[level-membership-for-anesthesiology-category]
Chapter 18 Uncommon Cardiac Diseases
CARDIAC TUMORS
Cardiac tumors are increasingly diagnosed before autopsy due to advancements in imaging, especially metastatic tumors of the heart and pericardium, which account for a majority of cardiac tumors. Data pooled from 22 large autopsy series show the prevalence of adult primary cardiac tumors as only about 0.02%, yet they are responsible for significant morbidity and mortality. Malignant tumors encompass about 20% of primary tumors in adults.1 Diagnosis can be elusive because these tumors may be associated with nonspecific symptoms mimicking other disease entities. Two-dimensional echocardiography (echo) modalities and magnetic resonance imaging (MRI) have allowed earlier, more frequent, and more complete assessment of cardiac tumors.
Myxoma
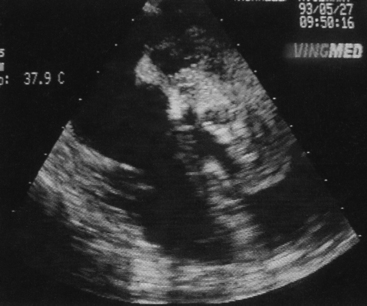
Rarely discovered by incidental echocardiography examination, myxomas may manifest a variety of symptoms. The classic triad includes embolism, intracardiac obstruction, and constitutional symptoms. Approximately 80% of individuals present with one component of the triad, yet up to 10% may be asymptomatic even with mitral myxomas, arising from both atrial and ventricular sides of the anterior mitral leaflet. The most common initial symptom, dyspnea on exertion, reflects mitral valve obstruction usually present with LA myxomas (Fig. 18-1). Because of the pedunculated nature of some myxomas, temporary obstruction of blood flow may cause hemolysis, hypotension, syncope, or sudden death. Other symptoms of mitral obstruction similar to mitral stenosis such as hemoptysis, systemic embolization, fever, and weight loss may also occur. The persistence of sinus rhythm in the presence of such symptoms may help distinguish atrial myxoma from mitral stenosis. Severe pulmonary hypertension without significant mitral valve involvement suggests obstruction of the tricuspid valve and recurrent pulmonary emboli known to occur with a myxoma in the RA or right ventricle (RV). Before echocardiography, angiography was used to identify all myxomas, but now it is only useful to confirm the diagnosis or determine coronary anatomy if considered necessary. TEE is 100% sensitive for diagnosis of myxoma. Specifically, it yields morphologic detail in the evaluation of cardiac tumors, including points of tumor attachment and degree of mobility. Computed tomography (CT) and MRI can help delineate the extent of the tumor and its relationships to surrounding cardiac and thoracic structures. MRI is especially valuable in the diagnosis of myxoma when masses are equivocal or suboptimal on echocardiography or if the tumor is atypical in presentation. Difficulty may arise in differentiating thrombus from myxoma because both are so heterogeneous.2
Tumors with Systemic Cardiac Manifestations
Anesthetic Considerations
Patients who have carcinoid heart disease and require cardiac surgery pose an anesthetic challenge.3 A carcinoid crisis with vasoactive mediator release can be provoked by stress, physical stimulation, or medications such as meperidine, morphine, or histamine-releasing muscle relaxants (atracurium). Preoperative control of carcinoid activity is a critical aspect of perioperative management, made considerably easier with the administration of octreotide, a synthetic analog of somatostatin that inhibits the vasoactive compounds that produce carcinoid syndrome. It reduces the occurrence of symptoms in more than 70% of patients. The longer half-life of octreotide than somatostatin allows subcutaneous injection of 150 μg three times daily to control symptoms. Intermittent intravenous doses of 50 to 200 μg or continuous infusions are given to stop severe hypotension and prevent further carcinoid symptoms. Severe hyperglycemia may occur with octreotide due to its inhibition of insulin secretion.
CARDIOMYOPATHY
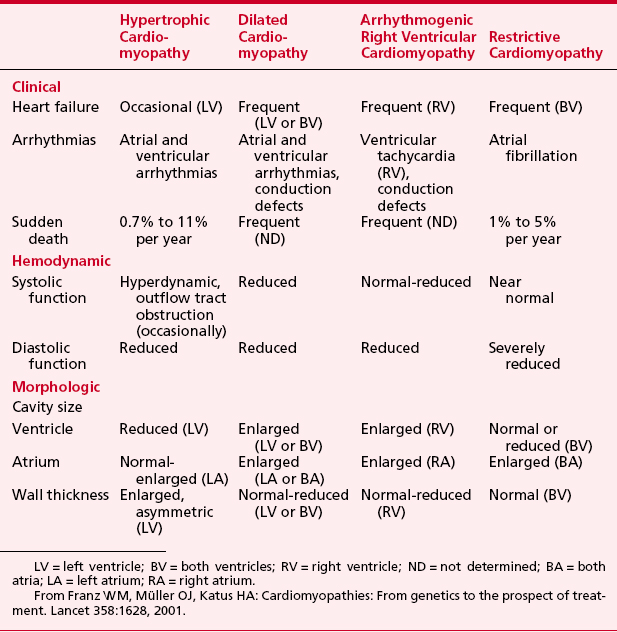
The annual incidence of cardiomyopathy in adults is 8.7 cases per 100,000 person-years. General characteristics of all four cardiomyopathies are displayed in Table 18-1.
