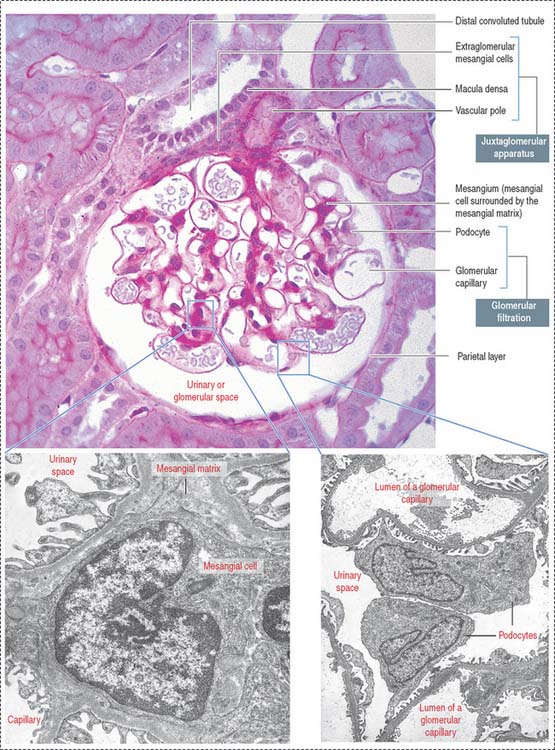
The renal corpuscle, or malpighian corpuscle (Figure 14-6), consists of the capsule of Bowman investing a capillary tuft, the glomerulus.
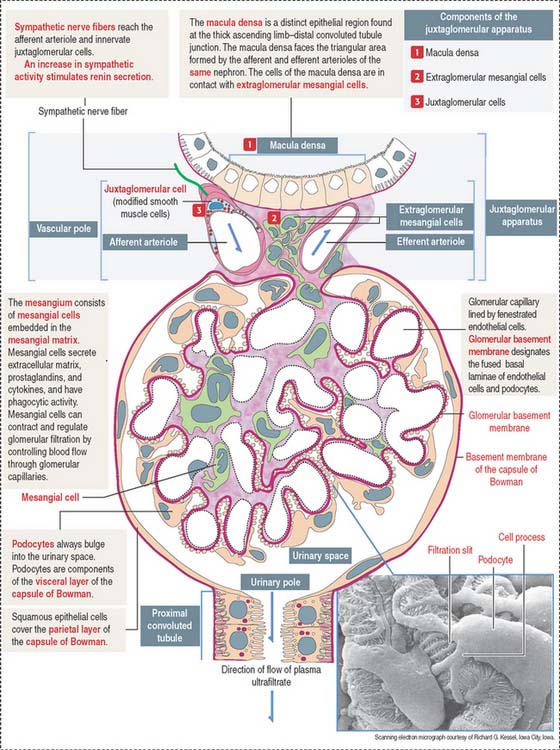
A urinary space (Bowman’s space or capsular space), containing the plasma ultrafiltrate (primary urine), exists between the visceral and parietal layers of the capsule. The plasma ultrafiltrate contains trace amounts of protein. The urinary space is continuous with the lumen of the proximal convoluted tubule at the urinary pole, the gate through which the plasma ultrafiltrate flows into the proximal convoluted tubule. The opposite pole, the site of entry and exit of the afferent and efferent glomerular arterioles, is called the vascular pole.
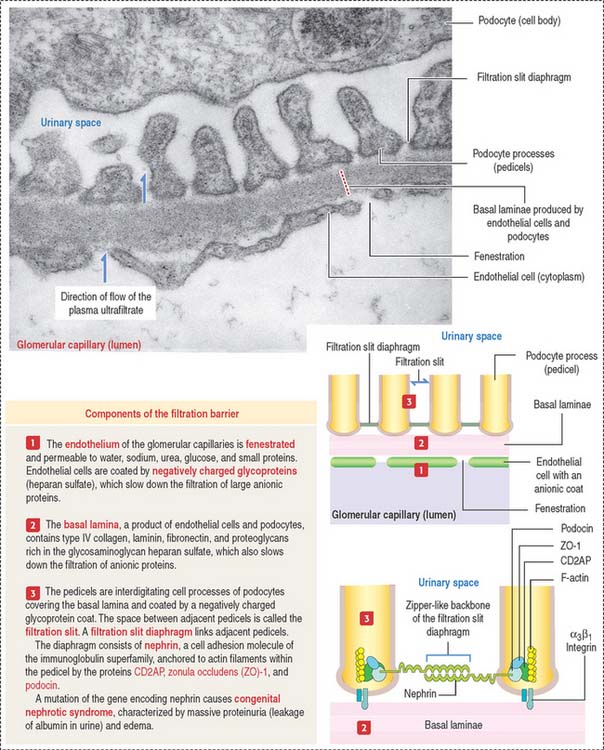
The renal corpuscle: Glomerular filtration barrier
The endings of the cell processes, the pedicels, from the same podocyte or adjacent podocytes, interdigitate to cover the basal lamina and are separated by gaps, the filtration slits. Filtration slits are bridged by a membranous material, the filtration slit diaphragm (Figure 14-8). Pedicels are attached to the basal lamina by α3β1 integrin.
