Chapter 44 Uncommon Thoracic Tumors
Thymic Tumors
The thymus gland is a bilobed lymphoepithelial organ located in the anterior mediastinum, behind the sternum and in front of the great vessels. In early life, the thymus functions in T-lymphocyte differentiation and maturation and releases T lymphocytes into the circulation. It weighs 12 to 15 g at birth and 40 g at puberty, and during adulthood, it slowly involutes and is largely replaced by adipose tissue. The thymus is composed of an outer cortex consisting primarily of epithelial cells, degenerated keratinized epithelial cells (Hassall’s corpuscles), myoid cells, thymic lymphocytes (“thymocytes”), and B lymphocytes, which form rare germinal centers.12
Although various tumors and cysts can arise in the thymus, tumors of the thymus are uncommon. Most thymic tumors arise in the epithelial cells of the thymus. They account for approximately 50% of anterior mediastinal masses.13,14 The most common thymic tumors are thymoma, thymic carcinoma, and thymic carcinoids. Thymomas, 90% of which are found in the anterosuperior mediastinum, are commonly associated with myriad paraneoplastic syndromes. They are the most common tumor found in the anterior mediastinum.13,15 Thymomas are associated with an exuberant lymphoid component composed of immature cortical thymocytes. Although they appear benign histologically, they may exhibit invasive behavior clinically.
Thymic carcinomas (i.e., type C thymomas) also arise in the thymic epithelium, but they have a higher propensity for capsular invasion and widespread metastases, especially those with features of high-grade malignancy. The histologic subtypes include clear cell carcinoma, sarcomatoid carcinoma, and anaplastic carcinoma. Low-grade thymic carcinomas (well-differentiated squamous cell carcinomas, basaloid carcinomas, and mucoepidermoid carcinomas) also exist and are characterized by a relatively more favorable clinical course, with a lower incidence of local recurrence and metastasis.16,17
Neuroendocrine tumors of the thymus are rare, accounting for less than 5% of all neoplasms of the anterior mediastinum. Like carcinoids at other sites, they are thought to originate from endodermal cell (or foregut cellular) precursors. Unlike carcinoids in other locations, most thymic carcinoids behave aggressively. They often invade locally and commonly metastasize to regional lymph nodes.18 Approximately 50% of patients may develop endocrine abnormalities.19
Etiology and Epidemiology
The etiology of thymic tumors is largely unknown. There is a reported association between Epstein-Barr virus (EBV) infection and tumors of the thymus. For instance, thymic diseases are more common in Far Eastern countries, where the incidence of EBV infection is high.20,21 Defective viral genomes have been isolated in patients with lymphoepithelioma-like thymic carcinoma.21–24 Childhood thymus irradiation has been linked to the development of thymic tumors,25 and familial cases have been reported, suggesting a possible relationship with cytogenetic abnormalities.26,27 In an analysis of secondary or concomitant neoplasms in 1495 patients with acute lymphoblastic leukemia enrolled in two consecutive multicenter protocols, an increased risk of solid tumors was identified.28 Although the histologic characteristics of these second malignant tumors varied, thymoma was among the solid tumors identified in an adult survivor of acute lymphoblastic leukemia. Among patients with primary thymic carcinoid tumors, up to 30% were reported in the setting of multiple endocrine neoplasia type 1 and 2.29,30 Other studies have reported a distinctive chromosome abnormality involving translocation of fragments of chromosomes 15 and 19 [t(15:19)(q15:p13)] in thymic carcinoma, particularly in young adults and pediatric patients with high-grade thymic carcinoma.31–33 Deletion of the short arm of chromosome 6 is associated with benign thymomas.34 However, the molecular mechanisms of thymic tumor oncogenesis are largely unknown. Aberrations involving chromosome 6 suggest that several putative tumor suppressor genes may contribute to the pathogenesis of thymoma.35
In a small study of 37 cases, most type A thymomas did not show any chromosomal aberration, whereas type B3 thymomas and thymic carcinoma (i.e., type C thymoma) shared some genetic aberrations, including the loss of chromosome 6 and the gain of chromosome 1q.36 The loss of chromosome 6, on which the human leukocyte antigen locus and some of the tumor suppressor genes have been identified, and the gain of chromosome 1q, to which growth promoter genes have been mapped,36–38 may play a role in tumorigenesis and pathogenesis of the paraneoplastic autoimmunity characteristics of thymoma. Such chromosomal aberrations have been implicated in a variety of human neoplasms.39,40
The true incidence of thymoma is unknown, and the annual incidence in the United States has not been well defined. The SEER program reported a thymoma incidence of 0.13 to 0.15 per 100,000 people.1 Although thymomas are uncommon, they are the most frequently diagnosed mediastinal masses, representing approximately 30% of anterior mediastinal lesions and 20% of all mediastinal tumors in adults.13–15,42–45 Most patients with thymoma are between the ages of 40 and 60 years, with a median age of 52 years and an equal overall gender ratio.46–49 Thymomas are less common in children, accounting for approximately 15% of anterior mediastinal masses.15,46–49
Thymic carcinomas are distinct from invasive thymomas both pathologically and clinically and carry a poorer prognosis.50 They account for 5% to 36% of all thymic neoplasms.51,52–54 The wide range in incidences from various studies reflects differences and changes in the pathologic classification of this rare tumor. Patients with thymic carcinoma are typically middle-aged or elderly, and there is a slight male predominance. Thymic carcinoids represent less than 5% of anterior mediastinum lesions55,56 and typically affect middle-aged men.16
Prevention and Early Detection
A few small subgroups of cases are associated with thymic irradiation during childhood, EBV infection (associated with lymphoepithelioid thymic carcinoma), or familial cytogenetic abnormalities; the etiologic factors for most thymic tumors are unknown, however, and there is no known mechanism for prevention. Thymomas are characterized by an indolent growth pattern with a tendency toward local invasion. Thymomas are associated with autoimmune disorders, particularly myasthenia gravis, in 30% to 45% of cases.57,58 Most thymic tumors are discovered incidentally because of abnormalities observed on routine radiographs or during evaluation for myasthenia gravis. Approximately 10% to 15% of patients with myasthenia gravis have benign or malignant thymomas.59,60,61–63 Thymic carcinomas and carcinoids are commonly detected incidentally. They are locally invasive and are found to have metastasized to regional lymph nodes and distant sites in up to 30% of patients at diagnosis.18,52,64
Biologic Characteristics and Molecular Biology
Thymomas are usually slow-growing tumors with an indolent natural history. Although histologically benign, they spread by direct extension and are capable of local and regional invasion and have a marked tendency for recurrence in the mediastinum and pleura. Intrathoracic recurrence can develop after complete surgical resection in up to 40% of those with invasive disease.65 Late relapse is not uncommon.60,66,67 Metastases are typically confined to the pleura, pericardium, or diaphragm.68 Nodal and hematogenous metastases may occur but are rare.45,69 An increased risk for second malignant tumors has been reported.70,71,72
Patients with thymoma may have dysregulation of the lymphocyte selection process associated with abnormal proliferation, autoimmunity, and immunodeficiency. Thymoma-associated autoimmune disease involves an alteration in circulating T-cell subsets.73–75 Up to 70% of thymomas may be associated with paraneoplastic syndromes.51 Various autoantibodies have been described.76,77 In addition to T-cell defects, B-cell lymphopenia has been observed in thymoma-related immunodeficiency along with hypogammaglobulinemia (i.e., Good’s syndrome).78,79
Factors influencing the different biologic behaviors of thymoma subtypes are poorly understood. Some pathologic features, such as altered expression of TP53 and BCL2,80,81 counts of argyrophilic nucleolar organizer regions,82,83 proliferating cell nuclear antigen, matrix metalloproteinases, and Ki-67 index,82,84 are associated with biologic behavior, clinical stage, and prognosis.85 There is a tendency for up-regulation of adhesion and co-stimulatory molecules in thymoma.86–89 Altered TP53 expression may be implicated in the initial stages of tumorigenesis, and increased expression of epidermal growth factor (EGF) and epidermal growth factor receptor (EGFR) may play a role in thymoma genesis.90 A decreased OS time is predicted by Src kinase and TP53 co-expression.91 Recently, it has been proposed that a deficiency in the autoimmune regulator (AIRE) gene has an oncogenic effect on thymomas. AIRE expression is lacking in approximately 95% of thymomas; therefore loss of AIRE expression may provide a potential mechanistic link for the well-recognized association of autoimmunity with thymoma.92,93
Thymic carcinomas possess overt features of malignancy similar to those of carcinoma arising in any other organ, with a higher propensity for capsular invasion and metastases than invasive thymomas. Associated paraneoplastic syndromes occasionally exist with well-differentiated lesions.94,95 Most variants of thymic carcinoma are highly lethal, producing frequent metastases to regional lymph nodes, bone, liver, and lung.96–98 Thymic carcinomas may express high levels of EGFR,90,99,100 but EGFR gene mutations are rare.101 Tumor and serum levels of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) can be elevated,102,103 and correlations among such growth factors, angiogenesis, and invasiveness have been observed.102,104 Thymic carcinoma has been associated with increased expression of epithelial membrane antigen, cytokeratin subtypes, and TP53 protein.80,105–109 Somatic KIT gene mutations are present in some thymic carcinomas but not in thymomas.110 Increased expression of CD70 may serve as a marker for thymic carcinoma.111 Other surface antigenic molecules, such as CD5 and CD99, have aberrant expression in thymic carcinoma.112–114 Cytoplasmic phosphorylated AKT serine 473 and insulin-like growth factor-1 receptor (IGF-1R) are expressed in a higher proportion of recurrent tumors and more aggressive subtypes according to the World Health Organization (WHO) staging system. IGF-1R was associated with a worse progression-free survival time.115 Thymic carcinomas can be distinguished from lung carcinomas by negative expression of thyroid transcription factor-1105 and a distinctly different cytokeratin profile.116 Additionally, although mutations in the TP53 gene are common in lung carcinoma, TP53 gene mutation was not found in any of seven examined thymic carcinomas.110
Thymic carcinoids have a similar malignant clinical course with a poor prognosis, frequently worse than carcinoids of the gastrointestinal tract. They may secrete peptides, amines, kinins, prostaglandins, or other substances. In a study of 80 patients with thymic carcinoids, 74% were male, 72% had related symptoms, 6% had Cushing’s syndrome, and 16% had other endocrine abnormalities.117 Even after complete resection, recurrence is common (range, 36% to 87% of patients).18,117,118 OS rates were only 28% and 10% at 5 and 10 years, respectively.
Pathology and Pathways of Spread
There is no direct correlation between the histopathology of thymomas and their malignant potential.49,119,120 However, an accurate histologic diagnosis is still crucial in patient management.
Grossly, thymomas are nodular, multilobulated, and firm. They may contain cystic spaces, calcification, or hemorrhage, and they may be neatly encapsulated, adherent to surrounding structures, or invasive. Thymomas generally display two cell types: epithelial and lymphatic cells. They are classified as predominantly epithelial, predominantly lymphocytic, mixed lymphoepithelial, or spindle cell type. Morphologically, these cells are rather large and may be round, oval, or spindle shaped with vesicular nuclei and small nucleoli. The cytoplasm is often eosinophilic or amphophilic. Thymic neoplasms arise from epithelial cells (Fig. 44-1). The lymphocytic component is mostly made up of normal-appearing mature lymphocytes. Some of the other microscopic features that may be seen in thymomas include Hassall’s corpuscles, keratinizing squamous epithelium, rosettes, glands, cysts, papillary structures, and germinal centers. Immunohistochemistry testing is often helpful in making the diagnosis. Thymomas typically stain positive for a number of thymic epithelial markers, including cytokeratin, thymosin ß3 and α1, and epithelial membrane antigen.
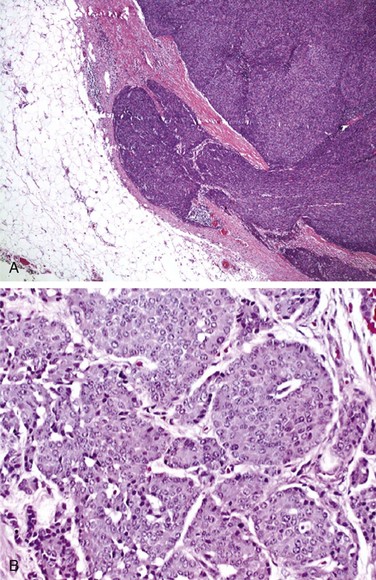
Thymic carcinoma histologic characteristics are more diverse. The tumor is cytologically malignant, and the largest proportion of cases have squamous cell histologic findings. Other subtypes include lymphoepithelioma-like carcinomas, clear cell carcinomas, sarcomatoid carcinomas, adenosquamous carcinomas, mucoepidermoid carcinomas, adenocarcinomas, and basaloid squamous cell carcinomas.16 Their histologic appearances are not different from carcinomas in other sites.121
The histologic features of thymic carcinoids are identical to carcinoid tumors in other organs. Unlike thymomas, they are rarely encapsulated. Immunohistochemically, they may stain positively with CAM 5.2, low-molecular-weight cytokeratins, chromogranins, synaptophysin, and leucine-7.117
The predominant pattern of spread of thymomas is by direct invasion into adjacent organs. The degree of encapsulation and the invasion of adjacent tissues define malignancy for these tumors rather than the histologic appearance.66 Approximately 50% of cases in surgical series are noninvasive.* Thymomas may metastasize as implants on pleural surfaces or pulmonary nodules, but they rarely metastasize to extrathoracic areas.45 When thymomas metastasize, the most common site is the pleural cavity, in which they form pleural plaques, malignant pleural effusions, and diaphragmatic masses. Invasion into the superior vena cava, brachiocephalic vein, lung, and pericardium may be observed.127
Thymic carcinomas invade locally, often involve the pleura and mediastinal nodes, and sometimes spread regionally to the cervical and axillary lymph nodes.128 Distant metastases to the lungs, liver, brain, and bone also occur. Distant metastases to bone, liver, or skin occur in 30% to 40% of cases of thymic carcinoids,19 and they may be seen in 70% of patients within 8 years from initial diagnosis.129
Clinical Manifestations and Patient Evaluation
Approximately 30% to 40% of patients with thymomas, thymic carcinomas, or carcinoids are asymptomatic and are discovered incidentally.45,48,58,130 Clinical symptoms vary greatly, depending on the size of the tumor and effect on adjacent structures, but they are usually those of a mediastinal mass producing cough, chest pain, dyspnea, hoarseness, superior vena cava syndrome, and symptoms related to tumor hemorrhage.16,131,132 Patients may also have dysphagia, fever, weight loss, and anorexia.
Some thymomas present with symptomatic paraneoplastic syndromes; the most common is myasthenia gravis, which is seen in approximately 45% of patients.58 Box 44-1 lists other syndromes.* Thymic carcinoids may also be associated with Cushing’s syndrome, Eaton-Lambert syndrome, syndrome of inappropriate secretion of antidiuretic hormone (SIADH), and hypercalcemia,19 but the classic carcinoid syndrome is rare. The causes of these syndromes remain obscure, although autoantibodies have been demonstrated, mostly in thymoma patients.76,77,137
Myasthenia gravis is an autoimmune neuromuscular junction disorder characterized by the presence of antiacetylcholine receptor antibodies, which cause an acetylcholine receptor deficiency at the motor end plate. The disease is characterized by rapid exhaustion of voluntary muscular contractions, with a slow return to the normal state.138,139 Myasthenia gravis is common with thymoma but rare in thymic carcinoma. Patients with thymoma and myasthenia gravis may have an increased operative mortality rate, and most surgical deaths are attributed to myasthenic crisis. Death of patients with thymoma and myasthenia gravis is commonly caused by complications of myasthenia gravis, whereas in patients without myasthenia gravis, death often is attributed to local progression of tumor.124 The overall long-term prognosis does not appear to be adversely affected by the presence of myasthenia gravis.60,124
Prognostic Factors
The two most important prognostic factors for thymoma are invasiveness (stage) and completeness of surgical resection.† Capsular invasion is commonly used as the basis for designation as benign or malignant. Tumor size (>10 cm) and the presence of symptoms also have prognostic value.43,143,144 Patients with complete or radical excision have significantly improved survival times over those with subtotal resection or biopsy only.141,145 Although almost all noninvasive thymomas can be totally resected, the ability to achieve a complete resection in invasive cases varies from 58% to 73%.60,125,141 Older series reported a poor prognosis associated with myasthenia gravis,46,47,126 but several modern series have failed to confirm this observation.* Myasthenia gravis may even confer a survival advantage because neuromuscular symptoms may lead to earlier discovery of a small thymoma.143,152,153 After thymectomy, patients with myasthenia gravis have attenuation of the severity of symptoms over time but not necessarily complete resolution of them.154–156 About 50% of patients with pure red cell aplasia have thymoma, and about 10% of patients with hypogammaglobulinemia have thymoma. Patients with red cell aplasia, hypogammaglobulinemia, and lupus erythematosus appear to have a poorer prognosis.124,143,152,157
Although the degree of tumor invasiveness is strongly related to stage and prognosis of thymoma, there are data to support the prognostic significance of the histologic findings, independent of tumor stage.146,158,159 The historical classification proposed by Marino and Muller-Hermelink categorized thymoma as cortical, mixed, and medullary types.160 Thymomas arising from the epithelial cells of the cortex were classified as cortical thymomas, and those arising from the medullary spindle cells were classified as medullary thymomas. According to the Marino and Muller-Hermelink data, cortical thymomas have a more aggressive course than medullary thymomas and are more likely to be associated with myasthenia gravis. Of note, thymic carcinomas were categorized as a separate entity.
In 1999, the World Health Organization (WHO) published a classification including six subtypes of thymic tumor, based on the relative proportion of epithelial and lymphocytic cells (Box 44-2). This is the most widely accepted histologic classification system, and data have shown that the WHO cell type is an independent prognostic factor.159,161,162,163 A retrospective study of 324 patients found that patients with WHO types A, AB, and B1 had a 100% disease-specific survival rate without RT and therefore did not benefit from adjuvant RT.164 There was no survival difference for cell types B2 and B3 with or without adjuvant RT. Another series, from Reiker and colleagues,165 noted that survival rates of patients with types A, AB, B1, and B2 were better than rates for type B3, and that type C had the lowest OS rates. The prognostic value of the subtype of type B thymomas has been examined by one series that found no significant differences in recurrence or survival rates among the three subtypes of type B, but all patients who recurred had stage III disease, indicating that the Masaoka stage was predictive.166 The risk of myasthenia gravis increases progressively with WHO type from A (~20%) to B3 (~60%).93
Box 44-2 World Health Organization Classification of Thymic Epithelial Tumors
| Type | Pathologic Classification | Prognosis |
|---|---|---|
| A | Medullary thymoma | Benign clinical course |
| Spindle cell thymoma | ||
| AB | Mixed thymoma | |
| B1 | Lymphocyte-rich thymoma | Moderately malignant clinical course |
| Lymphocytic thymoma | ||
| Predominately cortical thymoma | ||
| Organoid thymoma | ||
| B2 | Cortical thymoma | |
| B3 | Epithelial thymoma | |
| Atypical thymoma | ||
| Squamoid thymoma | ||
| Well-differentiated thymic carcinoma | ||
| C | Thymic carcinoma | Highly malignant clinical course |
Tumors that are mixed tend to have an intermediate prognosis.167 The 10-year survival rates are approximately 100% for medullary thymomas, 76% for mixed thymomas, and 45% for cortical and well-differentiated thymomas.167–170 In one series, no medullary and mixed thymomas (types A and AB) recurred, even though 30% had capsular invasion. Organoid and cortical thymomas (types B1 and B2) showed intermediate invasiveness and a low but significant risk of late relapse, even with minimal invasion.29,159
Associated paraneoplastic syndromes occasionally exist with well-differentiated thymic carcinomas.94,95 However, clinical features have not proved useful as prognostic indicators for thymic carcinomas.159 As with thymomas, total resection and stage at presentation are important prognostic factors for thymic carcinoma.18
Diagnostic Workup for Thymic Tumors
The diagnostic workup of a patient with an anterior mediastinal mass begins with a thorough history and physical examination. Particular focus should be given to detect signs and symptoms that may suggest myasthenia gravis, such as fatigue, diplopia, ptosis, and dysarthria. Constitutional symptoms such as fever, chills, and weight loss are more suggestive of lymphoma. Routine screening blood work and chemistry testing may give clues to the presence of associated syndromes. In the case of suspected Cushing’s syndrome, seen in 1% to 2% of patients, a dexamethasone suppression test and urinary cortisol level should be obtained., although some carcinoids can be suppressed, making diagnosis difficult. The differential diagnosis, in addition to thymic lesions, includes germ cell tumors, lymphomas, and thyroid proliferative disorders. Serum alpha-fetoprotein and beta-human chorionic gonadotropin levels should be obtained in young men if a nonseminomatous germ cell tumor is suspected.171–173 Exclusion of extrathymic primary tumors is important because carcinomas and carcinoids are rare in the thymus and may represent metastases.
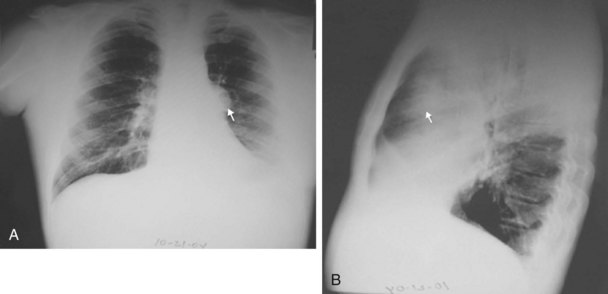
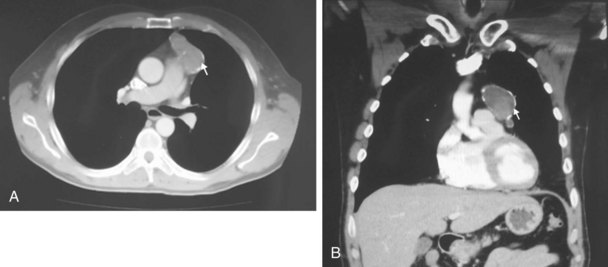
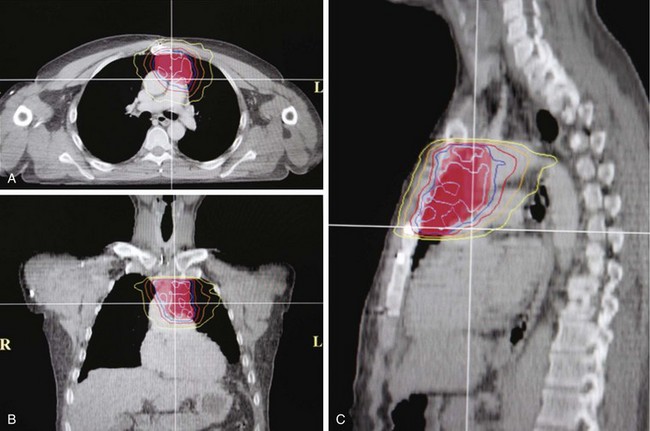
The chest radiograph often demonstrates a mass in the hilar region that may be mistaken for the heart border or pulmonary artery (Fig. 44-2). CT defines174,175 the size, contour, tissue density, and homogeneity of a mediastinal lesion, as well as its relation to or invasion of other mediastinal structures. A CT scan with intravenous contrast dye is preferred to delineate the relationship between the thymoma and surrounding structures and to define the degree of the tumor’s vascularity. Thymic tumors are usually homogeneous, well-demarcated lesions with a round or lobulated shape, and they vary in size and occasionally have calcification176 (Fig. 44-3). The presence of fat planes between the tumor and adjacent structures suggests localized disease. Pleural involvement may be seen in advanced disease. The radiographic presence of both an anterior compartment mass as well as “drop” metastases to the pleura is highly suggestive of the diagnosis. Thymic carcinomas often contain calcifications, cysts, or necrosis on imaging.177 In the absence of symptoms and signs, extensive radiographic imaging is unnecessary. Magnetic resonance imaging (MRI) has not been superior to CT scanning.178 The role of positron emission tomography (PET) has not been established, but early data suggest it can differentiate thymoma from thymic hyperplasia, and high uptake of fluorodeoxyglucose appears to correlate with the degree of tumor invasiveness.179–181
Patients presenting with a thymic mass need a histologic diagnosis before definitive therapy is instituted. CT- or ultrasound-guided fine-needle aspiration biopsy of the mass may be performed to establish the diagnosis preoperatively.182 Such procedures have a sensitivity and specificity of 87% to 90% and 88% to 100%, respectively, for thoracic neoplasms.183 When larger tumor samples are required to distinguish between lymphoma and lymphoid-predominant thymoma, core-needle biopsy provides sufficient specimens with an overall sensitivity of 96% and specificity of 100%.183 Bronchoscopy, video-assisted thoracoscopic surgery, mediastinoscopy, or anterior thoracoscopy may help yield the diagnosis before resection, especially if enlarged lymph nodes are present.172,184,185 The potential risk of breaching the capsule, leading to spillage and seeding of tumor cells during biopsy, has been debated and remains unsettled.136,186,187
Staging
Bergh and associates122 introduced the first clinical staging system for thymoma in 1978. Their staging system was subsequently modified and refined by Masaoka and associates in 1981141 and is the most widely accepted (Box 44-3). It is based on pathologic findings at time of surgery.
