Tumors and Tumorlike Conditions
Nonneoplastic Lesions
Overview: Benign lymphoid hyperplasia is believed to represent a benign condition of the bowel and is most frequently found in the distal ileum and in the colon. These lesions represent patches of lymphoid tissue and can be seen in both adults and children. The precise prevalence in children is unknown, but the entity has been found in up to 30% of symptomatic patients undergoing colonoscopy.1
Etiology: A number of possible causes have been postulated. The observation of benign lymphoid hyperplasia in families suggests that genetic or environmental factors could be pertinent.2 A recent study by Krauss et al.3 found that the prevalence of lymphoid hyperplasia at colonoscopy also is high in adults, and they postulated that it may relate to an enhanced immune response. In children, a number of theories have been proposed, including a local response to infection, immunodeficiency states, and local hypersensitivity reaction. Controversy also exists regarding the association of benign lymphoid hyperplasia and the autism spectrum disorder.4
Clinical Presentation: Benign lymphoid hyperplasia is usually discovered incidentally either on imaging or at endoscopy. As such, it is almost certainly underrecognized, and its true prevalence is unknown. Visual inspection at colonoscopy will demonstrate multiple raised, closely spaced areas along the bowel wall.
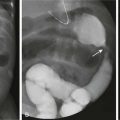
Imaging: The appearance on imaging studies is that of innumerable small filling defects, mostly uniform in size, at times umbilicated, and most commonly seen on double-contrast imaging of the colon (Fig. 109-1). These lesions are often too small to be detected on a single-contrast examination. One of the earliest imaging descriptions stressed their benign nature and the need to distinguish the lesion from true polyps,5 citing instances in which colectomies were performed because the benign nature of these lesions was not recognized. This pattern of innumerable small lesions (often in the range of 2 to 3 mm) also should be distinguished from benign lymphoid polyps, which are more common in adults and can become fairly large and pedunculated.6
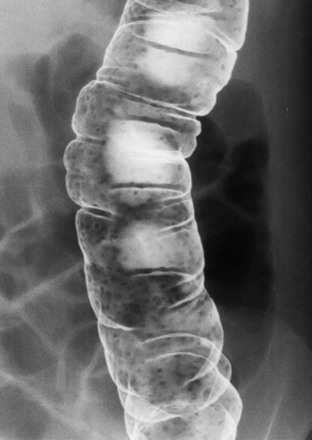
Figure 109-1 Benign lymphoid hyperplasia.
An 11-year-old girl had a double-contrast barium enema as part of the workup for rectal bleeding. A spot image of the descending colon from that examination demonstrates numerous filling defects that are uniform in size. The findings are consistent with benign lymphoid hyperplasia. Similar findings were seen throughout the colon.
Vascular Lesions
Overview: A number of vascular lesions and vascular tumors can arise within the colon. They are rare in children but can have specific clinical settings and imaging findings, which can allow for a specific diagnosis. Some vascular lesions will be found incidentally. Others can present as lower gastrointestinal (GI) tract bleeding.
Etiology: Vascular lesions of the colon represent a wide array of conditions. The etiology is known for some lesions and remains unknown for others. Colonic varices (particularly in the rectal region) can be seen as mural lesions and typically are seen with portal hypertension, providing a collateral pathway between the portomesenteric and the systemic venous systems. This collateral pathway has been reported to occur in nearly one third of children with portal hypertension. In that group of patients, significant rectal bleeding was seen in 7%.7
Other vascular lesions also can result in lower GI bleeding. Venous malformations, arteriovenous malformations, angiodysplasia, telangiectasias, and hemangiomas all have been described in the colon in children but are rare.8 Their etiology is not well understood.
Clinical Presentation: Because vascular lesions of the colon tend to come to light when they are symptomatic, the true incidence is unknown, but they are believed to be rare in children. The presentation can be that of lower GI bleeding or anemia. It should be noted that hemangiomas, which fall under the category of vascular tumors of the GI tract, can be associated with cutaneous hemangiomas in up to 50% of cases.
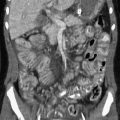
Imaging: The lesions are difficult to resolve with most imaging modalities. Multidetector computed tomography (CT) with imaging in the early arterial phase after contrast injection can demonstrate these lesions. Involved bowel will show intense enhancement (Fig. 109-2). Some patients will have abnormal supplying arteries or draining veins visible on CT. In a minority of cases with bleeding, active extravasation can be visualized.9
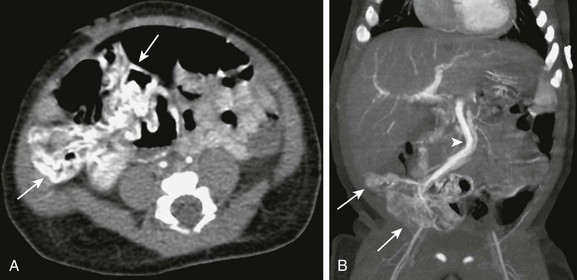
Figure 109-2 Colonic infantile hemangioma.
