Chapter 37 Tumors of the Pineal Region
• Pineal region tumors account for about 1% of adult and 3% to 8% of all pediatric intracranial tumors. They are rare but surgically treatable tumors that require surgical expertise, experience, and multimodality therapy.
• The histological variety of pineal region tumors consists of several major categories: (a) germ cell tumors (germinomas), (b) nongerminomatous tumors (embryonal carcinoma, choriocarcinoma, and teratoma), (c) pineal parenchymal tumors (pineoblastoma, pineocytoma, and papillary tumor), (d) tumors of the supporting structures (meningioma, ependymoma, astrocytoma, mixed glioma, choroid plexus neoplasm), and (e) other tumor types (metastases, cysts, lymphoma, variable).
• Diagnostic workup includes a thorough clinical and neurological examination, magnetic resonance imaging (MRI), and serum markers. Serum markers when positive are best used to follow the tumor response to therapy. Diagnostic approaches to pineal region tumors include stereotactic biopsy, endoscopic biopsy, and open surgery. In many cases, symptoms are caused by compression of the neural structures or obstructive hydrocephalus. Concomitant hydrocephalus can be treated by an endoscopic third ventriculostomy with biopsy, ventricular diversion/shunting, or surgical removal of the tumor.
• Pineal region tumors can be removed with relatively low morbidity using a variety or combination of approaches. The most popular ones are a posterior fossa approach (infratentorial-supracerebellar), supratentorial approaches (occipital transtentorial), and posterior and anterior transcallosal approaches. Surgeons should be familiar with the anatomical structures in the pineal region and with the indications, limitations, risks, and benefits of each approach. Each tumor requires an individualized surgical and treatment approach.
• Multimodal treatments such as radiotherapy, chemotherapy, and radiosurgery in combination with open microsurgery or stereotactic or endoscopic surgery should be incorporated into any treatment paradigm based on the extent of resection and histological type of tumor.
History
The mystique of the pineal gland’s function and the challenge of operating in this region make it worthwhile to provide a brief overview of the relevant historical facts. The anatomists of ancient times knew about the pineal body and named it konareion because of its cone-shaped appearance. It was first described by Herophilus of Alexandria (325-280 BC). Like all his contemporaries, he believed the ventricles to be the “seat of spirits” and thought that the pineal body acted like a sphincter, regulating the flow of thoughts between the third and the fourth ventricles. Galen (129-201 AD) was most likely the first to hypothesize that the pineal body served as a gland. During Galen’s time, the structures of the pineal region were compared with the external genital organs of men. They named the pineal body the “penis,” the superior colliculi of the quadrigeminal plate “testes,” and the inferior colliculli of the quadrigeminal plate “nates,” simply based on their comparable appearance to these structures. Gibson, in 1763, would later recapitulate these ideas in his Epitome of Anatomy.
Medical history therefore has three hypotheses about the function of the pineal gland:
Anatomy
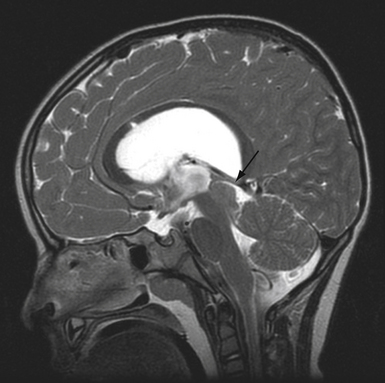
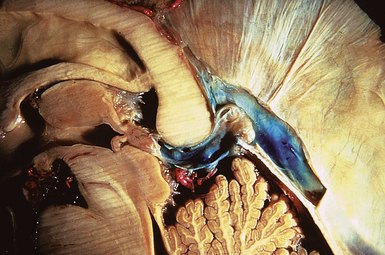
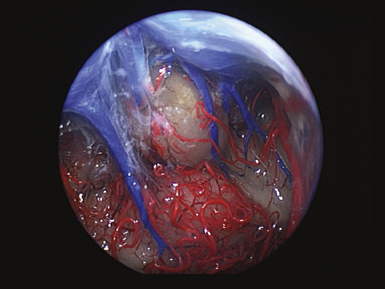
The pineal region is defined as the area of the brain bordered superiorly by the splenium of the corpus callosum and the tela choroidea, inferiorly by the quadrigeminal plate and midbrain tectum, anteriorly by the posterior parts of the third ventricle, and posteriorly by the cerebellar vermis as can be nicely shown in a midline sagittal section through the pineal gland and surrounding anatomical structures in an injected fixed human cadaver (Fig. 37.1). The pineal gland itself, as an invagination of the diencephalic ependymal roof between the habenular commissure and the posterior commissure, lies between the superior colliculi and basal to the splenium of the corpus callosum. It has a thin stalk to the epithalamus and developmentally is considered to be a paired structure. The neural connections with adjacent centers are the topic of discussion but have to be clarified. The pineal body itself has an average length of 7.97 mm (range 5.0-12.0 mm) and is 4.25 mm (2.5-7.0 mm) in height and width. It is oval in shape. The suprapineal recess projects posteriorly between the upper surface of the pineal gland and the lower layer of the tela choroidea in the roof. The stalk of the pineal body, from which the gland extends into the quadrigeminal cistern, has a cranial lamina and a caudal lamina. The habenular commissure, which connects the habenulae, crosses the midline over the cranial lamina, and the posterior commissure crosses the caudal lamina. The pineal recess projects posteriorly from the third ventricle into the pineal body between the two laminae. The posterior commissure forms the base of the triangular orifice of the aqueduct of Sylvius; the other two limbs are formed by the central gray matter of the midbrain. The quadrigeminal cistern is the subarachnoid space of the pineal region, and provides protection for the midbrain from the sharp edge of the tentorial notch. The arteries and veins inside the cistern are embedded in the firm arachnoidal septa. The quadrigeminal cistern communicates anteriorly with the cistern of the tela choroidea of the third ventricle, dorsally along the great vein of Galen with the dorsal cistern of the corpus callosum, laterally into the alae, caudally into the superior cistern of the cerebellum, which is formed by the medullary velum and the vermis, and anterodorsally into the ambient cistern. Through the latter cistern, the large arteries, the posterior cerebral artery and its branches, and the superior cerebellar artery approach the dorsal surface of the brainstem along the edge of the tentorium. The posterior border of the quadrigeminal cistern is formed by thickened opalescent arachnoid. Care must be taken when dissecting this area because the vein of Galen and its tributaries lie immediately rostral. The mesencephalon with the pineal body has its arterial blood supply from various sources. Small vessels supplying the parenchyma of the cerebral peduncles and the rest of the midbrain arise from the posterior communicating artery as perforating branches forming an internal or direct system. The vascularization in situ can be shown using neuroendoscopic techniques in injected human cadavers (arteries red, veins blue) and gives an impression of the enormous variety of the vascular structure (Fig. 37.2). All these arteries widely anastomose with each other, which explains the rarity of infarcts in the mesencephalon and the relative tolerance of the midbrain to direct surgical intervention. The pineal body is supplied by the pineal artery, which originates from the medial posterior choroidal artery, arising from the posterior cerebral artery (PCA), and enters the gland through its lateral portion from both sides. The great vein of Galen and its tributaries form a roof-like, dense venous network above the pineal body and the quadrigeminal plate, formed from the internal cerebral veins after they pass posteriorly through the tela choroidea and unite with the basal veins of Rosenthal. Other tributaries of the vein of Galen include the precentral cerebellar vein, internal occipital vein, posterior mesencephalic vein, and posterior ventricular vein. The straight sinus is formed by the joining of the great vein of Galen with the inferior sagittal sinus.
Histology
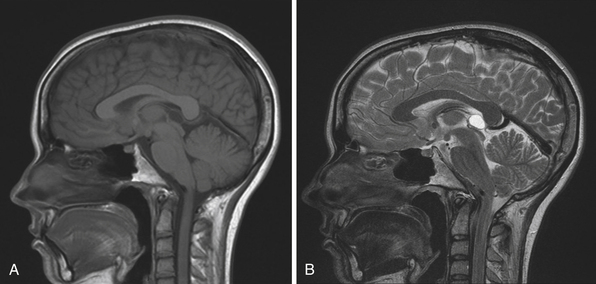
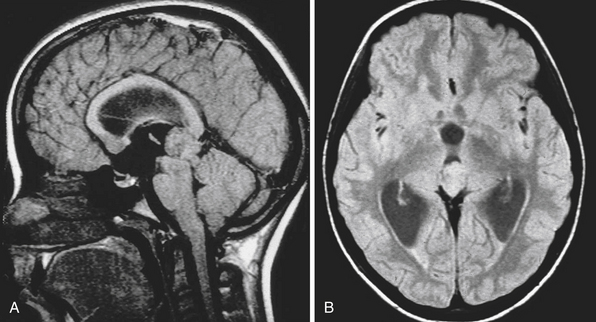
The normal pineal gland contains pinealocytes derived from amino precursor uptake and decarboxylation (APUD) cells and glial cells (astrocytes). The pineal gland is usually calcified by the age of 16 and appears so on routine computed tomography (CT) scans. Pineal cysts can be found incidentally in magnetic resonance imaging (MRI) studies in about 1.1% to 4.3% and this incidence seems to be higher in females (~2.4%) than in males (~1.5%) (Fig. 37.3). They can be identified in patients of all ages, with an increased prevalence found in older patients and in approximately 25% to 40% of autopsies, albeit many of the pineal cysts are microcystic. Their etiology remains obscure and may be the result of ischemic glial degeneration or of sequestration of the pineal diverticulum. Usually they are benign, non-neoplastic and may contain clear, xanthochromic, sometimes hemorrhagic, fluid. In the majority of cases, they are asymptomatic and may be followed on serial MRIs. Only in very rare cases do they show an enlargement, and may become symptomatic by causing hydrocephalus through aqueductal compression, headache, or gaze paresis, or hypothalamic symptoms. In those unusual situations with neurological symptoms or signs there is an indication to consider neurosurgical intervention.