Box 44-3
Masaoka Staging System for Thymoma
| Stage | Description |
|---|---|
| I | Macroscopically completely encapsulated with no microscopic detectable capsular invasion |
| II | Macroscopic invasion into surrounding mediastinal fatty tissue or mediastinal pleura or microscopic invasion into the capsule |
| III | Macroscopic invasion into surrounding organs (e.g., pericardium, great vessels, lung) or intrathoracic metastases, or both |
| IVA | Pleural or pericardial implants or dissemination |
| IVB | Lymphogenous or hematogenous metastases |
Adapted from Masaoka A, Monden Y, Nakahara K, Tanioka T: Follow-up study of thymomas with special reference to their clinical stages, Cancer 48:2485-2492, 1981.
A separate, simplified staging paradigm was proposed by Suster and Moran.188 Stage I lesions are localized and encapsulated; stage II lesions are locally invasive; and stage III lesions have nodal, visceral, or distant metastasis. A tumor-node-metastasis (TNM) type of staging system was proposed by Tsuchiya and associates189 but has not been proved clinically, and it is not widely accepted. There is no formal American Joint Committee on Cancer (AJCC) staging system for thymoma.
Treatment
Surgery for Thymoma
Surgery is the mainstay of therapy in resectable cases. However, adjuvant or neoadjuvant radiation therapy plays an important role in subtotally resected or unresectable cases. Many patients who present with symptoms of myasthenia gravis benefit from undergoing a thymectomy.151 Consequently, surgical resection alone for stage II thymomas, in the hands of an experienced team, is a reasonable approach.190,191 In general, surgical resection for thymoma carries low risks of morbidity and mortality; most surgical deaths can be attributed to a myasthenia gravis crisis, but the risk can be minimized with appropriate perioperative management, including plasmapheresis, in selected situations.124,192 After an encapsulated thymoma without associated myasthenia gravis is removed without disturbing the integrity of the capsule, recurrences have been observed, but they are rare.46,60,123,193
Successful surgical treatment of locally invasive thymoma depends on the completeness of resection.194 Consequently, the surgeon should remove as much of the lesion as possible, including surrounding mediastinal fat, possibly to include an extended thymectomy.195 However, resection of an involved phrenic nerve is controversial, and surgeons advocate debulking alone, leaving both phrenic nerves intact if there is bilateral phrenic nerve involvement, because of the respiratory morbidity that would result from resection. When complete resection is not possible, a debulking operation should be considered, because good long-term results can be achieved when such surgery is followed by postoperative radiation therapy.124 Only about one-fourth of patients with stage IVA disease have resectable disease.58 Surgical approaches have varied from discrete resection of pleural metastases to en bloc mediastinal dissection with EPP.196 A few small single-institution series have suggested that EPP may improve local control rates.196–198 The surgeon should delineate the extent of the tumor, specify the areas of invasion, and identify the areas of positive or questionable margins and residual disease with metallic clips to assist future radiation therapy planning.
Radiation Therapy for Thymoma
Although the role of adjuvant irradiation for invasive thymomas has never been tested in a prospective, randomized fashion, there is general agreement that RT is an effective adjuvant therapy for invasive thymomas,199 and most retrospective studies have reported improvements in local tumor control and survival rates after adjuvant irradiation.* However, patients with encapsulated, noninvasive stage I thymoma do not require postoperative RT after complete resection, because the recurrence rate is approximately 0% to 2%.†
For patients with Masaoka stage II disease, there is controversy about whether postoperative RT should be used for completely resected invasive thymomas. A meta-analysis of 22 studies with 592 patients with completely resected stage II or III thymoma found no reduction in recurrence after adjuvant RT,203 but it is difficult to draw firm conclusions from the available data, particularly given the selection bias in retrospective series as to which patients received RT. Berman and colleagues10 reported a single-institution retrospective cohort comparison of 74 stage II thymoma patients after complete surgical resection. Patients who were thought to be at high risk for local recurrence by the operating surgeon and the thoracic oncology team were referred for adjuvant RT. The rate of locoregional recurrence was 8% in patients who were observed versus 0% in the RT group (p = .15). The researchers concluded that the rates of recurrence after complete resection of stage II thymoma were low and that no benefit of postoperative RT was observed in stage II thymoma patients as a group; however, a higher-risk subset of these patients may benefit from adjuvant therapy.10 Although some studies showed no benefit from adjuvant RT,164,190,191,204 the recurrence rates of surgery alone, even with complete resection, may approach 30%.65,143 A SEER study of 901 patients showed a 10% absolute improvement in 5-year OS rates but no statistical difference in cause-specific survival rates in patients with stage II to III disease.204 In cases with gross fibrous adhesions of the tumor to the pleura at the time of surgery or microscopic invasion of the pleura on histologic studies, there is an increased risk for recurrence. Haniuda and colleagues202,206 found that patients with fibrous adhesion to the mediastinal pleura without microscopic invasion benefited most from postoperative radiation therapy. The recurrence rates, with and without adhesion to the mediastinal pleura, were 36% and 0%, respectively. In another study by Monden and colleagues,70 patients with resected stage II thymoma had recurrence rates of 8% and 29%, respectively, with and without adjuvant radiation therapy. Although postoperative radiation therapy may decrease local recurrence, it does not appear to decrease the incidence of subsequent pleural dissemination that may occur outside of radiation fields; in fact, the pleura is the most common site of failure after radiation.202 This may be a reflection of the natural pattern of spread of this disease, with pleural dissemination occurring before, during, or after the time of surgery. Extended irradiation to include the entire pleura significantly increases the normal tissue toxicity and is not routinely employed.
Locally Advanced Thymoma and Palliation
For patients with stage III to IV disease, the evidence supporting the use of postoperative radiation therapy is robust. Urgesi and colleagues140 reported no in-field recurrences in a study of 33 patients with completely resected stage III thymoma treated with postoperative irradiation. Curran and associates65 reported a 53% 5-year actuarial mediastinal relapse rate with stage II and III patients after surgery alone, compared with 0% after total resection and irradiation and 21% after subtotal resection or biopsy and irradiation. Similarly, in a study of 70 patients with Masaoka stage III to IV thymoma, the relapse rate for patients receiving postoperative radiation therapy was reduced from 50% to 20%, and most disease (80%) recurred outside of the irradiated field.207
Preoperative adjuvant radiation therapy has been advocated for unresectable or marginally resectable thymomas.46,71,72,208 Several small studies assessing preoperative radiation therapy for extensive disease found a decrease in tumor burden at the time of surgery, with response rates as high as 80%, and described a theoretical decrease in the potential for tumor seeding during surgery.65,124,209,210 Onuki and colleagues210 found that a dose of 12 to 20 Gy given to 21 patients with stage III thymomas resulted in a 76% response rate, with more aggressive WHO histologic subtypes having a less robust volume reduction. These series demonstrated that preoperative irradiation facilitated total or subtotal resection of the invasive thymoma mass by reducing the tumor volume.208
Primary radiation therapy alone as the definitive treatment has been advocated in nonsurgical candidates or patients with unresectable advanced disease. In a study of 12 patients who presented with unresectable tumors and were treated with primary radiation therapy, 7 patients were alive 1 year 8 months to 5 years 1 month later.211 Similar outcomes were reported for 31 patients with unresectable stage III or IV disease.212 Five-year survival rates of 53% to 87% and 10-year survival rates of 44% were reported for patients with advanced thymoma receiving irradiation after biopsy only or incomplete resection.213,214 Urgesi and associates215 reported the use of radiation therapy alone in 21 patients with intrathoracic recurrences of thymoma. The 7-year survival rate of 70% was similar for those treated with irradiation alone and those treated with surgery and adjuvant therapy. However, the retrospective nature of these studies, small number of patients, differing amounts of clinical disease, and variations in radiation doses and techniques are significant confounding variables.
Chemotherapy for Thymoma
In general, thymomas are chemosensitive tumors, and chemotherapy is reserved for locally advanced or metastatic disease. Numerous case reports and small series have reported antineoplastic activity for multiple and single agents in the treatment of patients with advanced thymoma.* However, there are no reported prospective, randomized trials comparing different chemotherapeutic agents.233 Platinum-based regimens have commonly been used over the past decade, with response rates ranging between 24% and 100%, and response rates of more than 50% have been found consistently with the application of cisplatin-based combination chemotherapy.234 Some of the commonly employed combinations are cisplatin, doxorubicin, and cyclophosphamide (PAC), with reported overall responses in excess of 70%,235–237 and cisplatin, doxorubicin, vincristine, and cyclophosphamide (ADOC).238 In a study by Fornasiero and colleagues,222 37 patients with stage III or IV thymoma were treated with ADOC, with an overall response rate of 92% and a 43% complete response rate. Loehrer and associates235 studied 23 patients with localized but unresectable thymomas treated with PAC followed by radiation therapy. The overall objective response rate was 70%, with five complete responders.239 The estimated 5-year disease-free survival rate was 54%, with a median survival time of 93 months. Multivariate analysis demonstrated that chemotherapy was associated with an improvement in OS rates for patients with stage III and IV disease. Similar results with combination PAC have been reported in Taiwan; all responders had overexpression of the topoisomerase 2α gene, but patients with no detectable expression progressed with treatment.239
Combined-Modality Therapy for Thymoma
The role of chemotherapy as an adjuvant therapy after resection has not been established, but there is anecdotal evidence that combining chemotherapy with irradiation with or without surgery can be effective with acceptable toxicity in selected cases.230,240,241,242 It has also been shown that neoadjuvant chemotherapy and then surgery, followed by additional chemotherapy and irradiation, may improve the resectability and survival rates of the patients with locally advanced disease.241
In a phase II study of 22 patients with unresectable thymoma treated with induction PAC resection, adjuvant irradiation, and consolidative chemotherapy, there was a 77% response rate to chemotherapy, a 95% 5-year OS rate, and a 79% 7-year OS rate.237 A trial from the Japanese Clinical Oncology Group reported on 23 patients with unresectable stage III thymoma treated with weekly dose-dense cisplatin, vincristine, doxorubicin, and etoposide and found no deaths from toxicity, but only 57% were able to complete chemotherapy as planned.243 Thirteen of the patients were able to undergo resection, with nine R0 resections. With a 62% response rate, efficacy of the dose-dense regimen was no better than with conventional chemotherapy.235,237
In a study by Macchiarini and colleagues,230 all seven patients with stage III invasive thymoma treated with neoadjuvant cisplatin, epirubicin, and etoposide were able to undergo surgical resection. Four had complete resections, and three had incomplete resections. Shin and colleagues240 reported that 9 of 11 patients with unresectable stage III and IV thymoma were able to undergo complete resection after induction chemotherapy consisting of cyclophosphamide, doxorubicin, and cisplatin. All nine patients were given additional postoperative radiation therapy and chemotherapy. Of these patients, seven were disease free at a median follow-up time of 43 months. Wright and associates244 reported on 10 patients with stage III to IVA thymoma treated neoadjuvantly with two cycles of cisplatin and etoposide and concurrent irradiation followed by surgery. Eight patients had R0 resections, and the 5-year survival rate was 69%.
Results of Thymoma Treatment
There are no large prospective, randomized, phase III clinical trials to evaluate the efficacy of primary or adjuvant radiation therapy in patients with invasive thymoma, but multiple retrospective reviews have suggested that RT reduces recurrence rates and improves outcomes for incompletely resected stage II to III thymomas. The role of postoperative RT for completely resected stage II to III thymomas is controversial. The local control rates after a complete resection and adjuvant radiation therapy have ranged from 65% to 100%, and rates are lower for incomplete resection and radiation therapy.* Curran and coworkers65 reported a retrospective study of patients with stage II or III thymoma. Twenty-six percent (20 of 78) of the patients treated with surgery alone developed local recurrences, compared with 5% (2 of 43) of similar patients who had received postoperative radiation therapy. There was no significant difference in relapse rates or survival rates between patients undergoing biopsy and radiation therapy and those having subtotal resection and radiation therapy. The investigators reported a 5-year actuarial mediastinal relapse rate of 53% for patients treated with total resection alone, compared with no relapses in patients who received postoperative irradiation after total resection. Contrary to most studies, Kondo and Monden18 reported a multi-institutional, retrospective study of 1320 patients with thymic epithelial tumors and found no significant difference in the recurrence rates between two groups with stage II or III thymoma, one of which underwent surgery alone and the other surgery plus adjuvant irradiation. This observation may reflect the fact that most of the patients underwent complete resection (100% with stage II disease and 85% with stage III disease); as has been pointed out by the study authors, the recurrence rates they found were lower than in previous reports.
Postoperative radiation therapy has resulted in improved survival rates for patients with invasive thymomas after complete and incomplete resections.233 In a study of 141 patients with thymoma, Nakahara and associates125 reported a 5-year survival rate of 92% for patients with stage II disease and 88% for patients with stage III disease who were treated with postoperative irradiation. Patients undergoing radical surgery with complete resection before adjuvant radiation therapy often had substantially better local control and 5-year survival rates compared with those undergoing biopsy alone or limited tumor resection.124,125,209,246
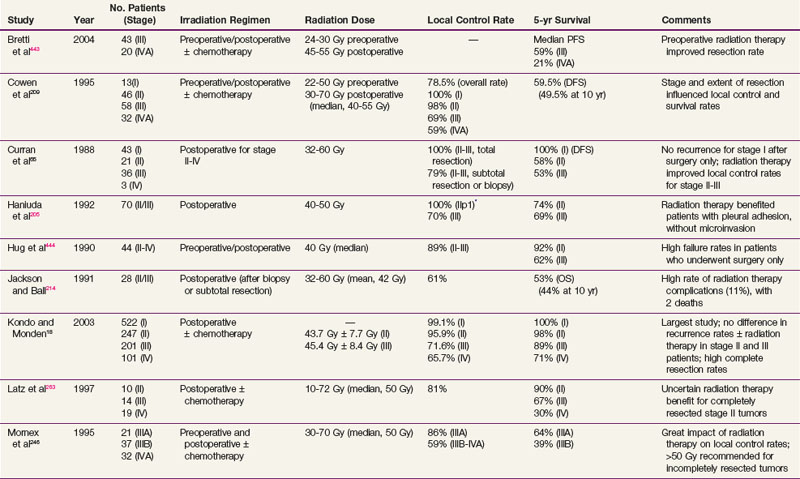
After complete resection, invasive thymomas still tend to carry a poorer prognosis than noninvasive tumors.47,233,247 Survival rates continue to fall with long-term follow-up, and 10-year survival rates may be considerably lower.149 The 5-year and 10-year survival rates for well-encapsulated thymomas without invasion are more than 90%, and for invasive thymomas, the rates range from 30% to 70%† The 5-year survival rates according to Masaoka stage are 83% to 100% for stage I disease, 86% to 98% for stage II disease, 68% to 89% for stage III disease, and 50% to 71% for stage IV disease.18,68,141,248 The approximate 10-year survival rates are 80%, 78%, 47%, and 30%, and the 15-year rates are 78%, 73%, 30%, and 8% for stages I to IV, respectively.68 Table 44-1 provides a summary of treatment results.
Anecdotal studies have demonstrated good local control of thymomas treated with primary irradiation. Of 23 patients treated with irradiation alone, 8 had complete regression, 10 had partial regression, and 5 had no regression of disease.46 In a report by Marks and colleagues,249 tumor was controlled in all nine cases treated with megavoltage irradiation. The average follow-up was 5.5 years (minimum, 30 months). Ariaratnam and associates250 observed tumor control in 8 of 11 patients with malignant thymoma with a minimum follow-up of 2 years. Two of the three patients who died had received only 30 Gy in 3 weeks to the mediastinum. Overall, irradiation as monotherapy may result in local control rates of approximately 65% and 5-year survival rates of 40% to 50%.213,214,233
In patients with mostly advanced thymoma, the results of combination chemotherapy with or without surgery or irradiation are encouraging, with an overall response rate of approximately 60%.222,224,236 All patients who achieved a pathologic complete response had received cisplatin-containing regimens. An intergroup study of 26 patients with unresectable disease treated with two to four cycles of PAC followed by RT (54 Gy) in those without progression was well tolerated. There was a 70% response rate to induction chemotherapy and a median survival rate of 93 months, and a 53% 5-year OS rate, which compares well with neoadjuvant chemotherapy followed by resection.229
Treatment of Thymic Carcinoma
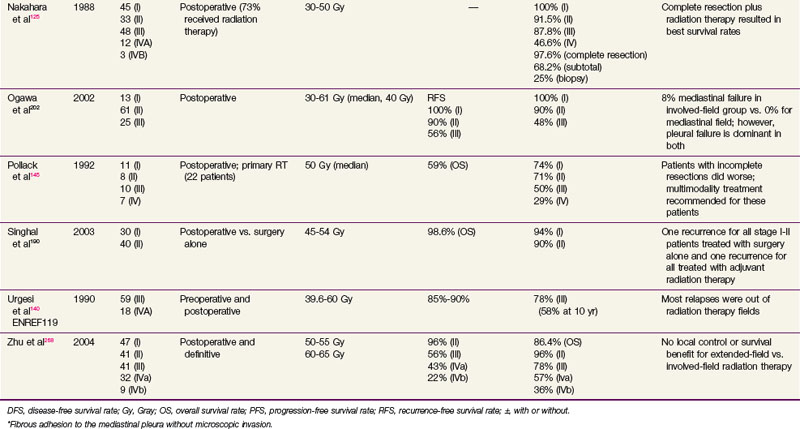
Although the optimal treatment of thymic carcinoma is unclear, surgical extirpation remains the cornerstone of therapy, and in most published studies, surgery has been followed by adjuvant radiation therapy.17,64,251–253 Because many series are gathered over decades, it has been difficult or impossible to control for the changes in pretreatment tumor imaging, surgical techniques, and thoracic radiation therapy planning and delivery, all of which may contribute to the inconsistent results in the literature. A prescriptive dose range has yet to be identified, with most studies using 40 to 70 Gy with a standard fractionation scheme (1.8 to 2 Gy per fraction), but in general, doses of 45 to 55 Gy are used in the adjuvant setting, whereas 60 Gy or more is used for definitive irradiation. Although a survival benefit has not been seen, there is a trend toward improved survival and local control rates with adjuvant therapy.17,252 No dose-related survival advantage has been established, although a relationship between dose and local control rates has been demonstrated.254
In a series of 26 patients treated with surgery and postoperative irradiation, Hsu and associates252 observed a 77% 5-year OS rate, with respective 82% and 66% survival rates for completely resected and subtotally resected cohorts. With a median dose of 60 Gy (range, 40 to 70 Gy), an excellent 5-year local control rate of 91% was observed. For a cohort of 40 patients receiving definitive or adjuvant radiation therapy, Ogawa and associates17 reported an absence of local recurrence for those with complete resection and radiation doses greater than 50 Gy. Kondo and Monden18 reported the largest retrospective, comparative study and found no statistically significant survival benefit from the addition of adjuvant irradiation to surgical resection, although the study authors stipulated that no definitive conclusions could be made regarding the role of adjuvant radiation therapy because of subgroup sample size limitations and the retrospective nature of the study.
Although local control rates are increased with irradiation, a survival benefit remains to be demonstrated. For patients with any question of clinical resectability, neoadjuvant platinum-based chemotherapy is a reasonable treatment consideration.255 Table 44-2 summarizes some of the treatment results of thymic carcinoma.
Treatment of Thymic Carcinoid
Complete surgical resection is the preferred method of treatment, although recurrence and distant metastases are common.18,256,257 A SEER analysis of 160 patients found that the median survival time was 79 months in patients who had surgical resection compared with 26 months in those who did not have surgery Patients who received irradiation had a worse OS rate, but patients who received irradiation were more likely to have advanced disease.257 In a study of 40 patients, the recurrence rate was 64%, even though 35 of the 40 patients had a complete resection,18 and the 5-year survival rate was 84%. Despite a lack of conclusive evidence, incomplete resections followed by irradiation or chemotherapy, or both, may improve local control rates without significantly increased morbidity and mortality rates.55,129,258–260 However, distant metastases occur in approximately 30% of patients.260 The long-term prognosis is poor, with an overall 5-year survival rate of 0% to 31%.256,261
Techniques of Irradiation
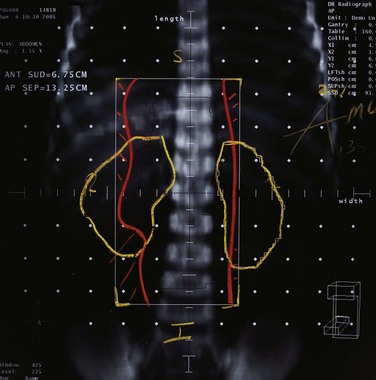
High-energy (>10-MV) x rays usually are preferred for irradiation. A CT scan is indispensable for adequate treatment planning. Clips placed at the time of surgery denoting the extent of resection in completely resected tumors or outlining regions of residual disease are useful in guiding postoperative irradiation. The planning target volume (PTV) should include the gross tumor volume (GTV) and the thymus, if any, and the tumor bed with a 1.5- to 2-cm margin. A four-dimensional breathing scan, if available, should be used to determine tumor motion. This information can be used to create an internal target volume (ITV) or to plan for radiation-coordinated breathing control, wherein the patient is treated only during a particular phase of the breathing cycle. Although the latter approach requires significant patient training and cooperation, it can provide superior sparing of normal lung.262 Depending on histologic and surgical findings, areas of suspicious subclinical disease and regional lymphatics are commonly included. However, treatment of the entire mediastinal and supraclavicular nodal basin prophylactically is not advised, because of the low incidence of lymph node involvement and a higher normal tissue complication rate when large target volumes are used.235,245,263,264 A SEER database analysis of 1334 patients from 1973 to 2005 by Fernandes and colleagues265 found no increase in long-term cardiac mortality rates or rates of secondary malignant tumors for patients treated with RT compared with patients treated with surgery alone.
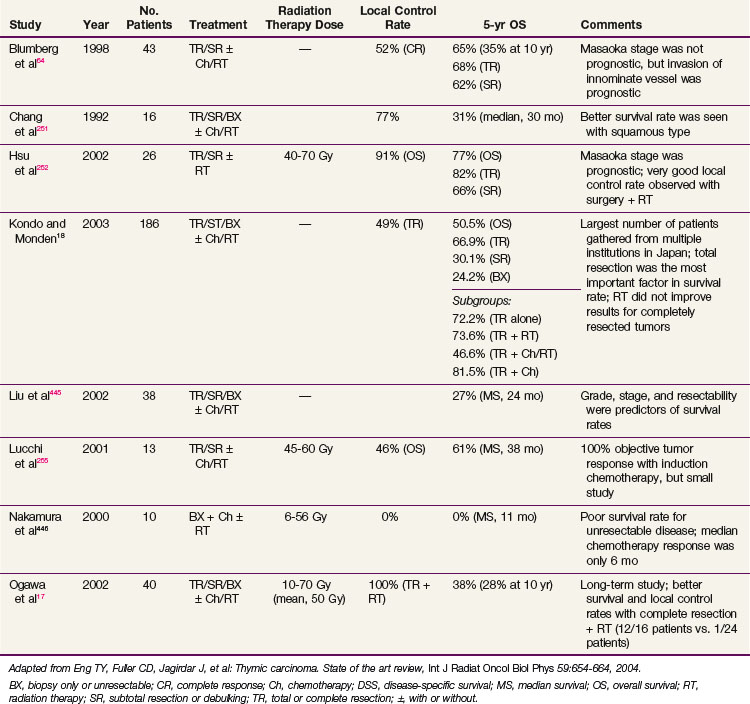
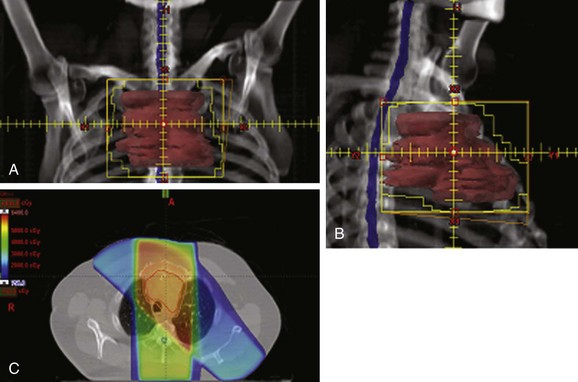
Conventional port arrangements may include a single anterior port, two opposed anteroposterior ports (weighted 2 : 1 or 3 : 2), wedged-pair techniques, and other multiple-field arrangements. Given the advanced technical capabilities available, three-dimensional conformal RT (3DCRT) or intensity-modulated radiation therapy (IMRT) should be considered to provide dose homogeneity and allow better sparing of normal critical structures. Figure 44-4 shows a postoperative radiation therapy field with multileaf collimators shaping the surgical tumor bed. Figure 44-5 shows a 3DCRT field with IMRT. The isodose lines more closely conform to the shape of the PTV. The risks of side effects depend on normal tissue tolerance, surrounding critical organs, prior treatments, irradiation dose, other concurrent therapies, and the general health of the patient.
Various dose and fractionation schemes for thymoma are reported in the literature, and it is unclear if there is a dose response.233,266 Although one retrospective study did not find any relationship between radiation dose and local control rates,209,254 others have found that radiation dose was a significant prognostic factor for local control rates, with good results reported when doses higher than 40 Gy were used for subclinical or microscopic disease.246,267 Retrospective data from Zhu and co-workers268 found a 5-year survival rate of 82% in patients given a dose greater than 50 Gy, compared with 70% for doses less than or equal to 50 Gy. The most important prognostic factors were Masaoka stage and extent of resection.
