59 Tumors of Bone and Soft Tissue
Radiographic Features
When a malignant condition of bone is suspected, plain film radiographic evaluation will provide, in most instances, a highly reliable presumptive diagnosis and should not be overlooked in the current era of cross-sectional imaging. The age of the patient, location of the tumor within the bone (epiphysis, metaphysis, or diaphysis), and skeletal site (axial or appendicular) set the foundation for radiographic diagnosis. Osteosarcomas commonly arise in the metaphyseal region and may extend into the diaphyseal or epiphyseal region, or both, of the affected bone.1 Cortical bone is destroyed in a disorganized fashion, and the periosteal reaction, which is the effort of the native bone to restore cortical integrity, results in the radiographic appearance of the classic Codman triangle and periosteal bone spicules (Fig. 59-1). The tumor itself images as a cloudlike density representing tumor osteoid. A soft tissue mass is usually present. Magnetic resonance imaging (MRI) is most useful for imaging soft tissue extension as well as identifying intramedullary skip metastases. The latter, which occur rarely, are also well-detected on bone scintigraphy.

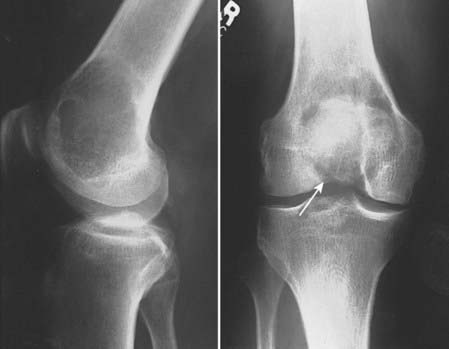
As most cartilaginous lesions are typically slow growing and commonly arise from pre-existing, benign cartilaginous lesions such as an enchondroma or an osteochondroma, the radiographic appearance of a chondrosarcoma may be only subtly different from that of a purely benign condition.2 Although the cartilaginous matrix typically mineralizes in a benign growth, development of lucent areas within the matrix are worrisome for malignant degeneration (Fig. 59-2). Similarly, an enchondroma typically induces endosteal scalloping and gradual cortical remodeling, but when this picture evolves to include cortical destruction and penetration, malignancy is likely. Serial radiographs are recommended in monitoring apparently benign lesions, especially those in an axial location.
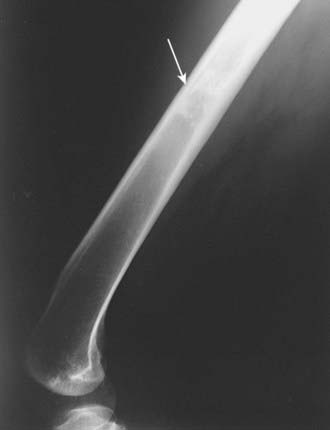
The most important radiographic features of giant cell tumors are their metaphyseal location and rather bland lytic appearance with little or no host response in the narrow transition zone between tumor and native bone.3 Extension to the subarticular cortex in the epiphysis is common (Fig. 59-3). This has given rise to some dispute as to the site of origin, with some investigators citing the epiphysis.4–6 These tumors usually cause cortical thinning, with frank breakthrough being a less common event.
Reports from institutions where the effects of radiation on giant cell tumors have been studied note that bone reparation may not be radiographically evident for up to 2 years after therapy.7,8 The short-term radiographic appearance may actually be confused with tumor progression as the original sclerotic rim involutes. Therefore, patience is required in the interpretation of radiographic studies in the short term after irradiation for giant cell tumors of bone.
Pathologic Features
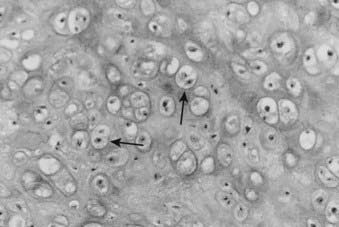
For example, in low-grade chondrosarcomas, mitoses are rare and the appearance of chondrocytes with a plump nucleus or moderate numbers of multinucleated cells may be diagnostic (Fig. 59-4). These histologic findings in association with an axial location in an adult would be more worrisome, whereas they would be of no particular importance in a child or in a distal location. As 10% of chondrosarcomas arise in pre-existing, benign cartilaginous growths, extensive specimen sampling is necessary to avoid overlooking areas of malignant degeneration. The periphery of the lesion is usually most revealing. Although the cartilaginous matrix may contain calcifications and ossifications, malignant osteoid is not seen.
Conversely, malignant osteoid is the hallmark of osteosarcoma. Although it is logical to assume that the tumor arises from osteoblasts, the presence of other malignant mesenchymal tissues within osteosarcomas suggests that the precursor cell is actually of a more primitive mesenchymal, pluripotential origin. Therefore, the microscopic appearance is that of a sarcomatous stroma directly forming tumor osteoid or bone (Fig. 59-5). A large amount of atypical cartilage or fibrosarcomatous matrix may also be evident.
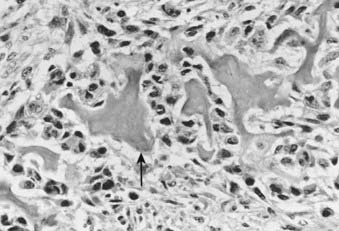
FIGURE 59-5 • Osteosarcoma. These highly atypical cells make up the sarcomatous stroma adjacent to malignant osteoid (arrow).
The physaliferous cell is the histologic hallmark of the chordoma. These so-called bubbly cells have abundant intracytoplasmic droplets of mucoid material and are usually seen in clusters surrounded by a sea of mucoid material (Fig. 59-6). They arise from the remnants of the notochord in the axial skeleton.9 During embryonic development, the notochord undergoes gradual obliteration within the vertebral bodies during the second month of gestation. However, the central portion of the intervertebral disk, which contains notochordal tissue in the form of nucleus pulposus, may persist for an indefinite period.
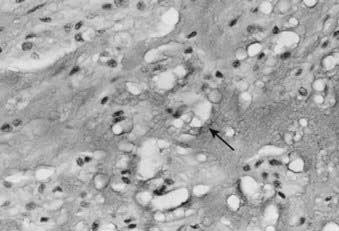
FIGURE 59-6 • Chordoma. The physaliferous cells (arrow) display a bubbly cytoplasm caused by copious intracytoplasmic mucoid droplets.
Experimentally, chordoma-like lesions have been produced in rabbits by piercing the anterior intervertebral ligaments with a needle and allowing the escape of nucleus pulposus.10 In fact, 50% of patients with sacrococcygeal chordomas have a history of previous lower back trauma.11
Radiotherapy Background, Role of Radiotherapy, Technique
Osteosarcoma
Background
True malignant bone tumors represent less than 1% of all malignant neoplasms.12 Osteosarcoma is the most prevalent primary malignancy of bone; it is twice as common as chondrosarcoma, three times as common as Ewing sarcoma, and 10 times more common than malignant fibrous histiocytoma (MFH) of bone.13
Although osteosarcoma can occur at any age, nearly 60% of patients are between 10 and 20 years of age. Not surprisingly, the age peak is younger in females (17 years) than in males (18 years), mirroring the period of most active skeletal growth for each sex. An additional 10% of cases occur in patients older than 60 years of age. There is a slightly greater incidence in males, 1.3 : 1 at Memorial Sloan-Kettering Cancer Center (MSKCC) and 1.6 : 1 at the Mayo Clinic, perhaps reflecting the longer duration of skeletal growth in males and the greater skeletal bulk. Similarly, the distal femur and proximal tibia account for more than 50% of cases in adolescents, as this is the site of most rapid skeletal growth. Until epiphyseal closure, the long bones are involved in nearly 80% of cases, whereas after skeletal maturity, the incidence is approximately equal between the long and flat bones, with the skull and axial skeleton representing 40% of cases.13
In patients older than 60, 56% of osteosarcomas are believed to arise as a consequence of other bone conditions, such as Paget disease and fibrous dysplasia, or following irradiation. In this setting, response to chemotherapy is less likely,14 with a 5-year survival rate of 15% in a review from MSKCC9 and 10% at the Mayo Clinic.15
In a cohort of 900 patients followed at the National Cancer Institute (NCI) Epidemiology Branch who had received repeated injections of radium-224 for the treatment of bone tuberculosis or ankylosing spondylitis, a 200-fold increased risk for developing bone sarcomas was noted.16 The estimated cumulative skeletal dose was 4.2 Gy.
Glicksman and Fabrikant reviewed the current literature regarding postirradiation second neoplasms.17 Bone sarcomas were the most common second malignancy reported. The authors noted that although doses ranged between less than 10 Gy and 80 Gy, only 21% of the secondary sarcomas were along the central axis of the radiation field and nearly 50% were on the field periphery. An additional 30% were located in the superficial tissues, superficial to maximum dose (dmax), causing the authors to speculate that the most likely regions of sarcoma induction received between 20% and 70% of the midplane dose.
Role of Radiotherapy
At least 50 years ago, it was evident that amputation alone was inadequate treatment for the majority of patients with osteosarcoma.18–21 The fact that 80% of patients developed lung metastases within 2 years of diagnosis led to efforts to approach this disease in consideration of its likely systemic nature. In the classic report by Cade in 1955,22 doses of 70 to 90 Gy in 8 to 12 weeks were delivered to the primary tumor at diagnosis and those patients who were free of systemic metastases at 6 to 9 months underwent amputation. Cade’s goal was to spare the children who were destined to develop pulmonary metastases from having an amputation. Among those patients who eventually underwent amputation after the course of radiation, many were found to have complete clearance of histologically viable tumor. Similarly, in 1973, Allen and Stevens23 reported on 10 patients with nonmetastatic osteosarcoma treated with 79 to 100 Gy with either 60-Co or 2-MV photons preoperatively. No histologic evidence of tumor was found in six of seven surgical specimens, and 6 of 10 patients were free of disease at a minimum follow-up of 30 months. The observation of improved survival compared with historic control subjects prompted the authors to speculate that tumors irradiated preoperatively were less likely to cause systemic metastases.
In 1975, Rosen and colleagues at MSKCC24 proposed using high-dose methotrexate with vincristine, doxorubicin (Adriamycin), and cyclophosphamide on the “T-4 protocol” as adjuvant therapy following surgical ablation of primary osteosarcoma. This study was based on their promising results in children with metastatic disease, in which they found a median survival of 3 months without therapy and 15 months with the chemotherapy regimen and an overall response rate of 77%. They argued that local radiation followed by amputation for those patients who had not developed pulmonary metastases was no longer a tenable approach; rather, immediate amputation followed by adjuvant systemic chemotherapy held more hope for the majority, who were assumed to have microscopic dissemination at diagnosis.25
The clear benefit of chemotherapy in an adjuvant setting led Rosen and colleagues at MSKCC to advocate using chemotherapy in a neoadjuvant fashion, thus permitting an “in vivo sensitivity test.”26,27 A good response, it was reasoned, would improve the opportunity to achieve better limb preservation with complete extirpation of the tumor and, moreover, could guide the selection of chemotherapy in the postoperative period.28,29
Recent publications have identified a small group of patients for whom local adjuvant radiation therapy may, in fact, be useful. Ozaki and colleagues,30 reporting for the Cooperative German/Austrian Osteosarcoma Study Group reviewed 1982 consecutive patients entered prospectively onto neoadjuvant chemotherapy studies between 1979 and 1998. Sixty-seven patients were identified with nonmetastatic, high-grade primary tumors of the pelvis (ilium, acetabulum, pubis, and ischium); sacral primaries were excluded. Eleven patients received radiation in some form to achieve local control: four postoperatively, and seven as definitive local management with doses between 56 and 68 Gy employed. The overall survival was statistically superior for this group of 11 patients when compared with 19 patients undergoing similar surgical management (intralesional surgery or no surgery) without radiation. The group of patients irradiated had a 29% 5-year overall survival compared to zero for the unirradiated group (P = 0.0033). The authors conclude that unresectable osteosarcomas or those operated on with inadequate margins should be treated with a regimen including radiotherapy.
Other investigators have noted that primary tumors of the pelvic bones pose a particular technical challenge in achieving a wide local resection while respecting the anatomic requirements to preserve structural and functional integrity.31 Tumors in the sacral and para-acetabular regions especially may benefit from preoperative, intraoperative, or postoperative radiation, either alone or in combination.32,33 Stea and colleagues34 at the NCI contend that the small cell variant of osteosarcoma may be as radiosensitive as Ewing sarcoma. They present both in vitro and clinical evidence for the radiation responsiveness of this tumor subtype and report 100% local control for five patients with gross disease treated with radiation alone.
Osteosarcoma of the axial skeleton presents considerable challenges in efforts toward local control. At the Mayo Clinic,21 patients with osteosarcoma of the spine were treated with radiation after biopsy or decompression and all but one were dead of disease at a median of 10 months.35 Only five patients received chemotherapy in this series. In a review from MSKCC of osteosarcoma of the spine, 13 patients underwent limited resection followed by radiation, whereas 11 patients, treated after 1978, underwent aggressive surgical resection, chemotherapy, and local radiation.36 In the latter group there were five long-term survivors, and aggressive multimodality therapy was recommended.
Investigators from Russia reported that fractionated external-beam radiation to a median dose of 60 Gy (range 40 to 68 Gy) was used after neoadjuvant chemotherapy in 31 patients who refused amputation for extremity osteosarcomas.37 The overall survival, progression-free survival, and metastasis-free survival at 5 years were 61%, 56%, and 61%, respectively. Survival was far higher in those that had a pronounced response to chemotherapy (91% versus 35%). Thus radiotherapy is an option for patients with excellent response to chemotherapy. Two recent reports indicate that postoperative radiotherapy improves results for patients with positive margins.38,39
Osteosarcomas of the facial bones appear to have different biologic features and natural history than those located elsewhere in the body. Numerous reports on small numbers of patients seem to consistently point to a lower tendency for distant metastases. Investigators from the Institut Gustave Roussy40 obtained a clinically complete response after treating a young patient with osteosarcoma involving the nasal cavity, ethmoid, and maxilla with 45 Gy local radiotherapy followed by three cycles of high-dose ifosfamide. Radical ethmoid-maxillectomy revealed 100% tumor necrosis. Suit reported that high-dose radiation followed by resection was used to treat mandibular osteosarcomas with a resultant 5-year disease-free survival of 73%.41 Giuffrida and colleagues42 reported on two patients with osteosarcoma of the mandible treated with preoperative external radiation to 60 Gy, preoperative chemotherapy, resection, and postoperative chemotherapy. Both patients were disease-free at 18 months.
Chondrosarcoma
Background
Chondrosarcoma is a tumor of cartilage cells. It is the second most common primary bone malignancy. This tumor is rare in people younger than 20. After age 20, the risk of getting a chondrosarcoma goes up until approximately age 75. Male/female ratios are 1:1. Chondrosarcomas can develop in any place where there is cartilage. Most develop in bones such as the pelvis, thigh, or humerus. Occasionally, chondrosarcoma develops in the trachea, larynx, and chest wall. Other sites are the scapula, spine, ribs, or skull. A review of the Survival, Epidemiology, and End Results (SEER) database revealed that 2890 cases of chondrosarcoma were reported in the United States between 1973 and 2003. In this group only grade and stage were independent prognostic factors for survival.43
Role of Radiotherapy
The M.D. Anderson Hospital (MDAH) experience with the use of radiotherapy in 20 patients with primary chondrosarcoma of bone for curative intent was reviewed.44 Of 11 patients treated with radiation alone, 9 were controlled for 26 to 156 months. Doses ranged from 40 to 70 Gy in standard fractionation. The investigators believe that radiation is indicated in two settings: (1) as primary treatment when surgery is not possible, and (2) postoperatively when the surgical margins are either grossly or microscopically inadequate. These concepts are in agreement with conclusions drawn from a 23-year experience in the irradiation of chondrosarcoma at the Princess Margaret Hospital (PMH).45,46 No patient had complete surgery, and the majority presented with pain and tumors in an axial site—all poor prognostic features. Despite these facts, 50% of those whose tumors were irradiated with curative intent were controlled locally following treatment, and 25% were free of disease at 15 years. The majority received 50 to 55 Gy in 2.5-Gy fractions. The investigators point out that tumors in the head and neck and truncal regions have an 85% local recurrence rate with surgery alone, supporting the value of radiation in an adjuvant setting where surgical resection is judged to be inadequate.
Huvos and colleagues46 reported a clinicopathologic analysis of 35 patients treated at Memorial Hospital for the mesenchymal variant of chondrosarcoma. They separated this group into patients with a predominant hemangiopericytomatoid component and those with a small cell undifferentiated pattern. These groups demonstrated responsiveness to both chemotherapy and radiation, in contrast to results with conventional chondrosarcoma.
Low-grade chondrosarcomas of the base of the skull have been treated with fractionated proton radiation therapy with a 5-year local control rate of 82%.47 Investigators from the Proton Therapy facility in Orsay, France, reported the results of combined proton-photon irradiation for 45 patients with base-of-the-skull chordomas or chondrosarcomas.48 The median dose to gross tumor was 67 cobalt gray equivalent (CGE) with one-third of the dose being delivered with protons. Three-year local control and survival rates of approximately 90% were reported. Two patients experienced serious late symptoms from treatment—one had memory decline and the other had bilateral vision loss.
The series from Massachusetts General Hospital (MGH) has published the results on treatment of 519 patients with chordomas and low-grade chondrosarcomas treated with proton therapy.49 Five-year local control rates of 73% for chordomas and 98% for chondrosarcomas were achieved with tumor doses from 66 to 83 CGE. Results from Loma Linda show similar success, with 5-year local control rates of 76% for chordomas and 92% for chondrosarcomas.50 Recently preoperative and postoperative radiotherapy has been used employing 20 Gy preoperatively and 50 Gy postoperatively. Local control was reached in 90% of initial cases.51
Technique
Proton beam or IMRT techniques should be employed for chondrosarcoma. Lesions in resectable locations with margins will not require radiotherapy. Thus lesions with close or positive margins located at the skull base, in the spine and sacrum will be the targets. Doses of 50 Gy to subclinical disease, 70 GyE to microscopic disease and 77 GyE to gross disease are advised. Using standard fractionation. Doses to the center of the cord and brainstem must be kept below 54 GyE in 1.8-GyE fractions.52
Chordoma
Background
According to Huvos,9 50% of chordomas arise in the sacrum, 35% in the clivus, and 15% in the true vertebrae. Between 5% and 43% eventually metastasize,9,53,54 with the lungs being the most common metastatic site. Prolonged survival, even with metastatic disease, is not uncommon. Keisch and associates,55 in their report on 21 patients treated at the Mallinckrodt Institute of Radiology, noted that no patient was controlled, regardless of initial therapy. The group treated with conventional radiation alone did most poorly, with no survivors. Patients with lumbosacral tumors treated with surgery plus radiation had a longer mean disease-free survival (6.6 years) than those treated with surgery alone (4.1 years). They recommend doses of 55 to 60 Gy in standard fractionation to respect normal tissue tolerance.
Role of Radiotherapy
Fagundes and associates54 reported a series of 204 patients with chordomas of the base of the skull and cervical spine treated with combined proton and photon radiation to a median dose of 70.1 CGE. Overall, 29% (60 patients) experienced local relapse. Although 10 of 60 who failed locally also recurred distantly, only 2 of 144 patients who were locally controlled developed distant metastases. Local control, therefore, proved to be a highly significant predictor of distant disease-free survival. Overall survival following local relapse was poor, with a 5% actuarial 5-year survival rate and no survivors at 7 years.
Complete surgical excision as the primary procedure with negative margins can be curative, but it is rarely possible. The poor long-term survival after local recurrence highlights the importance of a concerted approach by the surgical and radiation oncologists to achieve local control with combined modality therapy at the outset when negative margins cannot be attained. Local recurrence is the rule when the tumor is violated during resection, and a second radical resection will rarely achieve long-term local control. Heroic surgical procedures, such as hemicorporectomy,56,57 have been advocated for locally extensive growths. However, if positive margins or gross residual disease are known to exist after the primary surgery, postoperative radiation therapy is indicated.
Technique
Adequate delivery of high-dose conventional photon irradiation is virtually impossible owing to the proximity to normal neural tissues and, in the sacrococcygeal region, to the bowel. Therefore, results with conventional photon irradiation have been unimpressive.58 In contrast, proton beam therapy, using precise positioning and immobilization, is able to deliver doses as high as 74 CGE to chordomas and chondrosarcomas of the base of the skull and the cervical spine while respecting the normal tissue tolerance of the brainstem, spinal cord, optic structures, and temporal lobes. Investigators from MGH58,59 stress the importance of extreme positioning accuracy to extract the greatest advantage from the physical dosimetry characteristics of protons.