Function
In reptiles, the pineal gland functions as a photoreceptor to change skin color in response to light. In humans, it is involved with hormone secretion for circadian rhythms and is known to have a neurotransmitter secretory function. The pineal gland inhibits gonadal development and regulates menstruation, adrenal function, and thyroid function. It is innervated by sympathetic nerves from the superior cervical ganglion that release norepinephrine to increase the pineal gland’s melatonin secretion. Light stimuli thereby reach the pineal gland from the retina via a polysynaptic pathway and affect the production of the neuroendocrine substance, melatonin. Melatonin, a derivative of serotonin, is essential in regulating circadian rhythms in endocrine gland activity and produces an antigonadal effect through the hypothalamic-hypophyseal axis. The production of melatonin is inhibited by light. Apparently, in many animals the major function of the pineal gland is to regulate the reproductive cycle; in fall and winter when days are short, more melatonin is produced, causing a gonad-suppressing effect. The exact function of the pineal gland in humans remains unclear. Recent studies could prove a diagnostic value of the melatonin profile in case of tumors of the pineal region. These studies showed a dramatically reduced melatonin rhythm in cases of undifferentiated or invasive tumors as well as the absence of melatonin variation as a consequence of pineal distortion after surgery. Also, the evidence for melatonin deficiency is recognized as a predictive factor to prevent post-pinealectomy syndrome. The absence of production of melatonin, as after pinealectomy, causes a “jet-lag-like” syndrome consisting of a complete disturbance of circadian rhythms.
Pathology
Pineal region tumors are rare tumors, with an estimated overall incidence of about 1% of all intracranial tumors (ranging from 0.5% to 1.6% in different studies) and are more common in children (3-8%) than in adults (less than 1%). Also, the incidence of pineal region tumors is higher among the Japanese, at 4-6% of all intracranial tumors. A population-based study calculated the incidence of pineal region tumors at 0.06 to 0.07 per 100,000 persons per year. No significant differences in the incidence between races were noted. Other reports describe the incidence in children younger than 20 years of age to be about 0.061 per 100,000 children per year. They are uncommon tumors and occur in a wide variety of different histological types. Furthermore, many tumors are of mixed cell type. The pathological classification depends on the substrate in the pineal region from which the tumor may arise. There are tumors which arise from the pineal glandular tissue itself, such as pinealocytomas (WHO grade I), pineal parenchymal tumors of intermediate differentiation (WHO grades II or III), pinealoblastomas (WHO grade IV), and papillary tumors of the pineal region. Other tumors arise from the glial cells, such as astrocytomas, oligodrendrogliomas, and glial cysts. Arachnoid cells are the basis for pineal-region meningiomas and non-neoplastic arachnoid cysts. Ependymal cells give rise to ependymomas in this region. Sympathetic nerves serve as a substrate for chemodectomas. Germ cell remnants give rise to germ cell tumors such as germinoma, choriocarcinoma, embryonal carcinoma, endodermal sinus tumor (yolk sac tumor), and teratoma. Lastly, the absence of a blood-brain barrier in the pineal gland makes it a suitable site for hematogenous metastases mostly from breast cancer, stomach cancer, renal cancer, or melanomas (Table 37.1).
TABLE 37.1 Types of Pineal Tumors: 2007 WHO Classification of Tumors of the Central Nervous System
| Tumor | Origin | Frequency |
|---|---|---|
| Germ Cell Tumors | Rest of germ cells | ~60% |
| Germinoma Mature Teratoma Immature Teratoma Teratoma with Malignant Transformation Yolk sac tumor (endodermal sinus tumor) Embryonal carcinoma Choriocarcinoma |
||
| Pineal Parenchymal Tumors | Pineal glandular tissue | ~30% |
| Pineocytoma (WHO grade I) Pineal parenchymal tumor of intermediate differentiation (WHO grade II or III) Pinealoblastoma (WHO grade IV) Papillary tumor of pineal region |
||
| Tumors of Supportive and Adjacent Structures | ~10% | |
| Astrocytoma Glioma (glioblastoma or oligodendroglioma) Medulloepithelioma |
Glial cells | |
| Ependymoma Choroid plexus papilloma |
Ependymal lining | |
| Meningioma | Arachnoid cells | |
| Hemangioma Hemangiopericytoma or blastoma Chemodectoma Craniopharyngioma |
Vascular cells | |
| Non-neoplastic Tumor-like Conditions | <1% | |
| Arachnoid cyst | Arachnoid cells | |
| Degenerative cysts (pineal cysts) | Glial cells | |
| Cysticercosis | Parasites | |
| Arteriovenous malformations | Vascularization | |
| Cavernomas Aneurysms of the vein of Galen |
||
| Metastases | Absence of blood-brain barrier | <0.1% |
| Lung (most common), breast, stomach, kidney, melanoma | ||
Louis DN, Cavanee WK, Oghaki H. WHO Classification of Tumours of the Central Nervous System, 4th ed: WHO Classification of Tumours v. 1 (IARC WHO Classification of Tumours), World Health Org; 4th edition (May 2007).
Germ Cell Tumors
Germ cell tumors derive from pluripotential germ cells and span a wide range of differentiation and malignant characteristics. They constitute a unique class of rare tumors that affect mainly children and adolescents. Predominantly they occur in the midline when they arise in the central nervous system (CNS). Intracranial germ cell tumors, in general, account for 0.4% to 3.4% of intracranial neoplasms. Their average incidence is considered to be 0.1 per 100,000 persons per year. Pineal germ cell tumors occur primarily in males. Germ cell tumors in females are more common in the suprasellar region. Most germ cell tumors of the CNS occur in the first three decades (98%) with a peak in the second decade (65%) between 11 and 20 years. The histogenesis of germ cell tumors is a subject of controversy. For a long time, they have been assumed to represent the neoplastic offspring of primordial germ cells. During the last decade, however, a variety of speculative proposals have been published on this topic. As a result of the etiology, certain observations suggest that gonadotropins play a role in the development or progression of CNS germ cell tumors. These include the predilection with Klinefelter syndrome, a condition characterized by chronically elevated serum levels of gonadotropins. Due to the variety of possible origins, the following entities are distinguished: germinoma, embryonal carcinoma, yolk sac tumor (endodermal sinus tumor), choriocarcinoma, mature teratoma, immature teratoma, teratoma with malignant transformation, and mixed germ cell tumors. An accurate histological identification and subclassification of CNS germ cell tumors is critical for current treatment planning and prognostication. In fact, only the germinoma and teratoma are likely to be encountered as pure tumor types.