Clinical significance: Glomerular filtration defects
The fenestrated endothelial cells of the glomerular capillaries are covered by a basal lamina to which the foot processes of the podocytes attach (see Figure 14-8). Podocytes produce glomerular endothelial growth factor to stimulate the development of the endothelium and maintenance of its fenestrations.
Type IV collagens are directly involved in the pathogenesis of three diseases. (1) Goodpasture syndrome, an autoimmune disorder consisting in progressive glomerulonephritis and pulmonary hemorrhage, caused by anti-α3(IV) antibodies binding to the glomerular and alveolar basal laminae. (2) Alport’s syndrome, a progressive inherited nephropathy, characterized by irregular thinning, thickening, and splitting of the glomerular basal lamina. Alport’s syndrome is transmitted by an X-linked recessive trait, is predominant in males, and involves mutations of the COL4A5 gene. Patients with Alport’s syndrome—often associated with hearing loss (defective function of the stria vascularis of the cochlea) and ocular symptoms (defect of the lens capsule)—have hematuria (blood in the urine) and progressive glomerulonephritis leading to renal failure. The abnormal glomerular filtration membrane enables the passage of red blood cells and proteins. (3) Benign familial hematuria, caused by a dominant inherited mutation of the COL4A4 gene, which does not lead to renal failure.
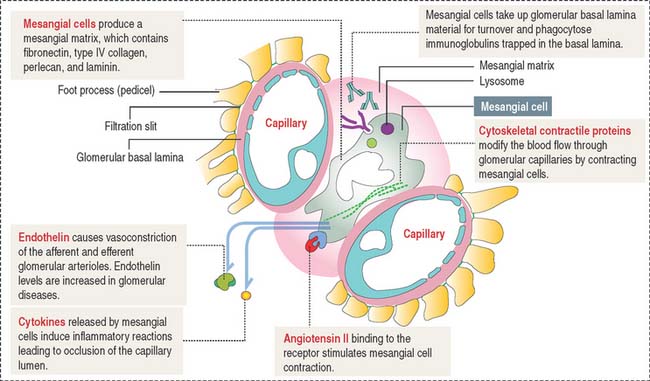
Mesangium
The mesangium, an intraglomerular structure interposed between the glomerular capillaries, consists of two components: (1) the mesangial cells and (2) the mesangial matrix. In addition, mesangial cells aggregate outside the glomerulus (extra-glomerular mesangial cells; see Figures 14-7 and 14-15) in a space limited by the macula densa and the afferent and efferent glomerular arterioles. Intraglomerular mesangial cells may be continuous with extraglomerular mesangial cells.
Mesangial cells participate indirectly in the glomerular filtration process by:
The glomerular filtration membrane does not completely surround the capillaries (Figure 14-9). Immunoglobulins and complement molecules, unable to cross the filtration barrier, can enter the mesangial matrix. The accumulation of immunoglobulin complexes in the matrix induces the production of cytokines by mesangial cells that trigger an immune response leading to the eventual occlusion of the glomerulus.
Clinical significance: Immuno mediated glomerular diseases
The damage to the glomerulus can be initiated by immune mechanisms. Antibodies against glomerular components (cells and basal lamina) and antibody-complement complexes circulating in blood in patients with systemic autoimmune diseases can cause glomerular injury such as membranoproliferative glomerulonephritis (Figure 14-10), membranous glomerulonephritis and immunoglobulin A nephropathy (Berger’s disease).
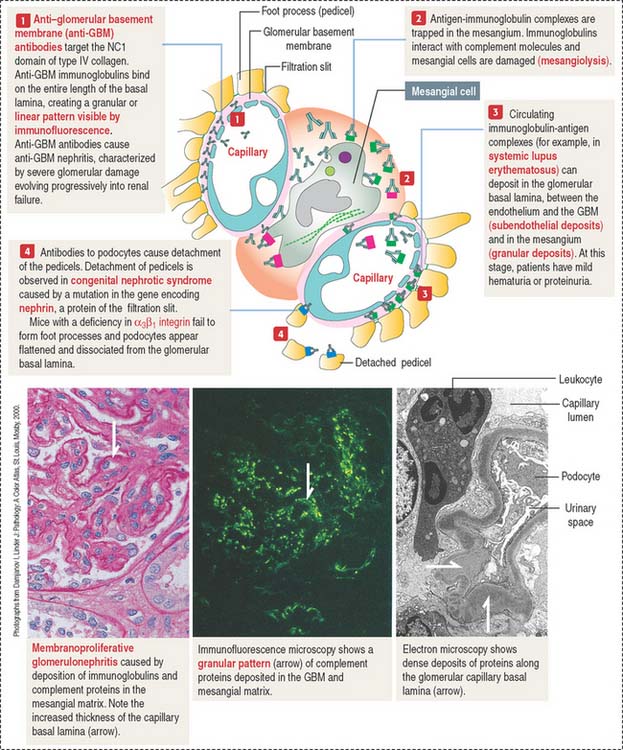
Antibody-antigen complexes are not immunologically targeted to glomerular components. They are trapped in the glomerulus because of the filtration properties of the glomerular filtration barrier. A complicating factor is that trapped antibody-antigen complexes provide binding sites to complement proteins, which also contribute to the glomerular damage (see Chapter 10, Immune-Lymphatic System, for a review of the complement cascade).
