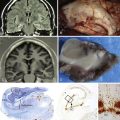
Chapter 10 Posterior Fossa and Brainstem Tumors in Children
• Presentation and Investigation
− Presentation of posterior fossa lesions is dictated by location, and aggressiveness of the lesion involved. The most common presenting symptom of a posterior fossa mass is headache related to mass effect, or obstruction of cerebrospinal fluid (CSF) pathways.
− Seizures from a posterior fossa mass are rare. If a patient with a posterior fossa tumor presents with seizures, consider the possibility of leptomeningeal spread.
− Magnetic resonance imaging (MRI) of the brain and spinal cord should be obtained to rule out disseminated disease. Occasionally lumbar puncture may be required for cytological examination.
− Pilocytic astrocytomas are the most common pediatric cerebellar neoplasm.
− The degree of mitotic activity, cellular atypia, and microvascular proliferation do not affect grade of pilocytic astrocytomas.
− Gross total resection of a pilocytic astrocytoma is considered curative. If the cyst wall enhances, it should be removed by the neurosurgeon.
− Desmoplastic medulloblastomas have a more favorable prognosis than classical medulloblastoma. Anaplastic/large cell histological finding has a poor prognosis and qualifies as high-risk medulloblastoma.
− The most common genetic anomaly in medulloblastoma is isochromosome 17q. In the near future, prognostic and risk stratification of medulloblastoma will be based on molecular profiling.
− “Second look” surgery should be considered when significant residual disease is evident on postoperative imaging.
− The current standard of care for average-risk medulloblastoma, age above 3 years, is gross total resection, postoperative craniospinal radiation of 23.4 Gy with a posterior fossa boost of 54 Gy, followed by 12 months of chemotherapy. Postoperative intensive chemotherapy can be used to delay radiation in children younger than 3 years of age.
− The 5-year survival rate for ependymoma is 60%.
− Classic histological features include ependymal rosettes and pseudorosettes.
− Ependymoma of the cerebellopontine (CP) angle can encase vital neurovascular structures.
− Extent of surgical resection is the only factor with significant prognostic value.
• Atypical Teratoid/Rhabdoid Tumor
− Loss of INI1 is found in 90% of atypical teratoid/rhabdoid tumors (AT/RTs).
− Owing to intermixed histopathological features, AT/RT can be confused with medulloblastoma or primitive neuroectodermal tumors (PNETs) if only small areas are biopsied.
− Gross total resection predicts for improved survival. Radiation therapy should not be used for children under 3 years of age. Intrathecal chemotherapy is being tested in recent clinical trials.
− Histologically, choroid plexus papillomas are difficult to distinguish from normal choroid epithelium.
− Choroid plexus tumors are more common in the very young.
− Choroid plexus tumors are extremely vascular and intraoperative blood loss can lead to morbidity and fatal outcome.
− Dermoids typically present in midline locations in association with a dermal sinus tract. The sinus tract should be completely excised to prevent recurrence.
− Epidermoid cysts occur laterally and are commonly found in the CP angle.
− High signal on diffusion-weighted imaging aids in differentiating an epidermoid from an arachnoid cyst.
− A third ventriculostomy prior to tumor resection in children over age 3 can be effective in eliminating CSF shunt requirements postoperatively.
− Not all brainstem tumors have an unfavorable prognosis. Subsets of focal brainstem gliomas are associated with long-term survival. Diffuse intrinsic pontine gliomas account for 60% to 80% of brainstem gliomas, with an average survival of 9 months. NF1 patients have brainstem tumors with a more favorable survival time.
− The use of neuronavigation and neurophysiological mapping and monitoring is essential for maximizing resection while minimizing harm to patients.
• Diffuse Intrinsic Pontine Glioma
− There is no current role for biopsy or surgical excision of these tumors.
− Palliative radiation will provide transient symptom relief in 75% of patients.
− More than 80% of these lesions have an indolent course.
− Large size (>2 cm), enhancement, and invasion of adjacent structures are signs of more aggressive tectal lesions.
− Benign tectal lesions in children older than 3 years can be managed with a third ventriculostomy and serial follow-up.
• Surgical Approaches to the Posterior Fossa
− The telovelar approach to the fourth ventricle offers an alternative to the midline approach in which the cerebellar vermis is split.
− Intraoperative mapping/monitoring helps the surgeon in identifying and preserving lower cranial nerves and the brainstem nuclei during surgery.
− Use corridors created by the lesion such as cysts, or presentation to the pial surface, as landmarks to begin the resection of posterior fossa tumors. Do not transgress the floor of the fourth ventricle when resecting brainstem tumors.
Pediatric brain tumors are the leading cause of solid cancer–related death in children. Approximately 60% of these tumors occur below the tentorium, including the brainstem, cerebellum, fourth ventricle, and cerebellopontine angle. In contrast, in the adult population the majority of these neoplasms occurs in the supratentorial compartment. The pathological features of these tumors are diverse, and prognosis ranges from excellent to dismal, depending on histopathological findings, extent of surgical resection, and use of adjunctive therapies (Table 10.1). Great technological strides have been made in regard to improving and understanding tumor biology, imaging, surgical techniques, and chemotherapeutic/radiation protocols, leading to increased survival time in these patients. For example, survival time for medulloblastoma in Cushing’s day in the 1920s averaged 17 months. The addition of craniospinal radiation in the 1950s yielded 3-year survival rates of 65% and in the modern era with combined surgery, radiation, and chemotherapy average-risk medulloblastoma patients can achieve a 5-year survival rate of 80% to 85%.1 However, these treatments can lead to significant morbidity to the developing brain and thus we still have more to learn from these complex and challenging tumors.
TABLE 10.1 Survival Data for Pediatric Posterior Fossa Tumors With Current Best Management
| Tumor Subtype | Survival with Current Best Management |
|---|---|
| Pilocytic astrocytoma | EFS > 90% at 5 years28 |
| Medulloblastoma | Average risk: EFS 80% at 5 years67 High risk: EFS 60% at 4 years71 |
| Ependymoma | GTR: 70-80% 5-year survival rate101 STR: 20-40% 5-year survival rate |
| AT/RT | 3-year EFS 50-78% with radiation122,123 3-year EFS 11% in young children with no radiation123 |
| Choroid plexus papilloma | OS 100% at 2 years132 Atypical tumor: OS 89% at 2 years132 |
| Choroid plexus carcinoma | OS 36% at 2 years132 |
| Diffuse pontine glioma | 9-month survival207, 228 |
| Focal brainstem glioma | 5-year survival rates 50-100%194, 226–228, 239 |
AT/RT, atypical teratoid/rhabdoid tumor; CPP, choroid plexus papilloma; EFS, event-free survival rate; GTR, gross total resection; OS, overall survival rate; STR, subtotal resection.
General Diagnostic Imaging Features
Once stabilized, patients with a posterior fossa lesion require magnetic resonance imaging (MRI) of both the brain and spinal cord to rule out leptomeningeal spread. Cerebrospinal fluid (CSF) may be acquired by lumbar puncture if deemed safe prior to surgery to look for malignant cells. If a metastatic workup cannot be performed prior to surgery, then a waiting period of 10 to 14 days prior to imaging or obtaining lumbar CSF should elapse after surgery to avoid false positive results from surgical intervention.2 MRI can also alert the surgeon to instances when the brainstem or floor of the fourth ventricle has been compromised by an invading tumor.
Despite the classic imaging of each tumor type, it can be very difficult to differentiate these tumors based on MRI or CT alone. Classically, medulloblastoma has a higher cellularity than either pilocytic astrocytoma or ependymoma and as such has a higher density on CT or is hypointense on T1 MRI. The solid portion of a pilocytic astrocytoma is hyperintense to CSF on T2 sequences in 50% of cases.3 Ependymoma can typically be seen exiting laterally or inferiorly from the foramen of Luschka or Magendie, respectively. However, imaging characteristics often overlap or are atypical, and diagnosis based on traditional imaging alone is not reliable. Recently, studies have utilized diffusion-weighted imaging (DWI) and magnetic resonance spectroscopy (MRS) to enhance predictive values. DWI measures the microscopic diffusion of water in tissues. Highly cellular tumors such as medulloblastoma have restricted diffusion and higher signals on apparent diffusion coefficient (ADC) maps. Proton MRS analyzes the metabolic composition from tissues such as choline (high in tumors), N-acetylaspartate (reduced in tumors), lactate, taurine (high in medulloblastoma), glutamine, myoinositol, and alanine. Utilizing these two methods in conjunction may help increase predictive values of imaging for pediatric posterior fossa tumors or help distinguish relapse from radiation necrosis.4
Individual Posterior Fossa Tumor Types
Pilocytic Astrocytoma
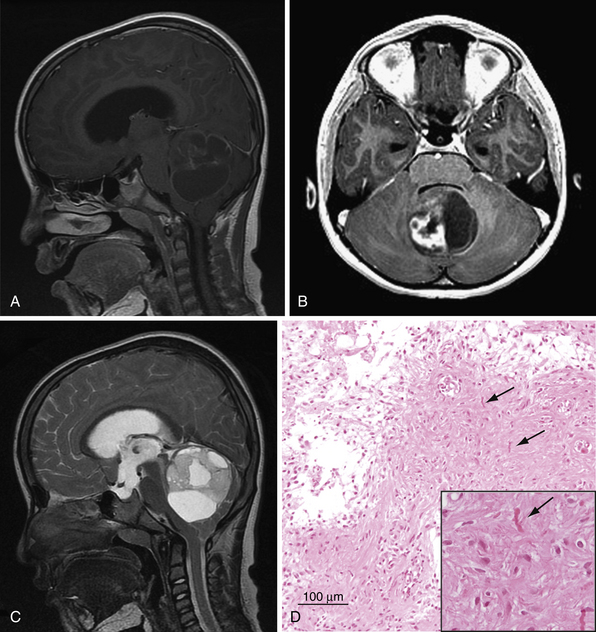
Cerebellar astrocytomas were first described in a series of 76 tumors by Harvey Cushing.5,6 Pilocytic astrocytomas (PAs) are the most common pediatric cerebellar neoplasm and occur at a mean age of 7 to 8 years.7,8 There is no gender predilection. Pilocytic astrocytomas are WHO (World Health Organization) grade I lesions, indicating their slow growth, indolent behavior, and high survival rate. They can occur in any location of the neuraxis, but the cerebellar hemispheres (~50%), optic pathways, thalamus, and hypothalamus are the most common sites.7 There are case reports of pilocytic tumors occurring within the CPA.9,10 The vast majority of cerebellar astrocytomas are pilocytic (WHO I), as was found in 88% of patients in the Hospital for Sick Children series.11 However, distinctions between fibrillary and pilocytic tumors of the cerebellum have not been useful in predicting prognosis.
Imaging
Typically, cerebellar pilocytic astrocytomas appear as well-circumscribed cystic lesions with a solid enhancing nodule (Fig. 10.1). Computed tomography (CT) imaging reveals a well-demarcated lesion with cystlike features, very occasional calcifications, and intense enhancement of the solid component with contrast agent administration.12,13 A pilocytic astrocytoma can present on imaging in four patterns: (1) An enhancing mural nodule or mass accompanied by a nonenhancing cyst; (2) an enhancing mural nodule with an intensely enhancing cyst; (3) a predominantly solid mass with no cyst component; and (4) a necrotic mass with a central nonenhancing zone.14 Pilocytic tumors typically arise from the vermis and the cerebellar hemispheres but can extend into the ventricular system.15 Pilocytic astrocytomas are isointense to hypointense relative to normal brain on T1 images and hyperintense to normal brain in T2 images.7 However, diagnosing pilocytic astrocytomas, medulloblastomas, and ependymomas with complete accuracy is not possible based on traditional imaging. Next-generation MRS and DWI techniques (described earlier) may be helpful in the future.