An infant presented at 4 weeks of age with gastrointestinal bleeding requiring multiple transfusions. An enhanced computed tomography scan of the abdomen and pelvis was done in the arterial phase. Axial (A) and coronal reconstruction (B) demonstrates intense enhancement of the wall of the distal ileum and cecum (arrows). Venous drainage is through a markedly dilated mesenteric vein (arrowhead in B). The affected segment was resected, and pathology confirmed that it was an infantile hemangioma.
Treatment: Much like vascular lesions elsewhere in the GI tract and indeed elsewhere in the body, the treatment of the lesion depends on the specific lesion, its location, the degree and extent of involvement, and the clinical status of the patient. These lesions can be treated medically or surgically, embolized via a catheter, or sclerosed endoscopically.
Although treatment very much depends on the type of lesion, some controversy exists with regard to the classification of intestinal vascular anomalies in children. Many persons advocate applying a classification system used for other vascular anomalies, such as the system proposed by Fishman and Mulliken,10 although other persons advocate use of a broader system.11
Neoplastic Lesions
Juvenile Polyps
Overview: Juvenile or hamartomatous polyps are the most common intestinal tumors in childhood, with a prevalence of 1% to 3%. They may be single or multiple, and most are found in the sigmoid colon and rectum, but they can arise anywhere along the GI tract.
Etiology: Histologically these lesions appear as mucus-filled glands, and they might be related to blocked, hyperplastic mucus glands. A dense infiltrate of inflammatory cells suggests an inflammatory inciting event, and thus the synonymous term “inflammatory polyp.” However, the underlying etiology of these isolated polyps remains to be determined.
Clinical Presentation: Although presentation is rare in the first year of life, most patients present in the first decade, typically between 2 and 5 years of age. The most common presenting symptom is painless bright red rectal bleeding. Pain is associated with the rare complication of colocolic intussusception. Some children present with iron deficiency anemia. Others can present with a prolapsing mass, which can be mistaken for rectal prolapse.12
Imaging: In the past, investigation for colonic neoplasms such as the juvenile polyp in a child with rectal bleeding often included a double-contrast enema. On such an examination, these lesions are typically smooth and can be sessile or pedunculated; most measure 3 cm or less. Currently the diagnosis is more frequently made endoscopically or on cross-sectional imaging, in which the lesions appear as nonspecific intraluminal masses (Fig. 109-3).
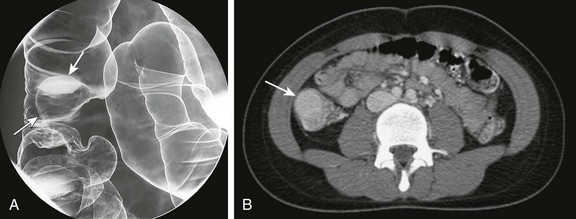
Figure 109-3 Juvenile polyp.
A 15-year-old girl presented with a 2-month history of abdominal pain. A, A double-contrast enema demonstrates a sessile mass consistent with a polyp arising from the wall of the ascending colon (arrows). B, A computed tomography scan in the same patient demonstrated an enhancing mass (arrow) within the lumen of the ascending colon.
Treatment: Because these solitary lesions carry no increased risk of malignancy, the treatment is polypectomy alone. Most commonly, a polypectomy is performed with snare resection during a colonoscopy. The anemia is treated with iron supplementation or transfusion if necessary. With resection of the polyp, the blood loss should cease. If multiple polyps are present or a family history of juvenile polyps is uncovered, then the patient should be evaluated for the juvenile polyposis syndrome (JPS), discussed later in this chapter.
Polyposis Syndromes
Although the inherited polyposis syndromes are rare, they have the potential to cause serious morbidity and mortality within affected families. A proper understanding of the various conditions is important for the primary clinician and consultant. Genetic screening and initiation of a surveillance plan is mandatory. Surveillance should include the GI tract as well as extraintestinal sites of potential disease.13
Syndromes Associated with Juvenile or Hamartomatous Polyps
Overview: First described in the literature in 1964,14 JPS is an autosomal-dominant condition that is characterized by a multiplicity of GI hamartomatous polyps. It is the most common of the hamartomatous syndromes.
Etiology: In approximately 75% of cases, a family history of the disease will be present. The condition also can occur sporadically as a result of new mutations. Mutations have been identified in the SMAD4 gene on chromosome 18 and the BMPR1A gene on chromosome 10.15
Clinical Presentation: From a clinical standpoint, the disease should be considered in any patient with five or more juvenile polyps in the colon, extracolonic juvenile polyps, or with any number of polyps when associated with a positive family history.16 The clinical presentation can be more variable than in the patient with nonsyndromic juvenile polyps. In addition to rectal bleeding, anemia, and intussusception, patients with JPS in whom there is involvement of a large segment of the GI tract can present with failure to thrive, malabsorption, or hypoalbuminemia. Associated congenital anomalies include hydrocephalus and hypertelorism. Small series have described JPS in association with other conditions, including hereditary hemorrhagic telangiectasia.17
Imaging: The imaging appearance of polyps in JPS differs in the number of polyps identified, but otherwise it is similar to that of isolated lesions.