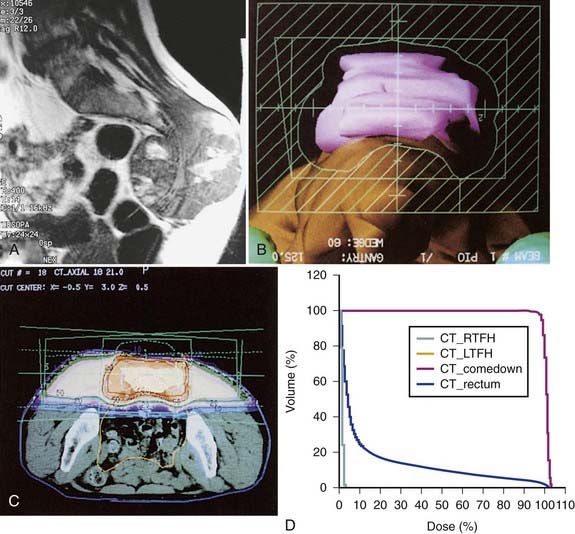
In addition, promising results with charged particle irradiation (helium and neon) in the treatment of sacral chordomas have been reported by the University of California Lawrence Berkeley Laboratory.60 It is not clear whether heavy charged particle therapy is superior to proton therapy, but there is no doubt that the physical characteristics and radiation biology of these modalities are superior to conventional photon irradiation. An approach using three-dimensional conformal radiation therapy may produce nearly equivalent dosimetric advantages (Fig. 59-7A-D). Such a technique requires fastidious attention to all phases of patient immobilization, fine cut–based computed tomography (CT) or MRI planning, and treatment delivery. For patients with these rare neoplasms, efforts to optimize local control when planning primary management might be best served by early collaboration at a center with such specialized equipment.
French workers have reported on 100 patients treated with combined photons and protons for tumors of the skull base and cervical spine. Local control rates at 2 and 4 years were 86% and 54%, respectively. Minimum dose level and homgeneity of dose were important outcome-related factors. Median total dose was 67 GyE.61 In a group of extracranial chordomas treated in Switzerland with proton scanning techniques, the median dose was 72 GyE and local control was 86%. Residual tumor greater than 30 cc was a poor prognostic factor.62 Recent results in sacral chordom reported form Boston indicate that local control is high in initial patients with doses of 73 GyE or higher.63 Technique therefore with either protons or IMRT must ensure even tumor coverage, low residual tumor volumes, doses of 72 GyE or higher, and limitation of dose to critical neural structures. Limited results are reported for radiosurgry of skull base chordoma with 5-year projected control in 18 patients of 63%.64
Giant Cell Tumor
Background
Giant cell tumors are more prevalent in females and rarely occur before skeletal maturity is reached, the majority occurring between 20 and 40 years of age. Approximately three-quarters of these tumors occur at the epiphysis of a long bone6 of the extremities, almost always extending to the articular cartilage. Only 3% are in vertebral bodies. Consequently, the majority are managed surgically, with local control rates in the range of 85% to 90%.6
Role of Radiotherapy
Dahlin and colleagues6 reported the treatment results of 195 cases of giant cell tumor of bone treated at the Mayo Clinic between 1910 and 1969. They found the local failure rate to be nearly identical (43%) in primary tumors treated with surgical techniques alone and those treated by surgical removal followed by radiation therapy. The series, collected before the advent of custom allographs and joint prostheses, reveals the necessity of a wide en bloc resection leaving no residual tumor. In the surgery-alone group, 2 of 40 patients developed malignant degeneration at failure, and in the irradiated group, 5 of 61 had this event. Although the authors conclude that radiation is an important factor in the malignant transformation of giant cell tumors, they clearly report the occurrence of (1) malignant giant cell tumors at diagnosis, (2) malignant transformation at recurrence in unirradiated patients, and (3) metastases from “benign” giant cell tumors. Therefore, owing to the inherent selection bias in the aggressive treatment group, the contention that radiation causes malignant transformation is probably untenable. Unfortunately, such reports, when considered along with the young age of most affected patients, contribute to a reluctance to use radiation in many cases.
Series reporting the efficacy of radiation in the management of giant cell tumors always reflect the sites and nature of involvement that are unusual. For example, in the series reported by Chen and associates,8 30 of the 35 cases reported occurred in the skull or spinal axis. In only two cases was radiation delivered after complete surgical excision; 33 patients had gross tumor at the time of irradiation. Patients who received less than 30 Gy experienced local failure in the majority of cases (5 of 8), whereas at doses of more than 35 Gy, the majority (14 of 17) were controlled. Although they present no further dose-response data, the authors do recommend 50 to 60 Gy when radiation will be the sole treatment modality and 30 to 40 Gy in the postoperative setting. In addition, they suggest that a 1- to 2-cm margin be used when designing radiation fields.
Similarly, investigators at the PMH65 report 21 patients treated for 42 years; 8 were treated at recurrence, but of the 13 primary cases, 11 were treated postoperatively for microscopic or grossly positive margins. Of these 11 patients, 10 were controlled with radiation doses between 10.8 and 50 Gy. For the eight patients treated at recurrence, seven were controlled with radiation doses between 35 and 55 Gy. The investigators concluded that even gross disease was readily controlled with moderate doses of radiation. They recommend 35 Gy in 15 fractions in situations in which the probability of recurrence is high and the potential morbidity of further relapse is high. They do not go so far as to recommend that radiation be routinely employed after surgery with resultant positive margins, and, in fact, the results in the recurrent setting were as good as those in the primary setting.
The MDAH group67 reported on 15 patients irradiated for giant cell tumors between 1948 and 1984, of whom 10 were evaluable. Of these 10, 3 patients died of uncontrolled local and distant disease and 5 had received chemotherapy as a component of their treatment course, attesting to the unrepresentative nature of the study population. Radiation doses ranged from 36 to 66 Gy, and the authors recommend 45 to 50 Gy when surgical excision cannot be performed.
In the University of Florida series spanning 15 years,66 all 14 patients were treated for gross unresectable disease; 10 were in the axial skeleton and 6 were recurrent lesions. Nevertheless, 75% were controlled with radiation doses between 35 and 55 Gy, and the remainder were controlled with surgical salvage. The authors recommend doses in excess of 40 Gy.
Aneurysmal Bone Cyst
Background
The aneurysmal bone cyst (ABC) is a solitary lytic lesion of bone arising most often in the second decade and more often in females. The most common locations are the metaphysic of the lower-extremity long bones, vertebral bodies or arches, and the flat bones of the pelvis.68 One theory of their origin is that they are caused by increased venous pressure; another is that they are secondary to trauma or an underlying, usually benign, tumor, often a giant cell tumor. Recently a translocation involving the 17p13 chromosome has been identified in primary ABC but not in secondary types associated with other tumors. These involve CDH11 or USP6 rearrangements and indicate that this is a true neoplasm.70
Role of Radiotherapy
Most lesions can be treated with resection using curettage and a high-speed burr and control achieved in 90% of cases. Rarely, radiotherapy can be used for resistant lesions that threaten to destroy function, such as lesions involving much of the hemipelvis. The University of Florida reported on nine ABC patients treated with megavoltage radiotherapy, most with 26 to 39 Gy. There were no local recurrences.69
Bone Metastases
Technique
Wu et al. performed a meta-analysis of trials published before 2001 and concluded that there was no significant difference in complete and overall pain relief between single and multifraction palliative radiotherapy for bone metastases.71 This conclusion was further strengthened by many other studies. See Chapter 3, “Fractionation Effects in Clinical Practice.”
Epidemiology
An estimated 8300 new cases of soft tissue sarcoma were diagnosed in the United States in 2003 and 3900 died of the disease.72 Soft tissue sarcomas constitute approximately 0.6% of all malignancies diagnosed yearly in this country (excluding skin cancers and carcinomas in situ). They can occur at any age, and, like carcinomas, are more common in older patients. Approximately 15% of affected persons are younger than age 16, and 40% are older than age 55. The male/female ratio is 1.12 : 1.
The pathogenesis of most soft tissue tumors is still unknown. Trauma or past injuries, frequently implicated in the development of sarcomas, appear to be events that call attention to the underlying neoplasm. Rare cases of these tumors arising from scar tissue following surgery, a burn, or the site of a foreign body implantation have been reported.73,74 Sarcomas (usually lymphangiosarcomas) have also been observed in the chronically edematous arm following breast cancer treatment as described in the Stewart-Treves syndrome. Chemical carcinogens, such as dioxin from herbicides and Agent Orange, have been linked to the development of soft tissue tumors. Yet none of the case control studies carried out since 1980 has been able to substantiate the claim.75–77 Olsson et al.78 showed a reduced risk of soft tissue sarcoma development with an odd ratio of 0.57 with the chronic use (>2 years) of oral contraceptives in a population-based study.
Past radiation exposure has been related to the development of soft tissue and bone sarcomas. The interval between radiation and tumor development ranges from 2 to 25 years. The frequency of neoplasm increases with higher radiation dose and longer follow-up.79 Taghian et al.80 reported a 0.2% rate of radiation-induced sarcoma in 7620 women treated for breast cancer. The estimated actuarial frequency of sarcoma development at 15 to 20 years is approximately 0.5%. This number may be slightly higher with the addition of chemotherapy. A U.S. population–based study of 274,245 breast cancer patients showed that those who received breast irradiation had a higher risk of developing soft tissue sarcoma than those not treated with radiation, although the excess risk with radiation was relatively small (incidence of 0.24 with radiation versus 0.14 without radiation at 15 years).81 The most common postradiation soft tissue sarcoma is pleomorphoric unfifferentiated sarcoma, previously called malignant fibrous histiocytoma (MFH)82 followed by fibrosarcoma and malignant nerve sheath tumor.
Genetics, Cytogenetics, and Molecular Biology
Both osseous and soft tissue sarcomas are included in the Li-Fraumeni cancer syndrome, which is characterized by a familial cluster of sarcomas, early breast cancers, brain tumors, leukemia, and adrenal carcinomas.83 Germ line abnormalities of the p53 tumor suppressor gene have been identified in the members of these families.84 Neurofibromatosis type 1 (NF1), characterized by café-au-lait spots and numerous neurofibromas, is another genetic disorder classically linked to the development of soft tissue tumors. Malignant peripheral nerve sheath tumors arise from malignant degeneration of neurofibromas in 1% to 5% of cases.85 The gene for NF1 has been cloned from the pericentromeric region of chromosome 17. The gene product (neurofibromin) appears to have tumor suppressor activities.86 A slight increase in frequency of soft tissue sarcomas has also been found in basal cell nevus syndrome, tuberous sclerosis, Werner syndrome, intestinal polyposis, and Gardner syndrome.
Cytogenetic studies have been carried out for many soft tissue tumors. Table 59-1 lists characteristic chromosomal aberrations and their frequency for certain sarcomas. On the molecular level, alterations in the Rb and p53 tumor suppressor genes have been found in soft tissue sarcomas. Wunder et al.87 reported Rb changes in 5 of 12 high-grade and 1 of 11 low-grade soft tissue tumors. Toguchida et al.88 discovered 42 somatic p53 alterations in 127 cases of osseous and soft tissue sarcomas. Twenty-one were gross gene arrangements, and 21 had subtle changes (missense or nonsense mutations). Others have reported abnormal p53 immunostaining in one-third of the cases.89,90 Of note, mutations of both Rb and p53 genes were frequently seen in the same tumors. MDM2 (located on chromosome 12q13-q14), an inhibitor of p53 transcriptional activities, was found to be amplified in 8 of 24 soft tissue tumors evaluated.91 This suggests that MDM2 amplification is an alternative mechanism for inactivating the cell cycle regulatory pathways. SAS, located in chromosome 12q13-q14 near a cyclin-dependent protein kinase gene, is thought to be involved in signal transduction and cell growth regulation. It was found to be amplified in 7 of 22 MFHs and in three of three liposarcomas.92 Its role as an oncogene has not been clearly established. More details of the cytogenetic changes and molecular biology of soft tissue tumors can be found in an excellent review by Cyril Fisher.93
Transcriptional profiling using either oligonucleotide or complementary deoxyribonucleic acid (cDNA) microarrays has been employed to characterize soft tissue tumors.94 The authors analyzed 41 soft tissue tumors, which provided more than 1.5 million data points for 5520 well-measured genes. Based on the level of gene expression, they found that these 41 specimens can be separated into five distinct groups: synovial sarcomas; gastrointestinal stromal tumors (GISTs); benign peripheral nerve sheath tumors; half of the leiomyosarcomas; and a broad group containing all the MFHs, the liposarcomas, and the other leiomyosarcomas. In addition, they found that c-Kit and protein kinase C are highly expressed in GIST, and cellular retinoic binding protein–1, retinoic acid receptor–γ, and the epidermal growth factor receptor are highly expressed in synovial sarcoma, suggesting that these can be exploited for molecular targeting of these tumors in the future.
Anatomy
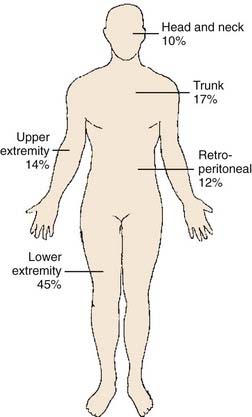
Embryologically, the soft tissue structures arise from the primitive mesenchyme of the mesoderm with some contribution from the neuroectoderm. Soft tissue structures consist of muscles, fat, fibrous tissues, blood vessels, and supporting cells of the peripheral nervous system. The frequency of involvement of different anatomic sites is shown in Table 59-2 and summarized in Fig. 59-8. Soft tissue sarcomas can arise in soft tissues in any part of the body.
Pathologic Conditions
Each of the soft tissues can give rise to a group of malignant sarcomas. Tumor histologic anatomy and their putative cells of origin (as suggested by Enzinger and Weiss85) are listed in Table 59-3. MFH and liposarcoma (Fig. 59-9 and Fig. 59-10) are the most common soft tissue neoplasms in adults, accounting for 35% to 45% of all sarcomas. The three most common sarcomas found in U.S. hospital registries in the 1980s were MFH (26%), liposarcoma (18%), and leiomyosarcoma (15%).95 Hashimoto and colleagues reported an incidence of 25% for MFH, 12% for liposarcoma, and 10% for rhabdomyosarcoma among 1116 cases evaluated.96 More recently, MFH has been renamed and the World Health Organization diagnosis is now called pleomorphic undifferentiated sarcoma (PUS).82
| Tissue | Sarcoma |
|---|---|
| I. Fibrous | Fibrosarcoma |
MPNST, Malignant peripheral nerve sheath tumor.
Modified from Enzinger FM, Weiss SW: Soft tissue tumors. In Greene LG, Page DL, Fleming ID, et al, eds: AJCC cancer staging manual, ed 6, New York, 2002, Springer, p 193.
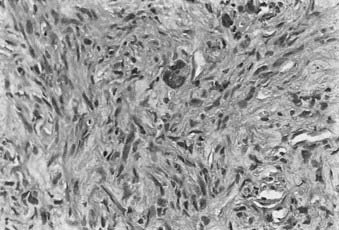
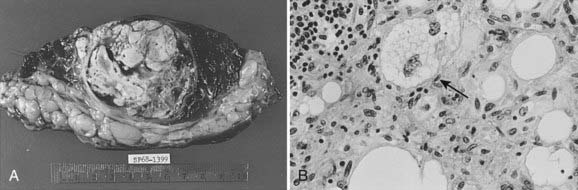
MFH was until recently considered the most common soft tissue sarcoma among adults. Histologically, karyotypically, and clinically, this is by far the most heterogeneous group of sarcomas, and the concept of MFH as a histogenetically separate entity was called into question. An alternative hypothesis is that MFH is a poorly differentiated sarcoma and might represent a common endpoint for various other sarcomas. Fletcher et al.82 performed histologic, immunohistochemical, and, where available, ultrastructural reanalysis of 100 MHF cases of the extremity and trunk. In 84 cases, a specific line of differentiation was strongly suggested. The most common diagnosis was myxofibrosarcoma (22 cases) and leiomyosarcoma (20 cases). They also showed that myogenic sarcomas had a worse prognosis than nonmyogenic sarcomas.82 Gene array analysis did not separate MFH from the liposarcomas, which strengthened the alternative view of MFH.94 Further work is needed to clarify this controversy. Even though MFH has been renamed PUS, the term MFH continues to be used and remains useful in diagnostic and clinical practice.
Sarcoma histologic anatomy alone does not provide sufficient evidence for predicting the clinical course. Grading and staging are essential for accurate prognosis and treatment of these neoplasms. Unlike past classifications, each histologic feature can be assigned a specific grade in the current system. When histologic grades are accounted for, with a few exceptions, most soft tissue sarcomas have common biologic behaviors irrespective of histologic type. In a series of 211 high-grade sarcomas, Potter and coworkers showed that histologic anatomy was not a significant determinant for either overall survival or disease-free survival.97
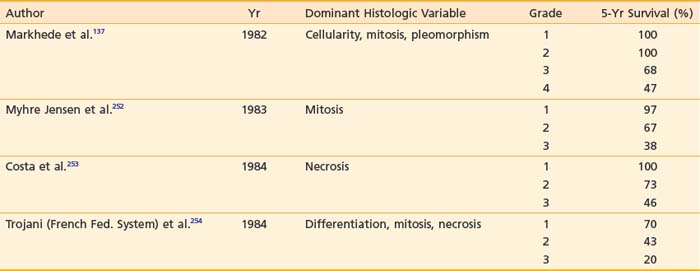
Traditionally, the designation of histopathologic grades depends on a combined assessment of several histologic features: degree of cellularity and pleomorphism, abundance of stroma, expansile or invasive growth, extent of differentiation, mitotic activity, and amount of necrosis. Mitotic count and necrosis appear to be the most significant prognostic factors in predicting the duration of survival and the time to developing distant metastasis.98 There are at least four different grading systems in the literature, as summarized in Table 59-4. Despite their differences, all showed a strong correlation between grade and survival. The two systems most favored by pathologists are those designated as the NCI and the French Federation of Cancer Centers Sarcoma Group (FNCLCC). A direct comparison between these two systems in 410 patients with nonmetastatic soft tissue sarcoma revealed that both systems were of prognostic value in predicting the risk of distant metastasis and tumor-related death, with the FNCLCC system providing a slightly better prediction.99 Unfortunately, there is poor reproducibility of histologic diagnosis and tumor grading among trained pathologists. Coindre et al.100 found a crude agreement rate of only 61% for histologic anatomy and 75% for tumor grade among 15 well-known sarcoma experts.
Clinical Presentation and Routes of Spread
The most common clinical presentation of soft tissue sarcoma is a slow-growing, painless mass. The median period between detection of the lesion and presentation to the clinician is approximately 4 months, and the clinician’s delay in establishing the diagnosis is approximately 1 month.47 Pain, numbness, and swelling may result from tumor invasion of bone or neurovascular bundles.
Approximately 6% to 10% of patients have metastatic disease at diagnosis. Rydholm et al.101 reviewed the records of 278 patients with soft tissue sarcomas of the extremities registered in the Southern District of the Swedish National Registry. They found 19 cases (6.8%) with metastatic disease at diagnosis. In our review of 65 head and neck soft tissue sarcomas treated primarily at the University of California, San Francisco (UCSF), four (6.1%) had distant metastases at presentation.
Hematogenous dissemination after diagnosis is common in high-grade lesions. Isolated pulmonary metastases are the most frequent, accounting for nearly 50% of all initial recurrence.102 Bone, liver, and skin involvement occurred in less than 5% of patients.103 Liposarcoma and retroperitoneal sarcomas display a different metastatic pattern. Although the lungs still predominate as a metastatic destination, liver involvement and peritoneal carcinomatosis do account for a small but significant number of deaths in these patients.102,104 Approximately 75% to 80% of distant metastases appear within 2 years of initial treatment.
Regional lymphatic dissemination is uncommon. In a comprehensive literature review, Weingrad observed a 5.8% incidence of nodal spread during the course of the disease.105 Mazeron found that 5.9% of 323 patients without distant metastatic disease at presentation had nodal involvement.106 There was a correlation between tumor grade and the frequency of nodal metastasis: 0% for grade 1, 2% for grade 2, and 12% for grade 3. Pooled data from published reports on 5257 patients treated for soft tissue tumors showed a higher incidence of nodal spread in certain histologic subtypes: clear cell sarcoma (28%), epithelioid sarcoma (20%), angiosarcoma (23%), rhabdomyosarcoma (15%), and synovial sarcoma (14%).106
Diagnostic and Staging Studies
Radiographic Imaging
Radiologic studies should preferentially be obtained before any surgical manipulation to avoid confusing postsurgical changes. MRI is the preferred imaging modality for soft tissue sarcomas because of its multiplanar imaging capability and superior soft tissue contrast. Most musculoskeletal tumors have intermediate to low signal intensity in T1-weighted images and appear bright on T2-weighted images. Exceptions to these rules are tumors with high fat or blood product content such as liposarcomas and angiosarcomas, which appear as masses of high signal intensities on T1-weighted images (Fig. 59-11).107 The use of contrast agents such as gadolinium has not been shown to improve the accuracy of tumor staging or lesion delineation.