Germinoma
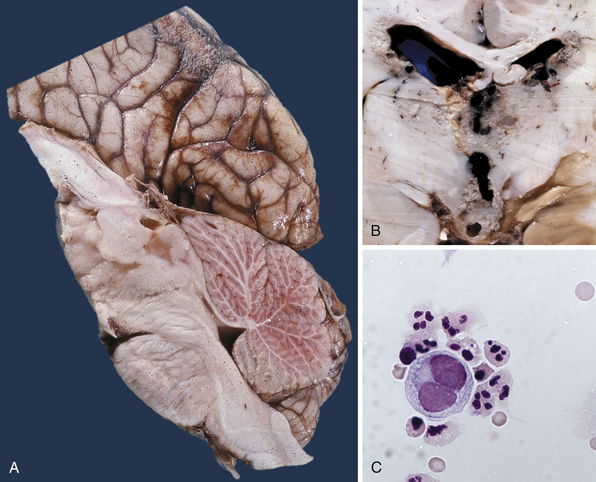
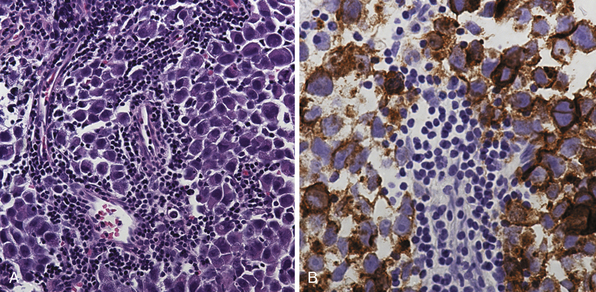
Pure germinomas (Fig. 37.4A to C) account for 65% to 72% of all intracranial germ cell tumors. These usually occur in the pineal region, although the second most common site is supra- and intrasellar, sometimes also parasellar. Germ cell tumors can be “synchronous,” and can be found both in the the suprasellar region and in the pineal region approximately 10% of the time. In fact, the discovery of a synchronous lesion on MRI is indicative of a germ cell tumor. The germ cell tumors are usually poorly circumscribed, light gray, granular, solid neoplasms that destroy the pineal gland and infiltrate the ventricular system and subarachnoid space early in their course. Hemorrhage into the tumor and necrosis or cystic degeneration are not often found. This tumor is composed of uniform cells resembling primitive germ cells, with large, vesicular nuclei, prominent nucleoli, and a clear, glycogen-rich cytoplasm. Additional features include lymphoid or lymphoplasma cellular infiltrates and, less frequently, scattered syncytiotrophoblastic giant cells (Fig. 37.5A and B).
Teratoma
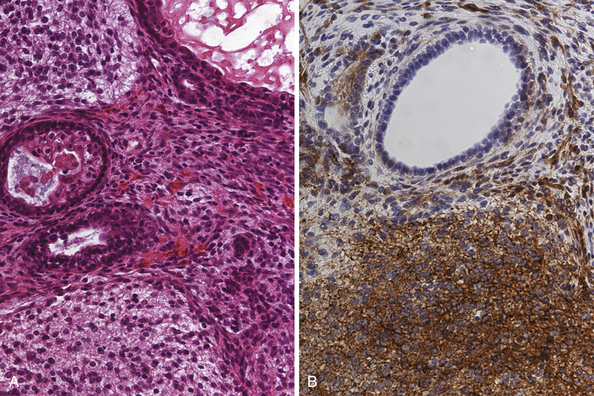
Teratomas (Fig. 37.6A and B) derive from all germ layers and are composed of well-differentiated tissues in an organ-like pattern. These tumors in the pineal region most often affect males. They are usually identified within the first two decades of life but occur more often in much younger children than other germ cell tumors (mostly in children younger than 9 years of age; about 20% occur between the ages of 16 and 18). By definition, the term teratoma can be used only when tumor elements derive from two or three germ layers. These tumors are usually well circumscribed, round or lobulated, and multicystic and compress the surrounding structures. The cystic component of the tumor may be watery, mucoid, or sebaceous. Sometimes bone, cartilage, hair, or teeth are present. Immature tumors are more frequently associated with a malignant course. Histologically, teratomas differentiate along ectodermal, endodermal, and mesodermal lines (e.g., they recapitulate somatic development from the three embryonic germ layers). Mature and immature variants as well as teratomas with malignant transformation must be distinguished from each other.
Immature Teratoma
This teratoma variant is composed of incompletely differentiated components resembling fetal tissue. Immature tumors are more frequently associated with a malignant course, but malignant potential may be explained by the presence of other germ cell tumor elements, such as choriocarcinoma or germinoma.
Pineal Parenchymal Tumors
Pinealocytoma (WHO Grade I)
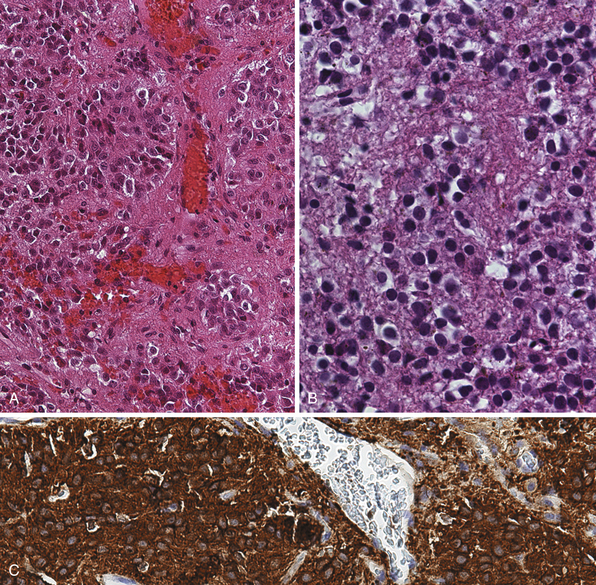
Pinealocytomas are defined as slow-growing pineal parenchymal neoplasms. They represent approximately 45% of all tumors with pineal parenchymal origin and occur throughout life, but most frequently in young adults (25-35 years). There is no sex predilection. Macroscopically, these tumors are well circumscribed with a gray-tan, homogeneous or granular cut surface. Degenerative changes, such as hemorrhage and small cystic cavities, can be present. They may disseminate widely along the CSF pathways. The histological appearance is of a well-differentiated neoplasm composed of small, uniform, mature cells resembling pinealocytes (Fig. 37.7A to C). They grow in sheets, but also feature large pinealocytomatous rosettes composed of abundant, delicate tumor cell processes. Pinealocytomas may contain astrocytic or neuronal components or both (so-called ganglioma of the pineal gland). Such tumors are the most benign of the pineal parenchymal neoplasms.
Pinealoblastoma (WHO Grade IV)
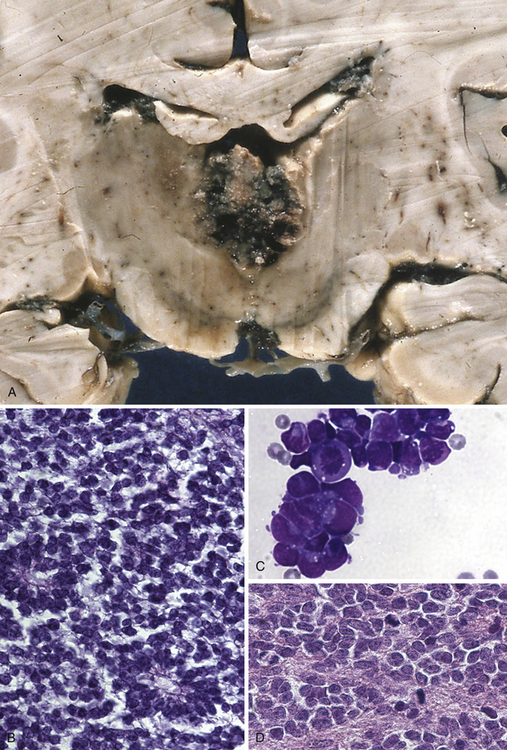
This type of tumor (Fig. 37.8A to D) is defined as a highly malignant, primitive embryonal tumor of the pineal gland itself and represents a true primitive neuroectodermal tumor (PNET). Although they are rare intracranial tumors, they constitute approximately 45% of all pineal parenchymal tumors. Usually they occur in the first two decades of life, more often in children, but principally can arise at any age. Some of these tumors may be present during the neonatal period. In larger series, pinealoblastomas are more common in males, with a male-female ratio of 2:1. The tumor usually replaces the tissue of the pineal gland. They are mostly soft, friable, and poorly demarcated and are pink, white, or gray, smooth or granular when cut, sometimes cystic, and frequently hemorrhagic or necrotic. Calcifications are rare and infiltration of the surrounding structures, including the meninges, is common. Constituting the most primitive of pineal parenchymal tumors, pinealoblastomas are composed of patternless sheets of densely packed small cells with round to irregular nuclei and scant cytoplasm. Pinealocytomatous rosettes are lacking, but Homer-Wright and Flexner-Wintersteiner rosettes may be seen. Retinoblastomatous differentiation of pinealoblastomas sometimes occurs and supports the theory of the photoreceptive origin of pineal gland cells. Pinealoblastomas may accompany bilateral retinoblastomas, in which case together they are called trilateral retinoblastoma. Pinealoblastomas sometimes contain melanin pigment. Because of its infiltrating nature into surrounding structures, dissemination in the CSF is not rare: according to the literature, between 8% and 24%. Metastases to bone, lung, and lymph nodes have been reported.