Immune complexes can deposit between the endothelial cells of the glomerular capillaries and the basal lamina (subendothelial deposits, see Figure 14-10), in the mesangium, and less frequently between the basal lamina and the foot processes of podocytes (subepithelial deposits).
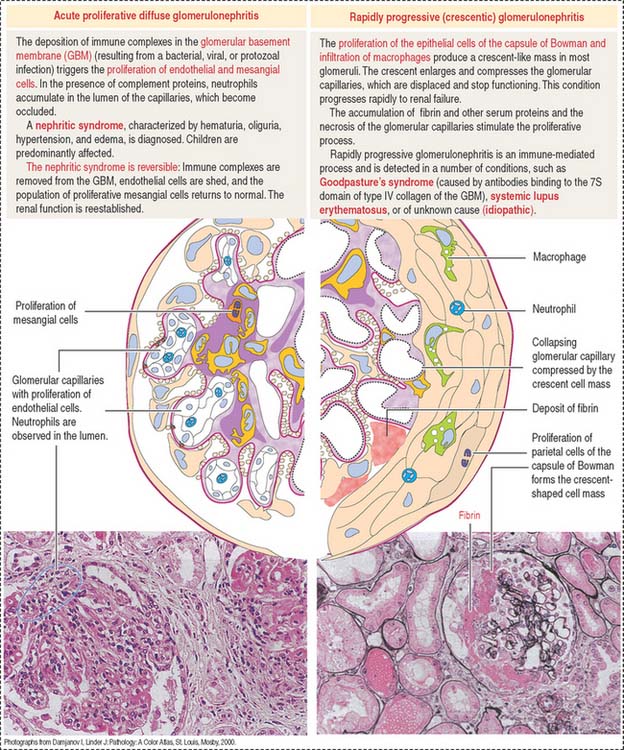
Immune complexes produced after bacterial infection can cause the proliferation of glomerular cells (endothelial and mesangial cells) and attract neutrophils and monocytes. This condition, known as acute proliferative glomerulonephritis, is observed in children and is generally reversible with treatment. This disease is more severe in adults: it can evolve into rapidly progressive (crescentic) glomerulonephritis (Figure 14-11).
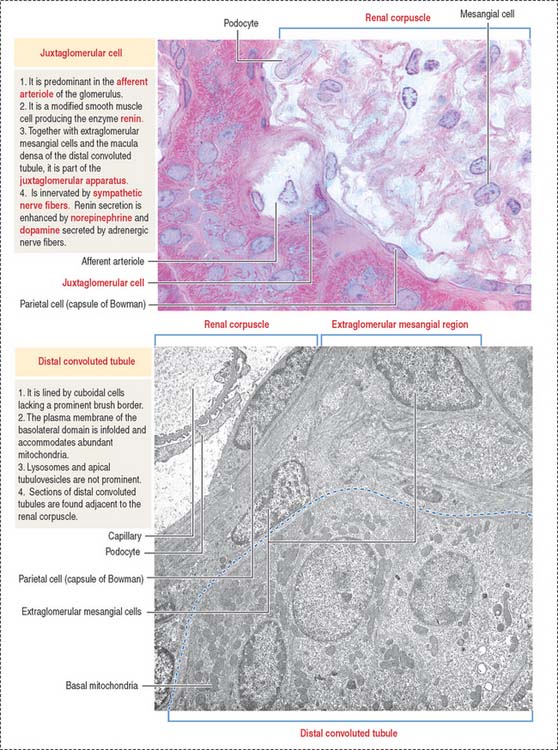
Juxtaglomerular apparatus
The juxtaglomerular apparatus is a small endocrine structure consisting of:
The macula densa is sensitive to changes in NaCl concentration and affects renin release by juxtaglomerular cells. Renin is secreted when the NaCl concentration or blood pressure falls. Extraglomerular mesangial cells (also called lacis cells) are connected to each other and to juxtaglomerular cells by gap junctions.
The juxtaglomerular apparatus is one of the components of the tubuloglomerular feedback mechanism involved in the autoregulation of renal blood flow and glomerular filtration.
The other component is the sympathetic nerve fibers (adrenergic) innervating the juxtaglomerular cells. Renin secretion is enhanced by norepinephrine and dopamine secreted by adrenergic nerve fibers. Norepinephrine binds to α1-adrenergic receptors in the afferent glomerular arteriole to cause vasoconstriction. There is no parasympathetic innervation.
We come back to the tubuloglomerular feedback mechanism when we discuss the renin-angiotensin-aldosterone regulatory mechanism (see Figure 14-18).