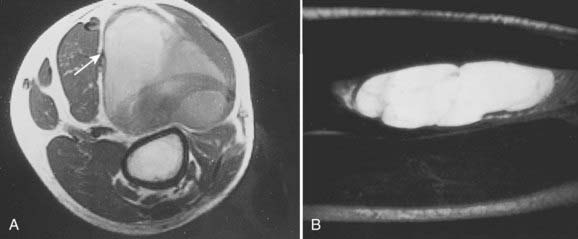
(Courtesy Dr. Charles Peterfly, Department of Radiology, University of California at San Francisco.)
CT remains the diagnostic modality of choice for detecting pulmonary metastases in high-grade tumors.105 Yet the cost-effectiveness of routine chest CT for metastasis screening in patients with T1 and T2 soft tissue sarcoma has been questioned. Fleming et al.108 showed that the routine use of chest CT in addition to a chest x-ray to screen for pulmonary metastasis in patients with T1 tumors of the extremity results in a substantial incremental cost of $2.9 million per case of pulmonary metastasis detected because less than 1% of the patients in this group presented with lung metastasis at diagnosis. Similarly Porter et al.109 found that the use of routine chest CT, when compared with selective CT, for evaluation of metastatic disease for T2 tumors is most cost-effective in patients with extremity and high-grade lesions. For primary tumor evaluation, CT is a useful supplement to plain radiographs to evaluate for suspected bony invasion. Finally, CT is also widely used to image the tumor for three-dimensional treatment planning in radiation therapy.
Bone scans are not useful in the diagnostic workup of soft tissue sarcomas. Bony metastases in the absence of visceral spread are rare. Bony invasion is best demonstrated by CT. Positron emission tomography (PET) scanning and magnetic resonance spectroscopy may help provide information on tumor metabolism and grade,110 although the role of these studies is still undefined. Dimitrakopoulou-Strauss et al.111 studied the use of 18F-FDG PET for the diagnosis of primary or recurrent soft tissue sarcoma and found a sensitivity of 76%, specificity of 43%, and an accuracy of 68%. It appeared to be most accurate in patients with high-grade tumors. Conrad et al.112 reported that the average standardized uptake values (SUV) obtained from PET scans were useful in differentiating high-grade and large tumors with high metabolism from low-grade and small tumors in 108 patients with bone sarcoma and soft tissue sarcoma. Finally, PET may serve as a useful predictor for response from neoadjuvant therapy. Schuetze et al.113 performed serial PET scans on 56 patients treated with two to three cycles of neoadjuvant doxorubicin-based chemotherapy. Patients with a greater than 50% reduction in maximal SUV had a longer time to recurrence (38 versus 18 months, P = 0.03) and overall survival (not yet reached versus 41 months, P = 0.02).
Biopsy Considerations
Biopsy is a critical step in the evaluation of these neoplasms. Incisional biopsy is the preferred technique for diagnosis of most soft tissue sarcomas. This technique involves a wedge removal of tissue with minimal surgical manipulation. The incision should be short, oriented in the same direction as the surrounding musculature, and situated so that it could easily be removed in future surgery without compromising limb-salvage or reconstructive options. Meticulous attention should be paid to hemostasis to prevent tumor dissemination by hematoma, and drains should not be used routinely. Occasionally, an excisional biopsy will suffice for lesions less than 3 cm in size, for which at least a 2-cm cuff of normal tissue around the tumor is resected. True-cut core needle biopsy is becoming an increasingly popular method for diagnosing soft tissue tumors, particularly in situations in which preoperative chemotherapy or radiotherapy or both are being considered for large accessible lesions.114–115 Core needle biopsy and immunohistochemistry often confirm the diagnosis of sarcoma and shed light on the tumor grade even if exact classification of the tumor is not possible.116 Many times this information is sufficient for clinicians to initiate preoperative therapy for high-grade tumors and resect the low-grade ones. Fine-needle aspiration is not routinely used to diagnose soft tissue sarcomas because of the paucity of available tissues for examination and the loss of tissue architecture.
Histopathologic Studies
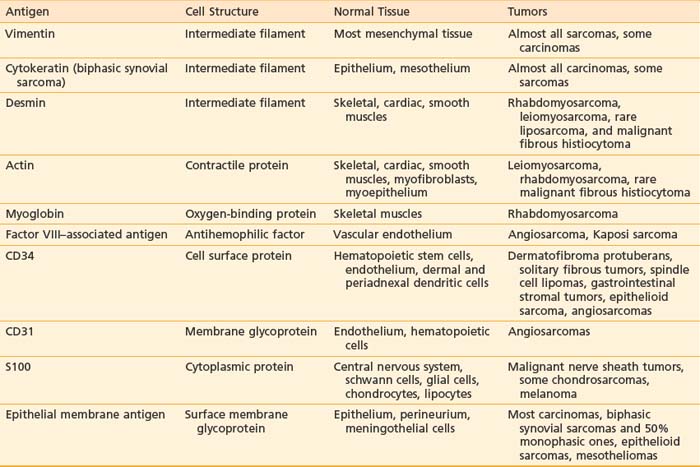
The pathologic evaluation of soft tissue tumors should be carried out by an experienced pathologist because of the vast diversity of sarcomas and their notoriety in mimicking other neoplasms. In addition to light microscopy, tools including electron microscopy, immunohistochemistry, cytogenetics, and molecular genetics are widely used to establish the diagnosis. Table 59-5 lists some commonly employed immunohistochemical antigens, their cellular origins, and distributions among certain types of soft tissue sarcomas.
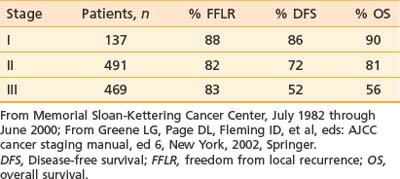
Staging
The most frequently used staging system is that proposed by the American Joint Committee on Cancer (AJCC)117 (Table 59-6). Table 59-7 summarizes the 5-year freedom from local recurrence, disease-free survival, and overall survival rates in more than 1000 patients treated at MSKCC by the new AJCC staging system. Histologic grade and tumor size are the primary determinants of clinical stage in the AJCC system. The T1 and T2 tumor sizes are further substaged based on tumor depth. Superficial tumors are defined as those located exclusively above the superficial fascia without fascial invasion and deep lesions are those that are deep to or invade or cross the investing fascia. For practical purposes, all retroperitoneal, visceral, mediastinal, intrathoracic, pelvic, and the majority of head and neck tumors are classified as deep lesions. The new staging system also accommodates a preferred “two-tiered” grading system (low versus high grade). In the three-tiered system, grade 1 is considered “low grade” and grades 2 to 3 are “high grade.” In the four-tiered system, grades 1 to 2 are considered “low grade” and grades 3 to 4 are “high grade.” The system is designed to stage extremity tumors optimally, but is also applicable to torso, head and neck, and retroperitoneal sarcomas. It should not be used to stage sarcomas arising within the confines of the dura mater, brain, parenchymatous organs, and hollow viscera. All soft tissue sarcoma histologic findings, including GISTs and Ewing sarcomas or primitive neuroectodermal tumors, are included, except Kaposi sarcomas, dermatofibrosarcoma protuberans, fibromatosis (desmoids), angiosarcomas, and malignant mesenchymomas.
Table 59-6 Classification of Soft-Tissue Sarcomas
Rights were not granted to include this table in electronic media. Please refer to the printed book.

A major pitfall of the current AJCC staging system is its failure to recognize the anatomic site of the tumor, which is an important determinant of prognosis. Although tumor site is not officially incorporated as a specific component of any present staging system, prospective data collection should include tumor site, and outcome data should be reported on a site-specific basis. The recommended groupings for tumor sites are head and neck; extremity and superficial trunk; gastrointestinal; genitourinary; visceral; retroperitoneal; gynecologic; breast; lung, pleural, and mediastinal; and others.117
Prognostic Factors
Different prognostic factors exist for local recurrence, distant recurrence, and survival in patients with soft tissue sarcoma. In a large analysis of prognostic factors in 1041 patients with extremity soft tissue sarcoma, tumor grade, tumor size, and deep location were the strongest prognostic factors for disease-specific survival (Table 59-8).118 These three factors were also confirmed independently by the FNCLCC study of 546 patients with soft tissue sarcoma of the extremities, head and neck, trunk, pelvis, and retroperitoneum.119 Other reported, unconfirmed prognostic factors for survival are gender, leiomyosarcoma or malignant peripheral nerve sheath tumor, microscopically involved margins, no adjuvant chemotherapy, local recurrence at presentation, and lower extremity site.
Table 59-8 Prognostic Factors for Mortality, Distant Metastasis, and Local Recurrence in Soft Tissue Sarcomas
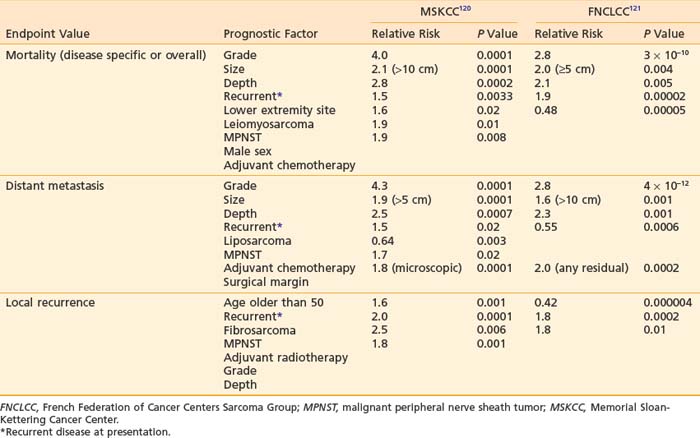
For distant recurrence, tumor grade, tumor size, and deep location were significant prognostic factors in both large studies mentioned earlier (see Table 59-8). In addition, leiomyosarcoma or nonliposarcoma histologic features, local recurrence, and no adjuvant chemotherapy also appeared to affect the risk of distant metastasis to a lesser degree.
Unlike other solid tumors, local control appears to be influenced by a separate set of prognostic factors, different from those that predict survival and distant metastasis. Pisters et al.118 found that age older than 50 or local recurrence at diagnosis, microscopically involved surgical margins, fibrosarcoma, or peripheral nerve sheath tumor histologic features adversely influenced local control in 1041 patients treated for extremity soft tissue sarcoma. Approximately 40% of these patients received adjuvant radiotherapy and 23% had microscopic residual disease. In a different group of patients, including those with tumors outside the extremities, of whom 58% had adjuvant radiotherapy and only 69% had complete surgery, Coindre et al.119 found that poor surgery (defined as local excision only), no adjuvant radiotherapy, grade 3, and deep location were adverse predictors for local control.
Although rare, the presence of nodal metastasis is associated with poorer survival. Mazeron and Suit found that only 32% of patients with nodal involvement at presentation were alive more than 58 months after treatment.106
The location of primary tumors often influences the resectability of the lesion and the ability to deliver high-dose radiation. Sarcomas of the head and neck, mediastinum, and retroperitoneum are more difficult to control locally because of their proximity to critical organs. Retroperitoneal sarcomas are often very large at the time of diagnosis. These adverse factors are reflected in the lower survival rates for patients with these tumors as compared with those found in the extremities.95,120
There is evidence that the status of surgical margins influences local control. Tanabe et al.121 reported a 5-year local disease-free survival of 91% for margin-negative patients, and 62% for margin-positive patients (P = 0.005). Similarly, Suit and Spiro122 found that the 5-year local control rate for patients with negative margins was 97% compared with 81% for those with positive margins. Interestingly, local control appears to be independent of the degree of margin negativity (negative at 1 mm or >1 mm). It is controversial whether tumor involvement at the surgical margins affects survival. Tanabe et al.121 reported similar 5-year survival rates for margin-negative (65%) and margin-positive patients (70%), despite a higher local failure rate in the latter group. However, Heslin et al.123 reported that a positive microscopic margin in patients with high-risk soft tissue sarcoma (high grade, deep, or ≥5 cm) is a predictor for biologically aggressive tumors and is associated with poorer survival.
It is controversial whether achievement of local control after the first treatment affects long-term survival. Tanabe et al.121 found that local failure did not adversely affect overall survival. They did emphasize that their number of local recurrences was too small to make a definite conclusion. In an NCI prospective randomized trial comparing amputation to limb-sparing surgery with radiotherapy, Rosenberg et al. found no difference in survivals between the two groups, despite a higher local recurrence rate in the combined modality therapy arm.124 In a prospective randomized study to evaluate the effectiveness of adjuvant brachytherapy (BRT) at MSKCC, Harrison et al.125 demonstrated an improvement in local control for patients with high-grade tumors receiving additional radiation. Unfortunately, this advantage in local control was not associated with an improvement in survival.125 However, many other authors have concluded that local recurrences have a negative affect on survival.102,126–128 Despite the difficulty in defining a relationship between local control and survival, every effort should be made to prevent local recurrences because salvage therapy often requires more mutilating surgery than would have been needed initially.
Attention is now focusing on molecular pathologic prognostic factors. Ki-67, an antigenic measurement for cell proliferation, was shown to be an independent prognostic factor for soft tissue sarcoma in at least three separate studies.129–131 Data on the underlying prognostic significance of P53 and MDM2 status are conflicting. Some investigators reported no independent adverse prognostic significance by regression analysis,129 whereas others found a highly significant correlation with outcomes.132 Plasma metalloprotease-9 (MMP) activity was shown to be elevated in patients with soft tissue sarcoma when compared with healthy volunteers or patients with benign tumors in a small study.133 In addition, MMP-9 circulating activity was decreased in nine patients after successful surgery and increased in three patients with relapsed tumor, suggesting that it may be a potential marker for tumor surveillance.
In synovial sarcomas, the t(X;18)(p11;q11) translocation fuses the SYT gene from chromosome 18 to either of two homologous genes at Xp11, SSX1, or SSX2. The fusion transcripts (SYT-SSX1 or SYT-SSX2) are believed to function as aberrant transcriptional regulators. In a study of 45 patients, there was a significant correlation between histologic subtype and fusion transcript; all 12 biphasic tumors had SYT-SSX1 fusion, whereas only 17 (52%) of 33 monophasic tumors had the same transcript. Moreover, the presence of STY-SSX1 transcript was associated with worse survival in this patient group.134 This is an exciting observation suggesting that these fusion transcripts can be used as a prognostic marker for synovial sarcoma; however, these findings need to be confirmed in larger studies.
General Management
Surgery
Enneking and colleagues have defined four categories of surgical procedures used in the treatment of soft tissue sarcomas based on surgical margins135:
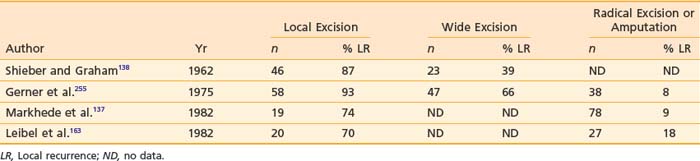
Traditionally, surgical resection has been the main treatment modality for soft tissue sarcoma of all sites. Until the early 1950s, the surgical approach was a local excision or amputation. The local failure rate for local excision ranged from 60% to 90%.136–138 Attempts to reduce local failures led to the introduction of radical soft tissue compartmental resection (including amputation) in the 1960s. The local control rate increased to the 80% to 90% range at the expense of limb function.139,140 Table 59-9 shows the local recurrence rates reported in the literature for different types of surgical procedures.
Radiation Alone
Historically, the use of radiation treatment was limited to patients with locally advanced, inoperable, recurrent, or metastatic disease. The dose of radiation was usually low, and was reserved mainly for palliation. In 1951, Cade reported long-term, disease-free survival (5 to 26 years) for 6 of 22 patients with unresectable tumors treated by radiation alone.141 Between 1935 and 1959, McNeer et al.142 treated 25 patients with radiation alone at MSKCC. Fourteen survived without disease for more than 5 years. Tepper and Suit reported on 51 patients treated with primary radiation at the MGH.143 The overall 5-year survival and local control rates were 25% and 33%, respectively. For those who received a dose of 64 Gy or more, the 5-year survival and local control rates were 28% and 44%, respectively. A 67% local control rate has also been observed in patients treated palliatively with fast neutron therapy.144 These results indicate that patients may be cured by radiation alone if high doses are employed. The results also imply that sarcomas are not as radioresistant as previously thought. Primary radiation should be reserved for patients with unresectable lesions because of anatomic location, medical inoperability, or refusal to undergo surgery.
Combined Conservative Surgery and Radiation Therapy
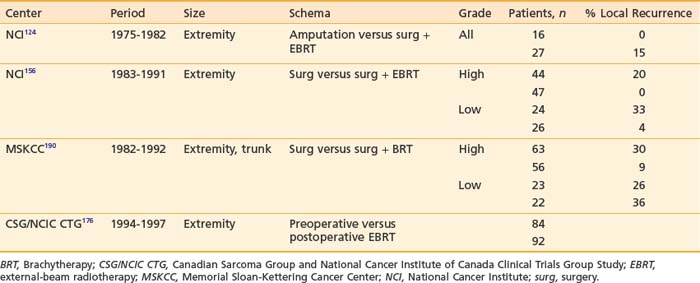
In 1951, Cade obtained a 61% survival rate in patients with wide excision and radiation therapy, compared with 27% with amputation with or without radiation.141 He advocated combining radiation with surgery. In the 1960s, Suit and colleagues investigated the role of radiation as an adjunct to limb-sparing surgery in an effort to preserve function and to reduce recurrence.145 For the first 57 patients treated, a local control rate of 87% was achieved, comparable to that with radical resections. Between 1975 and 1981, the NCI conducted a prospective randomized study in which 43 patients with high-grade lesions of the extremities were randomized to either amputation or limb-sparing surgery with adjuvant radiation therapy. The radiation dose was 50 Gy to the anatomic area at risk, and 60 to 70 Gy to the tumor bed, delivered via a shrinking-field technique. All patients had postoperative chemotherapy consisting of doxorubicin, cyclophosphamide (Cytoxan), and high-dose methotrexate. With the median follow-up of 4 years and 10 months, none of the 16 tumors of patients undergoing amputation recurred locally. Four of 27 patients randomized to the limb-sparing arm relapsed locally. However, there was no difference in disease-free survival and overall survival between the two arms. It was concluded that limb-sparing procedures in combination with radiation are effective for most patients with soft tissue sarcomas, a practice widely in use at present.124 The results of this small randomized trial, coupled with large reports from numerous institutions,146–149 provided the impetus for the National Institutes of Health to publish a consensus statement recommending limb-sparing procedures.150
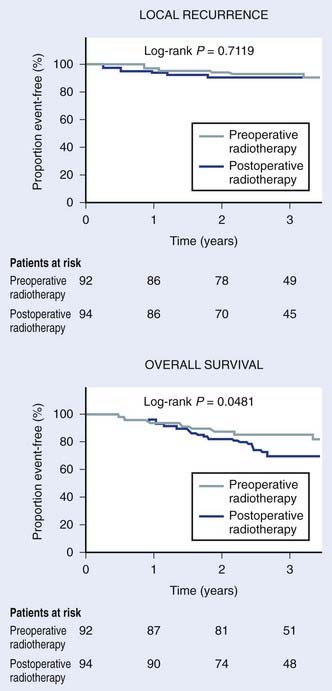
Many different strategies have been used to combine radiation and surgery. These included preoperative radiation, postoperative radiation, or intraoperative radiation with either BRT or electron-beam techniques (intraoperative electron-beam radiation therapy [IOERT]). The theoretic advantages and disadvantages of these treatment strategies are outlined in Table 59-10. Table 59-11 shows the results of the published randomized studies evaluating surgery alone versus surgery plus radiotherapy for local control in soft tissue sarcoma. Although there was no direct comparison between postoperative external-beam radiotherapy and BRT, the results suggest that both are equally effective for high-grade tumors, whereas external-beam irradiation appeared to be more useful for local control in low-grade tumors. Controversies exist regarding the timing of delivering external-beam irradiation in relation to surgery (i.e., whether it should be in the preoperative or postoperative settings). Recently, the National Cancer Institute of Canada (NCIC) conducted a randomized trial comparing these two approaches in patients with extremity soft tissue sarcoma who would need combined surgery and radiotherapy. The trial was closed when an interim analysis showed a higher rate of acute wound complication with preoperative radiotherapy (35% versus 17%; P = 0.01).151 However, there was an unexplained overall survival advantage with preoperative radiotherapy, but no difference in distant metastasis or disease-free survival, suggesting that there was an inbalence in the two arms leading to increased death from other causes in the postoperative arm (Fig. 59-12). With longer follow-up (median 76 months) there was a trend to higher grade 2 or high fibrosis in the higher dose postoperative arm (31% versus 48%; P = 0.07).152 To date, there has been no difference in local control between the two strategies. At UCSF, our policy is to employ postoperative external-beam radiation. For most small, resectable tumors, wide local excisions are performed followed by postoperative external-beam radiotherapy. For large (usually >5 cm) and deep tumors, for which surgical resection is difficult, we prefer IOERT followed by postoperative radiation. IEORT acts as a reverse boost and allows for a decrease in the dose of postoperative radiation needed, potentially leading to decreased fibrosis. We also use IOERT for recurrent disease in patients who have received radiation in the past. Chemotherapy is reserved for those with high-grade neoplasms.
