[level-membership-for-pediatrics-category]
Chapter 662 Tumors of the Skin
See also Chapters 499 and 643.
Epidermal Inclusion Cyst (Epidermoid Cyst)
Epidermoid cysts are the nodules most commonly seen in children. Such a cyst is a sharply circumscribed, dome-shaped, firm, freely movable, skin-colored nodule (see Fig. 662-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com  ), often with a central dimple or punctum that is a plugged, dilated pore of a pilosebaceous follicle. Epidermoid cysts form most frequently on the face, neck, chest, or upper back and may periodically become inflamed and infected secondarily, particularly in association with acne vulgaris. The cyst wall may also rupture and induce an inflammatory reaction in the dermis. The wall of the cyst is derived from the follicular infundibulum. A mass of layered keratinized material that may have a cheesy consistency fills the cavity. Epidermoid cysts may arise from occlusion of pilosebaceous follicles, from implantation of epidermal cells into the dermis as a result of an injury that penetrates the epidermis, and from rests of epidermal cells. Multiple epidermoid cysts may be present in Gardner syndrome and the nevoid basal cell carcinoma syndrome. Excision of the cysts with removal of the entire sac and its contents is indicated, particularly if the cyst becomes recurrently infected. A fluctuant, infected cyst should be treated with an antibiotic effective against Staphylococcus aureus. After the inflammation subsides, the cyst should be removed.
), often with a central dimple or punctum that is a plugged, dilated pore of a pilosebaceous follicle. Epidermoid cysts form most frequently on the face, neck, chest, or upper back and may periodically become inflamed and infected secondarily, particularly in association with acne vulgaris. The cyst wall may also rupture and induce an inflammatory reaction in the dermis. The wall of the cyst is derived from the follicular infundibulum. A mass of layered keratinized material that may have a cheesy consistency fills the cavity. Epidermoid cysts may arise from occlusion of pilosebaceous follicles, from implantation of epidermal cells into the dermis as a result of an injury that penetrates the epidermis, and from rests of epidermal cells. Multiple epidermoid cysts may be present in Gardner syndrome and the nevoid basal cell carcinoma syndrome. Excision of the cysts with removal of the entire sac and its contents is indicated, particularly if the cyst becomes recurrently infected. A fluctuant, infected cyst should be treated with an antibiotic effective against Staphylococcus aureus. After the inflammation subsides, the cyst should be removed.
Milium
Milium is a 1- to 2-mm, firm, pearly white or yellowish, subepidermal keratin cyst. Milia in newborns is discussed in Chapter 639. Secondary milia occur in association with subepidermal blistering diseases, after dermabrasion or other injury to the skin. They are retention cysts caused by hyperproliferation of injured epithelium and are indistinguishable histopathologically from primary milia. Those that develop after blistering usually arise from the eccrine sweat duct, but they may develop from the hair follicle, sebaceous duct, or epidermis. A milium body differs from an epidermoid cyst only in its small size and superficial location.
Pilar Cyst (Trichilemmal Cyst)
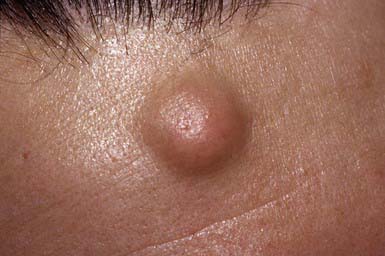
Pilar cyst may be clinically indistinguishable from an epidermoid cyst. It manifests as a smooth, firm, mobile nodule, predominantly on the scalp (Fig. 662-2). Pilar cysts occasionally develop on the face, neck, or trunk. A cyst may become inflamed and may occasionally suppurate and ulcerate. The cyst wall is composed of epithelial cells with indistinct intercellular bridges. The peripheral cell layer of the wall shows a palisade arrangement, which is not seen in an epidermoid cyst. No granular layer is present. The cyst cavity contains homogeneous eosinophilic keratinous material, and foci of calcification are seen in 25% of cases. The propensity for development of pilar cysts may be inherited in an autosomal dominant manner. More than one cyst generally develops in a patient. Numerous pilar and epidermoid cysts, desmoid tumors, fibromas, lipomas, or osteomas may be associated with colonic polyposis or adenocarcinoma in Gardner syndrome. Pilar cysts shell out easily from the dermis.
Pilomatricoma
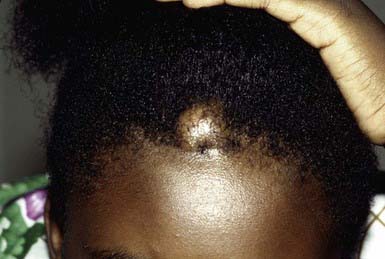
The second most common nodule seen in children, pilomatricoma is a benign tumor that manifests as a 3- to 30-mm, firm, solitary, deep dermal or subcutaneous tumor on the head, neck, or upper extremities. The overlying epidermis is usually normal. The tumor may occasionally be located more superficially, however, tinting the overlying skin blue-red (Fig. 662-3). Multiple pilomatricomas are seen in myotonic dystrophy, Gardner syndrome, Rubinstein-Taybi syndrome, and Turner syndrome. In general, however, pilomatricomas are not hereditary. Histopathologically, irregularly shaped islands of epithelial cells are embedded in a cellular stroma. Calcium deposits are found in 75% of tumors. Pilomatricomas are caused by mutations in beta-catenin.