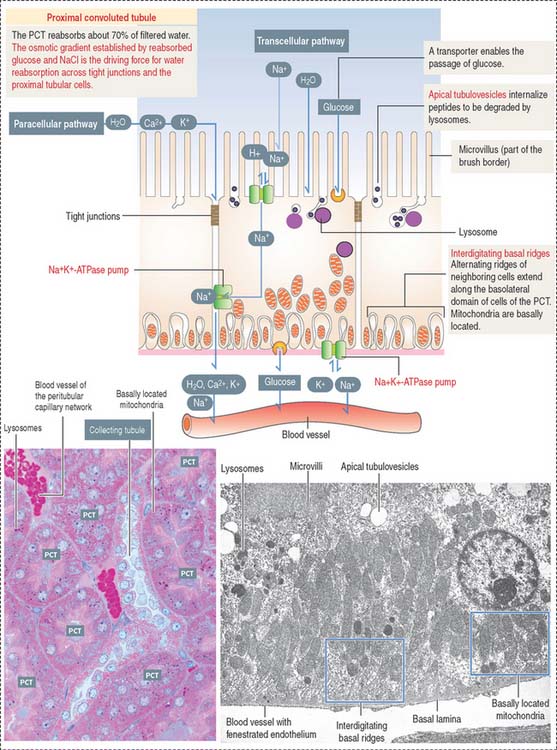
Proximal convoluted tubule: The reabsorption component
Cuboidal epithelial cells, held together by apical tight junctions, line the PCT and have structural characteristics suitable for reabsorption. They display the following features (Figure 14-12):
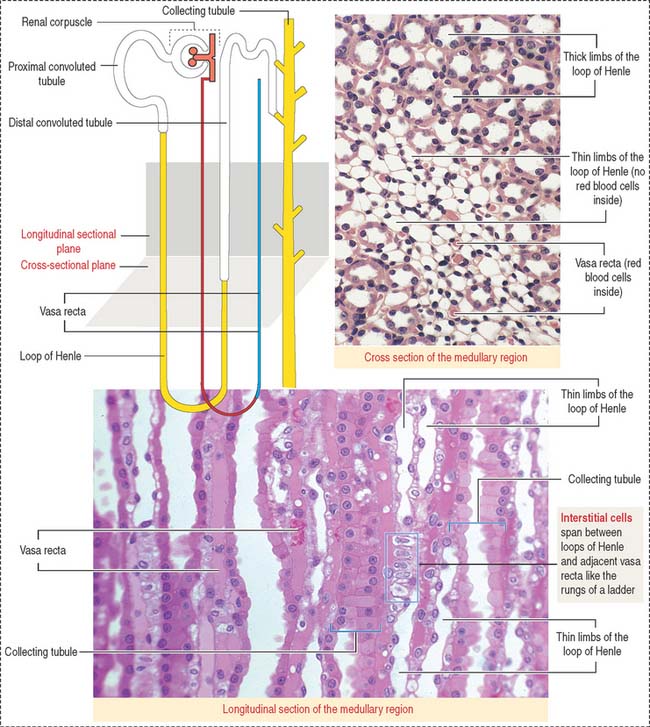
Loop of Henle
The loop of Henle consists of a descending limb and an ascending limb. Each limb is formed by a thick segment and a thin segment (Figure 14-13).
Distal convoluted tubule
NaCl enters the cell across the apical domain and leaves the cell by an Na+, K+-ATPase pump (Figure 14-14). The reabsorption of NaCl is reduced by thiazide diuretics that inhibit the apical domain transporting mechanism (see Figure 14-20).
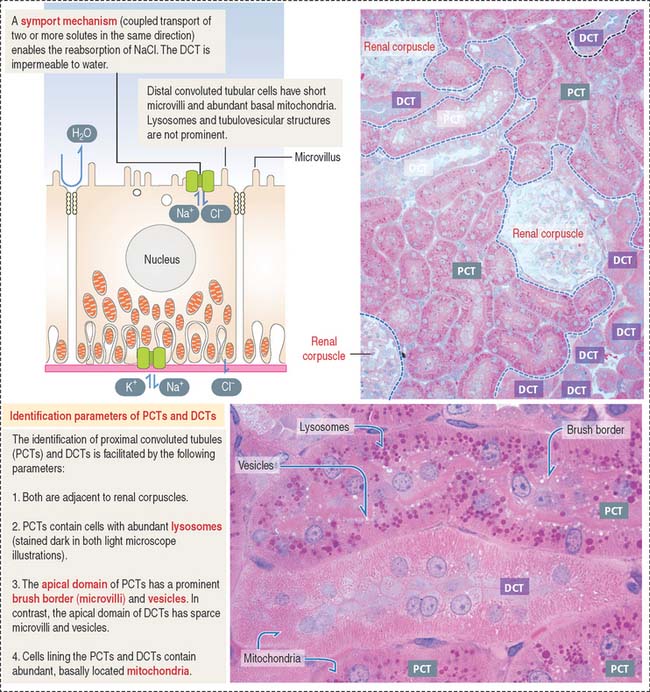
The cuboidal epithelial cell lining of the DCT has the following characteristics (Figure 14-15; see also Figure 14-14):
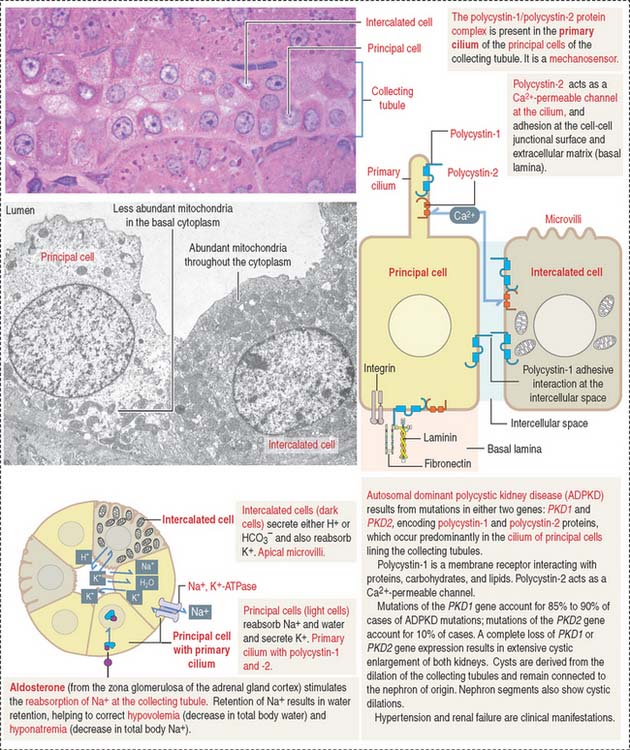
Collecting tubule (duct)
The collecting tubule (also called duct) is lined by a cuboidal epithelium composed of two cell types: principal cells and intercalated cells (Figure 14-16). Principal cells have an apical primary cilium and a basolateral domain with moderate infoldings and mitochondria. They reabsorb Na+ and water and secrete K+ in an Na+, K+-ATPase pump-dependent manner. Intercalated cells have apical microvilli and abundant mitochondria and secrete either H+ or HCO3−. Therefore, they are important regulators of acid-base balance. They also reabsorb K+.
Interstitial cells
We noted in Figure 14-13 the presence of vertical stacks of interstitial cells extending from the loops of Henle to adjacent vasa recta like the rungs of a ladder. There are two populations of interstitial cells: renal cortical and medullary fibroblasts. Their function is the maintenance of renal architecture and production of erythropoietin. Synthetic erythropoietin is used in the treatment of anemia resulting from chronic renal failure or cancer chemotherapy. We discussed in Chapter 6, Blood and Hematopoiesis, the mechanism by which erythropoietin stimulates the production of red blood cells.