Table 59-10 Advantages and Disadvantages of Different Combined Treatment Approaches
| Treatment | Advantages | Disadvantages |
|---|---|---|
| 1. Preoperative radiation |
IORT, Intraoperative radiotherapy.
The role of radiotherapy for small, low-grade tumors is controversial. A small, randomized study153 and at least one retrospective study154 suggested a local control benefit with the addition of postoperative radiotherapy, at least in larger tumors (>5 cm) with close surgical margins. Other studies indicated that in a subset of well-selected patients, radiotherapy may be omitted without compromising local control.155–157 We do not typically recommend postoperative irradiation for small lesions that have been widely removed with negative margins because many are cured with surgery alone. Radiation therapy is used in patients with positive or close margins or in those with large tumors in whom local recurrence would require amputation.
Radiation Therapy Techniques
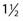 to 3 weeks.
to 3 weeks.Simulation and Field Size
During simulation, the optimal position for the patient is determined. This involves rotating the extremity to treat the affected compartment while minimizing dose to surrounding tissues. Part of the thigh can be treated in the frog leg position. Elevation of one leg above the other allows for treatment of the posterior compartment. The “throwing” position,158 which consists of abduction of the shoulder joint and flexion of the elbow joint, is an ideal position for treating part of the upper arms. Occasionally, patients may be treated in a sitting position while resting their extremities on the treatment couch. The lithotomy position is useful for treating perineal sarcomas, whereas prone positions are occasionally used for retroperitoneal lesions to minimize radiation exposure to the small bowel. Positioning of the patient can sometimes be difficult because of postsurgical fibrosis. To ensure treatment reproducibility, the area treated should be immobilized. We employ devices such as a foam cradle (Fig. 59-13) or thermoplastic cast to provide special immobilization for individual patients.

CT treatment planning should be used in treating soft tissue sarcoma. One must make sure that the CT is performed in the treatment position. In the preoperative setting, the gross tumor volume (GTV) typically represents the radiographically defined tumor. Peritumoral edema should be included when preoperative irradiation is given. Strictly speaking, there should not be a GTV for the postoperative setting. However, it is useful to designate a putative GTV for cone-down purposes. This GTV should encompass all the tissues handled during the surgery, including all tumor-related scars and draining sites. The optimal longitudinal margins that should be included around the GTV (i.e., the clinical target volume [CTV]) is an area of controversy. At the MDAH, a 5-cm longitudinal margin is used for grade 1 sarcomas, and a 7-cm margin is employed for grade 2 to 3 lesions (Matt Ballo, MD, personal communication).159 No attempt is made to encompass the entire anatomic compartment. The large field is treated to a total dose of 50 Gy, followed by a boost field encompassing the primary lesion with 3- to 4-cm margins. The practice at MGH is to use 5-cm margins for small grade 1 tumors, 5- to 10-cm margins for large grade 1 and small grade 2 to 3 tumors, and 10- to 15-cm margins for larger high-grade lesions.160 Again, a shrinking field technique is used with the large field size treated for the first 5 weeks, followed by field reductions at the sixth and the seventh week. At the NCI, initial radiation portals are designed to treat the muscle group from origin to insertion followed by shrinking volume as described.161 The treatment policy at the PMH has been to place a field margin of 5 cm around the GTV, including any peritumoral edema, regardless of grade or size of the tumor.162 At UCSF, we generally follow the recommendation from PMH with a 5-cm margin proximal-distally, 2-cm margin radially, and at least a 2-cm margin on the surgical scar and drain sites, without attempting to cover the entire muscle compartment. In all cases, we use three-dimensional treatment planning to guide our field arrangement (Fig. 59-14 and Fig. 59-15) and most cases use a field-within-field technique to allow for better homogeniety and improved coverage of the tumor volume (Fig. 59-16). Because the incidence of nodal involvement is low, we do not recommend routine nodal prophylaxis. However, in the case of epithelioid sarcoma, clear cell sarcoma, high grade rhabdomyosarcoma, and synovial sarcoma, regional nodal treatment should be considered.
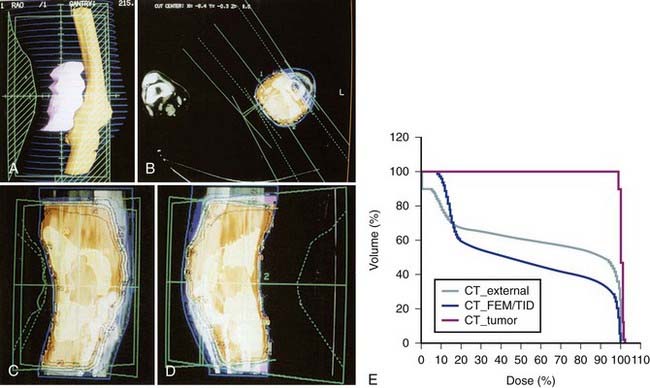
FIGURE 59-14 • Computed tomography-based three-dimensional treatment plan using a right anterior oblique (RAO) and a left posterior oblique field for the patient in Fig. 59-13. A, Beam’s-eye view of the RAO field showing the tumor volume (in pink), the femur, the knee joint, the tibia (yellow), and the computer-generated blocks (in green). C and D, Isodose distribution in relation to the tumor volume (white line) and surrounding structure—axial view (B), coronal view (C), and sagittal view (D). Note that the 95% isodose line (in red) encompasses all tumor volume. E, Dose-volume histogram for the tumor volume (white), the surrounding skin (green), and adjacent bones (yellow) generated by three-dimensional computed tomography treatment planning. By using the plan, 40% to 60% of adjacent normal tissues will receive a full dose and the rest will receive 0% to 20% of the treatment dose from scatter radiation.

FIGURE 59-15 • Simulation and port films of the right anterior oblique field for the patient in Figs. 59-13 and 59-14. Note the sparing of the anterior half of the skin, knee joint, and bones.
Circumferential irradiation of the extremity can result in severe fibrosis with pain, edema, and loss of function, which may necessitate amputation.145 This should be avoided by sparing as many of the soft tissue compartments as possible without missing the tumor bed. A portion of the circumference of the uninvolved bone should be spared to reduce the risk of a radiation-induced fracture. If the scar crosses the joint, part of the joint should be blocked out unless the joint space was violated surgically. A bolus should be placed over the surgical scar when it is not located in the tangential part of the beams to ensure adequate skin dose for the first part of treatment. Gonadal shielding should be considered in the treatment of thigh, groin, or pelvic lesions. If long fields are required, they must be gapped, and the match line needs to be moved weekly to avoid excessive hot or cold spots. Wedge filters and compensators should be employed regularly in treatment planning to enhance dose homogeneity.
Intensity-Modulated Radiation Therapy
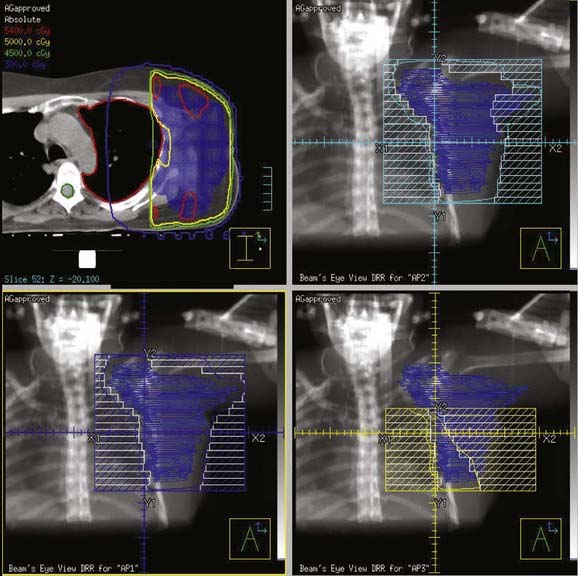
IMRT has been used to help spare normal tissue if the tumor is in close proximity to normal structures. As with squamous cell carimomas of the head and neck region, IMRT should be used in all sarcoma cases in this region. IMRT allows for better sparing of multiple critical structure within this region. For extremity sarcomas, IMRT has been used when conventional radiation techniques would encompass the entire bone. The group from MSKCC compared 10 patients’ plans using IMRT to the same 10 patients using a traditional three-dimensional technique. The CTV was defined as the GTV with a 1.5-cm margin axially, except the bone interface was used as the CTV if the 1.5-cm expansion extended beyond the bone interface. The PTV was defined as the CTV with a 5-mm margin all around. The results showed that IMRT could obtain equal coverage of both the CTV and PTV with a decreased dose to the femor.163 An example of an IMRT plan used at UCSF is shown in Fig. 59-17.
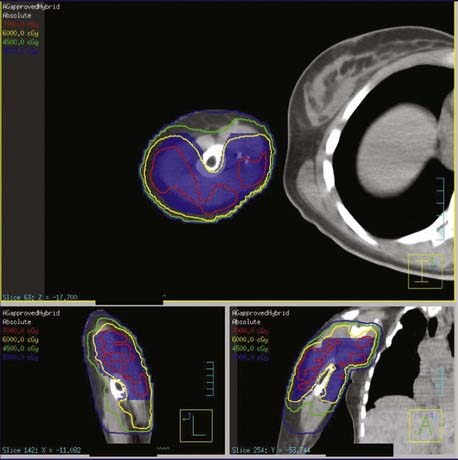
One of the concerns with IMRT for extremity sarcomas is that the tght dose distribution might compromse tumor coverage and lead to more local failures. The same group at MSKCC published their clinical results on 41 patients with primary soft tissue sarcomas of the extremity treated with limb-sparing surgery and adjuvant IMRT. Preoperative IMRT was given to seven patients (50 Gy) and postoperative IMRT was given to 34 patients (median dose, 63 Gy). With a median follow-up of 35 months, 2 of 41 patients developed a local failure. The 4-year actuarial local control rate was 94%. These data suggest that the tight dose distribution of IMRT does not compromise local control.164
Radiation Dose
In the past, sarcomas were thought to be radioresistant. Clinical and laboratory data have indicated otherwise. The radiation sensitivity of cell lines derived from human sarcomas appears to be the same as that for epithelial tumors.122 In a retrospective study of 65 primary head and neck soft tissue sarcomas at our institution, a dose-response relationship according to surgical margin status could not be demonstrated. Similarly, our review of extremity sarcomas failed to show a dose-response relationship.165 In the 1970s, the MDAH reduced its total postoperative dose from 70 Gy to 60 Gy for low-grade lesions, and 65 Gy for high-grade lesions without an increased incidence of local recurrence.148 At the NCI, a postoperative tumor dose of 63 Gy at 1.8 Gy per day is recommended. A dose of 70 to 75 Gy is used for gross residual disease.161 At the MGH, preoperative radiation is advocated for large tumors. A dose of 50 Gy in 5 weeks is administered, followed by a  – to 3-week rest period before surgical resection. A boost of 15 Gy is given for microscopically positive margins, and 25 Gy is used for gross residual disease, delivered by external-beam radiation, IOERT, or BRT. If administered postoperatively, 50 Gy is given to the large volume, followed by two field reductions to deliver a total dose of 60 Gy for negative margins, 66 Gy for microscopic residual disease, and 75 Gy for gross residual disease. The total dose is reduced by 10% in patients who also receive doxorubicin-based chemotherapy.160 Our treatment policy at UCSF and Stanford is similar to that of the MGH. If radiation is to be given with doxorubicin, the starting time is delayed for 3 to 4 days after drug administration. The doses used in the treatment of sites other than the extremities, such as the head and neck and the retroperitoneum, depend on the volume of treatment and radiation tolerance of surrounding critical tissues.
– to 3-week rest period before surgical resection. A boost of 15 Gy is given for microscopically positive margins, and 25 Gy is used for gross residual disease, delivered by external-beam radiation, IOERT, or BRT. If administered postoperatively, 50 Gy is given to the large volume, followed by two field reductions to deliver a total dose of 60 Gy for negative margins, 66 Gy for microscopic residual disease, and 75 Gy for gross residual disease. The total dose is reduced by 10% in patients who also receive doxorubicin-based chemotherapy.160 Our treatment policy at UCSF and Stanford is similar to that of the MGH. If radiation is to be given with doxorubicin, the starting time is delayed for 3 to 4 days after drug administration. The doses used in the treatment of sites other than the extremities, such as the head and neck and the retroperitoneum, depend on the volume of treatment and radiation tolerance of surrounding critical tissues.
Brachytherapy
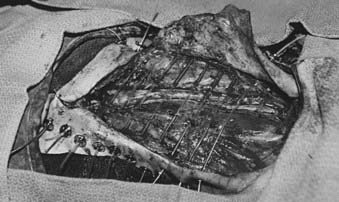
At many institutions, BRT is generally used as a boost or for treatment of recurrent tumors following surgical resection for patients who have failed prior external-beam irradiation. At MSKCC, BRT administered through afterloading catheters can be used either alone as definitive postoperative treatment for high-grade lesions or in combination with external-beam radiotherapy in patients with multiple close or involved surgical margins, suboptimal dosimetry, or gross residual disease. The American Brachytherapy Society has published a comprehensive guideline and recommendations on the use of BRT for soft tissue sarcoma.166 Fig. 59-18 shows catheter placements as recommended by the society. The target area, which includes the tumor bed plus a 2- to 3-cm margin, is defined intraoperatively by the radiation oncologist and the surgeon and should be delineated with radiopaque markers such as surgical clips. Afterloading catheters are placed percutaneously at 1- to 1.5-cm intervals, sutured in the target region to either underlying fascia or muscles, and fixed to skin with buttons and silk sutures (Fig. 59-19). The skin entry point should be at least 1 cm away from the incision with consideration given to the ease of catheter loading. Whenever possible, normal structures at risk such as nerve bundles should be identified and marked with radiopaque markers different from those used to identify the target volume. Efforts should also be made to minimize doses to the nearby critical structures not directly involved with tumor by placing biodegradable spacers such as gelfoam layers between the catheters and these structures. The surgeon then places a drain in the implanted area and closes the skin. Care should be taken to ensure that the incision can be closed without tension to the wound. Plastic or metal wire inserts should be maintained within the catheter lumens until the treatment time to minimize the risk of catheter bending. Intraoperative radiographic localization is necessary if catheter movement after placement is suspected.
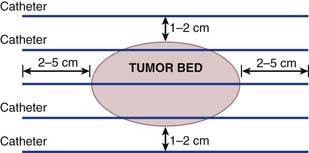
On the first postoperative day, localization films are taken with dummy seeds within the catheters and computerized dosimetry performed (Fig. 59-20). If the orthogonal radiographic technique is used, a third film at a different angle should be taken to verify and improve delineation of source localization. Placement of a skin grid or radiopaque skin mark helps to define the skin dose. Catheters are loaded with iridium-192 (192Ir) between day 5 and day 14 to permit adequate wound healing if BRT is used as adjuvant monotherapy. However, the radioactive source can be loaded sooner (2 to 3 days after surgery) if BRT is used as a boost and the total BRT dose is 20 Gy. Times longer than 7 days can be associated with a higher risk of infection. Approximately 15 to 25 Gy at 40 to 50 cGy/hr can be safely given as a boost treatment. Alternatively, a dose of 45 Gy for 4 to 6 days, which is equivalent to 60 Gy of external-beam radiation, is used for definitive postoperative radiation therapy or for the treatment of recurrent sarcomas. Loading of the catheters can be accomplished using standard iridium seeds, or afterloading iridium with the pulsed Selectron or fractionated high-dose rate (HDR) BRT approach. Table 59-12 shows the BRT modalities considered applicable by the American Brachytherapy Society in various clinical situations.
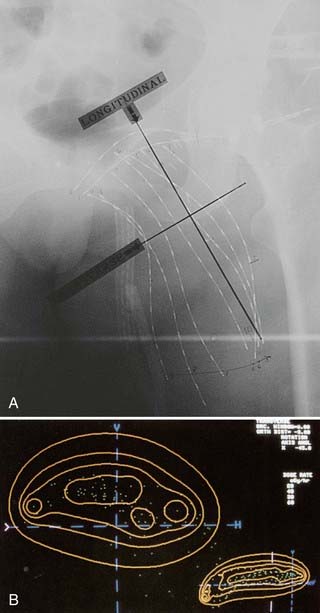
Table 59-12 Treatment Modalities Applicable for Brachytherapy of Soft Tissue Sarcomas in Various Clinical Situations
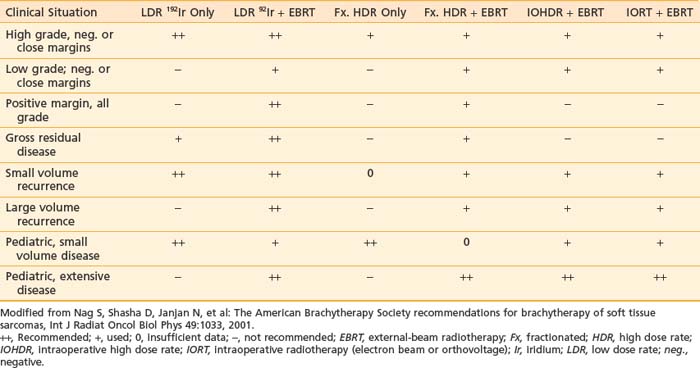
HDR BRT offers a number of theoretic advantages over LDR treatments. In addition to improved radiation protection for involved medical personnel, HDR can also facilitate the optimization of dose distribution. At present, the convenience of short treatment time for HDR is offset by the need for increased fractionation. To achieve clinical results comparable to LDR, most HDR treatments require multiple fractions, each in the range of 4 to 9 Gy.167 At present, there is no consensus on an optimal number of HDR fractions or the total dose.
Intraoperative Electron Radiation Therapy
Similar to IOHDR BRT, IOERT has been used for soft tissue sarcomas of the extremities in conjunction with postoperative radiation therapy. In one of the largest studies, 153 patients were treated at a single center in Heidelberg, Germany, with a median IOERT dose of 15 Gy and mean EBRT dose of 43 Gy (40-50.4 Gy). With a median follow-up of 33 months, the 5-year local control rate was 78%. Extremity salvage until death or time of last follow-up was achieved in 90% of the patients, 86% of whom showed excellent limb function without impairment in activities of daily life.168 In Pamplona, Spain, 45 patients, both primary and recurrent cases, were treated with IOERT and postoperative EBRT (45-50 Gy). Actuarial local control at 5 years was 88% for patients with negative or close margins and 57% for patients with positive margins (P = 0.04). Extremity preservation was achieved in 88% of the patients. The treatment was associated with a 11% neuropathy incidence.169 At UCSF, we have reported on 56 consecutive patients who underwent limb-salvage resection by a single orthopedic oncologist followed by IOERT. The median dose of IOERT was 12.5 Gy (range 10-17.9 Gy). With a median follow-up of 22 months, the 5-year local control rate was 79%. The 2-year local control rate for tumors with positive or close margins (≤1 mm) was 76%, compared with 83% for margins larger than 1 mm. The 2-year local control rate for patients who did not receive adjuvant EBRT was 65%, and 87% for those who did (median dose 56 Gy). The 2-year local control rate was 91% versus 47% for primary versus recurrent tumors (P = 0.01). Patients with primary tumors had a 2-year local control rate of 100% if the margin was larger than 1 mm, and 83% for positive or close margins. Eight patients (14.3%) developed grade 3 wound healing complications; seven of these patients had received adjuvant EBRT.170 At UCSF we use IOERT for extremity soft tissue sarcomas when a close or positive surgical margin is expected. In these cases, 15-Gy IOERT is delivered when major nerves can be excluded from the radiation field, and 12 Gy if not. Patients are routinely treated with postoperative radiation therapy. The dose of postoperative EBRT is 50 Gy for negative margins, 54 to 60 Gy for a positive margin included in the IOERT field, and 60 to 66 Gy for a positive margin in an area not included in the IOERT field.