Tumors of Supportive and Adjacent Structures
There are several other tumors that can also be considered pineal parenchymal tumors, such as astrocytomas, ependymomas, or less frequently gliomas (glioblastomas or oligodendrogliomas). Glial cell tumors can also occur in the pineal gland, because astrocytes are normally present there, or may arise from the pluripotential pineal parenchymal cells. However, almost all glial tumors may arise from the glial tissue elements intimately surrounding the pineal gland. Real, true astrocytomas of the pineal gland itself are extremely rare. Tumors of other histological types may also occur as a substrate from the surrounding tissue—for example, choroidus plexus papillomas or medulloepitheliomas. Tumors of mesenchymal origin that may occur include meningiomas, hemangiomas, and hemangiopericytomas or blastomas, which arise mostly from the falx and tentorium near their junction or from the velum interpositum at the roof of the third ventricle. Also reported are exceptionally rare cases of chemodectomas and craniopharyngiomas of the pineal region. Another not-so-well-known entity is malignant solitary fibrous tumors that also may occur in the pineal gland. Solitary fibrous tumors are rare fibroblastic tumors that most commonly arise in the pleura, sometimes in the orbit. In the CNS in general they are extremely rare with around 100 reports in the literature and just a handful from the pineal region.
Non-Neoplastic Tumor-Like Conditions
Sometimes of neurosurgical importance are non-neoplastic tumor-like malformations, for example, arachnoid cysts arising from the arachnoid cells of that region or “degenerative” cysts lined by fibrillary astrocytes (so-called pineal cysts). They occur in 1.1% to 4.3% but may be seen more often, as proved in autopsy series up to 25% to 40%. Cysticercosis and vascular lesions, such as arteriovenous malformations, cavernomas, and aneurysms of the vein of Galen, can also be included, but are very rare.
Signs and Symptoms
In the case of drop metastases from the CSF, seeding radiculopathy or myelopathy can occur, sometimes from a nonspecific appearance. Signs and symptoms can occur in very rare cases as a result of hematogenous metastases to several structures outside the CNS. The most typical clinical signs and symptoms are summarized in Table 37.2.
Diagnostic Studies
Imaging
Diagnostic tools such as MRI have revolutionized the management of pineal region tumors (Figs. 37.9 to 37.11). The ability to obtain high-resolution images and multidimensional views makes it possible to show tumor location and extension clearly and accurately. Today, ventriculography and pneumencephalography are primarily of historical value. Skull radiography, formerly an important diagnostic study, has fallen into disuse because of its low sensitivity in detecting tumors. The normal pineal gland is seen on plain films as a calcified mass in 60% of the population over 20 years of age and very rarely in children before the age of 6 years. Any calcified pineal gland that is larger than 1 cm in any dimension should be viewed with suspicion. The appearance of a calcified pineal gland in early childhood is usually abnormal, but can, in fact, be physiologically normal in up to 5% of cases.
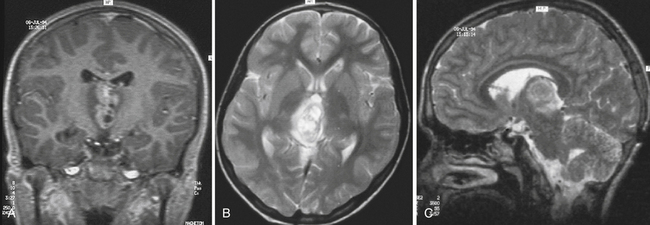
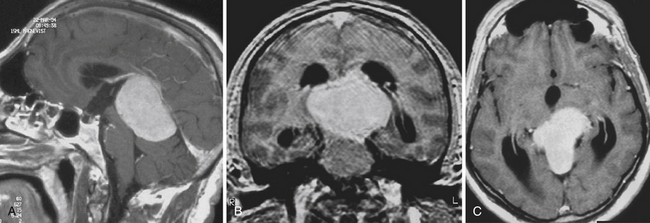
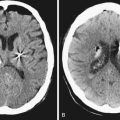
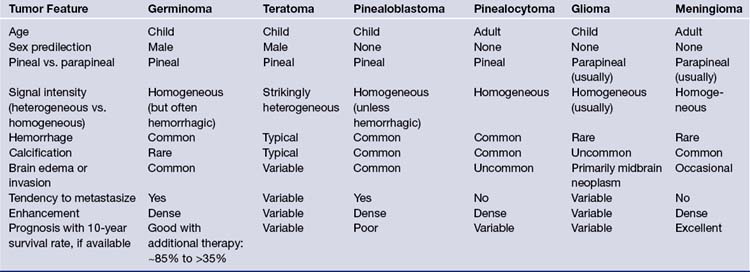
CT superseded all other radiological imaging methods for the detection of pineal region tumors in the early 1980s. Currently, high-resolution CT with or without intravenous contrast administration is used to examine the pineal region, but is often more important for planning the appropriate approach than as a diagnostic tool. Carotid and vertebral arteriography are only occasionally required, often in cases in which preoperative embolization may be considered; superselective angiography, in skilled hands, is used in this region for further endovascular treatment when a vascular tumor is encountered. In some institutions, magnetic resonance (MR) angiography or CT angiography is the angiographic study of choice for noninvasive evaluation. Both modalities provide accurate information on the anatomy of the arteries and veins in the region for surgical planning. Therefore, the current recommendation is to perform a high-resolution MRI examination, in combination with MR angiography and, when appropriate, in addition, CT scans with and without contrast, respectively, with or without CT angiography. Table 37.3 gives an overview and comparison of the neuroradiological appearance of pineal region tumors in correlation to their pineal versus parapineal appearance, signal intensity (heterogeneous versus homogeneous), appearance of hemorrhage, calcification, brain edema or invasion, and contrast enhancement.
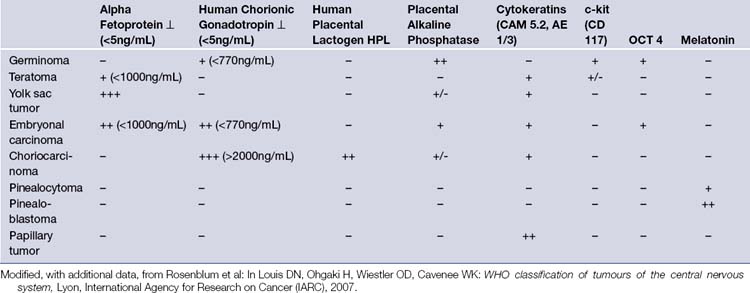
Tumor Markers
Certain pineal region tumors manifest tumor markers in the CSF and blood serum. The identification of markers is important not only for diagnostic purposes, when present, but also for monitoring response to treatment and relapse. However, it should be noted that these markers are not uniformly present in all patients with pineal tumors and their absence does not eliminate the diagnosis of a specific tumor.
Although the levels of markers in serum and CSF, when present, have been found to be extremely useful in assessing the efficacy of various treatment modalities and tumor recurrence, the tumor markers alone do not yield a definitive histological diagnosis. Furthermore, in a large number of patients these tumor markers are simply absent. Table 37.4 gives a brief overview of the serum and CSF levels occurring in pineal region tumors. Advanced neuroimaging (MRI, MRI spectroscopy, CSI and PET, high-resolution CT) and tumor marker evaluation, when present, improve diagnostic accuracy.
TABLE 37.4 Frequency of Typical Clinical Signs and Symptoms in Pineal Region Tumors
| Clinical Sign/Symptom | Frequency |
|---|---|
| Headache Vomiting Lethargy Memory disturbance Hydrocephalus |
Common (~80%) |
| Nystagmus | Common (~60-70%) |
| Parinaud’s sign Convergence Accommodation palsy Supranuclear upward glaze |
Frequent (~50-75%) |
| Lid retraction Setting sun sign (Collier’s sign) |
Less frequent (~10%) |
| Endocrine disturbance |
Choice of Treatments
Stereotactic Procedures
Stereotactic procedures, frame-base or frameless, have become increasingly more advanced and easy to use as a diagnostic tool for tumors that may not be amenable to resection. Most reports concerning this type of procedure indicate that the risks of intervention are minimal. However, caution is advised because the pineal region is surrounded by an important complex of arteries and veins, as described in the earlier section on anatomy. The vessels may be displaced from their normal position for several reasons, such as tumor mass, or hydrocephalus. Also, some tumors, such as pinealoblastomas or choriocarcinomas, are highly vascular. However, the complication rate in stereotactic biopsy of lesions in this location is low, approximately 1.3% mortality rate and 7% morbidity rate in skilled hands according to a large European series. The diagnostic accuracy and specificity is equivalently high (94%), although stereotactic biopsy may fail to disclose the histological heterogeneity of selected tumors due to sampling error, especially in the case of mixed malignant tumors.