Dissemination or leptomeningeal spread of pilocytic astrocytoma is rare and is more common in tumors arising from the hypothalamus, partially resected tumors, and tumors of the very young.16 In contrast with other more aggressive tumors, leptomeningeal dissemination of a pilocytic astrocytoma is not incompatible with long-term survival.17
Histology
Histologically, pilocytic astrocytomas are characterized by a classic biphasic pattern of loose glial tissue and compacted piloid tissue (see Fig. 10.1). The piloid component comprises dense sheets of bipolar cells with fibrillary process containing Rosenthal fibers. The loose glial component contains protoplasmic astrocytes and eosinophilic granular bodies.18 Macroscopically pilocytic tumors appear well circumscribed; however, on the microscopic level 64% show infiltration of the surrounding brain, making surgical extirpation difficult.13 In the cerebellar pilocytic astrocytoma, invasion of the leptomeninges is common.18 However, this invasion does not correlate with a poor prognosis. Interestingly, the degree of mitotic index, cellular atypia, and microvascular proliferation has no effect on event-free survival. However, histopathological evidence of vascular hyalinization, calcification, necrosis, or oligodendroglioma-like features may predict a poorer clinical outcome.19 Very rarely, pilocytic astrocytomas undergo malignant transformation to an anaplastic pilocytic astrocytoma.7 The majority of malignant transformation has occurred after administration of radiotherapy.20,21
Contrary to the supratentorial and brainstem pilocytic tumors, cerebellar pilocytic astrocytomas are rare in the neurofibromatosis type 1 (NF1) population. Recently, genetic investigations have revealed gains at 7q34 resulting in the discovery of two important fusion proteins: KIA1546–BRAF and SRGAP3-RAF1. These fusion proteins lead to constitutive activation of the ERK/MAP kinase pathway.22–25
Management
Gross total resection of a pilocytic astrocytoma is considered curative.8,15,16,26,27 Resection of the mural nodule is key in the surgical extirpation of pilocytic astrocytomas. Surgeons debate the need for removal of the cystic wall, but no statistical difference in survival has been noted between patients with cyst wall removal and those without.14 Postoperative MRI is imperative to evaluate degree of resection because direct neurosurgical evaluation is unreliable. With gross total resection 10-year survival rates are in excess of 90%.28 Subtotal resection increases rate of recurrence (7% vs. 27%) but not overall survival.7,28 Spontaneous regression after partial resection as measured by MRI occurs more frequently than growth. Thus, a good argument can be made for observation of residual tumor in cases in which reoperation for total resection carries a high morbidity rate, such as when the tumor invades the fourth ventricle (~10%).28,29 No adjunctive therapy is required for the treatment of pilocytic astrocytoma unless leptomeningeal spread is evident. Leptomeningeal dissemination can then be treated with chemotherapy or radiation, although no standard protocol exists.16,17,30,31
Medulloblastoma
Medulloblastoma is the most common malignant solid neoplasm of childhood. Medulloblastomas are related to primitive neuroectodermal tumors (PNETs) and occur exclusively in the posterior fossa. The median age at presentation is 9 years in the entire population and 7.3 years in the pediatric population.32 There is a slight male predominance of 1.6:1.32
Histology and Genetics
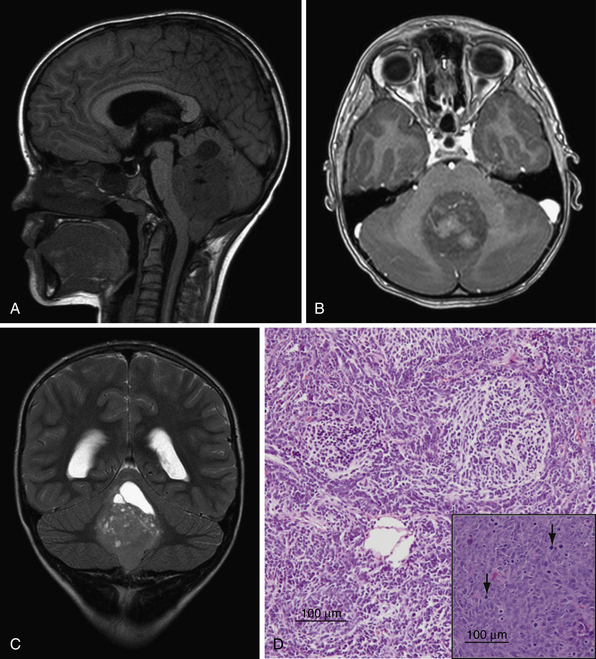
The WHO classifies medulloblastoma as a grade IV lesion and recognizes five subtypes: classic, desmoplastic/nodular, medulloblastoma with extreme nodularity, anaplastic, and large cell.33 Desmoplastic/nodular lesions have a more favorable prognosis as opposed to large cell and anaplastic lesions, which have a poorer prognosis when compared to classic medulloblastoma.34–36 Classic medulloblastoma appears as a small blue cell tumor, composed of densely packed undifferentiated oval cells with hyperchromatic nuclei and scant cytoplasm (Fig. 10.2). As with other malignant lesions, there is marked nuclear pleomorphism and brisk mitotic activity.37 Homer-Wright rosettes composed of neoplastic cells concentrically arranged around fibrillary processes are a common histological feature. The desmoplastic type of medulloblastoma contains “pale islands” of reticulin fibers surrounding a nodular reticulin-free zone.37 The large cell and anaplastic subtypes are characterized by large nuclei with prominent nucleoli, and a lower nuclear-to-cytoplasmic ratio than classic medulloblastoma.38,39
Genetic insights into medulloblastoma were gained by studying the familial tumor predisposition syndromes: Gorlin’s syndrome (a mutation in the PTCH gene in the sonic hedgehog signaling pathway), Turcot’s syndrome (a mutation in the APC gene affecting the beta-catenin/Wnt signaling pathway, and Li-Fraumeni syndrome (a mutation in p53 tumor suppressor gene).40 However, these familial syndromes account for only a small percentage of medulloblastomas.
The largest genetic analysis on sporadic medulloblastoma comes from a study by Northcott and associates in which they analyzed 212 medulloblastomas with high-resolution SNP (single nucleotide polymorphism) genotyping. Isochromosome 17q was confirmed as the most common genetic alteration in medulloblastomas, found in 28% of specimens.41 This large data set corroborated other genes associated with medulloblastoma such as amplification of MYC oncogenes, OTX2, TERT, PDGFRA, and CDK.41 Interestingly, this paper identified a novel pathway of medulloblastoma pathogenesis including histone lysine methylation genes, which regulate gene expression during development.41
The cell of origin in medulloblastoma remains elusive. Some groups have proposed medulloblastoma to arise from the external granule layer of the developing cerebellum and others have proposed that the cell of origin arises from a subventricular progenitor zone. Recent genetic subgroup analysis has classified medulloblastoma into one of four or five distinct genetic groups.42,43 In a series of over 400 patients which was linked to patient prognostic data, medulloblastomas could be segregated into one of four subgroups: group A (characterized by defects in WNT pathway signaling), group B (characterized by defects in SHH signaling), group C, and group D. Prognosis was correlated with subgroup and revealed that patients with group B medulloblastoma were at the extremes of age (infant or adult), have a desmoplastic phenotype, and have a more favorable prognosis. Group A medulloblastoma behaved in a similar fashion to classic medulloblastoma, whereas group C and D tumors had a poorer prognosis, and presented with disseminated disease. Of greatest interest, however, was the ability to predict genetic subgroup based on immunohistochemistry alone (Table 10.2). These distinct genetic and prognostic subgroups argue for a multiple cell of origin for medulloblastoma, but what is more important, they may change the way we risk stratify and treat patients with medulloblastoma in the future.
TABLE 10.2 Novel Stratification of Medulloblastoma Based on Genetic Subgroup Analysis
| Subtype | Genetic Background | Immunohistochemical Stain |
|---|---|---|
| A | WNT tumors | DKK1 |
| B | Sonic Hedgehog (SHH) tumors | SFRP1 |
| C | NPR3 | |
| D | KCNA1 |
Data from Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 2011;29(11):1408-1414.
Imaging
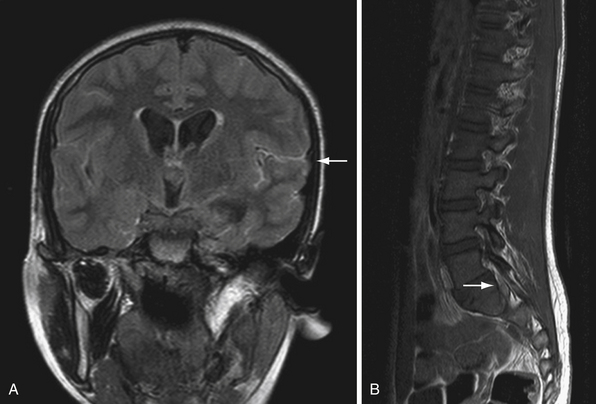
Medulloblastomas are typically midline cerebellar lesions arising from the vermis; however, in older children and adults they can arise from the cerebellar hemispheres.44 Classic CT imaging of medulloblastoma is a hyperdense lesion in the cerebellar vermis with surrounding vasogenic edema.45,46 Calcification (22%) and cyst formation (59%) may be observed in some cases.47 There is a high degree of variability of MR appearances of medulloblastoma. T1 sequences are usually iso-hypointense to white matter and hyperintense on T2 sequences (see Fig. 10.2). Tumor enhancement can be both homogeneous or heterogeneous.46 Similar to ependymoma, approximately14% of medulloblastomas may show foraminal extension.48 Leptomeningeal seeding on MRI is found in approximately 33% of patients. The spinal canal is the most common location of seeding; however, the supratentorial compartment may also be involved. Nodular or diffuse enhancement along the leptomeninges, nerve roots in the spinal canal, or cranial nerves in the CPA are common findings of CSF seeding (Fig. 10.3).46,49–51 Interestingly only 15% to 60% of patients with evidence of metastasis by MRI have positive CSF by cytological examination and only 70% of patients with positive cytological findings have evidence of metastasis by MRI. This highlights the importance of CSF cytological examination, especially in cases in which the MRI imaging is negative.52,53 Rare cases of extraneural spread have been reported to bone, lymph nodes, liver, and lung.54
Management
With combined surgical resection, radiation, and high-dose chemotherapy, 5-year progression-free survival rate in average-risk medulloblastoma is approximately 80%. Currently risk stratification is based on age (less than 3 years of age), presence of disseminated disease, and extent of surgical resection (Table 10.3). However, this stratification system fails to take into consideration the biology of medulloblastoma tumors, and a system based on molecular markers and genetic subgroup analysis may provide a better way of categorizing patients in the future. Current treatment studies are including favorable molecular markers such as nuclear beta-catenin and TrkC expression and unfavorable markers such as Myc genes and ERBB2 in their stratification paradigms.55
| Average-Risk Medulloblastoma | High-Risk Medulloblastoma |
|---|---|
| Age < 3 years ≤1.5 cm2 residual disease M0—no dissemination of tumor |
Age ≥ 3 years >1.5 cm2 residual disease M1—positive lumbar CSF cytological findings, negative MRI of brain and spine M2—macroscopic dissemination on MRI of brain and negative MRI of spine M3—macroscopic spinal dissemination on MRI M4—extraneural spread Anaplastic histological pattern |
CSF, cerebrospinal fluid; M0-M4, distant metastasis (staging); MRI, magnetic resonance imaging.
Surgery for Medulloblastoma
The goal of surgery in medulloblastoma is complete resection without causing neurological injuries. Several clinical trials have shown that extent of surgical resection correlates with recurrence-free survival.56–59 Postoperative imaging should be obtained 24 to 48 hours after surgery and compared to preoperative scans to determine extent of resection. In cases in which there is greater than 1.5 cm2 of residual tumor, a repeat procedure should be considered, if safe and anatomically feasible, to place the patient in the best prognostic category.
Treatment of Children Over Age 3 with Average-Risk Medulloblastoma
Management of medulloblastoma has evolved over time as new clinical trials are published on risk stratification and treatment paradigms. Medulloblastoma is a radiosensitive tumor and incorporation of radiotherapy has become a standard of care in treatment of children older than 3 years of age. The original radiation dose of 36 Gy to the neuroaxis and 54 Gy to the posterior fossa resulted in numerous detrimental side effects including cognitive decline, endocrine insufficiency, hearing loss, vascular complications, and secondary malignancies.60–65 Because of these detrimental effects of craniospinal radiation, efforts began to reduce the degree of craniospinal radiation in average-risk patients aimed at preserving neurological function. This was accomplished by increasing the intensity of chemotherapy. Chemotherapy had already proved an effective adjunct to radiation and surgery in earlier studies.66,67 Packer and colleagues reduced the degree of cranial spinal radiation to 23.4 Gy by adding chemotherapy during and after radiotherapy and still maintained excellent control with event-free-survival (EFS) of greater than 80% in nondisseminated medulloblastoma.68 Typical chemotherapeutics used in medulloblastoma include cisplatin, CCNU, vincristine, cyclophosphamide, and etoposide. There is some evidence to support a further reduction of craniospinal radiation to 18 Gy and the Children’s Oncology Group is currently examining this possibility.69 The current standard of care for average-risk medulloblastoma is gross total resection, postoperative craniospinal radiation of 23.4 Gy with a posterior fossa boost of 54 Gy, followed by 12 months of chemotherapy.
Treatment of Children Over Age 3 with High-Risk Medulloblastoma
The 5-year event-free survival rate across studies in high-risk medulloblastoma ranges from 30% to 70%.70 The best outcomes to date involve surgery, craniospinal radiation with 36 to 39.6 Gy, with a posterior fossa boost followed by intense cyclophosphamide, vincristine, cisplatin, and peripheral stem cell rescue. This regimen yielded a 5-year EFS rate of 70%.71 A 4-year EFS rate of 66% was reached by the Children’s Oncology Group (COG 99701) with surgery, concomitant daily vincristine/carboplatin and craniospinal radiation (36 Gy), and a posterior fossa boost followed by monthly cyclophosphamide and vincristine.72 These studies highlight the intensive regimens required to treat high-risk medulloblastoma. The adverse effects associated with these intensive treatments must be taken into consideration when one considers the enhanced survival rates for these patients.
Treatment of Infants and Young Children with Medulloblastoma
Treating infants and young children with medulloblastoma remains challenging and suboptimal. The developing brain is particularly susceptible to the toxicity of treatment regimes. These patients suffer from severe neurocognitive decline secondary to craniospinal irradiation. Thus, recent neuro-oncology clinical trials have promoted the use of chemotherapeutic strategies to delay or avoid craniospinal radiation. The Pediatric Oncology Group (POG) initiated a trial in the 1980s (the Baby POG study) treating infants with prolonged postoperative chemotherapy in an attempt to delay radiation until 3 years of age. The overall 5-year progression-free survival (PFS) rate was 31.8% but increased to 69% in those children with gross total resection.73 This study showed that craniospinal radiation could be delayed.73 The best results, to date, come from the Head Start 1 and 2 trials using induction chemotherapy of cisplatin, vincristine, etoposide, and cyclophosphamide followed by myeloablative chemotherapy and autologous stem cell rescue (methotrexate was added for disseminated disease). The 5-year PFS rates for infants was 64% in those with localized disease and 45% for disseminated disease. Interestingly, 50% of survivors never received radiation.74
Novel Therapeutics
As we begin to understand the molecular biology of medulloblastoma we have come to recognize and identify possible therapeutic targets. One such example is the use of a sonic hedgehog (SHH) pathway inhibitor GDC-0449. A 26-year-old man with recurrent resistant medulloblastoma with aberrant activation of SHH signaling was treated with the inhibitor and showed rapid (albeit transient) response.75
Proton beam therapy is a type of radiation therapy that utilizes charged beams that have a finite range in tissues and potentially allows sparing of normal tissues compared to conventional types of radiotherapy. To date, only a few patients with medulloblastoma have been treated with proton beam therapy; however, there is currently a phase II trial being undertaken at this time.76
Intrathecal (IT) chemotherapy to delay radiation threapy, salvage recurrent disease, or treat leptomeningeal seeding is being used now in several trials. IT chemotherapeutic regimens have been successfully used to avoid craniospinal radiation in the pediatric leukemia population.77 Case reports have shown moderate success and phase I and II trials for pediatric medulloblastoma are currently ongoing.78,79
Ependymoma
Ependymomas are the third most frequent brain tumor in the pediatric population. First described in 1926 by Cushing and Bailey, they account for 6.4% of primary brain tumors in children aged 0 to 14 years and for 30% of tumors in children less than 3 years of age.80 The mean age of presentation is 3.7 years.11 Unlike medulloblastoma, 5-year survival rates for ependymoma are approximately 60%.81
Imaging
The classic infratentorial ependymoma fills the fourth ventricle and extends laterally through the foramina of Luschka (15%) and inferiorly through the foramen of Magendie (60%). Typically, ependymoma demonstrates low T1, high T2, and intermediate to high fluid attenuated inversion recovery (FLAIR) signal intensity (Fig. 10.4).82 However, the lesions can be heterogeneous owing to cystic areas, calcifications (50%), and hemorrhage. Postcontrast imaging shows a heterogeneous enhancing tumor with areas of avid enhancement mixed with poorly enhancing regions. Diffusion-weighted imaging (DWI) is intermediate between pilocytic astrocytomas (low) and primitive neuroectodermal tumors (PNETs) (high).83 Ependymomas often encase neurovascular structures in the CPA, thus making surgical removal difficult without causing cranial nerve or vascular injury. Ikezaki and co-workers, classified posterior fossa ependymomas into three groups based on location: (1) the lateral type presenting in the CPA characterized by a poor prognosis secondary to involvement of cranial nerves and brainstem; (2) ependymomas localized to the floor of the fourth ventricle with an intermediate prognosis; and (3) those localized to the roof of the fourth ventricle with the most favorable outcome.84 Leptomeningeal dissemination occurs in 8% to 12% of patients by CSF cytological findings and occurs more frequently with anaplastic grades.85 Leptomeningeal disease with drop metastasis is most common in the lumbosacral region.