Postoperative doses of 45 to 55 Gy given by conventional fractionation have been used effectively in most cases.* The dose to the spinal cord should be limited to 45 Gy and can be accomplished using oblique mediastinal fields or IMRT. For patients with gross residual disease after resection or unresectable disease, doses of 60 Gy or more are probably required,145,212,246 but higher doses correlate with higher risks of complications.269 Sophisticated treatment planning with IMRT may facilitate delivery of high doses to gross tumor while respecting dose limits to organs at risk.270
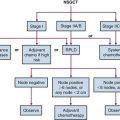
Treatment Algorithm and Controversies
Pulmonary Carcinoid Tumors
Neuroendocrine lung tumors have a spectrum of clinical behaviors and include typical carcinoids (the most indolent type), atypical carcinoids, large cell neuroendocrine tumors, and small cell lung carcinomas (the most aggressive type).271 The term carcinoid (from the German karzinoide) was originally defined as a carcinoma-like lesion without malignant characteristics by Oberndorfer in 1907.272 Pulmonary carcinoids used to be known as bronchial adenomas because they were believed to be benign, but they are now recognized as malignant because of their potential to metastasize. Pulmonary or bronchial carcinoid tumors are typically low-grade malignant neoplasms that arise from neuroendocrine cells of the amine precursor uptake and decarboxylation (APUD) system that have migrated from the embryonic neural crest. These cells are the Kulchitsky (enterochromaffin) cells, which are located in the basal layer of bronchial epithelium.273,274 Approximately 25% of carcinoids are located in the respiratory tract, which is the second most common site after the gastrointestinal tract. Pulmonary carcinoids are frequently centrally located and confined to the main or lobar bronchi.275 These tumors are rare, and their biologic behavior mostly depends on their histologic characteristics.
Etiology and Epidemiology
The cause of pulmonary carcinoid is unknown, and no environmental risk factors have been identified. Carcinogens that are associated with lung cancer have not been consistently associated with pulmonary carcinoids,276 although a few studies revealed a higher frequency of smokers with atypical carcinoids.277–279 Some of the neoplastic cells express mutant retinoblastoma (RB1) or mutant TP53 gene products, and they show occasional loss of heterozygosity, especially in atypical carcinoids.280 Most familial pulmonary carcinoids have been reported in patients with multiple endocrine neoplasia type 1 (MEN 1). In cases of atypical and typical lung carcinoids, there is a characteristic allelic loss within the region of 11q-13, which harbors the MEN1 gene, a tumor suppressor gene.281–285 Other frequently detected genetic alterations in lung carcinoids, especially atypical carcinoids, include losses of 3p, 5p, 9p, 10q, and 13q.283,284 Ki-67 expression in more than 5% of nucleoli is a poor prognostic factor for survival.286
The annual incidence of carcinoid tumors is one or two cases per 100,000 people in the United States.2,276 For bronchopulmonary carcinoids, the reported incidence is 0.6 per 100,000 people.3 Carcinoid tumor of the lung represents 1% to 2% of all lung malignant tumors, with an equal frequency of these tumors occurring in men and women276; approximately 3500 new cases were reported in 2004.287,288 Bronchial carcinoids are the most common primary lung tumor in children, typically presenting in late adolescence. Typical carcinoids are about four times more common than atypical carcinoids. Data from the SEER program of the National Cancer Institute have shown a trend of relative increase of pulmonary carcinoids.3,276
Prevention and Early Detection
There is no known preventive measure for pulmonary carcinoids. Carcinoids are detected like other space-occupying tumors. The most common findings on a chest radiograph include a hilar mass with occasional atelectasis. Functioning carcinoids are suspected on the basis of the symptoms and signs, and the diagnosis is confirmed by demonstrating increased urinary excretion of the serotonin metabolite 5-hydroxyindoleacetic acid (5-HIAA). Thyroid transcription factor-1 is expressed in 80% of metastatic pulmonary carcinoids but not in intestinal carcinoids, and it may be of value in the workup and diagnosis of pulmonary carcinoids.289,290 Sometimes, localization of the tumor may require an extensive evaluation, including laparotomy. If hepatic involvement is suspected, a specific liver scan may be sufficient to demonstrate metastases.
Biologic Characteristics and Molecular Biology
Carcinoid tumors often occur sporadically and are thought to arise from neuroendocrine cells, which are present in a wide range of organs. The WHO classification of lung neuroendocrine tumors describes two different subtypes of carcinoids, typical and atypical carcinoids, with distinctive clinical behaviors and different prognoses.288,291 Based on the criteria proposed by Travis and associates,271,291 the spectrum of neuroendocrine tumors also includes large cell neuroendocrine carcinoma and small cell lung carcinoma.
Although most pulmonary carcinoids are nonfunctional, some have the ability to synthesize and secrete a variety of physiologically active substances, leading to paraneoplastic syndromes, including carcinoid syndrome, Cushing’s syndrome, and acromegaly.274 Serotonin (5-hydroxytryptamine [5-HT]) is one of the frequently encountered vasoactive substances released by carcinoid tumors. Other substances, including corticotropin, histamine, dopamine, substance P, neurotensin, prostaglandins, and kallikrein, have been reported.292–297
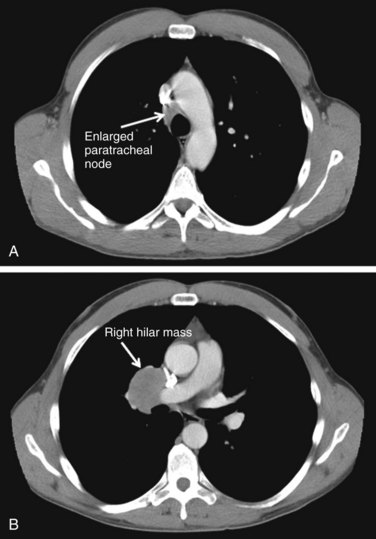
The biologic behavior of these tumors varies depending on the histologic characteristics. Typical carcinoid tumors of the lung generally have a better prognosis than other primary lung cancers, with a 5-year survival rate greater than 90%. For atypical carcinoids, 5-year survival rates range from 25% to 75%, depending on the series.298–303 A population-based study of over 1400 patients from several European countries showed a 5-year OS rate of 78% for pulmonary carcinoids as a whole.304 Although typical carcinoids have an indolent clinical course, atypical carcinoids behave more aggressively, with nodal involvement in 20% to 60% of cases and a higher rate of distant metastases. Figure 44-6 shows a right-sided hilar atypical carcinoid tumor with associated mediastinal adenopathy. The recurrence rates and stage at presentation frequently are higher than for typical carcinoids. In atypical carcinoids, there is a high frequency of molecular alterations and inactivation of tumor suppressor genes, such as the TP53, RB1, CDKN2A (previously designated p16), and CDKN2D (previously designated p19) genes. The proliferation rates, as assessed by MIB-1 and Ki-67, are also higher than for typical carcinoids.288,305,306
Pathology and Pathways of Spread
Carcinoids are thought to be derived from different embryonic divisions of the gut and are classified accordingly. Foregut carcinoids are often found in the lungs, bronchi, and stomach, and midgut and hindgut tumors are typically seen in the small and large intestines. Histologically, they may stain positive with silver or Grimelius stains, cytokeratin 7 and 11, neuron-specific enolase, synaptophysin, carcinoembryonic antigen, and chromogranin; there is no marker exclusive to carcinoids, however.289,307,308 Microscopically, they are characterized by small uniform cells and the presence of numerous membrane-bound, neurosecretory eosinophilic granules containing a variety of hormones and biogenic amines in the cytoplasm. Most pulmonary carcinoids (70% to 90%) are typical carcinoids or well-differentiated neuroendocrine tumors, which are characterized by small cells with well-rounded nuclei; atypical carcinoids (10% to 30%), or poorly differentiated neuroendocrine tumors, have malignant histologic features of increased nuclear atypia with high mitotic activity (>2 mitoses per 10 high-power fields), lymphovascular invasion, nuclear pleomorphism, and areas of necrosis.291,308–311
It is sometimes difficult to establish the specific diagnosis of carcinoid because of small biopsy samples or to differentiate atypical carcinoid from small cell lung carcinoma without immunohistochemical tests.291,312–314 Well-differentiated carcinoids are often indolent, with a relatively low risk of nodal or distant metastasis of 3% to 15%.287,311,315–317 Atypical carcinoid may have an aggressive clinical course, with frequent organ metastases.280,318,319 The risk of nodal metastasis, frequently to mediastinal lymph nodes, is 30% to 57%.287,320,321 Distant metastases occur in more than 20% of patients.287 The most common sites of distant metastasis include liver, bone, adrenal glands, brain, skin, and soft tissue.288,299,322–324
Clinical Manifestations, Patient Evaluation, and Staging
Most patients with pulmonary carcinoids present in their 50s.2,276,287,319 Carcinoids in children are rare.325 Between 70% and 80% of these tumors are located centrally near the hilum within the tracheobronchial tree and may produce a variety of clinical symptoms and neuroendocrine manifestations at the time of diagnosis.326,327 About 25% of patients present with symptoms such as recurrent pneumonia, cough, hemoptysis, or chest pain. Carcinoids are vascular tumors and can bleed secondary to bronchial irritation. Hyperparathyroidism may be seen. At least 50% of pulmonary atypical carcinoid tumors present in the periphery of the lung.328 Patients with peripheral lesions may be asymptomatic, and their lesions are usually discovered incidentally.300,329
Carcinoids may produce various amines and polypeptides with corresponding signs and symptoms, often precipitated by emotional upset, food ingestion, or alcohol. The serotonin metabolite 5-HIAA, which is excreted in urine, acts on smooth muscle to produce bronchoconstriction or diarrhea, colic, and malabsorption. 5-HIAA levels are often greater than five times the upper limit of normal. Histamine and bradykinin, through their vasodilator effects, cause flushing. The release of such vasoactive substances into the systemic circulation can cause the carcinoid syndrome, characterized by episodic flushing, typically of the head and neck; asthmatic wheezing; abdominal cramps with recurrent diarrhea resulting in malabsorption syndrome; and valvular heart disease, including right-sided endocardial fibrosis, leading to pulmonary stenosis and tricuspid regurgitation.330 Bronchial carcinoids generally have low serotonin content because the tumor cells lack the aromatic amino acid decarboxylase enzyme and cannot make serotonin and its metabolites. Hence, the secretion of 5-hydroxytryptamine (5-HTP) is increased and 5-HIAA urinary excretion is normal. Lesions of the left side of the heart, which have been reported with bronchial carcinoids, are rare because serotonin is destroyed during passage through the lung. Some patients complain of decreased libido and impotence. However, the incidence of carcinoid syndrome or other paraneoplastic syndromes, including Cushing’s syndrome and acromegaly, is usually no more than 3% to 5% of cases, and these syndromes mainly occur in patients with larger tumors (>5 cm) or metastatic disease.* Carcinoid syndrome is seen more commonly in gastrointestinal carcinoids than in pulmonary carcinoids. Carcinoid syndrome is seen in 1% to 5% of patients, usually in those with metastatic disease or large primary tumors.332 Paraneoplastic syndromes in the absence of lymph node or distant metastases do not affect the prognosis.329,333 Levels of human chorionic gonadotropin and pancreatic polypeptide are occasionally elevated.
Patients with a suspected lung mass require thorough history taking and a physical examination. Routine screening blood tests and chemistry tests may give clues to the presence of associated syndromes (Table 44-3). The differential diagnosis, in addition to bronchopulmonary carcinoid, includes lung carcinoma and metastasis from an extrapulmonary primary tumor. Conventional radiographs of the chest and comparison with older films are often helpful. CT and bronchoscopy are two of the most valuable diagnostic procedures. Up to 80% of carcinoids manifest with type 2 somatostatin receptors, and the receptors can be targeted by radioactive octreotide or pentetreotide. Somatostatin receptor scintigraphy with indium-111–radiolabeled octreotide (111In-octreotide) has demonstrated reliable uptake in primary tumors and been used to detect early recurrence.334–336 The result of FDG-PET is often negative.278,337–339 PET with copper-64 (64Cu) TETA-octreotide as a tracer has been investigated in carcinoids.340 Carbon-11 (11C) 5-HT PET scanning is more sensitive than somastostatin receptor scintigraphy, but the short half-life of the compound (20 minutes) has prevented its routine use.341
The prognostic factors are atypical histologic findings, nodal involvement, and the presence of symptoms at the time of presentation.† A high mitotic rate, tumor size larger than 3.5 cm, and female gender have also been implicated as prognostic factors for patients with atypical pulmonary carcinoids.277 There is no specific staging system for pulmonary carcinoids, and the American Joint Committee on Cancer (AJCC) lung cancer staging system (TNM) is commonly used.
Treatment
Primary Therapy
Curative resection is the treatment of choice.288,323,324,342,343 After complete resection, the long-term results are generally excellent.287,323,327,342 Conservative surgical resection, consisting of wedge or segmental resection, is the preferred therapy for localized pulmonary carcinoids.344,345 Patients with central lesions may require bronchial sleeve resection or sleeve lobectomy.343 Endoscopic laser ablation is not considered curative but may be used to alleviate tumor obstruction, improve atelectasis, and reduce inflammation before resection.316 Intraoperative nodal evaluation should be performed, and if the results are positive, complete nodal dissection is indicated. Because of higher rates of local recurrence in patients with atypical carcinoids, a more extensive surgical procedure, such as lobectomy or pneumonectomy with nodal dissection, should be considered.288,323,324,346 Patients may present with metastatic or unresectable disease, however, and curative resection may not be obtainable. Debulking and other cytoreduction procedures may be of benefit in cases of gastrointestinal carcinoids,347,348 but these procedures are of uncertain benefit for patients with pulmonary carcinoids.
Medical Treatment
Certain symptoms, including flushing, can be relieved by pharmacologic agents. Ondansetron, one of the specific 5-hydroxytryptamine-3 (5-HT3) antagonists, provides sustained symptomatic relief in patients with carcinoid syndrome.349 Somatostatin analogs are potent inhibitors of neuropeptide release and gut exocrine or endocrine function. These analogs are effective in relieving symptoms by binding to somatostatin receptors (mainly, receptor 2),350 which are present in approximately 80% of carcinoids, leading to inhibition of hormone secretion.351,352 Octreotide and lanreotide, long-acting analogs of somatostatin, are the drugs of choice for controlling diarrhea and flushing. The response rate is about 70% for symptomatic improvement, and there is a more than 50% reduction in urinary 5-HIAA secretion, with a median duration of response of 12 months. Data suggest that these somatostatin analogs may have a direct antitumoral effect, with stabilization of tumor growth and reduction of tumor size in patients with carcinoids of various sites.351,352
Radiation Therapy
There are no prospective trials that address the use of adjuvant therapy for patients with bronchopulmonary carcinoids. In general, typical carcinoids do not require adjuvant radiation therapy or chemotherapy after curative resection.287,323,327,342,353 A series of 25 patients with node-positive bronchial carcinoids treated at the Memorial Sloan-Kettering Cancer Center (MSKCC) (12 typical and 13 atypical tumors) found no survival benefit from postoperative radiation therapy.275 Recurrence and survival rates appeared to depend on cell type rather than nodal status. In another study of 163 patients (including three-fourths of patients with typical carcinoids, most of which were stage N0), however, the prognosis depended on the presence (N positive versus N negative) and extent (N1 versus N2) of nodal involvement rather than histologic subtype.298 In most studies, the role of adjuvant radiation therapy is unsettled because of the retrospective nature of studies and the small number of patients.277,318,354 Although prospective, randomized data are lacking, adjuvant radiation therapy has been used in patients at high risk for locoregional recurrence, including those with large tumor size (>3 cm), positive nodes, positive margins, atypical histologic findings, residual disease, and inoperable disease.311,318,354,355 Recently, Mutyala and colleagues356 reported a series of 59 patients who underwent resection with permanent iodine-125 (125I) interstitial seed implantation for a variety of thoracic malignant tumors, including pulmonary carcinoid with close or positive margins. They reported an excellent 2-year local control rate of 67% with minimal added morbidity from this procedure.356
Palliative radiation therapy can be considered in patients with metastatic typical or atypical pulmonary carcinoids. Tumor-targeted irradiation with radioactive somatostatin analogs (111In-octreotide or 131I-metaiodobenzylguanidine [131I-MIBG]) has been used, often as second- or third-line therapy.355,357,358 The results have been inconclusive, with occasional long-term survivors reported.
Chemotherapy
Unlike chemotherapy for patients with small cell lung cancer, systemic chemotherapy has produced only limited success in patients with metastatic carcinoid tumors.355 Although no effective chemotherapeutic regimen has been established, aggressive treatment with adjuvant systemic chemotherapy is recommended in high-risk patients with stage III atypical or metastatic carcinoid.320 Several trials have shown response rates of 21% to 33% with various combinations of doxorubicin, streptozocin, cyclophosphamide, etoposide, and fluorouracil in patients with metastatic carcinoids.320,359–362 There was no significant difference in survival rates between treatment groups, and the side effects were substantial. Patients with atypical carcinoids treated with a combination of cisplatin and etoposide appear to have a higher response rate than patients taking other regimens.
Locally Advanced Disease and Palliation
For patients with locally advanced or metastatic disease, surgery is generally limited to those whose metastases are confined to one site amenable to local resection, because aggressive debulking may enhance the outcome in terms of symptomatic relief.347,363 Radiation therapy has been used successfully to palliate symptoms in patients with locally advanced or metastatic disease.364 Local control and symptomatic relief can be achieved in most patients with 4000 to 5000 cGy. Endoscopic resection and laser photoablation have been used for palliation of symptoms.316 Somatostatin analogs alone or in combination with chemotherapy or interferon-alpha may be considered for symptomatic treatment or tumor stabilization.365
Results of Therapy
In general, patients with typical carcinoid tumors treated with surgery alone have a very good outcome. However, patients with atypical pulmonary carcinoid tumors and regional lymph node metastases are at high risk for recurrent disease and have worse survival rates when they are treated with surgical resection alone.319 The 5-year OS rate ranges from 78% to 100% for all resected pulmonary carcinoids, 90% to 100% for typical carcinoids, 25% to 69% for atypical carcinoids, and 18% to 38% for metastatic carcinoids.* Ten-year rates are 10% to 20% lower than 5-year rates. Some of the published results are summarized in Table 44-4. Although the role of adjuvant radiation therapy as part of multimodality treatment is undefined, adjuvant irradiation may improve the outcome of high-risk patients with atypical carcinoid tumors, positive nodes, or positive margins.322 The National Comprehensive Cancer Network (NCCN) recommends adjuvant therapy for stage II and III atypical carcinoids.368
Irradiation Techniques
The same general principles of thoracic irradiation apply to irradiation of pulmonary carcinoids. High-energy (>10-MV) x rays are preferred with a CT scan for treatment planning. As in treating other tumors, surgical clips denoting the extent of resection or outlining regions of residual disease are useful in guiding postoperative irradiation. The PTV should include the gross or residual tumor volume and tumor bed with a 1.5- to 2-cm margin. Depending on the histologic and surgical findings, areas of suspicious subclinical disease and regional lymphatics are included. Treatment field arrangements may resemble the typical lung tumor fields. There are no consistent guidelines regarding dose and fractionation regimens in the literature. Postoperative doses of 45 to 58 Gy delivered in conventional fractionation of 1.8 to 2 Gy per day have been reported (see Table 44-4). For patients with gross residual or unresectable disease, a higher total dose may be justified as long as the dose to other critical structures does not exceed the tolerance of normal tissues.
Treatment Algorithm and Controversies
Surgery is the primary treatment for typical and atypical pulmonary carcinoids. Although conservative resection has resulted in low recurrence rates and excellent long-term survival rates for patients with well-differentiated pulmonary carcinoids, the adequacy of such resection in patients with atypical carcinoids has not been settled. More extensive surgical procedures, with higher treatment-related morbidity rates, have been advocated by some investigators.320,369 Adjuvant radiation therapy or systemic chemotherapy may be considered even for patients with early-stage disease because of the high incidence of regional and distant failure.318 Palliative irradiation should be considered for those with symptomatic disease.
Mesothelioma
Malignant pleural mesothelioma (MPM) is an aggressive malignant disease, and local disease progression is the main cause of symptoms and death. Disseminated disease is usually reported only very late in the course of MPM.370–374 MPM spreads by direct extension and seeding throughout the pleural space, including fissures, diaphragmatic and pericardial surfaces, through the chest wall, and into the mediastinum, peritoneum, and lymph nodes. The major problem for patients with MPM is poor local disease control in the thorax. Aggressive surgery alone, even in carefully selected patients with early-stage disease, has not improved the 2-year survival rate of 10% to 33%.323–325 Combined-modality treatment suggests improved local control and survival rates,375–377 but local recurrence is still the most common site of first relapse.378
Even after EPP, the diffuse nature of most malignant mesotheliomas and the manipulation of the exposed tumor during surgery put the entire ipsilateral chest wall, diaphragm insertion, pericardium, mediastinum, and bronchial stump at very high risk for local recurrence. The hemithorax and mediastinum have an irregular shape and are adjacent to critical structures, such as the spinal cord, liver, kidneys, esophagus, heart, and contralateral lung. Conventional irradiation of the hemithorax and mediastinum is limited by the ability of these organs to tolerate radiation371,378,379 and by the total volume of tissue being irradiated.
Etiology and Epidemiology
MPM has an incidence of approximately 10 cases per million people per year in the United States.380 The incidence has been increasing and represents a significant clinical problem in populations exposed to asbestos. The Center for Lung Cancer and Related Disorders estimates approximately 3000 new cases of MPM per year, with a peak incidence of approximately 4000 per year expected to occur in about 2025. The incidence is rising due to the long latency between asbestos exposure and clinically apparent disease; most cases occur over 20 years after exposure. The median age at diagnosis is approximately 60 years.
Approximately 90% of mesothelioma cases can be attributed to prior asbestos exposure. This is typically an occupational exposure (e.g., construction work, automotive brake repair, boiler work, shipbuilding), and it explains the approximately 90% male predominance. However, other sources of exposure may include a spouse or parent asbestos worker. Even a history of living near asbestos-using businesses, such as automotive shops (from brake linings) or cement manufacturers (which may have added asbestos), can increase the risk of mesothelioma. Patients from certain regions of the world (e.g., central Turkey, Western Australia) also have endogenous environmental asbestos exposure. There is no direct association between mesothelioma and smoking, but smoking greatly increases the risk when there is also asbestos exposure.381 Radiation exposure has been shown to be causatively linked to mesothelioma, with an increased incidence identified in adults with previous thoracic radiation treatment.
Clinical Evaluation and Staging
Imaging
The utility of MRI in the staging of mesothelioma is controversial. MRI may slightly more accurately predict the extent of chest wall invasion. This finding should not preclude surgery, however, as long as the region of invasion can be removed with a limited chest wall resection. Invasion to the peritoneal surface of the diaphragm cannot always be appreciated without invasive staging. FDG-PET or PET/CT scanning has been applied to mesotheliomas382 because they tend to avidly take up FDG. Sensitivity and specificity of 90% or more has been reported and is helpful in distinguishing between benign pleural thickening and MPM.383,384 Furthermore, PET/CT, when compared with CT, in a series of 35 patients, upstaged tumors and therefore prevented unnecessary surgery in 40% of patients.385
Invasive Staging
The diagnosis of mesothelioma can be difficult because of the relative rarity of the disease and the cytologic similarity to more common metastatic neoplasms, such as adenocarcinoma. Thoracentesis is often the first procedure performed to obtain diagnosis, but it has a diagnostic sensitivity of only 32%.394 Percutaneous fine-needle aspiration biopsy similarly has low diagnostic sensitivity, although ultrasound-guided core-needle biopsy has been reported to improve accuracy.395 Before beginning multimodality treatment, accurate preoperative staging is necessary. An open pleural biopsy is the gold standard.386 Surgical intervention with open thoracotomy or video-assisted thoracoscopic surgery can also be done. Diagnostic thoracentesis is an alternative, though it has a lower yield than video-assisted thoracoscopic surgery.387
Staging
The AJCC seventh edition staging system for mesothelioma is shown in Box 44-4 and is based on the International Mesothelioma Interest Group (IMIG) staging system. The staging systems are useful only after surgical staging, and no clinical staging system has been developed or validated.