EXCRETORY PASSAGES OF URINE
The walls of the ureter and urinary bladder (Figure 14-17) contain folds (rugae). As the bladder fills with urine, the rugae flatten and the volume of the bladder increases with minimal increase in intravesical pressure. The renal calyces, pelvis, ureter, and urinary bladder are lined by a transitional epithelium, the urothelium, composed of basal and superficial cells. The epithelium and the subjacent lamina propria are surrounded by combined helical and longitudinal layers of smooth muscle fibers.
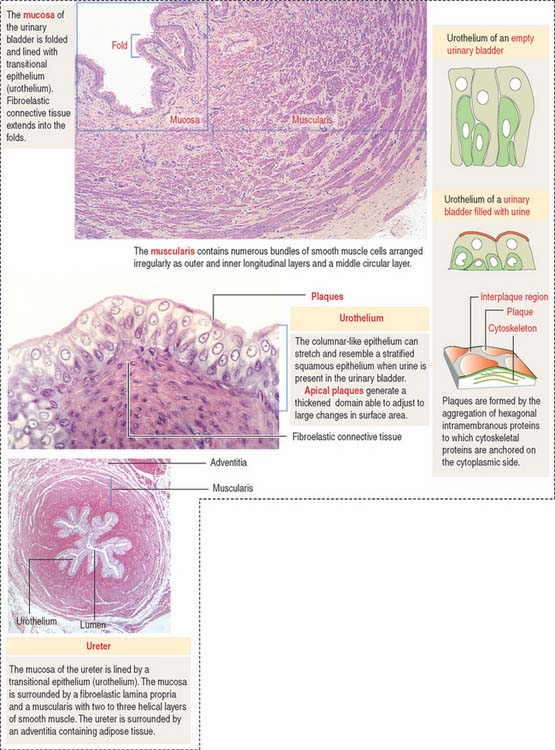
Nephrolithiasis is a condition in which kidneys stones, composed of calcium salts, uric acid, or magnesium-ammonium acetate, form by crystallization when urine is concentrated. When the ureter is blocked by a stone, the contraction of the smooth muscle generates severe pain in the flank.
The male urethra is 20 cm long and consists of three segments. Upon leaving the urinary bladder, the prostatic urethra—lined by transitional epithelium—crosses the prostate gland, continues as a short membranous urethra segment, and ends as the penile urethra, which is enclosed by the corpus spongiosum of the penis (see Figure 21-12 in Chapter 21, Sperm Transport and Maturation). Both the membranous and penile urethra are lined by pseudostratified or stratified columnar epithelium.
The female urethra is 4 cm long and its longitudinally microfolded mucosa is covered by a stratified squamous epithelium that becomes moderately keratinized stratified squamous epithelium near the urethral meatus. The lamina propria contains elastic fibers and a venous plexus. An inner smooth muscle layer and an external striated muscle layer (continuous with the internal sphincter) are present in the wall. Additional structural details of the male and female urethra can be found in Chapter 21, Sperm Transport and Maturation, and Chapter 22, Follicle Development and the Menstrual Cycle, respectively.