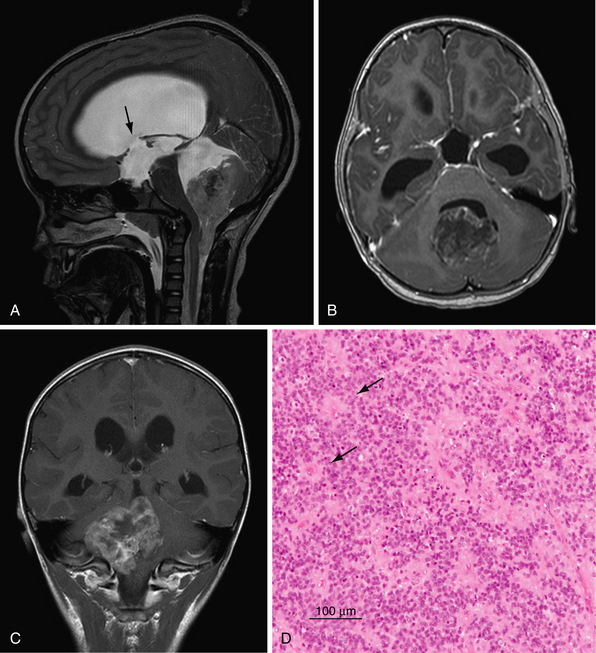
Histology
The WHO recognizes three grades of ependymoma and four histological variants (Table 10.4).33,80 The classic histological features of ependymoma are perivascular and ependymal rosettes. The former corresponds to ependymal cell processes radially arranged around a cell-free perivascular zone. The latter, ependymal rosettes, comprise tumor cells concentrically arranged to form a lumen.80 Ependymomas are given an anaplastic grade when there is evidence of (1) brisk mitotic activity; (2) increased cellularity; (3) microvascular proliferation; or (4) pseuodopalisading necrosis. However, there is debate in the literature as to the precise definition of anaplastic criteria. In part, this controversy has called into question whether or not histological grade predicts for survival.86–95
| WHO Grade | Histological Subtype(s) |
|---|---|
| Grade I (subependymoma and myxopapillary) | Cellular ependymoma (grade II) |
| Grade II (ependymoma) | Papillary ependymoma (grade II or III) |
| Grade III (anaplastic ependymoma) | Clear cell ependymoma (grade II or III) |
| Tanycytic ependymoma (grade II or III) |
WHO, World Health Organization.
The most common genetic alteration in ependymoma is loss of chromosome 22; however, in posterior fossa ependymoma the putative tumor suppressor at this locus remains elusive.96 Other genetic events common in posterior fossa ependymomas are 9q and 1q gain, loss of 6q, and monosomy 17p.96 Interestingly, DNA identical to portions of the SV40 virus has been isolated from ependymoma, and SV40 is capable of inducing ependymoma in rodents.
Surgical Management
The key to treatment of ependymoma remains complete macroscopic surgical resection, as it is known that the degree of resection is the most significant predictor of survival. The goals of surgery are tissue diagnosis, management of hydrocephalus, and cyotoreduction. Complete surgical resection leads to 5-year survival rates ranging around 70% to 80%,80 whereas patients with subtotal resection have significantly poorer outcomes with 5-year survival rates around 20% to 40%.91,97 Given the predictive value of resection on survival, patients should undergo a postoperative MRI within 48 hours to ensure macroscopic resection. “Second look” surgery is warranted in cases in which bulky residual disease is found after initial surgery.91,98–100 It should be stressed here that gross total resection can be made difficult with large ependymomas extending into the CPA encasing cranial nerves and vascular structures or when invasion of the floor of the fourth ventricle occurs. The morbidity of complete resection in these cases may be high (10-30%).101
Adjunctive Therapies
Postoperative radiation treatment is an established standard of care for patients with ependymoma because it is a radiosensitive tumor. A recent study from St. Jude Children’s Research Hospital showed a 7-year EFS rate and overall survival (OS) rate of 69% and 81%, respectively, with maximal surgical resection and local conformational radiation therapy. Craniospinal radiation was reserved for those cases with evidence of CSF dissemination by imaging or cytological appearance.102 Interestingly, baseline and longitudinal testing of cognitive outcome demonstrate most survivors within normal range despite a large portion of patients being 3 years or less at treatment.102
Currently, there are no proven established protocols for chemotherapy in the treatment of ependymoma. There has been moderate success in using chemotherapy to delay radiation therapy in young children with ependymoma.73,103,104 Current studies are being undertaken to evaluate molecularly targeted therapy such as the small molecule tyrosine kinase inhibitors (geftinib, erlotinib, baevacizumab).105
The rate of relapse following treatment ranges from 30% to 72%.97,102,104,,106,107 The vast majority of relapses occurs locally, and median survival time after relapse varies from 8.4 to 24 months.108 The literature supports reoperation for recurrence of ependymoma.98,107 For children who have not received radiation therapy, radiation therapy should be given. Re-irradiation is an option for children previously treated with radiotherapy, but morbidity rate from radiation necrosis may be high.109–111
Atypical Teratoid/Rhabdoid Tumor
Malignant rhabdoid tumors (MRTs) were first described as a highly malignant subtype of Wilms’ tumor.112 It is now recognized that MRTs occur throughout the body. Biggs and associates first described an intracranial MRT in 1987.113 These tumors were first termed atypical teratoid/rhabdoid tumors (AT/RTs) in a landmark paper in 1995 because of their histological characteristics, which showed neuroepithelial, peripheral epithelial, and mesenchymal elements.114 The WHO first recognized AT/RT as a separate tumor entity in 2000. It is predominantly a tumor of infants and young children with a median age of diagnosis at 26 months with a slight male predominance.114,115 Approximately 30% of AT/RTs occur infratentorially (CPA and cerebellum), and 22% have CSF dissemination at diagnosis.115 The overall survival time is 18 months; however, in patients presenting with signs of metastasis the prognosis is significantly worse at 8 months.115
Histology
Pathologically, AT/RT is composed of nests or sheets of rhabdoid cells intermixed with areas indistinguishable from PNET or medulloblastoma.114 The intermixed histological features of AT/RT underscores the issues with sampling errors in biopsies of the AT/RT. The histopathological diagnosis of AT/RT can be aided by staining for the loss of nuclear INI1.116 INI1 is a tumor suppressor gene found on chromosome 22q, and 60% to 90% of AT/RTs show monosomy or deletions of chromosome 22.117
Imaging
There are no distinguishing features on radiographic imaging that specifically distinguish AT/RTs from other fourth ventricular tumors. As with medulloblastomas, the solid portions of AT/RTs are hyperdense on CT, isointense on T1, and heterogeneous on T2, although the appearance can vary.118–122 With contrast administration there is heterogeneous enhancement (Fig. 10.5).
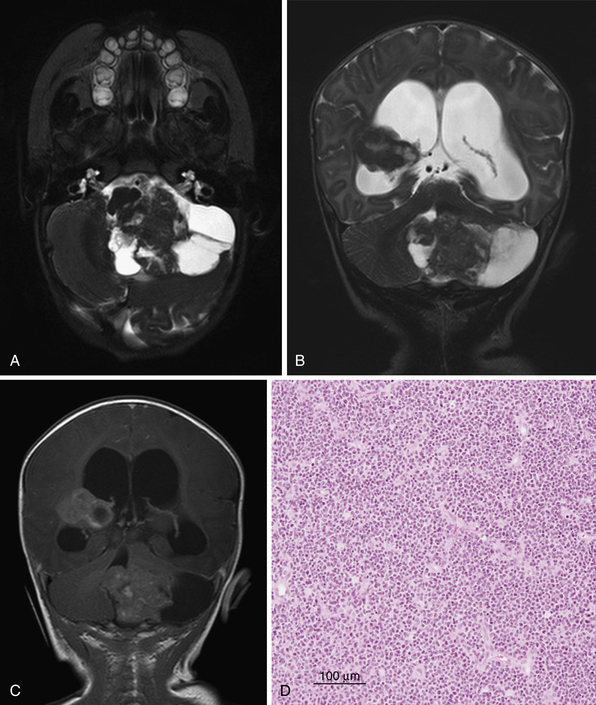
Management
Given the overlap of AT/RT with other tumor subtypes of the posterior fossa, it is important to take into consideration the diagnostic tools used to classify AT/RT in each study cohort, specifically whether loss of INI1 by immunohistochemistry was utilized. The St. Jude Children’s Research Hospital, Boston Children’s Hospital, and the central registry in Cleveland have all published their series of patients with AT/RT. All studies showed that gross total resection correlated with OS and PFS rates.123–125
Radiation therapy, either craniospinal or focal, is important in the control of AT/RT. In a meta-analysis by Athale and colleagues, children receiving radiation in combination with chemotherapy versus children not treated with radiotherapy had an overall survival time of 18.4 versus 8.5 months.116 The St. Jude group treated all patients with surgery and high-dose alkylator chemotherapy, and those over 3 years of age received craniospinal radiation. The 3-year EFS rate of those receiving chemotherapy was 78% versus 11% in children younger than 3 years who did not receive initial craniospinal radiation.124 However, a large number of patients with AT/RT present before the age of 3 years, increasing the morbidity of radiation.
The variability of chemotherapeutic regimens and the small series of AT/RT cases render conclusions regarding specific chemotherapeutic regimens difficult. Investigators have used cisplatin, high-dose alkylators plus stem cell rescue, variations of the Intergroup Rhabdomyosarcoma Study, IRS III regimens, and temozolamide. In a meta-analysis of chemotherapeutic regimens, intrathecal (IT) chemotherapy increased the probability of survival at 2 years to 64%.115 This survival benefit was maintained but was not as great in children younger than 3 years of age.115,123 The Boston Children’s Hospital recently published their prospective series of 25 children (mean 26 months) treated with intensive multimodal therapy consisting of surgical resection, chemoradiation, consolidation therapy, and IT chemotherapy. Two-year EFS rate was 53% and OS rate reached 70%.123 Whether IT therapy can substitute for craniospinal irradiation for CNS treatment or prophylaxis remains unclear.
Novel approaches to AT/RT treatment will be necessary to increase survival in this disease. A recent study on dendritic cell–based vaccinations was performed for three children with AT/RT (two at relapse and one at presentation with metastatic disease). The two patients who were treated with surgery, chemotherapy, radiation, and vaccination at relapse were still alive at 34- and 53-month follow-up. The patient with a metastatic presentation succumbed at 51 months.126 In conclusion, AT/RT is an aggressive CNS tumor of young children that remains a therapeutic challenge.
Choroid Plexus Papilloma and Carcinoma
Choroid plexus tumors are rare primary brain tumors arising from choroid plexus epithelium. Guerard first described these neoplasms in 1833 and Bielschowsky performed the first operative procedure in 1906. Choroid plexus neoplasms represent 0.4% to 0.8% of all primary neoplasms of the brain. Although these tumors can occur in all age groups, a greater number of them occur in childhood, with 70% being diagnosed before the age of 2 years. Studies have shown a skewed distribution slightly in favor of the male gender 1-1.3:1.127 The WHO recognizes three classes of choroid plexus neoplasms: the grade I choroid plexus papilloma (CPP), the grade II atypical CPP, and the grade III choroid plexus carcinoma (CPC).33
Anatomically, choroid plexus neoplasms occur more frequently in the lateral (50-70%), fourth (20-40%), and third (5-10%) ventricles.128 They have also occurred in the CPA and in biventricular locations (5%).128 However, the anatomical distribution differs markedly in relation to age. In young children, the vast majority of choroid plexus tumors present in the lateral ventricles, whereas fourth ventricular and CPA tumors are more common in older children and adults.127 Metastatic disease from CPC has been documented anywhere along the neuraxis including the leptomeninges.