Treatment: Patients with JPS have an increased risk of colonic malignancy that is reported to be as high as 50% on the basis of family studies.18 Screening of these children can be done with a combination of endoscopy, various imaging modalities, and capsule endoscopy. Polyps typically are removed via snare polypectomy.
Cowden Syndrome
Overview: Cowden syndrome is a rare autosomal-dominant syndrome with an estimated prevalence of 1 : 200,000 individuals.19 Features include hamartomatous polyps of the GI tract, hamartomatous lesions of the skin, hamartomas of other solid organs, and neoplasms of the breast, thyroid, and endometrium.19 GI tract polyps arising in these patients include inflammatory, hyperplastic, lipomatous, and even adenomatous lesions.
Etiology: Cowden syndrome is one of several disorders that fall under the category of the protein tyrosine phosphatase and tensin (PTEN) hamartoma tumor syndromes. Mutations in the tumor suppressor gene PTEN are present in up to 80% of patients with Cowden syndrome.19,20
Clinical Presentation: The presentation of patients with Cowden syndrome can be similar to the other conditions in which colonic polyps occur. Patients with Cowden syndrome also have an increased risk of neoplasms of the thyroid, endometrium, and breast. The lifetime risk of breast cancer in women with Cowden syndrome is between 25% and 50%. In these women, the onset of breast cancer is earlier than in sporadic cases. At present, the risk of GI malignancy in these patients is unknown.19
Imaging: The imaging appearance of polyps in Cowden syndrome is similar to that of polyps described earlier in the chapter.
Treatment: Polyps in patients with Cowden syndrome are treated with snare polypectomy; screening can include endoscopy, fluoroscopic and cross-sectional imaging modalities, and capsule endoscopy. Anemia is treated with iron supplementation and transfusion if necessary. Appropriate screening for the associated neoplasms also must be initiated. This screening consists of early breast self-examination and perhaps early initiation of mammography. Thyroid and uterine surveillance with screening ultrasound also is recommended.
Peutz-Jeghers Syndrome
Overview: Peutz-Jeghers syndrome is an autosomal-dominant syndrome with an incidence of approximately 1 : 120,000 people.21 It was described separately, in 1921 and 1949, by the authors after whom the syndrome was named. In this condition, hamartomatous polyps develop in the GI tract, in association with skin and mucosal hyperpigmented lesions.
Etiology: Peutz-Jeghers syndrome can be divided into two types. In the familial type, evidence indicates that the syndrome relates to mutations in the STK11 (serine threonine kinase) gene. In up to 50% of cases, no family history of the disease is present, which suggests a fairly high rate of new mutation.22
Clinical Presentation: The condition is characterized by pigmented lesions of the lips and buccal mucosa, as well as by hamartomatous polyps of the GI tract. The polyps can occur anywhere in the GI tract, although they are most commonly found in the small bowel, particularly in the jejunum. Polyps in the large and small bowel tend to be pedunculated. Those in the stomach tend to be more sessile (Fig. 109-4, A). Polyps also can arise in the gall bladder, bronchi, urinary bladder, and ureter.
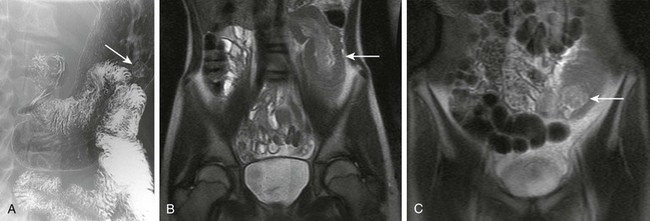
Figure 109-4 Peutz-Jeghers syndrome in a 14-year-old girl.
A, A double-contrast upper gastrointestinal series demonstrates a small polyp (arrow) arising in the proximal body of the stomach. B and C, Several years later, the patient presented with abdominal pain and was found to have a small bowel intussusception (arrow). B, A coronal magnetic resonance imaging single shot fast spin echo sequence demonstrates the intussusception along the left flank (arrow). C, What was believed to be a polyp functioning as a lead point (arrow) was seen lower in the pelvis on a more anterior image of the same sequence.
Imaging: The appearance of the polyps in Peutz-Jeghers syndrome does not differ significantly from that seen in other hamartomatous polyposes. It is not unusual for patients to present with intussusception. In that situation, the appearance is typical of an intussusception (see Chapter 108), with the polyp acting as a pathological lead point (Fig. 109-4, B and C).
Treatment: The treatment of the polyps is removal, either surgically or endoscopically, when necessary. Anemia is treated with iron supplementation and transfusion if necessary. Although it is not clear whether the polyps themselves have malignant potential, it is now accepted that there is an overall increased risk of neoplasms in patients with Peutz-Jeghers syndrome, typically in adulthood. Colorectal, breast, and ovarian tumors predominate.21
Polyposis Syndromes Associated with Adenomatous Polyps
Familial Adenomatous Polyposis and Variants
Overview: The prevalence of FAP is believed to be between 1:5000 and 1:17,000, depending on the series.23 Although the condition is defined by the presence of more than five adenomatous polyps, affected persons often will have hundreds, if not thousands, of polyps.