Dilated Cardiomyopathy
Management of acute decompensated CHF continues to evolve, but the onset of overt CHF is a poor prognostic indicator for patients with DCM. Treatment revolves around management of symptoms and progression of DCM, whereas other measures are designed to prevent complications such as pulmonary thromboembolism and arrhythmias. The mainstay of therapy for DCM is vasodilators combined with digoxin and diuretics.4 All patients receive angiotensin-converting enzyme inhibitors (ACEIs) to reduce symptoms, improve exercise tolerance, and reduce cardiovascular mortality without a direct myocardial effect. Perhaps more important than the hemodynamic effects, ACEIs suppress ventricular remodeling and endothelial dysfunction, accounting for the improvement in mortality noted with this medication in DCM. Other afterload-reducing agents, such as selective phosphodiesterase-3 inhibitors like milrinone, may improve quality of life but do not affect mortality, so they are rarely administered in chronic situations. Spironolactone has assumed a greater role in treatment as mortality was reduced by 30% from all causes in patients receiving standard ACEIs for DCM with the addition of spironolactone in a large double-blind randomized trial. The use of β-blockers in DCM has provided not only symptomatic improvement but also substantial reductions in sudden death and progressive death in patients with New York Heart Association (NYHA) class II and III heart failure. This is especially significant because almost 50% of deaths are sudden. High-grade ventricular arrhythmias are common with DCM. Approximately 12% of all patients with DCM die suddenly, but overall prediction of sudden death in an individual with DCM is poor. The best predictor of sudden death remains the degree of LV dysfunction. Patients who have sustained ventricular tachycardia or out-of-hospital ventricular fibrillation are at increased risk for sudden death, but more than 70% of patients with DCM have nonsustained ventricular tachycardia during ambulatory monitoring. Antiarrhythmic medications are hazardous in patients with poor ventricular function owing to their negative inotropic and sometimes proarrhythmic properties. Amiodarone is the preferred antiarrhythmic agent in DCM because its negative inotropic effect is less than that of other antiarrhythmic medications and its proarrhythmic potential is lowest. Implantable defibrillators reduce the risk of sudden death as well as reducing mortality. Evidence has indicated that with previous cardiac arrest or sustained ventricular tachycardia, more benefit was gained from use of an implantable defibrillator. This was based on a 27% reduction in the relative risk of death attributed to a 50% reduction in arrhythmia-related mortality compared with treatment with amiodarone.
Hypertrophic Cardiomyopathy
Referred to as idiopathic hypertrophic subaortic stenosis, hypertrophic obstructive cardiomyopathy, and asymmetric septal hypertrophy, among other names, the accepted term is now hypertrophic cardiomyopathy.5 In the past 40 years, advancements regarding the hemodynamics, systolic and diastolic abnormalities, electrophysiology, genetics, and clinical care of HCM have contributed to a greater understanding of this disease. HCM is the most common genetic cardiac disease, with marked heterogeneity in clinical expression, pathophysiology, and prognosis. The overall prevalence for adults in the general population is 0.2%, affecting men and women equally.
Two-dimensional echocardiography establishes the diagnosis of HCM easily and reliably. Classic echocardiography features are thickening of the entire ventricular septum from base to apex disproportional to that of the posterior wall, poor septal motion, and anterior displacement of the mitral valve without LV dilation (Fig. 18-2). Echocardiography has reduced the need for invasive catheterization procedures, unless coronary artery disease (CAD) or severe mitral valve disease is suspected or diagnostic problems are present. MRI has been useful in cases in which echocardiography is technically inadequate.