Histology and Genetics
Genetic syndromes predispose to the formation of CPP and CPC, such as Li-Fraumeni syndrome, neurofibromatosis type 2, Aicardi’s syndrome, Down syndrome, and von Hippel-Lindau disease.128 Germline mutations in TP53 and hSNF5/INI1 have been found in familial cases of choroid plexus tumors, and similar mutations have been found in tumors of sporadic cases of CPC and CPP.129,130 Multiple chromosomal imbalances have been described in CPP and CPC.131 The most clinically relevant cytogenetic abnormality was demonstrated in a small cohort of patients with a gain of 9p and loss of 10q, which provided a survival advantage.132
Histologically, CPPs are similar to normal choroid plexus in that they show many papillae covered by simple columnar or cuboidal epithelium, eosinophilic cytoplasm, round to oval nuclei situated basally, and papillary fronds consisting of vascular connective stroma.127,128 CPPs do not have necrosis, brain invasion, or mitotic figures. As with other cancers, CPC can show marked cytological atypia, nuclear pleomorphism, loss of polarity, high cellular density, frequent mitosis, necrosis, vascular proliferation, hemorrhage, and brain infiltration.127 Atypical CPPs show an intermediate degree of nuclear atypical and mitotic figures.133
Imaging
Imaging characteristics of CPPs often include a homogeneously enhancing tumor with vascular feeding pedicles along with a “frondlike” solid tumor and associated hydrocephalus. At times, however, it is difficult to differentiate between CPP and CPC on imaging studies except that CPCs will often demonstrate parenchymal invasion and peritumoral edema whereas CPPs do not. Choroid plexus lesions are iso- to hyperdense on CT with 25% showing calcification. They are typically isointense on T1 and heterogeneous on T2 MRI sequences (Fig. 10.6).127
Management
Extent of surgical resection is a significant prognostic factor in choroid plexus tumors especially the CPC.134–137 Choroid plexus tumors are highly vascular and perioperative blood loss may contribute to high perioperative morbidity.138,139 Preresection chemotherapy has been successfully used by some groups to diminish tumor vacularity prior to surgery.140 The CPT-SIOP-2000 trial recommends treating CPPs with gross total resection alone.133 Atypical CPPs were treated with surgery alone unless evidence of metastasis or residual disease was present, and all patients with CPCs were treated with gross total resection where possible, chemotherapy, and craniospinal radiation (if >3 years of age). An early analysis of 106 patients and 2.2-year follow-up shows 100% overall survival in the CPP group (n = 39), 89% (n = 24) in atypical CPP, and 36% in CPC (n = 29).133 Rates for complete surgical resection were 79%, 63%, and 47%, respectively; 9 of 24 patients with atypical CPP were treated with chemotherapy and 1 with radiation.133
Hemangioblastoma
Hemangioblastomas, WHO grade I, are highly vascular tumors seen in the cerebellum and spinal cord. They are the most common posterior fossa lesion in adults but are rarer in the pediatric population.141,142 They can occur sporadically or as part of the von Hippel-Lindau (VHL) familial syndrome. Any patient diagnosed with a hemangioblastoma should be assessed for signs or symptoms of other manifestations of this rare syndrome. VHL is an autosomal dominant disorder characterized by CNS hemangioblastomas, retinal angionatosis, pancreatic cysts, renal cell carcinoma, pheochromocytoma, and epididymal cysts.143 VHL patients have a germline mutation in the VHL gene that normally acts as a sensor of hypoxia to induce vascular remodeling and increased levels of vascular endothelial growth factor (VEGF).144 In VHL, this gene is constitutively active, leading to a highly vascular tumor. Sporadic hemangioblastomas are also found to have mutations to VHL.145
Hemangioblastomas are typically well-circumscribed cystic lesions with a small mural nodule abutting the pia; however, presentation can vary from a solid lesion to a lesion with a central cyst.146,147 The cyst is hyperintense on both T1 and T2 MRI sequences, and the mural nodule is isointense on T1 and hyperintense on T2. There is strong contrast enhancement of the solid component.146
Surgical excision is the treatment of choice and can be curative with gross total resection.148 In cases in which gross total resection cannot be achieved, gamma knife radiosurgery or fractionated radiotherapy can achieve tumor control.149,150
Dermoid/Epidermoid Cysts
Dermoid and epidermoid cysts are rare (<1%) intracranial congenital non-neoplastic lesions that arise from retained ectodermal cells and mesenchymal elements in dermoid cysts in the neural groove during embryonic neural tube closure.151,152 They typically present in the third decade of life with a male predominance and are rare in the pediatric population.153 In the adult population epidermoid cysts are significantly more common than dermoid cysts (4:1-10:1).153 Interestingly, in children, dermoids may account for a higher percentage than epidermoids.154 Dermoids and epidermoids are histologically benign but can present with mass effect, aseptic meningitis, infectious meningitis, or neurovascular compression.155–157
Dermoid cysts contain both dermal and epidermal elements, are typically midline in location, and are often associated with dermal sinus tracts.154 The dermal sinus tract of posterior fossa dermoid cysts is typically located at the inion and is associated with the usual cutaneous stigmata of hair tufts, cutaneous angioma, and fluid leakage. There may be an association with Klippel-Feil syndrome.158–162
Generally, dermoid and epidermoid cysts appear as well-circumscribed lesions with no edema and moderate mass effect. The cysts appear as hypo- and hyperintense on T1- and T2-weighted MRI sequences, respectively.154 Epidermoids commonly occur in the CPA and can mimic an arachnoid cyst on imaging with the exception that epidermoid cysts exhibit DWI changes on MRI sequences.163
Epidermoid and dermoid cysts are benign lesions for which surgical excision is curative.153,154,164 A thick capsule adherent to vital neuroanatomical structures can complicate complete surgical removal.165 In this case it is wise to perform a subtotal resection, and monitor for recurrence in the postoperative period. Malignant degeneration of dermoid or epidermoid cysts to squamous carcinoma has been reported but is extremely rare.166 Care must be taken to avoid spillage of cyst contents into the subarachnoid space during surgical excision because this may result in aseptic meningitis.
Surgical Management of Posterior Fossa Tumors
Management of Hydrocephalus
A majority of patients (83%) with posterior fossa lesions in and around the fourth ventricle will present with hydrocephalus.167–169 However, only about 30% of children will require postoperative shunting after tumor removal.14,167,170–172 This number may be reduced to 6% if an endoscopic third ventriculostomy (ETV) is performed as the first CSF diversion technique or in combination with a ventriculostomy.173 However, by performing an ETV in every child with hydrocephalus and a posterior fossa tumor, one would be exposing 70% of children to the morbidity of an extra surgical procedure. Riva-Cambrin and co-workers have developed a preoperative clinical grading system to aid in determining the need for postoperative CSF diversion.174 This grading system scores children based on age (<2 years), initial degree of hydrocephalus, tumor histological features, and presence of metastasis to predict probability of hydrocephalus at 6 months.174 This system can aid the surgeon in determining whether to perform CSF diversion (shunt or ETV) prior to surgical removal of tumor. Today there is an option to place an occipital or “Frazier” burr hole during the surgical procedure. This is for emergent decompression of the lateral ventricles should postoperative swelling result in obstruction of the fourth ventricle and acute hydrocephalus.
Surgical Approaches to Posterior Fossa Lesions
The midline suboccipital craniotomy has been the workhorse for resection of fourth ventricular tumors. Classically, it involves splitting the inferior vermis for large tumors within the fourth ventricle. However, the process of splitting the vermis followed by lateral retraction of the dentate nuclei and affecting the dentatonucleocortical projection has raised concerns about cerebellar mutism (see later discussion). Thus, modifications of the midline approach, including the telovelar approach with dissection of the cerebellomedullary fissure to reach the fourth ventricle without splitting the vermis, have been developed.175–177 In this procedure, after performing a standard suboccipital craniotomy, the cerebellomedullary fissure is opened by separating the tonsillo-uvular and tonsillomedullary spaces. The inferior roof of the fourth ventricle is exposed with retraction of the uvula superiorly and the tonsils laterally. The inferior medullary velum and the tela choroidea are then opened, exposing the fourth ventricle from aqueduct to obex. The opening of the tela can be continued laterally to expose the foramen of Luschka.175,177,178 Proponents of the telovelar approach argue that with combined removal of the posterior arch of C1, one can obtain a larger working area and more lateral access to the foramen of Luschka, facilitating removal of large lesions.179
The suboccipital retrosigmoid approach can be used to reach lesions in the CPA. This approach allows for good visualization of the lower cranial nerves and preserves hearing. A curvilinear incision is made 1 to 2 cm behind the mastoid. The craniotomy is performed medial to the sigmoid sinus. Once the dura is opened, the arachnoid over the cisterna magna or the superolateral cerebellum is opened to allow for CSF drainage, thereby facilitating the retraction of the cerebellum medially to expose the CPA.180,181 Care should be taken to monitor the seventh cranial nerve in these cases to aid in its preservation. Other more complex skull base approaches such as the posterior petrosal or far lateral approach can be utilized in conjunction or separately from the suboccipital retrosigmoid approach.182
Surgical Adjuncts
There are many tools available to the surgeon to assist in surgical removal of posterior fossa lesions. Image-guided surgery or neuronavigation based on presurgical MRI or CT scans can aid in planning craniotomy and identifying important surgical landmarks. These preoperative scans, however, can become less reliable as the surgical case proceeds, and thus the need for updated real-time imaging arises. This imaging may be more important in cases such as ependymoma and medulloblastoma in which degree of resection is critical. Many centers have reported on their case series of intraoperative MRI and have found it both helpful and safe.157,165 When intraoperative MRI is not available, intraoperative ultrasound may aid the surgeon in understanding the degree of tumor resection and identifying surgical landmarks.183,184
Physiological mapping and monitoring during surgery can be invaluable to surgeons operating near the brainstem and cranial nerves. Mapping is the physical stimulation of a brain region of interest and awaiting a response. Mapping the floor of the fourth ventricle for the facial, glossopharyngeal, vagal, and hypoglossal nuclei has been performed to enhance surgical removal of posterior fossa tumors with promising results.185,186 Monitoring is the ongoing activation and recording of neural circuits during surgery to provide “warnings” of a breach in pathway integrity. Electrodes can be placed in facial, pharyngeal, and tongue muscles, with sound-emitting electrodes in the ear, and stimulators of the gag reflex to monitor cranial nerves and their respective nuclei. Needle electrodes can be placed in the extremities to monitor the corticospinal tracts. Scalp electrodes can be placed in conjunction with stimulators to monitor evoked sensory potential activity from the extremities or auditory nerve.187 A 50% drop in amplitude or an increased latency of 10% is thought to be indicative of pathway injury.187 When this occurs, the neurosurgeon should stop operating until the potentials return to normal.
Complications of Therapy
Cerebellar mutism is one of the most feared complications following posterior fossa surgery and can complicate even the most anatomically successful of surgeries. Cerebellar mutism is defined as the complete absence of speech without impairment of consciousness. Other symptoms include hypotonia, ataxia, and emotional lability. Cerebellar mutism occurs in approximately 25% of cases and is more common after posterior fossa surgery for medulloblastoma.188 Speech is typically intact immediately following surgery with abrupt cessation 1 to 5 days after surgery. Resolution occurs up to 6 months later but some form of speech impairment may persist thereafter.189 The exact etiology of cerebellar mutism remains elusive; however, damage to the dentatothalamocortical outflow tracts is suggested by modern imaging modalities. Related to and perhaps an extension of cerebellar mutism is the cerebellar cognitive affective syndrome.188 This syndrome is the chronic impairment of cognitive abilities from disruption of the cerebellum and its interconnections and manifests as speech, executive function, visual-spatial, and personality dysfunction. Cerebellar cognitive affective syndrome can occur in the absence of posterior fossa radiation.188
The long-term neurocognitive side effects of craniospinal radiation are well known. Craniospinal radiation (36 Gy) is associated with a 20- to 30-point decrease in IQ, and 23.4 Gy is associated with a 10- to 15-point decline.190,191 These declines are more pronounced the younger the patient. Endocrine anomalies are strongly correlated with the degree of irradiation to the hypothalamus and commonly require hormone replacement therapy, such as growth hormone to increase stature in the pediatric population.192 Neurovascular complications are present in up to 5% of patients.193 Patients receiving cranial radiation are at risk for subsequent secondary malignancy. In a large cohort study of 1877 survivors of CNS tumors, 10.7% of patients had secondary tumors, most commonly nonmelanoma skin cancers and benign meningiomas. However, malignant neoplasms occurred in 4.1% of patients including malignant glioma, malignant meningioma, and PNET.194 Patients who have received radiation therapy have a significantly higher risk of mental health issues, unemployment, and remaining single.194
Brainstem Gliomas
Epidemiology and Classification
The brainstem is defined as the neural axis between the diencephalon and cervical spinal cord. Brainstem gliomas (BSGs) can occur anywhere in this neuraxis and account for 10% to 15% of primary pediatric intracranial neoplasms.195 Two decades ago, tumors were considered inoperable owing to their anatomical location and therefore were frequently fatal. Experience over the last several decades has demonstrated that brainstem gliomas comprise a heterogeneous group of tumors, some of which are amenable to long-term survival.196,197 BSGs are considered predominantly a pediatric entity with a mean age of presentation of 7 to 9 years.196–199 The most commonly utilized classification system segregates brainstem tumors based on their radiological appearance into three major categories: (1) diffuse, (2) focal, or (3) exophytic.196,200 In general, the focal and exophytic types are low-grade gliomas and carry a much better prognosis than the diffuse high-grade glioma. Diffuse intrinsic pontine gliomas (DIPGs) account for 60% to 80% of brainstem gliomas.201
Clinical Presentation
The specific signs and symptoms of brainstem tumors are dependent on their anatomical location. Patients with DIPGs typically have a rapidly progressive course of cranial neuropathies, with pyramidal tract and cerebellar signs. In contrast, the presentation of focal brainstem gliomas is more insidious with localizing signs such as isolated cranial nerve deficits and contralateral hemiparesis spanning months to years.202 Tumors of the cervicomedullary junction commonly present with lower cranial nerve palsies, pyramidal tract signs, ataxia, spinal cord dysfunction, and nystagmus.203–205 Signs and symptoms of hydrocephalus usually manifest later in the disease progression with the exception of tectal tumors to its location near the cerebral aqueduct.