Extraintestinal lesions also are well described. In nearly 1 in 5 patients, a desmoid tumor will develop (e-Fig. 109-5), which will be discussed further later in this chapter. Congenital hypertrophy of the retinal pigment epithelial cells is seen in 60% to 90% of patients and is present at birth and can be assessed with fundoscopy. Lesions that can develop later in life include osteomas (often in the skull and mandible), lipomas, fibromas, and epidermoid cysts. An increased risk exists of developing hepatoblastoma (e-Fig. 109-6), thyroid and pancreatic cancers, cholangiocarcinoma, and central nervous system (CNS) tumors, particularly medulloblastoma.24 It is believed that with better management of the intestinal manifestations of FAP and subsequent longer life spans of affected patients, the incidence of these extraintestinal manifestations will likely increase.
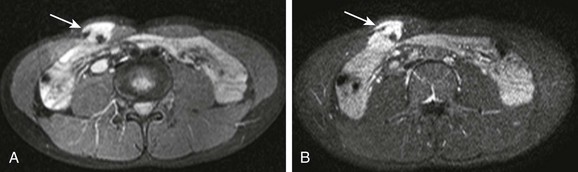
e-Figure 109-5 A desmoid tumor in a patient with familial adenomatous polyposis.
A 16-year-old boy with a history of familial adenomatous polyposis presented with a palpable mass of the abdominal wall. A, An axial T2-weighted spectral presaturation with inversion recovery image demonstrates a lesion with high signal (arrow) that has arisen in or infiltrated the right rectus abdominis muscle. B, A gadolinium-enhanced axial T1-weighted fat-saturated image demonstrates mostly avid enhancement. Some areas within the lesion on both sequences are low in signal and may represent areas of fibrosis. The lesion was resected, and a desmoid tumor was diagnosed at pathology.
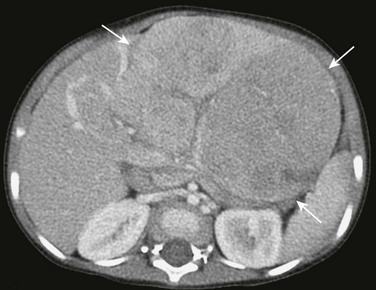
e-Figure 109-6 A hepatoblastoma in a patient with familial adenomatous polyposis (FAP).
A 22-month-old boy presented with an abdominal mass. The family history included FAP. An axial contrast-enhanced computed tomography image though the upper abdomen demonstrates a large mass (arrows) that arises from the left lobe of the liver. A biopsy was performed, and the findings were consistent with hepatoblastoma.
Gardner syndrome is believed to represent a variant of FAP, rather than a separate syndrome; some clinicians include these patients within the FAP group and agree that the term Gardner syndrome is now obsolete, although others continue to use the term in cases where the extraintestinal disease is particularly prominent. In Gardner syndrome, one sees the typical GI manifestations of FAP, including the plethora of polyps and the associated malignancies. Desmoid tumors often are cited as a classic extracolonic lesion in persons with Gardner syndrome; these tumors are a local aggressive form of fibromatosis and arise from the fascial tissue associated with muscle or from the mesentery. The tumors typically appear in the abdomen or in the abdominal wall.25 They may develop after trauma, such as after a prophylactic colectomy, and are a major cause of morbidity and mortality in these patients.25
Etiology: FAP is an autosomal-dominant condition in which a mutation is present in the adenomatous polyposis coli (APC) gene; approximately 30% of cases result from spontaneous mutations.26 This mutation results in a failure of apoptosis, and in uncontrolled cell growth, with the ultimate development of polyps—in this case, adenomatous ones.
Clinical Presentation: Polyps tend to develop in the second and late in the first decades of life, and often it is not until the third decade that symptoms arise. Therefore, unless screened, patients may not present with symptoms until adulthood. The most common presentation in childhood is rectal bleeding.23
Imaging: In classic FAP, the colon is carpeted with lesions (Fig. 109-7). The polyps tend to vary in size and lack the central umbilication often seen in benign lymphoid hyperplasia.
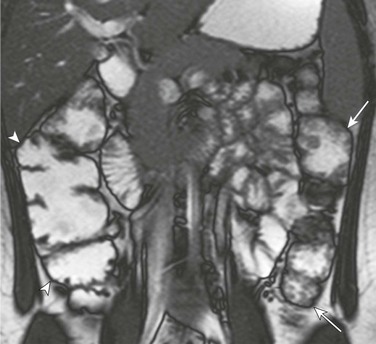
Figure 109-7 Familial adenomatous polyposis (FAP).
A 17-year-old girl with FAP. A coronal true fast imaging with steady state precession magnetic resonance series through the abdomen demonstrates multiple nodular lesions consistent with small polyps along the descending colon (arrows). The ascending colon (arrowheads) also was involved to a lesser degree.