REGULATION OF WATER AND NaCl ABSORPTION
Several hormones and factors regulate the absorption of water and NaCl (see Box 14-A for a review of terminology related to osmoregulation):
Box 14-A Review of terminology
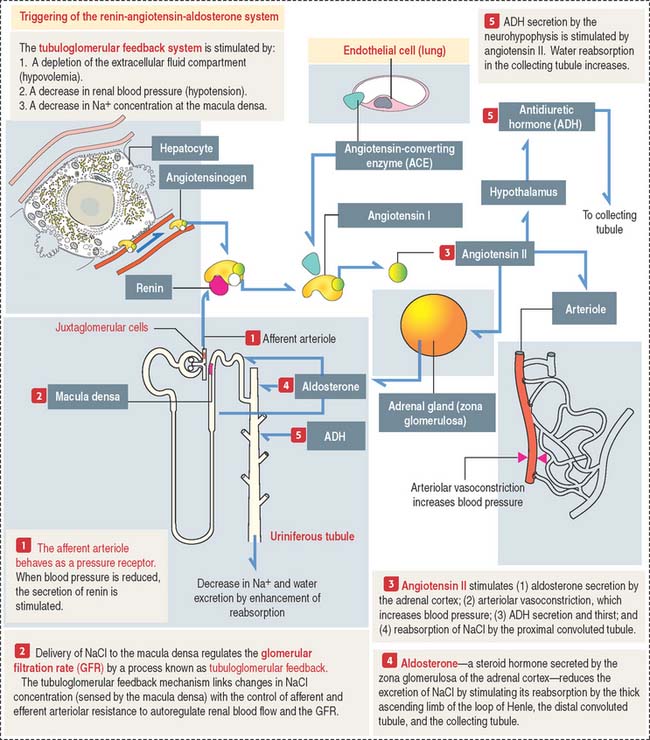
RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM
The tubuloglomerular feedback system consists of:
The renin-angiotensin-aldosterone system consists of the following components (Figure 14-18):
Angiotensin has several important functions:
Aldosterone acts primarily on principal cells of the collecting tubule and secondarily on the thick ascending limb of Henle to increase the entry of NaCl across the apical membrane. As with all steroid hormones, aldosterone enters the cell and binds to a cytosolic receptor. The aldosterone-receptor complex enters the nucleus and stimulates gene activity required for the reabsorption of NaCl.
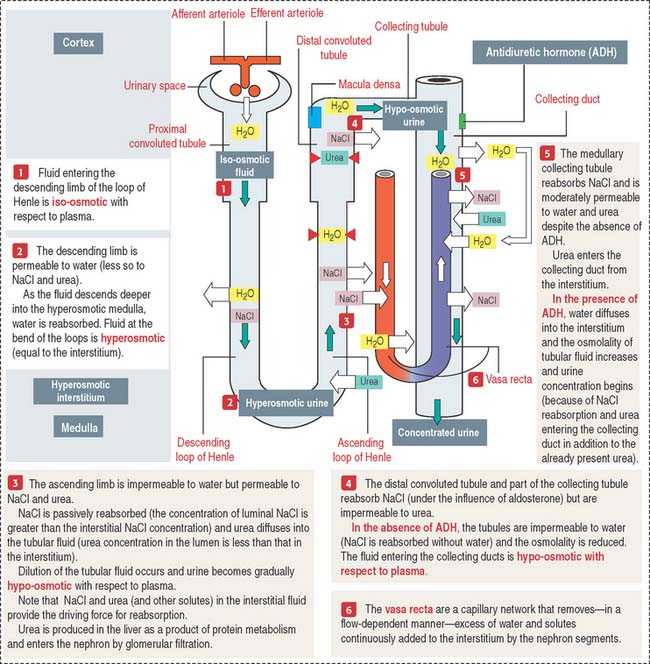
COUNTERCURRENT MULTIPLIER AND EXCHANGER
Figure 14-19 summarizes the essential steps of urine formation and excretion:
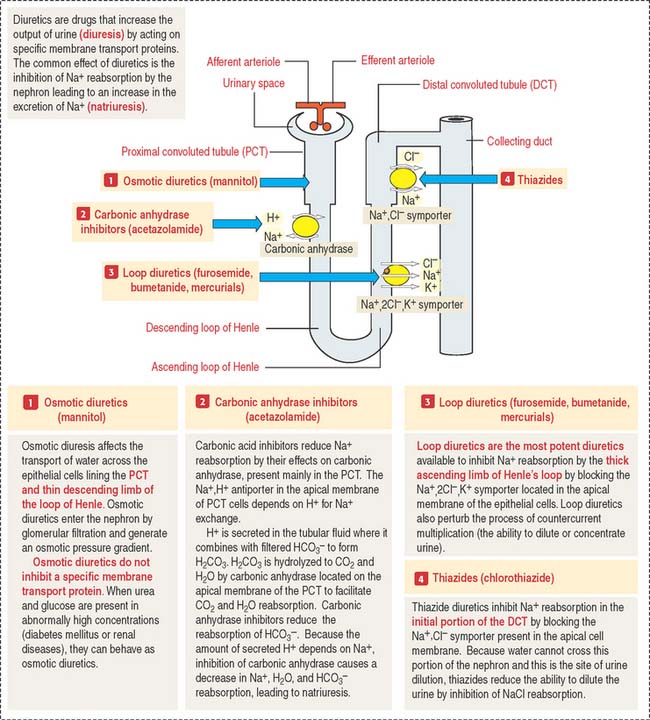
Clinical significance: Mechanism of action of diuretics
Figure 14-20 provides a summary of the mechanism of action of osmotic diuretics, carbonic anhydrase inhibitors, loop diuretics, and thiazide diuretics.