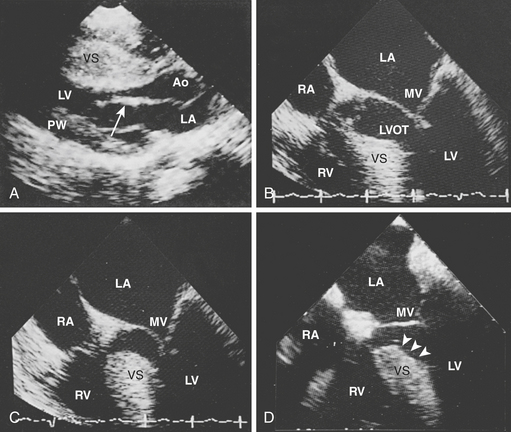
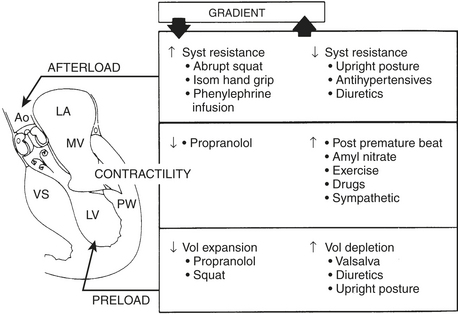
Two thirds of individuals with LV outflow tract obstruction become severely symptomatic and 10% die within 4 years of diagnosis. The outflow tract is narrowed from septal hypertrophy and anterior displacement of the papillary muscles and mitral leaflets, creating a dynamic LV outflow obstruction (see Fig. 18-2). Elongation of the mitral leaflets results in coaptation of the body of the leaflets instead of the tips. The part of the anterior leaflet distal to the coaptation is subjected to strong Venturi forces that provoke systolic anterior motion (SAM), mitral septal contact, and ultimately LV outflow obstruction. SAM of the anterior leaflet may also cause mitral regurgitation. The onset and duration of mitral leaflet-septal contact determine the magnitude of the gradient and the degree of mitral regurgitation. The pressure gradient between the aorta and LV is worsened by decreased end-diastolic volume, increased contractility, or decreased aortic outflow resistance (Fig. 18-3).
Surgical correction of HCM is directed primarily at relieving symptoms of LV obstruction in the 5% of patients who are refractory to medication.6 In general, these are individuals with subaortic gradients more than 50 mmHg and frequently associated with severe CHF. A myotomy-myectomy through a transaortic approach relieves the obstruction. The muscle is excised from the proximal septum extending just beyond the mitral valve leaflets to widen the LV outflow tract. This is a technically challenging operation due to the limited exposure and precise area to excise the muscle. It is usually reserved for centers with considerable experience. When myectomy is successful, the outflow tract of the LV is widened and SAM, mitral regurgitation, and outflow gradient are all decreased.
Restrictive Cardiomyopathy
RCM may be classified as myocardial (infiltrative, noninfiltrative, and storage) or endomyocardial (Box 18-1) according to etiology. RCM may be associated with another disease entity except pericardial disease. Restrictive myocardial disorders are characteristically atypical in presentation and hemodynamics at times, complicating perioperative management.
BOX 18-1 Classification of Types of Restrictive Cardiomyopathy According to Cause
From Kushwaha SS, Fallon JT, Fuster V: Medical progress: Restrictive cardiomyopathy. N Engl J Med 336:268, 1997.
Anesthetic Considerations
Adults with RCM rarely require cardiac surgery for reasons other than transplantation or mitral or tricuspid valve replacement. Occasionally, anesthesia is administered for a scheduled pericardiectomy only to find RCM instead of constrictive pericarditis. Despite essentially normal ventricular systolic function, diastolic dysfunction and filling abnormalities result in a poor CO and systemic perfusion. Aggressive preoperative diuretic therapy may contribute to the difficulty of maintaining adequate circulating blood volume. Elevated airway pressures from pulmonary congestion may further impair oxygen delivery to the tissues.7
Arrhythmogenic Right Ventricular Cardiomyopathy
Anesthetic Considerations
During the course of ARVC, arrhythmias may occur at any time. Presently, there are no guidelines for arrhythmia prevention. A family history of sudden death or syncope at an early age should heighten the awareness of ARVC.8 With ARVC, arrhythmias are more likely in the perioperative period. During or after anesthesia, the patient should be carefully observed to avoid noxious stimuli, hypovolemia, hypercarbia, and light anesthesia. Acidosis may be especially detrimental due to its effect on arrhythmia generation and myocardial function. General anesthesia alone does not appear to be arrhythmogenic because reports describe multiple exposures to general anesthesia without arrhythmias. More than 200 patients with ARVC had undergone general anesthesia without a single cardiac arrest. Nonetheless, any family history of sudden death elicited during preoperative assessment merits further investigation. Anesthesia has been successfully conducted with propofol, midazolam, and alfentanil. Amiodarone is the first line of antiarrhythmic medication during anesthesia.
MITRAL VALVE PROLAPSE
Mitral valve prolapse (MVP), often referred to as Barlow’s syndrome, is a structural and functional disorder affecting 2.5% to 5% of the population. As the most commonly diagnosed cardiac valve abnormality, it occurs in adults who are otherwise healthy or in association with many pathologic conditions (Box 18-2). Women, representing two thirds of adults with MVP, are more frequently affected during the third, fourth, and fifth decades of life but account for a decreasing prevalence beyond the third decade. The incidence of MVP in men is unrelated to age. MVP may be characterized as anatomic or functional (syndrome). Anatomic MVP includes individuals with a broad range of valvular abnormalities corresponding to symptoms of progressive mitral valve regurgitation. MVP syndrome consists of MVP with various symptoms reflecting a neuroendocrine or an autonomic basis. Approximately 80% of patients with MVP experience MVP syndrome instead of anatomic MVP.