A desmoid tumor is most often hypoechoic and at times nearly anechoic on ultrasound. On CT, the lesion often will be low attenuation, exhibit little enhancement in the portal venous phase, and may show increased enhancement in a delayed phase. Signal characteristics vary on MR. The lesion is usually of low signal intensity on T1-weighted sequences and of high signal on T2-weighted sequences (see e-Fig. 109-5).
Treatment: Colorectal carcinoma will develop in nearly all patients with FAP if it is left untreated. The accepted standard of care, therefore, is prophylactic colectomy. Polyps also can develop in other parts of the GI tract in patients with FAP. Periampullary polyps of the duodenum are well described; however, the development of duodenal carcinoma is rare overall, particularly in children.27 Nevertheless, surveillance of the upper GI tract is recommended after prophylactic colectomy. Any polyps that are found are removed endoscopically.
Turcot Syndrome
Overview: Turcot syndrome originally was described in 1959 in siblings found to have adenomatous polyps of the colon and malignancies of the CNS.28 It is now understood that the condition is one of adenomatous polyposis, with an increased risk of colorectal carcinoma and an increased risk of CNS tumors. Within the past decade, the syndrome has been reclassified to include two separate conditions with distinct genetic and molecular abnormalities and phenotype. One of these is termed the brain tumor-polyposis syndrome-1; the second group is termed brain tumor-polyposis syndrome-2. Myriad colonic polyps characteristically develop in patients, with carcinoma typically developing by 40 years of age. Medulloblastoma is the more common CNS tumor that develops in these patients.29
Etiology: Brain tumor-polyposis syndrome-1 is inherited as an autosomal-recessive condition and is the result of a defect in mismatch repair (MMR) genes, which correct deoxyribonucleic acid replication errors. This genetic abnormality is unusual in that, when heterozygous, the gene expresses a different phenotype, termed Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer syndrome. Brain tumor-polyposis syndrome-2 is inherited as an autosomal-dominant condition, with mutations in the APC (adenomatous polyposis coli) gene.29,30
Clinical Presentation: The clinical presentation is similar to that seen in FAP. Patients also can exhibit café au lait–type lesions and multiple lipomas. Patients with brain tumor-polyposis syndrome-1 are characterized by development of primarily astrocytomas and glioblastomas, as well as hematologic malignancies. Colorectal carcinomas arise at a mean age of 16 years; colon adenomas and carcinomas of the small bowel also develop in persons with this syndrome. Myriad colonic polyps characteristically develop in patients with brain tumor-polyposis syndrome-2, with carcinoma typically developing by 40 years of age; medulloblastoma is the more common CNS tumor that develops in these patients.29
Imaging: The imaging of colonic polyps has been described earlier. The appearance of the polyps in patients with Turcot syndrome does not differ from that seen in other polyposes. The CNS malignancies typically are investigated initially and followed up with MRI.
Treatment: The treatment in patients with Turcot syndrome depends on the individual clinical presentation and concerns. Polyps can be removed by snare polypectomy; however, given the risk of colorectal carcinoma, the patient may undergo a prophylactic colectomy. The CNS malignancies or other tumors are treated accordingly.
Other Colonic Neoplasms
Rarely, other benign neoplasms can arise in the pediatric colon; they are largely sporadic and are not associated with other conditions. Lipomas (e-Fig. 109-8), leiomyomas, and neurofibromas have all been described. Hemangiomas and vascular malformations were discussed earlier in this chapter.
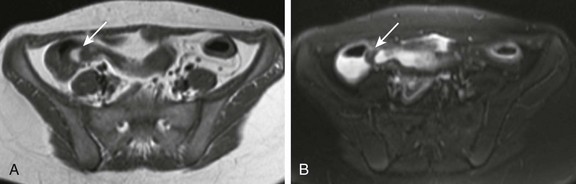
e-Figure 109-8 A lipoma at the ileocecal valve.
A 14-year-old girl presented with abdominal pain. T1-weighted (A) and fat saturated T2-weighted (B) magnetic resonance images through the pelvis demonstrate a small mass at the level of the ileocecal valve (arrows). The lesion is high in signal on the T1-weighted sequence and low in signal on the fat-saturated T2-weighted sequence.
Adenocarcinoma
Overview: Adenocarcinoma of the colon is exceedingly rare in children, with a prevalence of 1 in 1,000,000 persons younger than 19 years of age.31 Mucinous carcinoma is the most common subtype described in sporadic cases of pediatric colon cancer. The tumor is more often associated with underlying predisposing conditions, such as FAP and its variants, hereditary nonpolyposis colorectal cancer syndrome, Peutz-Jeghers syndrome, and JPS, as well as Crohn disease and ulcerative colitis.31
Etiology: The etiology of colorectal adenocarcinoma is not completely understood. What is known is that a number of risk factors relate to age, ethnicity, diet, other medical conditions, smoking, and alcohol consumption.