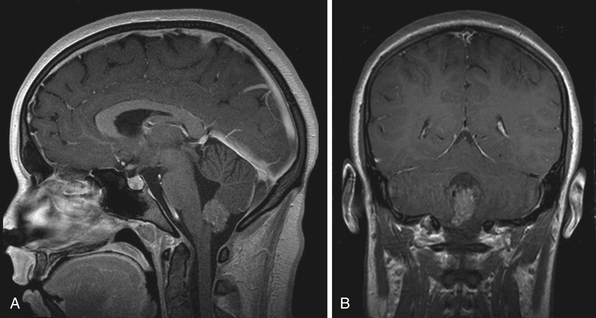
Diffuse Intrinsic Pontine Glioma
DIPGs are the most common brainstem tumors, accounting for approximately 60% to 80% of lesions.201 Unfortunately, DIPGs are the most devastating brainstem tumor with a median survival of 9 months despite treatment with multiple therapeutic modalities.206–208 Children usually present with rapid onset and progression of a triad of symptoms (cranial nerve palsies, long tract and cerebellar signs).209 Hydrocephalus occurs in the advanced stages of this disease.210
These lesions have a characteristic appearance on MRI. They appear hypointense on T1-weighted and hyperintense on T2-weighted images with indistinct margins, reflecting their infiltrative nature.208 On midsagittal imaging, their confinement to the pons is better delineated (Fig. 10.7). Gadolinium enhancement is variable and provides no additional prognostic information.211 Magnetic resonance spectroscopy (MRS) is an evolving imaging modality in brainstem tumors. DIPGs have increased metabolic ratios of choline to creatine and N-acetylaspartate, which is useful in delineating DIPG from demyelination, dysmyelination of NF1, encephalitis, and radionecrosis.212,213
Because the natural history and malignant course of DIPGs are well established, there is currently little clinical role for a diagnostic biopsy.201,214–216 A biopsy is reserved for indeterminate lesions on MRI, an unusual presentation, or when mandated by a study protocol.209 The rationale is that nearly all DIPGs are high-grade astrocytomas, outcomes are poor regardless of pathological grade, treatment strategies do not hinge on tumor grade, and biopsy is associated with significant morbidity and mortality rates. However, if we are to gain new insight into the biological and genetic behavior of these tumors, in the hopes of designing targeted therapies, biopsy may become an important strategy in the future.198
The mainstay of treatment for DIPGs is radiation therapy at a dose of 50 Gy. There is no role for surgical resection and very little long-term benefit from adjuvant chemotherapy.217 Surgery may be required to treat hydrocephalus; however, the majority of these cases are mild and symptomatic relief may be achieved with steroid administration. Palliative radiation may provide temporarily relief of symptoms in 75% of children, but unfortunately, all patients eventually suffer recurrence.218,219
Focal Brainstem Tumors
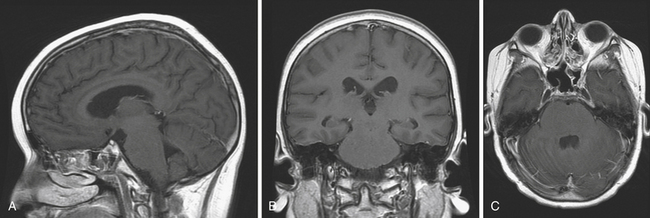
Tectal Tumors
Tectal tumors are intrinsic lesions in the region of the mesencephalon representing 5% of brainstem lesions and are typically WHO grade I or II.220 Given their close proximity to the cerebral aqueduct, tectal tumors cause hydrocephalus, rapid deterioration, and death at a small size.221 Despite the vast majority of tectal gliomas exhibiting an indolent benign course (>85%) there is a second subtype that behaves more aggressively.221 MRI may help distinguish this aggressive subtype, as they typically are greater than 2 cm; invade the adjacent tegmentum, thalamus, or pons; and demonstrate contrast enhancement (Fig. 10.8).
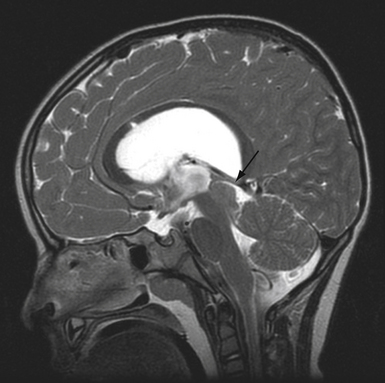
The more common benign intrinsic tectal region tumors generally follow an indolent course. These are typically low-grade astrocytomas that are well-circumscribed, nonenhancing lesions that present with signs and symptoms of hydrocephalus.197 Other signs include gait disturbances, ataxia, Parinaud syndrome, and strabismus. Treating the underlying obstructive hydrocephalus and follow-up with serial MRI is a safe approach to nonsuspicious tectal lesions.221 Some neurosurgeons favor a more aggressive surgical approach given that 18% to 30% of tectal tumors eventually progress.220 Hydrocephalus can be treated with a ventriculoperitoneal shunt or endoscopic third ventriculostomy (ETV), which has demonstrated to be safe and effective.220,222,223 It is important to continue to follow these patients because benign tectal gliomas may progress and ETVs can fail, leading to serious complications.220,224 Gamma knife radiosurgery has been recently utilized on 13 patients with tectal tumors and result in tumor stabilization; however, it is uncertain whether this treatment provides an improvement over a more conservative approach.225
The aggressive tectal tumors are typically larger on presentation compared to the indolent variety. In addition, they more frequently present with neurological symptoms, and may enhance and show evidence of invasion to adjacent neural structures on MRI. These aggressive lesions are rare and definitive management is debated in the literature with some authors supporting biopsy followed by radiotherapy and others supporting complete surgical resection followed by radiation therapy.221,226
Dorsally Exophytic Brainstem Tumors
The dorsally exophytic subtype accounts for approximately 10% to 20% of brainstem tumors.227,228 Children usually present with an insidious history of headaches, vomiting, ataxia, and cranial nerve dysfunction (usually sixth and seventh).227,228 Papilledema, torticollis, and long tract signs can be found on neurological examination.227,228 By definition, these tumors protrude into the fourth ventricle, but occasionally they can be dorsolaterally exophytic, projecting into the CPA.227 The hypointense signal on T1 and the hyperintense signal on T2 will generally display consistent tumor edges reflecting its less infiltrative nature. Contrast enhancement is typical. These tumors are predominantly pilocytic astrocytomas with occasional grade 2 and 3 astrocytomas and gangliogliomas, generally carrying a good prognosis.227,229 Dorsally exophytic tumors are the most surgically accessible of all brainstem gliomas. Similar to pilocytic astrocytomas elsewhere in the neuraxis, the primary modality of treatment is surgical debulking followed by serial imaging. Radio- and chemotherapy is reserved for recurrence. Gross total resection is usually not possible owing to the critical nature of adjacent structures. Neuronavigation, diffusion tensor MRI, white matter tractography, and brainstem monitoring are aids to maximize tumor resection and minimize morbidity.
Cervicomedullary Tumors
Children with cervicomedullary tumors commonly present with slowly progressive lower cranial nerve palsies, pyramidal tract signs, ataxia, spinal cord dysfunction, or nystagmus. Lower cranial nerve deficits may include dysphagia, nasal speech, nausea, vomiting, palate deviation, facial nerve palsy, head tilt, apnea, or irregular breathing patterns. The gradual onset of these symptoms reflects the slowly growing, relatively benign histological picture that is most commonly found, namely, the pilocytic astrocytoma.197
MRI typically demonstrates a hypointense lesion on T1 and hyperintense lesion on T2-weighted images.230 The craniocaudal extent can be best appreciated on the sagittal images. These tumors typically extend from the caudal two thirds of the medulla to the rostral portion of the cervical spinal cord. Some authors believe that cervicomedullary tumors may in fact be intramedullary tumors that expand rostrally.231 There is some evidence that in the more benign pathological subtypes, the tumor is less likely to penetrate the “anatomical barrier” of the pyramidal decussating fibers, medial lemniscus, efferent fibers from the inferior olivary complex, and inferior cerebellar peduncle rostrally.231
The prognosis for these lesions is generally favorable following neurosurgical resection, given the low-grade nature of the majority of these lesions. An aggressive surgical resection is usually undertaken given that these tumors are more likely to possess a defined surgical plane compared to other brainstem tumors. However, surgery in this location carries significant risks including quadriparesis, sleep apnea, cranial nerve palsy, proprioceptive deficits, and spasticity.232 Radiation therapy can be utilized after surgery, although most groups wait until evidence of recurrence.205,233 The role of chemotherapy in cervicomedullary tumors is ill-defined. Some advocate its use as front-line therapy in children younger than 10 years and for older patients who have demonstrated tumor progression despite radiation; others recommend it only for patients who have failed surgery or as an adjuvant therapy.230 Weiner and associates’ retrospective study has shown that the 5-year progression-free survival rate was about 60%, with 89% of the patients being alive after 5 years.205 Patients with good preoperative neurological status, early surgical intervention, and benign histological findings experience the longest progression-free survival.205
Other Focal Brainstem Tumors
Focal tumors in other location such as the medulla, midbrain tegmentum, or the pons are less common (<5%).234 These lesion are typically low-grade; however, anaplastic astrocytoma and glioblasstoma have been described. Non-neoplastic lesions such as vascular malformations, demyelination, and glangliosidoses need to be excluded by imaging or histological examination. If a biopsy is required, then surgical planning is crucial to minimize trajectories that will harm the patient; diffusion tensor imaging (DTI) or positron emission tomography (PET) may be useful in this situation. Surgical resection is the ideal treatment. However, owing to significant operative morbidity, conventional fractionated radiation therapy and radiosurgery have been tried with moderate results.201,225 Chemotherapy may have a role in delaying radiation or treatment of refractory progressive low-grade gliomas.197,235
Brainstem Gliomas in Neurofibromatosis Type 1
Brainstem gliomas should not be confused with the “unidentified bright objects” seen in NF1 patients. These bright spots are common in the brainstem of children and disappear spontaneously.236 Although less common than optic gliomas in the NF1 population, BSGs in NF1 may represent a distinct entity from non-NF1 brainstem gliomas. NF1 BSGs have a more favorable prognosis and survival rates of up to 90% at 5 years of age.236
Role of Surgery in Brainstem Gliomas
Biopsy of BSGs is reserved for lesions in which the diagnosis is indeterminate on MRI. Many authors have shown that biopsies can be accomplished with reasonable safety, with sufficient tissue for a histological diagnosis; complication rates range from 10% to 30%.237–239 There is a concern with sampling errors when stereotactic biopsies are performed.
Surgical resection of the more common DIPG is currently futile.208,229,232 The benefit of surgery is in the treatment of focal brainstem lesions. Sandri and colleagues reported their retrospective review of 17 focal BSGs treated with surgical excision and radiotherapy upon progression and achieved 4-year OS and EFS rates of 87% and 59%, respectively.240 Gross total resection was achieved in only four patients and correlated with improved EFS.240 Dorsal exophytic tumors have a good prognosis with long-term survival rates of 92% to 94%.227,228 Intrinsic medullary tumors tend to have good survival rates and stable progression after surgery; however, the perioperative risk of ventilation, tracheostomy, and gastrostomy was 41% in a 41-patient series by Jallo and co-workers. Almost 80% of these patients had eventual complete recovery.241 Patients with cervicomedullary BSGs can achieve favorable results with surgery, with 4- and 5-year survival rates of 72% to 100%.205,230 In cervicomedullary tumors, the degree of preoperative morbidity predicts postoperative function.197
The prone position is used in most cases of focal BSGs in children with the exception of midbrain lesions. The goals of surgery are to debulk the lesion and avoid neurological sequelae.229 Anterior focal pontine lesions can be accessed via the retrosigmoid approach, and dorsal focal intrinsic pontine lesions can be accessed via the midline suboccipital approach. “Safe” entry areas are regions of gliotic tissue presenting to the pial surface or in the zone around the facial colliculus when pial presentation does not occur.242 Dorsal exophytic tumors can be reached via a suboccipital craniotomy with high cervical laminectomy and exposure of the fourth ventricle via a telovelar approach.195 It is advisable not to follow the dorsal exophytic tumor into the brainstem as this will result in a high likelihood of cranial nerve nuclei injury. Cervicomedullary tumors can be approached via a laminectomy with or without suboccipital craniotomy for rostral extension. Intraoperative ultrasound can be utilized to define tumor margins.195,232,234 A dorsal myelotomy is performed to preserve the posterior columns. The tumor is located and debulked. However, extension into the medulla may limit gross total resection to avoid injury to the lower cranial nerves. When operating near the medulla it is essential to identify normal anatomical structures and the lower cranial nerves to avoid injuring these structures.
Conclusion
Despite improvements in understanding the biology and advances in treatment strategies (particularly radio- and chemotherapy), brainstem tumors continue to present a therapeutic challenge for neurosurgeons. Poor patient outcomes, especially for DIPGs, are still commonplace.197 Surgical advancements in techniques, neurophysiological monitoring, and neuroimaging have allowed many patients with focal and dorsally exophytic brainstem tumors to experience a prolonged tumor-free survival.
Please go to expertconsult.com to view the the complete list of references.
1. Rutka J.T., Hoffman H.J. Medulloblastoma: a historical perspective and overview. J Neurooncol. 1996;29(1):1-7.
2. Pang J., Banerjee A., Tihan T. The value of tandem CSF/MRI evaluation for predicting disseminated disease in childhood central nervous system neoplasms. J Neurooncol. 2008;87(1):97-102.
3. Arai K., Sato N., Aoki J., et al. MR signal of the solid portion of pilocytic astrocytoma on T2-weighted images: is it useful for differentiation from medulloblastoma? Neuroradiology. 2006;48(4):233-237.
4. Schneider J.F., Confort-Gouny S., Viola A., et al. Multiparametric differentiation of posterior fossa tumors in children using diffusion-weighted imaging and short echo-time 1H-MR spectroscopy. J Magn Reson Imaging. 2007;26(6):1390-1398.
5. Ilgren E.B., Stiller C.A. Cerebellar astrocytomas. Part II. Pathologic features indicative of malignancy. Clin Neuropathol. 1987;6(5):201-214.
6. Ilgren E.B., Stiller C.A. Cerebellar astrocytomas. Part I. Macroscopic and microscopic features. Clin Neuropathol. 1987;6(5):185-200.
7. Koeller K.K., Rushing E.J. From the archives of the AFIP: pilocytic astrocytoma: radiologic-pathologic correlation. Radiographics. 2004;24(6):1693-1708.
8. Abdollahzadeh M., Hoffman H., Blazer S., et al. Benign cerebellar astrocytoma in childhood: experience at the Hospital for Sick Children 1980-1992. Childs Nerv Syst. 1994;10(6):380-383.
9. Takada Y., Ohno K., Tamaki M., Hirakawa K. Cerebellopontine angle pilocytic astrocytoma mimicking acoustic schwannoma. Neuroradiology. 1999;41(12):949-950.
10. Beutler A.S., Hsiang J.K., Moorhouse D.F., et al. Pilocytic astrocytoma presenting as an extra-axial tumor in the cerebellopontine angle: case report. Neurosurgery. 1995;37(1):125-128.
11. Kulkarni A.V., Becker L.E., Jay V., et al. Primary cerebellar glioblastomas multiforme in children. Report of four cases. J Neurosurg. 1999;90(3):546-550.
12. Lee Y.Y., Van Tassel P., Bruner J.M., et al. Juvenile pilocytic astrocytomas: CT and MR characteristics. AJR Am J Roentgenol. 1989;152(6):1263-1270.
13. Coakley K.J., Huston J.3rd, Scheithauer B.W., et al. Pilocytic astrocytomas: well-demarcated magnetic resonance appearance despite frequent infiltration histologically. Mayo Clin Proc. 1995;70(8):747-751.