Eruptive Vellus Hair Cysts
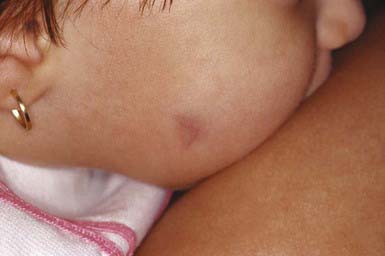
Eruptive vellus hair cysts are 1- to 3-mm, asymptomatic, soft, skin-colored follicular papules on the chest (Fig. 662-4). They may become crusted or umbilicated. Abnormal vellus hair follicles become occluded at the level of the infundibulum, resulting in retention of hairs within an epithelium-lined cystic dilatation of the proximal part of the follicle. Most cases are chronic, but spontaneous regression has been reported.
Syringoma
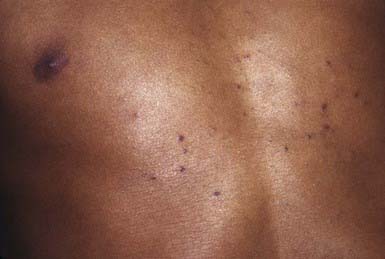
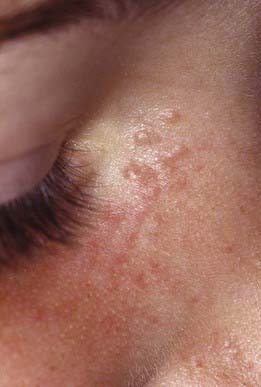
The benign tumors known as syringomas are soft, small, skin-colored or yellowish brown papules that develop on the face, particularly in the periorbital regions (Fig. 662-5). Other sites of predilection include the axillae and umbilical and pubic areas. They often develop during puberty and are more frequent in females. Eruptive syringomas develop in crops over the anterior trunk during childhood or adolescence. A syringoma is derived from an intraepidermal sweat gland duct. Syringomas are of cosmetic significance only. Sparse lesions may be excised, but they are often too numerous to remove.
Dermatofibroma (Histiocytoma)
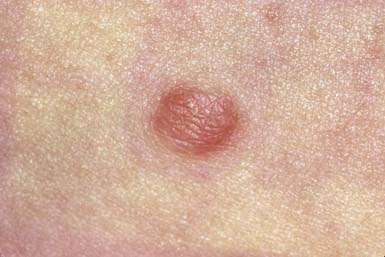
A benign dermal tumor, dermatofibroma may be pedunculated, nodular (Fig. 662-6), or flat and is usually well circumscribed and firm but occasionally feels soft on palpation. The overlying skin is usually hyperpigmented, may be shiny or keratotic, and dimples when the tumor is pinched. Dermatofibromas range in size from 0.5 to 10 mm, arise most frequently on the limbs, and are usually asymptomatic but may occasionally be pruritic. They are composed of fibroblasts, young and mature collagen, capillaries, and histiocytes in varying proportions, forming a nodule in the dermis that has poorly defined edges. The cause of these tumors is unknown, but trauma such as an insect bite or folliculitis appears to induce reactive fibroplasia. The differential diagnosis includes epidermal inclusion cyst, juvenile xanthogranuloma, hypertrophic scar, and neurofibroma. Dermatofibromas may be excised or left intact, according to the patient’s preference. They usually persist indefinitely.
Juvenile Xanthogranuloma
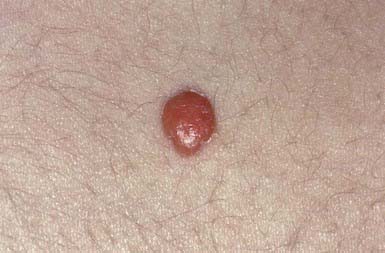
A firm, dome-shaped, yellow, pink, or orange papule or nodule (Fig. 662-7), juvenile xanthogranuloma varies from 5 mm to approximately 4 cm in diameter. The average age at onset is 2 years. These nodules are 10 times more common in white than in black individuals. Sites of predilection are the scalp, face, and upper trunk, where they may erupt in profusion or remain as solitary lesions. Nodular lesions may appear on the oral mucosa. Mature lesions are characterized histopathologically by a dermal infiltrate of lipid-laden histiocytes, admixed inflammatory cells, and Touton giant cells. The lesions may clinically resemble papulonodular urticaria pigmentosa, dermatofibromas, or xanthomas of hyperlipoproteinemia but can be distinguished from these entities histopathologically.
Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
Efron PA, Chen MK, Glavin FL, et al. Pediatric basal cell carcinoma: case reports and literature review. J Pediatr Surg. 2008;43:2277-2280.
Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004;6:530-539.