Wound Complications and Functional Outcome
Wound Complications
Quantitation of the individual effect of surgery, radiation, and chemotherapy on wound healing and limb function is quite difficult. In addition, there is a large variability among sarcoma patients with respect to anatomic site, tumor size, tumor grade, treatment age, premorbid conditions (e.g., diabetes, vascular diseases), and type of treatment. Surgery alone renders a total complication rate of 30% to 35%, of which 3% to 10% are considered severe.171,172 With the addition of external-beam radiation, the overall complication rate ranges between 17% and 37%, and the severe complication rate between 7% and 16.5%.165,173 Significant factors that have been reported to increase wound healing problems include lower extremity lesions, older age group, postoperative boost with interstitial implant, accelerated fractionation,173 large specimen size, the use of preoperative radiation, and history of smoking or vascular disease.174
In the past, the use of BRT was connected to increased wound complication: 44% with interstitial implant versus 14% with surgery alone.172 Further analysis of this problem showed a correlation between early loading time and the high incidence of wound breakdown. Delaying loading of radioactive seeds until after the fifth postoperative day resulted in a significant decrease in the complication rate in the BRT arm: 14% versus 10% for the surgery-alone group.175
Higher major acute wound complication rates have been observed with preoperative radiotherapy as compared with postoperative treatment in a phase III randomized study.176 A similar high wound complication rate was seen in a single institution study, which reported a 31% incidence of major wound complications in 78 patients with extremity soft tissue sarcoma treated with preoperative radiotherapy to 50 Gy.177 Table 59-13 summarizes the incidence of wound morbidity for different treatment modalities.
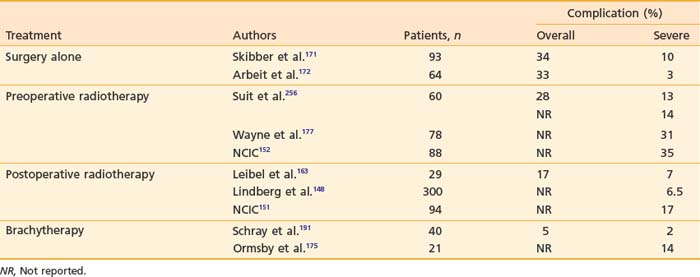
Preoperative chemotherapy does not appear to influence morbidity in patients undergoing radical surgery for soft tissue sarcoma. In a large series of 309 patients, of which 105 received neoadjuvant chemotherapy, there was no increase in the postoperative wound complication rates with neoadjuvant chemotherapy in either extremity (34% versus 41%, P = 0.37) or retroperitoneal or visceral sites (29% versus 34%, P = 0.66), even though patients treated with neoadjuvant chemotherapy had larger and more high-grade tumors.178 Prospective randomized studies are needed to confirm these observations.
Functional Outcome in Extremity Tumors
Commonly reported functional impairments after treatment for extremity soft tissue sarcoma are pain, loss of range of motion, muscle weakness, edema, and tissue fibrosis.179 Pain was noted in 10% to 50% of the cases with less than 10% of patients requiring pain medication on a regular basis. Joint motion restriction was noted in 33% to 50% of the patients, muscle weakness in 33%, moderate to severe edema in 25%, and soft tissue fibrosis in 50% to 60%.179 Radiation parameters that can increase post-treatment impairment include inclusion of the joint in radiation field (predictive of joint contracture), dose greater than 63 Gy at 1.8 Gy per fraction (predictive for pain, edema, muscle weakness), field greater than 35 cm, and lower extremity location (predictive for edema, muscle weakness).180 Other factors that have been shown to influence functional outcomes are tumor size, motor nerve sacrifice,181 the use of adjuvant radiotherapy, and an older age group.182 Finally, the IORT volume in patients treated with IORT and postoperative irradiation for extremity soft tissue sarcoma is associated with the risk of soft tissue fibrosis. The risk is 5% for IORT volumes 210 cm3, whereas it approaches 50% for volume of 420 cm3 or larger.183 Further details on the functional outcome after treatment of extremity soft tissue sarcoma can be found in an excellent review written by Davis for Seminars in Radiation Oncology.179
Treatment Results
Surgery Alone
Several studies have suggested that surgery alone can result in excellent local control and survival in highly selected patients. Fabrizio et al.155 reported no local failure or distant relapse and a 96% survival rate in 18 patients with low-grade tumors treated with surgery alone after a median follow-up of 55 months. Geer et al.157 reviewed 117 cases with T1 tumors treated with surgery alone and reported a local control rate of 92%. Investigators from the University of Chicago used a wide-margin excision approach in 62 patients with subcutaneous sarcomas of the extremity.184 The surgeons employed wide radial margins and resection of unviolated fascia. There were no local recurrences in the group of patients treated with surgery only. Alektiar et al.164 reviewed the records of 204 patients treated at MSKCC for stage IIB (high-grade histology and tumor size 5 cm) soft tissue sarcoma of the extremity. All had negative surgical margins and 43% received adjuvant radiotherapy (BRT in 26% and external-beam irradiation in 17%); 17% also had adjuvant chemotherapy. The patients who did or did not receive radiation were well balanced with regard to age, tumor size, and location, with the exception of tumor depth with more deep tumors in the radiotherapy group. There was no difference in 5-year local control rates (84% versus 80%) with the addition of radiotherapy. These data suggest that it is appropriate to consider withholding radiation in a select group of patients with low-grade tumors, widely negative surgical margins, and in whom a function-preserving surgical salvage is possible in the event of a local relapse. The role of radiotherapy in small, high-grade tumor is controversial; the findings from MSKCC are provocative and need confirmation in prospective randomized trials.
Preoperative Radiation
Many institutions advocate preoperative radiation treatment. Theoretic advantages of this approach are listed in Table 59-9. Nielsen et al.185 provided the first quantitative evidence that treatment field sizes can be smaller when radiation is given preoperatively. They resimulated 26 patients who had received preoperative radiation treatments in the postoperative settings. Preoperatively, margins of 5 cm around the tumor were used for grade 1 to 2 disease and 7 cm for grade 3 sarcomas. The same margins were employed postoperatively, but around the surgical bed. The average preoperative field size was significantly smaller than that of the postoperative setting: 241 cm2 versus 391 cm2 (P < 0.001). Furthermore, more joints were included in the postoperative treatment volumes.
The outcome of preoperative radiation has been impressive. Barkley et al.186 reported on 110 patients treated with preoperative radiation followed by wide-local excision at the MDAH between 1970 and 1984. Of the lesions treated, 89% were greater than 5 cm. With the minimal follow-up of 17 months, the local control and disease-free survival rates were 90% and 61%, respectively. Suit found a local control advantage for preoperative over postoperative radiation treatment, especially for larger lesions (Table 59-14).122 The local control rates for lesions greater than 15 cm were 56% for postoperative radiation and 86% for preoperative radiation. The conclusions from these studies can be subject to biases caused by the retrospective nature of the studies. The NCIC conducted a randomized study comparing preoperative and postoperative radiotherapy for soft tissue sarcoma, with wound complication, local control, and overall survival rates as major endpoints. In addition, quality of physical function, economic cost, radiation planning parameters, and treatment-related toxicity were evaluated. The study was closed when an interim analysis showed a higher acute wound complication rate with preoperative radiotherapy. With a median follow-up of more than 3.3 years, there was no difference in either the local control rates and in functional scores between the two treatment arms, but an unexplained survival advantage with preoperative radiotherapy.149
Table 59-14 Five-Year Actuarial Local Control for Primary Soft Tissue Sarcoma According to Size and Radiotherapy Sequencing: The MGH Experience
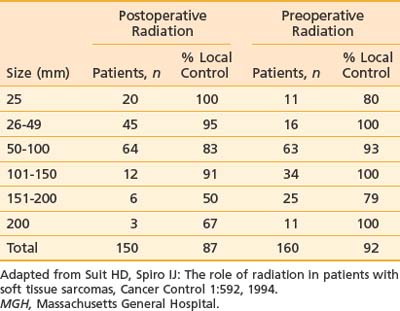
A number of investigators have studied preoperative chemotherapy either sequentially or in combination with preoperative radiotherapy. Based on promising results from single institution reports, the Radiation Therapy Oncology Group conducted an intergroup phase II trial of multiagent chemotherapy (a modified regimen with mesna, doxorubicin [Adriamycin], ifosfamide, and dacarbazine) delivered concurrently with preoperative radiotherapy (44 Gy given in split courses of 22 Gy) followed by resection in patients with high-risk, soft tissue sarcoma (grade ≥II and ≥8 cm in maximal diameter).187 This is followed by three cycles of postoperative chemotherapy with granulocyte colony-stimulating factor. Of these patients, 88% completed preoperative chemotherapy and 93% completed radiation therapy. Delayed wound healing was observed in 26% of the patients. Of 45 patients with at least 1 year of follow-up, the estimated 2-year survival was 95%, which was encouraging. Edmonson et al.188 treated 49 patients with two cycles of ifosfamide, mesna, mitomycin, doxorubicin, and cisplatin followed by reduced-dose ifosfamide, doxorubicin, and cisplatin chemotherapy concurrently with radiotherapy to 45 Gy, followed by limb-sparing surgery. An additional 10- to 20-Gy boost was delivered to the tumor bed via IORT, BRT, or external-beam radiotherapy at the completion of surgery. All but five patients had tumors greater than 5 cm. The estimated 5-year survival was 80%.
Eilber and colleagues have employed combined preoperative doxorubicin-based chemotherapy and radiation since 1974 at UCLA for the treatment of soft tissue and bone sarcomas.189 A list of the trials, the time frame, and the results for grade 3 lesions are shown in Table 59-15. In the initial trial, patients were treated with intra-arterial doxorubicin (30 mg/day × 3 days), followed by 35 Gy given in 10 fractions and conservative surgical resection. To reduce the 25% complication rate, a second trial, in which patients were given identical chemotherapy and surgery, but with a reduced radiation dose (17.5 Gy in five fractions), was opened. In the third trial, the radiation dose was raised to 28 Gy in eight fractions because of poor local control in the second study. At the same time, this protocol compared the efficacy of intravenous (IV) versus intra-arterial doxorubicin. Because there was no difference in the complication or local control rates with the route of administration, a fourth trial evaluating the efficacy of IV doxorubicin, IV cisplatin, and 28-Gy radiation was started. The results were comparable to that of IV doxorubicin alone. The final protocol involved two courses of high-dose ifosfamide followed by 28-Gy radiation, doxorubicin, cisplatin, and conservative surgery. The last regimen improved the local control rate to 95%. Treatment-induced pathologic necrosis, as determined from the tumor specimen after preoperative therapy, appeared to be an independent predictor for both local recurrence and overall survival in this group of 496 patients.117 Patients with less than 95% pathologic necrosis were 2.5 times more likely to develop local recurrence and 1.9 times more likely to die of their disease as compared with those with 95% or greater pathologic necrosis or a complete response.
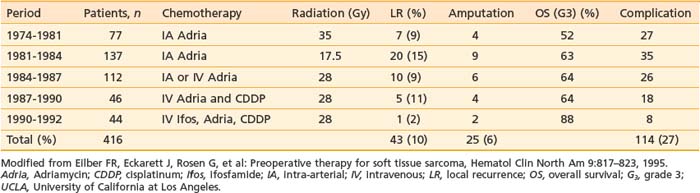
Postoperative Radiation
The largest reported series on postoperative radiation treatment came from the MDAH.148 Three hundred patients with soft tissue tumors of the extremities, head and neck, and the retroperitoneum were treated with conservative excision, followed by radiation to a total dose of 60 to 75 Gy in 6 to 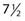 weeks. No attempts were made to cover the entire muscle or anatomic compartment. The local recurrence rate was 20% for extremity, 38% for abdominal, and 23% for head and neck sarcomas. One-third of the extremity failures occurred beyond margins of the irradiated field, and local control did not change with a 10-Gy dose reduction in the later period. The 5-year disease-free survival was 61% for all sites.
weeks. No attempts were made to cover the entire muscle or anatomic compartment. The local recurrence rate was 20% for extremity, 38% for abdominal, and 23% for head and neck sarcomas. One-third of the extremity failures occurred beyond margins of the irradiated field, and local control did not change with a 10-Gy dose reduction in the later period. The 5-year disease-free survival was 61% for all sites.
At UCSF, 29 patients with extremity sarcomas were treated with conservative surgery and postoperative radiotherapy.163 The majority of cases had grade 3 or larger than 5-cm tumors. The radiation dose ranged from 50 to 75 Gy. Of these patients, 82% received more than 55 Gy. The local control rate with surgery and postoperative radiation was 90%, with a 5-year relapse-free survival of 68%. These results were superior to excisional surgery alone and comparable to radical surgery on a stage-by-stage basis. This study also demonstrated the ability of postoperative radiotherapy to sterilize microscopic disease. In the simple excision with positive margin group, the local recurrence rates were 14% (3 of 22) for patients receiving postoperative radiation versus 79% (11 of 14) for those with surgery alone. At the MGH, Suit and Spiro160 treated 150 patients with postoperative irradiation. They achieved a local control rate of 87%. The results of these three series are summarized in Table 59-16.
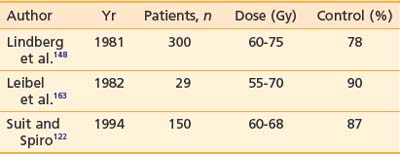
Preoperative versus Postoperative Radiation Therapy
The NCIC randomly allocated 94 patients to preoperative radiotherapy (50 Gy in 25 fractions) and 96 to postoperative radiotherapy (66 Gy in 33 fractions). Median follow-up was 3.3 years. Wound complications were recorded in 35% in the preoperative group and 17% in the postoperative group (P = 0.01). There was no difference in local control.151 In an update in 2005, 129 patients were evaluated; 48% in the postoperative arm compared with 31.5% in the preoperative arm had grade 2 or greater fibrosis (P = 0.07). Although not statistically significant, edema was more frequent in the postoperative arm at 23% versus 15.5% in the preoperative arm, as was joint stiffness, which found 23% in the postoperative arm versus 18% in the preoperative arm.152
Brachytherapy
Theoretic benefits for BRT include shorter treatment duration, relative sparing of normal tissues, and earlier delivery of radiation before fibrosis and hypoxia set in. Harrison and colleagues conducted a prospective randomized trial to evaluate the benefit of adjuvant BRT.125 One hundred and twenty-six patients with soft tissue sarcomas of the extremity and the superficial trunk underwent gross complete resection with limb-sparing procedures. During the resection, they were randomized to either observation or adjuvant BRT to a total dose of 42 to 45 Gy for 4 to 6 days via 192Ir afterloading implants. The target area was defined by adding a 2-cm margin in the cephalocaudal direction, and a 1.5- to 2-cm margin in the mediolateral direction around the tumor bed. No attempt was made to treat the surgical scar or drain site. With a median follow-up of 76 months, there was a significant improvement in local control in the high-grade tumor group with BRT; the 5-year local control rates were 82% and 69% in the BRT and no-BRT groups, respectively (P = 0.04).190 However, there was no difference in disease-specific survival or the rate of distant metastasis between the two groups. There was also no distinction in local control between the two arms for low-grade lesions.
Schray et al.191 evaluated the role of BRT as a boost to the tumor bed in addition to external-beam radiotherapy for nonretroperitoneal lesions treated at the Mayo Clinic. A dose of 15 to 20 Gy via 192Ir afterloading catheters was combined with 45 to 50 Gy of external-beam radiotherapy. With a median follow-up of 20 months, there were five local failures: one in the implant volume, two in the external-beam volume, and two outside the radiation field. Delannes et al.192 reported a 5-year local control rate of 89% in 58 patients treated with external-beam irradiation to 45 Gy with a 20 Gy LDR BRT boost for predominantly large tumors. Zelefsky and colleagues employed BRT and conservative resection at MSKCC for the treatment of 45 locally advanced tumors involving the neurovascular bundles.193 This was a group of poor-risk patients; more than 60% had high-grade lesions, positive margins, or tumor larger than 10 cm. In many cases, 192Ir afterloading catheters were placed directly over the neurovascular bundles. An average dose of 44 Gy for 5 days was administered, and a number of patients (21 of 45) also received additional external-beam irradiation. The 5-year actuarial local control and disease-free survival rates were 70% and 66%, respectively. After salvage treatments, 84% maintained functional extremities. Interestingly, they reported only a 9% incidence of neuritis, occurring in those who received a cumulative dose exceeding 90 Gy to the neurovascular bundles.
BRT has been advocated for retreatment of recurrent tumors after previous external-beam radiation. Nori et al.194 reported their experience with 40 patients treated with 45 Gy via 192Ir afterloading catheters for recurrent extremity sarcomas. Half had evidence of bone, joint, or neurovascular bundle involvement. The 5-year actuarial local control rate with BRT and conservative resection was 68%. All local failures occurred in patients with more than two prior recurrences.
The use of fractionated HDR as a boost has also been popularized by a number of institutions.195–197 Preliminary results suggested that local control and acute complications appear to be comparable to LDR BRT. Unfortunately, most series are small and the dose-fractionation regimens are not uniform. In addition, the follow-up period is too short to make any definitive statement on treatment results or complication rates with fractionated HDR.
Chemotherapy
Approximately 50% of patients with high-grade sarcomas die of distant disease despite adequate local control. To improve this rate, there have been intensive efforts in the last 3 decades to identify adjuvant systemic agents effective in preventing tumor dissemination. The most active single chemotherapeutic agents include ifosfamide and doxorubicin with single-agent response rates of 15% to 30%.198–200 The response rates with combination therapy are higher than single-agent doxorubicin; however, the overall survival remains the same. Cyclophosphamide, dactinomycin, dacarbazine (DTIC), methotrexate, and cisplatin are also effective against soft tissue sarcoma. These drugs have been used in combination with doxorubicin for metastatic disease as well as in the adjuvant setting for localized sarcomas. Temozolomide,201 gemcitabine,202 navelbine,203 and trabectedin (ET-743, Yondelis)—a cytotoxic alkaloid derived from a marine organism204—have also shown promising activity against soft tissue sarcoma.
A number of prospective, randomized studies have reported on the role of adjuvant chemotherapy for M0 soft tissue sarcomas. Five compared high-dose doxorubicin to untreated surgical controls (Table 59-17).205–209 Most of these studies did not have enough power to detect small differences in results, and many are open to criticism on methodologic grounds. Antman et al.206 reported on 42 patients with localized large (>5 cm) intermediate and high-grade sarcomas of all sites treated in an intergroup trial. After optimal local therapy, patients were randomized to either adjuvant doxorubicin (90 mg/m2 × 5 days) or observation. With the median follow-up of 44 months, no difference in survival was observed between the two arms (see Table 59-17); however, patients receiving chemotherapy had a delay in metastatic recurrence.