Carbonic anhydrase inhibitors inhibit Na+, HCO3−, and water reabsorption in the PCT.
Thiazide diuretics inhibit the reabsorption of NaCl in the DCT
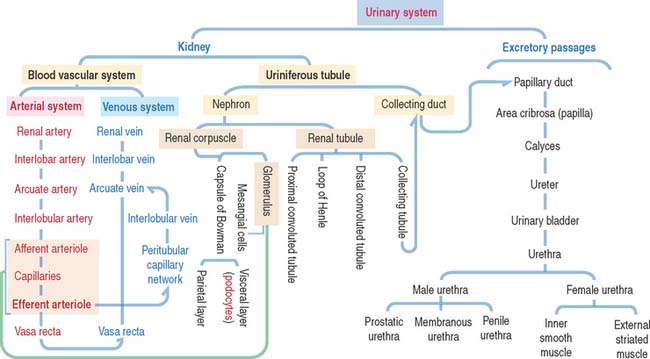
Urinary System
Blood vessels derived from the branching of the glomerular efferent arterioles form two different vascular networks: (1) a peritubular capillary network, surrounding the cortical segments of the uriniferous tubules, and (2) the vasa recta (straight vessels) with a descending arteriolar-capillary component and an ascending capillary-venous component, alongside the descending and ascending limbs of the loops of Henle, respectively. This vascular-tubular arrangement is essential for understanding the countercurrent multiplier and exchange mechanism of urine formation.
The major steps leading to the production of angiotensin II and its activities are:
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
14 URINARY SYSTEM
The urinary system has three critical functions: (1) to clear the blood of nitrogenous and other waste metabolic products by filtration and excretion; (2) to balance the concentration of body fluids and electrolytes, also by filtration and excretion; and (3) to recover by reabsorption small molecules (amino acids, glucose, and peptides), ions (Na+, C1−, Ca2+, PO3–), and water, in order to maintain blood homeostasis (Greek homoios, similar; stasis, standing).
The kidneys is also an endocrine organ. It produces erythropoietin, a stimulant of red blood cell production in bone marrow (for the role of erythropoietin, see Chapter 6, Blood and Hematopoiesis). It also activates 1,25-hydroxycholecalciferol, a vitamin D derivative involved in the control of calcium metabolism (see vitamin D metabolism in Chapter 19, Endocrine System).
Organization of the renal vascular system
We start our discussion by focusing on the vascularization of the kidneys (Figure 14-1).
At the corticomedullary junction, interlobar arteries give off several branches at right angles, changing their vertical path to a horizontal direction to form the arcuate arteries, running along the corticomedullary boundary. The renal arterial architecture is terminal. There are no anastomoses between interlobular arteries. This is an important concept in renal pathology for understanding focal necrosis as a consequence of an arterial obstruction. For example, renal infarct can be caused by atherosclerotic plaques in the renal artery or embolization of atherosclerotic plaques in the aorta.
Vertical branches emerging from the arcuate arteries, the interlobular arteries, penetrate the cortex. As interlobular arteries ascend toward the outer cortex, they branch several times to form the afferent glomerular arterioles (see Figure 14-1).
The afferent glomerular arteriole, in turn, forms the glomerular capillary network, enveloped by the two-layered capsule of Bowman, and continues as the efferent glomerular arteriole. This particular arrangement, a capillary network flanked by two arterioles (instead of an arteriole and a venule) is called the glomerulus or arterial portal system. As discussed in Chapter 12, Cardiovascular System, the glomerular arterial portal system (Figure 14-2) is structurally and functionally distinct from the venous portal system of the liver.
Vasa recta
Difference between lobe and lobule
A renal lobule is a cortical structure that can be defined in two different ways (see Figure 14-1): (1) The renal lobule is a portion of the cortex flanked by two adjacent ascending interlobular arteries. Each interlobular artery gives rise to a series of glomeruli, each consisting of an afferent glomerular arteriole, a capillary network, and the efferent glomerular arteriole. (2) The renal lobule consists of a single collecting duct (of Bellini) and the surrounding nephrons that drain into it. The straight portions of the nephrons, together with the single collecting duct, is called a medullary ray (of Ferrein). A medullary ray is the axis of the lobule (Figure 14-3).
Note that the cortex has many lobules and that each lobule has a single medullary ray.