Box 44-4
TNM Staging of Mesothelioma
| Primary Tumor (T) | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor limited to the ipsilateral parietal pleura, with or without mediastinal pleura and with or without diaphragmatic pleural involvement |
| T1a | No involvement of the visceral pleura |
| T1b | Tumor also involving the visceral pleura |
| T2 | Tumor involves each of the ipsilateral pleural surfaces (parietal, mediastinal, diaphragmatic, and visceral pleura) with at least one of the following: |
| (1)involvement of diaphragmatic muscle | |
| (2)extension of tumor from visceral pleura into underlying pulmonary parenchyma | |
| T3 | Locally advanced but potentially resectable tumor. Tumor involving all of the ipsilateral pleural surfaces (parietal, mediastinal, diaphragmatic, and visceral pleura) with at least one of the following: |
| (1)Invasion of the endothoracic fascia | |
| (2)Invasion into mediastinal fat | |
| (3)Solitary completely resectable focus of tumor invading the soft tissues of the chest wall | |
| (4)Nontransmural involvement of the pericardium | |
| T4 | Locally advanced, technically unresectable tumor. Tumor involving all of the ipsilateral pleural surfaces (parietal, mediastinal, diaphragmatic, and visceral pleura) with at least one of the following: |
| (1)Diffuse extension or multifocal masses of tumor in the chest wall, with or without associated rib destruction | |
| (2)Direct transdiaphragmatic extension to the peritoneum | |
| (3)Direct extension to mediastinal organs | |
| (4)Direct extension to the contralateral pleura | |
| (5)Direct extension into the spine | |
| (6)Extension to the internal surface of the pericardium with or without a pericardial effusion or tumor involving the myocardium | |
| Regional Lymph Nodes (N) | |
| NX | Regional nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1 | Metastases in the ipsilateral bronchopulmonary and/or hilar lymph node(s) |
| N2 | Metastases in the subcarinal or ipsilateral mediastinal lymph nodes (including ipsilateral internal mammary and peridiaphragmatic nodes) |
| N3 | Metastases in the contralateral mediastinal, internal mammary, or hilar lymph node(s) and/or the ipsilateral or contralateral supraclavicular or scalene lymph node(s) |
| Distant Metastasis (M) | |
| MX | Distant metastasis cannot be assessed |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| Stage Groupings | |
| I | T1N0M0 |
| IA | T1aN0M0 |
| IB | T1bN0M0 |
| II | T2N0M0 |
| III | T1-2N1M0 |
| T1-2N2M0 | |
| T3N0-2M0 | |
| IV | T4 Any NM0 |
| Any TN3M0 | |
| Any T Any NM1 | |
| Residual Tumor (R) | |
| RX | Presence of residual tumor cannot be assessed |
| R0 | No residual tumor |
| R1 | Microscopic residual tumor |
| R2 | Macroscopic residual tumor |
From Edge SB, Byrd D, Compton CC, et al, editors, for the American Joint Committee on Cancer: AJCC Cancer Staging Manual, ed 7. New York, 2010, Springer.
Several important prognostic markers have been identified for MPM. Metastases to any nodal station predict a worse outcome. However, unlike non-small cell lung cancer, it is not clear that mediastinal metastases reflect a worse prognosis than hilar or intrapulmonary nodal metastases. The completeness of resection is also prognostic. Patients with R1 or R2 resections have been reported to have a worse outcome in some series.388
Another poor prognostic marker is nonepithelioid histologic findings. Epithelioid histologic characteristics are found in 50% of cases. Pure epithelioid mesothelioma may be difficult to distinguish from metastatic adenocarcinoma of the lung, so immunohistochemistry or electron microscopy is essential.389 Patients with sarcomatoid (10% of patients), mixed (30% of patients), and desmoplastic tumors fare much worse than patients with epithelioid histologic types.390,391,392 Thrombocytosis and high levels of platelet-derived growth factor predict a worse outcome. Several reports suggest that serum levels of mesothelin are elevated in most patients at diagnosis and that they fall after surgery. Serum mesothelin may be a marker for tumor recurrence.393 Good prognostic factors include young age, early-stage disease, performance status of 0 to 1, lack of chest pain, and presence of symptoms for longer than 6 months.392
Treatment
Surgery
Pleurectomy
Pleurectomy for treatment of MPM has been advocated by many surgeons, although some believe it offers inferior disease control than EPP. Local recurrences are significantly higher than after EPP. Because the ipsilateral lung is preserved by pleurectomy, the procedure is often preferred for patients with poor contralateral lung function. Pleurectomy is effective in allowing expansion of the trapped lung and reduces the likelihood of recurrent pleural effusion. Unfortunately, mesothelioma usually involves the visceral and parietal pleural surfaces of the lung, and these extend into the pleura-lined pulmonary fissures. Cytoreduction is not as complete as that achieved with EPP. A large retrospective study of 663 patients actually showed improved OS rates with pleurectomy as compared with EPP. Patients treated with pleurectomy and decortication had a higher rate of local recurrence of disease, whereas patients undergoing EPP had greater surgical morbidity and mortality rates.396 Because the lung and hemidiaphragm remain after pleurectomy, tumoricidal postoperative external beam radiation therapy cannot be effectively administered, so some centers have been exploring the addition of photodynamic therapy in lieu of external beam radiation therapy.
Extrapleural Pneumonectomy
Most experts consider EPP to be the operation of choice for mesothelioma because it achieves the greatest397 degree of cytoreduction possible and allows adjunctive therapies such as chemoperfusion, photodynamic therapy, or hemithoracic irradiation to be given without risk to the remaining vital organs. A review of 34 studies of EPP for patients with MPM found a perioperative mortality rate of nil to 11.8% and a median OS time of 9.4 to 27.5 months. Without adjunctive therapy, EPP has been associated with recurrence rates as high as 80%, but with current multimodality techniques, studies have reported local recurrence rates of between 40% and 50%. One study398 reported a local recurrence rate of 13% (7 of 55 patients) for patients treated with hemithoracic irradiation to 54 Gy after EPP.
EPP is an extensive surgical procedure, removing the ipsilateral lung along with the parietal pleura, pericardium, and most of the hemidiaphragm, so patients require careful assessment preoperatively to exclude the presence of locally advanced or distant disease (which would preclude resection) and meticulous cardiopulmonary screening to ensure that the operation can be tolerated physiologically. We perform EPP only after surgical staging of the mediastinum and abdomen (with washings). In general, patients should have adequate cardiac function (i.e., ejection fraction >40% and no reversible ischemia on radionuclide imaging) and have an estimated postoperative forced expiratory volume in 1 second (FEV1) value of greater than 0.8 L/min, or 30% of predicted. The ideal patient is young, with epithelial histologic typing and a good performance status and without lymph node involvement. Even in centers with extensive experience with carefully selected patients, EPP has a high rate of complications and is not considered the standard of care.399 Due to the extremely poor prognosis of the sarcomatoid subtype, some have argued against EPP for this histologic group.400
Chemotherapy
Chemotherapy for mesothelioma has not met with much success. Reviews401,402 describe modest success with several chemotherapeutic agents. Reported response rates of more than 25% include the use of methotrexate (37%),403 gemcitabine plus cisplatin (48%),404 pemetrexed plus carboplatin (50%),405 pemetrexed plus cisplatin (38%),406 mitomycin plus cisplatin (26%),407 raltitrexed plus oxaliplatin (26%),408 and doxorubicin plus cisplatin (28%).409 At the M.D. Anderson Cancer Center, hyperthermic intraoperative cisplatin has been used following EPP, with an MPM recurrence rate of 51% and an ipsilateral recurrence rate of 17%.410
Perhaps the most exciting development in chemotherapy for mesothelioma is the use of cisplatin plus the antifolate pemetrexed.411 A total of 456 patients were randomized to cisplatin alone versus cisplatin and pemetrexed. The median survival time was 12.1 months for the pemetrexed plus cisplatin arm and 9.3 months for the cisplatin alone arm (p = .020). Toxicity was dramatically reduced by the addition of vitamin supplementation, but this did not influence the effectiveness of therapy. Time to progression was improved from 3.9 to 5.7 months by the addition of pemetrexed (p = .001). The response rate was 17% for cisplatin alone compared with 41% for combined treatment.405 Krug and colleagues405a recently reported the results of a multicenter phase II trial of neoadjuvant pemetrexed plus cisplatin followed by EPP and irradiation for MPM, with a promising median survival time of 29.1 months and a 2-year survival rate of 61.2%.
The British Thoracic Society performed a randomized trial to assess the role of chemotherapy in palliation.412 Of the 242 patients registered, 109 were randomized to active symptom control or one of two chemotherapy regimens (i.e., mitomycin C, vinblastine, and cisplatin or vinorelbine alone). They found that active symptom control with chemotherapy provided better palliation of chest pain and shortness of breath than active symptom control alone.
Rationale for Irradiation
Although technical challenges remain, several lines of evidence suggest that radiation therapy can be effective in treating mesothelioma. First, mesothelioma cell lines are not particularly radioresistant in vitro, and they have radiosensitivities similar to those of non–small cell lung cancer.413,414 Second, there is a clinically meaningful dose-response relationship for symptom palliation, such that doses greater than 40 Gy seem more effective than lower doses.370,415 This dose-response relationship suggests that clinically significant cell killing results from modest radiation doses. Third, modest radiation doses can dramatically reduce the local failure rate at thoracotomy or other instrumentation sites. These data demonstrate that irradiation can effectively kill mesothelioma cells in regions where the tumor burden is low. Because MPM is moderately radiosensitive, it follows that postoperative irradiation can be effective in preventing locoregional recurrence and potentially improve survival rates.
Radiation Therapy after Pleurectomy or Decortication
Thirty-four patients with pathologically proven MPM were treated between 1982 and 1988 at the Helsinki University Central Hospital.416,417 Twenty-nine patients had partial pleurectomy, and the others had only biopsy. Patients were irradiated with one of three schedules that delivered between 55 Gy and 70 Gy to the hemithorax, and significant pulmonary damage occurred in many patients. A similar study at the MSKCC418 treated 41 patients with partial pleurectomy, and all had residual gross disease that was given a boost dose with permanent 125I implantation at surgery. Photon and electron fields were combined to treat the superficial chest wall and spare the underlying lung. Radiation doses were 45 Gy to the pleural surface, but the dose to “most of the lung” was kept below 20 Gy. Local recurrence developed in 29 of 41 patients, with 22 having distant failures. The median time to local failure was 9 months. Radiation-related toxicity occurred in four patients (one each of radiation pneumonitis, symptomatic pulmonary fibrosis, pericardial effusion, and esophagitis), but the severity of toxicity was not described.
These results were updated with results for 105 patients treated with pleurectomy or decortication from 1976 to 1988.371,374 Forty-one patients received external beam radiation treatment only, and 53 patients received external beam irradiation with an intraoperatively placed 125I boost to gross disease. The median survival time was 12.6 months, but 12 patients developed radiation pneumonitis and 8 developed radiation pericarditis. The severity of these reactions and the local control rates were not described.
One article419 described the results of 24 patients treated with pleurectomy or decortication, intraoperative radiation therapy (IORT), and postoperative external beam radiation therapy. Fourteen patients were irradiated using three-dimensional conformal radiation therapy, and IMRT was used to treat 10 patients. IORT was targeted mainly to the intrapulmonary fissures, pericardium, and diaphragm, with doses ranging from 5 to 15 Gy. The target volumes for the external beam treatments were not described, but the doses ranged from 30.1 to 48.8 Gy (median, 41.4 Gy). Twelve patients also received chemotherapy (i.e., cyclophosphamide, doxorubicin, and cisplatin). Despite the relatively early stage of disease, the outcome was relatively poor, with a median OS time of 18 months and a progression-free survival rate of 12 months. Major complications included radiation pneumonitis in four patients, pericarditis in one patient, and esophageal stricture in one patient who had received previous radiation therapy to the esophagus. Tumor recurrence was “mostly locoregional” at sites of previous gross tumor. Only the number of IORT sites was predictive of the OS rates, suggesting that higher external beam doses might be required for this approach to be widely used. The MSKCC group also reported their experience with high-dose-rate IORT after pleurectomy or EPP. There were serious complications in three of six patients (50%) of the EPP/IORT group and in one of five patients (20%) of the PD/IORT group, and they have abandoned IORT after EPP.420
Pleurectomy or decortication followed by irradiation can reduce locoregional failure rates compared with historical controls. However, local failure is common, and survival rates are disappointing even when modern IMRT techniques are applied. Although the overall median survival time in these pleurectomy or decortication studies tends to be about 2 years, carefully selected patients with early-stage disease treated with EPP can expect median survival times of more than 4 years.377 Unfortunately, there are no prospective, randomized trials to guide therapy.
Intrapleural Photodynamic Therapy
The Food and Drug Administration (FDA) has approved photodynamic therapy for non–small cell lung cancer and cancer of the esophagus. However, photodynamic therapy is being investigated for treatment of mesothelioma. Photodynamic therapy uses light activation of sensitized tumor cells to achieve sterilization. It is delivered intraoperatively immediately following pleurectomy or EPP with the goal of sterilizing microscopic disease throughout the thorax.396
The theory being investigated at the University of Pennsylvania is that if photodynamic therapy effectively eradicates residual disease after a meticulous pleurectomy, it is not necessary to remove the lung itself. Prior to surgery, the patient is infused with a photosensitizer that accumulates in malignant tissue. This sensitizer is activated by 630 nm of red light in the operating room, producing reactive oxygen species. The singlet oxygen destroys tumor via direct damage to malignant cells as well as the tumor miscorvasculature.421
A phase I trial of Foscan-mediated photodynamic therapy found that the maximum tolerated dose of photosensitizer was 0.1 mg/kg 6 days prior to surgery.422 Dose-limiting toxicity at the next dose level consisted of systemic capillary leak syndrome and led to death in two of three patients treated. Other photodynamic therapy-related toxicities included wound burns and skin photosensitivity. Fourteen patients were treated at the maximum tolerated dose (MTD) without significant complications.
One advantage of photodynamic therapy is direct visualization of regions at risk for recurrence. Light sensors placed in the chest allow measurements of light, permitting delivery of a uniform measured dose throughout the hemithorax. The depth of penetration of the light is a few millimeters, so it poses a low risk of damage to the lung parenchyma. Photodynamic therapy does not preclude the use of other adjuvant therapies such as external beam radiation therapy and chemotherapy.424 Studies validating the use of photodynamic therapy after pleurectomy are ongoing.
Radiation Therapy after Extrapleural Pneumonectomy
The first large report of combined-modality therapy that included EPP was described in 1997.378 In this series, 49 patients underwent EPP. Four to six cycles of chemotherapy were given postoperatively, followed by irradiation in 35 patients. The target volumes, stage, and technique were not described. The prescribed dose was 30.6 Gy to the “hemithorax,” followed by a boost to bring the dose to about 50 Gy. Sixteen irradiated patients had a “local” recurrence. However, of the patients with “abdominal” failures, five had a chest mass extending into the abdomen, two had ascites, and four had a “retroperitoneal mass.” This latter fact is potentially important because the diaphragm is reconstructed much higher than the insertion of the diaphragm, “abdominalizing” the retroperitoneal posterior chest wall (Fig. 44-7). Failure to irradiate this region, which can occasionally be below the ipsilateral kidney, can result in apparent retroperitoneal recurrences. The true rate of locoregional failure was at least 21 of 49 cases and possibly as high as 25 of 49 cases. Despite this finding, the 3-year OS rate was 34%. Radiation-related morbidity was tolerable, including esophagitis371 and thrombocytopenia.374 No radiation pneumonitis was described, but five patients experienced “respiratory compromise,” one of whom died of pneumonia. These data suggest that postoperative radiation therapy can reduce local recurrence rates and result in the long-term survival of well-selected patients.
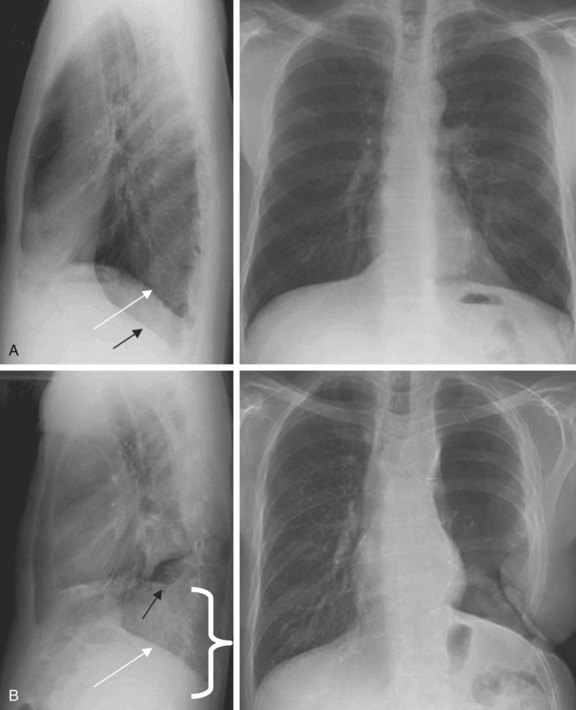
Encouraging results have also been reported from the MSKCC.374 From 1995 to 1998, 54 patients underwent EPP followed by radiation therapy.425 Two-thirds of the patients had stage III disease. The target volume was described as the “hemithorax” and drain sites, and the inferior field edge “rarely” included the ipsilateral kidney. The treatment technique involved photon irradiation to a dose of 54 Gy in 30 fractions. The spinal cord was shielded after 41.4 Gy, and the liver, heart, and stomach were appropriately shielded. The chest wall in the shielded regions was irradiated with matched electron fields so that the goal dose of 54 Gy could be more safely achieved. The median survival time was about 18 months. Only seven patients had local recurrences; two had only local recurrences. Another 22 patients had “peritoneal” or “ipsilateral visceral” recurrences. It is not clear whether any of these were in the radiation fields or whether some might have been marginal misses. It is critical to distinguish marginal failures (which require better definition of the clinical target volume [CTV]) from in-field failures (which require more dose or perhaps less inhomogeneity in dose).
The results of 28 patients treated with EPP followed by IMRT have been reported426–428 from the M.D. Anderson Cancer Center. Twenty-six had stage III disease, most had involved lymph nodes, and all patients required partial chest wall resection. During EPP, radiopaque surgical clips were placed at the insertion of the diaphragm, including the crus, and at the anteromedial pleural extension, which often crosses the midline over the heart. The target volume included the entire hemithorax and all surgical clips, all sites of instrumentation, and the ipsilateral mediastinum. Boosts were given to any close or positive resection margins. All volumes were reviewed with the surgeon. The goal dose was 45 to 50 Gy to the hemithorax and 60 Gy to the boost volume. All irradiation was completed in 25 fractions. The 2-year OS rate was 62%. There were no in-field failures. Two marginal misses occurred, one near the crus and one across the midline anterior to the heart. Major toxicities included nausea and vomiting, fatigue, and skin irritation. This series has been extended to 100 patients; EPP followed by IMRT to 45 or 50 Gy resulted in a 13% local or regional recurrence rate.429 There were three (5%) in-field failures. For patients with stage N0 disease and epithelioid histologic typing (about half of patients), the 3-year OS rate was 41% and the median survival time was 28 months.
An update of the MSKCC experience has been published, regarding their radiation therapy technique and outcome.430 Thirty-five patients underwent radiation therapy after EPP, with the goal of delivering 54 Gy in 30 fractions. The target volume was the ipsilateral hemithorax from the top of vertebra T1, ideally to the bottom of vertebra L2, and laterally to flash the skin. All drain sites were included. The medial field edge was the contralateral edge of the vertebral bodies if mediastinal lymph nodes were negative; otherwise, the field was extended 1.5 to 2 cm beyond the contralateral edge of the vertebral bodies. For right-sided lesions, the liver and ipsilateral kidney were blocked. For left-sided lesions, the heart was blocked after 19.8 Gy. The blocked regions were boosted to the target dose with electrons. After 41.4 cGy, the medial field edge was moved to the ipsilateral edge of the vertebral bodies. Patients tolerate this therapy well, with the main toxicities being nausea, vomiting, and dysphagia.
Perrot and colleagues431 reviewed 60 patients treated with induction chemotherapy, EPP, and hemithoracic RT (≥50 Gy) and found that in stage N0 patients, the median survival time was 59 months, with a 5-year disease-free survival rate of 53%.
These data demonstrate several important points (Table 44-5). First, high-dose postoperative irradiation can dramatically reduce ipsilateral thoracic failures. Second, irradiation after pleural decortication results in a much higher rate of locoregional failure than irradiation after EPP. Consequently, the use of hemithoracic photodynamic therapy after pleurectomy is being studied to decrease the local failure rate with less morbidity than EPP. Third, true in-field failure needs to be well documented. Fourth, postoperative irradiation is well tolerated. Fifth, postoperative radiation therapy changes the pattern of relapse so that distant metastases become more prevalent, requiring the integration of systemic therapy into the treatment regimen.
Palliative Radiation Therapy
Pain Control
Irradiation can provide effective palliation for patients with MPM. A radiation dose-response relationship has been reported after the review of outcomes of 29 courses of palliative external beam radiation therapy delivered to 17 patients with MPM.374 Four of six patients treated with more than 40 Gy achieved significant relief of symptoms. Only one patient treated with lower doses achieved significant palliation of any symptom, and this was for a painful chest wall mass.
In another study,432 26 radiation therapy courses were reviewed for symptomatic improvement. Pain was improved in 13 of 18 cases, and the response was similar regardless of dose or fractionation (i.e., 20 Gy in 5 fractions or 30 Gy in 10 fractions). Although the sample size is small, palliation was achieved in about half of the cases in this very mixed patient group. The duration of response was not assessed in either study. These data demonstrate that symptoms from mesothelioma can usually be palliated effectively with brief courses of irradiation.
Therapy for Drain Sites
Unlike most malignant tumors, MPM has a tendency to recur along tracks of previous chest wall instrumentation.433,434 There is randomized evidence both in support of and against use of local irradiation to drain and biopsy sites.435 One group took 40 consecutive patients with pathologically proven MPM and randomized them to immediate irradiation to each site of instrumentation or to observation. Elective treatment was 21 Gy in three fractions delivered using en face electron fields. There were no local failures in the irradiated patients compared with eight failures (40%) in the untreated patients.433 A slightly larger randomized trial found no reduction in the incidence of tumor seeding in the irradiated patients.436 Because such recurrences typically are painful, elective irradiation is a reasonable means of maintaining the patients’ quality of life.
Irradiation Techniques
Patient Immobilization and Setup
Two basic techniques have been described for thoracic irradiation following EPP.428,430 A description of the IMRT techniques for the postoperative treatment of MPM has been described.427,428 This level of detail is required to ensure that the very complex target volumes can be identified and reproducibly treated for 5 weeks.
Target Volume Delineation
The ipsilateral mediastinum should be included in the target volume,427,435,437 even for node-negative patients. The superior border should be placed at the thoracic inlet. The medial border includes the ipsilateral nodal regions, the trachea and subcarinal regions,428 or the vertebral body.430 The posterior mediastinal structures behind the heart need not be included, because no failures in this region have been recorded (unpublished data) despite their omission from the CTV.427
The anteromedial pleural reflection is a potential problem for target volume delineation. As shown in Figure 44-8, A, the medial pleural space sometimes can cross the midline. This anatomic relationship can be lost after surgery (see Fig. 44-8, B). When possible, this region should be marked intraoperatively with radiopaque clips. Alternatively, the medial extent of the pleura could be identified on preoperative CT scans and then estimated on the treatment-planning CT scan. This has been the site of a marginal miss in our series (see Fig. 44-8, C, arrow).
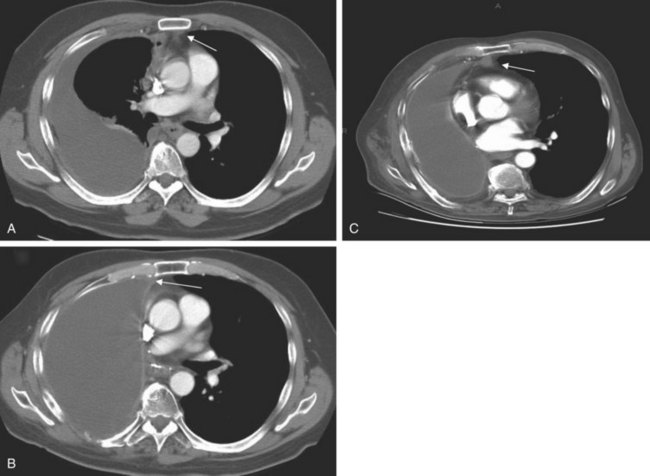
The inferior border should be the insertion of the diaphragm. The location of this is quite variable, ranging from vertebrae L1 to L4. Because of this variability, the diaphragm insertion should be marked by intraoperative placement of radiopaque clips426 or by suturing the neodiaphragm in this location.430 When the border of the intrathoracic contents and the abdominal contents is well marked, the radiation therapy margins can be maximally reduced. Because there can be great difficulty in differentiating liver from thoracic fluid, the use of radiopaque Gore-Tex patches for diaphragm reconstructions is helpful.
When the entire region is extensively clipped, a pattern such as that seen in Figure 44-9 emerges, with regions of potential pitfall highlighted. These potential problem areas include the anterior medial pleural reflection, the crus of the diaphragm, and the inferior aspect of the diaphragmatic insertion.
Treatment Planning
Three-Dimensional Conformal Radiation Therapy
The three-dimensional conformal radiation therapy approach has been described best in two reports from the MSKCC.425,430 The technique applies anteroposterior-posteroanterior beam geometry to the hemithorax using the volumes described previously as the CTV. For right-sided cases, an abdominal block is present throughout treatment, and the region is boosted with electrons at 1.53 Gy per day, which accounts for scatter under the block. For left-sided cases, the kidney and heart are blocked. The kidney block is present throughout treatment, and the heart block is added after 19.8 Gy. The spinal cord is shielded after 41.4 Gy in all cases. The goal dose to the target volume is 54 Gy in 30 fractions, with the dose calculated at the midplane with equally weighted beams. Patients were treated with arms akimbo.
Intensity-Modulated Radiation Therapy
The target doses and the dose-volume limits for the critical structures are listed in Table 44-6. Treatment is delivered with 13 to 27 intensity-modulated fields using 8 to 11 gantry angles, typically with 100 segments per intensity-modulated field. Because patients have only one lung after EPP, the Duke group recommends contralateral lung irradiation limited to a mean lung dose of less than 9.5 Gy.438 This is consistent with the results of whole-lung irradiation.439 The University of Pennsylvania dose constraints are slightly more conservative, with a mean lung dose of less than 9 Gy, a V5 of less than 60%, and a V20 of less than 20%.440 With experience, the mean lung dose can be kept to well under 8 Gy.440,439
TABLE 44-6 Goal Dose or Constraint Dose of the Target or Organ
| Target or Organ | Goal Dose or Constraint Dose |
|---|---|
| CTV | 50 Gy in 25 fractions |
| bCTV | 60 Gy in 25 fractions |
| Lung | <20% to receive >20 Gy and mean <9.5 Gy |
| Liver | <30% to receive >30 Gy |
| Contralateral kidney | <20% to receive >15 Gy |
| Heart | <50% to receive >45 Gy |
| Spinal cord | <10% to receive >45 Gy |
| No portion to receive >50 Gy | |
| Esophagus | <30% to receive >55 Gy |
bCTV, boost clinical target volume; CTV, clinical target volume; Gy, Gray.