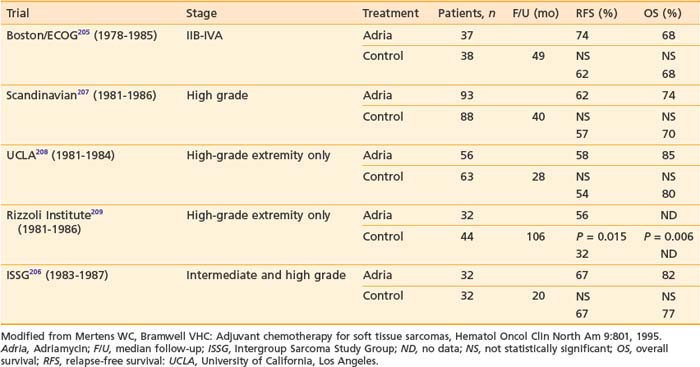
Table 59-18 summarizes the results of six randomized studies using combination chemotherapy.210–215 The MDAH trial results were initially reviewed with a median follow-up of 18 months, unacceptable toxicity and a lower disease-free survival rate for the experimental arm led to its early termination. However, after 10 years of follow-up, a significantly larger number of patients in the chemotherapy group were still alive and disease free compared with the controls.210 The initial poor results could be accounted for by an imbalance in the treatment arms, particularly for tumor grades. At the NCI, 67 patients were randomized to either adjuvant chemotherapy or no further therapy after local treatments.212 Chemotherapy consisted of high-dose doxorubicin, cytoxan, and methotrexate. There was a superior disease-free survival (75% versus 54%, P = 0.04) and local control (one local failure versus four local failures, P < 0.05) in the chemotherapy arm. Toxicity was significant, however; 15% of the patients developed drug-induced congestive heart failure. In a subsequent study, 88 patients were randomized to receive either the same chemotherapy regimen (total doxorubicin dose: 500 to 550 mg/m2) or a lower dose regimen of doxorubicin (350 mg/m2). There was no difference in relapse-free survival and overall survival between the two arms. The largest published trial of adjuvant chemotherapy came from the European Organization for Research and Treatment of Cancer (EORTC).211 Three hundred and seventeen patients were randomized to adjuvant cytophosphamide, vincristine, doxorubicin, and DTIC (CYVADIC) chemotherapy or no chemotherapy. With a median follow-up of 80 months, the 7-year relapse-free survival was higher (56% versus 43%, P = 0.007) and the local failure was lower (17% versus 31%, P = 0.004) in the chemotherapy arm. The reduction in local recurrence was limited to tumors of the head, neck, and trunk. Criticisms of this study included a high ineligibility rate (33%) and a poor compliance rate (only 52% completed the planned eight courses of chemotherapy). Accrual spanned 11.5 years, during which new diagnostic and therapeutic techniques were developed. A study that showed an advantage in survival came from Fondation Bergonie in France.216 The chemotherapy used was identical to, but in a more intense regimen than, that of the EORTC. With a median follow-up of 4.4 years, there was a significant improvement in the chemotherapy arm with respect to local control (97% versus 85%, P = 0.03), freedom from metastases (84% versus 43%, P = 0.0003), and overall survival (87% versus 54%, P = 0.002). The study, however, failed to stratify patients according to known prognostic factors, resulting in a high proportion of patients with extremity sarcomas (a better prognostic group) in the treatment arm. Frustaci et al.217 conducted a phase III randomized study of 104 patients with T2 and grade 3 to 4 soft tissue sarcoma treated with either five cycles of high-dose epirubicin (60 mg/m2 on days 1 and 2) and ifosfamide (1.8 mg/m2 on days 1 through 5) or no adjuvant chemotherapy. There were three different primary treatment strategies (radical surgery versus surgery and postoperative EBRT versus preoperative EBRT and surgery) performed in at least six different treatment centers. Although the patients appear to be well-balanced between the two groups, there were larger tumors in the control arm and more patients in the control arm required amputation, suggesting worse tumor biologic features. The trial was terminated early when an interim analysis revealed a significant disease-free and overall survival advantage with the addition of chemotherapy. With a median follow-up of 59 months, the median disease-free and overall survivals were 16 months and 46 months, respectively, for the control group and they were 48 months (P = 0.04) and 75 months for the treatment group (P = 0.03). The absolute survival benefit was 19% at 4 years with the addition of chemotherapy. These results have been updated with a median follow-up of 89.6 months.217 The 5-year overall survival in the chemotherapy arm was 66% versus 46% in the observation arm (P = 0.04).
Table 59-18 Adjuvant Chemotherapy: Summary of Randomized Trials of Combination Chemotherapy versus Control
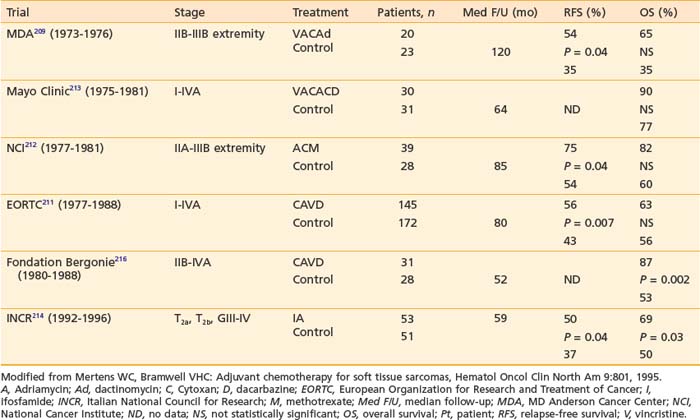
In an attempt to overcome the sample size limitations observed in the randomized studies, the Sarcoma Meta-Analysis Collaboration performed an individual data meta-analysis of the outcome for 1568 patients included in 14 randomized trials. The group compared adjuvant doxorubicin-based chemotherapy with no chemotherapy.218 Significant improvements in local relapse-free interval (HR 0.73, P = 0.016), distant relapse-free interval (HR 0.70, P = 0.0003) and overall relapse-free interval (HR 0.75, P = 0.0001) were observed with adjuvant chemotherapy. In addition, there was a trend toward improved overall survival with an absolute survival benefit of 4% at 10 years (HR 0.89, P = 0.12). However, there was a significant 7% absolute survival benefit at 10 years (HR 0.80, P = 0.029) for soft tissue sarcoma of the extremity with the addition of chemotherapy. The treatment could delay distant recurrence and possibly prolong survival.
An updated meta-analysis has been reported by Pervaiz et al.219 with 18 randomized adjuvant trials (n = 1953). The odds ratio for distant and overall recurrence was 0.67, in favor of chemotherapy (95% CI 0.56-0.82; P = 0.0001). The odds ratio was 0.56 for doxorubicin and ifosfamide combination therapies (95% CI, 0.36-0.85; P = 0.01), again in favor for chemotherapy. These results were not seen in single-agent doxorubicin trials. This meta-analysis did not include the recently reported EORTC 62931 trial.220 This study randomized high-grade sarcomas to chemotherapy with doxorubicin 75 mg/m2 in combination with ifosfamide 5 g/m2 every 3 weeks for 5 cycles versus observation. Patients had radiation therapy after completion of chemotherapy, if indicated. A total of 351 patients were randomized, with 81% of patients with extremity or limb girdle sarcomas. The 5-year relapse-free survival was 51% versus 53% in favor of observation (P = 0.497). The 5-year overall survival was 65% for chemotherapy versus 69% for the observation arm (P = 0.935). This study is criticized for the lower dose of ifosfamide compared with the Frustaci trial and for delay in radiation therapy. Because of these conflicting results, additional well-conducted, carefully planned, multi-institutional prospective randomized trials will be needed before adjuvant chemotherapy can be considered the standard of care. It is reasonable to discuss the option of adjuvant systemic chemotherapy with patients younger than 65 who have high-grade, large soft tissue sarcomas.
Surgical Management of Isolated Pulmonary Metastases
The most frequent site of distant metastasis in soft tissue sarcomas is the lung. Despite a lack of randomized data on the use of neoadjuvant or adjuvant chemotherapy in the treatment of isolated pulmonary metastases, the most common approach today is systemic chemotherapy followed by surgical resection when the disease has stabilized for large lesions, or surgery followed by chemotherapy for smaller nodules. Candidates for pulmonary metastasectomy are patients who have (1) control of the primary tumor and no evidence of extrapulmonary metastases, (2) resectable lesions, (3) four or fewer nodules on imaging, (4) tumor doubling time of more than 40 days, and (5) a disease-free interval of more than 12 months. Numerous studies have shown that these patients have benefitted from surgical resection.221,222 The 5-year survival for patients with complete resection is approximately 25% (range: 15% to 35%).223 When surgery is not possible, stereotactic body radiosurgery has been used with high control of treated lesions, ranging from 80% to 95%.224
Some Special Sarcoma Subgroups
Retroperitoneal Sarcomas
These rare tumors account for less than 15% of all adult soft tissue sarcoma. Most retroperitoneal sarcomas are large (>10 cm) and locally infiltrative at diagnosis, making resection quite difficult. In a large series from MSKCC, only 49% of 158 lesions were amenable to complete resection.225 The 5-year overall survival was 40% for patients with gross-total resection, and only 3% for those with incomplete tumor removal. Inability to obtain wide resection, high-grade lesions, and local recurrence negatively affected survival. There was a suggestion that adjuvant radiation improved 5-year survival (53% with radiation versus 30% without); however, the gain was not statistically significant. The NCI prospectively evaluated the role of adjuvant radiation and chemotherapy to improve outcome.226 They found no survival advantage with adjuvant chemotherapy. There was only one (3%) in-field failure with external-beam radiation; however, the complication rate with this approach was high, with 22% of patients developing signs and symptoms of late radiation enteritis. To reduce bowel injury, a prospective randomized trial was conducted comparing IORT plus low-dose external-beam irradiation versus high-dose external-beam irradiation alone for resectable cases.227 Fifteen patients received 20-Gy IORT followed by external-beam irradiation to 35 to 40 Gy postoperatively. Twenty had external-beam irradiation alone to 50 to 55 Gy. In an updated analysis, the IORT arm had fewer local regional failures (40% versus 80%, P = 0.08) and gastrointestinal complications (13% versus 50%, P < 0.05) when compared with the control arm.228 Peripheral neuropathy was higher with the use of IORT at 60% versus 5%. To reduce the potential neurotoxicity, MSKCC, UCSF, and others have reduced the IORT dose in the range of 12 to 15 Gy. This is typically given with postoperative radiation therapy to doses of 45 to 50 Gy. Using the technique of HDR-IORT, Alektiar et al.229 treated 32 patients (12 with primary and 20 with recurrent tumors) with IOHD. The 5-year local control rate was 62% for the entire group, 74% for patients with primary tumors, and 54% for patients with recurrent tumors. Treatment complications included gastrointestinal obstruction (18%), fistula formation (9%), peripheral neuropathy (6%), hydronephrosis (3%), and wound complication (3%).
Gieschen et al.230 retrospectively compared 17 patients treated with preoperative external-beam irradiation followed by surgery and IORT to the same number treated with external-beam irradiation and surgery alone. In patients who had gross total resections, the local control (83% versus 61%) and overall survival rates (74% versus 30%) were superior with the addition of IORT. Petersen et al.231 reported on 43 primary and 44 recurrent retroperitoneal sarcomas treated with 8.75 to 30 Gy IORT in addition to external-beam irradiation (median dose 48.6 Gy) after partial or complete tumor resection. There were 20 local failures: four within the IORT fields, three within IORT and external-beam fields, and 13 within external-beam fields alone. Ten patients had gastrointestinal complications (seven fistulae and three severe proctitis) related to surgery, external-beam irradiation, or both. Nine patients (10%) developed grade 3 peripheral neuropathy, but none had pain as a component of their neuropathy. In view of current results, IORT in conjunction with external-beam radiotherapy is recommended for the treatment of retroperitoneal sarcomas.
Head and Neck Soft Tissue Sarcomas
Soft tissue sarcomas of the head and neck areas rarely occur in adults. They account for less than 1% of all malignant neoplasms and approximately 10% of all soft tissue tumors.232 Historically, they have been associated with poorer outcome when compared with sarcomas from other anatomic sites.95,120 Reported 5-year, disease-specific survival and local-control rates ranged from 45% to 70%.233–236 Tumor size, tumor grade, and the extent of surgical resection are important prognostic factors for survival and local control.235,237–239 Age, tumor site, histologic features, and bony involvement have also been suggested to affect tumor control; however, their significance independent of other prognostic variables has not been firmly established. In reviewing 65 patients treated at UCSF for head and neck sarcomas, we found age, tumor grade, surgical margin status, and the extent of surgical resection to significantly influence disease-specific survival.239 High tumor grade and large size also had an adverse effect on local control. The use of adjuvant radiation appeared to decrease local failure; however, it did not reach statistical significance because of the small sample size.
Technically, this is a challenging group of tumors to manage because of the difficulty in achieving a complete resection in this region without causing significant cosmetic impairment or functional morbidity. High local recurrence rates (up to 50%) have been reported with surgery alone.239,240 Adjuvant radiotherapy, therefore, plays an essential part in the management of these sarcomas, particularly in cases of marginal surgical excision. Tran et al.234 reported a notable improvement in local control with radiation for 94 patients treated at UCLA. Local failure was observed in 48% of patients treated with surgery alone compared with 10% of those treated with surgery and radiotherapy. Eeles and colleagues also found the use of adjuvant radiation to be superior to surgery alone.242 The role of chemotherapy in the treatment of head and neck soft tissue sarcoma remains undefined. The EORTC reported an improvement in local control with the addition of CYVADIC chemotherapy.240 McKenna et al.243 treated 16 patients with aggressive surgery, high-dose postoperative radiation (60 to 63 Gy) and doxorubicin-based drugs. With the median follow-up of 43 months, 64% were disease free, and 75% maintained local control. These encouraging results, however, have not been duplicated in other series.236,242
Fibromatosis (Desmoid Tumors)
Desmoid tumors—also known as aggressive fibromatoses—are rare, slow–growing, benign fibrous neoplasms that arise from fascial or musculoaponeurotic structures. They are commonly associated with Gardner syndrome. Although first described as tumors of the anterior abdominal wall in postpartum women, they may arise from any anatomic location, most commonly the shoulder girdle and thigh areas.244 Histologically, they appear as well-differentiated fibrous neoplasms without associated cellular pleomorphism or mitotic activity (Fig. 59-21). These tumors rarely metastasize. Their natural history consists of slow, progressive invasion and destruction of contiguous structures by fingerlike projections. This infiltrative behavior results in a high incidence of local recurrence (occasionally fatal) after conservative surgery.241,244 Posner and colleagues described 11 deaths from locally uncontrolled tumors of 138 patients treated at MSKCC.245 Factors predictive of local failure were age between 18 and 30, recurrent disease, incomplete resection, close or microscopically positive margins, and no adjuvant radiation for gross residual disease.
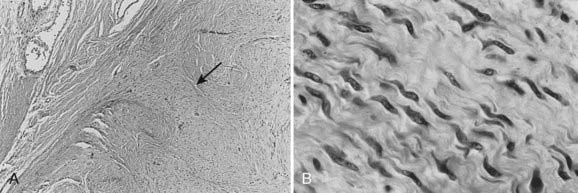
The traditional treatment of desmoid tumors is aggressive surgical resection in an effort to obtain a wide margin. However, radiation alone has been shown effective for the control of unresectable or recurrent lesions. Leibel et al.246 reviewed 19 cases of recurrent or residual tumors treated with radiation at UCSF. At the time of treatment, eight patients had tumors larger than 10 cm, five with residuals between 4 and 6 cm. The majority received a tumor dose of 50 to 55 Gy at 1.6 to 1.8 Gy per fraction. With a median follow-up of 8 years, 13 patients remained free of disease. The 5-year relapse-free survival and overall survival rates were 73% and 88%, respectively. Complete tumor regression after radiation required 8 months to 2 years. There was only one true in-field recurrence. Other centers have reported comparable results.247,248 Adjuvant radiotherapy has been shown to improve local control, especially in patients with recurrent or involved surgical margins. Jelinek et al.249 showed that adjuvant radiotherapy was the only significant factor affecting local control in 54 patients treated for desmoid tumors at the University of Washington Medical Center. The 5-year local control rate was 81% for patients receiving adjuvant radiotherapy compared with 53% for those treated with surgery alone. Nuyttens et al.250 reviewed the literature and concluded that the best local control rates were observed in patients treated with surgery and radiotherapy compared with surgery alone even after stratifying patients by surgical margin status and by primary versus recurrent tumors.
1 Bloem JL, Kroon HM. Imaging of bone and soft tissue tumors: osseous lesions. Radiol Clin North Am. 1993;31:261.
2 Giudici MA, Moser RP, Kransdorf MJ. Imaging of bone and soft tissue tumors: Cartilaginous bone tumors. Radiol Clin North Am. 1993;31:237.
3 Manaster BJ, Doyle AJ. Imaging of bone and soft tissue tumors: giant cell tumors of bone. Radiol Clin North Am. 1993;31:299.
4 Hutter RVP, Worchester JNJr, Francis KC, et al. Benign and malignant giant cell tumors of bone: a clinicopathological analysis of the natural history of the disease. Cancer. 1962;15:653.
5 Catto ME. Diseases of bone. In: Anderson JR, editor. Muir’s textbook of pathology. ed 12. Baltimore: Edward Arnold; 1985:1.
6 Dahlin DC, Cupps RE, Johnson EWJr. Giant cell tumor: a study of 195 cases. Radiology. 1986;161:537.
7 Bennett CJJr, Marcus RBJr, Million RR, et al. Radiation therapy for giant cell tumor of bone. Int J Radiat Oncol Biol Phys. 1993;26:299.
8 Chen ZX, Gu DZ, Yu ZH, et al. Radiation therapy of giant cell tumor of bone: analysis of 35 patients. Int J Radiat Oncol Biol Phys. 1986;12:329.
9 Huvos AG. Chordoma. In Bone tumors: diagnosis, treatment and prognosis, ed 2, Philadelphia: WB Saunders; 1991:599.
10 Congdon CC. Proliferative lesions resembling chordoma following puncture of nucleus pulposus in rabbits. J Natl Cancer Inst. 1952;12:892.
11 Krayenbuhl H, Yasargil MG. Cranial chordomas. Prog Neurol Surg. 1975;6:380.
12 Stanfill P, Pratt C. Bone tumors. In: Hockenberry MJ, Coody DK, editors. Pediatric oncology and hematology: perspectives on care. St Louis: Mosby; 1987:104.
13 Huvos AG. Osteogenic sarcoma. In Bone tumors: diagnosis, treatment and prognosis, ed 2, Philadelphia: WB Saunders; 1991:85.
14 Healey JH, Buss D. Radiation and pagetic osteogenic sarcomas. Clin Orthop. 1991;270:128.
15 Frassica FJ, Sim FH, Frassica DA, et al. Survival and management considerations in postirradiation osteosarcoma and Paget’s osteosarcoma. Clin Orthop. 1991;270:120.
16 Mays CW, Spiess H, Chmelevsky D. Cancers in patients injected with 224 Ra (meeting abstract). Health Phys. 1989;56(suppl 1):S44.
17 Glicksman AS, Fabrikant J. Analysis of post-radiation sarcomas: correlation of clinical data with experimental results (meeting abstract). Proc Ann Meet Am Assoc Cancer Res. 1987;28:88.
18 Dahlin DC, Coventry MB. Osteogenic sarcoma: a study of six hundred cases. J Bone Joint Surg (Am). 1967;49:101.
19 Lindbom A, Soderberg G, Spjut HJ. Osteosarcoma: a review of 96 cases. Acta Radiol. 1961;56:1.
20 Marcove RC, Mike V, Hajek JV, et al. Osteogenic sarcoma under the age of twenty-one: a review of 145 operative cases. J Bone Joint Surg (Am). 1970;52:411.
21 Weinfeld MS, Dudley HR. Osteogenic sarcoma: a follow-up study of the 94 cases observed at the Massachusetts General Hospital from 1920 to 1960. J Bone Joint Surg (Am). 1962;44:269.
22 Cade S. Osteogenic sarcoma: a study based on 133 patients. J R Coll Surg Edinb. 1955;1:79.
23 Allen CV, Stevens KR. Preoperative irradiation for osteogenic sarcoma. Cancer. 1973;31:1364.
24 Rosen G, Tan C, Sanmaneechai A, et al. The rationale for multiple drug chemotherapy in the treatment of osteogenic sarcoma. Cancer. 1975;35:936.
25 Rosen G, Tefft M, Martinez A, et al. Combination chemotherapy and radiation therapy in the treatment of metastatic osteogenic sarcoma. Cancer. 1975;35:622.
26 Rosen G, Caparros B, Huvos AG, et al. Preoperative chemotherapy for osteogenic sarcoma: selection of postoperative adjuvant chemotherapy based on the response of the primary tumor to preoperative chemotherapy. Cancer. 1982;49:1221.
27 Rosen G, Marcove RC, Caparros B, et al. Primary osteogenic sarcoma: the rationale for preoperative chemotherapy and delayed surgery. Cancer. 1979;43:2163.
28 Craft AW. Management of osteogenic sarcoma (abstract). Br J Radiol. 1988;61:730.
29 Meskens M, Burssens A, Hoogmartens M, et al. Osteogenic sarcoma in children: a retrospective study of 58 cases. Acta Orthop Belg. 1993;59:64.
30 Ozaki T, Flege S, Kevric M, et al. Osteosarcoma of the pelvis: experience of the cooperative osteosarcoma study group. J Clin Oncol. 2003;21:334.
31 Fahey M, Spanier SS, Vander Griend RA. Osteosarcoma of the pelvis: a clinical and histopathological study of twenty-five patients. J Bone Joint Surg (Am). 1992;74:321.
32 Monticelli G, Santori FS, Ghera S, et al. Surgical problems in the treatment of pelvic tumors. Monogr Ser Eur Org Res Treat Cancer. 1986;16:101.