The uriniferous tubule consists of a nephron and a collecting duct
Each kidneys has about 1.3 million uriniferous tubules surrounded by a stroma containing loose connective tissue, blood vessels, lymphatics, and nerves. Each uriniferous tubule consists of two embryologically distinct segments (Figure 14-4): (1) the nephron and (2) the collecting duct.
Depending on the distribution of renal corpuscles, nephrons can be either cortical or juxtamedullary. Renal tubules derived from cortical nephrons have a short loop of Henle that penetrates just up to the outer medulla. Renal tubules from juxtamedullary nephrons have a long loop of Henle projecting into the inner medulla (Figure 14-5).
The renal corpuscle
The renal corpuscle, or malpighian corpuscle (Figure 14-6), consists of the capsule of Bowman investing a capillary tuft, the glomerulus.
A urinary space (Bowman’s space or capsular space), containing the plasma ultrafiltrate (primary urine), exists between the visceral and parietal layers of the capsule. The plasma ultrafiltrate contains trace amounts of protein. The urinary space is continuous with the lumen of the proximal convoluted tubule at the urinary pole, the gate through which the plasma ultrafiltrate flows into the proximal convoluted tubule. The opposite pole, the site of entry and exit of the afferent and efferent glomerular arterioles, is called the vascular pole.
The renal corpuscle: Glomerular filtration barrier
The endings of the cell processes, the pedicels, from the same podocyte or adjacent podocytes, interdigitate to cover the basal lamina and are separated by gaps, the filtration slits. Filtration slits are bridged by a membranous material, the filtration slit diaphragm (Figure 14-8). Pedicels are attached to the basal lamina by α3β1 integrin.
Clinical significance: Glomerular filtration defects
The fenestrated endothelial cells of the glomerular capillaries are covered by a basal lamina to which the foot processes of the podocytes attach (see Figure 14-8). Podocytes produce glomerular endothelial growth factor to stimulate the development of the endothelium and maintenance of its fenestrations.
Type IV collagens are directly involved in the pathogenesis of three diseases. (1) Goodpasture syndrome, an autoimmune disorder consisting in progressive glomerulonephritis and pulmonary hemorrhage, caused by anti-α3(IV) antibodies binding to the glomerular and alveolar basal laminae. (2) Alport’s syndrome, a progressive inherited nephropathy, characterized by irregular thinning, thickening, and splitting of the glomerular basal lamina. Alport’s syndrome is transmitted by an X-linked recessive trait, is predominant in males, and involves mutations of the COL4A5 gene. Patients with Alport’s syndrome—often associated with hearing loss (defective function of the stria vascularis of the cochlea) and ocular symptoms (defect of the lens capsule)—have hematuria (blood in the urine) and progressive glomerulonephritis leading to renal failure. The abnormal glomerular filtration membrane enables the passage of red blood cells and proteins. (3) Benign familial hematuria, caused by a dominant inherited mutation of the COL4A4 gene, which does not lead to renal failure.
Mesangium
The mesangium, an intraglomerular structure interposed between the glomerular capillaries, consists of two components: (1) the mesangial cells and (2) the mesangial matrix. In addition, mesangial cells aggregate outside the glomerulus (extra-glomerular mesangial cells; see Figures 14-7 and 14-15) in a space limited by the macula densa and the afferent and efferent glomerular arterioles. Intraglomerular mesangial cells may be continuous with extraglomerular mesangial cells.
Mesangial cells participate indirectly in the glomerular filtration process by:
The glomerular filtration membrane does not completely surround the capillaries (Figure 14-9). Immunoglobulins and complement molecules, unable to cross the filtration barrier, can enter the mesangial matrix. The accumulation of immunoglobulin complexes in the matrix induces the production of cytokines by mesangial cells that trigger an immune response leading to the eventual occlusion of the glomerulus.
Clinical significance: Immuno mediated glomerular diseases
The damage to the glomerulus can be initiated by immune mechanisms. Antibodies against glomerular components (cells and basal lamina) and antibody-complement complexes circulating in blood in patients with systemic autoimmune diseases can cause glomerular injury such as membranoproliferative glomerulonephritis (Figure 14-10), membranous glomerulonephritis and immunoglobulin A nephropathy (Berger’s disease).
Antibody-antigen complexes are not immunologically targeted to glomerular components. They are trapped in the glomerulus because of the filtration properties of the glomerular filtration barrier. A complicating factor is that trapped antibody-antigen complexes provide binding sites to complement proteins, which also contribute to the glomerular damage (see Chapter 10, Immune-Lymphatic System, for a review of the complement cascade).
Immune complexes can deposit between the endothelial cells of the glomerular capillaries and the basal lamina (subendothelial deposits, see Figure 14-10), in the mesangium, and less frequently between the basal lamina and the foot processes of podocytes (subepithelial deposits).
Immune complexes produced after bacterial infection can cause the proliferation of glomerular cells (endothelial and mesangial cells) and attract neutrophils and monocytes. This condition, known as acute proliferative glomerulonephritis, is observed in children and is generally reversible with treatment. This disease is more severe in adults: it can evolve into rapidly progressive (crescentic) glomerulonephritis (Figure 14-11).