Clinical Presentation: Presenting symptoms include abdominal pain, an abdominal mass, “constipation,” weight loss, and GI bleeding. Whereas these symptoms might lead to immediate consideration of a GI malignancy in adults, that might not be an early consideration in a child. As a consequence, diagnosis may be delayed.32
Imaging: Radiographs may be normal or suggest a bowel obstruction. Rarely one might see a mass or mass effect. Calcifications can be seen in the mucinous forms, both in the primary tumor and in the metastatic lesions (Fig. 109-9). Contrast enema may show irregularity of the bowel wall, a mural lesion, or circumferential narrowing (Fig. 109-10). CT and MRI are the modalities used to evaluate for local and distant spread. The most common sites of metastases are lymph nodes, liver, lung, and adrenal glands.
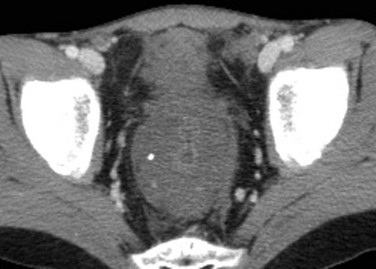
Figure 109-9 Rectal mucinous adenocarcinoma.
A 12-year-old boy presented with several weeks of rectal bleeding and an 11-lb weight loss. Computed tomography with intravenous contrast material shows marked thickening of the rectal wall with internal calcifications. At surgery, diffuse carcinomatosis with peritoneal implants was also found. (Figure courtesy Marta Hernanz-Schulman, MD, Nashville, TN.)
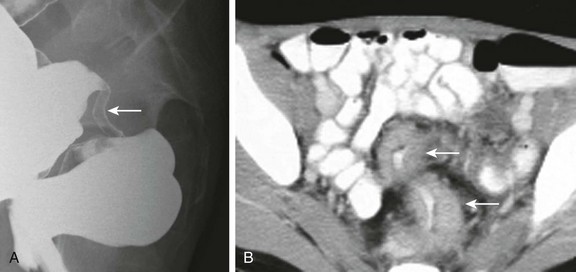
Figure 109-10 Rectosigmoid adenocarcinoma.
A 14-year-old boy presented with rectal bleeding. A, Lateral projection from a double-contrast barium enema demonstrates an area of circumferential narrowing (arrow) near the junction of the sigmoid colon and rectum. B, A contrast-enhanced computed tomography scan also demonstrates circumferential soft tissue narrowing the lumen (arrows). The lesion was resected and found to be an adenocarcinoma.
Carcinoid
Overview: Carcinoid tumors are neoplasms of epithelial origin, which are most commonly periappendiceal (Fig. 109-11). Attempts have been made to establish a staging system that would help predict prognosis.33 The most important prognostic criteria appear to be patient age, tumor size, histology, and the presence or absence of lymph node involvement or distant metastases. In larger series, approximately 40% of lesions are well differentiated. The remainder are almost equally split between moderately and poorly differentiated lesions.33
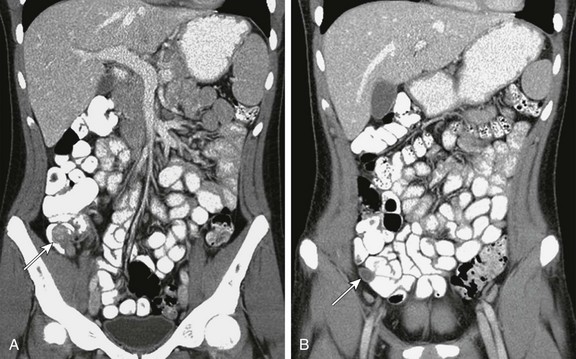
Figure 109-11 An appendiceal carcinoid tumor.
A 16-year-old boy presented with intermittent right lower quadrant pain. A, A contrast-enhanced computed tomography coronal reformat demonstrates a mass at the base of the appendix invaginating into the cecum (arrow). B, The obstructed appendix is distended to its tip with fluid (arrow).
Etiology: The inciting event for the development of carcinoid tumor is not yet clear. These tumors may be seen in patients who have other malignant neoplasms.
Clinical Presentation: When a carcinoid tumor involves the GI tract, it most commonly arises in the appendix. Patients can present with abdominal pain. Commonly the lesion is discovered only at appendectomy in a patient thought to have acute appendicitis. In some cases the lesion may act as a lead point for an intussusception. Patients with liver metastases can manifest the carcinoid syndrome, although it is extremely rare in the pediatric age group.
Imaging: The imaging findings depend on the size of the mass, its location, and the extent of disease. If it is sufficiently small, the lesion can present by occluding the appendiceal lumen, resulting in a distended/obstructed appendix (see Fig. 109-11). In such a case it may mimic acute appendicitis. Otherwise a nonspecific mass is seen with or without metastatic spread. As in cases of adenocarcinoma, CT and MRI can be used to assess for extent of disease. The tumor can metastasize to the liver, lungs, and bone.