Surgical Approaches
The efficacy of open microsurgery on pineal region tumors is undisputed in the twenty-first century. The main goal is complete tumor removal with minimal morbidity whenever possible. Even if radical resection cannot be achieved for several reasons, histological verification, maximal cytoreduction, and more often, restoration of CSF pathways may be achieved. For the benign pineal region tumor, surgery alone is often curative. Although about 80% of pediatric tumors are malignant, open surgery is recommended in most adult cases and selected pediatric cases in which resection is considered safe based on the appearance of the lesion by MRI. Radical resection is especially helpful in cases of the malignant nongerminoma germ cell tumors, as the extent of resection influences the prognosis of the patient. Because the pineal region is located in the geometric center of the intracranial cavity, operative approaches from every conceivable angle and direction have been developed (Fig. 37.12). The five most common surgical approaches are as follows:
1. The posterior transcallosal approach pioneered by Dandy
2. The transventricular approach pioneered by Van Wagenen
3. The occipital transtentorial approach pioneered by Foerster and Poppen
4. The infratentorial-supracerebellar approach pioneered by Krause and popularized by Stein
5. The three-quarter prone, operated side down, occipital transtentorial approach described by Ausman
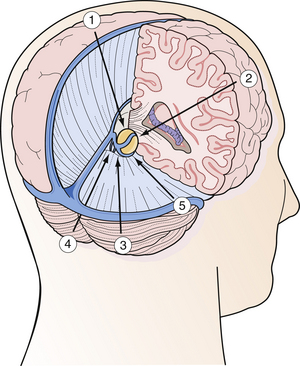
FIGURE 37.12 Schematic drawing of the various operative approaches to the pineal region from every direction.
(Modified from Day JD, Koos WT, Matula C, Lang J, editors. Color Atlas of Microneurosurgical Approaches. Stuttgart: Thieme; 1997.)
The Posterior Transcallosal Approach
In 1921, Walter Dandy was one of the first to propose this approach to the pineal region. The primary anatomical structures exposed are the splenium of the corpus callosum, internal cerebral veins, basal vein of Rosenthal, vein of Galen, pineal body, posterior commissure, and quadrigeminal plate. Patient position is similar to the position for the classical anterior transcallosal approach. However, a higher angle is required to provide the appropriate trajectory of about 40 degrees. We prefer a semilunar skin incision over the midline. Craniotomy should include the lambdoid at the posterior margin of the bone flap (Fig. 37.13). A semilunar dural flap is elevated to expose the cortical surface of the posterior parietal lobe. Entry point is the interhemispheric fissure (Fig. 37.14). Bridging veins are rare posteriorly but should be preserved, whenever possible. Retraction of the mesial parietal lobe exposes the splenium of the corpus callosum and after splitting the splenium, the internal cerebral veins can be seen as they drain into the vein of Galen. The pineal gland is located inferior to the venous complex (Fig. 37.15). The limitations of this approach include difficulty in visualization of the quadrigeminal plate and cerebellum, and laterally the mesial occipital lobe, and the requirement to split the splenium of the corpus callosum and retract the mesial occipital lobe. Advantages of this approach are the superior access superior to the venous complex (internal cerebral veins) and the ability to visualize these veins in the velum interpositum so that they are not sacrificed. We recommend this approach for tumors spreading above the venous complex and expanding anteriorly into the third ventricle as well as those extending upward into the corpus callosum (Fig. 37.16A-C). It is an approach that still has indications and should be a part of the surgeon’s armamentarium.
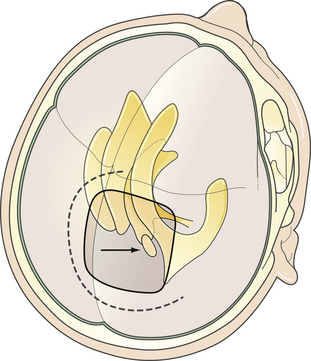
FIGURE 37.13 The posterior transcallosal approach. Direction of the approach, skin incision, and position of craniotomy.
(Modified from Day JD, Koos WT, Matula C, Lang J, editors. Color Atlas of Microneurosurgical Approaches. Stuttgart: Thieme; 1997.)
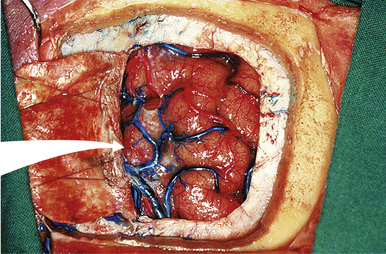
FIGURE 37.14 Dura opening and entrance into the interhemispheric fissure in an anatomical cadaver.
(Modified from Day JD, Koos WT, Matula C, Lang J, editors. Color Atlas of Microneurosurgical Approaches. Stuttgart: Thieme; 1997.)
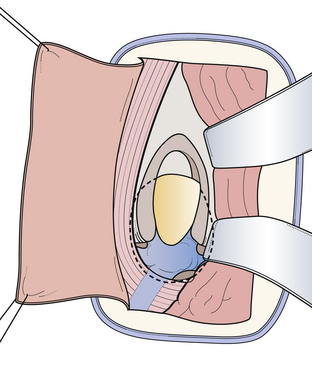
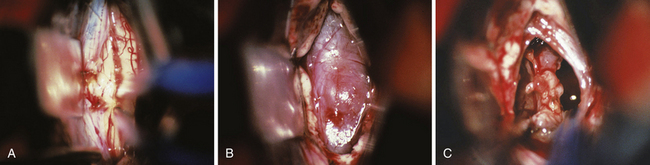
FIGURE 37.16 Intraoperative findings in the case of a pinealoblastoma as presented in Figure 37.6. Situation during splitting the splenium of the corpus callosum using a nonstick bipolar forceps (A). Appearance of the tumor located in the posterior part of the third ventricle before resection (B) and after complete tumor removal (C).
The Occipital Transtentorial Approach
This approach was first proposed by Foerster in 1928 and described in detail by Poppen in 1966. Primary anatomical structures exposed are the splenium of the corpus callosum, internal cerebral veins, basal vein of Rosenthal, vein of Galen, precentral cerebellar vermian vein, pineal body, posterior commissure, and quadrigeminal plate. This approach comes from a more lateral direction and we prefer to have the patient in the recumbent, semisitting position with the chin tucked. Skin incision is an inverted L-shaped incision to expose the occipital region. The inion should be clearly exposed and the bone flap is made with its superior margin approximately 1 to 2 cm below the lambdoid suture. The inferior margin is below the inion, thus placing the torcular within the inferior portion of the dural exposure (Fig. 37.17). After opening the dura, the occipital lobe is retracted laterally to expose the posterior incisural edge and its junction with the falx cerebri. After the tentorium is incised, the lateral entry point has been reached and the venous complex can be explored (Fig. 37.18). Sharp dissection of the arachnoid exposes the vein of Galen and the ipsilateral basal vein of Rosenthal. The pineal gland is most clearly visible inferior to the venous complex (Fig. 37.19).
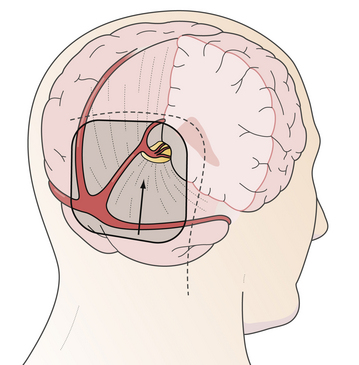
FIGURE 37.17 The occipital transtentorial approach: trajectory of the approach, skin incision, and position of craniotomy.
(Modified from Day JD, Koos WT, Matula C, Lang J, editors. Color Atlas of Microneurosurgical Approaches. Stuttgart: Thieme; 1997.)
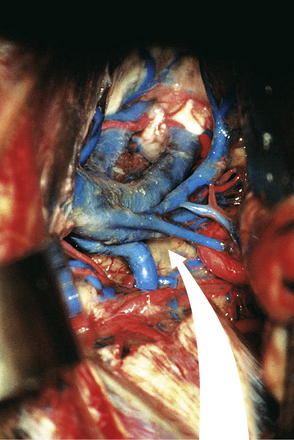
FIGURE 37.18 Dura opening and entrance after incision of the tentorium in an anatomical cadaver.
(Modified from Day JD, Koos WT, Matula C, Lang J, editors. Color Atlas of Microneurosurgical Approaches. Stuttgart: Thieme; 1997.)
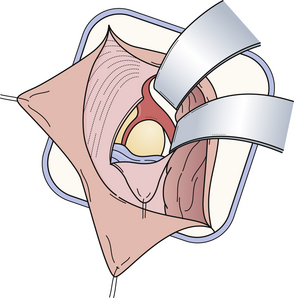
Limitations of the approach include poor visualization of the internal cerebral veins anteriorly, the quadrigeminal plate inferiorly, the falx cerebri and vein of Galen medially, and the occipital lobe laterally. In addition, it is not uncommon to compress and temporarily affect the visual cortex through the extremes of retraction that are required to lift the occipital lobe. An advantage of this approach is the superb view, both above and below the tentorium. This approach is recommended for tumors growing through the tentorial hiatus with a supra- and infracerebellar extension, and especially in the case of meningiomas (Fig. 37.20A-C). This approach is used in about 20% of the cases in our institution and makes this the second most common approach for dealing with tumors in the pineal region.