BOX 18-2 Conditions Associated with Mitral Valve Prolapse
From O’Rourke RA (ed): Current Problems in Cardiology, May 1991, p 333.
Anatomic MVP, inherited in an autosomal dominant manner, is believed to result from myxomatous degeneration of the mitral valve, elongation and thinning of the chordae tendineae, and the presence of redundant and excessive valve tissue. The posterior leaflet is affected more frequently than the anterior leaflet. Changes are often observed at the site of chordal insertion, leading to rupture of the chordae and tethering of the valve leaflet. The degenerative changes of the mitral valve that are responsible for progression from an asymptomatic condition with murmurs and systolic clicks to dyspnea with severe mitral regurgitation occur over an average of 25 years. With the onset of severe mitral regurgitation, PAH, left atrial enlargement, and atrial fibrillation frequently emerge. Subsequently, mitral valve repair or replacement is usually necessary within 1 year.9
ACUTE PULMONARY EMBOLISM
Pulmonary embolism (PE) has an incidence in the United States of 1 per 1000 and a mortality rate of more than 15% at 3 months after diagnosis.10 Approximately two thirds of deaths occur within the first hour of a PE, and many of the remaining occur within 4 to 6 hours. Because 5% of patients present in cardiogenic shock, treatment involves not only prevention of a recurrence but also hemodynamic support. Because a correct diagnosis can alter the outcome of PE measurably, it is unfortunate that 60% to 80% of cases of fatal PE in the hospital setting are clinically unsuspected.
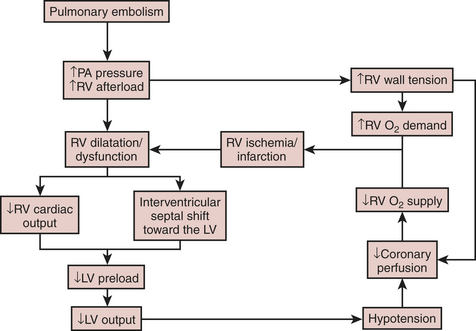
Massive PE represents 5% of cardiac arrests, with more than 60% of those noted to be pulseless electrical activity. Symptoms include severe dyspnea, cyanosis, tachycardia, and elevated CVP. Massive PE is defined as 50% obstruction of pulmonary blood flow that usually leads to RV failure. On mechanical obstruction, humoral mediators are released, augmenting pulmonary vasoconstriction and PAH. This sudden increase in RV afterload results in RV dilation and dysfunction that displaces the interventricular septum, causing underfilling of the LV (Fig. 18-4). Low CO and severe hypotension follow, ultimately leading to circulatory collapse and death. Increased RV pressure also compresses the right coronary artery, causing RV ischemia and contributing to RV failure. Mortality is 40% to 80% within 2 hours of the onset of a PE.
Anesthesia Considerations for Pulmonary Embolectomy and Pulmonary Embolism
The first surgical embolectomy without CPB was described by Trendelenburg in 1908, and the first with CPB was described by Cooley in 1961. Its frequency has decreased, partly due to the success of thrombolytic measures for massive PE; however, thrombolysis is unsuccessful in 15% to 30% of cases. Of 3000 patients with documented PE over a 20-year period, 3% underwent pulmonary embolectomy.11 In general, it has been reserved for patients with PE and refractory circulatory compromise, failed medical management, or contraindications to thrombolysis. Contraindication to thrombolysis accounts for more than one third of embolectomies. The overall mortality of pulmonary embolectomy varies between 20% and 90%. The patient’s preoperative status is predictive of survival. Preoperative cardiac arrest increases operative mortality by more than 50%. Age older than 60 years and a long history of dyspnea also increase the mortality of embolectomy. Echocardiography has greatly decreased the time to surgery by enabling a rapid and reliable diagnosis to be made without angiography. This has improved not only initial survival rate of embolectomy but also long-term survival.