Lymphoma
Overview: The colon can be a site of involvement in patients with lymphoma, although the small bowel is more commonly involved. Primary colorectal lymphoma accounts for less than 1% of all colorectal malignancies and occurs most often in the cecum.34 Disorders predisposing developing bowel lymphoma include ataxia-telangiectasia, Wiskott-Aldrich syndrome, agammaglobulinemia, severe combined immunodeficiency, and solid organ or bone marrow transplantation. It is becoming understood that patients with inflammatory bowel disease (IBD) also can have an increased risk of developing lymphoma. IBD per se may result in chronic antigenic stimulation,34 but data suggest that patients with IBD undergoing treatment with immunosuppressive and biologic agents are at greater risk of developing lymphoma.35 The risk of developing a neoplasm must be weighed against the risks associated with not adequately treating the primary disease.
Etiology: Lymphoma in the colon may arise as a result of conditions such as immunosuppression after transplantation, as noted above. Some investigators have postulated a genetic component, because in some cases a family history can be elicited. Others have postulated a role for infectious agents, particularly the Epstein-Barr virus in patients with underlying IBD.36
Clinical Presentation: The presenting symptoms in patients with GI lymphoma are often nonspecific and include abdominal pain and weight loss. Bleeding and a change in bowel habits occur less frequently. If the lesion is large enough, it can present as a palpable mass. In some cases the presentation is one of a colocolic intussusception (Fig. 109-12).37
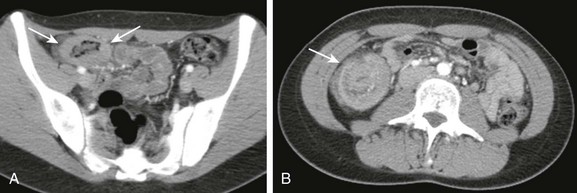
Imaging: If contrast enema is performed, the appearance can be one of irregularity of the bowel wall, a smooth or lobulated mural mass, or circumferential narrowing. A long segment of the bowel can be affected. On ultrasound the lesion is typically, although not always, hypoechoic, but is proved to be a solid mass by demonstration of flow on Doppler imaging. On CT or MRI an enhancing soft tissue mass is seen (e-Fig. 109-13), which can be associated with adenopathy and/or solid visceral involvement.
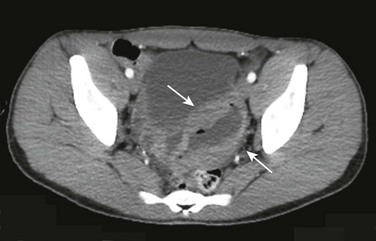
e-Figure 109-13 Burkitt lymphoma involving the sigmoid colon.
A 17-year-old boy presented with a palpable mass in the neck. As part of his neoplastic workup, a contrast-enhanced computed tomography scan of the abdomen was performed, which demonstrates a focal area of circumferential thickening in the sigmoid colon (arrows). Pathologic findings confirmed Burkitt lymphoma.
Alkhouri, N, Franciosi, JP, Mamula, P. Familial adenomatous polyposis in children and adolescents. J Pediatr Gastroenterol Nutr. 2010;51:727–732.
Barnard, J. Screening and surveillance recommendations for pediatric gastrointestinal polyposis syndromes. J Pediatr Gastroenterol Nutr. 2009;48(suppl 2):S75–S78.
Hill, DA, Furman, WL, Billups, CA, et al. Colorectal carcinoma in childhood and adolescence: a clinicopathologic review. J Clin Oncol. 2007;25:5808–5814.
Krauss, E, Konturek, P, Maiss, J, et al. Clinical significance of lymphoid hyperplasia of the lower gastrointestinal tract. Endoscopy. 2010;42:334–337.
Landry, CS, Woodall, C, Scoggins, CR, et al. Analysis of 900 appendiceal carcinoid tumors for a proposed predictive staging system. Arch Surg. 2008;143:664–670.
Pickhardt, PJ, Kim, DH, Menias, CO, et al. Evaluation of submucosal lesions of the large intestine: part 2. Nonneoplastic causes. Radiographics. 2007;27:1693–1703.
Shih, SL, Liu, YP, Tsai, YS, et al. Evaluation of arterial phase MDCT for the characterization of lower gastrointestinal bleeding in infants and children: preliminary results. AJR Am J Roentgenol. 2010;194:496–499.
Wong, MT, Eu, KW. Primary colorectal lymphomas. Colorectal Dis. 2006;8:586–591.
References
1. Iacono, G, et al. Colonic lymphoid nodular hyperplasia in children: relationship to food hypersensitivity. Clin Gastroenterol Hepatol. 2007;5(3):361–366.
2. Kleinman, R. Nodular lymphoid hyperplasia. In: Kleinman R, ed. Atlas of pediatric gastrointestinal disease. Hamilton, Canada: B.C. Becker; 1998:205–207.
3. Krauss, E, et al. Clinical significance of lymphoid hyperplasia of the lower gastrointestinal tract. Endoscopy. 2010;42(4):334–337.
4. Galiatsatos, P, Gologan, A, Lamoureux, E. Autistic enterocolitis: fact or fiction? Can J Gastroenterol. 2009;23(2):95–98.
5. Capitanio, MA, Kirkpatrick, JA. Lymphoid hyperplasia of the colon in children. Roentgen observations. Radiology. 1970;94(2):323–327.