IMRT is more complicated and takes longer to deliver than three-dimensional conformal radiation therapy, but very good coverage of the target can be achieved. IMRT has dosimetric advantages over three-dimensional conformal therapy due to improved coverage and homogeneity, but there is more dose spillage into the contralateral lung and an increase in the mean lung dose.441,442 A combined electron and 3DCRT to a dose of 54 Gy had significant reduction to the contralateral lung, contralateral kidney, and heart when compared with IMRT to a dose of 45 Gy.441 Figure 44-10 shows field and block arrangements employed with the use of a combined electron-photon technique. The IMRT plan of 50 Gy to the isodose line in Figure 44-11 encompasses most of the CTV. Review of the dose-volume histogram for this case (Fig. 44-12) demonstrates that the target volume is well covered and the normal tissue constraints are met. The liver and contralateral lung are spared with this technique. In this patient, it was possible to spare the ipsilateral kidney because the organ was particularly low. The ipsilateral kidney usually receives a high dose because the CTV typically abuts its posterior edge. Adequate contralateral renal function is ensured by pretreatment renal scans. For left-sided lesions, the spleen is likely to receive a high radiation dose and pneumococcal prophylaxis is recommended.
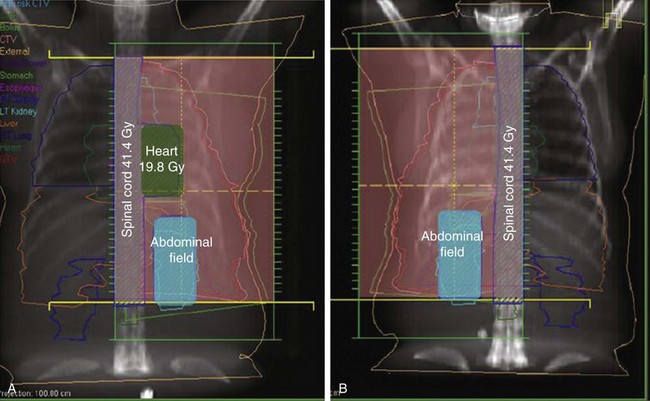
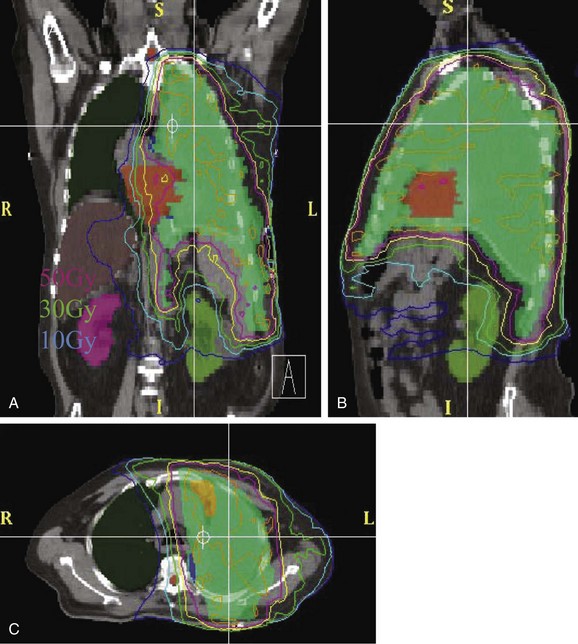
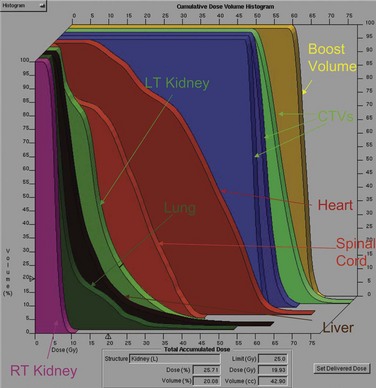
Summary
A review of the limited patient studies (see Table 44-5) in MPM suggests that EPP followed by irradiation gives better local control than pleurectomy followed by irradiation in selected patients. The addition of photodynamic therapy to pleurectomy may improve local control rates, but randomized data are lacking. Postoperative irradiation should include the entire volume immediately adjacent to the pleural space that is at highest risk for microscopic residual disease. This volume should include all sites of pleural reflection, the ipsilateral mediastinum, and the insertion of the diaphragm. The technique by which irradiation is delivered (traditional technique or IMRT) is probably not as important as the target volume delineation and coverage. Prospective trials integrating systemic therapy with EPP and postoperative irradiation are being conducted. Treatment of mesothelioma requires a multidisciplinary approach by an experienced team consisting of a pulmonary specialist, thoracic surgeon, thoracic radiation oncologist, and medical oncologist.
3 Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-959.
10 Berman A, Litzky L, LiVolsi VA, et al. Adjuvant radiotherapy for completely resected stage II thymoma cancer. Cancer. 2011. Epub ahead of print February 1
18 Kondo K, Monden Y. Therapy for thymic epithelial tumors. A clinical study of 1,320 patients from Japan. Ann Thorac Surg. 2003;76:878-884. discussion Ann Thorac Surg 76:884-885, 2003
50 Kundel Y, Yellin A, Popovtzer A, et al. Adjuvant radiotherapy for thymic epithelial tumor. Treatment results and prognostic factors. Am J Clin Oncol. 2007;30:389-394.
51 Suster S, Rosai J. Thymic carcinoma. A clinicopathologic study of 60 cases. Cancer. 1991;67:1025-1032.
60 Maggi G, Giaccone G, Donadio M, et al. Thymomas. A review of 169 cases, with particular reference to results of surgical treatment. Cancer. 1986;58:765-776.
65 Curran WJJr, Kornstein MJ, Brooks JJ, et al. Invasive thymoma. The role of mediastinal irradiation following complete or incomplete surgical resection. J Clin Oncol. 1988;6:1722-1727.
68 Regnard JF, Magdeleinat P, Dromer C, et al. Prognostic factors and long-term results after thymoma resection. A series of 307 patients. J Thorac Cardiovasc Surg. 1996;112:376-384.
70 Monden Y, Nakahara K, Iioka S, et al. Recurrence of thymoma. Clinicopathological features, therapy, and prognosis. Ann Thorac Surg. 1985;39:165-169.
117 Moran CA, Suster S. Neuroendocrine carcinomas (carcinoid tumor) of the thymus. A clinicopathologic analysis of 80 cases. Am J Clin Pathol. 2000;114:100-110.
124 Maggi G, Casadio C, Cavallo A, et al. Thymoma. Results of 241 operated cases. Ann Thorac Surg. 1991;51:152-156.
141 Masaoka A, Monden Y, Nakahara K, et al. Follow-up study of thymomas with special reference to their clinical stages. Cancer. 1981;48:2485-2492.
148 Murakawa T, Nakajima J, Kohno T, et al. Results from surgical treatment for thymoma. 43 years of experience. Jpn J Thorac Cardiovasc Surg. 2000;48:89-95.
159 Chen G, Marx A, Wen-Hu C, et al. New WHO histologic classification predicts prognosis of thymic epithelial tumors. A clinicopathologic study of 200 thymoma cases from China. Cancer. 2002;95:420-429.
160 Marino M, Muller-Hermelink HK. Thymoma and thymic carcinoma. Relation of thymoma epithelial cells to the cortical and medullary differentiation of thymus. Virchows Arch A Pathol Anat Histopathol. 1985;407:119-149.
163 Wright CD, Wain JC, Wong DR, et al. Predictors of recurrence in thymic tumors. Importance of invasion, World Health Organization histology, and size. J Thorac Cardiovasc Surg. 2005;130:1413-1421.
188 Suster S, Moran CA. Thymic carcinoma. Spectrum of differentiation and histologic types. Pathology. 1998;30:111-122.
190 Singhal S, Shrager JB, Rosenthal DI, et al. Comparison of stages I-II thymoma treated by complete resection with or without adjuvant radiation. Ann Thorac Surg. 2003;76:1635-1641. discussion Ann Thorac Surg 76:1641-1642, 2003
191 Mangi AA, Wright CD, Allan JS, et al. Adjuvant radiation therapy for stage II thymoma. Ann Thorac Surg. 2002;74:1033-1037.
201 Zhang H, Lu N, Wang M, et al. Postoperative radiotherapy for stage I thymoma. A prospective randomized trial in 29 cases. Chin Med J (Engl). 1999;112:136-138.
202 Ogawa K, Uno T, Toita T, et al. Postoperative radiotherapy for patients with completely resected thymoma. A multi-institutional, retrospective review of 103 patients. Cancer. 2002;94:1405-1413.
203 Korst RJ, Kansler AL, Christos PJ, et al. Adjuvant radiotherapy for thymic epithelial tumors. A systematic review and meta-analysis. Ann Thorac Surg. 2009;87:1641-1647.
204 Forquer JA, Rong N, Fakiris AJ, et al. Postoperative radiotherapy after surgical resection of thymoma. Differing roles in localized and regional disease. Int J Radiat Oncol Biol Phys. 2010;76:440-445.
206 Haniuda M, Miyazawa M, Yoshida K, et al. Is postoperative radiotherapy for thymoma effective? Ann Surg. 1996;224:219-224.
209 Cowen D, Richaud P, Mornex F, et al. Thymoma. Results of a multicentric retrospective series of 149 non-metastatic irradiated patients and review of the literature. FNCLCC trialists. Federation Nationale des Centres de Lutte Contre le Cancer. Radiother Oncol. 1995;34:9-16.
212 Ciernik IF, Meier U, Lutolf UM. Prognostic factors and outcome of incompletely resected invasive thymoma following radiation therapy. J Clin Oncol. 1994;12:1484-1490.
222 Fornasiero A, Daniele O, Ghiotto C, et al. Chemotherapy for invasive thymoma. A 13-year experience. Cancer. 1991;68:30-33.
229 Loehrer PJSr, Jiroutek M, Aisner S, et al. Combined etoposide, ifosfamide, and cisplatin in the treatment of patients with advanced thymoma and thymic carcinoma. An intergroup trial. Cancer. 2001;91:2010-2015.
240 Shin DM, Walsh GL, Komaki R, et al. A multidisciplinary approach to therapy for unresectable malignant thymoma. Ann Intern Med. 1998;129:100-104.
241 Venuta F, Rendina EA, Pescarmona EO, et al. Multimodality treatment of thymoma. A prospective study. Ann Thorac Surg. 1997;64:1585-1591. discussion Ann Thorac Surg 64:1591-1592, 1997
244 Wright CD, Choi NC, Wain JC, et al. Induction chemoradiotherapy followed by resection for locally advanced Masaoka stage III and IVA thymic tumors. Ann Thorac Surg. 2008;85:385-389.
246 Mornex F, Resbeut M, Richaud P, et al. Radiotherapy and chemotherapy for invasive thymomas. A multicentric retrospective review of 90 cases. The FNCLCC trialists. Federation Nationale des Centres de Lutte Contre le Cancer. Int J Radiat Oncol Biol Phys. 1995;32:651-659.
287 Fink G, Krelbaum T, Yellin A, et al. Pulmonary carcinoid. Presentation, diagnosis, and outcome in 142 cases in Israel and review of 640 cases from the literature. Chest. 2001;119:1647-1651.
291 Travis WD, Rush W, Flieder DB, et al. Survival analysis of 200 pulmonary neuroendocrine tumors with clarification of criteria for atypical carcinoid and its separation from typical carcinoid. Am J Surg Pathol. 1998;22:934-944.
319 Thomas CFJr, Tazelaar HD, Jett JR. Typical and atypical pulmonary carcinoids. Outcome in patients presenting with regional lymph node involvement. Chest. 2001;119:1143-1150.
320 Marty-Ane CH, Costes V, Pujol JL, et al. Carcinoid tumors of the lung. Do atypical features require aggressive management? Ann Thorac Surg. 1995;59:78-83.
327 Schrevens L, Vansteenkiste J, Deneffe G, et al. Clinical-radiological presentation and outcome of surgically treated pulmonary carcinoid tumours. A long-term single institution experience. Lung Cancer. 2004;43:39-45.
342 Hurt R, Bates M. Carcinoid tumours of the bronchus. A 33 year experience. Thorax. 1984;39:617-623.
366 Vadasz P, Palffy G, Egervary M, et al. Diagnosis and treatment of bronchial carcinoid tumors. Clinical and pathological review of 120 operated patients. Eur J Cardiothorac Surg. 1993;7:8-11.
390 Curran D, Sahmoud T, Therasse P, et al. Prognostic factors in patients with pleural mesothelioma. The European Organization for Research and Treatment of Cancer experience. J Clin Oncol. 1998;16:145-152.
391 Herndon JE, Green MR, Chahinian AP, et al. Factors predictive of survival among 337 patients with mesothelioma treated between 1984 and 1994 by the Cancer and Leukemia Group B. Chest. 1998;113:723-731.
396 Flores RM, Pass HI, Seshan VE, et al. Extrapleural pneumonectomy versus pleurectomy/decortication in the surgical management of malignant pleural mesothelioma. Results in 663 patients. J Thorac Cardiovasc Surg. 2008;135:620-626. e3
398 Rusch VW, Rosenzweig K, Venkatraman E, et al. A phase II trial of surgical resection and adjuvant high-dose hemithoracic radiation for malignant pleural mesothelioma. J Thorac Cardiovasc Surg. 2001;122:788-795.
411 Vogelzang NJ, Rusthoven JJ, Symanowski J, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21:2636-2644.
429 Rice DC, Stevens CW, Correa AM, et al. Outcomes after extrapleural pneumonectomy and intensity-modulated radiation therapy for malignant pleural mesothelioma. Ann Thorac Surg. 2007;84:1685-1692. discussion Ann Thorac Surg 84:1692-1693, 2007
435 Sugarbaker DJ, Garcia JP. Multimodality therapy for malignant pleural mesothelioma. Chest. 1997;112:272S-275S.
441 Hill-Kayser CE, Avery S, Mesina CF, et al. Hemithoracic radiotherapy after extrapleural pneumonectomy for malignant pleural mesothelioma. A dosimetric comparison of two well-described techniques. J Thorac Oncol. 2009;4:1431-1437.
446 Nakamura Y, Kunitoh H, Kubota K, et al. Platinum-based chemotherapy with or without thoracic radiation therapy in patients with unresectable thymic carcinoma. Jpn J Clin Oncol. 2000;30:385-388.
447 Wirth LJ, Carter MR, Janne PA, et al. Outcome of patients with pulmonary carcinoid tumors receiving chemotherapy or chemoradiotherapy. Lung Cancer. 2004;44:213-220.
449 Mezzetti M, Raveglia F, Panigalli T, et al. Assessment of outcomes in typical and atypical carcinoids according to latest WHO classification. Ann Thorac Surg. 2003;76:1838-1842.
1 Engels EA, Pfeiffer RM. Malignant thymoma in the United States. Demographic patterns in incidence and associations with subsequent malignancies. Int J Cancer. 2003;105:546-551.
2 Modlin IM, Sandor A. An analysis of 8305 cases of carcinoid tumors. Cancer. 1997;79:813-829.
3 Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-959.
4 Peto J, Seidman H, Selikoff IJ. Mesothelioma mortality in asbestos workers. Implications for models of carcinogenesis and risk assessment. Br J Cancer. 1982;45:124-135.
5 Wilkins EWJr, Edmunds LHJr, Castleman B. Cases of thymoma at the Massachusetts General Hospital. J Thorac Cardiovasc Surg. 1966;52:322-330.
6 Kattach H, Hasan S, Clelland C, et al. Seeding of stage I thymoma into the chest wall 12 years after needle biopsy. Ann Thorac Surg. 2005;79:323-324.
7 Detterbeck FC. Does an anecdote substantiate dogma? Ann Thorac Surg. 2006;81:1182. author reply Ann Thorac Surg 81:1182-1183, 2006
8 Oertel YC. Thymoma mimicking thyroid papillary carcinoma. Another pitfall in fine-needle aspiration. Diagn Cytopathol. 1997;17:61-63.
9 Daniels CE, Lowe VJ, Aubry MC, et al. The utility of fluorodeoxyglucose positron emission tomography in the evaluation of carcinoid tumors presenting as pulmonary nodules. Chest. 2007;131:255-260.
10 Berman A, Litzky L, LiVolsi VA, et al. Adjuvant radiotherapy for completely resected stage II thymoma. Cancer. 2011. Epub ahead of print February 1
11 Diaco DS, Hajarizadeh H, Mueller CR, et al. Treatment of metastatic carcinoid tumors using multimodality therapy of octreotide acetate, intra-arterial chemotherapy, and hepatic arterial chemoembolization. Am J Surg. 1995;169:523-528.
12 von Gaudecker B. Functional histology of the human thymus. Anat Embryol (Berl). 1991;183:1-15.
13 Davis RDJr, Oldham HNJr, Sabiston DCJr. Primary cysts and neoplasms of the mediastinum. Recent changes in clinical presentation, methods of diagnosis, management, and results. Ann Thorac Surg. 1987;44:229-237.
14 Whooley BP, Urschel JD, Antkowiak JG, et al. Primary tumors of the mediastinum. J Surg Oncol. 1999;70:95-99.
15 Mullen B, Richardson JD. Primary anterior mediastinal tumors in children and adults. Ann Thorac Surg. 1986;42:338-345.
16 Ritter JH, Wick MR. Primary carcinomas of the thymus gland. Semin Diagn Pathol. 1999;16:18-31.
17 Ogawa K, Toita T, Uno T, et al. Treatment and prognosis of thymic carcinoma. A retrospective analysis of 40 cases. Cancer. 2002;94:3115-3119.
18 Kondo K, Monden Y. Therapy for thymic epithelial tumors. A clinical study of 1,320 patients from Japan. Ann Thorac Surg. 2003;76:878-884. discussion Ann Thorac Surg 76:884-885, 2003
19 Wick MR, Rosai J. Neuroendocrine neoplasms of the mediastinum. Semin Diagn Pathol. 1991;8:35-51.
20 McGuire LJ, Huang DP, Teoh R, et al. Epstein-Barr virus genome in thymoma and thymic lymphoid hyperplasia. Am J Pathol. 1988;131:385-390.
21 Teoh R, McGuire L, Wong K, et al. Increased incidence of thymoma in Chinese myasthenia gravis. Possible relationship with Epstein-Barr virus. Acta Neurol Scand. 1989;80:221-225.
22 Leyvraz S, Henle W, Chahinian AP, et al. Association of Epstein-Barr virus with thymic carcinoma. N Engl J Med. 1985;312:1296-1299.
23 Patton DF, Ribeiro RC, Jenkins JJ, et al. Thymic carcinoma with a defective Epstein-Barr virus encoding the BZLF1 trans-activator. J Infect Dis. 1994;170:7-12.
24 Dimery IW, Lee JS, Blick M, et al. Association of the Epstein-Barr virus with lymphoepithelioma of the thymus. Cancer. 1988;61:2475-2480.
25 Jensen MO, Antonenko D. Thyroid and thymic malignancy following childhood irradiation. J Surg Oncol. 1992;50:206-208.
26 Lam WW, Chan FL, Lau YL, et al. Paediatric thymoma. Unusual occurrence in two siblings. Pediatr Radiol. 1993;23:124-126.
27 Matani A, Dritsas C. Familial occurrence of thymoma. Arch Pathol. 1973;95:90-91.
28 Tavernier E, Le QH, de Botton S, et al. Secondary or concomitant neoplasms among adults diagnosed with acute lymphoblastic leukemia and treated according to the LALA-87 and LALA-94 trials. Cancer. 2007;110:2747-2755.
29 Rosai J, Levine G. Tumors of the thymus. In: Atlas of Tumor Pathology. Washington, DC: Armed Forces Institute of Pathology; 1976:55-59.
30 Teh BT, Zedenius J, Kytola S, et al. Thymic carcinoids in multiple endocrine neoplasia type 1. Ann Surg. 1998;228:99-105.
31 Kubonishi I, Takehara N, Iwata J, et al. Novel t(15;19)(q15;p13) chromosome abnormality in a thymic carcinoma. Cancer Res. 1991;51:3327-3328.
32 Lee AC, Kwong YI, Fu KH, et al. Disseminated mediastinal carcinoma with chromosomal translocation (15;19). A distinctive clinicopathologic syndrome. Cancer. 1993;72:2273-2276.
33 Kees UR, Mulcahy MT, Willoughby ML. Intrathoracic carcinoma in an 11-year-old girl showing a translocation t(15;19). Am J Pediatr Hematol Oncol. 1991;13:459-464.
34 Herens C, Radermecker M, Servais A, et al. Deletion (6)(p22p25) is a recurrent anomaly of thymoma. Report of a second case and review of the literature. Cancer Genet Cytogenet. 2003;146:66-69.
35 Inoue M, Marx A, Zettl A, et al. Chromosome 6 suffers frequent and multiple aberrations in thymoma. Am J Pathol. 2002;161:1507-1513.
36 Zettl A, Strobel P, Wagner K, et al. Recurrent genetic aberrations in thymoma and thymic carcinoma. Am J Pathol. 2000;157:257-266.
37 Abdollahi A, Roberts D, Godwin AK, et al. Identification of a zinc-finger gene at 6q25. A chromosomal region implicated in development of many solid tumors. Oncogene. 1997;14:1973-1979.
38 Miozzo M, Pierotti MA, Sozzi G, et al. Human TRK proto-oncogene maps to chromosome 1q32-q41. Oncogene. 1990;5:1411-1414.
39 Knuutila S, Bjorkqvist AM, Autio K, et al. DNA copy number amplifications in human neoplasms. Review of comparative genomic hybridization studies. Am J Pathol. 1998;152:1107-1123.
40 Knuutila S, Aalto Y, Autio K, et al. DNA copy number losses in human neoplasms. Am J Pathol. 1999;155:683-694.
41 Reference deleted in proofs.
42 Azarow KS, Pearl RH, Zurcher R, et al. Primary mediastinal masses. A comparison of adult and pediatric populations. J Thorac Cardiovasc Surg. 1993;106:67-72.
43 Cohen AJ, Thompson L, Edwards FH, et al. Primary cysts and tumors of the mediastinum. Ann Thorac Surg. 1991;51:378-384. discussion Ann Thorac Surg 51:385-386, 1991
44 Strollo DC, Rosado de Christenson ML, Jett JR. Primary mediastinal tumors. Part 1. Tumors of the anterior mediastinum. Chest. 1997;112:511-522.
45 Lewis JE, Wick MR, Scheithauer BW, et al. Thymoma. A clinicopathologic review. Cancer. 1987;60:2727-2743.
46 Batata MA, Martini N, Huvos AG, et al. Thymomas. Clinicopathologic features, therapy, and prognosis. Cancer. 1974;34:389-396.
47 Bernatz PE, Khonsari S, Harrison EGJr, et al. Thymoma. Factors influencing prognosis. Surg Clin North Am. 1973;53:885-892.
48 LeGolvan DP, Abell MR. Thymomas. Cancer. 1977;39:2142-2157.
49 Salyer WR, Eggleston JC. Thymoma. A clinical and pathological study of 65 cases. Cancer. 1976;37:229-249.
50 Kundel Y, Yellin A, Popovtzer A, et al. Adjuvant radiotherapy for thymic epithelial tumor. Treatment results and prognostic factors. Am J Clin Oncol. 2007;30:389-394.
51 Suster S, Rosai J. Thymic carcinoma. A clinicopathologic study of 60 cases. Cancer. 1991;67:1025-1032.
52 Hsu CP, Chen CY, Chen CL, et al. Thymic carcinoma. Ten years’ experience in twenty patients. J Thorac Cardiovasc Surg. 1994;107:615-620.
53 Wick MR, Scheithauer BW, Weiland LH, et al. Primary thymic carcinomas. Am J Surg Pathol. 1982;6:613-630.
54 Kuo TT, Chang JP, Lin FJ, et al. Thymic carcinomas. Histopathological varieties and immunohistochemical study. Am J Surg Pathol. 1990;14:24-34.
55 Economopoulos GC, Lewis JWJr, Lee MW, et al. Carcinoid tumors of the thymus. Ann Thorac Surg. 1990;50:58-61.
56 Moran CA, Suster S. Spindle-cell neuroendocrine carcinomas of the thymus (spindle-cell thymic carcinoid). A clinicopathologic and immunohistochemical study of seven cases. Mod Pathol. 1999;12:587-591.
57 Lara PNJr. Malignant thymoma. Current status and future directions. Cancer Treat Rev. 2000;26:127-131.
58 Detterbeck FC, Parsons AM. Thymic tumors. Ann Thorac Surg. 2004;77:1860-1869.
59 Morgenthaler TI, Brown LR, Colby TV, et al. Thymoma. Mayo Clin Proc. 1993;68:1110-1123.
60 Maggi G, Giaccone G, Donadio M, et al. Thymomas. A review of 169 cases, with particular reference to results of surgical treatment. Cancer. 1986;58:765-776.