PULMONARY ARTERY HYPERTENSION
Pulmonary artery hypertension, according to the 2004 WHO classification, is defined as a group of diseases characterized by virtually identical obstructive pathologic changes of the pulmonary microcirculation and by a favorable response to the long-term administration of prostacyclin.12 The WHO classifies PAH into the following groups:
Secondary Pulmonary Hypertension
Anesthetic management of individuals with PAH is challenging because perioperative increases in PVR readily occur and may provoke right-sided heart failure, resulting in death. The tolerance of the RV is a major concern. The RV is acutely sensitive to increases in PVR (afterload). Factors that increase PVR, such as hypoxia, acidosis, hypercapnia, hypothermia, and α-adrenergic stimulation, should be minimized. Furthermore, a decrease in blood pressure or increase in RV pressure impairs coronary perfusion of the right side of the heart. A PA catheter allows perioperative detection and monitoring of PVR and therapy, enabling hyperventilation to reduce PVR. In general, intravenous anesthetics have less effect on hypoxic pulmonary vasoconstriction, PVR, and oxygenation than do volatile agents. Nitrous oxide has been reported to increase PVR, but it is not contraindicated in these patients. Isoflurane may be beneficial by decreasing PAP and has been frequently used during noncardiac procedures. Fentanyl may be given as an adjunct or a primary anesthetic agent in these patients because it causes little myocardial depression and excellent circulatory stability.13
Pulmonary endarterectomy (PEA) is the accepted treatment today for chronic thromboembolic pulmonary hypertension. The operation is not an embolectomy but rather a true endarterectomy, removing the fibrosis obstructive tissue from the pulmonary arteries. Extracorporeal circulation and periods of circulatory arrest under deep hypothermia are essential for successful endarterectomy.14
PERICARDIAL HEART DISEASE
Cardiac Tamponade
Tamponade exists when fluid accumulation in the pericardial sac limits filling of the heart. Hemodynamic manifestations are mainly due to atrial rather than ventricular compression. Initially, with mild tamponade, diastolic filling is limited, causing reduced stroke volume that stimulates sympathetic reflexes to increase heart rate and contractility to maintain CO.15 The rising RA pressure reflexly stimulates tachycardia and peripheral vasoconstriction. Blood pressure is supported by vasoconstriction, but CO begins to fall as pericardial fluid continues to increase. Subsequently, diastolic filling begins to disappear so the jugular venous pulse has no prominent Y-descent but a prominent X-descent. Eventually, the pericardial pressure-volume curve becomes almost vertical so any additional fluid greatly restricts cardiac filling and reduces diastolic compliance. Ultimately, the RA pressure, pulmonary artery diastolic pressure, and pulmonary capillary wedge pressure equilibrate. Equilibration of pressures (within 5 mmHg of each other) merits immediate action to rule out acute tamponade. Once the blood pressure begins to fall, it is a precipitous drop that reduces coronary artery blood flow, leading to ischemia, especially subendocardially.
CARDIAC SURGERY DURING PREGNANCY
The nonphysiologic nature of CPB combines with the changes of pregnancy for an unpredictable response and tolerance by mother and fetus.16 Initiation of CPB activates a whole-body inflammatory response, with multiple effects on coagulation, autoregulation, release of vasoactive substances, hemodilution, and other physiologic processes that may adversely affect the fetus and mother. Maternal blood pressure may fall immediately after or within 5 minutes of initiation of CPB, lowering placental perfusion secondary to low SVR, hemodilution, and release of vasoactive agents. Fetal heart rate variability is often lost and fetal bradycardia (<80 beats per minute) also may occur at this time. Because uterine blood flow is not autoregulated and relies on maternal blood flow, decreases in maternal blood pressure cause fetal hypoxia and bradycardia. Increasing CPB flows (>2.5 L/min/m2) or perfusion pressure (>70 mmHg) will raise maternal blood flow and usually returns the heart rate to 120 beats per minute. A compensatory catecholamine-driven tachycardia (170 beats per minute) may ensue that suggests an oxygen debt existed. Nonetheless, increasing CPB flow and mean arterial pressure does not always correct fetal bradycardia; if not, other causes must be considered. Problems with venous return or other mechanical aspects of extracorporeal circulation may also limit systemic flow, causing reduced placental perfusion. If acidosis persists throughout CPB, other factors may be responsible for it, such as maternal hypothermia, uterine contractions, or medications that are transferable to the fetus, rather than low maternal blood pressure. Monitoring the fetal heart rate is important to assess fetal viability and subsequent therapeutic initiatives. It partially reduces fetal mortality by early recognition of problems.
RENAL INSUFFICIENCY AND CARDIAC SURGERY
Anesthesia Considerations
A rapid-sequence induction with cricoid pressure is recommended in those with CRF in response to the likelihood of delayed gastric emptying. Significant extracellular volume contraction may also be present before induction of anesthesia due to a 6- to 8-hour fast before surgery and dialysis within 24 hours of surgery that may lead to hypotension on induction. Because fluid requirements are usually significant with CPB, a PA catheter is especially useful to manage fluid requirements. TEE may complement fluid management by assessment of LV volume and function. Before the initiation of CPB, fluid administration should be limited, especially if the patient is dialysis dependent. Otherwise, fluid should be given to maintain adequate urine output but avoid excessive cardiovascular filling pressures risking pulmonary edema. However, restricting fluids too aggressively in these patients may lead to acute renal failure superimposed on CRF. Low-dose dopamine has been recommended for patients with CRF, but its value is indeterminate. Fenoldopam, a new dopamine-1-receptor agonist, may reduce the incidence of renal dysfunction in patients with multiple risk factors for renal failure undergoing CPB. Patients with preoperative creatinine levels above 1.5 mg/dL were given renal-dose dopamine or fenoldopam perioperatively.17 Postoperative parameters were only improved in those receiving fenoldopam, suggesting a renal protective effect.