6. Pickhardt, PJ, et al. Evaluation of submucosal lesions of the large intestine: part 2. Nonneoplastic causes. Radiographics. 2007;27(6):1693–1703.
7. Heaton, ND, Davenport, M, Howard, ER. Incidence of haemorrhoids and anorectal varices in children with portal hypertension. Br J Surg. 1993;80(5):616–618.
8. Turck, D, Michaud, L. Lower gastrointestinal Bleeding. In: Kleinman R, ed. Walker’s pediatric gastrointestinal disease. Hamilton, Canada: B.C. Decker; 2008:1309–1319.
9. Shih, SL, et al. Evaluation of arterial phase MDCT for the characterization of lower gastrointestinal bleeding in infants and children: preliminary results. AJR Am J Roentgenol. 2010;194(2):496–499.
10. Fishman, SJ, Mulliken, JB. Hemangiomas and vascular malformations of infancy and childhood. Pediatr Clin North Am. 1993;40(6):1177–1200.
11. Defreyne, L, et al. Colonic arteriovenous malformation in a child misinterpreted as an idiopathic colonic varicosis on angiography: remarks on current classification of childhood intestinal vascular malformations. Eur Radiol. 2003;13(suppl 4):L138–L141.
12. Durno, CA. Colonic polyps in children and adolescents. Can J Gastroenterol. 2007;21(4):233–239.
13. Barnard, J. Screening and surveillance recommendations for pediatric gastrointestinal polyposis syndromes. J Pediatr Gastroenterol Nutr. 2009;48(suppl 2):S75–S78.
14. McColl, I, et al. Juvenile polyposis coli. Proc R Soc Med. 1964;57:896–897.
15. Schreibman, IR, et al. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol. 2005;100(2):476–490.
16. Chow, E, Macrae, F. A review of juvenile polyposis syndrome. J Gastroenterol Hepatol. 2005;20(11):1634–1640.
17. Schwenter, F, et al. Juvenile polyposis syndrome, SMAD4 mutations, and hereditary hemorrhagic telangiectasia. J Pediatr Gastroenterol Nutr. 2012;54(1):120–122.
18. Howe, JR, Mitros, FA, Summers, RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol. 1998;5(8):751–756.
19. Gustafson, S, et al. Cowden syndrome. Semin Oncol. 2007;34(5):428–434.
20. Eng, C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22(3):183–198.
21. McGarrity, TJ, Kulin, HE, Zaino, RJ. Peutz-Jeghers syndrome. Am J Gastroenterol. 2000;95(3):596–604.
22. Beggs, AD, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59(7):975–986.
23. Alkhouri, N, Franciosi, JP, Mamula, P. Familial adenomatous polyposis in children and adolescents. J Pediatr Gastroenterol Nutr. 2010;51(6):727–732.
24. Groen, EJ, et al. Extra-intestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol. 2008;15(9):2439–2450.
25. Sturt, NJ, et al. Evidence for genetic predisposition to desmoid tumours in familial adenomatous polyposis independent of the germline APC mutation. Gut. 2004;53(12):1832–1836.
26. Erdman, SH, Barnard, JA. Gastrointestinal polyps and polyposis syndromes in children. Curr Opin Pediatr. 2002;14(5):576–582.
27. Groves, CJ, et al. Duodenal cancer in patients with familial adenomatous polyposis (FAP): results of a 10 year prospective study. Gut. 2002;50(5):636–641.
28. Turcot, J, Despres, JP, St Pierre, F. Malignant tumors of the central nervous system associated with familial polyposis of the colon: report of two cases. Dis Colon Rectum. 1959;2:465–468.
29. Villani, A, Malkin, D, Tabori, U. Syndromes predisposing to pediatric central nervous system tumors: lessons learned and new promises. Curr Neurol Neurosci Rep. 2012;12(2):153–164.
30. Hamilton, SR, et al. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995;332(13):839–847.
31. Donoghue, L, Klein, M. Tumors of the digestive tract. In: Kliegman R, ed. Nelson textbook of pediatrics. Philadelphia: Saunders; 2007:1641–1644.
32. Hill, DA, et al. Colorectal carcinoma in childhood and adolescence: a clinicopathologic review. J Clin Oncol. 2007;25(36):5808–5814.
33. Landry, CS, et al. Analysis of 900 appendiceal carcinoid tumors for a proposed predictive staging system. Arch Surg. 2008;143(7):664–670.
34. Wong, MT, Eu, KW. Primary colorectal lymphomas. Colorectal Dis. 2006;8(7):586–591.
35. Jones, JL, Loftus, EV, Jr. Lymphoma risk in inflammatory bowel disease: is it the disease or its treatment? Inflamm Bowel Dis. 2007;13(10):1299–1307.
36. Wong, NA, et al. Epstein-Barr virus infection in colorectal neoplasms associated with inflammatory bowel disease: detection of the virus in lymphomas but not in adenocarcinomas. J Pathol. 2003;201(2):312–318.
37. Gollub, MJ. Colonic intussusception: clinical and radiographic features. AJR Am J Roentgenol. 2011;196(5):W580–W585.