14. Pencalet P., Maixner W., Sainte-Rose C., et al. Benign cerebellar astrocytomas in children. J Neurosurg. 1999;90(2):265-273.
15. Hayostek C.J., Shaw E.G., Scheithauer B., et al. Astrocytomas of the cerebellum. A comparative clinicopathologic study of pilocytic and diffuse astrocytomas. Cancer. 1993;72(3):856-869.
16. Mamelak A.N., Prados M.D., Obana W.G., et al. Treatment options and prognosis for multicentric juvenile pilocytic astrocytoma. J Neurosurg. 1994;81(1):24-30.
17. Aryan H.E., Meltzer H.S., Lu D.C., et al. Management of pilocytic astrocytoma with diffuse leptomeningeal spread: two cases and review of the literature. Childs Nerv Syst. 2005;21(6):477-481.
18. Burger P.C., Cohen K.J., Rosenblum M.K., Tihan T. Pathology of diencephalic astrocytomas. Pediatr Neurosurg. 2000;32(4):214-219.
19. Tibbetts K.M., Emnett R.J., Gao F., et al. Histopathologic predictors of pilocytic astrocytoma event-free survival. Acta Neuropathol. 2009;117(6):657-665.
20. Tomlinson F.H., Scheithauer B.W., Hayostek C., et al. The significance of atypia and histologic malignancy in pilocytic astrocytoma of the cerebellum: a clinicopathologic and flow cytometric study. J Child Neurol. 1994;9(3):301-310.
21. Dirks P.B., Jay V., Becker L.E., et al. Development of anaplastic changes in low-grade astrocytomas of childhood. Neurosurgery. 1994;34(1):68-78.
22. Forshew T., Tatevossian R.G., Lawson A.R., et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol. 2009;218(2):172-181.
23. Jones D.T., Kocialkowski S., Liu L., et al. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549: BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009;28(20):2119-2123.
24. Sievert A.J., Jackson E.M., Gai X., et al. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol. 2009;19(3):449-458.
25. Bar E.E., Lin A., Tihan T., et al. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol. 2008;67(9):878-887.
26. Schneider J.H.Jr., Raffel C., McComb J.G. Benign cerebellar astrocytomas of childhood. Neurosurgery. 1992;30(1):58-62. discussion 62–63
27. Forsyth P.A., Shaw E.G., Scheithauer B.W., et al. Supratentorial pilocytic astrocytomas. A clinicopathologic, prognostic, and flow cytometric study of 51 patients. Cancer. 1993;72(4):1335-1342.
28. Due-Tonnessen B.J., Helseth E., Scheie D., et al. Long-term outcome after resection of benign cerebellar astrocytomas in children and young adults (0-19 years): report of 110 consecutive cases. Pediatr Neurosurg. 2002;37(2):71-80.
29. Palma L., Celli P., Mariottini A. Long-term follow-up of childhood cerebellar astrocytomas after incomplete resection with particular reference to arrested growth or spontaneous tumour regression. Acta Neurochir (Wien). 2004;146(6):581-588. discussion 588
30. Pollack I.F., Hurtt M., Pang D., Albright A.L. Dissemination of low grade intracranial astrocytomas in children. Cancer. 1994;73(11):2869-2878.
31. Perilongo G., Garre M.L., Giangaspero F. Low-grade gliomas and leptomeningeal dissemination: a poorly understood phenomenon. Childs Nerv Syst. 2003;19(4):197-203.
32. Roberts R.O., Lynch C.F., Jones M.P., Hart M.N. Medulloblastoma: a population-based study of 532 cases. J Neuropathol Exp Neurol. 1991;50(2):134-144.
33. Louis D.N., Ohgaki H., Wiestler O.D., et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97-109.
34. Rutkowski S., Bode U., Deinlein F., et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med. 2005;352(10):978-986.
35. McManamy C.S., Lamont J.M., Taylor R.E., et al. Morphophenotypic variation predicts clinical behavior in childhood non-desmoplastic medulloblastomas. J Neuropathol Exp Neurol. 2003;62(6):627-632.
36. McManamy C.S., Pears J., Weston C.L., et al. Nodule formation and desmoplasia in medulloblastomas—defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol. 2007;17(2):151-164.
37. Gulino A., Arcella A., Giangaspero F. Pathological and molecular heterogeneity of medulloblastoma. Curr Opin Oncol. 2008;20(6):668-675.
38. Giangaspero F., Perilongo G., Fondelli M.P., et al. Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg. 1999;91(6):971-977.
39. Eberhart C.G., Kepner J.L., Goldthwaite P.T., et al. Histopathologic grading of medulloblastomas: a Pediatric Oncology Group study. Cancer. 2002;94(2):552-560.
40. Carlotti C.G.Jr., Smith C., Rutka J.T. The molecular genetics of medulloblastoma: an assessment of new therapeutic targets. Neurosurg Rev. 2008;31(4):359-368. discussion 368–369
41. Northcott P.A., Nakahara Y., Wu X., et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009;41(4):465-472.
42. Northcott P.A., Korshunov A., Witt H., et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29(11):1408-1414.
43. Kool M., Koster J., Bunt J., et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3(8):e3088.
44. Maleci A., Cervoni L., Delfini R. Medulloblastoma in children and in adults: a comparative study. Acta Neurochir (Wien). 1992;119(1-4):62-67.
45. Sandhu A., Kendall B. Computed tomography in management of medulloblastomas. Neuroradiology. 1987;29(5):444-452.
46. Koeller K.K., Rushing E.J. From the archives of the AFIP: medulloblastoma: a comprehensive review with radiologic-pathologic correlation. Radiographics. 2003;23(6):1613-1637.
47. Nelson M., Diebler C., Forbes W.S. Paediatric medulloblastoma: atypical CT features at presentation in the SIOP II trial. Neuroradiology. 1991;33(2):140-142.
48. Mueller D.P., Moore S.A., Sato Y., Yuh W.T. MRI spectrum of medulloblastoma. Clin Imaging. 1992;16(4):250-255.
49. Rippe D.J., Boyko O.B., Friedman H.S., et al. Gd-DTPA-enhanced MR imaging of leptomeningeal spread of primary intracranial CNS tumor in children. AJNR Am J Neuroradiol. 1990;11(2):329-332.
50. George R.E., Laurent J.P., McCluggage C.W., Cheek W.R. Spinal metastasis in primitive neuroectodermal tumors (medulloblastoma) of the posterior fossa: evaluation with CT myelography and correlation with patient age and tumor differentiation. Pediatr Neurosci. 1985;12(3):157-160.
51. Stanley P., Senac M.O.Jr., Segall H.D. Intraspinal seeding from intracranial tumors in children. AJR Am J Roentgenol. 1985;144(1):157-161.
52. Heinz R., Wiener D., Friedman H., Tien R. Detection of cerebrospinal fluid metastasis: CT myelography or MR? AJNR Am J Neuroradiol. 1995;16(5):1147-1151.
53. Fouladi M., Gajjar A., Boyett J.M., et al. Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumor. J Clin Oncol. 1999;17(10):3234-3237.
54. Rochkind S., Blatt I., Sadeh M., Goldhammer Y. Extracranial metastases of medulloblastoma in adults: literature review. J Neurol Neurosurg Psychiatry. 1991;54(1):80-86.
55. Dhall G. Medulloblastoma. J Child Neurol. 2009;24(11):1418-1430.
56. Albright A.L., Wisoff J.H., Zeltzer P.M., et al. Effects of medulloblastoma resections on outcome in children: a report from the Children’s Cancer Group. Neurosurgery. 1996;38(2):265-271.
57. Gajjar A., Kühl J., Epelman S., et al. Chemotherapy of medulloblastoma. Childs Nerv Syst. 1999;15(10):554-562.
58. Packer R.J., Sutton L.N., Elterman R., et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg. 1994;81(5):690-698.
59. Tomita T. Surgical management of cerebellar peduncle lesions in children. Neurosurgery. 1986;18(5):568-575.
60. Grau C., Overgaard J. Postirradiation sensorineural hearing loss: a common but ignored late radiation complication. Int J Radiat Oncol Biol Phys. 1996;36(2):515-517.
61. Plowman P.N. Post-radiation sensorineuronal hearing loss. Int J Radiat Oncol Biol Phys. 2002;52(3):589-591.
62. Merchant T.E., Kiehna E.N., Li C., et al. Modeling radiation dosimetry to predict cognitive outcomes in pediatric patients with CNS embryonal tumors including medulloblastoma. Int J Radiat Oncol Biol Phys. 2006;65(1):210-221.
63. Ricardi U., Corrias A., Einaudi S., et al. Thyroid dysfunction as a late effect in childhood medulloblastoma: a comparison of hyperfractionated versus conventionally fractionated craniospinal radiotherapy. Int J Radiat Oncol Biol Phys. 2001;50(5):1287-1294.
64. Bowers D.C., Liu Y., Leisenring W., et al. Late-occurring stroke among long-term survivors of childhood leukemia and brain tumors: a report from the Childhood Cancer Survivor Study. J Clin Oncol. 2006;24(33):5277-5282.
65. Neglia J.P., Robison L.L., Stovall M., et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2006;98(21):1528-1537.
66. Taylor R.E., Bailey C.C., Robinson K., et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: the International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 Study. J Clin Oncol. 2003;21(8):1581-1591.
67. Oyharcabal-Bourden V., Kalifa C., Gentet J.C., et al. Standard-risk medulloblastoma treated by adjuvant chemotherapy followed by reduced-dose craniospinal radiation therapy: a French Society of Pediatric Oncology Study. J Clin Oncol. 2005;23(21):4726-4734.
68. Packer R.J., Gajjar A., Vezina G., et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24(25):4202-4208.
69. Goldwein J.W., Radcliffe J., Johnson J., et al. Updated results of a pilot study of low dose craniospinal irradiation plus chemotherapy for children under five with cerebellar primitive neuroectodermal tumors (medulloblastoma). Int J Radiat Oncol Biol Phys. 1996;34(4):899-904.
70. Mueller S., Chang S. Pediatric brain tumors: current treatment strategies and future therapeutic approaches. Neurotherapeutics. 2009;6(3):570-586.
71. Gajjar A., Chintagumpala M., Ashley D., et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St. Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7(10):813-820.
72. Jakacki R.I. Treatment strategies for high-risk medulloblastoma and supratentorial primitive neuroectodermal tumors. Review of the literature. J Neurosurg. 2005;102(Suppl 1):44-52.
73. Duffner P.K., Horowitz M.E., Krischer J.P., et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328(24):1725-1731.
74. Dhall G., Grodman H., Ji L., et al. Outcome of children less than three years old at diagnosis with non-metastatic medulloblastoma treated with chemotherapy on the “Head Start” I and II protocols. Pediatr Blood Cancer. 2008;50(6):1169-1175.
75. Rudin C.M., Hann C.L., Laterra J., et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361(12):1173-1178.
76. Fossati P., Ricardi U., Orecchia R. Pediatric medulloblastoma: toxicity of current treatment and potential role of protontherapy. Cancer Treat Rev. 2009;35(1):79-96.
77. Pui C.H., Campana D., Pei D., et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360(26):2730-2741.
78. Blaney S.M., Balis F.M., Berg S., et al. Intrathecal mafosfamide: a preclinical pharmacology and phase I trial. J Clin Oncol. 2005;23(7):1555-1563.
79. Conroy S., Garnett M., Vloeberghs M., et al. Medulloblastoma in childhood: revisiting intrathecal therapy in infants and children. Cancer Chemother Pharmacol. 2010;65(6):1173-1189.
80. Godfraind C. Classification and controversies in pathology of ependymomas. Childs Nerv Syst. 2009;25(10):1185-1193.
81. Tamburrini G., D’Ercole M., Pettorini B.L., et al. Survival following treatment for intracranial ependymoma: a review. Childs Nerv Syst. 2009;25(10):1303-1312.
82. Yuh E.L., Barkovich A.J., Gupta N. Imaging of ependymomas: MRI and CT. Childs Nerv Syst. 2009;25(10):1203-1213.
83. Rumboldt Z., Camacho D.L., Lake D., et al. Apparent diffusion coefficients for differentiation of cerebellar tumors in children. AJNR Am J Neuroradiol. 2006;27(6):1362-1369.
84. Ikezaki K., Matsushima T., Inoue T., et al. Correlation of microanatomical localization with postoperative survival in posterior fossa ependymomas. Neurosurgery. 1993;32(1):38-44.
85. Qian X., Goumnerova L.C., De Girolami U., Cibas E.S. Cerebrospinal fluid cytology in patients with ependymoma: a bi-institutional retrospective study. Cancer. 2008;114(5):307-314.
86. Figarella-Branger D., Civatte M., Bouvier-Labit C., et al. Prognostic factors in intracranial ependymomas in children. J Neurosurg. 2000;93(4):605-613.
87. Horn B., Heideman R., Geyer R., et al. A multi-institutional retrospective study of intracranial ependymoma in children: identification of risk factors. J Pediatr Hematol Oncol. 1999;21(3):203-211.
88. Jaing T.H., Wang H.S., Tsay P.K., et al. Multivariate analysis of clinical prognostic factors in children with intracranial ependymomas. J Neurooncol. 2004;68(3):255-261.
89. Korshunov A., Neben K., Wrobel G., et al. Gene expression patterns in ependymomas correlate with tumor location, grade, and patient age. Am J Pathol. 2003;163(5):1721-1727.
90. Merchant T.E. Current management of childhood ependymoma. Oncology (Williston Park). 2002;16(5):629-644. discussion 645–648
91. Perilongo G., Massimino M., Sotti G., et al. Analyses of prognostic factors in a retrospective review of 92 children with ependymoma: Italian pediatric neuro-oncology group. Med Pediatr Oncol. 1997;29(2):79-85.
92. Sala F., Talacchi A., Mazza C., et al. Prognostic factors in childhood intracranial ependymomas: the role of age and tumor location. Pediatr Neurosurg. 1998;28(3):135-142.
93. Snuderl M., Chi S.N., De Santis S.M., et al. Prognostic value of tumor microinvasion and metalloproteinases expression in intracranial pediatric ependymomas. J Neuropathol Exp Neurol. 2008;67(9):911-920.