HEMATOLOGIC PROBLEMS IN PATIENTS UNDERGOING CARDIAC SURGERY
Antithrombin
Various blood products have been tried as an alternative to fresh frozen plasma (FFP) to rapidly raise the AT level. Cryoprecipitate and FFP have similar amounts of AT, but the infectious risk is greater for cryoprecipitate. AT concentrate preparations are derived from human plasma pools but are subjected to fractionation procedures and heating to inactivate viral contaminants. This process does not reduce biologic activity and viral transmission has not been reported. One bottle of AT contains approximately 500 units or the equivalent of 2 units of FFP and can be safely administered over 10 to 20 minutes. The baseline AT activity of the heparin-resistant patients was 56 ± 25%, which improved to 75 ± 31% after administration of AT concentrate.18
Cold Agglutinins
If hypothermic CPB is necessary despite the presence of CAs, the choices are preoperative plasmapheresis, standard hemodilution, and maintenance of CPB temperature above the CA thermal amplitude.19 Cold cardioplegia may be used without first undergoing plasmapheresis if normothermic CPB is used and 37°C cardioplegic solution is injected before administration of 4°C cardioplegic solution, clearing all potentially reactive cells. The risk of hemolysis is still high in patients with high-thermal-amplitude CAs. If CAs are particularly malignant, all of the patient’s blood from the venous reservoir is drained and discarded. It is replaced entirely by donor blood, unfortunately exposing the patient to allogeneic blood products. Today, normothermic CPB and antegrade or retrograde warm blood cardioplegia may be the best option. If CAs should go undetected, postoperative end-organ damage or low CO may occur. Subsequently, plasma exchange, corticosteroids, elevated urine output, and maintenance of a good CO are the best treatments.
SUMMARY
1. Bakaeen F.G., Reardon M.J., Coselli J.S., et al. Surgical outcome in 85 patients with primary cardiac tumors. Am J Surg. 2003;186:641.
2. Amano J., Kono T., Wada Y., et al. Cardiac myxoma: Its origin and tumor characteristics. Ann Thorac Cardiovasc Surg. 2003;9:215.
3. Weingarten T.N., Abel M.D., Connolly H.M., et al. Anesthetic management for cardiac surgery in carcinoid heart disease: A review of 84 patients. Anesth Analg. 2003;96:SCA 138.
4. Tang W.H., Francis G.S. Novel pharmacological treatments for heart failure. Exp Opin Investig Drugs. 2003;12:1791.
5. Maron B.J., McKenna W.J., Danielson G.K., et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687.
6. Roberts R., Sigwart U. New concepts in hypertrophic cardiomyopathies: II. Circulation. 2001;104:2249.
7. Chatterjee K., Alpert J. Constrictive pericarditis and restrictive cardiomyopathy: Similarities and differences. Heart Failure Monit. 2003;3:118.
8. Fontaine G., Gallais Y., Fornes P., et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy. Anesthesiology. 2001;95:250.
9. Hanson E.W., Neerhut R.K., Lynch C.3rd:. Mitral valve prolapse. Anesthesiology. 1996;85:178.
10. Goldhaber S.Z., Elliott C.G. Acute pulmonary embolism: I. Epidemiology, pathophysiology, and diagnosis. Circulation. 2003;108:2726.
11. Aklog L., Williams C.S., Byrne J.G., et al. Acute pulmonary embolectomy: A contemporary approach. Circulation. 2002;105:1416.
12. Simonneau G., Galie N., Rubin L.J., et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43(suppl S):55.
13. Fischer L.G., Van Aken H., Burkle H. Management of pulmonary hypertension: Physiological and pharmacological considerations for anesthesiologists. Anesth Analg. 2003;96:1603.
14. Manecke G.R., Wilson W., Auger W.R., et al. Anesthesia for pulmonary thromboendarterectomy. Semin Cardiothorac Vasc Anesth. 2005;9:189-204.
15. Maisch B., Ristic A.D. The classification of pericardial disease in the age of modern medicine. Curr Cardiol Rep. 2002;4:13.
16. Mahli A., Izdes S., Coskun D. Cardiac operations during pregnancy: Review of factors influencing fetal outcome. Ann Thorac Surg. 2000;69:1622.