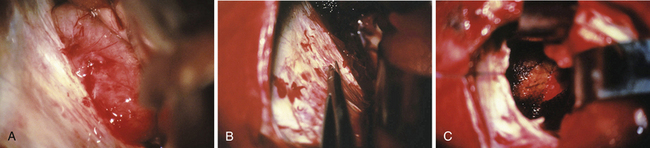
FIGURE 37.20 Intraoperative findings in case of a meningioma as presented in Figure 37.7. Photograph before the incision of the tentorium with the tumor in the background (A). Incision of the tentorium, as one of the key steps in performing the approach, (B) and the situation after complete tumor removal. The surrounding tissue is covered with Surgicel (C).
The Infratentorial-Supracerebellar Approach
The infratentorial-supracerebellar approach was first described by Krause in 1913 and then modified and popularized by Stein in 1971. Performing this kind of approach in the sitting position means that gravity assists the cerebellum in falling down from the undersurface of the tentorium. Primary anatomical structures exposed are the cerebellum, cerebellar veins, cerebellar vermian veins, internal cerebellar veins, basal veins of Rosenthal, vein of Galen, pineal body, posterior commissure, quadrigeminal plate, splenium of the corpus callosum, and posterior third ventricle. We prefer the patient to be placed in the sitting position with the head flexed anteriorly. The amount of flexion depends on the relationship of the tumor to the straight sinus. As an alternative position, we recommend the so-called modified concord position that allows the surgeon to sit comfortably behind the patient. One of the main advantages of the concord position is that it decreases the risk of air embolism. One of the disadvantages is that it is sometimes impossible when the tentorium is very steep. The skin incision is made in the midline, and we use either an S-shaped or a straight midline incision. The craniotomy is centered over the external occipital protuberance and includes the torcular herophili at the superior portion of dural exposure (Fig. 37.21). This allows some upward retraction of the sinus complex, decreasing the degree of necessary downward retraction of the cerebellum. In most of the cases it is not necessary to open the foramen magnum to drain the cisterna magnum so that the cerebellum requires very little retraction. The preserved rim of bone inferiorly provides support for the cerebellum, decreasing traction on its superior connecting elements to the brainstem. Arachnoid dissection and division of the superomedial cerebellar bridging veins allow subsequent inferior retraction of the cerebellar hemispheres (Fig. 37.22). In most cases, a very thick arachnoid membrane is found spanning the interval between the cerebellar vermis and the central posterior incisura. After dissection of the arachnoid membrane the venous complex surrounding the pineal gland can be explored (Fig. 37.23). Limitations of the approach are its restriction to the midline and limited extension to the lateral side. The most powerful advantage is the superior access inferior to the venous complex. We recommend this approach in most of the cases (more than 72%) in which the tumors are restricted to the midline and inferior to the venous complex. It also represents a perfect opportunity to use a rigid angled endoscope as an adjuvant during surgery. This so-called endoscope-assisted or endoscope-guided procedure allows not only perfect illumination (which gives the anatomical structures more plasticity), but also enables the surgeon to have a “look around a corner” without even touching any anatomical structure. This helps the surgeon to detect any residual tumor hiding in the recesses of the pineal region, thus increasing the chance for a radical tumor resection (Fig. 37.24A-D).
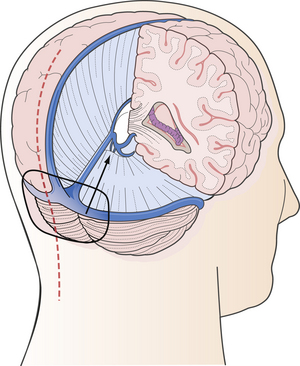
FIGURE 37.21 The infratentorial-supracerebellar approach: trajectory of the approach, skin incision, and position of craniotomy.
(Modified from Day JD, Koos WT, Matula C, Lang J, editors. Color Atlas of Microneurosurgical Approaches. Stuttgart: Thieme; 1997.)
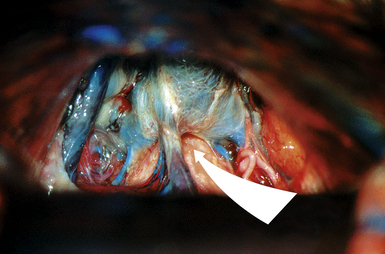
FIGURE 37.22 The infratentorial-supracerebellar entry into the pineal region in an anatomical cadaver.
(Modified from Day JD, Koos WT, Matula C, Lang J, editors. Color Atlas of Microneurosurgical Approaches. Stuttgart: Thieme; 1997.)
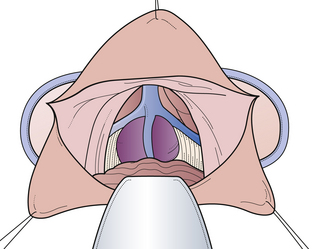
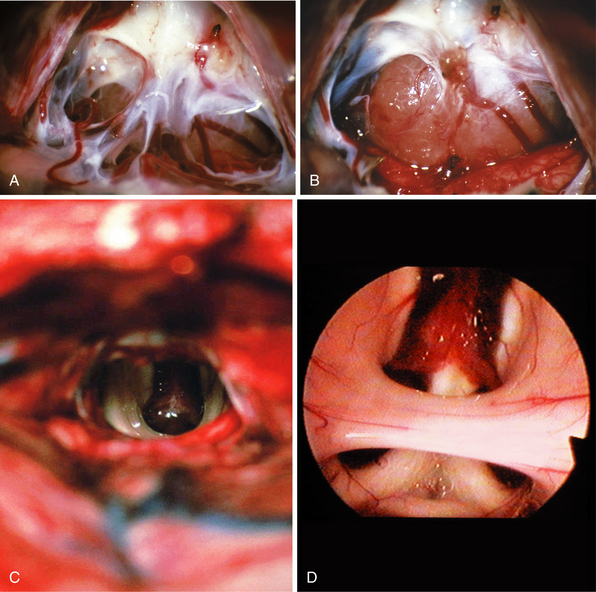
FIGURE 37.24 Intraoperative findings in case of a germinoma as presented in Figure 37.8. Photograph during the opening of the arachnoid membranes in the pineal region with the tumor in the background (A). Appearance of the tumor before resection (B) and after complete tumor removal (C), and endoscope-assisted approach with the view into the third ventricle presenting the intermediate mass, the choroid plexus at the roof, both fornices, and the anterior commissure (D).
Transverse Sinus–Tentorium Splitting Approach
As mentioned previously, depending on the individual situation, it can be very helpful to chose some combination, especially in huge tumors. One of the modifications is the so-called transverse sinus–tentorium splitting approach described by Beck and modified by Sekhar. The key point is the splitting of the transverse sinus and dissection of the tentorium to get the best of both worlds, supratentorial and infratentorial, as schematically drawn in Figure 37.25.
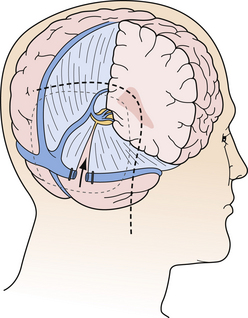
Other Therapeutic Modalities
Radiotherapy
Although more than 70% of tumors in the pineal region and the posterior third ventricle are highly radiosensitive, radiation therapy is still a subject of controversy. Select tumors respond to adequate courses of radiation therapy within 3 to 6 months. The current neurosurgical paradigm of the treatment of acute hydrocephalus followed by diagnostic biopsy or radical resection of tumor has obviated the need for preoperative empirical radiation therapy prior to diagnosis. However, it has been clinically proved that germinomas, like seminomas, are highly sensitive to radiation therapy. Radiotherapy is not a benign form of treatment, especially to the developing central nervous system, such as in the pediatric population. Serious consequences of such treatment include endocrine deficiencies, severe vasculitis, and significant intellectual impairment. All of these risks limit the dosage and modalities used in radiotherapy of pineal region tumors in young children. The application of preradiation chemotherapy and stereotactic radiation techniques such as the linear particle accelerator (linac) and gamma knife radiosurgery may provide some relief from the potential detrimental side effects of radiation on the developing brain. Despite the well-established radiosensitivity of germinomas, about 10% of the patients may experience tumor recurrence. Radiosurgery (linac, gamma knife, or CyberKnife) is increasingly being used to treat tumors in the pineal region, either as an additional therapy after conventional treatments or as a primary treatment. The optimal treatment of huge tumors can be obtained when radiosurgery is used in conjunction with open surgery and conventional radiotherapy.
Conclusion
In the Western hemisphere, tumors of the pineal region constitute about 1% of all intracranial neoplasms and are more common in children (3-8% of all pediatric brain tumors). However, in Japan, pure germ cell tumors are more common for unknown reasons, and pineal region tumors constitute 4% to 7% of all intracranial neoplasms. The author’s own data show a predominance of males (67%) to females (33%) in all cases. About 65% of all the cases involve patients who are younger than 20 years of age and about 35% who are aged between 21 and 70. Pathologically, primary tumors of the pineal region can be divided into germ cell tumors (about 60%), pineal parenchymal tumors (about 30%), tumors of supportive and adjacent structures (about 10%), non-neoplastic tumor-like conditions (less than 1%), and metastases (less than 0.1%). Symptoms of tumors in the pineal region are caused either by obstruction of CSF pathways or by local involvement (compression or invasion) of adjacent structures. Characteristic local signs include visual disturbances, the most common being Parinaud syndrome.