94. Tihan T., Zhou T., Holmes E., et al. The prognostic value of histological grading of posterior fossa ependymomas in children: a Children’s Oncology Group study and a review of prognostic factors. Mod Pathol. 2008;21(2):165-177.
95. Zamecnik J., Snuderl M., Eckschlager T., et al. Pediatric intracranial ependymomas: prognostic relevance of histological, immunohistochemical, and flow cytometric factors. Mod Pathol. 2003;16(10):980-991.
96. Mack S.C., Taylor M.D. The genetic and epigenetic basis of ependymoma. Childs Nerv Syst. 2009;25(10):1195-1201.
97. Pollack I.F., Gerszten P.C., Martinez A.J., et al. Intracranial ependymomas of childhood: long-term outcome and prognostic factors. Neurosurgery. 1995;37(4):655-666. discussion 666–667
98. Goldwein J.W., Glauser T.A., Packer R.J., et al. Recurrent intracranial ependymomas in children. Survival, patterns of failure, and prognostic factors. Cancer. 1990;66(3):557-563.
99. Massimino M., Giangaspero F., Garrè M.L., et al. Salvage treatment for childhood ependymoma after surgery only: pitfalls of omitting “at once” adjuvant treatment. Int J Radiat Oncol Biol Phys. 2006;65(5):1440-1445.
100. Grundy R.G., Wilne S.A., Weston C.L., et al. Primary postoperative chemotherapy without radiotherapy for intracranial ependymoma in children: the UKCCSG/SIOP prospective study. Lancet Oncol. 2007;8(8):696-705.
101. Sanford R.A., Gajjar A. Ependymomas. Clin Neurosurg. 1997;44:559-570.
102. Merchant T.E., Li C., Xiong X., et al. Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study. Lancet Oncol. 2009;10(3):258-266.
103. Timmermann B., Kortmann R.D., Kühl J., et al. Combined postoperative irradiation and chemotherapy for anaplastic ependymomas in childhood: results of the German prospective trials HIT 88/89 and HIT 91. Int J Radiat Oncol Biol Phys. 2000;46(2):287-295.
104. Grill J., Le Deley M.C., Gambarelli D., et al. Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: a multicenter trial of the French Society of Pediatric Oncology. J Clin Oncol. 2001;19(5):1288-1296.
105. Wright K.D., Gajjar A. New chemotherapy strategies and biological agents in the treatment of childhood ependymoma. Childs Nerv Syst. 2009;25(10):1275-1282.
106. Robertson P.L., Zeltzer P.M., Boyett J.M., et al. Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children: a report of the Children’s Cancer Group. J Neurosurg. 1998;88(4):695-703.
107. Vinchon M., Leblond P., Noudel R., Dhellemmes P. Intracranial ependymomas in childhood: recurrence, reoperation, and outcome. Childs Nerv Syst. 2005;21(3):221-226.
108. Sangra M., Thorp N., May P., et al. Management strategies for recurrent ependymoma in the paediatric population. Childs Nerv Syst. 2009;25(10):1283-1291.
109. Merchant T.E., Boop F.A., Kun L.E., Sanford R.A. A retrospective study of surgery and reirradiation for recurrent ependymoma. Int J Radiat Oncol Biol Phys. 2008;71(1):87-97.
110. Stafford S.L., Pollock B.E., Foote R.L., et al. Stereotactic radiosurgery for recurrent ependymoma. Cancer. 2000;88(4):870-875.
111. Liu A.K., Foreman N.K., Gaspar L.E., et al. Maximally safe resection followed by hypofractionated re-irradiation for locally recurrent ependymoma in children. Pediatr Blood Cancer. 2009;52(7):804-807.
112. Beckwith J.B., Palmer N.F. Histopathology and prognosis of Wilms’ tumors: results from the First National Wilms’ Tumor Study. Cancer. 1978;41(5):1937-1948.
113. Biggs P.J., Garen P.D., Powers J.M., Garvin A.J. Malignant rhabdoid tumor of the central nervous system. Hum Pathol. 1987;18(4):332-337.
114. Rorke L.B., Packer R., Biegel J. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. J Neurooncol. 1995;24(1):21-28.
115. Athale U.H., Duckworth J., Odame I., Barr R. Childhood atypical teratoid rhabdoid tumor of the central nervous system: a meta-analysis of observational studies. J Pediatr Hematol Oncol. 2009;31(9):651-663.
116. Wharton S.B., Wardle C., Ironside J.W., et al. Comparative genomic hybridization and pathological findings in atypical teratoid/rhabdoid tumour of the central nervous system. Neuropathol Appl Neurobiol. 2003;29(3):254-261.
117. Janson K., Nedzi L.A., David O., et al. Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation. Pediatr Blood Cancer. 2006;47(3):279-284.
118. Arslanoglu A., Aygun N., Tekhtani D., et al. Imaging findings of CNS atypical teratoid/rhabdoid tumors. AJNR Am J Neuroradiol. 2004;25(3):476-480.
119. Fenton L.Z., Foreman N.K. Atypical teratoid/rhabdoid tumor of the central nervous system in children: an atypical series and review. Pediatr Radiol. 2003;33(8):554-558.
120. Martinez-Lage J.F., Nieto A., Sola J., et al. Primary malignant rhabdoid tumor of the cerebellum. Childs Nerv Syst. 1997;13(7):418-421.
121. Caldemeyer K.S., Smith R.R., Azzarelli B., Boaz J.C. Primary central nervous system malignant rhabdoid tumor: CT and MR appearance simulates a primitive neuroectodermal tumor. Pediatr Neurosurg. 1994;21(4):232-236.
122. Hanna S.L., Langston J.W., Parham D.M., Douglass E.C. Primary malignant rhabdoid tumor of the brain: clinical, imaging, and pathologic findings. AJNR Am J Neuroradiol. 1993;14(1):107-115.
123. Chi S.N., Zimmerman M.A., Yao X., et al. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol. 2009;27(3):385-389.
124. Tekautz T.M., Fuller C.E., Blaney S., et al. Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol. 2005;23(7):1491-1499.
125. Hilden J.M., Meerbaum S., Burger P., et al. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. 2004;22(14):2877-2884.
126. Ardon H., De Vleeschouwer S., Van Calenbergh F., et al. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr Blood Cancer. 2010;54(4):519-525.
127. Gupta N. Choroid plexus tumors in children. Neurosurg Clin North Am. 2003;14(4):621-631.
128. Rickert C.H., Paulus W. Tumors of the choroid plexus. Microsc Res Tech. 2001;52(1):104-111.
129. Taylor M.D., Gokgoz N., Andrulis I.L., et al. Familial posterior fossa brain tumors of infancy secondary to germline mutation of the hSNF5 gene. Am J Hum Genet. 2000;66(4):1403-1406.
130. Zakrzewska M., Wojcik I., Zakrzewski K., et al. Mutational analysis of hSNF5/INI1 and TP53 genes in choroid plexus carcinomas. Cancer Genet Cytogenet. 2005;156(2):179-182.
131. Kamaly-Asl I.D., Shams N., Taylor M.D. Genetics of choroid plexus tumors. Neurosurg Focus. 2006;20(1):E10.
132. Rickert C.H., Wiestler O.D., Paulus W. Chromosomal imbalances in choroid plexus tumors. Am J Pathol. 2002;160(3):1105-1113.
133. Wrede B., Hasselblatt M., Peters O., et al. Atypical choroid plexus papilloma: clinical experience in the CPT-SIOP-2000 study. J Neurooncol. 2009;95(3):383-392.
134. McGirr S.J., Ebersold M.J., Scheithauer B.W., et al. Choroid plexus papillomas: long-term follow-up results in a surgically treated series. J Neurosurg. 1988;69(6):843-849.
135. Wrede B., Liu P., Wolff J.E. Chemotherapy improves the survival of patients with choroid plexus carcinoma: a meta-analysis of individual cases with choroid plexus tumors. J Neurooncol. 2007;85(3):345-351.
136. Packer R.J., Perilongo G., Johnson D., et al. Choroid plexus carcinoma of childhood. Cancer. 1992;69(2):580-585.
137. Pencalet P., Sainte-Rose C., Lellouch-Tubiana A., et al. Papillomas and carcinomas of the choroid plexus in children. J Neurosurg. 1998;88(3):521-528.
138. Berger C., Thiesse P., Lellouch-Tubiana A., et al. Choroid plexus carcinomas in childhood: clinical features and prognostic factors. Neurosurgery. 1998;42(3):470-475.
139. Ellenbogen R.G., Winston K.R., Kupsky W.J. Tumors of the choroid plexus in children. Neurosurgery. 1989;25(3):327-335.
140. St Clair S.K., Humphreys R.P., Pillay P.K., et al. Current management of choroid plexus carcinoma in children. Pediatr Neurosurg. 1991;17(5):225-233.
141. Kaderali Z., Lamberti-Pasculli M., Rutka J.T. The changing epidemiology of paediatric brain tumours: a review from the Hospital for Sick Children. Childs Nerv Syst. 2009;25(7):787-793.
142. Constans J.P., Meder F., Maiuri F., et al. Posterior fossa hemangioblastomas. Surg Neurol. 1986;25(3):269-275.
143. Bradley S., Dumas N., Ludman M., Wood L. Hereditary renal cell carcinoma associated with von Hippel-Lindau disease: a description of a Nova Scotia cohort. Can Urol Assoc J. 2009;3(1):32-36.
144. Kaelin W.G.Jr. The von Hippel-Lindau tumor suppressor protein: an update. Methods Enzymol. 2007;435:371-383.
145. Woodward E.R., Wall K., Forsyth J., et al. VHL mutation analysis in patients with isolated central nervous system haemangioblastoma. Brain. 2007;130(Pt 3):836-842.
146. Lee S.R., Sanches J., Mark A.S., et al. Posterior fossa hemangioblastomas: MR imaging. Radiology. 1989;171(2):463-468.
147. Papaioannou G., Sebire N.J., McHugh K. Imaging of the unusual pediatric “blastomas”. Cancer Imaging. 2009;9:1-11.
148. Vougioukas V.I., Gläsker S., Hubbe U., et al. Surgical treatment of hemangioblastomas of the central nervous system in pediatric patients. Childs Nerv Syst. 2006;22(9):1149-1153.
149. Kano H., Niranjan A., Mongia S., et al. The role of stereotactic radiosurgery for intracranial hemangioblastomas. Neurosurgery. 2008;63(3):443-450. discussion 450–451
150. Koh E.S., Nichol A., Millar B.A., et al. Role of fractionated external beam radiotherapy in hemangioblastoma of the central nervous system. Int J Radiat Oncol Biol Phys. 2007;69(5):1521-1526.
151. Cobbs C.S., Pitts L.H., Wilson C.B. Epidermoid and dermoid cysts of the posterior fossa. Clin Neurosurg. 1997;44:511-528.
152. Alvord E.C.Jr. Growth rates of epidermoid tumors. Ann Neurol. 1977;2(5):367-370.
153. Yasargil M.G., Abernathey C.D., Sarioglu A.C. Microneurosurgical treatment of intracranial dermoid and epidermoid tumors. Neurosurgery. 1989;24(4):561-567.
154. Caldarelli M., Massimi L., Kondageski C., Di Rocco C. Intracranial midline dermoid and epidermoid cysts in children. J Neurosurg. 2004;100(Suppl 5):473-480.
155. Bucciero A., Del Basso De Caro M.L., Carraturo S., et al. Supratentorial dermoid cysts. Presentation and management of five cases. J Neurosurg Sci. 1995;39(1):7-11.
156. Hayek G., Mercier P., Fournier H.D., et al. [Dermal sinus and dermoid cyst revealed by abscess formation in posterior fossa. Report of 2 pediatric cases and review of the literature.]. Neurochirurgie. 2001;47(2-3 Pt 1):123-127.
157. Tekkok I.H., Baeesa S.S., Higgins M.J., Ventureyra E.C. Abscedation of posterior fossa dermoid cysts. Childs Nerv Syst. 1996;12(6):318-322.
158. Pai V.V., Lowe L.H., Castillo M., et al. Posterior fossa dermoid cysts in association with Klippel-Feil syndrome: report of three cases. AJNR Am J Neuroradiol. 2007;28(10):1926-1928.
159. Turgut M. Klippel-Feil syndrome in association with posterior fossa dermoid tumour. Acta Neurochir (Wien). 2009;151(3):269-276.
160. Dickey W., Hawkins S.A., Kirkpatrick D.H., et al. Posterior fossa dermoid cysts and the Klippel-Feil syndrome. J Neurol Neurosurg Psychiatry. 1991;54(11):1016-1017.
161. Muzumdar D., Goel A. Posterior cranial fossa dermoid in association with craniovertebral and cervical spinal anomaly: report of two cases. Pediatr Neurosurg. 2001;35(3):158-161.
162. Chandra P.S., Gupta A., Mishra N.K., Mehta V.S. Association of craniovertebral and upper cervical anomalies with dermoid and epidermoid cysts: report of four cases. Neurosurgery. 2005;56(5):E1155. discussion E1155
163. Chen S., Ikawa F., Kurisu K., et al. Quantitative MR evaluation of intracranial epidermoid tumors by fast fluid-attenuated inversion recovery imaging and echo-planar diffusion-weighted imaging. AJNR Am J Neuroradiol. 2001;22(6):1089-1096.
164. Yamakawa K., Shitara N., Genka S., et al. Clinical course and surgical prognosis of 33 cases of intracranial epidermoid tumors. Neurosurgery. 1989;24(4):568-573.
165. Samii M., Tatagiba M., Piquer J., Carvalho G.A. Surgical treatment of epidermoid cysts of the cerebellopontine angle. J Neurosurg. 1996;84(1):14-19.
166. Hamlat A., Hua Z.F., Saikali S., et al. Malignant transformation of intra-cranial epithelial cysts: systematic article review. J Neurooncol. 2005;74(2):187-194.
167. Raimondi A.J., Tomita T. Hydrocephalus and infratentorial tumors. Incidence, clinical picture, and treatment. J Neurosurg. 1981;55(2):174-182.
168. Taylor W.A., Todd N.V., Leighton S.E. CSF drainage in patients with posterior fossa tumours. Acta Neurochir (Wien). 1992;117(1-2):1-6.