61 Drachman DB. Myasthenia gravis. N Engl J Med. 1994;330:1797-1810.
62 Willcox N. Myasthenia gravis. Curr Opin Immunol. 1993;5:910-917.
63 Souadjian JV, Enriquez P, Silverstein MN, et al. The spectrum of diseases associated with thymoma. Coincidence or syndrome? Arch Intern Med. 1974;134:374-379.
64 Blumberg D, Burt ME, Bains MS, et al. Thymic carcinoma. Current staging does not predict prognosis. J Thorac Cardiovasc Surg. 1998;115:303-308. discussion Ann Thorac Surg 115:308-309, 1998
65 Curran WJJr, Kornstein MJ, Brooks JJ, et al. Invasive thymoma. The role of mediastinal irradiation following complete or incomplete surgical resection. J Clin Oncol. 1988;6:1722-1727.
66 Cohen DJ, Ronnigen LD, Graeber GM, et al. Management of patients with malignant thymoma. J Thorac Cardiovasc Surg. 1984;87:301-307.
67 Ruffini E, Mancuso M, Oliaro A, et al. Recurrence of thymoma. Analysis of clinicopathologic features, treatment, and outcome. J Thorac Cardiovasc Surg. 1997;113:55-63.
68 Regnard JF, Magdeleinat P, Dromer C, et al. Prognostic factors and long-term results after thymoma resection. A series of 307 patients. J Thorac Cardiovasc Surg. 1996;112:376-384.
69 Strobel P, Bauer A, Puppe B, et al. Tumor recurrence and survival in patients treated for thymomas and thymic squamous cell carcinomas. A retrospective analysis. J Clin Oncol. 2004;22:1501-1509.
70 Monden Y, Nakahara K, Iioka S, et al. Recurrence of thymoma. Clinicopathological features, therapy, and prognosis. Ann Thorac Surg. 1985;39:165-169.
71 Sellors TH, Thackray AC, Thomson AD. Tumours of the thymus. A review of 88 operation cases. Thorax. 1967;22:193-220.
72 Weissberg D, Goldberg M, Pearson FG. Thymoma. Ann Thorac Surg. 1973;16:141-147.
73 Hoffacker V, Schultz A, Tiesinga JJ, et al. Thymomas alter the T-cell subset composition in the blood. A potential mechanism for thymoma-associated autoimmune disease. Blood. 2000;96:3872-3879.
74 Buckley C, Douek D, Newsom-Davis J, et al. Mature, long-lived CD4+ and CD8+ T cells are generated by the thymoma in myasthenia gravis. Ann Neurol. 2001;50:64-72.
75 Strobel P, Helmreich M, Menioudakis G, et al. Paraneoplastic myasthenia gravis correlates with generation of mature naive CD4(+) T cells in thymomas. Blood. 2002;100:159-166.
76 Agarwala SS. Paraneoplastic syndromes. Med Clin North Am. 1996;80:173-184.
77 Mygland A, Vincent A, Newsom-Davis J, et al. Autoantibodies in thymoma-associated myasthenia gravis with myositis or neuromyotonia. Arch Neurol. 2000;57:527-531.
78 Tarr PE, Sneller MC, Mechanic LJ, et al. Infections in patients with immunodeficiency with thymoma (Good syndrome). Report of 5 cases and review of the literature. Medicine (Baltimore). 2001;80:123-133.
79 Montella L, Masci AM, Merkabaoui G, et al. B-cell lymphopenia and hypogammaglobulinemia in thymoma patients. Ann Hematol. 2003;82:343-347.
80 Tateyama H, Eimoto T, Tada T, et al. p53 protein expression and p53 gene mutation in thymic epithelial tumors. An immunohistochemical and DNA sequencing study. Am J Clin Pathol. 1995;104:375-381.
81 Chen FF, Yan JJ, Jin YT, et al. Detection of bcl-2 and p53 in thymoma. Expression of bcl-2 as a reliable marker of tumor aggressiveness. Hum Pathol. 1996;27:1089-1092.
82 Tateyama H, Mizuno T, Tada T, et al. Thymic epithelial tumours. Evaluation of malignant grade by quantification of proliferating cell nuclear antigen and nucleolar organizer regions. Virchows Arch A Pathol Anat Histopathol. 1993;422:265-269.
83 Pich A, Chiarle R, Chiusa L, et al. Long-term survival of thymoma patients by histologic pattern and proliferative activity. Am J Surg Pathol. 1995;19:918-926.
84 Yang WI, Efird JT, Quintanilla-Martinez L, et al. Cell kinetic study of thymic epithelial tumors using PCNA (PC10) and Ki-67 (MIB-1) antibodies. Hum Pathol. 1996;27:70-76.
85 Takahashi E, Tateyama H, Akatsu H, et al. Expression of matrix metalloproteinases 2 and 7 in tumor cells correlates with the World Health Organization classification subtype and clinical stage of thymic epithelial tumors. Hum Pathol. 2003;34:1253-1258.
86 Marx A, Schomig D, Schultz A, et al. Distribution of molecules mediating thymocyte-stroma-interactions in human thymus, thymitis and thymic epithelial tumors. Thymus. 1994;23:83-93.
87 Ruco LP, Paradiso P, Pittiglio M, et al. Tissue distribution of very late activation antigens-1/6 and very late activation antigen ligands in the normal thymus and in thymoma. Am J Pathol. 1993;142:765-772.
88 Schultz A, Greiner A, Nenninger R, et al. [CD40 as a mediator of proliferation in normal ans neoplastic thymic epithelium]. Verh Dtsch Ges Pathol. 1996;80:250-255.
89 Gilhus NE, Willcox N, Harcourt G, et al. Antigen presentation by thymoma epithelial cells from myasthenia gravis patients to potentially pathogenic T cells. J Neuroimmunol. 1995;56:65-76.
90 Hayashi Y, Ishii N, Obayashi C, et al. Thymoma. Tumour type related to expression of epidermal growth factor (EGF), EGF-receptor, p53, v-erb B and ras p21. Virchows Arch. 1995;426:43-50.
91 Khoury T, Arshad A, Bogner P, et al. Apoptosis-related (survivin, Bcl-2), tumor suppressor gene (p53), proliferation (Ki-67), and non-receptor tyrosine kinase (Src) markers expression and correlation with clinicopathologic variables in 60 thymic neoplasms. Chest. 2009;136:220-228.
92 Marx A, Hohenberger P, Hoffmann H, et al. The autoimmune regulator AIRE in thymoma biology. Autoimmunity and beyond. J Thoracic Oncol. 2010;5:S266-S272.
93 Strobel P, Chuang WY, Chuvpilo S, et al. Common cellular and diverse genetic basis of thymoma-associated myasthenia gravis. Role of MHC class II and AIRE genes and genetic polymorphisms. Ann N Y Acad Sci. 2008;1132:143-156.
94 Negron-Soto JM, Cascade PN. Squamous cell carcinoma of the thymus with paraneoplastic hypercalcemia. Clin Imaging. 1995;19:122-124.
95 Suzuki K, Tanaka H, Shibusa T, et al. Parathyroid-hormone-related-protein-producing thymic carcinoma presenting as a giant extrathoracic mass. Respiration. 1998;65:83-85.
96 Jung KJ, Lee KS, Han J, et al. Malignant thymic epithelial tumors. CT-pathologic correlation. AJR Am J Roentgenol. 2001;176:433-439.
97 Zhang Z, Cui Y, Li B, et al. Thymic carcinoma (report of 14 cases). Chin Med Sci J. 1997;12:252-255.
98 Shimosato Y, Kameya T, Nagai K, et al. Squamous cell carcinoma of the thymus. An analysis of eight cases. Am J Surg Pathol. 1977;1:109-121.
99 Henley JD, Koukoulis GK, Loehrer PJSr. Epidermal growth factor receptor expression in invasive thymoma. J Cancer Res Clin Oncol. 2002;128:167-170.
100 Gilhus NE, Jones M, Turley H, et al. Oncogene proteins and proliferation antigens in thymomas. Increased expression of epidermal growth factor receptor and Ki67 antigen. J Clin Pathol. 1995;48:447-455.
101 Yoh K, Nishiwaki Y, Ishii G, et al. Mutational status of EGFR and KIT in thymoma and thymic carcinoma. Lung Cancer. 2008;62:316-320.
102 Tomita M, Matsuzaki Y, Edagawa M, et al. Correlation between tumor angiogenesis and invasiveness in thymic epithelial tumors. J Thorac Cardiovasc Surg. 2002;124:493-498.
103 Sasaki H, Yukiue H, Kobayashi Y, et al. Elevated serum vascular endothelial growth factor and basic fibroblast growth factor levels in patients with thymic epithelial neoplasms. Surg Today. 2001;31:1038-1040.
104 Tomita M, Matsuzaki Y, Edagawa M, et al. Clinical and immunohistochemical study of eight cases with thymic carcinoma. BMC Surg. 2002;2:3.
105 Fukai I, Masaoka A, Hashimoto T, et al. The distribution of epithelial membrane antigen in thymic epithelial neoplasms. Cancer. 1992;70:2077-2081.
106 Fukai I, Masaoka A, Hashimoto T, et al. Cytokeratins in normal thymus and thymic epithelial tumors. Cancer. 1993;71:99-105.
107 Oyama T, Osaki T, Mitsudomi T, et al. p53 alteration, proliferating cell nuclear antigen, and nucleolar organizer regions in thymic epithelial tumors. Int J Mol Med. 1998;1:823-826.
108 Kuo TT, Chan JK. Thymic carcinoma arising in thymoma is associated with alterations in immunohistochemical profile. Am J Surg Pathol. 1998;22:1474-1481.
109 Hino N, Kondo K, Miyoshi T, et al. High frequency of p53 protein expression in thymic carcinoma but not in thymoma. Br J Cancer. 1997;76:1361-1366.
110 Girard N, Shen R, Guo T, et al. Comprehensive genomic analysis reveals clinically relevant molecular distinctions between thymic carcinomas and thymomas. Clin Cancer Res. 2009;15:6790-6799.
111 Hishima T, Fukayama M, Hayashi Y, et al. CD70 expression in thymic carcinoma. Am J Surg Pathol. 2000;24:742-746.
112 Hishima T, Fukayama M, Fujisawa M, et al. CD5 expression in thymic carcinoma. Am J Pathol. 1994;145:268-275.
113 Tateyama H, Eimoto T, Tada T, et al. Immunoreactivity of a new CD5 antibody with normal epithelium and malignant tumors including thymic carcinoma. Am J Clin Pathol. 1999;111:235-240.
114 Dorfman DM, Shahsafaei A, Chan JK. Thymic carcinomas, but not thymomas and carcinomas of other sites, show CD5 immunoreactivity. Am J Surg Pathol. 1997;21:936-940.
115 Zucali PA, Petrini I, Lorenzi E, et al. Insulin-like growth factor-1 receptor and phosphorylated AKT-serine 473 expression in 132 resected thymomas and thymic carcinomas. Cancer. 2010;116:4686-4695.
116 Fukai I, Masaoka A, Hashimoto T, et al. Differential diagnosis of thymic carcinoma and lung carcinoma with the use of antibodies to cytokeratins. J Thorac Cardiovasc Surg. 1995;110:1670-1675.
117 Moran CA, Suster S. Neuroendocrine carcinomas (carcinoid tumor) of the thymus. A clinicopathologic analysis of 80 cases. Am J Clin Pathol. 2000;114:100-110.
118 Fukai I, Masaoka A, Fujii Y, et al. Thymic neuroendocrine tumor (thymic carcinoid). A clinicopathologic study in 15 patients. Ann Thorac Surg. 1999;67:208-211.
119 Rosai J, Higa E, Davie J. Mediastinal endocrine neoplasm in patients with multiple endocrine adenomatosis. A previously unrecognized association. Cancer. 1972;29:1075-1083.
120 Quintanilla-Martinez L, Wilkins EWJr, Choi N, et al. Thymoma. Histologic subclassification is an independent prognostic factor. Cancer. 1994;74:606-617.
121 Eng TY, Fuller CD, Jagirdar J, et al. Thymic carcinoma. State of the art review. Int J Radiat Oncol Biol Phys. 2004;59:654-664.
122 Bergh NP, Gatzinsky P, Larsson S, et al. Tumors of the thymus and thymic region. I. Clinicopathological studies on thymomas. Ann Thorac Surg. 1978;25:91-98.
123 Fujimura S, Kondo T, Handa M, et al. Results of surgical treatment for thymoma based on 66 patients. J Thorac Cardiovasc Surg. 1987;93:708-714.
124 Maggi G, Casadio C, Cavallo A, et al. Thymoma. Results of 241 operated cases. Ann Thorac Surg. 1991;51:152-156.
125 Nakahara K, Ohno K, Hashimoto J, et al. Thymoma. Results with complete resection and adjuvant postoperative irradiation in 141 consecutive patients. J Thorac Cardiovasc Surg. 1988;95:1041-1047.
126 Wilkins EWJr, Castleman B. Thymoma. A continuing survey at the Massachusetts General Hospital. Ann Thorac Surg. 1979;28:252-256.
127 Akaogi E, Ohara K, Mitsui K, et al. Preoperative radiotherapy and surgery for advanced thymoma with invasion to the great vessels. J Surg Oncol. 1996;63:17-22.
128 Lee JD, Choe KO, Kim SJ, et al. CT findings in primary thymic carcinoma. J Comput Assist Tomogr. 1991;15:429-433.
129 Asbun HJ, Calabria RP, Calmes S, et al. Thymic carcinoid. Am Surg. 1991;57:442-445.
130 Legg MA, Brady WJ. Pathology and clinical behavior of thymomas. A survey of 51 cases. Cancer. 1965;18:1131-1144.
131 Patterson GA. Thymomas. Semin Thorac Cardiovasc Surg. 1992;4:39-44.
132 Konstantinov IE, Saxena P, Koniuszko M, et al. Superior vena cava obstruction by tumour thrombus in invasive thymoma. Diagnosis and surgical management. Heart Lung Circ. 2007;16:462-464.
133 Marchevsky A, Kaneko M. Surgical Pathology of the Mediastinum. New York: Raven Press; 1984.
134 Chahinian AP, Bhardwaj S, Meyer RJ, et al. Treatment of invasive or metastatic thymoma. Report of eleven cases. Cancer. 1981;47:1752-1761.
135 LeBlanc J, Wood D. Diagnosis of mediastinal tumors. In: Wood DE, Thomas CRJr, editors. Mediastinal Tumors: Update 1995. Medical Radiology–Diagnostic Imaging and Radiation Oncology. Heidelberg, Germany: Springer-Verlag; 1995:1-10.
136 Wilkins EWJr, Grillo HC, Scannell JG, et al. J. Maxwell Chamberlain Memorial Paper. Role of staging in prognosis and management of thymoma. Ann Thorac Surg. 1991;51:888-892.
137 Vincent A. Autoimmunity to acetylcholine receptors in myasthenia gravis. Biochem Soc Trans. 1991;19:180-183.
138 Drachman DB. Myasthenia gravis (first of two parts). N Engl J Med. 1978;298:136-142.
139 Vincent A, Palace J, Hilton-Jones D. Myasthenia gravis. Lancet. 2001;357:2122-2128.
140 Urgesi A, Monetti U, Rossi G, et al. Role of radiation therapy in locally advanced thymoma. Radiother Oncol. 1990;19:273-280.
141 Masaoka A, Monden Y, Nakahara K, et al. Follow-up study of thymomas with special reference to their clinical stages. Cancer. 1981;48:2485-2492.
142 Rea F, Marulli G, Girardi R, et al. Long-term survival and prognostic factors in thymic epithelial tumours. Eur J Cardiothorac Surg. 2004;26:412-418.
143 Blumberg D, Port JL, Weksler B, et al. Thymoma. A multivariate analysis of factors predicting survival. Ann Thorac Surg. 1995;60:908-913. discussion Ann Thorac Surg 60:914, 1995
144 Pescarmona E, Rendina EA, Venuta F, et al. Analysis of prognostic factors and clinicopathological staging of thymoma. Ann Thorac Surg. 1990;50:534-538.
145 Pollack A, Komaki R, Cox JD, et al. Thymoma. Treatment and prognosis. Int J Radiat Oncol Biol Phys. 1992;23:1037-1043.
146 Lardinois D, Rechsteiner R, Lang RH, et al. Prognostic relevance of Masaoka and Muller-Hermelink classification in patients with thymic tumors. Ann Thorac Surg. 2000;69:1550-1555.
147 McCart JA, Gaspar L, Inculet R, et al. Predictors of survival following surgical resection of thymoma. J Surg Oncol. 1993;54:233-238.
148 Murakawa T, Nakajima J, Kohno T, et al. Results from surgical treatment for thymoma. 43 years of experience. Jpn J Thorac Cardiovasc Surg. 2000;48:89-95.
149 Park HS, Shin DM, Lee JS, et al. Thymoma. A retrospective study of 87 cases. Cancer. 1994;73:2491-2498.
150 Wilkins KB, Sheikh E, Green R, et al. Clinical and pathologic predictors of survival in patients with thymoma. Ann Surg. 1999;230:562-572. discussion Ann Surg 230:572-574, 1999
151 Venuta F, Rendina EA, De Giacomo T, et al. Thymectomy for myasthenia gravis. A 27-year experience. Eur J Cardiothorac Surg. 1999;15:621-624. discussion Eur J Cardiothorac Surg 15:624-625, 1999
152 Kohman LJ. Controversies in the management of malignant thymoma. Chest. 1997;112:296S-300S.
153 Verstandig AG, Epstein DM, Miller WTJr, et al. Thymoma—report of 71 cases and a review. Crit Rev Diagn Imaging. 1992;33:201-230.
154 El-Medany Y, Hajjar W, Essa M, et al. Predictors of outcome for myasthenia gravis after thymectomy. Asian Cardiovasc Thorac Ann. 2003;11:323-327.
155 Goldstein SD, Yang SC. Assessment of robotic thymectomy using the Myasthenia Gravis Foundation of America Guidelines. Ann Thorac Surg. 2010;89:1080-1085. discussion Ann Thorac Surg 89:1085-1086, 2010
156 Bulkley GB, Bass KN, Stephenson GR, et al. Extended cervicomediastinal thymectomy in the integrated management of myasthenia gravis. Ann Surg. 1997;226:324-334. discussion Ann Surg 226:334-335, 1997
157 Murakawa T, Nakajima J, Sato H, et al. Thymoma associated with pure red-cell aplasia. Clinical features and prognosis. Asian Cardiovasc Thorac Ann. 2002;10:150-154.
158 Rios A, Torres J, Galindo PJ, et al. Prognostic factors in thymic epithelial neoplasms. Eur J Cardiothorac Surg. 2002;21:307-313.
159 Chen G, Marx A, Wen-Hu C, et al. New WHO histologic classification predicts prognosis of thymic epithelial tumors. A clinicopathologic study of 200 thymoma cases from China. Cancer. 2002;95:420-429.
160 Marino M, Muller-Hermelink HK. Thymoma and thymic carcinoma. Relation of thymoma epithelial cells to the cortical and medullary differentiation of thymus. Virchows Arch A Pathol Anat Histopathol. 1985;407:119-149.
161 Detterbeck FC. Clinical value of the WHO classification system of thymoma. Ann Thorac Surg. 2006;81:2328-2334.
162 Lucchi M, Basolo F, Ribechini A, et al. Thymomas. Clinical-pathological correlations. J Cardiovasc Surg (Torino). 2006;47:89-93.
163 Wright CD, Wain JC, Wong DR, et al. Predictors of recurrence in thymic tumors. Importance of invasion, World Health Organization histology, and size. J Thorac Cardiovasc Surg. 2005;130:1413-1421.
164 Utsumi T, Shiono H, Kadota Y, et al. Postoperative radiation therapy after complete resection of thymoma has little impact on survival. Cancer. 2009;115:5413-5420.
165 Rieker RJ, Hoegel J, Morresi-Hauf A, et al. Histologic classification of thymic epithelial tumors. Comparison of established classification schemes. Int J Cancer. 2002;98:900-906.
166 Kim HK, Choi YS, Kim J, et al. Type B thymoma. Is prognosis predicted only by World Health Organization classification? J Thorac Cardiovasc Surg. 2010;139:1431-1435. e1
167 Ricci C, Rendina EA, Pescarmona EO, et al. Correlations between histological type, clinical behaviour, and prognosis in thymoma. Thorax. 1989;44:455-460.
168 Ho FC, Fu KH, Lam SY, et al. Evaluation of a histogenetic classification for thymic epithelial tumours. Histopathology. 1994;25:21-29.
169 Tan PH, Sng IT. Thymoma—a study of 60 cases in Singapore. Histopathology. 1995;26:509-518.
170 Verley JM, Hollmann KH. Thymoma. A comparative study of clinical stages, histologic features, and survival in 200 cases. Cancer. 1985;55:1074-1086.
171 Hoffman OA, Gillespie DJ, Aughenbaugh GL, et al. Primary mediastinal neoplasms (other than thymoma). Mayo Clin Proc. 1993;68:880-891.
172 Shields TW. Primary tumors and cysts of the mediastinum. In: Shields TW, editor. General Thoracic Surgery. Philadelphia: Lea and Febiger; 1983:927.
173 Dahlgren S, Sandstedt B, Sundstrom C. Fine needle aspiration cytology of thymic tumors. Acta Cytol. 1983;27:1-6.
174 Baron RL, Lee JK, Sagel SS, et al. Computed tomography of the abnormal thymus. Radiology. 1982;142:127-134.
175 Levitt RG, Husband JE, Glazer HS. CT of primary germ-cell tumors of the mediastinum. AJR Am J Roentgenol. 1984;142:73-78.
176 Nasseri F, Eftekhari F. Clinical and radiologic review of the normal and abnormal thymus: pearls and pitfalls. Radiographics. 2010;30:413-428.
177 Quagliano PV. Thymic carcinoma. Case reports and review. J Thorac Imaging. 1996;11:66-74.
178 Casamassima F, Villari N, Fargnoli R, et al. Magnetic resonance imaging and high-resolution computed tomography in tumors of the lung and the mediastinum. Radiother Oncol. 1988;11:21-29.
179 Kubota K, Yamada S, Kondo T, et al. PET imaging of primary mediastinal tumours. Br J Cancer. 1996;73:882-886.
180 Liu RS, Yeh SH, Huang MH, et al. Use of fluorine-18 fluorodeoxyglucose positron emission tomography in the detection of thymoma. A preliminary report. Eur J Nucl Med. 1995;22:1402-1407.
181 Sasaki M, Kuwabara Y, Ichiya Y, et al. Differential diagnosis of thymic tumors using a combination of 11C-methionine PET and FDG PET. J Nucl Med. 1999;40:1595-1601.
182 Shabb NS, Fahl M, Shabb B, et al. Fine-needle aspiration of the mediastinum. A clinical, radiologic, cytologic, and histologic study of 42 cases. Diagn Cytopathol. 1998;19:428-436.
183 Morrissey B, Adams H, Gibbs AR, et al. Percutaneous needle biopsy of the mediastinum: review of 94 procedures. Thorax. 1993;48:632-637.
184 Cirino LM, Milanez de Campos JR, Fernandez A, et al. Diagnosis and treatment of mediastinal tumors by thoracoscopy. Chest. 2000;117:1787-1792.
185 Demmy TL, Krasna MJ, Detterbeck FC, et al. Multicenter VATS experience with mediastinal tumors. Ann Thorac Surg. 1998;66:187-192.
186 Moran CA, Travis WD, Rosado-de-Christenson M, et al. Thymomas presenting as pleural tumors. Report of eight cases. Am J Surg Pathol. 1992;16:138-144.
187 Shih DF, Wang JS, Tseng HH, et al. Primary pleural thymoma. Arch Pathol Lab Med. 1997;121:79-82.
188 Suster S, Moran CA. Thymic carcinoma. Spectrum of differentiation and histologic types. Pathology. 1998;30:111-122.
189 Tsuchiya R, Koga K, Matsuno Y, et al. Thymic carcinoma. Proposal for pathological TNM and staging. Pathol Int. 1994;44:505-512.
190 Singhal S, Shrager JB, Rosenthal DI, et al. Comparison of stages I-II thymoma treated by complete resection with or without adjuvant radiation. Ann Thorac Surg. 2003;76:1635-1641. discussion Ann Thorac Surg 76:1641-1642, 2003
191 Mangi AA, Wright CD, Allan JS, et al. Adjuvant radiation therapy for stage II thymoma. Ann Thorac Surg. 2002;74:1033-1037.
192 Yeh JH, Chen WH, Huang KM, et al. Prethymectomy plasmapheresis in myasthenia gravis. J Clin Apher. 2005;20:217-221.
193 Reddy RH, Shah R, Kumar B, et al. Recurrence of stage I thymoma in sternum, 13 years after “complete” excision. Eur J Cardiothorac Surg. 2003;23:134-135.
194 Yagi K, Hirata T, Fukuse T, et al. Surgical treatment for invasive thymoma, especially when the superior vena cava is invaded. Ann Thorac Surg. 1996;61:521-524.
195 Mussi A, Lucchi M, Murri L, et al. Extended thymectomy in myasthenia gravis. A team-work of neurologist, thoracic surgeon and anesthetist may improve the outcome. Eur J Cardiothorac Surg. 2001;19:570-575.
196 Okereke IC, Kesler KA, Morad MH, et al. Prognostic indicators after surgery for thymoma. Ann Thorac Surg. 2010;89:1071-1077. discussion Ann Thorac Surg 89:1077-1079, 2010
197 Ishikawa Y, Matsuguma H, Nakahara R, et al. Multimodality therapy for patients with invasive thymoma disseminated into the pleural cavity. The potential role of extrapleural pneumonectomy. Ann Thorac Surg. 2009;88:952-957.