Blakeley J.O., Grossman S.A. Management of pineal region tumors. Curr Treat Options Oncol. 2006;7(6):505-516.
Bruce J.N., Ogden A.T. Surgical strategies for treating patients with pineal region tumors. J Neurooncol. 2004;69(1-3):221-236.
Day J.D., Koos W.T., Matula C., Lang J., editors. Color Atlas of Microneurosurgical Approaches. Cranial Base and Intracranial Midline. Stuttgart: Thieme, 1997.
De Girolami U., Fèvre-Montange M., Seilhean D., Jouvet A. Pathology of tumors of the pineal region. Rev Neurol (Paris). 2008;164(11):882-895.
Radovanovic I., Dizdarevic K., de Tribolet N., et al. Pineal region tumors—neurosurgical review. Med Arh. 2009;63(3):171-173.
Please go to expertconsult.com to view the complete list of references.
1. Abay E.O., Laws E.R.Jr., Grado G.L., et al. Pineal tumors in children and adolescents: treatment by CSF shunting and radiotherapy. J Neurosurg. 1981;55:889-895.
2. Allen J.C., Nisselbaum J., Epstein F., et al. Alpha-fetoprotein and human chorionic gonadotropin determination in cerebrospinal fluid: an aid to the diagnosis and management of intracranial germ-cell tumors. J Neurosurg. 1979;51:368-374.
3. Al-Holou W.N., Garton H.J., Muraszko K.M., et al. Prevalence of pineal cysts in children and young adults. Clinical article. J Neurosurg Pediatr. 2009;4(3):230-236.
4. Al-Hussaini M., Sultan I., Abuirmileh N., et al. Pineal gland tumors: experience from the SEER database. J Neurooncol. 2009;94(3):351-358.
5. Arita N., Ushio V., Hayakawa T., et al. Tumor markers: their role and limit for management of pineal tumor. In: Samii M., editor. Surgery in and Around the Brain Stem and the Third Ventricle. Berlin: Springer-Verlag; 1986:318-325.
6. Ausman J.I., Malik G.M., Pearce J., Rogers J.S. A new operative approach to the pineal region. In: Samii M., editor. Surgery in and Around the Brain Stem and the Third Ventricle. Berlin: Springer-Verlag; 1986:326-328.
7. Beck H., Moriyama E. Transverse sinus-tentorium splitting approach for pineal region tumors—case report. Neurol Med Chir (Tokyo). 2001;41(4):217-221.
8. Berhouma M., Jemel H., Ksira I., Khaldi M. Transcortical approach to a huge pineal mature teratoma. Pediatr Neurosurg. 2008;44(1):52-54.
9. Blakeley J.O., Grossman S.A. Management of pineal region tumors. Curr Treat Options Oncol. 2006;7(6):505-516.
10. Bruce J.N., Stein S.M. lnfratentorial approach to pineal tumors. In: Wilson C.B., editor. Neurosurgical Procedures: Personal Approaches to Classic Operations. Baltimore: Williams & Wilkins; 1992:63-76.
11. Bruce J.N., Ogden A.T. Surgical strategies for treating patients with pineal region tumors. J Neurooncol. 2004;69(1-3):221-236.
12. Budka H., Schmidbauer M. Pathology of tumors of the pineal region [Pathologie der Tumoren der Pinealisregion]. In: Bamberg M., Sack H., editors. Therapy of Primary Brain Tumors [Therapie Primärer Hirntumoren]. Munich: W. Zuckerschwerdt; 1988:334-342.
13. Cho B.K., Wang K.C., Nam D.H., et al. Pineal tumors: experience with 48 cases over 10 years. Childs Nerv Syst. 1998;14:53-58.
14. Citow J.S., Wollmann R.L., Macdonald R.L., editors. Neuropathology and Neuroradiology. A Review. Stuttgart: Thieme, 2001.
15. Clark W.K. Occipital transtentorial approach. In: Apuzzo M.U., editor. Surgery of the Third Ventricle. Baltimore: Williams & Wilkins; 1987:591-610.
16. Coello A.F., Torres A., Acebes J.J., Boluda S. Papillary tumor of the pineal region. Neurology. 2009;73(6):486.
17. Connolly E.S., McKhann G.M.II, Huang J., Choudhri T.F. Fundamentals of Operative Techniques in Neurosurgery. Stuttgart: Thieme; 2002.
18. Di Ieva A., Tschabitscher M., Rodriguez y Baena R. Lancisi’s nerves and the seat of the soul. Neurosurgery. 2007;60(3):563-568.
19. Dandy W.E. An operation for the removal of pineal tumors. Surg Gynecol Obstet. 1921;33:113-119.
20. Day J.D., Koos W.T., Matula C., Lang J., editors. Color Atlas of Microneurosurgical Approaches. Cranial Base and Intracranial Midline. Stuttgart: Thieme, 1997.
21. De Girolami U., Fèvre-Montange M., Seilhean D., Jouvet A. Pathology of tumors of the pineal region. Rev Neurol (Paris). 2008;164(11):882-895.
22. Edwards M.S.B., Levin V. Chemotherapy of tumors of the third ventricular region. In: Apuzzo M.U., editor. Surgery of the Third Ventricle. Baltimore: Williams & Wilkins; 1987:838-842.
23. Fukui M., Natori Y., Matsushima T., et al. Operative approaches to the pineal region tumors. Childs Nerv Syst. 1998;14:49-52.
24. Gangemi M., Maiuri F., Colella G., Buonamassa S. Endoscopic surgery for pineal region tumors. Minim Invasive Neurosurg. 2001;44:70-73.
25. Ganti S.R., Hilal S.K., Stein S.M., et al. CT of pineal region tumors. AJNR. 1986;7:97-104.
26. Greenberg M.S., editor. Handbook of Neurosurgery, 6th ed., Stuttgart: Thieme, 2006.
27. Grimoldi N., Tomei G., Stankov B., et al. Neuroendocrine, immunohistochemical, and ultrastructural study of pineal region tumors. J Pineal Res. 1998;25:147-158.
28. Haimovich I.E., Sharer L., Hyman R.A., Beresford H.R. Metastasis of intracranial germinoma through a ventriculoperitoneal shunt. Cancer. 1981;48:1033-1036.
29. Herrmann H.O., Winkler D., Westphal M. Treatment of tumours of the pineal region and posterior part of the third ventricle. Acta Neurochir (Wien). 1992;116:137-146.
30. Hirato J., Nakazato Y. Pathology of pineal region tumors. J Neurooncol. 2001;54:239-249.
31. Jakacki R.I. Pineal and nonpineal supratentorial primitive neuroectodermal tumors. Childs Nerv Syst. 1999;15:586-591.
32. Jooma R., Kendall B.E. Diagnosis and treatment of pineal tumors. J Neurosurg. 1983;58:654-665.
33. Julow J., Viola A., Major T. Review of radiosurgery of pineal parenchymal tumors. Long survival following 125-iodine brachytherapy of pineoblastomas in 2 cases. Minim Invasive Neurosurg. 2006;49(5):276-281.
34. Kalhor N., Ramirez P.T., Deavers M.T., et al. Immunohistochemical studies of trophoblastic tumors. Am J Surg Pathol. 2009;33(4):633-638.
35. Kang J.K., Jeun S.S., Hang Y.K., et al. Experience with pineal region tumors. Childs Nerv Syst. 1998;14:63-68.
36. Kobayashi T., Lunsford L.D. Progress in Neurological Surgery: Pineal Region Tumors: Diagnosis and Treatment Options. Basel: Karger; 2009.
37. Konovalov A.N., Pitskhelauri D.I. Principles of treatment of the pineal region tumors. Surg Neurol. 2003;59(4):250-268.
38. Koos W.T., Pendl G. Lesions of the cerebral midline. Acta Neurochir (Wien). 1985;35(Suppl):622.
39. Koos W.T., Spetzler R.F., Lang J., editors. Color Atlas of Microneurosurgery, Vol 1. Stuttgart: Thieme, 1993.
40. Korogi Y., Takahashi M., Ushio Y. MRI of pineal region tumors. J Neurooncol. 2001;54:251-261.
41. Kurisaka M., Arisawa M., Mori T., et al. Combination chemotherapy (cisplatin, vinblastin) and low-dose irradiation in the treatment of pineal parenchymal cell tumors. Childs Nerv Syst. 1998;14:564-569.
42. Lapras C., Patet J.D. Controversies, techniques, and strategies for pineal tumor surgery. In: Apuzzo M.U., editor. Surgery of the Third Ventricle. Baltimore: Williams & Wilkins; 1987:649-662.