17. Landoni G., Zoccai G., Marino G., et al. Fenoldopam reduces the need for renal replacement therapy and in-hospital death in cardiovascular surgery: A meta-analysis. J Cardiothorac Vasc Anesth. 2008;22:27.
18. Lemmer J.H.Jr., Despotis G.J. Antithrombin III concentrate to treat heparin resistance in patients undergoing cardiac surgery. J Thorac Cardiovasc Surg. 2002;123:213.
19. Agarwal S.K., Ghosh P.K., Gupta D. Cardiac surgery and cold-reactive proteins. Ann Thorac Surg. 1995;60:1143.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 18 Uncommon Cardiac Diseases
CARDIAC TUMORS
Cardiac tumors are increasingly diagnosed before autopsy due to advancements in imaging, especially metastatic tumors of the heart and pericardium, which account for a majority of cardiac tumors. Data pooled from 22 large autopsy series show the prevalence of adult primary cardiac tumors as only about 0.02%, yet they are responsible for significant morbidity and mortality. Malignant tumors encompass about 20% of primary tumors in adults.1 Diagnosis can be elusive because these tumors may be associated with nonspecific symptoms mimicking other disease entities. Two-dimensional echocardiography (echo) modalities and magnetic resonance imaging (MRI) have allowed earlier, more frequent, and more complete assessment of cardiac tumors.
Myxoma
Rarely discovered by incidental echocardiography examination, myxomas may manifest a variety of symptoms. The classic triad includes embolism, intracardiac obstruction, and constitutional symptoms. Approximately 80% of individuals present with one component of the triad, yet up to 10% may be asymptomatic even with mitral myxomas, arising from both atrial and ventricular sides of the anterior mitral leaflet. The most common initial symptom, dyspnea on exertion, reflects mitral valve obstruction usually present with LA myxomas (Fig. 18-1). Because of the pedunculated nature of some myxomas, temporary obstruction of blood flow may cause hemolysis, hypotension, syncope, or sudden death. Other symptoms of mitral obstruction similar to mitral stenosis such as hemoptysis, systemic embolization, fever, and weight loss may also occur. The persistence of sinus rhythm in the presence of such symptoms may help distinguish atrial myxoma from mitral stenosis. Severe pulmonary hypertension without significant mitral valve involvement suggests obstruction of the tricuspid valve and recurrent pulmonary emboli known to occur with a myxoma in the RA or right ventricle (RV). Before echocardiography, angiography was used to identify all myxomas, but now it is only useful to confirm the diagnosis or determine coronary anatomy if considered necessary. TEE is 100% sensitive for diagnosis of myxoma. Specifically, it yields morphologic detail in the evaluation of cardiac tumors, including points of tumor attachment and degree of mobility. Computed tomography (CT) and MRI can help delineate the extent of the tumor and its relationships to surrounding cardiac and thoracic structures. MRI is especially valuable in the diagnosis of myxoma when masses are equivocal or suboptimal on echocardiography or if the tumor is atypical in presentation. Difficulty may arise in differentiating thrombus from myxoma because both are so heterogeneous.2
Tumors with Systemic Cardiac Manifestations
Anesthetic Considerations
Patients who have carcinoid heart disease and require cardiac surgery pose an anesthetic challenge.3 A carcinoid crisis with vasoactive mediator release can be provoked by stress, physical stimulation, or medications such as meperidine, morphine, or histamine-releasing muscle relaxants (atracurium). Preoperative control of carcinoid activity is a critical aspect of perioperative management, made considerably easier with the administration of octreotide, a synthetic analog of somatostatin that inhibits the vasoactive compounds that produce carcinoid syndrome. It reduces the occurrence of symptoms in more than 70% of patients. The longer half-life of octreotide than somatostatin allows subcutaneous injection of 150 μg three times daily to control symptoms. Intermittent intravenous doses of 50 to 200 μg or continuous infusions are given to stop severe hypotension and prevent further carcinoid symptoms. Severe hyperglycemia may occur with octreotide due to its inhibition of insulin secretion.
CARDIOMYOPATHY
The annual incidence of cardiomyopathy in adults is 8.7 cases per 100,000 person-years. General characteristics of all four cardiomyopathies are displayed in Table 18-1.