198 Huang J, Rizk NP, Travis WD, et al. Feasibility of multimodality therapy including extended resections in stage IVA thymoma. J Thorac Cardiovasc Surg. 2007;134:1477-1483. discussion J Thorac Cardiovasc Surg 134:1483-1484, 2007
199 Ohara K, Tatsuzaki H, Fuji H, et al. Radioresponse of thymomas verified with histologic response. Acta Oncol. 1998;37:471-474.
200 Fechner RE. Recurrence of noninvasive thymomas. Report of four cases and review of literature. Cancer. 1969;23:1423-1427.
201 Zhang H, Lu N, Wang M, et al. Postoperative radiotherapy for stage I thymoma. A prospective randomized trial in 29 cases. Chin Med J (Engl). 1999;112:136-138.
202 Ogawa K, Uno T, Toita T, et al. Postoperative radiotherapy for patients with completely resected thymoma. A multi-institutional, retrospective review of 103 patients. Cancer. 2002;94:1405-1413.
203 Korst RJ, Kansler AL, Christos PJ, et al. Adjuvant radiotherapy for thymic epithelial tumors. A systematic review and meta-analysis. Ann Thorac Surg. 2009;87:1641-1647.
204 Forquer JA, Rong N, Fakiris AJ, et al. Postoperative radiotherapy after surgical resection of thymoma. Differing roles in localized and regional disease. Int J Radiat Oncol Biol Phys. 2010;76:440-445.
205 Haniuda M, Morimoto M, Nishimura H, et al. Adjuvant radiotherapy after complete resection of thymoma. Ann Thorac Surg. 1992;54:311-315.
206 Haniuda M, Miyazawa M, Yoshida K, et al. Is postoperative radiotherapy for thymoma effective? Ann Surg. 1996;224:219-224.
207 Gripp S, Hilgers K, Wurm R, et al. Thymoma. Prognostic factors and treatment outcomes. Cancer. 1998;83:1495-1503.
208 Ohara K, Okumura T, Sugahara S, et al. The role of preoperative radiotherapy for invasive thymoma. Acta Oncol. 1990;29:425-429.
209 Cowen D, Richaud P, Mornex F, et al. Thymoma. Results of a multicentric retrospective series of 149 non-metastatic irradiated patients and review of the literature. FNCLCC trialists. Federation Nationale des Centres de Lutte Contre le Cancer. Radiother Oncol. 1995;34:9-16.
210 Onuki T, Ishikawa S, Yamamoto T, et al. Pathologic radioresponse of preoperatively irradiated invasive thymomas. J Thorac Oncol. 2008;3:270-276.
211 Arakawa A, Yasunaga T, Saitoh Y, et al. Radiation therapy of invasive thymoma. Int J Radiat Oncol Biol Phys. 1990;18:529-534.
212 Ciernik IF, Meier U, Lutolf UM. Prognostic factors and outcome of incompletely resected invasive thymoma following radiation therapy. J Clin Oncol. 1994;12:1484-1490.
213 Ichinose Y, Ohta M, Yano T, et al. Treatment of invasive thymoma with pleural dissemination. J Surg Oncol. 1993;54:180-183.
214 Jackson MA, Ball DL. Post-operative radiotherapy in invasive thymoma. Radiother Oncol. 1991;21:77-82.
215 Urgesi A, Monetti U, Rossi G, et al. Aggressive treatment of intrathoracic recurrences of thymoma. Radiother Oncol. 1992;24:221-225.
216 Boston B. Chemotherapy of invasive thymoma. Cancer. 1976;38:49-52.
217 Campbell MG, Pollard R, Al-Sarraf M. A complete response in metastatic malignant thymoma to cis-platinum, doxorubicin and cyclophosphamide. A case report. Cancer. 1981;48:1315-1317.
218 Chahinian AP, Holland JF, Bhardwaj S. Chemotherapy for malignant thymoma. Ann Intern Med. 1983;99:736.
219 Daugaard G, Hansen HH, Rorth M. Combination chemotherapy for malignant thymoma. Ann Intern Med. 1983;99:189-190.
220 Dy C, Calvo FA, Mindan JP, et al. Undifferentiated epithelial-rich invasive malignant thymoma. Complete response to cisplatin, vinblastine, and bleomycin therapy. J Clin Oncol. 1988;6:536-542.
221 Evans WK, Thompson DM, Simpson WJ, et al. Combination chemotherapy in invasive thymoma role of COPP. Cancer. 1980;46:1523-1527.
222 Fornasiero A, Daniele O, Ghiotto C, et al. Chemotherapy for invasive thymoma. A 13-year experience. Cancer. 1991;68:30-33.
223 Fornasiero A, Daniele O, Sperandio P, et al. Chemotherapy of invasive or metastatic thymoma. Report of 11 cases. Cancer Treat Rep. 1984;68:1205-1210.
224 Giaccone G, Ardizzoni A, Kirkpatrick A, et al. Cisplatin and etoposide combination chemotherapy for locally advanced or metastatic thymoma. A phase II study of the European Organization for Research and Treatment of Cancer Lung Cancer Cooperative Group. J Clin Oncol. 1996;14:814-820.
225 Green JD, Forman WH. Response of thymoma to steroids. Chest. 1974;65:114-116.
226 Highley MS, Underhill CR, Parnis FX, et al. Treatment of invasive thymoma with single-agent ifosfamide. J Clin Oncol. 1999;17:2737-2744.
227 Hu E, Levine J. Chemotherapy of malignant thymoma. Case report and review of the literature. Cancer. 1986;57:1101-1104.
228 Jaffrey IS, Denefrio JM, Chahinian P. Response to maytansine in a patient with malignant thymoma. Cancer Treat Rep. 1980;64:193-194.
229 Loehrer PJSr, Jiroutek M, Aisner S, et al. Combined etoposide, ifosfamide, and cisplatin in the treatment of patients with advanced thymoma and thymic carcinoma. An intergroup trial. Cancer. 2001;91:2010-2015.
230 Macchiarini P, Chella A, Ducci F, et al. Neoadjuvant chemotherapy, surgery, and postoperative radiation therapy for invasive thymoma. Cancer. 1991;68:706-713.
231 Oshita F, Kasai T, Kurata T, et al. Intensive chemotherapy with cisplatin, doxorubicin, cyclophosphamide, etoposide and granulocyte colony-stimulating factor for advanced thymoma or thymic cancer. Preliminary results. Jpn J Clin Oncol. 1995;25:208-212.
232 Sickles EA, Belliveau RE, Wiernik PH. Primary mediastinal choriocarcinoma in the male. Cancer. 1974;33:1196-1203.
233 Thomas CR, Wright CD, Loehrer PJ. Thymoma. State of the art. J Clin Oncol. 1999;17:2280-2289.
234 Hejna M, Haberl I, Raderer M. Nonsurgical management of malignant thymoma. Cancer. 1999;85:1871-1884.
235 Loehrer PJSr, Chen M, Kim K, et al. Cisplatin, doxorubicin, and cyclophosphamide plus thoracic radiation therapy for limited-stage unresectable thymoma. An intergroup trial. J Clin Oncol. 1997;15:3093-3099.
236 Loehrer PJSr, Kim K, Aisner SC, et al. Cisplatin plus doxorubicin plus cyclophosphamide in metastatic or recurrent thymoma. Final results of an intergroup trial. The Eastern Cooperative Oncology Group, Southwest Oncology Group, and Southeastern Cancer Study Group. J Clin Oncol. 1994;12:1164-1168.
237 Kim ES, Putnam JB, Komaki R, et al. Phase II study of a multidisciplinary approach with induction chemotherapy, followed by surgical resection, radiation therapy, and consolidation chemotherapy for unresectable malignant thymomas. Final report. Lung Cancer. 2004;44:369-379.
238 Berruti A, Borasio P, Gerbino A, et al. Primary chemotherapy with adriamycin, cisplatin, vincristine and cyclophosphamide in locally advanced thymomas. A single institution experience. Br J Cancer. 1999;81:841-845.
239 Liu JM, Wang LS, Huang MH, et al. Topoisomerase 2alpha plays a pivotal role in the tumor biology of stage IV thymic neoplasia. Cancer. 2007;109:502-509.
240 Shin DM, Walsh GL, Komaki R, et al. A multidisciplinary approach to therapy for unresectable malignant thymoma. Ann Intern Med. 1998;129:100-104.
241 Venuta F, Rendina EA, Pescarmona EO, et al. Multimodality treatment of thymoma. A prospective study. Ann Thorac Surg. 1997;64:1585-1591. discussion Ann Thorac Surg 64:1591-1592, 1997
242 Goldel N, Boning L, Fredrik A, et al. Chemotherapy of invasive thymoma. A retrospective study of 22 cases. Cancer. 1989;63:1493-1500.
243 Kunitoh H, Tamura T, Shibata T, et al. A phase II trial of dose-dense chemotherapy, followed by surgical resection and/or thoracic radiotherapy, in locally advanced thymoma. Report of a Japan Clinical Oncology Group trial (JCOG 9606). Br J Cancer. 2010;103:6-11.
244 Wright CD, Choi NC, Wain JC, et al. Induction chemoradiotherapy followed by resection for locally advanced Masaoka stage III and IVA thymic tumors. Ann Thorac Surg. 2008;85:385-389.
245 Krueger JB, Sagerman RH, King GA. Stage III thymoma. Results of postoperative radiation therapy. Radiology. 1988;168:855-858.
246 Mornex F, Resbeut M, Richaud P, et al. Radiotherapy and chemotherapy for invasive thymomas. A multicentric retrospective review of 90 cases. The FNCLCC trialists. Federation Nationale des Centres de Lutte Contre le Cancer. Int J Radiat Oncol Biol Phys. 1995;32:651-659.
247 Penn CR, Hope-Stone HF. The role of radiotherapy in the management of malignant thymoma. Br J Surg. 1972;59:533-539.
248 Whooley BP, Urschel JD, Antkowiak JG, et al. A 25-year thymoma treatment review. J Exp Clin Cancer Res. 2000;19:3-5.
249 Marks RDJr, Wallace KM, Pettit HS. Radiation therapy control of nine patients with malignant thymoma. Cancer. 1978;41:117-119.
250 Ariaratnam LS, Kalnicki S, Mincer F, et al. The management of malignant thymoma with radiation therapy. Int J Radiat Oncol Biol Phys. 1979;5:77-80.
251 Chang HK, Wang CH, Liaw CC, et al. Prognosis of thymic carcinoma. Analysis of 16 cases. J Formos Med Assoc. 1992;91:764-769.
252 Hsu HC, Huang EY, Wang CJ, et al. Postoperative radiotherapy in thymic carcinoma. Treatment results and prognostic factors. Int J Radiat Oncol Biol Phys. 2002;52:801-805.
253 Takeda S, Sawabata N, Inoue M, et al. Thymic carcinoma. Clinical institutional experience with 15 patients. Eur J Cardiothorac Surg. 2004;26:401-406.
254 Arriagada R, Bretel JJ, Caillaud JM, et al. Invasive carcinoma of the thymus. A multicenter retrospective review of 56 cases. Eur J Cancer Clin Oncol. 1984;20:69-74.
255 Lucchi M, Mussi A, Basolo F, et al. The multimodality treatment of thymic carcinoma. Eur J Cardiothorac Surg. 2001;19:566-569.
256 de Montpreville VT, Macchiarini P, Dulmet E. Thymic neuroendocrine carcinoma (carcinoid). A clinicopathologic study of fourteen cases. J Thorac Cardiovasc Surg. 1996;111:134-141.
257 Gaur P, Leary C, Yao JC. Thymic neuroendocrine tumors. A SEER database analysis of 160 patients. Ann Surg. 2010;251:1117-1121.
258 Lin FC, Lin CM, Hsieh CC, et al. Atypical thymic carcinoid and malignant somatostatinoma in type I multiple endocrine neoplasia syndrome. Case report. Am J Clin Oncol. 2003;26:270-272.
259 Tiffet O, Nicholson AG, Ladas G, et al. A clinicopathologic study of 12 neuroendocrine tumors arising in the thymus. Chest. 2003;124:141-146.
260 Wang DY, Chang DB, Kuo SH, et al. Carcinoid tumours of the thymus. Thorax. 1994;49:357-360.
261 Chaer R, Massad MG, Evans A, et al. Primary neuroendocrine tumors of the thymus. Ann Thorac Surg. 2002;74:1733-1740.
262 Gomez D, Komaki R. Technical advances of radiation therapy for thymic malignancies. J Thorac Oncol. 2010;5:S336-S343.
263 Latz D, Schraube P, Oppitz U, et al. Invasive thymoma. Treatment with postoperative radiation therapy. Radiology. 1997;204:859-864.
264 Kersh CR, Eisert DR, Hazra TA. Malignant thymoma. Role of radiation therapy in management. Radiology. 1985;156:207-209.
265 Fernandes AT, Shinohara ET, Guo M, et al. The role of radiation therapy in malignant thymoma. A Surveillance, Epidemiology, and End Results database analysis. J Thorac Oncol. 2010;5:1454-1460.
266 Johnson SB, Eng TY, Giaccone G, et al. Thymoma. Update for the new millennium. Oncologist. 2001;6:239-246.
267 Mayer R, Beham-Schmid C, Groell R, et al. Radiotherapy for invasive thymoma and thymic carcinoma. Clinicopathological review. Strahlenther Onkol. 1999;175:271-278.
268 Zhu G, He S, Fu X, et al. Radiotherapy and prognostic factors for thymoma. A retrospective study of 175 patients. Int J Radiat Oncol Biol Phys. 2004;60:1113-1119.
269 Marcus RBJr, Million RR. The incidence of myelitis after irradiation of the cervical spinal cord. Int J Radiat Oncol Biol Phys. 1990;19:3-8.
270 Fuller CD, Ramahi EH, Aherne N, et al. Radiotherapy for thymic neoplasms. J Thorac Oncol. 2010;5:S327-S335.
271 Travis WD, Linnoila RI, Tsokos MG, et al. Neuroendocrine tumors of the lung with proposed criteria for large-cell neuroendocrine carcinoma. An ultrastructural, immunohistochemical, and flow cytometric study of 35 cases. Am J Surg Pathol. 1991;15:529-553.
272 Oberndorfer S. Karzinoide Tumoren des Dunndarms. Frankf Z Pathol. 1907;1:425-429.
273 Gould VE, Memoli V, Chejfec G, et al. The APUD cell system and its neoplasms. Observations on the significance and limitations of the concept. Surg Clin North Am. 1979;59:93-108.
274 Mendonca C, Baptista C, Ramos M, et al. Typical and atypical lung carcinoids. Clinical and morphological diagnosis. Microsc Res Tech. 1997;38:468-472.
275 Martini N, Zaman MB, Bains MS, et al. Treatment and prognosis in bronchial carcinoids involving regional lymph nodes. J Thorac Cardiovasc Surg. 1994;107:1-6. discussion J Thorac Cardiovasc Surg 107:6-7, 1994
276 Godwin JD2nd. Carcinoid tumors. An analysis of 2,837 cases. Cancer. 1975;36:560-569.
277 Beasley MB, Thunnissen FB, Brambilla E, et al. Pulmonary atypical carcinoid. Predictors of survival in 106 cases. Hum Pathol. 2000;31:1255-1265.
278 Erasmus JJ, McAdams HP, Patz EFJr, et al. Evaluation of primary pulmonary carcinoid tumors using FDG PET. AJR Am J Roentgenol. 1998;170:1369-1373.
279 Kayser K, Kayser C, Rahn W, et al. Carcinoid tumors of the lung. Immuno- and ligandohistochemistry, analysis of integrated optical density, syntactic structure analysis, clinical data, and prognosis of patients treated surgically. J Surg Oncol. 1996;63:99-106.
280 Laitinen KL, Soini Y, Mattila J, et al. Atypical bronchopulmonary carcinoids show a tendency toward increased apoptotic and proliferative activity. Cancer. 2000;88:1590-1598.
281 Debelenko LV, Swalwell JI, Kelley MJ, et al. MEN1 gene mutation analysis of high-grade neuroendocrine lung carcinoma. Genes Chromosomes Cancer. 2000;28:58-65.
282 Dong Q, Debelenko LV, Chandrasekharappa SC, et al. Loss of heterozygosity at 11q13. Analysis of pituitary tumors, lung carcinoids, lipomas, and other uncommon tumors in subjects with familial multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. 1997;82:1416-1420.
283 Lai SL, Brauch H, Knutsen T, et al. Molecular genetic characterization of neuroendocrine lung cancer cell lines. Anticancer Res. 1995;15:225-232.
284 Onuki N, Wistuba II, Travis WD, et al. Genetic changes in the spectrum of neuroendocrine lung tumors. Cancer. 1999;85:600-607.
285 Walch AK, Zitzelsberger HF, Aubele MM, et al. Typical and atypical carcinoid tumors of the lung are characterized by 11q deletions as detected by comparative genomic hybridization. Am J Pathol. 1998;153:1089-1098.
286 Liao H, Rao H, Zhang X, et al. [Retrospective study of clinicopathological characteristics in bronchopulmonary carcinoid]. Zhongguo Fei Ai Za Zhi. 2010;13:591-597.
287 Fink G, Krelbaum T, Yellin A, et al. Pulmonary carcinoid. Presentation, diagnosis, and outcome in 142 cases in Israel and review of 640 cases from the literature. Chest. 2001;119:1647-1651.
288 Hage R, de la Riviere AB, Seldenrijk CA, et al. Update in pulmonary carcinoid tumors. A review article. Ann Surg Oncol. 2003;10:697-704.
289 Cai YC, Banner B, Glickman J, et al. Cytokeratin 7 and 20 and thyroid transcription factor 1 can help distinguish pulmonary from gastrointestinal carcinoid and pancreatic endocrine tumors. Hum Pathol. 2001;32:1087-1093.
290 Oliveira AM, Tazelaar HD, Myers JL, et al. Thyroid transcription factor-1 distinguishes metastatic pulmonary from well-differentiated neuroendocrine tumors of other sites. Am J Surg Pathol. 2001;25:815-819.
291 Travis WD, Rush W, Flieder DB, et al. Survival analysis of 200 pulmonary neuroendocrine tumors with clarification of criteria for atypical carcinoid and its separation from typical carcinoid. Am J Surg Pathol. 1998;22:934-944.
292 Feldman JM. Increased dopamine production in patients with carcinoid tumors. Metabolism. 1985;34:255-260.
293 Feldman JM, O’Dorisio TM. Role of neuropeptides and serotonin in the diagnosis of carcinoid tumors. Am J Med. 1986;81:41-48.
294 Limper AH, Carpenter PC, Scheithauer B, et al. The Cushing syndrome induced by bronchial carcinoid tumors. Ann Intern Med. 1992;117:209-214.
295 Lucas KJ, Feldman JM. Flushing in the carcinoid syndrome and plasma kallikrein. Cancer. 1986;58:2290-2293.
296 Sandler M, Karim SM, Williams ED. Prostaglandins in amine-peptide-secreting tumours. Lancet. 1968;2:1053-1054.
297 Skrabanek P, Cannon D, Kirrane J, et al. Substance P secretion by carcinoid tumours. Ir J Med Sci. 1978;147:47-49.
298 Cardillo G, Sera F, Di Martino M, et al. Bronchial carcinoid tumors. Nodal status and long-term survival after resection. Ann Thorac Surg. 2004;77:1781-1785.
299 Skuladottir H, Hirsch FR, Hansen HH, et al. Pulmonary neuroendocrine tumors. Incidence and prognosis of histological subtypes. A population-based study in Denmark. Lung Cancer. 2002;37:127-135.
300 Soga J, Yakuwa Y. Bronchopulmonary carcinoids. An analysis of 1,875 reported cases with special reference to a comparison between typical carcinoids and atypical varieties. Ann Thorac Cardiovasc Surg. 1999;5:211-219.
301 Divisi D, Crisci R. Carcinoid tumors of the lung and multimodal therapy. Thorac Cardiovasc Surg. 2005;53:168-172.
302 Gustafsson BI, Kidd M, Chan A, et al. Bronchopulmonary neuroendocrine tumors. Cancer. 2008;113:5-21.
303 Asamura H, Kameya T, Matsuno Y, et al. Neuroendocrine neoplasms of the lung. A prognostic spectrum. J Clin Oncol. 2006;24:70-76.
304 Gatta G, Ciccolallo L, Kunkler I, et al. Survival from rare cancer in adults. A population-based study. Lancet Oncol. 2006;7:132-140.
305 Lohmann DR, Fesseler B, Putz B, et al. Infrequent mutations of the p53 gene in pulmonary carcinoid tumors. Cancer Res. 1993;53:5797-5801.
306 Sugio K, Osaki T, Oyama T, et al. Genetic alteration in carcinoid tumors of the lung. Ann Thorac Cardiovasc Surg. 2003;9:149-154.
307 Hasleton PS, Bostanci G. Pulmonary carcinoid and related tumours. Rocz Akad Med Bialymst. 1997;42(Suppl 1):28-42.
308 Kulke MH, Mayer RJ. Carcinoid tumors. N Engl J Med. 1999;340:858-868.
309 Garcia-Yuste M, Matilla JM, Alvarez-Gago T, et al. Prognostic factors in neuroendocrine lung tumors. A Spanish Multicenter Study. Spanish Multicenter Study of Neuroendocrine Tumors of the Lung of the Spanish Society of Pneumonology and Thoracic Surgery (EMETNE-SEPAR). Ann Thorac Surg. 2000;70:258-263.
310 Grote TH, Macon WR, Davis B, et al. Atypical carcinoid of the lung. A distinct clinicopathologic entity. Chest. 1988;93:370-375.
311 McCaughan BC, Martini N, Bains MS. Bronchial carcinoids. Review of 124 cases. J Thorac Cardiovasc Surg. 1985;89:8-17.
312 Crapanzano JP, Zakowski MF. Diagnostic dilemmas in pulmonary cytology. Cancer. 2001;93:364-375.
313 Nicholson SA, Ryan MR. A review of cytologic findings in neuroendocrine carcinomas including carcinoid tumors with histologic correlation. Cancer. 2000;90:148-161.
314 Thomas JS, Lamb D, Ashcroft T, et al. How reliable is the diagnosis of lung cancer using small biopsy specimens? Report of a UKCCCR Lung Cancer Working Party. Thorax. 1993;48:1135-1139.
315 Okike N, Bernatz PE, Woolner LB. Carcinoid tumors of the lung. Ann Thorac Surg. 1976;22:270-277.
316 Schreurs AJ, Westermann CJ, van den Bosch JM, et al. A twenty-five-year follow-up of ninety-three resected typical carcinoid tumors of the lung. J Thorac Cardiovasc Surg. 1992;104:1470-1475.
317 Torre M, Barberis M, Barbieri B, et al. Typical and atypical bronchial carcinoids. Respir Med. 1989;83:305-308.
318 Kaplan B, Stevens CW, Allen P, et al. Outcomes and patterns of failure in bronchial carcinoid tumors. Int J Radiat Oncol Biol Phys. 2003;55:125-131.
319 Thomas CFJr, Tazelaar HD, Jett JR. Typical and atypical pulmonary carcinoids. Outcome in patients presenting with regional lymph node involvement. Chest. 2001;119:1143-1150.
320 Marty-Ane CH, Costes V, Pujol JL, et al. Carcinoid tumors of the lung. Do atypical features require aggressive management? Ann Thorac Surg. 1995;59:78-83.
321 Smolle-Juttner FM, Popper H, Klemen H, et al. Clinical features and therapy of “typical” and “atypical” bronchial carcinoid tumors (grade 1 and grade 2 neuroendocrine carcinoma). Eur J Cardiothorac Surg. 1993;7:121-124. discussion Eur J Cardiothorac Surg 7:125, 1993
322 Choi N, Mohiuddin M, Fidias P: Atypical Carcinoids of the Thorax. Proceedings of the 90th Radiological Society of North America Conference, Chicago, November 28 to December 3, 2004.
323 Filosso PL, Rena O, Donati G, et al. Bronchial carcinoid tumors. Surgical management and long-term outcome. J Thorac Cardiovasc Surg. 2002;123:303-309.
324 Scott WJ. Surgical treatment of other bronchial tumors. Chest Surg Clin North Am. 2003;13:111-128.
325 Moraes TJ, Langer JC, Forte V, et al. Pediatric pulmonary carcinoid. A case report and review of the literature. Pediatr Pulmonol. 2003;35:318-322.
326 Rosado de Christenson ML, Abbott GF, Kirejczyk WM, et al. Thoracic carcinoids. Radiologic-pathologic correlation. Radiographics. 1999;19:707-736.
327 Schrevens L, Vansteenkiste J, Deneffe G, et al. Clinical-radiological presentation and outcome of surgically treated pulmonary carcinoid tumours. A long-term single institution experience. Lung Cancer. 2004;43:39-45.
328 Chong S, Lee KS, Chung MJ, et al. Neuroendocrine tumors of the lung. Clinical, pathologic, and imaging findings. Radiographics. 2006;26:41-57. discussion Radiographics 26:57-58, 2006
329 Harpole DHJr, Feldman JM, Buchanan S, et al. Bronchial carcinoid tumors. A retrospective analysis of 126 patients. Ann Thorac Surg. 1992;54:50-54. discussion Ann Thorac Surg 54:54-55, 1992
330 Anderson AS, Krauss D, Lang R. Cardiovascular complications of malignant carcinoid disease. Am Heart J. 1997;134:693-702.