33 Calvo FA, Ortiz de Urbina D, Sierrasesumaga L, et al. Intraoperative radiotherapy in the multidisciplinary treatment of bone sarcomas in children and adolescents. Med Pediatr Oncol. 1991;19:478.
34 Stea B, Cavazzana A, Kinsella TJ. Small-cell osteosarcoma: correlation of in vitro and clinical radiation response. Int J Radiat Oncol Biol Phys. 1988;15:1233.
35 Shives TC, Dahlin DC, Sim FH, et al. Osteosarcoma of the spine. J Bone Joint Surg (Am). 1986;68:660.
36 Sundaresan N, Rosen G, Huvos AG, et al. Combined treatment of osteosarcoma of the spine. Neurosurgery. 1988;23:714.
37 Machak GN, Tkachev SI, Solovyev YN, et al. Neoadjuvant chemotherapy and local radiotherapy for high grade osteosarcoma of the extremities. Mayo Clin Proc. 2003;78:147.
38 Laskar S, Basu A, Muckaden MA, et al. Osteosarcoma of the head and neck region: lessons learned from a single-institution experience of 50 patients. Head Neck. 2008;30(8):1020-1026.
39 Guadagnolo BA, Zagars GK, Raymond AK, et al. Osteosarcoma of the jaw/craniofacial region: outcomes after multimodality treatment. Cancer. 2009;115(14):3262-3270.
40 Gandia D, Domenge C, Schwaab G, et al. Radiotherapy followed by high-dose ifosfamide (HDI) in osteosarcoma of the facial bones (meeting abstract). Ann Oncol. 1992;3(suppl 5):169.
41 Suit HD. Role of therapeutic radiology in cancer of bone. Cancer. 1975;35:930.
42 Giuffrida AY, Burgueno JE, Koniaris LG, et al. Chondrosarcoma in the United States (1973 to 2003): an analysis of 2890 cases from the SEER database. J Bone Joint Surg Am. 2009;91(5):1063-1072.
43 McNaney D, Lindberg RD, Ayala AG, et al. Fifteen-year radiotherapy experience with chondrosarcoma of bone. Int J Radiat Oncol Biol Phys. 1982;8:187.
44 Harwood AR, Krajbich JI, Fornasier VL. Radiotherapy of chondrosarcoma of bone. Cancer. 1980;45:2769.
45 Krochak R, Harwood AR, Cummings BJ, et al. Results of radical radiation for chondrosarcoma of bone. Radiother Oncol. 1983;1:109.
46 Huvos AG, Rosen G, Dabska M, et al. Mesenchymal chondrosarcoma: a clinicopathologic analysis of 35 patients with emphasis on treatment. Cancer. 1983;51:1230.
47 Austin-Seymour M, Munzenrider J, Linggood R, et al. Fractionated proton radiation therapy of cranial and extracranial tumors. Am J Clin Oncol. 1990;13:327.
48 Noel G, Habrand JL, Mammar H, et al. Combination of photon and proton radiation therapy for chordomas and chondrosarcomas of the skull base: the Centre de Protontherapie D’Orsay Experience. Int J Radiat Oncol Biol Phys. 2001;51:392.
49 Munzingrider JE, Liebsch NJ. Proton therapy for tumors of the skull base. Strahlenther Onkol. 1999;175(suppl II):57.
50 Hug EB, Loredo LN, Slater JD, et al. Proton radiation therapy for chordomas and chondrosarcomas of the skull base. J Neurosurg. 1999;91:432.
51 Wagner TD, Kobayashi W, Dean S, et al. Combination short-course preoperative irradiation, surgical resection, and reduced-field high-dose postoperative irradiation in the treatment of tumors involving the bone. Int J Radiat Oncol Biol Phys. 2009;73(1):259-266.
52 DeLaney TF, Liebsch NJ, Pedlow FX, et al. Phase II study of high-dose photon/proton radiotherapy in the management of spine sarcomas. Int J Radiat Oncol Biol Phys. 2009;74(3):732-739.
53 Wang CC, James AEJr. Chordoma: Brief review of the literature and report of a case with widespread metastases. Cancer. 1968;22:162.
54 Fagundes MA, Hug EB, Liebsch NJ, et al. Radiation therapy for chordomas of the base of skull and cervical spine: patterns of failure and outcome after relapse. Int J Radiat Oncol Biol Phys. 1995;33:579.
55 Keisch ME, Garcia DM, Shibuya RB. Retrospective long-term follow-up analysis in 21 patients with chordomas of various sites treated at a single institution. J Neurosurg. 1991;75:374.
56 Maroske D, Hupe K. Sacrococcygeal chordoma: radical operation, a problem. Chirurg. 1977;48:118.
57 Miller TR, Mackenzie AR, Randal HT, et al. Hemicorporectomy. Surgery. 1966;59:988.
58 Watkins L, Khudados ES, Kaleoglu M, et al. Skull base chordomas: a review of 38 patients, 1958–1988. Br J Neurosurg. 1993;7:241.
59 Tatsuzaki H, Urie MM. Importance of precise positioning for proton beam therapy in the base of skull and cervical spine. Int J Radiat Oncol Biol Phys. 1991;21:757.
60 Niemierko A, Urie M, Gotein M. Optimization of 3D radiation therapy with both physical and biological end points and constraints. Int J Radiat Oncol Biol Phys. 1992;23:99.
61 Schoenthaler R, Castro JR, Petti PL, et al. Charged particle irradiation of sacral chordomas. Int J Radiat Oncol Biol Phys. 1993;26:291.
62 Noël G, Feuvret L, Calugaru V, et al. Chordomas of the base of the skull and upper cervical spine. One hundred patients irradiated by a 3D conformal technique combining photon and proton beams. Acta Oncol. 2005;44(7):700-708.
63 Rutz HP, Weber DC, Sugahara S, et al. Extracranial chordoma: outcome in patients treated with function-preserving surgery followed by spot-scanning proton beam irradiation. Int J Radiat Oncol Biol Phys. 2007;67(2):512-520.
64 Park L, Delaney TF, Liebsch NJ, et al. Sacral chordomas: impact of high-dose proton/photon-beam radiation therapy combined with or without surgery for primary versus recurrent tumor. Int J Radiat Oncol Biol Phys. 2006;65(5):1514-1521.
65 Martin JJ, Niranjan A, Kondziolka D, et al. Radiosurgery for chordomas and chondrosarcomas of the skull base. J Neurosurg. 2007;107(4):758-764.
66 Malone S, O’Sullivan B, Catton C, et al. Long-term follow-up of efficacy and safety of megavoltage radiotherapy in high-risk giant cell tumors of bone. Int J Radiat Oncol Biol Phys. 1995;33:689.
67 Seider MJ, Rich TA, Ayala AG, et al. Giant cell tumors of bone: treatment with radiation therapy. Radiology. 1986;161:537.
68 Feigenberg SJ, Marcus RBJr, Zlotecki RA, et al. Megavoltage radiotherapy for aneurysmal bone cysts. Int J Radiat Oncol Biol Phys. 2001;49(5):1243-1247.
69 Mendenhall WM, Zlotecki RA, Gibbs CP, et al. Aneurysmal bone cyst. Am J Clin Oncol. 2006;29(3):311-315. Review
70 Oliveira AM, Perez-Atayde AR, Inwards CY, et al. USP6 and CDH11 oncogenes identify the neoplastic cell in primary aneurysmal bone cysts and are absent in so-called secondary aneurysmal bone cysts. Am J Pathol. 2004;165(5):1773-1780.
71 Wu JS, Wong R, Johnston M, et al. Meta-analysis of dose-fractionation radiotherapy trials for the palliation of painful bone metastases. Int J Radiat Oncol Biol Phys. 2003;55:594.
72 Jemal A, Murray T, Samuels A, et al. Cancer statistics, 2002. CA Cancer J Clin. 2003;53:5.
73 Brand KG. Foreign body induced sarcomas. In: Becker FF, editor. Cancer. New York: Plenum Press; 1975:485.
74 Dijkstra MD, Balm AJ, Gregor RT, et al. Soft tissue sarcomas of the head and neck associated with surgical trauma. J Laryngol Otol. 1995;109:126.
75 Bailar JC. How dangerous is dioxin? [editorial; comment] [see comments]. N Engl J Med. 1991;324:260.
76 Kang H, Enzinger FM, Breslin P, et al. Soft tissue sarcoma and military service in Vietnam: a case control study. J Natl Cancer Inst. 1987;79:693.
77 Suruda AJ, Ward EM, Fingerhut MA. Identification of soft tissue sarcoma deaths in cohorts exposed to dioxin and to chlorinated naphthalenes. Epidemiology. 1993;4:14.
78 Olsson HL, Perfekt R, Alvergard T, et al. Reduced risk for soft tissue sarcoma after oral contraceptive use—results from population based studies. Proceedings of ASCO. 2001;20:363a.
79 Kim JH. Radiation induced soft tissue sarcoma and bone sarcoma. Radiology. 1978;129:501.
80 Taghian A, DeVathaire F, Terrier M, et al. Long-term risk of sarcoma following radiation treatment for breast cancer. Int J Radiat Oncol Biol Phys. 1991;21:361.
81 Yap J, Chuba PJ, Williams J, et al. Breast cancer and the incidence rates of sarcomas as subsequent malignant neoplasms: a US population-based analysis. Proc ASCO. 1999;18:549a.
82 Fletcher CD, Gustafson P, Rydholm A, et al. Clinicopathologic re-evaluation of 100 malignant fibrous histiocytomas: prognostic relevance of subclassification. J Clin Oncol. 2001;19:3035.
83 Li FP, Fraumeni JFJ. Soft-tissue sarcomas, breast cancer, and other neoplasms. Ann Intern Med. 1969;71:747.
84 Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233.
85 Enzinger EM, Weiss SW. General considerations. In: Enzinger EM, Weiss SW, editors. Soft tissue tumors. St Louis: Mosby; 1995:4.
86 Basu TN, Gutmann DH, Fletcher JA, et al. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients [see comments]. Nature. 1992;356:713.
87 Wunder JS, Czitron AA, Kandel R, et al. Analysis of alterations in the retinoblastoma gene and tumor grade in bone and soft tissue sarcomas. J Natl Cancer Inst. 1991;83:194.
88 Toguchida J, Yamaguchi T, Ritchie B, et al. Mutation spectrum of the p53 gene in bone and soft tissue sarcomas. Cancer Res. 1992;52:6194.
89 Porter PL, Gown AM, Kramp SG, et al. Widespread p53 overexpression in human malignant tumors. Am J Pathol. 1992;140:145.
90 Soini Y, Vahakangas K, Nuorva K, et al. p53 Immunohistochemistry in malignant fibrous histiocytomas and other mesenchymal tumors. J Pathol. 1992;168:29.
91 Leach FS, Tokino T, Meltzer P, et al. p53 Mutation and MDM2 amplification in human soft tissue sarcomas. Cancer Res. 1993;53:2231.
92 Smith SA, Weiss SW, Jankowski SA, et al. SAS amplification in soft tissue sarcomas. Cancer Res. 1992;52:3746.
93 Fisher C. Current aspects of the pathology of soft tissue sarcomas. Semin Radiat Oncol. 1999;9:315.
94 Nielsen TO, West RB, Linn SC, et al. Molecular characterisation of soft tissue tumours: a gene expression study. Lancet. 2002;359:1301.
95 Lawrence W, Donegan WL, Natarajan N, et al. Adult soft tissue sarcomas: pattern of care survey of the American College of Surgeons. Ann Surg. 1987;205:349.
96 Hashimoto H, Daimaru Y, Takeshita S, et al. Prognostic significance of histologic parameters of soft tissue sarcomas. Cancer. 1992;70:2816.
97 Potter DA, Kinsella T, Glatstein E, et al. High grade soft tissue sarcomas of the extremities. Cancer. 1986;58:190.
98 van Unnik JAM, Coindre JM, Contesso C, et al. Grading of soft tissue sarcomas: experience of the EORTC soft tissue and bone sarcoma group. Eur J Cancer. 1993;29A:2089.
99 Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol. 1997;15:350.
100 Coindre JM, Trojani M, Contesso G, et al. Reproducibility of a histopathologic grading system for adult soft tissue sarcoma. Cancer. 1986;58:306.
101 Rydholm A, Berg NO, Gullberg B, et al. Epidemiology of soft-tissue sarcoma in the locomotor system. Acta Path Microbiol Immunol Scand. 1984;92:363.
102 Potter DA, Glenn J, Kinsella T, et al. Patterns of recurrence in patients with high-grade soft-tissue sarcomas. J Clin Oncol. 1985;3:353.
103 Vezeridis MP, Moore R, Karakousis CP. Metastatic patterns in soft-tissue sarcomas. Arch Surg. 1983;118:915.
104 Cheng EY, Springfield DS, Mankin HJ. Frequent incidence of extrapulmonary sites of initial metastasis in patients with liposarcoma. Cancer. 1995;75:1120.
105 Weingrad DN, Rosenberg SA. Early lymphatic spread of osteogenic and soft-tissue sarcomas. Surgery. 1978;84:231.
106 Mazeron JJ, Suit HD. Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer. 1987;60:1800.
107 Massengill AD, Seeger LL, Eckardt JJ. The role of plain radiography, computed tomography, and magnetic resonance imaging in sarcoma evaluation. Hematol Oncol Clin North Am. 1995;9:571.
108 Fleming JB, Holtz D, Cantor SB, et al. The utility and cost-effectiveness of computerized tomography (CT) to screen for pulmonary metastases in patients presenting with T1 extremity soft tissue sarcoma (STS). Proc ASCO. 1998;17:516a.
109 Porter GA, Ahmad SA, Cantor SB, et al. Cost-effectiveness of routine chest computed tomography (CT) in patients with T2 soft tissue sarcoma. Proc ASCO. 2000;19:553a.
110 Kern KA, Brunetti A, Norton JA, et al. Metabolic imaging of human extremity musculoskeletal tumors by PET. J Nucl Med. 1988;29:181.
111 Dimitrakopoulou-Strauss A, Strauss LG, Heichel T, et al. The role of quantitative (18)F-FDG PET studies for the differentiation of malignant and benign bone lesions. J Nucl Med. 2002;43:510.
112 Conrad EU, Eary JF, Van Linge J. FDG PET and “clinical grade” for treatment planning in sarcoma. Proc ASCO. 1999;18:548a.
113 Schuetze S, Conrad E, Bruckner J, et al. FDG PET response to neoadjuvant chemotherapy predicts survival in patients with soft tissue sarcoma. Proc ASCO. 2001;20:348a.
114 Ayala AG, Ro JY, Fanning CV, et al. Core needle biopsy and fine-needle aspiration in the diagnosis of bone and soft-tissue lesions. Hematol Oncol Clin North Am. 1995;9:633.
115 Serpell JW, Fish SH, Fisher C, et al. The diagnosis of soft tissue tumors. Ann R Coll Surg Engl. 1992;74:277.
116 Kempson RL, Fletcher CD, Evans HL, et al. Tumor of the soft tissues. In: Rosai J, Sobin LH, editors. Atlas of tumor pathology, Vol. 30. Washington DC: Armed Forces Institute of Pathology; 2001:17.
117 Soft tissue sarcoma. In: Greene FI, Page DL, Fleming ID, et al, editors. AJCC cancer staging manual. Chicago: Springer, 2002.
118 Pisters PW, Leung DH, Woodruff J, et al. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol. 1996;14:1679.
119 Coindre JM, Terrier P, Bui NB, et al. Prognostic factors in adult patients with locally controlled soft tissue sarcoma. A study of 546 patients from the French Federation of Cancer Centers Sarcoma Group. J Clin Oncol. 1996;14:869.
120 Ruka W, Emrich LJ, Driscoll DL, et al. Clinical factors and treatment parameters affecting prognosis in adult high-grade soft tissue sarcomas: a retrospective review of 267 cases. Eur J Surg Oncol. 1989;15:411.
121 Tanabe KK, Pollock RE, Ellis LM, et al. Influence of surgical margins on outcome in patients with preoperatively irradiated extremity soft tissue sarcomas. Cancer. 1994;73:1652.
122 Suit HD, Spiro IJ. The role of radiation in patients with soft tissue sarcomas. Cancer Control. 1994:592.
123 Heslin MJ, Woodruff J, Brennan MF. Prognostic significance of a positive microscopic margin in high-risk extremity soft tissue sarcoma: implications for management. J Clin Oncol. 1996;14:473.
124 Rosenberg SA, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemo therapy. Ann Surg. 1982;196:305.
125 Harrison LB, Franzese F, Gaynor JJ, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in the management of completely resected soft tissue sarcomas of the extremity and superficial trunk. Int J Radiat Oncol Biol Phys. 1993;27:259.
126 Huth JF, Eilber FR. Patterns of metastatic spread following resection of extremity soft-tissue sarcomas and strategies for treatment. Semin Surg Oncol. 1988;4:20.
127 Williard WC, Hajdu SI, Casper ES, et al. Comparison of amputation with limb-sparing operations for adult soft tissue sarcomas of the extremity. Ann Surg. 1992;215:269.
128 Bramwell VHC, Rouesse J, Santoro A. Combined modality management of local and disseminated adult soft tissue sarcomas: a review of 257 cases seen over 10 years at the Christie Hospital and Holt Radium Institute, Manchester. Br J Surg. 1985;51:301.
129 Heslin MJ, Cordon-Cardo C, Lewis JJ, et al. Ki-67 detected by MIB-1 predicts distant metastasis and tumor mortality in primary, high grade extremity soft tissue sarcoma. Cancer. 1998;83:490.
130 Levine EA, Holzmayer T, Bacus S, et al. Evaluation of newer prognostic markers for adult soft tissue sarcomas. J Clin Oncol. 1997;15:3249.
131 Rudolph P, Kellner U, Chassevent A, et al. Prognostic relevance of a novel proliferation marker, Ki-S11, for soft-tissue sarcoma. A multivariate study. Am J Pathol. 1997;150:1997.
132 Wurl P, Meye A, Schmidt H, et al. High prognostic significance of Mdm2/p53 co-overexpression in soft tissue sarcomas of the extremities. Oncogene. 1998;16:1183.
133 Pallotta MG, Lastiri JM, Varela MS, et al. Plasma MMP-9 activity in soft tissue sarcomas. Proc ASCO. 2000;19:559a.
134 Kawai A, Woodruff J, Healey JH, et al. SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma. N Engl J Med. 1998;338:153.
135 Enneking WF, Spanier SS, Malawer MM. The effect of the anatomic setting on the results of surgical procedures for soft parts sarcoma of the thigh. Cancer. 1981;47:1005.
136 Brennhovd IO. The treatment of soft tissue sarcomas—a plea for a more urgent and aggressive approach. Acta Chir Scand. 1966;131:483.
137 Markhede G, Angervall L, Stener B. A multivariate analysis of the prognosis after surgical treatment of malignant soft tissue tumors. Cancer. 1982;49:1721.
138 Shieber W, Graham P. An experience with sarcomas of the soft tissues in adults. Surgery. 1962;52:295.
139 Simon MA, Enneking WF. The management of soft-tissue sarcomas of the extremities. J Bone Joint Surg. 1976;58:317.
140 Shiu MH, Hajdu SI. Management of soft tissue sarcoma of the extremity. Semin Oncol. 1981;8:172.
141 Cade SS. Soft tissue tumours: their natural history and treatment. Proc R Soc Med. 1951:19.
142 McNeer GP, Cantin J, Chu F, et al. Effectiveness of radiation therapy in the management of sarcoma of the soft somatic tissues. Cancer. 1968;22:391.
143 Tepper JE, Suit HD. Radiation therapy alone for sarcoma of soft tissue. Cancer. 1985;56:475.
144 Schwartz DL, Einck J, Bellon J, et al. Fast neutron radiotherapy for soft tissue and cartilaginous sarcomas at high risk for local recurrence. Int J Radiat Oncol Biol Phys. 2001;50:449.
145 Suit HD, Russell WO, Martin RG. Management of patients with sarcoma of soft tissue in an extremity. Cancer. 1973;31:1247.
146 Hintz BL, Charyulu KK, Miller WE, et al. Adjuvant role of radiation in soft tissue sarcoma in adults. J Surg Oncol. 1977;9:329.
147 Eilber FR, Mirra JJ, Grant TT, et al. Is amputation necessary for sarcomas? A seven-year experience with limb salvage. Ann Surg. 1980;192:431.
148 Lindberg RD, Martin RG, Romsdahl MM, et al. Conservative surgery and postoperative radiotherapy in 300 adults with soft-tissue sarcomas. Cancer. 1981;47:2391.
