Chapter 337 Tumors of the Digestive Tract
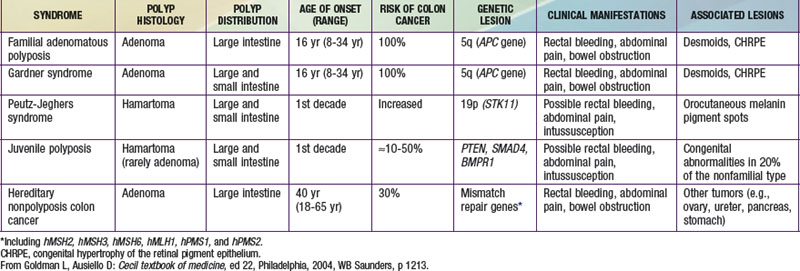
Tumors of the digestive tract are mostly polypoid. They are also commonly syndromic tumors and tumors with known genetic identification (see  Table 337-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com). They usually manifest as painless rectal bleeding, but they can serve as lead points for intussusception.
Table 337-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com). They usually manifest as painless rectal bleeding, but they can serve as lead points for intussusception.
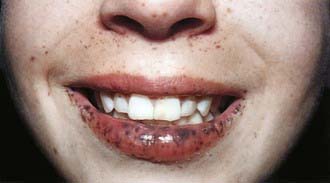
Peutz-Jeghers Syndrome
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant disorder (incidence, ∼1 : 120,000) characterized by mucocutaneous pigmentation and extensive GI hamartomatous polyposis. Macular pigmented lesions may be dark brown to dark blue and are found primarily around the lips and oral mucosa, although these lesions may also be found on the hands, feet, or perineum (Fig. 337-1). Lesions can fade by puberty or adulthood.
Attard TM, Tajouri T, Peterson KD, et al. Familial adenomatous polyposis in children younger than age 10 years: a multidisciplinary clinic experience. Dis Colon Rectum. 2008;51:207-212.
Erdman SH. Pediatric adenomatous polyposis syndromes: an update. Curr Gastroenterol Reports. 2007;9:237-244.
Hill DA, Furman WL, Billups CA, et al. Colorectal carcinoma in childhood and adolescence: a clinicopathologic review. J Clin Onco. 2007;25:5808-5814.
Ladd AP, Grosfeld J. Gastrointestinal tumors in adolescents and children. Sem Pediatr Surg. 2006;15:37-47.
Lynch HT, Lynch JF, Lynch PM, et al. Hereditary colorectal cancer syndromes: molecular genetics, genetic counseling, diagnosis and management. Familial Cancer. 2008;7:27-39.
Moglia A, Menciassi A, Dario P. Clinical update: endoscopy for small-bowel tumours. Lancet. 2007;370:114-116.
Pappo AS, Janeway KA. Pediatric gastrointestinal stromal tumors. Hematol Oncol Clin North Am. 2009;23:15-34.
Rubin BP, Heinrich MC, Corless CL. Gastrointestinal stromal tumour. Lancet. 2007;369:1731-1740.
Sidhu R, Sanders DS, McAlindon ME, et al. Capsule endoscopy and enteroscopy: modern modalities to investigate the small bowel in paediatrics. Arch Dis Child. 2008;93:154-159.
Vidal I, Podevin G, Piloquet H, et al. Followup and surgical management of Peutz-Jeghers syndrome in children. J Pediatr Gastroenterol Nutr. 2009;48:419-425.
von Allmen D. Intestinal polyposis syndromes: progress in understanding and treatment. Curr Opin Pediatr. 2006;18:316-320.