331 Lorcy Y, Perdu S, Sevray B, et al. [Acromegaly due to ectopic GH RH secretion by a bronchial carcinoid tumor. A case report]. Ann Endocrinol (Paris). 2002;63:536-539.
332 Fischer S, Kruger M, McRae K, et al. Giant bronchial carcinoid tumors. A multidisciplinary approach. Ann Thorac Surg. 2001;71:386-393.
333 Ducrocq X, Thomas P, Massard G, et al. Operative risk and prognostic factors of typical bronchial carcinoid tumors. Ann Thorac Surg. 1998;65:1410-1414.
334 Kaltsas G, Korbonits M, Heintz E, et al. Comparison of somatostatin analog and meta-iodobenzylguanidine radionuclides in the diagnosis and localization of advanced neuroendocrine tumors. J Clin Endocrinol Metab. 2001;86:895-902.
335 Lamberts SW, Bakker WH, Reubi JC, et al. Somatostatin receptor imaging in vivo localization of tumors with a radiolabeled somatostatin analog. J Steroid Biochem Mol Biol. 1990;37:1079-1082.
336 Musi M, Carbone RG, Bertocchi C, et al. Bronchial carcinoid tumours. A study on clinicopathological features and role of octreotide scintigraphy. Lung Cancer. 1998;22:97-102.
337 Marom EM, Sarvis S, Herndon JE2nd, et al. T1 lung cancers. Sensitivity of diagnosis with fluorodeoxyglucose PET. Radiology. 2002;223:453-459.
338 Squerzanti A, Basteri V, Antinolfi G, et al. Bronchial carcinoid tumors. Clinical and radiological correlation. Radiol Med. 2002;104:273-284.
339 West WM. Image and diagnosis. Carcinoid tumor of the lung. West Indian Med J. 2002;51:200-204.
340 Anderson CJ, Dehdashti F, Cutler PD, et al. 64Cu-TETA-octreotide as a PET imaging agent for patients with neuroendocrine tumors. J Nucl Med. 2001;42:213-221.
341 Orlefors H, Sundin A, Garske U, et al. Whole-body (11)C-5-hydroxytryptophan positron emission tomography as a universal imaging technique for neuroendocrine tumors. Comparison with somatostatin receptor scintigraphy and computed tomography. J Clin Endocrinol Metab. 2005;90:3392-3400.
342 Hurt R, Bates M. Carcinoid tumours of the bronchus. A 33 year experience. Thorax. 1984;39:617-623.
343 Terzi A, Lonardoni A, Falezza G, et al. Sleeve lobectomy for non-small cell lung cancer and carcinoids. Results in 160 cases. Eur J Cardiothorac Surg. 2002;21:888-893.
344 Dusmet ME, McKneally MF. Pulmonary and thymic carcinoid tumors. World J Surg. 1996;20:189-195.
345 Stamatis G, Freitag L, Greschuchna D. Limited and radical resection for tracheal and bronchopulmonary carcinoid tumour. Report on 227 cases. Eur J Cardiothorac Surg. 1990;4:527-532. discussion Eur J Cardiothorac Surg 4:533, 1990
346 Ruggieri M, Scocchera F, Genderini M, et al. Therapeutic approach of carcinoid tumours of the lung. Eur Rev Med Pharmacol Sci. 2000;4:43-46.
347 Makridis C, Rastad J, Oberg K, et al. Progression of metastases and symptom improvement from laparotomy in midgut carcinoid tumors. World J Surg. 1996;20:900-906. discussion World J Surg 20:907, 1996
348 Nave H, Mossinger E, Feist H, et al. Surgery as primary treatment in patients with liver metastases from carcinoid tumors. A retrospective, unicentric study over 13 years. Surgery. 2001;129:170-175.
349 Platt AJ, Heddle RM, Rake MO, et al. Ondansetron in carcinoid syndrome. Lancet. 1992;339:1416.
350 Kubota A, Yamada Y, Kagimoto S, et al. Identification of somatostatin receptor subtypes and an implication for the efficacy of somatostatin analogue SMS 201-995 in treatment of human endocrine tumors. J Clin Invest. 1994;93:1321-1325.
351 Reichlin S. Somatostatin. N Engl J Med. 1983;309:1495-1501.
352 Reubi JC, Kvols LK, Waser B, et al. Detection of somatostatin receptors in surgical and percutaneous needle biopsy samples of carcinoids and islet cell carcinomas. Cancer Res. 1990;50:5969-5977.
353 Mackley HB, Videtic GM: Primary carcinoid tumors of the lung. A role for radiotherapy, Oncology (Williston Park) 20:1537-1543, 2006; discussion Oncology (Williston Park) 20:1544-1545, 1549, 2006.
354 Carretta A, Ceresoli GL, Arrigoni G, et al. Diagnostic and therapeutic management of neuroendocrine lung tumors. A clinical study of 44 cases. Lung Cancer. 2000;29:217-225.
355 Granberg D, Eriksson B, Wilander E, et al. Experience in treatment of metastatic pulmonary carcinoid tumors. Ann Oncol. 2001;12:1383-1391.
356 Mutyala S, Stewart A, Khan AJ, et al. Permanent iodine-125 interstitial planar seed brachytherapy for close or positive margins for thoracic malignancies. Int J Radiat Oncol Biol Phys. 2010;76:1114-1120.
357 Filosso PL, Ruffini E, Oliaro A, et al. Long-term survival of atypical bronchial carcinoids with liver metastases, treated with octreotide. Eur J Cardiothorac Surg. 2002;21:913-917.
358 Krenning EP, de Jong M, Kooij PP, et al. Radiolabelled somatostatin analogue(s) for peptide receptor scintigraphy and radionuclide therapy. Ann Oncol. 1999;10(Suppl 2):S23-S29.
359 Bukowski RM, Johnson KG, Peterson RF, et al. A phase II trial of combination chemotherapy in patients with metastatic carcinoid tumors. A Southwest Oncology Group Study. Cancer. 1987;60:2891-2895.
360 Engstrom PF, Lavin PT, Moertel CG, et al. Streptozocin plus fluorouracil versus doxorubicin therapy for metastatic carcinoid tumor. J Clin Oncol. 1984;2:1255-1259.
361 Moertel CG, Hanley JA. Combination chemotherapy trials in metastatic carcinoid tumor and the malignant carcinoid syndrome. Cancer Clin Trials. 1979;2:327-334.
362 Oberg K, Eriksson B. The role of interferons in the management of carcinoid tumours. Br J Haematol. 1991;79(Suppl 1):74-77.
363 Que FG, Nagorney DM, Batts KP, et al. Hepatic resection for metastatic neuroendocrine carcinomas. Am J Surg. 1995;169:36-42. discussion Am J Surg 169:42-43, 1995
364 Schupak KD, Wallner KE. The role of radiation therapy in the treatment of locally unresectable or metastatic carcinoid tumors. Int J Radiat Oncol Biol Phys. 1991;20:489-495.
365 Oberg K, Astrup L, Eriksson B, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine tumours (including bronchopulmonary and thymic neoplasms). Part I. General overview. Acta Oncol. 2004;43:617-625.
366 Vadasz P, Palffy G, Egervary M, et al. Diagnosis and treatment of bronchial carcinoid tumors. Clinical and pathological review of 120 operated patients. Eur J Cardiothorac Surg. 1993;7:8-11.
367 Machuca TN, Cardoso PF, Camargo SM, et al. Surgical treatment of bronchial carcinoid tumors. A single-center experience. Lung Cancer. 2010;70:158-162.
368 Clark O. NCCN Clinical Practice Guidelines in Oncology. Neuroendocrine tumors. J Natl Compr Canc Netw. 2009;7:712-747.
369 Chughtai TS, Morin JE, Sheiner NM, et al. Bronchial carcinoid—twenty years’ experience defines a selective surgical approach. Surgery. 1997;122:801-808.
370 Gordon WJr, Antman KH, Greenberger JS, et al. Radiation therapy in the management of patients with mesothelioma. Int J Radiat Oncol Biol Phys. 1982;8:19-25.
371 Rusch VW. Pleurectomy/decortication and adjuvant therapy for malignant mesothelioma. Chest. 1993;103:382S-384S.
372 Butchart EG, Ashcroft T, Barnsley WC, et al. Pleuropneumonectomy in the management of diffuse malignant mesothelioma of the pleura. Experience with 29 patients. Thorax. 1976;31:15-24.
373 Jaklitsch MT, Grondin SC, Sugarbaker DJ. Treatment of malignant mesothelioma. World J Surg. 2001;25:210-217.
374 Rusch VW, Piantadosi S, Holmes EC. The role of extrapleural pneumonectomy in malignant pleural mesothelioma. A Lung Cancer Study Group trial. J Thorac Cardiovasc Surg. 1991;102:1-9.
375 Calavrezos A, Koschel G, Husselmann H, et al. Malignant mesothelioma of the pleura. A prospective therapeutic study of 132 patients from 1981-1985. Klin Wochenschr. 1988;66:607-613.
376 Huncharek M, Kelsey K, Mark EJ, et al. Treatment and survival in diffuse malignant pleural mesothelioma. A study of 83 cases from the Massachusetts General Hospital. Anticancer Res. 1996;16:1265-1268.
377 Sugarbaker DJ, Flores RM, Jaklitsch MT, et al. Resection margins, extrapleural nodal status, and cell type determine postoperative long-term survival in trimodality therapy of malignant pleural mesothelioma. Results in 183 patients. J Thorac Cardiovasc Surg. 1999;117:54-63. discussion J Thorac Cardiovasc Surg 117:63-65, 1999
378 Baldini EH, Recht A, Strauss GM, et al. Patterns of failure after trimodality therapy for malignant pleural mesothelioma. Ann Thorac Surg. 1997;63:334-338.
379 Bricout PB, Engler MJ. Computerized tomography scanning and the planning of high-dose radiotherapy for pleural mesothelioma. A report of five patients. Int J Radiat Oncol Biol Phys. 1981;7:821-826.
380 Connelly RR, Spirtas R, Myers MH, et al. Demographic patterns for mesothelioma in the United States. J Natl Cancer Inst. 1987;78:1053-1060.
381 Muscat JE, Wynder EL. Cigarette smoking, asbestos exposure, and malignant mesothelioma. Cancer Res. 1991;51:2263-2267.
382 Steinert HC, Santos Dellea MM, Burger C, et al. Therapy response evaluation in malignant pleural mesothelioma with integrated PET-CT imaging. Lung Cancer. 2005;49(Suppl 1):S33-S35.
383 Yildirim H, Metintas M, Entok E, et al. Clinical value of fluorodeoxyglucose-positron emission tomography/computed tomography in differentiation of malignant mesothelioma from asbestos-related benign pleural disease. An observational pilot study. J Thorac Oncol. 2009;4:1480-1484.
384 Benard F, Sterman D, Smith RJ, et al. Prognostic value of FDG PET imaging in malignant pleural mesothelioma. J Nucl Med. 1999;40:1241-1245.
385 Wilcox BE, Subramaniam RM, Peller PJ, et al. Utility of integrated computed tomography-positron emission tomography for selection of operable malignant pleural mesothelioma. Clin Lung Cancer. 2009;10:244-248.
386 Bueno R, Reblando J, Glickman J, et al. Pleural biopsy. A reliable method for determining the diagnosis but not subtype in mesothelioma. Ann Thorac Surg. 2004;78:1774-1776.
387 Boutin C, Rey F, Gouvernet J, et al. Thoracoscopy in pleural malignant mesothelioma. A prospective study of 188 consecutive patients. Part 2. Prognosis and staging. Cancer. 1993;72:394-404.
388 Flores RM, Zakowski M, Venkatraman E, et al. Prognostic factors in the treatment of malignant pleural mesothelioma at a large tertiary referral center. J Thorac Oncol. 2007;2:957-965.
389 Yaziji H, Battifora H, Barry TS, et al. Evaluation of 12 antibodies for distinguishing epithelioid mesothelioma from adenocarcinoma. Identification of a three-antibody immunohistochemical panel with maximal sensitivity and specificity. Mod Pathol. 2006;19:514-523.
390 Curran D, Sahmoud T, Therasse P, et al. Prognostic factors in patients with pleural mesothelioma. The European Organization for Research and Treatment of Cancer experience. J Clin Oncol. 1998;16:145-152.
391 Herndon JE, Green MR, Chahinian AP, et al. Factors predictive of survival among 337 patients with mesothelioma treated between 1984 and 1994 by the Cancer and Leukemia Group B. Chest. 1998;113:723-731.
392 Edwards JG, Abrams KR, Leverment JN, et al. Prognostic factors for malignant mesothelioma in 142 patients. Validation of CALGB and EORTC prognostic scoring systems. Thorax. 2000;55:731-735.
393 Robinson BW, Creaney J, Lake R, et al. Mesothelin-family proteins and diagnosis of mesothelioma. Lancet. 2003;362:1612-1616.
394 Renshaw AA, Dean BR, Antman KH, et al. The role of cytologic evaluation of pleural fluid in the diagnosis of malignant mesothelioma. Chest. 1997;111:106-109.
395 Heilo A, Stenwig AE, Solheim OP. Malignant pleural mesothelioma. US-guided histologic core-needle biopsy. Radiology. 1999;211:657-659.
396 Flores RM, Pass HI, Seshan VE, et al. Extrapleural pneumonectomy versus pleurectomy/decortication in the surgical management of malignant pleural mesothelioma. Results in 663 patients. J Thorac Cardiovasc Surg. 2008;135:620-626. J Thorac Cardiovasc Surg 135:626.e1-3, 2008
397 Cao CQ, Yan TD, Bannon PG, et al. A systematic review of extrapleural pneumonectomy for malignant pleural mesothelioma. J Thorac Oncol. 2010;5:1692-1703.
398 Rusch VW, Rosenzweig K, Venkatraman E, et al. A phase II trial of surgical resection and adjuvant high-dose hemithoracic radiation for malignant pleural mesothelioma. J Thorac Cardiovasc Surg. 2001;122:788-795.
399 Sugarbaker DJ, Jaklitsch MT, Bueno R, et al. Prevention, early detection, and management of complications after 328 consecutive extrapleural pneumonectomies. J Thorac Cardiovasc Surg. 2004;128:138-146.
400 Balduyck B, Trousse D, Nakas A, et al. Therapeutic surgery for nonepithelioid malignant pleural mesothelioma. Is it really worthwhile? Ann Thorac Surg. 2010;89:907-911.
401 Kindler HL. Malignant pleural mesothelioma. Curr Treat Options Oncol. 2000;1:313-326.
402 Stewart DJ, Edwards JG, Smythe WR, et al. Malignant pleural mesothelioma—an update. Int J Occup Environ Health. 2004;10:26-39.
403 Solheim OP, Saeter G, Finnanger AM, et al. High-dose methotrexate in the treatment of malignant mesothelioma of the pleura. A phase II study. Br J Cancer. 1992;65:956-960.
404 Byrne MJ, Davidson JA, Musk AW, et al. Cisplatin and gemcitabine treatment for malignant mesothelioma. A phase II study. J Clin Oncol. 1999;17:25-30.
405 Calvert A, Hughes A, Calvert P. Alimta in combination with carboplatin demonstrates clinical activity against malignant mesothelioma in a phase I trial. Proc Am Soc Clin Oncol. 2000;19:495a.
405a Krug LM, Pass HI, Rusch VW, et al. Multicenter phase II trial of neoadjuvant permetrexed plus cisplatin followed by extrapleural pneumonectomy and radiation for malignant pleural mesothelioma. J Clin Oncol. 2009;27:3007-3013.
406 Reck M, Gatzemeier U. Pemetrexed-cisplatin combination in mesothelioma. Expert Rev Anticancer Ther. 2005;5:231-237.
407 Chahinian AP, Antman K, Goutsou M, et al. Randomized phase II trial of cisplatin with mitomycin or doxorubicin for malignant mesothelioma by the Cancer and Leukemia Group B. J Clin Oncol. 1993;11:1559-1565.
408 Fizazi H, Doubre H, Viala J. The combination of raltitrexed (“tomudex”) and oxaliplatin is an active regimen in malignant mesothelioma. Results of a phase II stud. Proc Am Soc Clin Oncol. 2000;19:578a.
409 Berghmans T, Paesmans M, Lalami Y, et al. Activity of chemotherapy and immunotherapy on malignant mesothelioma. A systematic review of the literature with meta-analysis. Lung Cancer. 2002;38:111-121.
410 Tilleman TR, Richards WG, Zellos L, et al. Extrapleural pneumonectomy followed by intracavitary intraoperative hyperthermic cisplatin with pharmacologic cytoprotection for treatment of malignant pleural mesothelioma. A phase II prospective study. J Thorac Cardiovasc Surg. 2009;138:405-411.
411 Vogelzang NJ, Rusthoven JJ, Symanowski J, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21:2636-2644.
412 Muers MF, Rudd RM, O’Brien ME, et al. BTS randomised feasibility study of active symptom control with or without chemotherapy in malignant pleural mesothelioma. ISRCTN 54469112. Thorax. 2004;59:144-148.
413 Carmichael J, Degraff WG, Gamson J, et al. Radiation sensitivity of human lung cancer cell lines. Eur J Cancer Clin Oncol. 1989;25:527-534.
414 Hakkinen AM, Laasonen A, Linnainmaa K, et al. Radiosensitivity of mesothelioma cell lines. Acta Oncol. 1996;35:451-456.
415 Allen AM, Den R, Wong JS, et al. Influence of radiotherapy technique and dose on patterns of failure for mesothelioma patients after extrapleural pneumonectomy. Int J Radiat Oncol Biol Phys. 2007;68:1366-1374.
416 Maasilta P. Deterioration in lung function following hemithorax irradiation for pleural mesothelioma. Int J Radiat Oncol Biol Phys. 1991;20:433-438.
417 Maasilta P, Kivisaari L, Holsti LR, et al. Radiographic chest assessment of lung injury following hemithorax irradiation for pleural mesothelioma. Eur Respir J. 1991;4:76-83.
418 Hilaris BS, Nori D, Kwong E, et al. Pleurectomy and intraoperative brachytherapy and postoperative radiation in the treatment of malignant pleural mesothelioma. Int J Radiat Oncol Biol Phys. 1984;10:325-331.
419 Lee TT, Everett DL, Shu HK, et al. Radical pleurectomy/decortication and intraoperative radiotherapy followed by conformal radiation with or without chemotherapy for malignant pleural mesothelioma. J Thorac Cardiovasc Surg. 2002;124:1183-1189.
420 Rosenzweig KE, Fox JL, Zelefsky MJ, et al. A pilot trial of high-dose-rate intraoperative radiation therapy for malignant pleural mesothelioma. Brachytherapy. 2005;4:30-33.
421 Dougherty TJ, Gomer CJ, Henderson BW, et al. Photodynamic therapy. J Natl Cancer Inst. 1998;90:889-905.
422 Friedberg JS, Mick R, Stevenson J, et al. A phase I study of Foscan-mediated photodynamic therapy and surgery in patients with mesothelioma. Ann Thorac Surg. 2003;75:952-959.
423 Reference deleted in proofs.
424 Friedberg JS. Photodynamic therapy as an innovative treatment for malignant pleural mesothelioma. Semin Thorac Cardiovasc Surg. 2009;21:177-187.
425 Kutcher GJ, Kestler C, Greenblatt D, et al. Technique for external beam treatment for mesothelioma. Int J Radiat Oncol Biol Phys. 1987;13:1747-1752.
426 Ahamad A, Stevens CW, Smythe WR, et al. Promising early local control of malignant pleural mesothelioma following postoperative intensity modulated radiotherapy (IMRT) to the chest. Cancer J. 2003;9:476-484.
427 Ahamad A, Stevens CW, Smythe WR, et al. Intensity-modulated radiation therapy. A novel approach to the management of malignant pleural mesothelioma. Int J Radiat Oncol Biol Phys. 2003;55:768-775.
428 Forster KM, Smythe WR, Starkschall G, et al. Intensity-modulated radiotherapy following extrapleural pneumonectomy for the treatment of malignant mesothelioma. Clinical implementation. Int J Radiat Oncol Biol Phys. 2003;55:606-616.
429 Rice DC, Stevens CW, Correa AM, et al. Outcomes after extrapleural pneumonectomy and intensity-modulated radiation therapy for malignant pleural mesothelioma. Ann Thorac Surg. 2007;84:1685-1692. discussion Ann Thorac Surg 84:1692-1693, 2007
430 Yajnik S, Rosenzweig KE, Mychalczak B, et al. Hemithoracic radiation after extrapleural pneumonectomy for malignant pleural mesothelioma. Int J Radiat Oncol Biol Phys. 2003;56:1319-1326.
431 de Perrot M, Feld R, Cho BC, et al. Trimodality therapy with induction chemotherapy followed by extrapleural pneumonectomy and adjuvant high-dose hemithoracic radiation for malignant pleural mesothelioma. J Clin Oncol. 2009;27:1413-1418.
432 van Ooijen B, Eggermont AM, Wiggers T. Subcutaneous tumor growth complicating the positioning of Denver shunt and intrapleural port-a-cath in mesothelioma patients. Eur J Surg Oncol. 1992;18:638-640.
433 Boutin C, Rey F, Viallat JR. Prevention of malignant seeding after invasive diagnostic procedures in patients with pleural mesothelioma. A randomized trial of local radiotherapy. Chest. 1995;108:754-758.
434 Low EM, Khoury GG, Matthews AW, et al. Prevention of tumour seeding following thoracoscopy in mesothelioma by prophylactic radiotherapy. Clin Oncol (R Coll Radiol). 1995;7:317-318.
435 Sugarbaker DJ, Garcia JP. Multimodality therapy for malignant pleural mesothelioma. Chest. 1997;112:272S-275S.
436 O’Rourke N, Garcia JC, Paul J, et al. A randomised controlled trial of intervention site radiotherapy in malignant pleural mesothelioma. Radiother Oncol. 2007;84:18-22.
437 Rusch VW. Surgical techniques for pulmonary metastasectomy. Semin Thorac Cardiovasc Surg. 2002;14:4-9.
438 Miles EF, Larrier NA, Kelsey CR, et al. Intensity-modulated radiotherapy for resected mesothelioma. The Duke experience. Int J Radiat Oncol Biol Phys. 2008;71:1143-1150.
439 Della Volpe A, Ferreri AJ, Annaloro C, et al. Lethal pulmonary complications significantly correlate with individually assessed mean lung dose in patients with hematologic malignancies treated with total body irradiation. Int J Radiat Oncol Biol Phys. 2002;52:483-488.
440 Du KL, Both S, Friedberg JS, et al. Extrapleural pneumonectomy, photodynamic therapy and intensity modulated radiation therapy for the treatment of malignant pleural mesothelioma. Cancer Biol Ther. 2010;10:425-429.
441 Hill-Kayser CE, Avery S, Mesina CF, et al. Hemithoracic radiotherapy after extrapleural pneumonectomy for malignant pleural mesothelioma. A dosimetric comparison of two well-described techniques. J Thorac Oncol. 2009;4:1431-1437.
442 Krayenbuehl J, Oertel S, Davis JB, et al. Combined photon and electron three-dimensional conformal versus intensity-modulated radiotherapy with integrated boost for adjuvant treatment of malignant pleural mesothelioma after pleuropneumonectomy. Int J Radiat Oncol Biol Phys. 2007;69:1593-1599.
443 Bretti S, Berruti A, Loddo C, et al. Multimodal management of stages III-IVa malignant thymoma. Lung Cancer. 2004;44:69-77.
444 Hug E, Sobczak M, Choi N. The role of radiotherapy in the management of invasive thymoma. Int J Radiat Oncol Biol Physs. 1990;19:161-162.
445 Liu HC, Hsu WH, Chen YJ, et al. Primary thymic carcinoma. Ann Thorac Surg. 2002;73:1076-1081.
446 Nakamura Y, Kunitoh H, Kubota K, et al. Platinum-based chemotherapy with or without thoracic radiation therapy in patients with unresectable thymic carcinoma. Jpn J Clin Oncol. 2000;30:385-388.
447 Wirth LJ, Carter MR, Janne PA, et al. Outcome of patients with pulmonary carcinoid tumors receiving chemotherapy or chemoradiotherapy. Lung Cancer. 2004;44:213-220.
448 Ferguson MK, Landreneau RJ, Hazelrigg SR, et al. Long-term outcome after resection for bronchial carcinoid tumors. Eur J Cardiothorac Surg. 2000;18:156-161.
449 Mezzetti M, Raveglia F, Panigalli T, et al. Assessment of outcomes in typical and atypical carcinoids according to latest WHO classification. Ann Thorac Surg. 2003;76:1838-1842.
* See references 13, 46, 47, 49, 65, 122, 123, 124, 125, 126.
* See references 15, 58, 63, 66, 133–136.
† See references 46–48, 122, 140, 141, 142.
* See references 125, 140, 141, 144, 146, 147, 148, 149–151.
* See references 65, 125, 140, 141, 145, 147.
† See references 65, 124, 200, 201, 202.
* See references 216–221, 222, 223–228, 229, 230–232.
* See references 65, 140, 145, 205, 209, 211, 214, 240, 245.
† See references 65, 122, 125, 126, 140, 141, 149.
* See references 65, 143, 206, 209, 211, 204, 246, 263.
* See references 291, 294, 311, 315, 320, 326, 331.
† See references 318, 319, 320, 327, 329, 336.
* See references 287, 311, 315–317, 319, 321, 324, 329, 366, 367.