43. Leston J., Mottolese C., Champier J., et al. Contribution of the daily melatonin profile to diagnosis of tumors of the pineal region. J Neurooncol. 2009;93(3):387-394.
44. Lekovic G.P., Gonzalez L.F., Shetter A.G., et al. Role of gamma knife surgery in the management of pineal region tumors. Neurosurg Focus. 2007;23(6):E12.
45. Little K.M., Friedman A.H., Fukushima T. Surgical approaches to pineal region tumors. J Neurooncol. 2001;54(3):287-299.
46. Louis D.N., Ohgaki H., Wiestler O.D., Cavenee W.K., editors. World Health Organization WHO Classification of Tumors of the Central Nervous System. Lyon: IARC Press, 2007.
47. Luther N., Stetler W.R.Jr., Dunkel I.J., et al. Subarachnoid dissemination of intraventricular tumors following simultaneous endoscopic biopsy and third ventriculostomy. J Neurosurg Pediatr. 2010;5(1):61-67.
48. Medinger M., Kleinschmidt M., Mross K., et al. C-kit (CD117) expression in human tumors and its prognostic value: an immunohistochemical analysis. Pathol Oncol Res. 2010;16:295-301.
49. Mori Y., Kobayashi T., Hasegawa T., et al. Stereotactic radiosurgery for pineal and related tumors. Prog Neurol Surg. 2009;23:106-118.
50. Neuwelt E.A., editor. Diagnosis and Treatment of Pineal Region Tumors. Baltimore: Williams & Wilkins, 1984.
51. Nomura K. Epidemiology of germ cell tumors in Asia of pineal region tumor. J Neurooncol. 2001;54:211-217.
52. Oi S., Shibata M., Tominaga J., et al. Efficacy of neuroendoscopic procedures in minimally invasive preferential management of pineal region tumors: a prospective study. J Neurosurg. 2000;93:245-253.
53. Oi S., Kamio M., Joki T., Abe T. Neuroendoscopic anatomy and surgery in pineal region tumors: role of neuroendoscopic procedure in the “minimally-invasive preferential” management. J Neurooncol. 2001;54:277-286.
54. Pendl G., editor. Pineal and Midbrain Lesions. Berlin: Springer, 1985.
55. Radovanovic I., Dizdarevic K., de Tribolet N., et al. Pineal region tumors—neurosurgical review. Med Arh. 2009;63(3):171-173.
56. Regis J., Bouillot P., Rouby-Volot F., et al. Pineal region tumors and the role of stereotactic biopsy: review of the mortality, morbidity, and diagnostic rates in 370 cases. Neurosurgery. 1996;39:907-912. discussion 912-914
57. Raco A., Raimondi A.J., O’Alonzo A., et al. Radiosurgery in the management of pediatric brain tumors. Childs Nerv Syst. 2000;16:287-295.
58. Regis J., Bouillot P., Rouby-Volot F., et al. Microsurgery of the third ventricle, 2: operative approaches. Neurosurgery. 1981;8:357-373.
59. Rieger A., Rainov N.G., Brucke M., et al. Endoscopic third ventriculostomy is the treatment of choice for obstructive hydrocephalus due to pediatric pineal tumors. Minim Invasive Neurosurg. 2000;43:83-86.
60. Robertson P.L., DaRosso R.C., Allen J.C. Improved prognosis of intracranial non-germinoma germ cell tumors with multimodality therapy. J Neurooncol. 1997;32:71-80.
61. Robinson S., Cohen A.R. The role of neuroendoscopy in the treatment of pineal region tumors. Surg Neurol. 1997;48:360-365. discussion 365-367
62. Sano K. Pineal region tumors: problems in pathology and treatment. Clin Neurosurg. 1983;30:59-91.
63. Sano K. Treatment of tumors in the pineal and posterior third ventricular region. In: Samii M., editor. Surgery in and Around the Brain Stem and the Third Ventricle. Berlin: Springer-Verlag; 1986:309-317.
64. Santagata S., Maire C.L., Idbaih A., et al. CRX is a diagnostic marker of retinal and pineal lineage tumors. PLoS One. 2009;4(11):e7932.
65. Satoh H., Uozumi 1, Kiya K., et al. MRI of pineal region tumours: relationship between tumours and adjacent structures. Neuroradiology. 1995;37:624-630.
66. Sawaya R., Hawley D.K., Tobler W.D., et al. Pineal and third ventricle tumors. In: Youmans J.R., editor. Neurological Surgery. 3rd ed. Philadelphia: WB Saunders; 1990:3171-3203.
67. Sharma M.C., Jain D., Sarkar C., et al. Papillary tumor of the pineal region—a recently described entity: a report of three cases and review of the literature. Clin Neuropathol. 2009;28(4):295-302.
68. Scheithauer B.W. Neuropathology of pineal region tumors. Clin Neurosurg. 1985;32:351-383.
69. Schindler E., editor. Tumors of the Pineal Region [Die Tumoren der Pinealisregion]. Berlin: Springer-Verlag, 1985.
70. Schmidek H.H., editor. Pineal Tumors. New York: Massan Publishing, 1977.
71. Schmidek H.H., Waters A. Pineal masses: clinical features and management. In: Wilkins R.H., Rengachary S.S., editors. Neurosurgery. New York: McGraw-Hill; 1985:688-693.
72. Shinoda J., Yamada H., Sakai N., et al. Placental alkaline phosphatase as a tumor marker for primary intracranial germinoma. J Neurosurg. 1988;68:710-720.
73. Stein B.M., Fetell M.R. Therapeutic modalities for pineal region tumors. Clin Neurosurg. 1985;32:445-455.
74. Takakura K. Intracranial germ cell tumors. Clin Neurosurg. 1985;32:429-444.
75. Takakura K., Matsutani M. Therapeutic modality selection in management of germ cell tumors. In: Apuzzo M.U., editor. Surgery of the Third Ventricle. Baltimore: Williams & Wilkins; 1987:843-846.
76. Tien R.D., Barkovich A.J., Edwards M.S.B. MR imaging of pineal tumors. AJNR Am J Neuroradiol. 1990;11:557-565.
77. Timmermann B., Kortmann R.D., Kuhl J., et al. Role of radiotherapy in the treatment of supratentorial primitive neuroectodermal tumors in childhood: results of the prospective German brain tumor trials HIT 88/89 and 91. J Clin Oncol. 2002;20:842-849.
78. Tomita T. Pineal region tumors. In: Albright A.L., Pollack I.F., Adelson P.D., editors. Principles and Practice of Pediatric Neurosurgery. Stuttgart: Thieme, 2001.
79. Tribolet N., Barrelet l. Successful chemotherapy for pinealoma. Lancet. 1977;2:1228-1229.
80. Tsugu H., Oshiro S., Ueno Y., et al. Primary yolk sac tumor within the lateral ventricle. Neurol Med Chir (Tokyo). 2009;49(11):528-531.
81. Van Wagenen W.P. A surgical approach for the removal of certain pineal tumors: report of a case. Surg Gynecol Obstet. 1931;53:216-220.
82. Vorkapic P., Waldhauser F., Bruckner R., et al. Serum melatonin level: a new diagnostic tool in pineal region tumors. Neurosurgery. 1987;21:817-824.
83. Wara W., Gutin P.H. Radiotherapy of pineal and suprasellar tumors. In: Apuzzo M.U., editor. Surgery of the Third Ventricle. Baltimore: Williams & Wilkins; 1987:831-837.
84. Williams D.L., Warwick R., Dyson M., Bannister L.H., editors. Gray’s Anatomy, 37th ed., Edinburgh: Churchill Livingstone, 1989.
85. Yamamoto I. Pineal region tumor: surgical anatomy and approach. J Neurooncol. 2001;54:263-275.
86. Yamini B., Refai D., Rubin C.M., Frim D.M. Initial endoscopic management of pineal region tumors and associated hydrocephalus: clinical series and literature review. J Neurosurg (Pediatr). 2004;100(Suppl 5):437-441.
87. Yazici N., Varan A., Söylemezoğlu F., et al. Pineal region tumors in children: a single center experience. Neuropediatrics. 2009;40(1):15-21.
88. Youssef A.S., Keller J.T., van Loveren H.R. Novel application of computer-assisted cisternal endoscopy for the biopsy of pineal region tumors: cadaveric study. Acta Neurochir (Wien). 2007;149(4):399-406.
89. Zhang J., Cheng H., Qiao Q., et al. Malignant solitary fibrous tumor arising from the pineal region: case study and literature review. Neuropathology. 2009;30:294-298.
90. Zimmerman R.A. Pineal region masses: radiology. In: Wilkins R.H., Rengachary S.S., editors. Neurosurgery. New York: McGraw-Hill; 1985:680-687.
91. Zülch K.J. Reflections on the surgery of the pineal gland (a glimpse into the past). Neurosurg Rev. 1981;4:159-162.