169. Culley D.J., Berger M.S., Shaw D., Geyer R. An analysis of factors determining the need for ventriculoperitoneal shunts after posterior fossa tumor surgery in children. Neurosurgery. 1994;34(3):402-407. discussion 407–408
170. Gnanalingham K.K., Lafuente J., Thompson D., et al. MRI study of the natural history and risk factors for pseudomeningocoele formation following postfossa surgery in children. Br J Neurosurg. 2003;17(6):530-536.
171. Kumar V., Phipps K., Harkness W., Hayward R.D. Ventriculo-peritoneal shunt requirement in children with posterior fossa tumours: an 11-year audit. Br J Neurosurg. 1996;10(5):467-470.
172. Morelli D., Pirotte B., Lubansu A., et al. Persistent hydrocephalus after early surgical management of posterior fossa tumors in children: is routine preoperative endoscopic third ventriculostomy justified? J Neurosurg. 2005;103(Suppl 3):247-252.
173. Sainte-Rose C., Cinalli G., Roux F.E., et al. Management of hydrocephalus in pediatric patients with posterior fossa tumors: the role of endoscopic third ventriculostomy. J Neurosurg. 2001;95(5):791-797.
174. Riva-Cambrin J., Detsky A.S., Lamberti-Pasculli M., et al. Predicting postresection hydrocephalus in pediatric patients with posterior fossa tumors. J Neurosurg Pediatr. 2009;3(5):378-385.
175. Mussi A.C., Rhoton A.L.Jr. Telovelar approach to the fourth ventricle: microsurgical anatomy. J Neurosurg. 2000;92(5):812-823.
176. Shimoji K., Miyajima M., Karagiozov K., et al. Surgical considerations in fourth ventricular ependymoma with the transcerebellomedullary fissure approach in focus. Childs Nerv Syst. 2009;25(10):1221-1228.
177. Matsushima T., Kawashima M., Masuoka J., et al. Transcerebellomedullary fissure approach with special reference to methods of dissecting the fissure. J Neurosurg. 2001;94(2):257-264.
178. Gok A., Alptekin M., Erkutlu I. Surgical approach to the fourth ventricle cavity through the cerebellomedullary fissure. Neurosurg Rev. 2004;27(1):50-54.
179. Deshmukh V.R., Figueiredo E.G., Deshmukh P., et al. Quantification and comparison of telovelar and transvermian approaches to the fourth ventricle. Neurosurgery. 58(4 Suppl 2), 2006. ONS-202-206; discussion ONS-206-207
180. Zuccaro G., Sosa F. Cerebellopontine angle lesions in children. Childs Nerv Syst. 2007;23(2):177-183.
181. Ciric I., Zhao J.C., Rosenblatt S., et al. Suboccipital retro-sigmoid approach for removal of vestibular schwannomas: facial nerve function and hearing preservation. Neurosurgery. 2005;56(3):560-570. discussion 560–570
182. Klimo P.Jr., Browd S.R., Pravdenkova S., et al. The posterior petrosal approach: technique and applications in pediatric neurosurgery. J Neurosurg Pediatr. 2009;4(4):353-362.
183. Roth J., Beni Adani L., Biyani N., Constantini S. Intraoperative portable 0.12-tesla MRI in pediatric neurosurgery. Pediatr Neurosurg. 2006;42(2):74-80.
184. Roth J., Biyani N., Beni-Adani L., Constantini S. Real-time neuronavigation with high-quality 3D ultrasound SonoWand in pediatric neurosurgery. Pediatr Neurosurg. 2007;43(3):185-191.
185. Morota N., Deletis V., Epstein F.J., et al. Brainstem mapping: neurophysiological localization of motor nuclei on the floor of the fourth ventricle. Neurosurgery. 1995;37(5):922-929. discussion 929–930
186. Strauss C., Romstöck J., Nimsky C., Fahlbusch R. Intraoperative identification of motor areas of the rhomboid fossa using direct stimulation. J Neurosurg. 1993;79(3):393-399.
187. Abbott R. Intraoperative neurophysiologic monitoring: its impact on the practice of a pediatric neurosurgeon. Childs Nerv Syst. 2010;26(2):237-240.
188. Wells E.M., Walsh K.S., Khademian Z.P., et al. The cerebellar mutism syndrome and its relation to cerebellar cognitive function and the cerebellar cognitive affective disorder. Dev Disabil Res Rev. 2008;14(3):221-228.
189. Charalambides C., Dinopoulos A., Sgouros S. Neuropsychological sequelae and quality of life following treatment of posterior fossa ependymomas in children. Childs Nerv Syst. 2009;25(10):1313-1320.
190. Packer R.J., Vezina G. Management of and prognosis with medulloblastoma: therapy at a crossroads. Arch Neurol. 2008;65(11):1419-1424.
191. Packer R.J. Childhood brain tumors: accomplishments and ongoing challenges. J Child Neurol. 2008;23(10):1122-1127.
192. Merchant T.E., Pollack I.F., Loeffler J.S. Brain tumors across the age spectrum: biology, therapy, and late effects. Semin Radiat Oncol. 2010;20(1):58-66.
193. Keene D.L., Johnston D.L., Grimard L., et al. Vascular complications of cranial radiation. Childs Nerv Syst. 2006;22(6):547-555.
194. Armstrong G.T., Liu Q., Yasui Y., et al. Long-term outcomes among adult survivors of childhood central nervous system malignancies in the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2009;101(13):946-958.
195. Jallo G.I., Freed D., Epstein F. Intramedullary spinal cord tumors in children. Childs Nerv Syst. 2003;19(9):641-649.
196. Frazier J.L., Lee J., Thomale U.W., et al. Treatment of diffuse intrinsic brainstem gliomas: failed approaches and future strategies. J Neurosurg Pediatr. 2009;3(4):259-269.
197. Laigle-Donadey F., Doz F., Delattre J.Y. Brainstem gliomas in children and adults. Curr Opin Oncol. 2008;20(6):662-667.
198. Leach P.A., Estlin E.J., Coope D.J., et al. Diffuse brainstem gliomas in children: should we or shouldn’t we biopsy? Br J Neurosurg. 2008;22(5):619-624.
199. Littman P., Jarrett P., Bilaniuk L.T., et al. Pediatric brainstem gliomas. Cancer. 1980;45(11):2787-2792.
200. Choux M., Lena G., Do L. Brainstem tumors. In: Choux M., Di Rocco C., Hockley A., editors. Pediatric Neurosurgery. New York: Churchill Livingstone; 2000:479-491.
201. Freeman C.R., Farmer J.P. Pediatric brainstem gliomas: a review. Int J Radiat Oncol Biol Phys. 1998;40(2):265-271.
202. Donaldson S.S., Laningham F., Fisher P.G. Advances toward an understanding of brainstem gliomas. J Clin Oncol. 2006;24(8):1266-1272.
203. Behnke J., Christen H.J., Mursch K., Markakis E. Intra-axial endophytic tumors in the pons and/or medulla oblongata. II. Intraoperative findings, postoperative results, and 2-year follow up in 25 children. Childs Nerv Syst. 1997;13(3):135-146.
204. Farmer J.P., Montes J.L., Freeman C.R., et al. Brainstem gliomas. A 10-year institutional review. Pediatr Neurosurg. 2001;34(4):206-214.
205. Weiner H.L., Freed D., Woo H.H., et al. Intra-axial tumors of the cervicomedullary junction: surgical results and long-term outcome. Pediatr Neurosurg. 1997;27(1):12-18.
206. Walker M., Storrs B. Surgical therapy for intrinsic brainstem gliomas. Concepts Pediatr Neurosurg. 1985;5:178-186.
207. Korones D.N. Treatment of newly diagnosed diffuse brainstem gliomas in children: in search of the Holy Grail. Expert Rev Anticancer Ther. 2007;7(5):663-674.
208. Albright L. Biopsy of brainstem masses. J Neurosurg. 1996;84(2):304-306.
209. Nejat F., El Khashab M., Rutka J.T. Initial management of childhood brain tumors: neurosurgical considerations. J Child Neurol. 2008;23(10):1136-1148.
210. Lassman L.P. Tumours of the pons and medulla oblongata. In: Amador L.V., editor. Brain Tumours in the Young. Springfield, IL: Charles C Thomas; 1983:546-564.
211. Sposto R., Ertel I.J., Jenkin R.D., et al. The effectiveness of chemotherapy for treatment of high grade astrocytoma in children: results of a randomized trial. A report from the Children’s Cancer Study Group. J Neurooncol. 1989;7(2):165-177.
212. Thakur S.B., Karimi S., Dunkel I.J., et al. Longitudinal MR spectroscopic imaging of pediatric diffuse pontine tumors to assess tumor aggression and progression. AJNR Am J Neuroradiol. 2006;27(4):806-809.
213. Porto L., Hattingen E., Pilatus U., et al. Proton magnetic resonance spectroscopy in childhood brainstem lesions. Childs Nerv Syst. 2007;23(3):305-314.
214. Guillamo J.S., Doz F., Delattre J.Y. Brainstem gliomas. Curr Opin Neurol. 2001;14(6):711-715.
215. Epstein F., McCleary E.L. Intrinsic brainstem tumors of childhood: surgical indications. J Neurosurg. 1986;64(1):11-15.
216. Albright A.L., Packer R.J., Zimmerman R., et al. Magnetic resonance scans should replace biopsies for the diagnosis of diffuse brainstem gliomas: a report from the Children’s Cancer Group. Neurosurgery. 1993;33(6):1026-1029. discussion 1029–1030
217. Levin V.A., Edwards M.S., Wara W.M., et al. 5-Fluorouracil and 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea (CCNU) followed by hydroxyurea, misonidazole, and irradiation for brainstem gliomas: a pilot study of the Brain Tumor Research Center and the Childrens Cancer Group. Neurosurgery. 1984;14(6):679-681.
218. Hargrave D., Chuang N., Tariq N., Bouffet E. Can radiological characteristics predict the length of survival in pontine glioma. Neuro-Oncology. 2003;5:43.
219. Mandell L.R., Kadota R., Freeman C., et al. There is no role for hyperfractionated radiotherapy in the management of children with newly diagnosed diffuse intrinsic brainstem tumors: results of a Pediatric Oncology Group phase III trial comparing conventional vs. hyperfractionated radiotherapy. Int J Radiat Oncol Biol Phys. 1999;43(5):959-964.
220. Ternier J., Wray A., Puget S., et al. Tectal plate lesions in children. J Neurosurg. 2006;104(Suppl 6):369-376.
221. Daglioglu E., Cataltepe O., Akalan N. Tectal gliomas in children: the implications for natural history and management strategy. Pediatr Neurosurg. 2003;38(5):223-231.
222. Wellons J.C.3rd, Tubbs R.S., Banks J.T., et al. Long-term control of hydrocephalus via endoscopic third ventriculostomy in children with tectal plate gliomas. Neurosurgery. 2002;51(1):63-67. discussion 67–68
223. Squires L.A., Allen J.C., Abbott R., Epstein F.J. Focal tectal tumors: management and prognosis. Neurology. 1994;44(5):953-956.
224. Drake J., Chumas P., Kestle J., et al. Late rapid deterioration after endoscopic third ventriculostomy: additional cases and review of the literature. J Neurosurg. 2006;105(Suppl 2):118-126.
225. Yen C.P., Sheehan J., Steiner M., et al. Gamma knife surgery for focal brainstem gliomas. J Neurosurg. 2007;106(1):8-17.
226. Vandertop W.P., Hoffman H.J., Drake J.M., et al. Focal midbrain tumors in children. Neurosurgery. 1992;31(2):186-194.
227. Khatib Z.A., Heideman R.L., Kovnar E.H., et al. Predominance of pilocytic histology in dorsally exophytic brainstem tumors. Pediatr Neurosurg. 1994;20(1):2-10.
228. Pollack I.F., Hoffman H.J., Humphreys R.P., Becker L. The long-term outcome after surgical treatment of dorsally exophytic brainstem gliomas. J Neurosurg. 1993;78(6):859-863.
229. Jallo G.I., Biser-Rohrbaugh A., Freed D. Brainstem gliomas. Childs Nerv Syst. 2004;20(3):143-153.
230. Young Poussaint T., Yousuf N., Barnes P.D., et al. Cervicomedullary astrocytomas of childhood: clinical and imaging follow-up. Pediatr Radiol. 1999;29(9):662-668.
231. Epstein F.J., Farmer J.P. Brainstem glioma growth patterns. J Neurosurg. 1993;78(3):408-412.
232. Epstein F., Wisoff J. Intra-axial tumors of the cervicomedullary junction. J Neurosurg. 1987;67(4):483-487.
233. Pierre-Kahn A., Hirsch J.F., Vinchon M., et al. Surgical management of brainstem tumors in children: results and statistical analysis of 75 cases. J Neurosurg. 1993;79(6):845-852.
234. Recinos P.F., Sciubba D.M., Jallo G.I. Brainstem tumors: where are we today? Pediatr Neurosurg. 2007;43(3):192-201.
235. Khaw S.L., Coleman L.T., Downie P.A., et al. Temozolomide in pediatric low-grade glioma. Pediatr Blood Cancer. 2007;49(6):808-811.
236. Ullrich N.J., Raja A.I., Irons M.B., et al. Brainstem lesions in neurofibromatosis type 1. Neurosurgery. 2007;61(4):762-766. discussion 766–767
237. Steck J., Friedman W.A. Stereotactic biopsy of brainstem mass lesions. Surg Neurol. 1995;43(6):563-567. discussion 567–568
238. Boviatsis E.J., Kouyialis A.T., Stranjalis G., et al. CT-guided stereotactic biopsies of brainstem lesions: personal experience and literature review. Neurol Sci. 2003;24(3):97-102.
239. Rajshekhar V., Chandy M.J. Computerized tomography-guided stereotactic surgery for brainstem masses: a risk-benefit analysis in 71 patients. J Neurosurg. 1995;82(6):976-981.
240. Sandri A., Sardi N., Genitori L., et al. Diffuse and focal brainstem tumors in childhood: prognostic factors and surgical outcome. Experience in a single institution. Childs Nerv Syst. 2006;22(9):1127-1135.
241. Jallo G.I., Danish S., Velasquez L., Epstein F. Intramedullary low-grade astrocytomas: long-term outcome following radical surgery. J Neurooncol. 2001;53(1):61-66.
242. Bricolo A., Turazzi S., Cristofori L., Talacchi A. Direct surgery for brainstem tumours. Acta Neurochir Suppl (Wien). 1991;53:148-158.