Laskin WB, Miettinen M, Fetsch JF. Infantile digital fibroma/fibromatosis: a clinicopathologic and immunohistochemical study of 69 tumors from 57 patients with long-term follow-up. Am J Surg Pathol. 2009;33:1-13.
Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Orphanet J Rare Dis. 2008;3:32.
Pires-Fraga MF, Mello D, Jorge D, et al. Congenital infiltrating lipomatosis. J Plast Reconstr Aesthet Surg. 2009;62:e561-e564.
Price HN, Zaenglein AL. Diagnosis and management of benign lumps and bumps of childhood. Curr Opin Pediatr. 2007;19:420-424.
Raue F, Frank-Raue K. Genotype-phenotype relationship in multiple endocrine neoplasia type2: implications for clinical management. Hormones. 2009;8:23-28.
Van der Geer S, Ostertag JU, Krekels GA. Treatment of basal cell carcinomas in patients with nevoid basal cell carcinoma syndrome. J Eur Acad Dermatol Venereol. 2008;23:308-313.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 662 Tumors of the Skin
See also Chapters 499 and 643.
Epidermal Inclusion Cyst (Epidermoid Cyst)
Epidermoid cysts are the nodules most commonly seen in children. Such a cyst is a sharply circumscribed, dome-shaped, firm, freely movable, skin-colored nodule (see Fig. 662-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com ), often with a central dimple or punctum that is a plugged, dilated pore of a pilosebaceous follicle. Epidermoid cysts form most frequently on the face, neck, chest, or upper back and may periodically become inflamed and infected secondarily, particularly in association with acne vulgaris. The cyst wall may also rupture and induce an inflammatory reaction in the dermis. The wall of the cyst is derived from the follicular infundibulum. A mass of layered keratinized material that may have a cheesy consistency fills the cavity. Epidermoid cysts may arise from occlusion of pilosebaceous follicles, from implantation of epidermal cells into the dermis as a result of an injury that penetrates the epidermis, and from rests of epidermal cells. Multiple epidermoid cysts may be present in Gardner syndrome and the nevoid basal cell carcinoma syndrome. Excision of the cysts with removal of the entire sac and its contents is indicated, particularly if the cyst becomes recurrently infected. A fluctuant, infected cyst should be treated with an antibiotic effective against Staphylococcus aureus. After the inflammation subsides, the cyst should be removed.
Milium
Milium is a 1- to 2-mm, firm, pearly white or yellowish, subepidermal keratin cyst. Milia in newborns is discussed in Chapter 639. Secondary milia occur in association with subepidermal blistering diseases, after dermabrasion or other injury to the skin. They are retention cysts caused by hyperproliferation of injured epithelium and are indistinguishable histopathologically from primary milia. Those that develop after blistering usually arise from the eccrine sweat duct, but they may develop from the hair follicle, sebaceous duct, or epidermis. A milium body differs from an epidermoid cyst only in its small size and superficial location.
Pilar Cyst (Trichilemmal Cyst)
Pilar cyst may be clinically indistinguishable from an epidermoid cyst. It manifests as a smooth, firm, mobile nodule, predominantly on the scalp (Fig. 662-2). Pilar cysts occasionally develop on the face, neck, or trunk. A cyst may become inflamed and may occasionally suppurate and ulcerate. The cyst wall is composed of epithelial cells with indistinct intercellular bridges. The peripheral cell layer of the wall shows a palisade arrangement, which is not seen in an epidermoid cyst. No granular layer is present. The cyst cavity contains homogeneous eosinophilic keratinous material, and foci of calcification are seen in 25% of cases. The propensity for development of pilar cysts may be inherited in an autosomal dominant manner. More than one cyst generally develops in a patient. Numerous pilar and epidermoid cysts, desmoid tumors, fibromas, lipomas, or osteomas may be associated with colonic polyposis or adenocarcinoma in Gardner syndrome. Pilar cysts shell out easily from the dermis.
Pilomatricoma
The second most common nodule seen in children, pilomatricoma is a benign tumor that manifests as a 3- to 30-mm, firm, solitary, deep dermal or subcutaneous tumor on the head, neck, or upper extremities. The overlying epidermis is usually normal. The tumor may occasionally be located more superficially, however, tinting the overlying skin blue-red (Fig. 662-3). Multiple pilomatricomas are seen in myotonic dystrophy, Gardner syndrome, Rubinstein-Taybi syndrome, and Turner syndrome. In general, however, pilomatricomas are not hereditary. Histopathologically, irregularly shaped islands of epithelial cells are embedded in a cellular stroma. Calcium deposits are found in 75% of tumors. Pilomatricomas are caused by mutations in beta-catenin.
Eruptive Vellus Hair Cysts
Eruptive vellus hair cysts are 1- to 3-mm, asymptomatic, soft, skin-colored follicular papules on the chest (Fig. 662-4). They may become crusted or umbilicated. Abnormal vellus hair follicles become occluded at the level of the infundibulum, resulting in retention of hairs within an epithelium-lined cystic dilatation of the proximal part of the follicle. Most cases are chronic, but spontaneous regression has been reported.