Dilated Cardiomyopathy
Management of acute decompensated CHF continues to evolve, but the onset of overt CHF is a poor prognostic indicator for patients with DCM. Treatment revolves around management of symptoms and progression of DCM, whereas other measures are designed to prevent complications such as pulmonary thromboembolism and arrhythmias. The mainstay of therapy for DCM is vasodilators combined with digoxin and diuretics.4 All patients receive angiotensin-converting enzyme inhibitors (ACEIs) to reduce symptoms, improve exercise tolerance, and reduce cardiovascular mortality without a direct myocardial effect. Perhaps more important than the hemodynamic effects, ACEIs suppress ventricular remodeling and endothelial dysfunction, accounting for the improvement in mortality noted with this medication in DCM. Other afterload-reducing agents, such as selective phosphodiesterase-3 inhibitors like milrinone, may improve quality of life but do not affect mortality, so they are rarely administered in chronic situations. Spironolactone has assumed a greater role in treatment as mortality was reduced by 30% from all causes in patients receiving standard ACEIs for DCM with the addition of spironolactone in a large double-blind randomized trial. The use of β-blockers in DCM has provided not only symptomatic improvement but also substantial reductions in sudden death and progressive death in patients with New York Heart Association (NYHA) class II and III heart failure. This is especially significant because almost 50% of deaths are sudden. High-grade ventricular arrhythmias are common with DCM. Approximately 12% of all patients with DCM die suddenly, but overall prediction of sudden death in an individual with DCM is poor. The best predictor of sudden death remains the degree of LV dysfunction. Patients who have sustained ventricular tachycardia or out-of-hospital ventricular fibrillation are at increased risk for sudden death, but more than 70% of patients with DCM have nonsustained ventricular tachycardia during ambulatory monitoring. Antiarrhythmic medications are hazardous in patients with poor ventricular function owing to their negative inotropic and sometimes proarrhythmic properties. Amiodarone is the preferred antiarrhythmic agent in DCM because its negative inotropic effect is less than that of other antiarrhythmic medications and its proarrhythmic potential is lowest. Implantable defibrillators reduce the risk of sudden death as well as reducing mortality. Evidence has indicated that with previous cardiac arrest or sustained ventricular tachycardia, more benefit was gained from use of an implantable defibrillator. This was based on a 27% reduction in the relative risk of death attributed to a 50% reduction in arrhythmia-related mortality compared with treatment with amiodarone.
Hypertrophic Cardiomyopathy
Referred to as idiopathic hypertrophic subaortic stenosis, hypertrophic obstructive cardiomyopathy, and asymmetric septal hypertrophy, among other names, the accepted term is now hypertrophic cardiomyopathy.5 In the past 40 years, advancements regarding the hemodynamics, systolic and diastolic abnormalities, electrophysiology, genetics, and clinical care of HCM have contributed to a greater understanding of this disease. HCM is the most common genetic cardiac disease, with marked heterogeneity in clinical expression, pathophysiology, and prognosis. The overall prevalence for adults in the general population is 0.2%, affecting men and women equally.
Two-dimensional echocardiography establishes the diagnosis of HCM easily and reliably. Classic echocardiography features are thickening of the entire ventricular septum from base to apex disproportional to that of the posterior wall, poor septal motion, and anterior displacement of the mitral valve without LV dilation (Fig. 18-2). Echocardiography has reduced the need for invasive catheterization procedures, unless coronary artery disease (CAD) or severe mitral valve disease is suspected or diagnostic problems are present. MRI has been useful in cases in which echocardiography is technically inadequate.
Two thirds of individuals with LV outflow tract obstruction become severely symptomatic and 10% die within 4 years of diagnosis. The outflow tract is narrowed from septal hypertrophy and anterior displacement of the papillary muscles and mitral leaflets, creating a dynamic LV outflow obstruction (see Fig. 18-2). Elongation of the mitral leaflets results in coaptation of the body of the leaflets instead of the tips. The part of the anterior leaflet distal to the coaptation is subjected to strong Venturi forces that provoke systolic anterior motion (SAM), mitral septal contact, and ultimately LV outflow obstruction. SAM of the anterior leaflet may also cause mitral regurgitation. The onset and duration of mitral leaflet-septal contact determine the magnitude of the gradient and the degree of mitral regurgitation. The pressure gradient between the aorta and LV is worsened by decreased end-diastolic volume, increased contractility, or decreased aortic outflow resistance (Fig. 18-3).
Surgical correction of HCM is directed primarily at relieving symptoms of LV obstruction in the 5% of patients who are refractory to medication.6 In general, these are individuals with subaortic gradients more than 50 mmHg and frequently associated with severe CHF. A myotomy-myectomy through a transaortic approach relieves the obstruction. The muscle is excised from the proximal septum extending just beyond the mitral valve leaflets to widen the LV outflow tract. This is a technically challenging operation due to the limited exposure and precise area to excise the muscle. It is usually reserved for centers with considerable experience. When myectomy is successful, the outflow tract of the LV is widened and SAM, mitral regurgitation, and outflow gradient are all decreased.
Restrictive Cardiomyopathy
RCM may be classified as myocardial (infiltrative, noninfiltrative, and storage) or endomyocardial (Box 18-1) according to etiology. RCM may be associated with another disease entity except pericardial disease. Restrictive myocardial disorders are characteristically atypical in presentation and hemodynamics at times, complicating perioperative management.