149 Suit HD, Proppe KH, Mankin HJ, et al. Preoperative radiation therapy for sarcoma of soft tissue. Cancer. 1981;47:2269.
150 National Institutes of Health. Limb-sparing treatment of adult soft-tissue sarcomas and osteosarcomas. Natl Inst Health Consens Dev Conf Consens Statement. 1985;5:18.
151 O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet. 2002;359:2235.
152 Davis AM, O’Sullivan B, Turcotte R, et al. Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol. 2005;75:48.
153 Yang JC, Chang AE, Baker AR, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol. 1998;16:197.
154 Choong PF, Petersen IA, Nascimento AG, et al. Is radiotherapy important for low-grade soft tissue sarcoma of the extremity? Clin Orthop. 2001:191.
155 Fabrizio PL, Stafford SL, Pritchard DJ. Extremity soft-tissue sarcomas selectively treated with surgery alone. Int J Radiat Oncol Biol Phys. 2000;48:227.
156 Karakousis CP, Emrich LJ, Rao U, et al. Limb salvage in soft tissue sarcomas with selective combination of modalities. Eur J Surg Oncol. 1991;17:71.
157 Geer RJ, Woodruff J, Casper ES, et al. Management of small soft-tissue sarcoma of the extremity in adults. Arch Surg. 1992;127:1285.
158 Lawrence TS, Lichter AS. Soft tissue sarcomas (excluding retroperitoneum. In: Perez CA, Brady LW, editors. Principles and practice of radiation oncology. Philadelphia: JB Lippincott; 1992:1399.
159 Lindberg RD. Soft tissue sarcoma. In: Fletcher GF, editor. Textbook of radiotherapy. Philadelphia: Lea & Febiger; 1980:922.
160 Suit HD, Spiro I. Role of radiation in the management of adult patients with sarcoma of soft tissue. Semin Surg Oncol. 1994;10:347.
161 Yang JC, Rosenberg SA, Glatstein EJ, et al. Sarcomas of soft tissues. In: DeVita VTJ, Hellman S, Rosenberg SA, editors. Cancer: principles and practice of oncology. Philadelphia: JB Lippincott; 1993:1436.
162 O’Sullivan B, Wylie J, Catton C, et al. The local management of soft tissue sarcoma. Semin Radiat Oncol. 1999;9:328.
163 Leibel SA, Tranbaugh RF, Wara WM, et al. Soft tissue sarcomas of the extremities: survival and patterns of failure with conservative surgery and postoperative irradiation compared to surgery alone. Cancer. 1982;50:1076.
164 Alektiar KM, Leung D, Zelcfsky MJ, et al. Adjuvant radiation for stage IIB soft tissue sarcoma of the extremity. J Clin Oncol. 2002;20:1642.
165 Hong LH, Alektiar KM, Hunt M, et al. Intensity modulated radiotherapy for soft tissue sarcoma of the thigh. Int J Radiat Oncol Biol Phys. 2004;59:752.
166 Nag S, Shasha D, Janjan N, et al. The American Brachytherapy Society recommendations for brachytherapy of soft tissue sarcomas. Int J Radiat Oncol Biol Phys. 2001;49:1033.
167 Hilaris BS, Bodner WR, Mastoras CA. Role of brachytherapy in adult soft tissue sarcomas. Semin Surg Oncol. 1997;13:196.
168 Oertel S, Treiber M, Zahlten-Hinguranage A, et al. Intraoperative electron boost radiation followed by moderate doses of external beam radiotherapy in limb-sparing treatment of patients with extremity soft-tissue sarcoma. Int J Radiat Oncol Biol Phys. 2006;64:1416.
169 Azinovic I, Martinez-Monge R, Javier-Artistu J, et al. Intraoperative radiotherapy electron boost followed by moderate doses of external beam radiotherapy in resected soft-tissue sarcoma of the extremity. Radiother Oncol. 2003;67:331.
170 Tsuji SY, O’Donnell RJ, Haas-Kogan D, et al. Intraoperative radiotherapy in the treatment of extremity and limb-girdle soft tissue sarcoma. Int J Radiat Oncol Biol Phys. 2007;69:S751.
171 Skibber JM, Lotze MT, Seipp CA, et al. Limb-sparing surgery for soft tissue sarcomas: wound related morbidity in patients undergoing wide local excision. Surgery. 1987;102:447.
172 Arbeit JM, Hilaris BS, Brennan MF. Wound complications in the multimodality treatment of extremity and superficial truncal sarcomas. J Clin Oncol. 1987;5:480.
173 Bujko K, Suit HD, Springfield DS, et al. Wound healing after surgery and preoperative radiation for sarcoma of soft tissues. Surg Gynecol Obstet. 1992;176:124.
174 Peat BG, Bell RS, Davis A, et al. Wound healing complications after soft tissue sarcoma surgery. Washington, D.C.: American Academy of Orthopedic Surgeons; 1992.
175 Ormsby MV, Hilaris BS, Nori D, et al. Wound complications of adjuvant radiation therapy in patients with soft-tissue sarcomas. Ann Surg. 1989;210:93.
176 O’Sullivan B, Davis A, Bell R, et al. Phase III randomized trial of pre-operative versus post-operative radiotherapy in the curative management of extremity sarcoma. A Canadian sarcoma group and NCI Canada clinical trials group study. Proc ASCO. 1999;18:535a.
177 Wayne JD, Langstein HN, Pollack A, et al. Preoperative radiotherapy for extremity soft tissue sarcoma (STS): site specific wound complication rates and the impact of reconstructive surgery. Proc ASCO. 2000;19:558a.
178 Meric F, Milas M, Hunt KK, et al. Impact of neoadjuvant chemotherapy on postoperative morbidity in soft tissue sarcomas. J Clin Oncol. 2000;18:3378.
179 Davis AM. Functional outcome in extremity soft tissue sarcoma. Semin Radiat Oncol. 1999;9:360.
180 Stinson SF, DeLaney TF, Greenberg J, et al. Acute and long-term effects on limb function of combined modality limb sparing therapy for extremity soft tissue sarcoma. Int J Radiat Oncol Biol Phys. 1991;21:1493.
181 Bell RS, O’Sullivan B, Davis A, et al. Functional outcome in patients treated with surgery and irradiation for soft tissue tumours. J Surg Oncol. 1991;48:224.
182 Robinson MH, Spruce L, Eeles R, et al. Limb function following conservation treatment of adult soft tissue sarcoma. Eur J Cancer. 1991;27:1567.
183 Van Kampen M, Eble MJ, Lehnert T, et al. Correlation of intraoperatively irradiated volume and fibrosis in patients with soft-tissue sarcoma of the extremities. Int J Radiat Oncol Biol Phys. 2001;51:94.
184 Gibbs CP, Peabody TD, Mundt AJ, et al. Oncological outcomes of operative treatment of subcutaneous soft-tissue sarcomas of the extremities. J Bone Joint Surg Am. 1997;79:888.
185 Nielsen OS, Cummings B, O’Sullivan B, et al. Preoperative and postoperative irradiation of soft tissue sarcomas: effect on radiation field size. Int J Radiat Oncol Biol Phys. 1991;21:1595.
186 Barkley HT, Martin RG, Romsdahl MM, et al. Treatment of soft tissue sarcomas by preoperative radiation and conservative surgical resection. Int J Radiat Oncol Biol Phys. 1988;14:693.
187 Kraybill WG, Spiro I, Harris J, et al. Radiation Therapy Oncology Group (RTOG) 95-14: a phase II study of neoadjuvant chemotherapy (CT) and radiation therapy (RT) in high risk (HR), high grade, soft tissue sarcomas (STS) of the extremities and body wall: a preliminary report. Proc ASCO. 2001;20:348a.
188 Edmonson JH, Petersen IA, Shives TC, et al. Chemotherapy, irradiation, and surgery for function-preserving therapy of primary extremity soft tissue sarcomas: initial treatment with ifosfamide, mitomycin, doxorubicin, and cisplatin plus granulocyte macrophage-colony-stimulating factor. Cancer. 2002;94:786.
189 Eilber F, Eckardt J, Rosen G, et al. Preoperative therapy for soft tissue sarcoma. Hematol Oncol Clin North Am. 1995;9:817.
190 Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol. 1996;14:859.
191 Schray MF, Gunderson LL, Sim FH, et al. Soft tissue sarcoma: integration of brachytherapy, resection, and external irradiation. Cancer. 1990;66:451.
192 Delannes M, Thomas L, Martel P, et al. Low-dose-rate intraoperative brachytherapy combined with external beam irradiation in the conservative treatment of soft tissue sarcoma. Int J Radiat Oncol Biol Phys. 2000;47:165.
193 Zelefsky MJ, Nori D, Shiu MH, et al. Limb salvage in soft tissue sarcomas involving neurovascular structures using combined surgical resection and brachytherapy. Int J Radiat Oncol Biol Phys. 1990;19:913.
194 Nori D, Shupak K, Shiu MH, et al. Role of brachytherapy in recurrent extremity sarcoma in patients treated with prior surgery and irradiation. Int J Radiat Oncol Biol Phys. 1991;20:1229.
195 Koizumi M, Inoue T, Yamazaki H, et al. Perioperative fractionated high-dose rate brachytherapy for malignant bone and soft tissue tumors. Int J Radiat Oncol Biol Phys. 1999;43:989.
196 Crownover RL, Marks KE. Adjuvant brachytherapy in the treatment of soft-tissue sarcomas. Hematol Oncol Clin North Am. 1999;13:595.
197 Chun M, Kang S, Kim BS, et al. High dose rate interstitial brachytherapy in soft tissue sarcoma: technical aspects and results. Jpn J Clin Oncol. 2001;31:279.
198 Blum RH. An overview of studies of Adriamycin (NSC-123127) in the United States. Cancer Chemother Rep. 1975;6:247.
199 O’Bryan RM, Luce JK, Talley RW, et al. Phase II evaluation of Adriamycin in human neoplasia. Cancer. 1973;32:1.
200 Borden EC, Amato D, Enterline HT, et al. Randomized comparison of Adriamycin regimens for treatment of metastatic soft tissue sarcomas. J Clin Oncol. 1987;5:840.
201 Taub RN, Keohan M, Plitsas M, et al. Phase II study of temozolomide in advanced sarcomas. Proc ASCO. 2000;19:555a.
202 Spath-Schwalbe E, Koschuth A, Grunewald R, et al. Gemcitabine (GEM) in pretreated patients with advanced soft tissue sarcomas—preliminary results from a phase II trial. Proc ASCO. 1998;17:513a.
203 Fidias P, Demetri G, Harmon DC. Navelbine shows activity in previously treated sarcoma patients: phase II results from MGH/ Dana Farber/Partner’s Cancercare study. Proc ASCO. 1998;17:513a.
204 Demetri GD, Seiden M, Garcia-Carbonero R, et al. Ecteinascidin (ET-743) shows promising activity in distinct populations of sarcoma patients: summary of 3 U.S.-based phase II trials. Proc ASCO. 2000;19:553a.
205 Wilson RE, Wood WC, Lerner HL, et al. Doxorubicin chemotherapy in the treatment of soft-tissue sarcoma. Arch Surg. 1986;121:1354.
206 Antman K, Suit H, Amato D, et al. Preliminary results of a randomized trial of adjuvant doxorubicin for sarcomas: lack of apparent difference between treatment groups. J Clin Oncol. 1984;2:601.
207 Alvegard TA, Sigurdsson H, Mouridsen H, et al. Adjuvant chemotherapy with doxorubicin in high-grade soft tissue sarcoma: a randomized trial of the Scandinavian Sarcoma Group. J Clin Oncol. 1989;7:1504.
208 Eilber FR, Giuliano AE, Huth JF, et al. A randomized prospective trial using postoperative adjuvant chemotherapy (Adriamycin) in high-grade extremity soft-tissue sarcoma. Am J Clin Oncol. 1988;11:39.
209 Picci P, Bacci G, Gherlinzoni F, et al. Results of a randomized trial for the treatment of localized soft tissue tumors (STS) of the extremities in adult patients. In: Ryan HR, Baker LO, editors. Recent concepts in sarcoma treatment. Dordrecht, The Netherlands: Kluwer Academic; 1988:144.
210 Gherlinzoni F, Picci P, Bacci G, et al. Late results of a randomized trial for the treatment of soft tissue sarcomas (STS) of the extremities in adult patients [abstract]. Proc Am Soc Clin Oncol. 1993;12:468.
211 Benjamin RS, Terjanian TO, Genoglio CJ, et al. The importance of combination chemotherapy for adjuvant treatment of high risk patients with soft tissue sarcomas of the extremities. In: Salmon SE, editor. Adjuvant therapy of cancer. New York: Grune & Stratton; 1987:735.
212 Bramwell V, Rouesse J, Steward W, et al. Adjuvant CYVADIC chemotherapy for adult soft tissue sarcoma—reduced local recurrence but no improvement in survival: a study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol. 1994;12:1137.
213 Chang AE, Kinsella T, Glatstein E, et al. Adjuvant chemotherapy for patients with high-grade soft-tissue sarcomas of the extremity. J Clin Oncol. 1988;6:1491.
214 Edmonson JH, Fleming TR, Ivins JC, et al. Randomized study of systemic chemotherapy following complete excision of nonosseous sarcomas. J Clin Oncol. 1984;2:1390.
215 Frustaci S, Gherlinzoni F, De Paoli A, et al. Adjuvant chemotherapy for adult soft tissue sarcomas of the extremities and girdles: results of the Italian randomized cooperative trial. J Clin Oncol. 2001;19:1238.
216 Ravaud A, Bui NB, Coindre JM, et al. Adjuvant chemotherapy with CYVADIC in high risk soft tissue sarcoma: a randomized prospective trial. In: Salmon SE, editor. Adjuvant therapy of cancer VI. Philadelphia: WB Saunders; 1990:556.
217 Frustaci S, De Paoli A, Bidoli E, et al. Ifosfamide in the adjuvant therapy of soft tissue sarcomas. Oncology. 2003;65:80.
218 , Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data, Sarcoma Meta-analysis Collaboration; Lancet; 350; 1997:1647.
219 Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer. 2008;113:573.
220 Woll PJ, van Glabbeke M, Hohenberger A, et al. Adjuvant chemotherapy (CT) with doxorubicin and ifosfamide in resected soft tissue sarcoma (STS): interim analysis of a randomised phase III trial. J Clin Oncol. 2007;25:10008.
221 Casson AG, Putnam JB, Natarajan G, et al. Five-year survival after pulmonary metastasectomy for adult soft tissue sarcoma. Cancer. 1992;69:662.
222 Jablons D, Steinberg SM, Roth J, et al. Metastasectomy for soft tissue sarcoma. J Thorac Cardiovasc Surg. 1989;97:695.
223 Frost DB. Pulmonary metastasectomy of soft tissue sarcomas: is it justified? J Surg Oncol. 1995;59:110.
224 Sampson MC, Katz A, Consine LS. Stereotactic body radiation therapy for extracranial oligometastases: does the sword have a double edge? Semin Radiat Oncol. 2006;16:67-76.
225 Cody HSI, Turnbull AD, Fortner JG, et al. The continuing challenge of retroperitoneal sarcomas. Cancer. 1981;47:2147.
226 Glenn J, Sindelar WF, Kinsella T, et al. Results of multimodality therapy of resectable soft-tissue sarcomas of the retroperitoneum. Surgery. 1985;97:316.
227 Kinsella TJ, Sindelar WF, Lack E, et al. Preliminary results of a randomized study of adjuvant radiation therapy in resectable adult retroperitoneal soft tissue sarcomas. J Clin Oncol. 1988;6:18.
228 Sindelar WF, Kinsella TJ, Chen PW, et al. Intraoperative radiotherapy in retroperitoneal sarcomas. Final results of a prospective, randomized, clinical trial. Arch Surg. 1993;128:402.
229 Alektiar KM, Hu K, Anderson L, et al. High-dose-rate intraoperative radiation therapy (HDR-IORT) for retroperitoneal sarcomas. Int J Radiat Oncol Biol Phys. 2000;47:157.
230 Gieschen HL, Spiro IJ, Suit HD, et al. Long-term results of intraoperative electron beam radiotherapy for primary and recurrent retroperitoneal soft tissue sarcoma. Int J Radiat Oncol Biol Phys. 2001;50:127.
231 Petersen IA, Haddock MG, Donohue JH, et al. Use of intraoperative electron beam radiotherapy in the management of retroperitoneal soft tissue sarcomas. Int J Radiat Oncol Biol Phys. 2002;52:469.
232 Hajdu SI. Pathology of soft tissue tumors. Philadelphia: Lea & Febiger; 1979. p 14
233 Kraus DH, Dubner S, Harrison LB, et al. Prognostic factors for recurrence and survival in head and neck soft tissue sarcomas. Cancer. 1994;74:697.
234 Tran LM, Mark R, Meier R, et al. Sarcomas of the head and neck. Cancer. 1992;70:169.
235 Willers H, Hug EB, Spiro IJ, et al. Adult soft tissue sarcomas of the head and neck treated by radiation and surgery or radiation alone: patterns of failure and prognostic factors. Int J Radiat Oncol Biol Phys. 1995;33:585.
236 Farhood AI, Hajdu SI, Shiu MH, et al. Soft tissue sarcomas of the head and neck in adults. Am J Surg. 1990;160:365.
237 Le Vay J, O’Sullivan B, Catton C, et al. An assessment of prognostic factors in soft-tissue sarcoma of the head and neck. Arch Otolaryngol Head Neck Surg. 1994;120:981.
238 Le QT, Fu KK, Kroll S, et al. Prognostic factors in adult soft-tissue sarcomas of the head and neck. Int J Radiat Oncol Biol Phys. 1997;37:975.
239 Kowalski LP, San CI. Prognostic factors in head and neck soft tissue sarcomas: analysis of 128 cases. J Surg Oncol. 1994;56:83.
240 Weber RS, Benjamin RS, Peters LJ, et al. Soft tissue sarcoma of the head and neck in adolescents and adults. Am J Surg. 1986;152:386.
241 Conley J, Healy WV, Stout AP. Fibromatosis of the head and neck. Am J Surg. 1966:112.
242 Eeles RA, Fisher C, A’Hern RP, et al. Head and neck sarcomas: prognostic factors and implications for treatment. Br J Cancer. 1993;68:201.
243 McKenna WG, Barnes MM, Kinsella TJ, et al. Combined modality treatment of adult soft tissue sarcomas of the head and neck. Int J Radiat Oncol Biol Phys. 1987;13:1127.
244 Das Gupta TK, Brasfield RD, O’Hara J. Extraabdominal desmoids: a clinicopathological study. Ann Surg. 1969;170:109.
245 Posner MC, Shiu MH, Newsome JL, et al. The desmoid tumor—not a benign disease. Arch Surg. 1989;124:191.
246 Leibel SA, Wara WM, Hill DR, et al. Desmoid tumors: local control and patterns of relapse following radiation therapy. Int J Radiat Oncol Biol Phys. 1983;9:1167.
247 Bataini JP, Belloir C. Desmoid tumors in adults: the role of radiotherapy in their management. Am J Surg. 1988;155:754.
248 Karakousis CP, Mayordomo J, Zografos GC, et al. Desmoid tumors of the trunk and extremity. Cancer. 1993;72:1637.
249 Jelinek JA, Stelzer KJ, Conrad E, et al. The efficacy of radiotherapy as postoperative treatment for desmoid tumors. Int J Radiat Oncol Biol Phys. 2001;50:121.
250 Nuyttens JJ, Rust PF, Thomas CRJr, et al. Surgery versus radiation therapy for patients with aggressive fibromatosis or desmoid tumors: a comparative review of 22 articles. Cancer. 2000;88:1517.
251 Russel WO, Cohen J, Enzinger F, et al. A clinical and pathological staging system for soft tissue sarcomas. Cancer. 1977;40:1562.
252 Myhre Jensen O, Høgh J, Østgaard SE, et al. Histopathological grading of soft tissue tumors. Prognostic significance in a prospective study of 278 consecutive cases. J Pathol. 1991;163:19.
253 Costa J, Wesley RA, Glatstein E, et al. The grading of soft tissue sarcomas. Results of a clinicohistopathologic correlation in a series of 163 cases. Cancer. 1984;53:530.
254 Trojani M, Contesso G, Coindre JM, et al. Soft-tissue sarcomas of adults: study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer. 1984;33:37.
255 Gerner RE, Moore GE, Pickren JW. Soft tissue sarcomas. Ann Surg. 1975;181:803.
256 Suit HD, Mankin HJ, Wood WC, et al. Treatment of the patient with stage M0 soft tissue sarcoma. J Clin Oncol. 1988;6:854.